

Abstract
Opioids are powerful analgesics acting via the human μ‐opiate receptor (hMOR). Opioid use is associated with adverse effects such as tolerance, addiction, respiratory depression, and constipation. Two synthetic opioids, AH‐7921 and U‐47700 that were developed in the 1970s but never marketed, have recently appeared on the illegal drug market and in forensic toxicology reports. These agents were initially characterized for their analgesic activity in rodents; however, their pharmacology at hMOR has not been delineated. Thus, we synthesized over 50 chemical analogs based on core AH‐7921 and U‐47700 structures to assess for their ability to couple to Gαi signaling and induce hMOR internalization. For both the AH‐7921 and U‐47700 analogs, the 3,4‐dichlorobenzoyl substituents were the most potent with comparable EC50 values for inhibition of cAMP accumulation; 26.49 ± 11.2 nmol L−1 and 8.8 ± 4.9 nmol L−1, respectively. Despite similar potencies for Gαi coupling, these two compounds had strikingly different hMOR internalization efficacies: U‐47700 (10 μmol L−1) induced ~25% hMOR internalization similar to DAMGO while AH‐7921 (10 μmol L−1) induced ~5% hMOR internalization similar to morphine. In addition, the R, R enantiomer of U‐47700 is significantly more potent than the S, S enantiomer at hMOR. In conclusion, these data suggest that U‐47700 and AH‐7921 analogs have high analgesic potential in humans, but with divergent receptor internalization profiles, suggesting that they may exhibit differences in clinical utility or abuse potential.
Keywords: biased signaling, Gαi signaling, internalization, synthetic opioids, μ‐opioid receptor (OPRM1)
Abbreviations
- GPCRs
G protein‐coupled receptors
- GRK
G protein-coupled receptor kinase
- hMOR
human μ‐opiate receptor
- HRMS
high resolution mass spectrometry
- HPLC
high-performance liquid chromatography
- MOR
μ‐opiate receptor
- NLX
naloxone
- NMR
nuclear magnetic resonance
1. INTRODUCTION
The ability of opiates to suppress painful stimuli is undisputed. Opiates are a class of small molecules or peptides regardless of structure that can bind and activate opiate receptors. Morphine is an opioid extracted from the opium poppy that has been used as early as the third century BC to treat dysentery, pain, and suffering.1 It relieves pain by binding to and activating any of the three gene products encoding for cell surface G protein‐coupled receptors (GPCRs). These receptors are the μ‐, δ‐, or κ‐opiate receptors and they are encoded by the OPRM1, OPRD1, and OPRK1 genes, respectively.2, 3 Opioid analgesic properties stem from opioid receptor gene expression in sensory neurons of the brain and peripheral CNS and their coupling to intracellular heterotrimeric G proteins. Opiate binding induces a conformational change in opiate receptors and signals to rapidly suppress neuronal excitability by G protein‐dependent modulation of Ca2+, K+, and Na+ currents resulting in a profound reduced perception of pain. Of the three main opiate receptor subtypes, only compounds with relatively high selectivity for the μ‐opiate receptor (MOR) have achieved widespread clinical utility due, in part, to increased adverse effects such as dysphoria, convulsions, or poor selectivity of agents that have been developed to selectively target the δ‐ or κ‐ opiate receptors.1, 2
Agonist binding‐induced conformational changes of the MOR, in addition to activating inhibitory Gαi proteins, cause the phosphorylation of intracellular residues such as Ser375 by a number of kinases (eg G protein‐coupled receptor kinases [GRKs], PKC).4 The MOR phosphorylation sites and the efficacy of phosphorylation can differ based on the agonist structure. MOR phosphorylation leads to β‐arrestin recruitment, receptor desensitization, and internalization which are all regulatory processes central to the development of opiate tolerance.5, 6 While many synthetic and naturally occurring MOR agonists have high potency and efficacy for coupling to Gαi, they can diverge significantly in their ability to promote β‐arrestin recruitment and receptor internalization.7, 8, 9 For example, despite morphine's high potency and efficacy for Gαi coupling and widespread clinical use, it has very low efficacy for β‐arrestin recruitment and causes very little MOR internalization.7 Conversely, endogenously produced opioids, such as the enkephalins and β‐endorphins, are within a 10‐fold range of morphine in terms of Gαi coupling potency; however, they are far more efficacious than morphine for recruiting β‐arrestin and inducing receptor internalization.9
The concept that agonists could be designed to preferentially couple to Gαi vs β‐arrestin recruitment, referred to as “biased signaling,” was a driving force behind the development of new opiates such as oliceridine.10 This idea was bolstered by reports showing that β‐arrestin‐2 knockout mice display increased analgesia, decreased tolerance, and have less respiratory depression after morphine administration.11, 12, 13 Hence new opiates, such as oliceridine, were designed and selected for the ability to couple strongly to the Gαi pathway but with low efficacies for β‐arrestin recruitment and internalization, similar to morphine. However, recent events, such as the failure of FDA approval for oliceridine in 2018 and new studies using mouse models with targeted mutations in the OPRM1 gene that prevent MOR internalization, suggest that this may not be the best approach for developing safer opiates with fewer side effects. Mutations of carboxyl tail serine and threonine residues, including Ser375, that are phosphorylated by GRKs reveal that respiratory depression and constipation are significantly exacerbated when MOR receptors fail to internalize after agonist binding14 while analgesic tolerance is reduced. Thus, perhaps the pursuit of new opioids that better mimic the endogenous system might result in novel more “balanced” agents with improved clinical utility over morphine‐like derivatives.
In light of this, we sought to synthesize and pharmacologically characterize a series of structural analogs based on the ethylenediamine structural analogs AH‐7921 and U‐47700 and compare their pharmacology to morphine and the endogenous opioid mimetic DAMGO. These two synthetic opioids, first synthesized and patented in the early 1970s, have naloxone (NLX)‐reversible analgesic potential in rodent models15, 16 in the potency range of morphine. However, their pharmacological properties, including their ability to cause internalization of human μ‐opioid receptors (hMORs), are unknown. Thus, they represent a potentially useful series of core structures that are relatively easy to synthesize from where new “balanced” or “biased” opiates could be designed.
The design of analogs was to assess both compounds described in the patents, as well as related novel analogs, for their pharmacological selectivity and efficacy for causing hMOR internalization. Additionally, while the prior literature on the U‐series compounds indicated that stereoisomers differ in their selectivity for κ‐ vs μ‐opioid receptors, we sought to more clearly define the impacts of these differences on hMOR pharmacology by synthesizing and testing single stereoisomers of the U‐series compounds. We present herein findings related to structure activity relationships of these two compounds and over 50 structural analogous to provide new insights on how chemical structures affect potency for Gαi signaling and efficacy for hMOR internalization.
2. MATERIALS AND METHODS
2.1. Synthesis of compounds
The achiral amine precursor 1‐aminomethyl‐1‐cyclohexanedimethylamine for the AH‐series compounds, A01‐17, was prepared from cyclohexanone according to the original patent US4049663 Example 1b.17 (1R,2R)‐N,N,N′‐trimethyl‐1,2‐diaminocyclohexane, the precursor for the R,R enantiomer of the U‐series compounds, U01‐17, (1R,2R)‐N,N‐dimethyl‐1,2‐diaminocyclohexane, the precursor for the amide desmethyl U‐series compounds, Udes01‐09, and (1S,2S)‐N,N,N'‐trimethyl‐1,2‐diaminocyclohexane, the precursor for the S,S enantiomer of the U‐series compounds, US01‐09, were obtained from LabNetwork (San Diego, CA). The acid chlorides used were purchased from Fisher Scientific (analog code numbers 01‐04, 07‐17) or Sigma Aldrich (analog code numbers 05 and 06).
2.2. Acylation of starting amines
The respective acid chloride (1.05 eq) was added to a solution of the appropriate precursor amine (1.0 eq) and triethylamine (1.0 eq) in 5 mL of dry diethyl ether and stirred at room temperature for 16 hour. The reaction mixture was extracted with ethyl acetate (3×), washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude product was recrystallized from dichloromethane or precipitated with ethyl acetate upon sonication to give desired product as a solid. Reported hydrochloride salts of final compounds were made using 2.0 N HCl in diethyl ether solution.
2.3. Validation of synthesized compound purity and structure
Nuclear magnetic resonance (NMR) spectra were recorded on a Varian 400MR spectrometer (proton frequency 399.765 MHz) equipped with an AutoX DB broadband probe. Pulse sequences, acquisition, and data processing were accomplished using VnmrJ software (VnmrJ 4.2, Agilent Technologies, Santa Clara, CA). The spectrometer was locked on to D2O and spectra were acquired at 28°C without spinning. Water suppression suitability studies were carried out using the presaturation (presat), WET (WET1D), and excitation sculpting (water_ES) pulse sequences (VnmrJ 4.2, Agilent Technologies, Santa Clara, CA) with automatic suppression of the tallest peak (water at δ 4.86 ppm), an observation pulse of 90° (10.8 μs), a spectral width of 6410.3 Hz, a relaxation time of 30 second, and an acquisition time of 5.112 second. Eight scans were taken. Three replicates were taken for each sample.
High Resolution Mass Spectrometry spectra were collected on a Agilent Technologies 6530 Accurate‐Mass Q‐TOF LC/MS spectrometer using direct infusion into the nanoelectronspray source. Samples were dissolved in high‐performance liquid chromatography (HPLC)‐grade methanol to a final concentration of ∼0.01 mg mL−1. Spectra were run with 0.1% (v/v) formic acid/ HPLC‐grade methanol as solvent.
Purity determinations were performed by GCMS using a Shimadzu GC/MS 2010 SE with an Rtx‐5MS column (a DB‐5 MS equivalent); 30 m × 0.25 mm × 0.25 m. The carrier gas was helium at 1 mL min−1, with the injector at 280°C, MSD transfer line at 280°C, and ion source at 200°C. Injection Parameters: Split Ratio = 1:15, 1 μL injected. MS Parameters: Mass scan range: 34‐550 amu & Threshold: 100. Acquisition mode: scan. The oven programs were as follows: (a) 90°C initial temperature for 2.0 minutes; (b) Ramp to 300°C at 14°C min−1; (c) Hold final temperature for 10.0 minutes.
2.4. DNA constructs
Human µ‐opioid receptor, OPRM1, with three sequential hemagglutinin antigen (3xHA) tags at the N‐terminus (cDNA Resource Center OPRM10TN00) was subcloned into pENTR/D‐TOPO vector (Thermo Fisher K240020). The subcloned plasmid underwent a three‐way Gateway LR recombination along with pENTR‐EF1α vector containing EF1α promoter and the lentiviral 2k7bsd destination vector with blasticidin resistance coding sequence, as described previously.18 The resulting lentiviral plasmid vector is referred to herein as EF1α‐3xHA‐OPRM1‐2k7bsd.
2.5. Cell lines
Human fibrosarcoma HT1080 cells (ATCC CCL‐121) are maintained in DMEM high glucose, supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 1% L‐glutamine, 1% nonessential amino acid, and 1% sodium pyruvate. Wild type (WT) HT1080 cells were transduced with lentivirus generated with EF1α‐3xHA‐OPRM1‐2k7bsd lentiviral plasmid and transduced cells were selected for using DMEM supplemented media with 10 µg mL−1 blasticidin.
2.6. Western blot
5 × 105 OPRM1‐expressing HT1080 cells were lysed with 1xLDS buffer directly and ~25 μg of protein lysate is loaded into NuPAGE 4%‐12% Bis‐Tris Protein Gels (Thermo Fisher NP0335BOX) and transferred onto PVDF membrane. Membrane was blocked in 1X TBST with 5% w/v nonfat dry milk for 1 hour and probed with either rabbit anti‐HA antibody (Cell Signaling 3724S) or rabbit anti‐GAPDH (Cell Signaling 2118S) overnight at 4°C. The membrane was washed 3x with 1X TBST and probed with goat anti‐rabbit HRP antibody (Santa Cruz SC2004), then visualized using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher 34095) in Azure Biosystems c500 imaging system.
2.7. Immunocytochemistry
OPRM1‐expressing or WT HT1080 cells were seeded at 7.5 × 105 onto cover glass precoated with 0.1 mg mL−1 Poly‐D‐Lysine (Sigma P7280). Cells were first washed with Hank's Balanced Salt Solution (Fisher Scientific MT21022CV), and then fixed with 4% paraformaldehyde/PBS for 15 minutes followed by quenching with 0.1 mol L−1 glycine for 15 minutes. Afterward, the cells are washed 3x with 1X PBS and then blocked in 1%BSA/TBST for 1 hour. Rabbit monoclonal anti‐HA antibody (Cell Signaling 3724S) is used to stain the cells overnight, followed by goat anti‐rabbit Alexa Fluor 488 (Thermo Fisher A11070) and counterstained with Draq5 (Biolegend 424101) at 1:200 dilution. Slides were mounted and visualized using Leica DMI6000 confocal microscope using 63× oil‐immersion objective, with image processing using ImageJ software.
2.8. Catchpoint cAMP assay
OPRM1‐expressing or WT HT1080 cells were seeded at 2 × 104 in 96‐well plates in DMEM high glucose, supplemented with 5% fetal bovine serum, 1% penicillin/streptomycin, 1% L‐glutamine, 1% nonessential amino acid, and 1% sodium pyruvate. Culture media was removed and cells were washed with Krebs‐Ringer Bicarbonate Buffer (Sigma K4002), pH 7.4, then 1 mmol L−1 IBMX (Sigma I5879) was added to cells, and incubated at 37°C for 30 minutes. Compounds were added and incubated at 37°C for 15 minutes, followed by addition of 100 µmol L−1 forskolin (FSK) (Sigma F3917). Selected samples were treated with 10 μmol L−1 NLX (Sigma N7758) in addition to analogs that significantly altered FSK‐induced cAMP levels to validate that any observed changes in cAMP levels were mediated directly by hMOR. Cells were further incubated at 37°C for 15 minutes. A solution of 1X PBS was used for untreated cells in place of opioid compounds, FSK, and NLX as a vehicle control. All solutions were removed and the cells were lysed with Catchpoint Cyclic‐AMP Fluorescent Assay Kit Lysis Buffer (Molecular Devices R8088) and shaken on a plate shaker for 15 minutes at room temperature. Lysed cells are processed according to manufacturer's protocol in the Catchpoint Cyclic‐AMP Fluorescent Assay Kit and the cAMP levels were measured by SpectraMax Gemini EM Microplate Reader.
2.9. Quantification of agonist‐mediated receptor internalization
OPRM1‐expressing cells were seeded at 7.5 × 105 in 6‐well plates. Culture media was removed and drug compounds diluted in 10% FBS DMEM high glucose media (fully supplemented) were added to the cells. The drug compound media was removed following 60 minutes incubation at 37°C. Cells were washed with ice‐cold PBS (Ca2+ and Mg2+ free) and kept cold (4°C) for the remainder of the assay to prevent further receptor trafficking. Washed cells were detached from the plate by incubating with 2.9 mmol L−1 EDTA in PBS for 30 minutes. Detached cells were pelleted and washed with cold PBS before incubation with rabbit anti‐HA antibody (Cell Signaling 3724S) at 4°C. Cells are centrifuged down and washed with cold PBS before incubation with goat anti‐rabbit Alexa Fluor 488 antibody (Invitrogen A11070) at 4°C. Cells are then centrifuged and washed with cold PBS before analysis on BD Accuri C6 Flow Cytometer. Flow cytometry data were analyzed using FlowJo V10 software and gated for single, viable cells.
2.10. Data analysis and statistics
All data are presented as mean ± SEM. Due to the high number of compounds tested, for the initial screening of compounds, each compound was tested at three different concentrations (0.01, 0.1, and 1 μmol L−1) in triplicate using the Catchpoint cAMP assay on two independent experiments (n = 2). In addition, a morphine sulfate (MS) group was included as an internal positive control in each independent experiment (data not shown) in addition to a 1 μmol L−1 compound plus 10 μmol L−1 NLX group to validate that any changes in cAMP levels were mediated by hMOR. Cyclic AMP responses for each of the three single doses were compared to FSK alone by one‐way ANOVA with Dunnett multiple comparison (GraphPad Prism 7). An adjusted P < 0.05 being considered significant. All high potency agonists, that is, those which showed significant suppression of cAMP at the 0.01 µmol L−1 compared to FSK‐only (Dunnett's adjusted P < 0.05) dose, were then tested with expanded dose ranges (in triplicate) to compute the average EC50 value ± SEM from three independent expanded dose range experiments (n = 3). The EC50 values for each experimental replicate were computed by fitting dose response data for each experimental replicate to a variable‐slope sigmoidal curve (GraphPad Prism 7). Receptor internalization data were analyzed in GraphPad Prism 7 using unpaired t tests.
3. RESULTS
3.1. Chemical syntheses, stereochemical considerations, and structural analog design
AH‐7921 (Figure 1) is an achiral compound made from cyclohexanone via a Strecker reaction as described in the original patents and in (Figure 2A).17, 19, 20 The key intermediate of aminomethyl‐1‐cyclohexanedimethylamine was used to synthesize AH‐7921 (Compound A01) and derivatives. U‐47700 (Figure 1) has two chiral centers but is commercially available only as a racemic trans‐1,2‐diaminocyclohexane mixture. The goal of this study was to study the stereoselective pharmacology of single (R,R) or (S,S) isomers of U‐47700 and related analogs, which had not previously been determined. Thus, for the synthesis of (R,R)‐isomer U‐series (U01‐17, Figure 2B) and the Udes‐series (Udes01‐09, Figure 2B) compounds, the (1R,2R)‐N,N,N'‐trimethyl‐1,2‐diaminocyclohexane and (1R,2R)‐N,N‐dimethyl‐1,2‐diaminocyclohexane were used, respectively, as the starting material. The (S,S)‐isomer US‐series (US01‐09) was synthesized using (1S,2S)‐N,N,N'‐trimethyl‐1,2‐diaminocyclohexane as the starting material. (Analytical data of all compounds available in Supplementary information).
Figure 1.
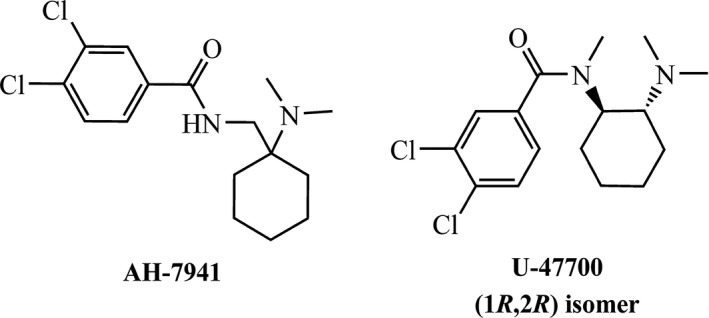
Structures of AH‐7921 and U‐47700
Figure 2.
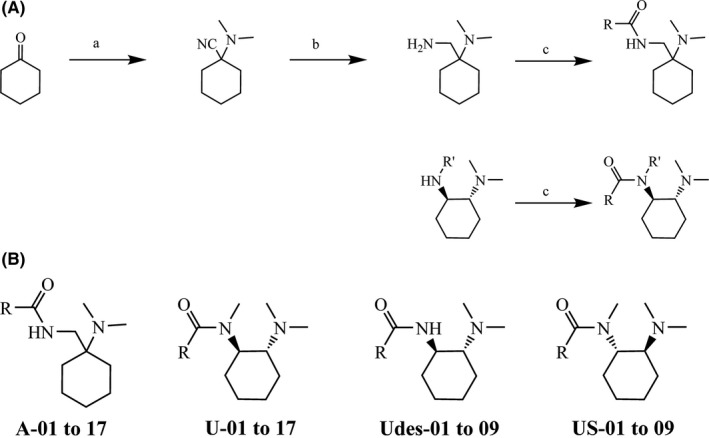
Core analog synthesis and structure overview (A) Synthetic routes to all analogs (R′ = Me for U‐ & US‐series, R′ = H for Udes‐series): (a) KCN, Me2NH.HCl, H2O, rt, 16 hour; (b) LiAlH4, Et2O, rt, 16 hour; (c) Et3N, RCOCl, Et2O, rt, 16 hour. US starts with the (1S,2S) diamine. (B) General structures of the analogs in the four series tested: A, U, Udes, and US
3.2. Validation of 3xHA‐tagged hMOR expression in HT1080 fibrosarcoma cells
A lentiviral construct (Figure S1) was used to generate stable transduced human 3x HA‐tagged OPRM1‐expressing HT1080 cells as described in Materials and Methods. Western blot was used to facilitate identification of antibodies suitable for immunohistochemistry and receptor internalization. The hMOR receptor was detected at the expected 65 kDa size in the hMOR but not in WT cells (Figure 3A). Confocal microscopy was also performed to further assess for hMOR expression at the subcellular level in the stable cell line. Human MOR expression was detected to be enriched at the cell membrane and in some endosomal compartments (Figure 3B).
Figure 3.
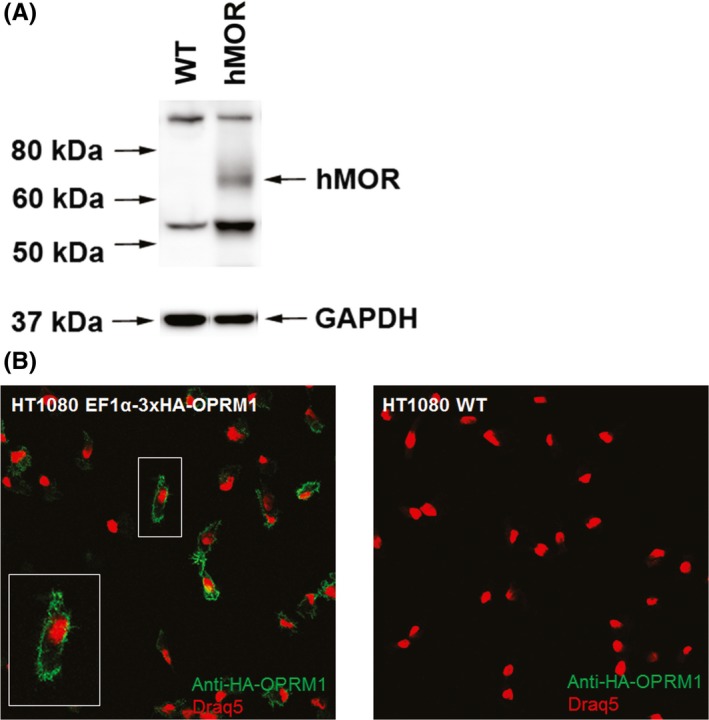
Validation of hMOR expression in HT1080 cells. (A) Western blot of HT1080 Wild Type (WT) and 3xHA‐OPRM1 lentivirus transduced cells as detected with an anti‐HA monoclonal antibody. (B) Confocal microscopy images detecting amino‐terminal 3x HA‐tagged hMOR using an anti‐HA primary antibody and Alexa488 conjugated secondary. Nucleus is counterstained with Draq5 and shown in red
3.3. Pharmacological validation of EF1α‐3xHA‐OPRM1‐expressing HT1080 cell (hMOR) line
Human MOR‐expressing and WT cells were assessed for opiate‐mediated inhibition of FSK‐stimulated adenylyl cyclase activity to assess for hMOR Gαi coupling in response to the prototypic opioid agonist MS to ensure that the 3x HA amino terminal tag did not interfere with receptor function. Treatment with MS (1 µmol L−1) resulted in a significant (P < 0.005) decrease in FSK‐induced cAMP accumulation in hMOR‐expressing cells but not in WT cells, validating surface expression and functional coupling of the epitope‐tagged hMOR in HT1080 cells and the lack of endogenous hMOR expression in WT HT1080 cells. Coadministration of MS with NLX completely inhibited the hMOR‐mediated Gαi response and restored cAMP to FSK‐only levels (Figure 4).
Figure 4.
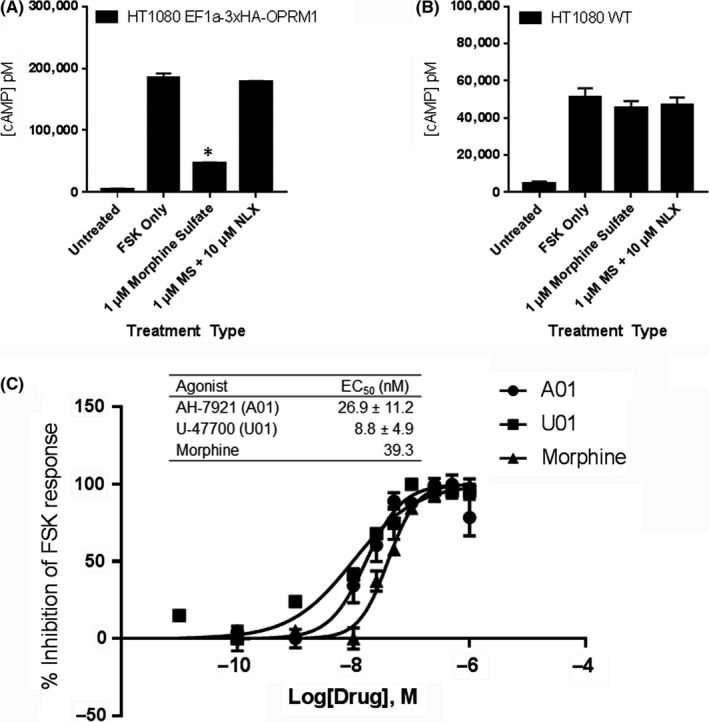
Pharmacological validation of HT1080 hMOR cells. (A) HT1080 hMOR cells are treated with 100 μmol L−1 of forskolin (FSK) only, FSK with 1 μmol L−1 morphine sulfate (MS), FSK with MS plus 10 μmol L−1 NLX, or PBS only (Untreated). (B) HT1080 WT is treated similarly. Data from representative experiment are shown. Statistical analysis is performed using one‐way ANOVA with Dunnett multiple comparison test with FSK only as standard. *P < 0.05. (C) Dose response curve and EC50 values table for morphine, AH‐7921 (A01), and U‐47700 (U01) in the HT1080 hMOR cell line. Each data point was performed in triplicate and repeated three times (n = 3). Data from representative experiment are shown
Expanded MS dose ranges were assessed to determine an EC50 value for MS (39.3 nmol L−1), which is comparable to previously published EC50 values of MS in heterologous OPRM1‐expressing cells.7, 8, 21 The EC50 values for AH‐7921 (A01) and U‐47700 (U01) found to be 26.9 ± 11.2 nmol L−1 and 8.8 ± 4.9 nmol L−1, respectively, (Figure 4C) in hMOR‐expressing cells.
3.4. Potency of AH‐7921 and structural analogs at hMOR
Seventeen analogs based on the AH‐7921 core structure were synthesized and assessed for hMOR pharmacological activity (Table S1). In addition to A01 (AH‐7921), two analogs were found to result in significant suppression of FSK‐induced cAMP accumulation that was reversed by NLX coadministration (Figure 5). A02 significantly decreased FSK‐induced cAMP levels at (0.1 µmol L−1) and showed further increasing dose‐dependent decrease of cAMP levels. Thus, of the AH‐series analogs we synthesized and tested, A01 (Figure 4C; EC50 26.9 ± 11.2 nmol L−1) and A02 (Figure S6; EC50 59.3 ± 2.0 nmol L−1) were classified as high potency hMOR agonists followed only by A04, which we classified as a low potency agonist (Figure 5). The remainder of the AH‐7921 compound series demonstrated either no activity or activity that was not reversible by NLX treatment (Table S1, Figure S2A).
Figure 5.
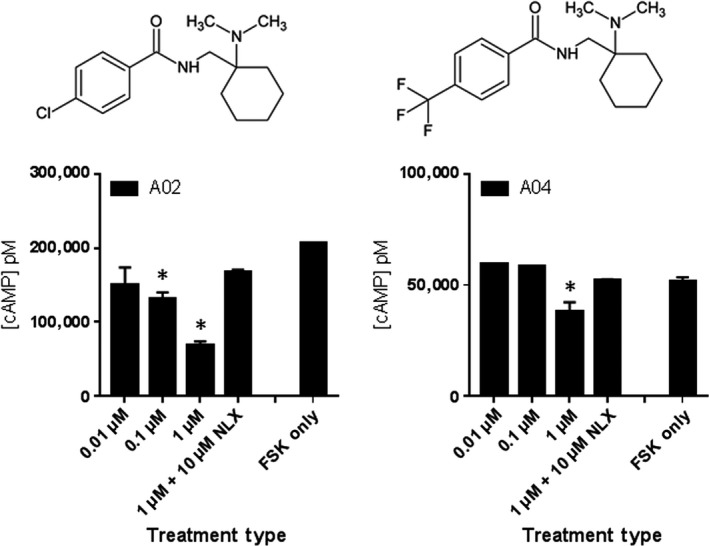
Drug potency of active AH‐7921 series compounds. HT1080 hMOR cells are treated with 100 μmol L−1 FSK only, or FSK with 0.01 μmol L−1 (low), 0.1 μmol L−1 (mid), 1 μmol L−1 (high) of the indicated compound, and high concentration dose of the compound with 10 μmol L−1 NLX. Each treatment dose was performed in triplicate (n = 2; data from representative experiment shown) and data were analyzed using one‐way ANOVA with Dunnett multiple comparison test and FSK only as standard. *P < 0.05
3.5. Potency of U‐47700 and structural analogs at hMOR
Sixteen different U‐47700‐related analogs with (R,R) stereochemistry were synthesized and assessed for hMOR activity (Table S2). Of these, U01 and U04 (Figure 6) were classified as hMOR selective high potency agonists with EC50 values 8.8 ± 4.9 nmol L−1 and 26.0 ± 11.1 nmol L−1, respectively (Figure 4C, Figure S6). Pharmacological profiling also revealed U02, U05, and U08 as low potency agonists. Analogous to findings with AH‐7921 analogs, most of the U‐47700 analogs were found to have no activity at hMOR at 1 μmol L−1 or lower concentrations. U06‐07 and U10‐17 were inactive based on comparison to FSK‐only control (Figure S3A). U03 and U09 showed significant decreases in cAMP levels in the range of concentrations tested, but their regulation of cAMP was not NLX reversible (Figure S3B). The summary of the U‐47700 series of opioid compound analogs is shown in Table S2.
Figure 6.
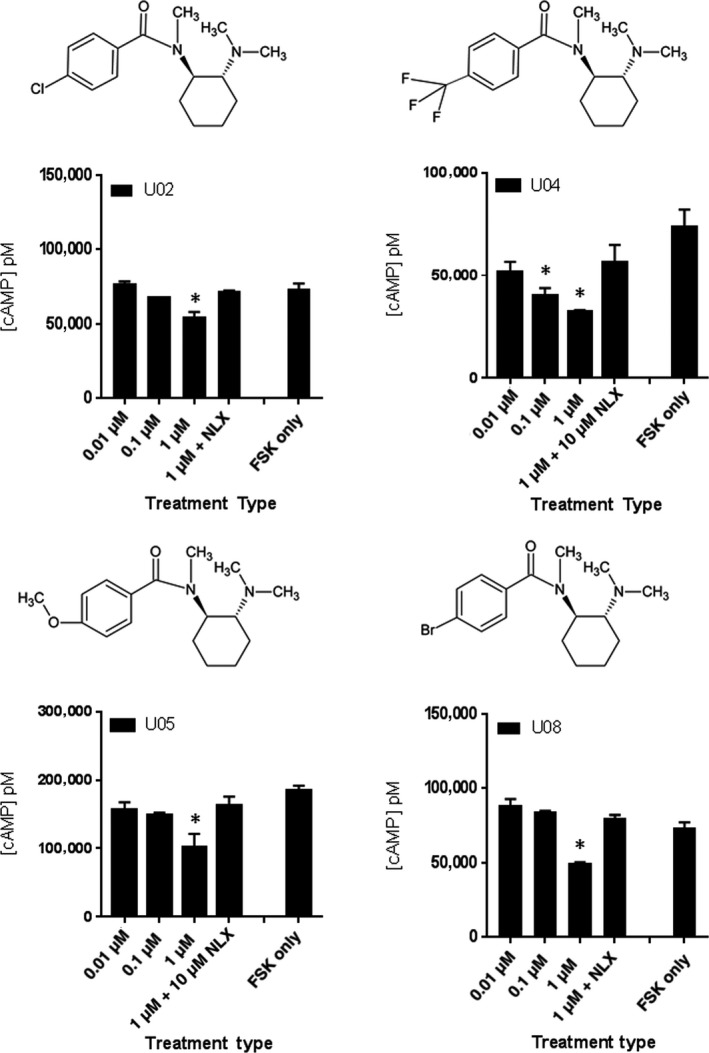
Drug potency of active U‐47700 series compounds. HT1080 hMOR cells are treated with 100 μmol L−1 FSK only, or FSK with 0.01 μmol L−1 (low), 0.1 μmol L−1 (mid), 1 μmol L−1 (high) of the indicated compound, and high concentration dose of the compound with 10 μmol L−1 NLX. Each treatment dose was performed in triplicate (n = 2; data from representative experiment shown) and data were analyzed using one‐way ANOVA with Dunnett multiple comparison test and FSK only as standard. *P < 0.05
3.6. Potency of Udes‐ and US‐series of analogs at hMOR
To study the effect of removing N‐methyl group on the amide in (R,R)‐U‐series, Udes‐series (compounds Udes01‐09) was synthesized and screened for hMOR signaling (Figure S4 and Table S3). Initial screening of Udes01 resulted in a significant decrease of FSK‐induced cAMP levels at 10 nmol L−1, and specificity for hMOR was confirmed by NLX reversibility (Figure 7). The EC50 value of Udes01 was subsequently determined to be 3.0 ± 0.3 nmol L−1 (Figure S6) characterizing it as the most potent analog that we discovered. The summary of the pharmacological activity of Udes‐series of compound analogs at the hMOR is shown in Table S3. The remaining Udes‐series (Udes02‐09) did not significantly alter FSK‐induced cAMP accumulation at any of the tested concentrations (Figure S4).
Figure 7.
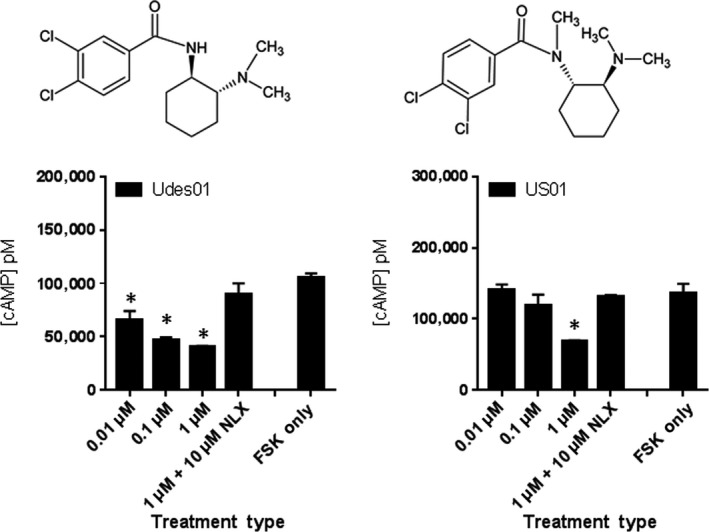
Drug potency of Udes01 and US01. HT1080 hMOR cells are treated with 100 μmol L−1 FSK only, or FSK with 0.01 μmol L−1 (low), 0.1 μmol L−1 (mid), 1 μmol L−1 (high) of the indicated compound, and high concentration dose of the compound with 10 μmol L−1 NLX. Each treatment dose was performed in triplicate (n = 2; data from representative experiment shown) and data were analyzed using one‐way ANOVA with Dunnett multiple comparison test and FSK only as standard. *P < 0.05
The US‐series was synthesized to assess for pharmacological activity of the S,S enantiomer of the U‐47700 analogs. The nine US‐series analogs were synthesized and screened for hMOR agonism (Table S4, Figure S5). Of these nine, only US01 (3,4‐dichlorobenzoyl substituent) demonstrated significant decrease in cAMP levels at the highest screening dose of 1 μmol L−1 but not at the lower (0.1 or 0.01 µmol L−1) concentration ranges (Figure 7, Figure S5) indicating that the S,S enantiomer of U‐47700 was significantly less potent than the R,R enantiomer.
3.7. Efficacy of hMOR internalization by high potency AH‐7921, U‐47700 analogs
Agonist‐induced receptor endocytosis remains a hallmark feature of GPCR activation and regulation and provides insights into the level of receptor desensitization that occurs after agonist stimulation.8 Thus, the AH‐7921, U‐47700, and Udes‐series compounds that demonstrated high potency were tested for their ability to induce hMOR internalization. The hMOR‐expressing cells were treated with saturating concentrations of 10 µmol L−1 [D‐Ala2,N‐MePhe4,Gly5‐ol] enkephalin (DAMGO) or 10 µmol L−1 morphine, which are reported to cause high levels or low levels of hMOR internalization, respectively.8, 21, 22 Similar results were found for DAMGO and morphine in our hMOR‐expressing cells (Figure 8). Interestingly, A01 and A02 (10 µmol L−1) resembled morphine treatment with only ~5% of cell‐surface receptors internalizing after 1 hour. In contrast, U01 (10 µmol L−1) resulted in 6‐fold more hMOR internalization than morphine and A01, to levels similar to DAMGO (10 µmol L−1) treatment (~30% of cell‐surface receptors). U04 and Udes01 demonstrated hMOR internalization similar to morphine and the two AH‐series compounds (Figure 8).
Figure 8.
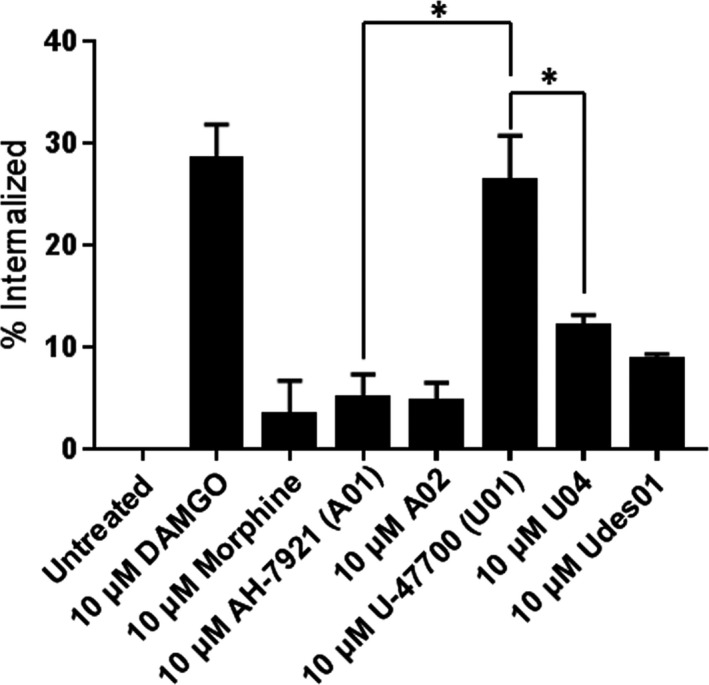
Measuring hMOR internalization levels after treatment with the high potency compounds. HT1080 hMOR cells were treated with the indicated concentration of drugs for 1 hour, then analyzed using flow cytometry to evaluate the changes in surface hMOR compared to untreated cells. The average internalization percentage is shown (n = 3). Data were analyzed using unpaired t test. *P < 0.05
4. DISCUSSION
We present a synthesis and screening strategy that enabled us to identify the pharmacological activity of over 50 ethylenediamine‐containing compounds at the hMOR. Of the high potency hMOR agonists that we found, the rank order of potencies for functional Gαi coupling and cAMP inhibition is as follows: Udes01 > U01 > U04 = A01 ≥ morphine sulfate > A02. We also classified A04, U02, U05, U08, and US01 as low potency hMOR agonists. Our findings are somewhat aligned with findings detailed in the original patents and early studies, however, with some key differences. In the literature, compounds A01 (AH‐7921), A03, and A13 demonstrated 100% inhibition in the mouse hot plate test when dosed at 100 mg kg−1 sc, but only A01 and A03 demonstrated activity greater than that of aspirin in a phenylquinone test when orally dosed at 100 mg kg−1.15 The determination of ED50 (mg kg−1 sc) gave the following rank order or potency: A01 2.5 ≥ morphine 2.8 > A02 5.0 > A15 9.5 > A13 15.5.23 They also demonstrated that AH‐7921 had a high addictive liability in rats and rhesus monkeys. In our system, A03, A13, and A15 failed to significantly suppress FSK‐induced cAMP accumulation based at 0.01, 0.1, or 1 μmol L−1 dosages in human OPRM1‐expressing cells (Figure S2) possibly reflecting differences in human and rodent OPRM1 receptor structures and underscoring the importance of validating SAR with human receptors.
In 1973, a group at Upjohn began looking at conformationally restrained analogs of phenampromide and by 1975 had developed the κ‐selective opioid analog, U‐50,488.24 Moving away from the N‐arylpropanamide structure, they developed the chemical series to which U‐47700 belongs. Rat tail flick (ED50 mg kg−1) and MOR‐related behavioral data (Straub tail, arched back, and increased locomotor activity) were reported.25, 26 A number of the compounds originally described are functionally characterized in this current study. The rank order of potency for analgesia (tail flick response time‐ED50 mg kg−1) is as follows: U01 (U‐47700) 0.2 > U04 0.4 > U02 1.0 > morphine sulfate 1.5 > Udes01 11.25, 26 Each of these analogs demonstrated MOR selectivity based on NLX reversibility. Our rank order potency data for cAMP inhibition are similar to these trends showing U01 to be the most potent U‐series analog followed by U04, then U02, U05, and U08; however, our data indicate that Udes01 was ~3X more potent than U01, which is much more potent than indicated in the aforementioned studies. This could be explained by differences in human vs rodent MOR protein‐coding sequences or if the Udes01 tested contained a racemic mixture, which is quite likely and could explain its much lower potency.
In light of this, our U‐series analog synthesis approach benefitted from the availability of the single stereoisomers of the advanced amine intermediates. This allowed for the synthesis of single stereoisomer analogs rather than the racemic versions of the analogs. Our findings, the (1R,2R) stereoisomer of U‐47700 (U01), were substantially more potent than the (1S,2S) stereoisomer (US01). The influence of stereochemistry in the U‐series compounds on μ‐ vs κ‐opioid receptor selectivity has been reported25, 27 indicating that the (1S,2S) isomer has high κ‐opioid receptor selectivity, while the (1R,2R) isomer has high μ‐opioid receptor selectivity.
Differentiating the effects of single isomers vs racemic U‐series compounds may help delineate in vivo effects in humans mediated by μ‐ and κ‐opioid receptors. One report of a seized sample of U‐49900, a diethyl amine version of U‐47700 which is a dimethyl amine, demonstrated that it was a racemic mixture, as determined by circular dichroism.28 A study reporting the murine μ‐opioid receptor affinity and activity of U‐47700 provided by Cayman Chemicals, reports a binding affinity of 57 nmol L−1 for U‐47700 at the mouse μ‐opioid receptor vs 5 nmol L−1 for MS and an EC50 of 214 ± 23 nmol L−1 in a [35S]‐GTPγS binding assay,29 which is less potent than we found for (1R,2R)‐U‐47700 (U01). To date, Cayman Chemicals has confirmed that they supply racemic trans isomers of U‐47700 and not the single (1R,2R) stereoisomer (personal communication with Cayman Chemical customer service 12/10/2018).
It has been proposed that hMOR endocytosis is closely associated with the development of drug tolerance in the opiate user8, 30 and in mouse models.14 Our data reveal that A01 and A02 demonstrated low levels (~15%) of hMOR internalization with similar drug potency for Gαi coupling. However, U01 and U04/Udes01 significantly (P < 0.05) diverged in their desensitization capacities: U01 induced high levels of internalization (~26%) while U04 and Udes01 induced between 8% and 12% of cell surface hMORs to internalize. Thus, the levels of hMOR internalization for A01, A02, U04, and Udes01 are similar to the reported internalization levels of well‐characterized opioids, such as morphine and fentanyl, while U01 is much more similar to the endogenous opioids, the endorphins and the enkephalins.7, 9, 31 Thus, if the goal was to design new opiates with safer and improved pharmacodynamic properties that mimic endogenously produced “balanced/natural” opiate responses at the hMOR, further assessment of the (1R,2R) single isomer U01 is likely to produce internalization and potentially receptor desensitization effects more aligned with endogenous opiates although further studies are needed.
In summary, our results demonstrate that this in vitro functional assay for hMOR pharmacology provides a foundation from which effects of these agents in humans can be anticipated and they provide a scaffold for the rational design of potentially superior analgesics that better mimic the endogenous opioid system.14, 32 Through our combinatorial approach, we discovered some novel moderately potent hMOR agonists. Whether these novel compounds have different tolerance profiles or addictive potential needs to be further investigated in vivo.
DISCLOSURE
None to declare.
AUTHOR CONTRIBUTIONS
ACZ, JLK, TH participated in research design; ACZ, JLK, TH, JRM, KY participated in writing and preparation of the manuscript, KY, VB, TL, TH, and JRM contributed to data acquisition.
Supporting information
ACKNOWLEDGEMENTS
This project was supported by Award No. [2016‐R2‐CX‐0059], awarded by the National Institute of Justice, Office of Justice Programs, US Department of Justice. The opinions, findings, and conclusions or recommendations expressed in this publication are those of the authors and do not necessarily reflect those of the Department of Justice.
Hsu T, Mallareddy JR, Yoshida K, et al. Synthesis and pharmacological characterization of ethylenediamine synthetic opioids in human μ‐opiate receptor 1 (OPRM1) expressing cells. Pharmacol Res Perspect. 2019;e00511 10.1002/prp2.511
Contributor Information
John L. Krstenansky, Email: john_krstenansky@kgi.edu.
Alexander C. Zambon, Email: azambon@kgi.edu.
REFERENCES
- 1. Goodman LS, Brunton LL, Chabner B, Knollmann BRC. Goodman & Gilman's Pharmacological Basis of Therapeutics, 12th edn New York: McGraw‐Hill; 2011. [Google Scholar]
- 2. Stein C. Opioid receptors. Annu Rev Med. 2016;67:433‐451. [DOI] [PubMed] [Google Scholar]
- 3. Minami M, Satoh M. Molecular biology of the opioid receptors: structures, functions and distributions. Neurosci Res. 1995;23:121‐145. [DOI] [PubMed] [Google Scholar]
- 4. Bailey CP, Smith FL, Kelly E, Dewey WL, Henderson G. How important is protein kinase C in mu‐opioid receptor desensitization and morphine tolerance? Trends Pharmacol Sci. 2006;27:558‐565. [DOI] [PubMed] [Google Scholar]
- 5. Schulz S, Mayer D, Pfeiffer M, Stumm R, Koch T, Hollt V. Morphine induces terminal micro‐opioid receptor desensitization by sustained phosphorylation of serine‐375. EMBO J. 2004;23:3282‐3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raehal KM, Schmid CL, Groer CE, Bohn LM. Functional selectivity at the mu‐opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol Rev. 2011;63:1001‐1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Keith DE, Anton B, Murray SR, et al. mu‐Opioid receptor internalization: opiate drugs have differential effects on a conserved endocytic mechanism in vitro and in the mammalian brain. Mol Pharmacol. 1998;53:377‐384. [PubMed] [Google Scholar]
- 8. Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737‐746. [DOI] [PubMed] [Google Scholar]
- 9. McPherson J, Rivero G, Baptist M, et al. μ‐opioid receptors: correlation of agonist efficacy for signalling with ability to activate internalization. Mol Pharmacol. 2010;78:756‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Siuda ER, Carr R, Rominger DH, Violin JD. Biased mu‐opioid receptor ligands: a promising new generation of pain therapeutics. Curr Opin Pharmacol. 2017;32:77‐84. [DOI] [PubMed] [Google Scholar]
- 11. Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu‐opioid receptor desensitization by beta‐arrestin‐2 determines morphine tolerance but not dependence. Nature. 2000;408:720‐723. [DOI] [PubMed] [Google Scholar]
- 12. Raehal KM, Walker JK, Bohn LM. Morphine side effects in beta‐arrestin 2 knockout mice. J Pharmacol Exp Ther. 2005;314:1195‐1201. [DOI] [PubMed] [Google Scholar]
- 13. Violin JD, Crombie AL, Soergel DG, Lark MW. Biased ligands at G‐protein‐coupled receptors: promise and progress. Trends Pharmacol Sci. 2014;35:308‐316. [DOI] [PubMed] [Google Scholar]
- 14. Kliewer A, Schmiedel F, Sianati S, et al. Phosphorylation‐deficient G‐protein‐biased mu‐opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat Commun. 2019;10:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harper NJ, Veitch GB, Wibberley DG. 1‐(3,4‐Dichlorobenzamidomethyl)cyclohexyldimethylamine and related compounds as potential analgesics. J Med Chem. 1974;17:1188‐1193. [DOI] [PubMed] [Google Scholar]
- 16. Hayes AG, Tyers MB. Determination of receptors that mediate opiate side effects in the mouse. Br J Pharmacol. 1983;79:731‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harper JN, Veitch G; Inventors ; Allen & Hanburys Limited, assignee . Ethylene Diamine Derivatives. 1977.
- 18. Suter DM, Cartier L, Bettiol E, et al. Rapid generation of stable transgenic embryonic stem cell lines using modular lentivectors. Stem Cells. 2006;24:615‐623. [DOI] [PubMed] [Google Scholar]
- 19. Szmuszkovicz J, Inventor ; The Upjohn Company, assignee . Analgesic N‐(2‐Aminocycloaliphatic)Benzamides. 1978.
- 20. Harper JN, Veitch G, Inventors ; Allen & Hanburys Limited, assignee . 1‐(3,4‐Dichlorobenzamidomethyl)Cyclohexyldimethylamine. 1976.
- 21. Ravindranathan A, Joslyn G, Robertson M, Schuckit MA, Whistler JL, White RL. Functional characterization of human variants of the mu‐opioid receptor gene. Proc Natl Acad Sci USA 2009;106:10811‐10816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tanowitz M, Hislop JN, von Zastrow M. Alternative splicing determines the post‐endocytic sorting fate of G‐protein‐coupled receptors. J Biol Chem. 2008;283:35614‐35621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brittain RT, Kellett DN, Neat ML, Stables R. Proceedings: Anti‐nociceptive effects in N‐substituted cyclohexylmethylbenzamides. Br J Pharmacol. 1973;49:158P‐159P. [PMC free article] [PubMed] [Google Scholar]
- 24. Szmuszkovicz J. U‐50,488 and the kappa receptor: a personalized account covering the period 1973 to 1990. Prog Drug Res. 1999;52:167‐195. [DOI] [PubMed] [Google Scholar]
- 25. Cheney BV, Szmuszkovicz J, Lahti RA, Zichi DA. Factors affecting binding of trans‐N‐[2‐(methylamino)cyclohexyl]benzamides at the primary morphine receptor. J Med Chem. 1985;28:1853‐1864. [DOI] [PubMed] [Google Scholar]
- 26. Szmuszkovicz J, Von Vigtlander PF. Benzeneacetamide amines: structurally novel non‐m mu opioids. J Med Chem. 1982;25:1125‐1126. [DOI] [PubMed] [Google Scholar]
- 27. Clark CR, Halfpenny PR, Hill RG, et al. Highly selective kappa opioid analgesics. Synthesis and structure‐activity relationships of novel N‐[(2‐aminocyclohexyl)aryl]acetamide and N‐[(2‐aminocyclohexyl)aryloxy]acetamide derivatives. J Med Chem. 1988;31:831‐836. [DOI] [PubMed] [Google Scholar]
- 28. Fabregat‐Safont D, Carbón X, Ventura M, et al. Updating the list of known opioids through identification and characterization of the new opioid derivative 3,4‐dichloro‐N‐(2‐(diethylamino)cyclohexyl)‐N‐methylbenzamide (U‐49900). Sci Rep. 2017;7:6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baumann MH, Majumdar S, Le Rouzic V, et al. Pharmacological characterization of novel synthetic opioids (NSO) found in the recreational drug marketplace. Neuropharmacology. 2018;134:101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. He L, Fong J, von Zastrow M, Whistler JL. Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell. 2002;108:271‐282. [DOI] [PubMed] [Google Scholar]
- 31. Koch T, Widera A, Bartzsch K, et al. Receptor endocytosis counteracts the development of opioid tolerance. Mol Pharmacol. 2005;67:280‐287. [DOI] [PubMed] [Google Scholar]
- 32. Johnson TA, Milan‐Lobo L, Che T, et al. Identification of the first marine‐derived opioid receptor "Balanced" agonist with a signaling profile that resembles the endorphins. ACS Chem Neurosci. 2017;8:473‐485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials