

Abstract
Persistently high plasma levels of serum amyloid A (SAA) may induce AA amyloidosis in various organs causing their dysfunction. Although SAA isoforms share a high degree of homology, only the SAA1.1 isoform is found in amyloid deposits. SAA1.1 misfolding is a nucleation-dependent process with dimer and trimer formation playing a major role in SAA fibril formation through self-catalyzed recruitment of native SAA molecules. Yet, a structural model of initial SAA oligomerization is still missing. In this study, we constructed a loosely associated model for murine SAA1.1 and SAA2.2 dimers in the presence or absence of hyaluronic acid as an exemplary glycosaminoglycan, a factor known to facilitate SAA fibril formation. Molecular dynamics simulations predicted that hyaluronic acid finally stabilized in a different binding pocket of the pathogenic SAA1.1 dimer compared to the nonpathogenic SAA2.2 dimer. Besides, Markov state modeling points to dynamic behavioral differences between the linker region of SAA1.1 and SAA2.2 and identifies a state unique to pathogenic SAA1.1 while bound to hyaluronic acid. The presence or absence of hyaluronic acid, as well as the dimer interface switch, affects dynamic behavior and possible oligomeric states, proposing a conceivable clue to the deviant pathogenicity of the two SAA isoforms.
Introduction
Serum amyloid A (SAA) is one of the most abundant acute phase proteins. It binds with high affinity to high-density lipoprotein (HDL) and modulates cholesterol homeostasis. Besides, it can regulate the immune response and facilitate bacterial clearance.1 In response to inflammatory stimuli caused by pathogen infection or tissue injury, SAA serum levels can rise up to 1 mg/mL, which is 1000 times higher than normal levels. Persistently high levels of SAA may cause accumulation of amyloid deposits consisting of highly ordered β-sheets of misfolded insoluble proteins in major organs. Such amyloid deposits might lead to organ dysfunction and life-threatening complications.1,2 Diseases characterized by persistently high plasma concentrations of SAA, such as rheumatoid arthritis, tuberculosis, ankylosing spondylitis, psoriatic arthritis, familial Mediterranean fever, and Crohn’s disease, are also commonly associated with the development of AA amyloidosis. Likewise, some tumors can be accompanied by AA amyloidosis.2
Mature human SAA is a 104-amino-acid protein synthesized by liver hepatocytes and released into the circulation in response to stimulation with proinflammatory cytokines.2 SAA protein found in amyloid deposits is mainly a C-terminus-truncated SAA1 variant, which contains a more hydrophobic N-terminal region compared to other SAA isoforms.1 After truncation, the remaining N-terminal portion of SAA is unstable and can easily convert from an α-helix to a β-sheet structure, which is characteristic for amyloid deposits indicating that the N-terminus represents the amyloidogenic portion of SAA.2 The structure of human SAA1 (SAA1.1) was only recently elucidated and shows a hexamer with two positively charged clusters formed by monomers folded into a unique antiparallel four-helix bundle with a well-organized C-terminal tail wrapped around one side of the helix bundle.3
Mature mouse SAA1.1 protein with 103 amino acids shares 76% sequence identity with the human SAA1 and is often used for in vivo studies.1,4,5 Similar to human SAA1, which has a tendency to form pathogenic amyloid fibrils, mouse SAA1.1 is pathogenic probably due to a proline-rich 13-amino-acid segment in the C-terminus, whereas mice expressing SAA2.2 are resistant to AA amyloidosis.1
In addition to HDL, SAA interacts also with carbohydrates known as glycosaminoglycans (GAGs). GAGs are unbranched polysaccharides of the extracellular matrix containing repeating motifs of hexosamine and hexuronic acid disaccharides. GAGs, mostly heparan sulfate, but also heparin and hyaluronic acid, are commonly found in AA amyloid deposits.2,6 SAA has specific binding sites for GAGs. Thus, residues 78–104 of the C-terminus of human SAA bind heparan sulfate.2 Animals overexpressing heparanase are resistant to develop AA amyloidosis, pointing to a causative role of GAGs in SAA aggregation.2 It has been suggested that due to highly anionic moieties, GAGs may serve as scaffolds for SAA proteins, facilitating their misfolding.7 Indeed, it has been shown that heparan sulfate displaces SAA from HDL particles2 and all GAGs enhance the SAA1.1 aggregation in vitro.7
Amyloid fibrils represent the terminal state of a complex self-assembly process of initial monomers. It is generally accepted that formation of amyloid fibrils is a nucleation-dependent event, and the aggregation of SAA begins with the formation of small oligomers, or the primary nucleus.2 In solution, human SAA1, murine SAA1.1, and SAA2.2 adopt diverse oligomeric states, such as monomeric, dimeric, trimeric, tetrameric (preferentially adopted by murine SAA1.1), and hexameric (preferentially adopted by murine SAA2.2 and human SAA1).8,9 Among the small oligomers, particularly, dimer levels correlate positively with the later formation of partially misfolded oligomers. Thus, in mice, increased levels of amyloidogenic SAA1.1 dimers in serum are closely linked to the development of AA amyloidosis.10 Furthermore, dimers or dimeric intermediates were proven to play a role in aggregation and fiber formation of other amyloidogenic proteins, such as amyloid β,11 transthyretin,12 and insulin.13 Taken together, the dimeric forms of SAA are likely to serve as a nucleus, leading to SAA fibril formation. However, available information about the SAA dimeric interface, especially the structural information, is still rather limited.
Based on the structural similarity between the human and murine SAA monomers and the available crystal structure of human SAA1, we built structural models of murine SAA1.1 and SAA2.2 dimers. Furthermore, we analyzed three phases of SAA oligomer formation: (i) possible formation of loosely associated SAA1.1 and SAA2.2 dimeric intermediates, (ii) transformation of SAA dimeric intermediates to stable dimers in solution, and (iii) reassembling of dimers into larger oligomers. In addition, Markov state modeling (MSM) was employed to analyze the potential effect of the interface on dynamic behavior of key residues and linker regions.
Methods
System Construction
The murine SAA1.1 and the SAA2.2 models were built by homology modeling. Coordinates of human SAA1 (PDB ID: 4IP8) were used for model construction. By expanding its asymmetric unit, a dimeric structure with the minimum interaction area was found and used as a template for murine SAA dimer construction. The interaction area was calculated in PyMol (Schrödinger 2015) by
where SASA is the solvent-accessible surface area.
The sequences of murine SAA1.1 and SAA2.2 were retrieved from the UniProt database with the UniProt IDs P05367 and P05366, respectively. The signaling peptide was removed, and the residues from G20-Y122 were used for homology modeling. The residue indexes of both sequences were renumbered to G1-Y103 for the following simulation. The sequence of murine SAA variants was aligned to the template by ClustalW214,15 and visualized by ESPript (http://espript.ibcp.fr),16 and the model was constructed by Modeller v9.12.17 Fifty models were generated, and each model was evaluated by the discrete optimized protein energy score, as well as the Ramachandra plot in Procheck.18 The top-scored structures were used for molecular docking and molecular dynamics (MD) simulations.
Model of the SAA–Hyaluronic Acid Complex Constructed by Molecular Docking
SAA1.1 and SAA2.2 were processed with Protein Preparation Wizard in Schrödinger 2014 and parameterized with all-atom optimized potentials for liquid simulation force field.19 A position-restrained energy minimization was performed by default setting. The three-dimensional (3D) structure of a hyaluronic acid fragment was built by the LEaP module in AmberTools (AMBER 2016, University of California, San Francisco) according to the two-dimensional (2D) structure from the PubChem database. Different conformations of hyaluronic acid were generated by the LigPrep module in Schrödinger 2014 suit, and the hyaluronic acid molecule was docked to the cleft between two SAA monomers by Induced Fit application in Schrödinger 2014 (Induced Fit Docking protocol, Glide and Prime software packages).20 The top-scored structures were used for the following MD simulation.
System Preparation and Molecular Dynamics Simulations
Protein and hyaluronic acid molecules were parameterized with Amber14SB21 and GLYCAM-06 force field,22 respectively. The protein or complex was dissolved within the TIP3P water box. The total atom number of SAA1.1 alone, the SAA1.1–hyaluronic acid complex, and the SAA2.2–hyaluronic acid complex was around 37 500, and the initial dimension of the system was around 71.8 Å × 71.8 Å × 90.2 Å.
SAA and SAA–hyaluronic acid systems were energy-minimized in three stages: first, all heavy atoms of the protein or the protein–hyaluronic acid complex were position-restrained for 500 steps, then the backbone atoms of the protein and heavy atoms of hyaluronic acid were position-restrained for 500 steps, and finally, the restraints were removed and the system was energy-minimized for another 500 steps. The energy-minimized system was gradually heated to 310 K in 100 ps with all heavy atoms of the protein or the protein–hyaluronic acid complex restrained. Finally, the restraints were gradually removed in a constant-pressure environment NPT for a total of 2.5 ns. The production NPT run was performed at 310 K and 1 atm for at least 1.2 μs for every system.
The simulation time step was set to 2 fs, and the van der Waals interactions were calculated at every step. The long-range electrostatic interactions were calculated every five steps by the particle mesh Ewald method23 with a grid size of about 1 Å and a tolerance of 10–6. The cutoff for the van der Waals interactions was set to 9 Å. The temperature and pressure were controlled using Langevin dynamics and the Langevin piston barostat method, respectively. The SHAKE method was used to restrain hydrogen atoms, and the tolerance was set to 10–8. Atomic coordinates of all atoms were saved every 1 ps. Energy minimization and MD simulations were performed with NAMD2.24
Trajectory Analysis
Gromacs,25 CARMA,26,27 and Plumed28 were used for all analyses. Amber topology was converted to Gromacs with Acpype.29 The root-mean-square deviations and root-mean-square fluctuations corresponding to the reference structure, the interaction area, the distance between two monomers, and the hydrogen bonds (as well as hydrogen-bond distribution) were calculated with Gromacs. The secondary structure of the protein was calculated by DSSP30 and visualized with do_dssp in Gromacs. Dihedrals and angle parameters were calculated by Plumed.
The free-energy landscape of the conformational change was calculated following principal component analysis of the whole trajectory. The first two eigenvectors were chosen to reconstruct the free-energy landscape,31,32 and for a given conformation (X), the free-energy was calculated by
where kB is the Boltzmann constant, T is the temperature, and P(X) is the probability of the above-mentioned conformation.
The analysis of the interaction between and within SAA monomers was conducted by PyEMMA.33 For interaction analysis between SAA monomers, the minimum distance between Cα atoms of each residue was calculated. The interaction was counted as 1 when the distance was shorter than 0.6 nm, otherwise 0. Markov state models (MSMs) were employed to analyze the trajectories of three different models according to settings recommended by PyEMMA. As an input, the nearest-neighbor heavy-atom contacts between the residues 4, 6, 7, 11, 16, 20, 21, 22, 28, 29, 30, 40, 46, 60, 63, 66, 67, 68, 70, 89, 92, 95, 101, and corresponding surrounding residues were calculated. The above residues were selected as they either differ between SAA1.1 and SAA2.2 or they are located within the linker region between helices. The interaction was counted as 1 when the distance was shorter than 0.4 nm, otherwise 0. The dimension of input coordinates was reduced by time-lagged independent component analysis (TICA) on two dimensions. To converge the maximum likelihood estimation, a time step was set to 2.0 ns (40 steps) (Figure S1A). Furthermore, the output of TICA was clustered into microstates using k-means clustering. The microstates were grouped into nine metastable states by Perron-Cluster Cluster Analysis (PCCA++). The number of metastable states was selected according to spectral analysis and the Chapman–Kolmogorov test (Figure S1B). The occupation of metastable states in each model was calculated by normalizing the frame numbers assigned to each metastable state with the total frame numbers.
Graphic Visualization
The 3D coordinates of snapshots were visualized with PyMOL, (Schrödinger 2015) and the electrostatic surface of SAA was calculated by APBS.34 Line plots and histograms were plotted with Grace (XMGRACE, version 5.1.19), whereas the free-energy landscapes were calculated and visualized by R package (https://www.R-project.org). Interaction between SAA monomers was visualized by Circos.35
Results and Discussion
Structural Dimeric SAA Model Adopts a Loosely Associated Conformation with a Potential Binding Site for Hyaluronic Acid
The resolved crystal structures of human SAA1 indicated a variety of possible interacting interfaces, therefore providing a useful tool to study formation of SAA dimers.3 Hence, 10 different SAA structural dimer models were constructed (Figure S2) and the area of interaction interfaces was evaluated. The human SAA1 dimer shown in Figure 1A generated by extending the asymmetric unit exhibits the lowest interaction area between the monomers (A = 0 Å2). It was chosen to mimic the intermediate states of the monomer-to-dimer transformation. In addition, the interface between the SAA monomers is linearly shaped and positively charged (Figure 1A, center panel), which enables the binding of a linearly shaped and negatively charged GAG molecule. Indeed, in vitro studies demonstrated that the GAGs heparin and heparan sulfate interact with the C-terminus of murine SAA1.1 and SAA2.2,36 which is in agreement with the SAA dimer model presented in Figure 1A. The major interaction force between the SAA monomers in solution is electrostatic force being long-range and, hence, contributing to a greater extent compared to other forces.37 Considering the differences in the amino acid sequences involved in the interactions between the SAA1.1 and SAA2.2 variants (Ile6Val, Gly7His, Gly27Asn, Asp30Asn, Gly31Ser, Ala60Gly, Ser63Ala, Met76Ile, and Ala101Asp), which are largely neutral, we assumed that SAA1.1 and SAA2.2 share similar loose binding interfaces.
Figure 1.

Generation of a loosely bound dimeric model of mouse SAA1.1 and SAA2.2 dimers based on the human SAA1 crystal structure. (A) Three-dimensional structure of human SAA1 (PDB ID: 4IP8) dimer. Left-hand panel: human SAA1 oligomer constructed by asymmetric unit manipulation (the dimeric SAA1 model is highlighted in green). Center panel: surface charge distribution. Right-hand panel: interacting residues. (B) Sequence alignment of human SAA1 and murine SAA1.1 and SAA2.2 reveals high homology with more than 70% sequence identity. (C) Overlay of homology models of two interacting monomers of murine SAA1.1 (pink) and murine SAA2.2 (cyan) shows structural similarities between the SAA1.1 and SAA2.2 variants. (D) Hyaluronic acid (green sticks) binds to the cleft between two murine SAA monomers.
Sequence alignment of human SAA1, murine SAA1.1, and murine SAA2.2 revealed over 70% sequence identity and 87% sequence similarity (Figure 1B). Although the crystal structures of murine SAA1.1 and SAA2.2 are not available, that of human SAA1 provided a good template for homology model construction of murine SAA1.1 and SAA2.2 (Figure 1C).
We have chosen hyaluronic acid, a linear high-molecular-weight glycosaminoglycan composed of disaccharide unit repeats [(1,3)-O-(2-acetamido-2-deoxy-β-d-glucopyranosyl)-(1,4)-O-β-d-glucopyranuronosyl]n, to evaluate the GAG effect on the formation of SAA1.1 and SAA2.2 oligomers. To enable molecular modeling, a hyaluronic acid dimer (tetrasaccharide) [(1,3)-O-(2-acetamido-2-deoxy-β-d-glucopyranosyl)-(1,4)-O-β-d-glucopyranuronosyl]2 was computed. As shown in Figure 1D, hyaluronic acid falls into the same binding pocket in SAA1.1 and SAA2.2 dimeric models, with SAA2.2 exhibiting a slightly higher binding affinity to hyaluronic acid. The predicted binding free energies of hyaluronic acid to SAA1.1 and SAA2.2 are −13.809 and −15.396 kcal/mol, respectively. According to the results from molecular docking, the binding structures are stabilized by electrostatic interactions between the surrounding Arg89, Asp90, Tyr103, and Arg95, as well as Arg46 located adjacent to the dimeric interface.
To further validate the model, two additional GAGs with different degrees of sulfation, heparin and chondroitin sulfate A, were docked into the proposed model and two local minima referring to two binding states, I and II, were identified by free-energy analysis and compared to that of nonsulfated GAG hyaluronic acid (Figure S3). Previous experimental data demonstrated that all three GAGs facilitate formation of thioflavin-T-positive SAA1.1 aggregates in vitro with heparin being the most efficient catalyst of SAA1.1 aggregation and chondroitin sulfate A the least potent one.7 Interestingly, different from heparin and hyaluronic acid, chondroitin sulfate A promoted formation of spherical particles and short protofibrils.7 Our model shows a slightly different binding pose for chondroitin sulfate A in state I and decreased binding affinity to the partially unfolded N-terminus in state II compared to heparin and hyaluronic acid (Figure S3), which might provide a feasible explanation for different effects the GAGs exert on SAA1.1 fibril morphology.
Dimeric Interface Formation and Alterations in Secondary Structures of SAA Dimers
SAA1.1, the SAA1.1–hyaluronic acid complex, and the SAA2.2–hyaluronic acid complex, all form dimeric structures during the simulation (Figure 2). The models might explain recent in vitro data that have demonstrated the existence of lipid-free SAA1.1 dimer in solution.38 However, the interaction interfaces in all structures differed. Thus, in SAA1.1 in the hyaluronic acid-free form, a symmetrical binding interface was detected around H3 and H4 helices (Figure 2A, left-hand panel). By contrast, in the SAA1.1 hyaluronic acid-bound form, an asymmetric interface was detected around H1 of one monomer and H4 of another monomer (Figure 2A, center panel, and Figure S4). The interface of two monomers in the SAA2.2–hyaluronic acid complex is located around the C-terminus of one monomer and the N-terminus of another monomer (Figure 2A, right-hand panel). At variance to SAA1.1, the interaction between two monomers in SAA2.2–hyaluronic acid is partially mediated by hyaluronic acid.
Figure 2.

Interaction between SAA1.1 monomers forming a dimer leads to partial unfolding and kink formation in the N-terminus and in parts of the H3 and H4 helices. (A) Scheme of interactions between two monomers for SAA1.1 in the hyaluronic acid-free form (left-hand panel), SAA1.1 in the hyaluronic acid-bound form (center panel), and SAA2.2 in the hyaluronic acid-bound form (right-hand panel). (B) Secondary structural changes for SAA1.1 in the hyaluronic acid-free form (left-hand panels), SAA1.1 in the hyaluronic acid-bound form (center panels), and SAA2.2 in the hyaluronic acid-bound form (right-hand panels). Labeling: monomer I, blue; monomer II, black. Representative conformational changes of the SAA1.1 N-terminus of the hyaluronic acid-free (C) and the hyaluronic acid-bound forms (D).
SAA aggregation and fibril formation are accompanied by secondary structure alterations. In response to low pH, acidic lysophospholipid environment, GAG binding, or high temperature, the α-helical structure of SAA1.1 is converted to random coil or β-strand structures,2,39 resulting in decreased solubility as well as decreased stability. Importantly, the amyloidogenicities of different SAA regions are different: the highly amyloidogenic segments are more sensitive to environmental changes and exhibit an increased rate of losing their helical structures. The identified amyloidogenic regions of human SAA1 include the N-terminal (residues 1–27) and central regions (residues 43–63).40 Two residues, residue 52 and residue 57 in human SAA1 (V51 and V56 in murine SAA1.1), are key determinants of the segment amyloidogenicity.41
MD simulations show that interaction between SAA1.1 monomers results in a secondary structure switch from α-helix to turn or coil in the N-terminus (residues 1–10) and parts of the H3 (residues 59–66) and H4 (residues 71–80) helices as shown in Figure 2B. Similar behavior is also seen in the SAA2.2–hyaluronic acid complex. However, the presence of a partially unfolded conformation in SAA2.2 is less frequent than in SAA1.1.
According to the simulation, the first 10 residues of the N-termini of SAA1.1 and SAA2.2 are least stable (Figure 2B). This finding is in accordance with a previous in silico study42 and with a recent publication supporting in vivo amyloidogenicity of the N-terminal residues of murine SAA1.1 and of human SAA1 lacking the N-terminal Arg.43 Interestingly, also monomeric SAA1.1 shows secondary structural transformation in the above-mentioned regions. Thus, the N-terminus of the H1 helix of a monomer is unfolded during the simulation, indicating the intrinsic instability of the N-terminus similar to that observed in the dimeric model. Occasionally, unfolded H3 and H4 helices are refolded to helical structures at the end of the simulation (Figure S5), which indicates that the destabilization of the amyloidogenic region might be partially dependent on dimerization.
Hyaluronic Acid Facilitates SAA1.1 Dimer Assembly by Reducing Nonspecific Interactions
Dynamical cross-correlation matrix (DCCM) is a commonly used tool for MD simulation analysis, which provides insights into the correlated motion of atoms in the whole system.44 In this study, the cross-correlation maps are visualized with a color map, with the color gradient going from dark blue (−1, anticorrelated motion), through yellow (0, noncorrelated motion), to dark red (1, cooperative motion). As shown in Figure 3A, the SAA1.1–hyaluronic acid complex shows more cooperative motion, especially in the off-diagonal regions. For example, a cooperative motion is observed between the H1 helix (residues 1–20) of monomer I and H2 (residues 20–40) or the H4 (residues 80–90) helix of monomer II. These cooperative motions are not observed in the hyaluronic acid-free form of the SAA1.1 dimer. On the other hand, hyaluronic acid changes the trend of motion between the H1 helix of monomer II and the H4 helix of monomer I from anticorrelated (hyaluronic acid-free form) to cooperative (hyaluronic acid-bound form), indicating that hyaluronic acid affects the SAA1.1 dimer formation.
Figure 3.

Hyaluronic acid binding stabilizes the SAA1.1 dimer by increasing cooperative motion and by decreasing nonspecific interactions. (A) Dynamic cross-correlation matrices (DCCMs) of hyaluronic acid-free and hyaluronic acid-bound forms of the SAA1.1 dimer reveal the increased off-diagonal cooperative motion of atoms in the hyaluronic acid-bound form (red squares). (B) Free-energy surface (FES) of SAA1.1 dimer in the hyaluronic acid-free and -bound forms; t = 0, starting point. The protein structures of SAA1.1 dimers exhibiting the lowest free energy as indicated in (B) are shown for the hyaluronic acid-free form (C) and for the hyaluronic acid-bound form (D). Hyaluronic acid is visualized as green sticks. State I, early time point; state II, later time point.
After examination of the MD trajectories of hyaluronic acid-free and -bound SAA1.1 dimers, we found that one monomer searches for a preferred binding site mainly through rotating around another monomer. To measure the rotation direction, two dihedrals were defined (Figure 3B). The dihedral I is used to measure the rotation along the axis of the H2 helix of the monomer II (denoted “b” in the left-hand panel of Figure 3B), and the dihedral II is used to measure the rotation perpendicular to the above-mentioned axis (denoted “a” in the left-hand panel of Figure 3B). The 2D density plot demonstrates the possible range of dihedrals and their corresponding free energy.
The free-energy surface (FES) of the hyaluronic acid-free form of SAA1.1 dimer shows one local minimum and a broad basin over a large space, whereas the SAA1.1 dimer complex with hyaluronic acid exhibits two local minima connected by a narrow deep basin (Figure 3B). The distance between the starting point (t = 0) and the local minima in the hyaluronic acid-bound form is much shorter than in the hyaluronic acid-free form. The closely positioned local minima and the short distance between the starting point and the local minima, as well as a narrow deep basin connecting the two local FES minima, indicate for hyaluronic acid an easier swap between the two potential ligand binding sites. Moreover, the movement of hyaluronic acid to another binding site might be used by the SAA1.1 dimer to attract a new SAA1.1 monomer by utilizing hyaluronic acid as a “bait”. On the other hand, the broad and shallow free-energy basins and high number of minima indicate that the hyaluronic acid-free SAA1.1 dimer is stabilized largely by nonspecific interactions, an observation supported also by others.45,46 On the contrary, a free-energy landscape with a narrow and deep basin indicates that hyaluronic acid facilitates the SAA1.1 dimer formation by decreasing nonspecific interactions of flexible regions located on the surface of the protein. Protein structures of SAA1.1 dimers exhibiting the lowest free energy, i.e., state I of the hyaluronic-free form and states I and II of the hyaluronic-bound form, are presented in Figure 3C,D, respectively.
SAA1.1 and SAA2.2 Possess Different Binding Interfaces for Hyaluronic Acid
To assess the binding site transition for hyaluronic acid in SAA1.1 and SAA2.2, the angle between either SAA dimer and hyaluronic acid and the distance between the monomer C-termini were plotted along with the simulation (Figure 4A). The relatively small angle (Figure 4A, left-hand panel) and a more symmetric hydrogen-bond distribution in the SAA2.2 dimer (Figure 4C) indicate that hyaluronic acid does not leave the cleft near the C-terminus of SAA2.2 dimer. The different paradigms in SAA1.1 and SAA2.2 dimerization in the presence of hyaluronic acid were further confirmed by additional simulations using different binding poses and starting velocities. As shown in Figure S6, a symmetrical distribution of hydrogen bonds in hyaluronic acid–SAA2.2 dimer and a generally asymmetrical distribution of hydrogen bonds in hyaluronic acid–SAA1.1 dimer were observed.
Figure 4.

SAA1.1 and SAA2.2 use different interfaces to bind and stabilize the hyaluronic acid molecule. (A) Angle between either SAA1.1 or SAA2.2 dimer and hyaluronic acid and the distance between the center of mass (COM) of the SAA C-termini are plotted along with the simulation. Angles were built using COM of SAA monomer I and hyaluronic acid as rays and COM of the monomer II as a vertex. The number of hydrogen bonds between SAA1.1 and hyaluronic acid (B) and between SAA2.2 and hyaluronic acid (C) along with the simulation. The N-terminus movement (arrows) and hydrogen bonds of SAA1.1 (D) and SAA2.2 (E) in the presence of hyaluronic acid during the first 50 ns of simulations.
On the other hand, the hydrogen bonds between hyaluronic acid and the SAA1.1 monomer II vanish after 750 ns (Figure 4B, right-hand panel). Unexpectedly, hydrogen bonds suddenly appear at about 1000 ns, indicating a remarkable binding site switch by hyaluronic acid on the SAA1.1 dimer. The trajectory shows that hyaluronic acid falls into the cleft between the H1 of monomer I and the H2, H3, and H4 of monomer II. Because of repulsion between the negatively charged cleft and hyaluronic acid, it moves quickly out of the binding site as reflected by the disappearance of the hydrogen bonds between hyaluronic acid and monomer II between 750 and 1000 ns (Figure 4B, right-hand panel). Differently, the repulsion in SAA2.2 is not sufficient to initiate the hyaluronic acid transition.
A comparison of snapshots from 0 to 50 ns during the simulation (Figure 4D,E) shows that the stability of the H1 helix as well as the difference in the residue 101 (Ala101Asp, SAA1.1 vs SAA2.2) between SAA1.1 and SAA2.2 might be the key to their binding site difference toward hyaluronic acid. Indeed, substitution of aspartic acid with alanine at position 101 of SAA2.2 increased the distance between the N-terminal helix and A101 in the C-terminus compared to wild-type protein in the SAA2.2 dimer model, supporting the hypothesis and the role of D101 in SAA2.2 dimer formation (Figure S7). Accordingly, computational studies demonstrate that the residues in the center of the helix play a role in either stabilizing or destabilizing the helical structure. Thus, His, Ala, and Glu contribute to the stabilization of a helix,47 whereas Pro, Gly, and Trp contribute to the unfolding or kink formation in an α-helix.48 In our simulation, the residue difference in position 7 (Gly in SAA1.1 and His in SAA2.2) results in a hydrogen-bond switch (Figure 4D,E, right-hand panels). Thus, the hydrogen bonds between the backbone atoms of Phe2, Phe3, Val6, and His7 in SAA2.2 are disrupted in SAA1.1. As a result, in SAA1.1, a kink in the helix is formed around Ile6 and Gly7. Hence, the partial unfolding and kink formation of the N-terminus adjacent to the H1 helix slightly increases the distance between the N- and C-termini in the SAA1.1 dimer, and hyaluronic acid leaves the original binding site through rotation of the SAA1.1 monomer and decreased steric effects. On the contrary, the electrostatic attraction between Asp101 and the positively charged sequence adjacent to the N-terminus reduces the distance between the N- and C-termini of two SAA2.2 monomers (Figure 4E), suggesting that the N-terminal helix and Asp101 in the C-terminus of SAA2.2 might “lock” hyaluronic acid in a cleft inside the dimer.
SAA1.1 Exhibits Three Different Binding Sites for Hyaluronic Acid
Because of the negatively charged properties of hyaluronic acid, its binding to amyloidogenic proteins is mediated largely by electrostatic attraction to polar residues. Accordingly, we plotted the hydrogen-bond number, as well as the distribution of hydrogen bonds to different residues. The hydrogen-bond-distribution pattern (Figure 5A) indicates three different binding sites for hyaluronic acid on the SAA1.1 dimer besides the one identified on the initial conformation. With the rotation of the SAA1.1 monomer I around monomer II, hyaluronic acid leaves the original binding site and binds to a shallow pocket located within the C-terminus of the SAA1.1 monomer II and a loop region connecting H3 and H4 helices in the SAA1.1 monomer I (180 ns snapshot in Figure 5B). Then, hyaluronic acid binds to a pocket formed by the H1, H3, and H4 helices in the SAA1.1 monomer I along with the rotation of monomer I (447 and 910 ns snapshots in Figure 5B). Interestingly, hyaluronic acid is finally stabilized by the partially unfolded N-terminus of SAA1.1 (1470 ns snapshot in Figure 5B). Besides, hydrogen bonds are mainly distributed around polar residues located on the surface of SAA1.1, suggesting that electrostatic forces and not hydrophobic interactions might play the major role in hyaluronic acid binding.
Figure 5.

SAA1.1 dimer exhibits three different binding sites for hyaluronic acid. (A) Total number of hydrogen bonds in the hyaluronic acid-bound SAA1.1 dimer (upper panel), and the hydrogen-bond-existence map (lower panel) showing three different patterns. (B) Residues stabilizing the interaction between hyaluronic acid and SAA1.1 demonstrate partial unfolding of the N-terminus stabilizing the structure.
Additional 800 ns simulations performed on SAA1.1 and SAA2.2 in the presence of hyaluronic acid confirmed the convergence and reproducibility of our molecular dynamics simulations (Figure S8).
Model of SAA1.1 in the Hyaluronic Acid-Bound Form Exhibits Specific Metastable State Distribution
As mentioned above, murine SAA1.1 and SAA2.2 differ only by the nine residues, Ile6Val, Gly7His, Gly27Asn, Asp30Asn, Gly31Ser, Ala60Gly, Ser63Ala, Met76Ile, and Ala101Asp. However, the two SAA isomers exhibit remarkably different fibril formation kinetics and pathogenic properties with only SAA1.1, but not SAA2.2, found in murine amyloid deposits.1 To find long-time statistical conformational dynamics of above residues in three models, SAA1.1, the SAA1.1–hyaluronic acid complex, and the SAA2.2–hyaluronic acid complex, Markov state models (MSMs) were constructed. The MSM analysis shows that the dynamic behavior of the key residues could be partially related to the formation of different interfaces.
Thus, MSM analysis has identified nine metastable states in the above three models (Table S1 and Figure 6A). The representative snapshots of each metastable state are shown in Figure 6B. According to the probability distribution (Table S1), a highly populated metastable state IX is detected in all three models. The conformation of the state IX shows no contact between linker I and linker III in all three models, which represent the conformation of the starting point (Figure 1B). However, the distribution of the other less populated metastable states varies in each model. In the SAA1.1–hyaluronic acid model, a broader distribution among all nine metastable states is observed with frequencies ranging between 3.5 and 29.1%, except for the state VI. However, the SAA1.1–hyaluronic acid-free model and the SAA2.2–hyaluronic acid models have only two or three highly populated models that capture around 95% of the population. In addition, MSM analysis has identified three states, I, II, and V, which are unique to the SAA1.1–hyaluronic acid model (Tables S1 and S2).
Figure 6.

Markov state modeling (MSM) for SAA1.1, the SAA1.1–hyaluronic acid complex, and the SAA2.2–hyaluronic acid complex identifies nine metastable states characterized by contact between linker I, linker III, helix I, and helix III. (A) Time-lagged independent component analysis (TICA) plot with microstates is shown in the left-hand panel. Microstates from k-means clustering are plotted with dots. The microstates are clustered into nine metastable states (I, light sea-green; II, olive; III, deep sky-blue; IV, aqua; V, lime; VI, medium sea-green; VII, dodger-blue; VIII, light sky-blue, and IX, blue). An illustration of linker I, linker III, helix I, and helix III is shown in the right-hand panel. (B) Representative structures of each metastable state. N-terminus, pink; linker I, marine; linker III, orange. (C) Interface and hyaluronic acid (green sticks) binding site for groups 3 and 4 of SAA1.1.
Metastable states identified by MSM can be further combined into four groups according to their conformation (Table 1). Group I is highly populated in all three models, which contain states III, VIII, and IX. Group 1 is characterized by few or even absent contacts between linker I and linker III. Group 2 contains states II, IV, V, and VI, which are characterized by a kink on the H3 helix as well as the contact between linker I and linker III. Both SAA1.1 models are highly populated in this group. Hence, residues 60 and 63 that are different in SAA1.1 and SAA2.2 (Ala60Gly, Ser63Ala) might contribute to the additional flexibility of the H3 helix in the SAA1.1 models. The role of the interface in group 2 cannot be derived from this analysis.
Table 1. Distribution of Conformational Groups as Analyzed by MSM.
| group | description | metastates | SAA1.1 % | SAA1.1 + hyaluronic acid % | SAA2.2 + hyaluronic acid % |
|---|---|---|---|---|---|
| 1 | no contact between linker I and linker III | IX, VIII, III | 43.9 | 55.5 | 63.5 |
| 2 | contact between linker I and linker III | II, IV, V, VI | 36.6 | 34.6 | 3.1 |
| 3 | contact between helix I and helix III | VII | 19.5 | 6.5 | 33.4 |
| 4 | loop III approaching HI helix | I | 0 | 3.4 | 0 |
In group 3, the modeling indicates a possible contact between the H1 and the H3 helix. In addition, SAA1.1–hyaluronic acid-free and SAA2.2–hyaluronic acid-bound models are highly populated in this group. Considering the fact that the interface is located around H1 and H3, the increased contact may result from a repulse between two monomers (Figure 6C). A decreased volume and decreased water exposure of residues on the H1 helix in group 3 might facilitate stabilization of the secondary structure of the protein.
Group 4 contains only one state, state I, and is unique to the SAA1.1–hyaluronic acid-bound model. As shown in Figure 6C, state I exhibits a characteristic partially unfolded H3 helix inserted into the cleft inside a SAA1.1 monomer. Further computational analysis shows that hyaluronic acid also participates in the formation of the partially unfolded H3 helix. Hence, we hypothesized that the increased freedom caused by the binding interface switch may contribute to the formation of this state. Besides, the increased unfolded structure and water exposure of hydrophobic residues are considered to be energetically unfavorable and less stable.
Possible Oligomeric States of SAA1.1 and SAA2.2 as Determined by Free-Energy Analysis of the Dimers
Both SAA1.1 and SAA2.2 complexes with hyaluronic acid exhibit two local minima according to the free-energy landscape (Figure 7A,B). States I of either complex, which appear at about 50–100 ns of the simulation, share a similar conformation: the binding site of hyaluronic acid is still located between SAA monomers, and the C-termini are far from each other. This model is in agreement with our hypothesis that SAA1.1 and SAA2.2 share similar initial binding modes of the hyaluronic acid molecule (Figure 7A,B, state I).
Figure 7.

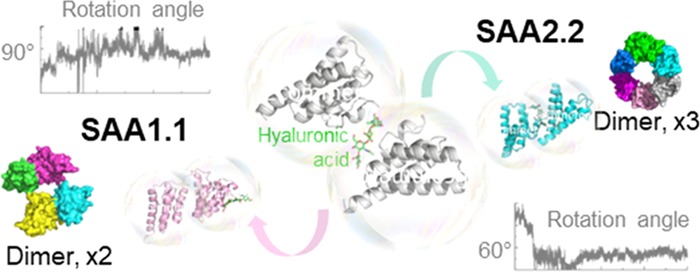
(A) SAA1.1 and (B) SAA2.2 oligomer formation. Conformations of SAA with the lowest free energy govern the proposed oligomeric states of SAA1.1 and SAA2.2. Upper panels: the free-energy landscape of SAA and the position of hyaluronic acid (green sticks) in SAA dimers (state I, early time point; state II, later time point). Lower panels: kinetics of the dihedral angel changes between the C-termini.
However, states II of SAA1.1 and SAA2.2 are different. As shown previously, SAA rotates a certain angle during the transformation from a loosely to a tightly bound structure. Accordingly, the dihedral of the C-termini of two SAA monomers (the most stable SAA regions) in the SAA1.1–hyaluronic acid complex stabilizes at around 85° (Figure 7A), whereas the dihedral of the SAA2.2–hyaluronic acid complex stabilizes at around 60° (Figure 7B). If we consider the oligomer formation as a continuous progress, a new SAA1.1 monomer binds to the cleft and also turns at about 85° to achieve the stabilized conformation, resulting in tetramer formation as a block for a larger oligomer structure (Figure 7A). Similarly, the SAA2.2 monomer will rotate 60° and form a hexamer with a central channel, as a building block for a larger oligomer structure (Figure 7B). This computational model offers a plausible explanation of literature data demonstrating that in solution SAA2.2 forms a hexamer with a central channel.8,9 The potential role of hyaluronic acid in oligomerization of SAA1.1 and SAA2.2 leading to formation of SAA1.1 tetramer and SAA2.2 hexamer building blocks is described in the Supporting Information and is illustrated in Figure S9.
SAA C-Terminus is a Stable Structural Motif with Diverse Functions
The C-terminus of human SAA1 is stabilized by hydrogen bonds and a salt bridge between Arg96, Lys103, Asn93, and Tyr104. In addition, the proline residues 93, 97, and 101 contribute to the stabilization of the C-terminus by stereoelectronic and steric effects.3 An NMR study showed that the C-terminal fragment is able to preserve its conformation in solution.49 Although the transition of the SAA dimeric interface happens rapidly during the first 100 ns of the simulation, the configuration of the C-terminal loop is preserved along with the simulation, which might be due to the relatively low B-factor of this region in the crystal structure and in accordance with in vitro findings.3,49
Functions of the SAA C-terminus include GAGs binding as well as modulation of the SAA fibril formation.2,50 Still, the mechanism of the C-terminus-mediated fibril formation is ambiguous. In humans, a C-terminal-truncated SAA1 shows an increased aggregation and fibril formation rate in the presence of GAGs compared to full-length SAA1, indicating that the C-terminus may function as a stabilizer of SAA.1 Differently, in vitro studies demonstrated that a C-terminal-truncated murine SAA2.2 exhibited a slower fibril formation rate and different oligomeric states compared to full-length SAA2.2.50 The results from our simulation suggest that the SAA C-terminus might not only form the binding pocket for hyaluronic acid in a loosely bound SAA dimeric model but might also participate in the formation of a more stable dimeric structure obtained from the free-energy landscape. This observation indicates that the function of the C-terminus of SAAs might be closely related to its dimeric interface. Further studies of the oligomeric states, particularly the oligomeric interfaces, might help us to better understand the function of the C-terminus in SAA proteins.
Conclusions
Formation of dimers and small oligomers is an important initial step of SAA fibril formation and subsequent aggregation that is affected by hyaluronic acid binding. Here, we built a loosely bound dimeric intermediate shared by murine SAA1.1 and SAA2.2 and revealed distinct dimeric interfaces formed by amyloidogenic SAA1.1 and nonamyloidogenic SAA2.2 in three distinct models, the SAA1.1–hyaluronic acid-free form, the SAA1.1–hyaluronic acid-bound form, and the SAA2.2 hyaluronic acid-bound form. The model suggests that in SAA1.1 binding of hyaluronic acid might promote oligomer formation by transient interaction, which decreases nonspecific interactions between SAA1.1 monomers and promotes the formation of an asymmetric interface involving the H1/H2 helices and the H4/C-terminus. In SAA2.2, hyaluronic acid is shown to bind to the cleft between two SAA2.2 monomers formed by H1 and H2 helices of one monomer and the C-terminus of another monomer, forming a scaffold for larger oligomer assembly.
Besides, Markov state modeling indicated that SAA1.1 and SAA2.2 exhibit distinct dynamic behavior of the linker region adjacent to H1 and H3 helices, which may be related to the differences in binding interfaces. A unique state in the SAA1.1–hyaluronic acid model shows a partially unfolded H3 helix.
The model and the interfaces elaborated by computational modeling suggest that the pathogenic SAA isomer, SAA1.1, might employ a different mechanism for oligomer formation compared to nonpathogenic SAA2.2, which might offer a plausible explanation why only the SAA1.1 isoform is found in SAA amyloid deposits. These findings might assist researchers working with mouse models of AA amyloidosis. Hence, the study might facilitate the discovery and design of novel SAA aggregation inhibitors targeting the interfaces in early steps of amyloid formation.
Acknowledgments
The authors acknowledge support by the State of Baden-Württemberg through the High Performance Computing Program (bwHPC) and by the Chinese Scholarships Council (No. 201708080165).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01590.
Lag time estimation and Chapman–Kolmogorov test for Markov state modeling; selection of the initial model and comparison of interaction interfaces; predicted binding conformations of heparin and chondroitin sulfate A in SAA1.1 dimeric model; additional models to validate the possibility of proposed hyaluronic binding site; analysis of binding interfaces in SAA1.1–hyaluronic acid-bound and SAA1.1–hyaluronic acid-free models; short simulation of the SAA2.2 D101A structure model in the hyaluronic acid-bound form; analysis of key observations by another independent simulation; hypothetical models demonstrating how GAG might facilitate oligomer formation; identified metastable states and their distribution; and a list of systems used in Markov state modeling (PDF)
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Sun L.; Ye R. D. Serum amyloid A1: Structure, function and gene polymorphism. Gene 2016, 583, 48–57. 10.1016/j.gene.2016.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark G. T.; Fandrich M.; Westermark P. AA amyloidosis: pathogenesis and targeted therapy. Annu. Rev. Pathol.: Mech. Dis. 2015, 10, 321–344. 10.1146/annurev-pathol-020712-163913. [DOI] [PubMed] [Google Scholar]
- Lu J.; Yu Y.; Zhu I.; Cheng Y.; Sun P. D. Structural mechanism of serum amyloid A-mediated inflammatory amyloidosis. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 5189–5194. 10.1073/pnas.1322357111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus S.; Meinhardt K.; Aumuller T.; Puscalau-Girtu I.; Linder J.; Haupt C.; Walther P.; Syrovets T.; Simmet T.; Fandrich M. Cellular mechanism of fibril formation from serum amyloid A1 protein. EMBO Rep. 2017, 18, 1352–1366. 10.15252/embr.201643411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmer M.; Meinhardt K.; Haupt C.; Liberta F.; Wulff M.; Linder J.; Handl L.; Heinrich L.; Loos C.; Schmidt M.; Syrovets T.; Simmet T.; Westermark P.; Westermark G. T.; Horn U.; Schmidt V.; Walther P.; Fandrich M. Electron tomography reveals the fibril structure and lipid interactions in amyloid deposits. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 5604–5609. 10.1073/pnas.1523496113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson S. R.; Lyon M.; Gallagher J. T.; Johnson E. A.; Pepys M. B. Isolation and characterization of the integral glycosaminoglycan constituents of human amyloid A and monoclonal light-chain amyloid fibrils. Biochem. J. 1991, 275, 67–73. 10.1042/bj2750067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera J. J.; Zhang F.; Beaudet J. M.; Linhardt R. J.; Colon W. Divergent effect of glycosaminoglycans on the in vitro aggregation of serum amyloid A. Biochimie 2014, 104, 70–80. 10.1016/j.biochi.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S.; Patke S.; Wang Y.; Ye Z.; Litt J.; Srivastava S. K.; Lopez M. M.; Kurouski D.; Lednev I. K.; Kane R. S.; Colon W. Pathogenic serum amyloid A 1.1 shows a long oligomer-rich fibrillation lag phase contrary to the highly amyloidogenic non-pathogenic SAA2.2. J. Biol. Chem. 2013, 288, 2744–2755. 10.1074/jbc.M112.394155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Lashuel H. A.; Walz T.; Colon W. Murine apolipoprotein serum amyloid A in solution forms a hexamer containing a central channel. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 15947–15952. 10.1073/pnas.252508399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K.; Uchida K.; Chambers J. K.; Ushio N.; Nakayama H. Deposition, clearance, and reinduction of amyloid A amyloid in interleukin 1 receptor antagonist knockout mice. Vet. Pathol. 2017, 54, 99–110. 10.1177/0300985816658772. [DOI] [PubMed] [Google Scholar]
- Shankar G. M.; Li S.; Mehta T. H.; Garcia-Munoz A.; Shepardson N. E.; Smith I.; Brett F. M.; Farrell M. A.; Rowan M. J.; Lemere C. A.; Regan C. M.; Walsh D. M.; Sabatini B. L.; Selkoe D. J. Amyloid β-protein dimers isolated directly from Alzheimer brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson A.; Ippel H. J.; Baranov V.; Horstedt P.; Wijmenga S.; Lundgren E. Capture of a dimeric intermediate during transthyretin amyloid formation. J. Biol. Chem. 2001, 276, 39592–39599. 10.1074/jbc.M103599200. [DOI] [PubMed] [Google Scholar]
- Cole H.; Porrini M.; Morris R.; Smith T.; Kalapothakis J.; Weidt S.; Mackay C. L.; MacPhee C. E.; Barran P. E. Early stages of insulin fibrillogenesis examined with ion mobility mass spectrometry and molecular modelling. Analyst 2015, 140, 7000–7011. 10.1039/C5AN01253H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliam H.; Li W.; Uludag M.; Squizzato S.; Park Y. M.; Buso N.; Cowley A. P.; Lopez R. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M. A.; Blackshields G.; Brown N. P.; Chenna R.; McGettigan P. A.; McWilliam H.; Valentin F.; Wallace I. M.; Wilm A.; Lopez R.; Thompson J. D.; Gibson T. J.; Higgins D. G. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Robert X.; Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N.; Webb B.; Marti-Renom M. A.; Madhusudhan M. S.; Eramian D.; Shen M. Y.; Pieper U.; Sali A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinf. 2006, 15, 5.6.1. 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; Rullmannn J. A.; MacArthur M. W.; Kaptein R.; Thornton J. M. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Beard H.; Cholleti A.; Pearlman D.; Sherman W.; Loving K. A. Applying physics-based scoring to calculate free energies of binding for single amino acid mutations in protein-protein complexes. PLoS One 2013, 8, e82849 10.1371/journal.pone.0082849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farid R.; Day T.; Friesner R. A.; Pearlstein R. A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. 10.1016/j.bmc.2005.12.032. [DOI] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner K. N.; Yongye A. B.; Tschampel S. M.; Gonzalez-Outeirino J.; Daniels C. R.; Foley B. L.; Woods R. J. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolafa J.; Perram J. W. Cutoff errors in the Ewald summation formulas for point-charge systems. Mol. Simul. 1992, 9, 351–368. 10.1080/08927029208049126. [DOI] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Koukos P. I.; Glykos N. M. Grcarma: A fully automated task-oriented interface for the analysis of molecular dynamics trajectories. J. Comput. Chem. 2013, 34, 2310–2312. 10.1002/jcc.23381. [DOI] [PubMed] [Google Scholar]
- Glykos N. M. Software news and updates. Carma: a molecular dynamics analysis program. J. Comput. Chem. 2006, 27, 1765–1768. 10.1002/jcc.20482. [DOI] [PubMed] [Google Scholar]
- Bonomi M.; Branduardi D.; Bussi G.; Camilloni C.; Provasi D.; Raiteri P.; Donadio D.; Marinelli F.; Pietrucci F.; Broglia R. A.; Parrinello M. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. 10.1016/j.cpc.2009.05.011. [DOI] [Google Scholar]
- da Silva A. W. S.; Vranken W. F. ACPYPE - AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. 10.1186/1756-0500-5-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touw W. G.; Baakman C.; Black J.; te Beek T. A.; Krieger E.; Joosten R. P.; Vriend G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. 10.1093/nar/gku1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauenfelder H.; Sligar S. G.; Wolynes P. G. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603. 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- Tsai C. J.; Ma B.; Nussinov R. Folding and binding cascades: Shifts in energy landscapes. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 9970–9972. 10.1073/pnas.96.18.9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer M. K.; Trendelkamp-Schroer B.; Paul F.; Perez-Hernandez G.; Hoffmann M.; Plattner N.; Wehmeyer C.; Prinz J. H.; Noe F. PyEMMA 2: A software package for estimation, validation, and analysis of Markov models. J. Chem. Theory Comput. 2015, 11, 5525–5542. 10.1021/acs.jctc.5b00743. [DOI] [PubMed] [Google Scholar]
- Jurrus E.; Engel D.; Star K.; Monson K.; Brandi J.; Felberg L. E.; Brookes D. H.; Wilson L.; Chen J.; Liles K.; Chun M.; Li P.; Gohara D. W.; Dolinsky T.; Konecny R.; Koes D. R.; Nielsen J. E.; Head-Gordon T.; Geng W.; Krasny R.; Wei G. W.; Holst M. J.; McCammon J. A.; Baker N. A. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. 10.1002/pro.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M.; Schein J.; Birol I.; Connors J.; Gascoyne R.; Horsman D.; Jones S. J.; Marra M. A. Circos: an information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancsin J. B.; Kisilevsky R. The heparin/heparan sulfate-binding site on apo-serum amyloid A. Implications for the therapeutic intervention of amyloidosis. J. Biol. Chem. 1999, 274, 7172–7181. 10.1074/jbc.274.11.7172. [DOI] [PubMed] [Google Scholar]
- Zhe Z.; Shawn W.; Emil A. On the role of electrostatics in protein–protein interactions. Phys. Biol. 2011, 8, 035001 10.1088/1478-3975/8/3/035001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman S.; Gantz D. L.; Haupt C.; Gursky O. Serum amyloid A forms stable oligomers that disrupt vesicles at lysosomal pH and contribute to the pathogenesis of reactive amyloidosis. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E6507–E6515. 10.1073/pnas.1707120114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M.; Nishimura A.; Takeshita H.; Takase H.; Yamada T.; Mukai T. Effect of lipid environment on amyloid fibril formation of human serum amyloid A. Chem. Phys. Lipids 2017, 202, 6–12. 10.1016/j.chemphyslip.2016.11.004. [DOI] [PubMed] [Google Scholar]
- Egashira M.; Takase H.; Yamamoto I.; Tanaka M.; Saito H. Identification of regions responsible for heparin-induced amyloidogenesis of human serum amyloid A using its fragment peptides. Arch. Biochem. Biophys. 2011, 511, 101–106. 10.1016/j.abb.2011.04.019. [DOI] [PubMed] [Google Scholar]
- Takase H.; Tanaka M.; Miyagawa S.; Yamada T.; Mukai T. Effect of amino acid variations in the central region of human serum amyloid A on the amyloidogenic properties. Biochem. Biophys. Res. Commun. 2014, 444, 92–97. 10.1016/j.bbrc.2014.01.029. [DOI] [PubMed] [Google Scholar]
- Nordling E.; Abraham-Nordling M. Colonic amyloidosis, computational analysis of the major amyloidogenic species, serum amyloid A. Comput. Biol. Chem. 2012, 39, 29–34. 10.1016/j.compbiolchem.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Liberta F.; Loerch S.; Rennegarbe M.; Schierhorn A.; Westermark P.; Westermark G. T.; Hazenberg B. P. C.; Grigorieff N.; Fandrich M.; Schmidt M. Cryo-EM fibril structures from systemic AA amyloidosis reveal the species complementarity of pathological amyloids. Nat. Commun. 2019, 10, 1104 10.1038/s41467-019-09033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan S.; Harte W. E.; Beveridge D. L. Investigation of domain structure in proteins via molecular dynamics simulation: application to HIV-1 protease dimer. J. Am. Chem. Soc. 1991, 113, 2717–2721. 10.1021/ja00007a054. [DOI] [Google Scholar]
- Cardone A.; Bornstein A.; Pant H. C.; Brady M.; Sriram R.; Hassan S. A. Detection and characterization of nonspecific, sparsely populated binding modes in the early stages of complexation. J. Comput. Chem. 2015, 36, 983–995. 10.1002/jcc.23883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone A.; Pant H.; Hassan S. A. Specific and non-specific protein association in solution: Computation of solvent effects and prediction of first-encounter modes for efficient configurational bias Monte Carlo simulations. J. Phys. Chem. B 2013, 117, 12360–12374. 10.1021/jp4050594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Bansal M. Geometrical and sequence characteristics of α-helices in globular proteins. Biophys. J. 1998, 75, 1935–1944. 10.1016/S0006-3495(98)77634-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilman H. R.; Shi J.; Deane C. M. Helix kinks are equally prevalent in soluble and membrane proteins. Proteins 2014, 82, 1960–1970. 10.1002/prot.24550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maszota M.; Karska N.; Spodzieja M.; Ciarkowski J.; Kolodziejczyk A. S.; Rodziewicz-Motowidlo S.; Czaplewska P. Structural studies of the C-terminal 19-peptide of serum amyloid A and its Pro→Ala variants interacting with human cystatin C. J. Mol. Recognit. 2015, 28, 413–426. 10.1002/jmr.2457. [DOI] [PubMed] [Google Scholar]
- Patke S.; Maheshwari R.; Litt J.; Srinivasan S.; Aguilera J. J.; Colon W.; Kane R. S. Influence of the carboxy terminus of serum amyloid A on protein oligomerization, misfolding, and fibril formation. Biochemistry 2012, 51, 3092–3099. 10.1021/bi201903s. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.