

Abstract
The BCL-2 family of proteins orchestrates a complex signaling network that governs the balance between cellular survival and death. A comprehensive understanding of the mechanistic interactions between these proteins continues to evolve in normal and malignant cells. The functional variation by individual BCL-2 proteins in different cell types has driven clinical therapeutic development in targeting individual BCL-2 members with the goal of fine-tuning cell death in diseased cells. Given the importance of understanding and validating the effect of activating or inhibiting BCL-2 protein interactions in individual cells, the methods used to measure apoptotic cell death have undergone increased scrutiny. Here, we describe two in vitro flow cytometry-based methods that are useful in measuring BCL-2 proteins and mitochondrial-based cell death in complex cell populations.
Keywords: BCL-2 proteins, Intracellular staining, Flow cytometry, Mitochondria outer membrane permeabilization (MOMP), Apoptosis
1. Introduction
Since ancient times, there has been great interest in the study of cell death. While Hippocrates first used the term “apoptosis” in the fourth century B.C., it was not until hundreds of years later that a more complete understanding of the cell death pathway was gleaned and appropriate tools to study it were developed. In the 1970s, apoptosis was formally described as a unique variation of cell death distinct from necrosis [1,2]. Apoptosis is a form of altruistic cell suicide and is a crucial homeostatic process essential for the clearance of superfluous cells, especially in the context of development, immunity, and malignancy [3]. Although molecular hallmarks of apoptosis were known for many years, only over the past 30 years have the proteins governing apoptosis been characterized. How these proteins regulate cell death in normal and diseased cells continues to be an active area of research.
Apoptosis can be initiated by two related but nonredundant pathways that ultimately coalesce at the level of the mitochondrion and result in the activation of caspases that mediate cell destruction [4]. The extrinsic pathway is mediated through cell membrane receptors (FAS or other members of the TNFR family) where ligand binding causes adaptor protein association and caspase-8-activation [5]. The intrinsic pathway operates in response to various intracellular stressors like cytoskeletal damage, ER stress, and growth factor withdrawal, and is mediated by the BCL-2 family of proteins [6]. The first identified member of this family, BCL-2, was found near the anomalous junction of chromosomes 14 and 18 (t(14;18)) in follicular lymphoma [7, 8]. Unlike other previously identified oncogenes that act by increasing cellular proliferation, overexpression of BCL-2 was found to block cell death and hence promote cell survival [9]. Rapid discovery of other BCL-2 family proteins ensued and continues to expand. There are now at least sixteen members of the BCL-2 family that have been identified and characterized to regulate apoptosis at some level [10].
The BCL-2 family of proteins can be divided into three functional categories, each containing up to four BCL-2 homology (BH) domains (Fig. 1). Despite sharing amino acid similarity, these homologs have both proapoptotic and antiapoptotic functions and provide their own series of checks and balances on one another to exquisitely regulate cell death. Multidomain proapoptotic effector proteins (BAK and BAX) coalesce at the mitochondria when activated and form a pore that leads to mitochondrial outer membrane permeabilization (MOMP) [11]. Multidomain antiapoptotic proteins (BCL-2, BCL-Xl, BCL-W, BLF-1, MCL-1) sequester BAX and BAK, preventing their activation and subsequent MOMP. The interactions between the proapoptotic and antiapoptotic multidomain proteins are regulated by proteins containing only the BH3 domain (BH3-only proteins; BIM, BID, PUMA, BAD, BIK, BMF, HRK, NOXA) [12]. BH3-only proteins are further divided into “direct activators” or “sensitizers” based on their ability to target antiapoptotics as well as directly bind and activate BAK and BAX or only bind the antiapoptotic proteins respectively (Fig. 2). MOMP is considered a “point-of-no-return” for cell viability and can be measured once completed through classical apoptotic morphological and cellular changes like cell shrinkage, plasma membrane blebbing, cellular detachment, nuclear condensation, nuclear DNA fragmentation, and externalization of phosphatidylserine (PS) on the outer cellular membrane [13]. However, the real-time measurement of apoptosis is more difficult given variations in apoptotic resistance, the speed of cell death following MOMP, and complexity of the interactions between BCL-2 proteins within individual cells.
Fig. 1.
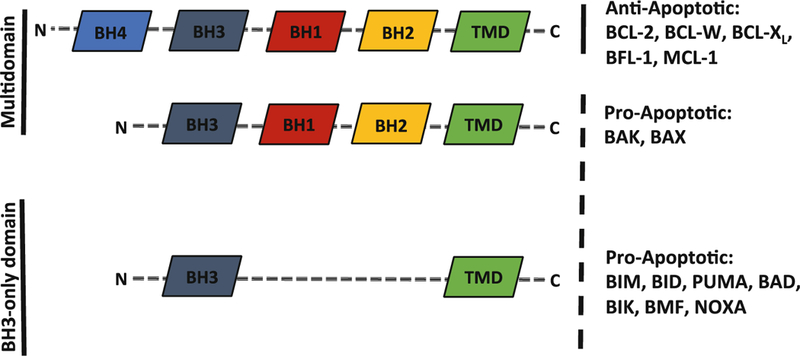
BCL-2 family members are characterized by the BH motifs they contain (labeled) and functions they perform. BH3-only BCL-2 proteins (e.g., BAD, BID, BIM) generally have proapoptotic functions as either activators or sensitizers while multidomain BCL-2 proteins can be either anti-apoptotic (e.g., BCL-2, BCL-W, BCL-XL) or proapoptotic (e.g., BAK, BAX). Members of each domain and functional group differ in size (indicated by dashed lines)
Fig. 2.
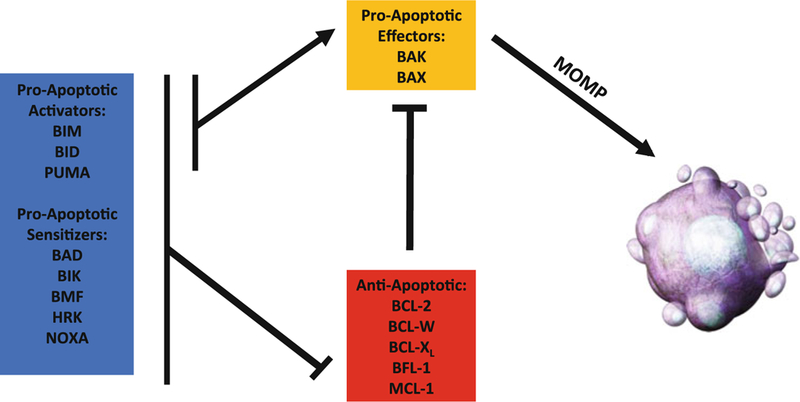
BH3-only proteins can act either as sensitizers by inhibiting the anti-apoptotic multidomain proteins (e.g., BCL-2, BCL-W, BCL-XL) or as activators by directly activating proapoptotic multidomain proteins (BAK and BAX). Ultimately, BAK/BAX activation and pore formation leads to mitochondrial outer membrane permeabi-lization (MOMP), cytochrome c release, activation of the caspase network, and cell death. BCL-2 family member functional roles and relationships are indicated as either activating (arrow) or inhibitory (crossed)
Many different techniques have been used to study cell death with varying specificity to apoptotic and nonapoptotic mechanisms [14]. Techniques range from light, electron, and fluorescence microscopy for identification of cellular morphological changes and fluorescently labeled cell death-associated proteins (e.g. PS exposure and annexin V positivity), to immunoblotting for identification of mitochondrial-released or caspase-cleaved proteins in cell extracts, to a wide-range of commercially available kits intended to measure aspects of cell death like loss of membrane integrity, caspase activity, and ATP/ADP ratio [15]. Given the complexity of cell death, a single “end point” assay is unable to accurately and specifically differentiate apoptosis from autophagy or necrosis. This is of particular importance, especially with the emphasis on BCL-2 mimetic drug discovery and validation [4,16,17]. Here we discuss two assays able to measure intracellular BCL-2 family protein content and mitochondrial-based cell death using flow cytometry. These techniques have become important validation tools in examining activation of the intrinsic apoptotic pathway.
1.1. Intracellular Flow Cytometry for BCL-2 Proteins
The real time activation of cell death can occur quickly or slowly. Understanding the quantitative BCL-2 family protein abundance as a function of time and treatment are valuable tools to comprehensively evaluate the mechanism(s) responsible for apoptotic triggering. Intracellular flow cytometry confers many advantages on the study of apoptosis. There are many commercially available kits that enable detection of caspase activity, and techniques like Western blotting that can probe for different members of the BCL-2 family of proteins, cleavage of caspases, or release of cytochrome c from the mitochondria to the cytoplasm. However, intracellular flow cytometry allows for rapid, multiparametric analysis of cells at multiple time points. Flow cytometry also allows the isolation of specific cell populations based on their cellular membrane protein expression profile and can be used to detect small populations of cells that would otherwise be missed using other techniques.
1.2. Mitochondrial Membrane Depolarization
A proton gradient exists across the inner mitochondrial membrane as a result of proton pumping by the respiratory chain apparatus embedded within the membrane [18, 19]. Conformational changes and homooligomerization of activated BAK and BAX lead to opening of the outer mitochondrial membrane through the formation of a pore [20, 21]. Once formed, the BAK/BAX pore permeabilizes the mitochondrial outer membrane leading to release of proteins normally sequestered within the intermembrane space, such as cytochrome c. Leakage of protons depolarizes the mitochondrion and can be visualized using flow cytometry. This can help differentiate apoptosis from necrosis [22, 23]. Detection of MOMP relies on the use of cationic dyes, such as rhodamine 123, tetramethylrosamine ethyl ester (TMRE), 5,5′,6,6-′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1), or diOC carbocyanine family probes that congregate within the negatively charged mitochondrial matrix [24–26]. An intact membrane results in dye accumulation and a positive fluorescent signal. If the outer membrane loses its integrity and negative charge, the dye will no longer associate with the mitochondria, resulting in a loss of fluorescence intensity. Many dyes start with a positive signal in a single fluorescent channel and lose that signal, however there are dyes that undergo a shift from one emission to another (JC-1 is an example of this) [27]. Therefore it is important to consider the expected emission of each dye and how it will overlap with any additional fluorophores being used in the experiment. Pros and cons of many dyes used to assess mitochondrial membrane potential can be found in a thorough review by Cottet-Rousselle et al. [25].
2. Materials
Prepare all solutions using analytical grade reagents. Use and store all reagents at 4 °C/ice, unless otherwise indicated.
2.1. Intracellular Flow for BCL-2 Proteins
FACS buffer: PBS, 1% FBS, (Optional: 0.1% sodium azide) (see Note 1).
Cellular permeabilization kit/buffers. We use the FOXP3 Fix/Perm Kit (ThermoFisher, formerly eBioScience): 10× Perm Buffer, Fixation/Permeabilization Concentrate, Diluent.
-
BCL-2 antibodies (see Note 2):
Murine and Human BCL-2 proteins commonly detected:
-
(a)BCL-2:
-
•PE Hamster Anti-Mouse Bcl-2 Set (3F11; comes with separate isotype control) BD 556537.
-
•PE Mouse Anti-Human Bcl-2 Set (Bcl-2/100; comes with separate isotype control) BD 556535.
-
•
-
(b)BCL-XL:
-
•(54H6) Rabbit mAb (PE conjugate) CST 13835S (detects human and murine isoforms).
-
•
-
(c)MCL-1:
-
•(Y37) Rabbit mAb (Alexa Fluor 647 conjugate) Abcam ab197035 (detects human and murine isoforms).
-
•
-
(d)BIM:
-
•(C34C5) Rabbit mAb (PE conjugate) CST 12186S (detects human and murine isoforms).
-
•
-
(e)BAX:
-
•(6A7) mAb (requires a secondary antibody conjugated to a fluorophore) ThermoFisher MA5–14003 (detects human and murine activated BAX isoforms).
-
•
-
(f)BAK:
-
•(Ab-1) mAb (requires a secondary antibody conjugated to a fluorophore) Millipore TC-100 (detects human activated BAK isoform).
-
•(NT) mAb (requires a secondary antibody conjugated to a fluorophore) Millipore ABC12 (detects human and murine activated BAK isoforms).
-
•
-
(a)
- IgG control antibodies:
-
(a)Rabbit (DA1E) mAb IgG Isotype Control Alexa(R)647 CST 2985S.
-
(b)Rabbit (DA1E) mAb IgG XP(R) Isotype Control (PE Conjugate) CST 5742S.
-
(c)PE Hamster Anti-Mouse BCL-2 Isotype Control (in kit listed above).
-
(a)
Optional: Secondary antibodies (if primary BCL-2 antibodies are used that are not conjugated to fluorophores) (see Note 3).
Optional: Viability Marker such as LIVE/DEAD Fixable Far Red Dead Cell Stain Kit (ThermoFisher L10120). We routinely use this to gate out late apoptotic/necrotic cells.
1.2 mL FACS tubes.
Flow cytometer (LSRII or similar).
2.2. Mitochondrial Outer Membrane Permeabilization (MOMP)
Cationic dyes such as Rhodamine 123, tetramethylrosamine ethyl ester (TMRE), 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolcarbocyanine iodide (JC-1), or DiOC family dyes can be used for this protocol. Rhodamine 123 at a working solution of450 nM will be used as detailed below (see Note 4).
FCCP (carbonyl cyanide-p-trifluoromethoxyphenylhydra-zone), 1 mM stock.
DMSO or other suitable negative control treatment.
Optional: Viability marker such as LIVE/DEAD Fixable Far Red Dead Cell Stain Kit (ThermoFisher L10120). We routinely use this to gate out late apoptotic/necrotic cells.
1.2 mL FACS tubes.
Flow cytometer (LSRII or similar).
3. Methods
Carry out all procedures at room temperature but keep reagents cold on ice, unless otherwise specified. All washes should be performed by spinning the cells down at 300–400 × g for 5 min and flicking the FACS tubes to dispose of the supernatant.
This protocol is optimized for cells in suspension and not for adherent cells. If adherent cells must be used, trypsinization should be avoided or at least minimized if possible to prevent anoikis-induced cell death [28, 29]. Although trypsinization usually does not affect traditional cell death assays (e.g., MTT, XTT assays), it can affect proteins bound to the cellular membrane and change the permeability profile of cells.
3.1. Intracellular Flow Cytometry for BCL-2 Proteins
Count cells and aliquot up to 1 × 106 cells per treatment into FACS tubes. Save two aliquots of cells for a fixed and unfixed unstained control (see Note 5).
Spin down the cells at 300–400 × g for 5 min and flick the tubes to dispose of the supernatant. Wash cells once with 200 μL PBS.
Optional: Stain cells for extracellular antibodies (see Note 6). After incubation, add 200 μL PBS and spin down, then wash once with 200 μL PBS.
Optional: Add Live/Dead stain. Check the manufacturer’s protocol to ensure that the Live/Dead stain is compatible with permeabilization, such as the LIVE/DEAD Fixable Far Red Stain Kit. For this kit, dilute the Live/Dead stain 1:10,000 in PBS. Add 100 μL to the cells and incubate for 15 min on ice. After incubating, add 400 μL of PBS and spin down and flick to remove the supernatant. Wash cells once with 200 μL PBS and proceed to the fixation step.
Prepare the Fixation/Permeabilization working solution: one part concentrate to three parts diluent (see Note 7).
Add 100 μL of Fixation/Permeabilization solution to the cells. Incubate for 60 min on ice. Keep cells in the dark if they have been stained for extracellular antigens or live/dead.
Dilute the 10× permeabilization buffer with Millipore water. After incubation, add 200 μL of permeabilization buffer to each sample and spin down. Wash once with 200 μL permeabilization buffer.
Prepare antibody solutions for the BCL-2 proteins of interest. In this protocol, antibody solutions are prepared specifically for detection of BCL-XL, MCL-1, BIM, and BCL-2, however, there are other BCL-2 proteins that have validated antibodies for use in flow cytometry (listed in the methods). A typical dilution is 1:50 in permeabilization buffer, but an antibody titration may need to be performed for best results. Follow manufacturer’s protocol closely as some antibody solutions may already be optimized and should not be diluted. For example, the anti-murine BCL-2 antibody listed above comes in a kit that has been optimized and prediluted along with its own isotype control. For this antibody, add 20 μL of the antibody solution straightaway to each sample. Additionally, an IgG isotype control should be prepared for each protein as well, using the same dilutions as the primary antibodies.
Add 50 μL of antibody to each sample (with the exception of the BCL-2 antibody and its control, see previous step). Incubate with intracellular antibodies for 60 min in the dark on ice. After incubation, add 200 μL permeabilization buffer and spin down (see Note 8).
Wash once with 200 μL permeabilization buffer. Resuspend cells in 200 μL FACS buffer.
Analyze via flow cytometry. Start with the unstained controls and adjust voltages so that there is no positive signal from the cells. The cells should be in a roughly uniform population between 0 and 102 in the unstained samples. If staining cells with multiple colors at a time, run single stain or compensation bead controls and adjust compensation accordingly.
Once the voltage and compensation has been set, run the samples. Vortex each sample before loading it onto the sample injection port. Run the samples on low if possible. Try to be consistent with the number of events collected for each sample; a value of 10,000 events per sample is typically sufficient.
Data analysis can be performed on FlowJo, FACSDiva, or a similar flow analysis software. Start with compensation if looking at multiple colors in one sample. For FlowJo, a compensation matrix can typically be generated automatically through the software. Start with the IgG controls and gate out the following: cell type of interest >single cells >live cells (if applicable) (Fig. 3). Once the population of interest has been gated, switch the flow plot to a histogram with the fluorophore of interest on the x-axis. For positive cells, draw a gate that only includes ~1% of the isotype control (Fig. 4). Click and drag the entire gating scheme to the rest of the samples. Repeat this gating strategy for all proteins/fluorophores. (Fig. 4) (see Note 9).
Fig. 3.
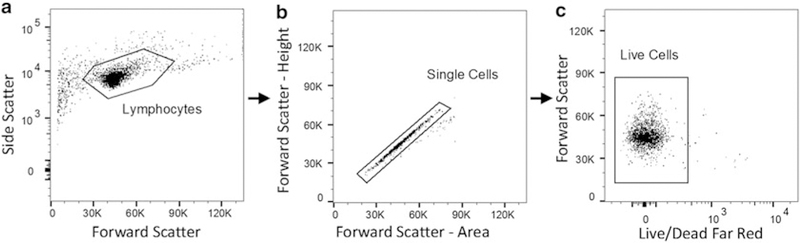
Example gating strategy using FlowJo software. (a) Start with forward scatter by side scatter and draw a gate around the cell type of interest, such as lymphocytes shown here. (b) Next analyze “forward scatter – area” by “forward scatter – height” to exclude any cell doublets. Make a gate around events that fall along the line y = x; these are single cells. If a live/dead stain has been used, draw a final gate around live cells. (c) In this example, dead cells fluoresce in the far red (APC-Cy7) channel, and therefore a gate has been drawn around the live (APC-Cy7Neg) cells
Fig. 4.
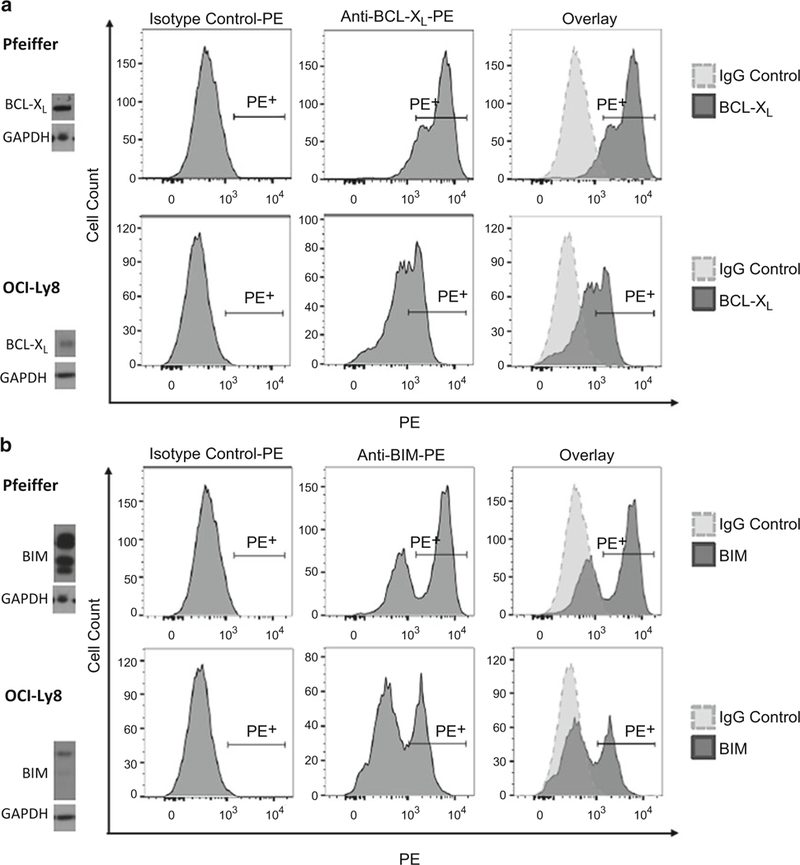
Representative examples of intracellular flow for (a) BCL-XL and (b) BIM in human diffuse large B cell lymphoma cell lines Pfeiffer and OCI-Ly8. The positive gate is drawn using the IgG isotype control (left column). This gate is copied over to the samples incubated with either BCL-XL-PE or BIM-PE antibodies (center column). An overlay of the samples with their isotype controls is shown in the right column. Western blotting of each cell line reveals the relative protein levels of BCL-XL and BIM. As reflected in protein lysate and intracellular flow, Pfeiffer has higher relative amounts of BCL-XL and BIM compared to OCI-Ly8
3.2. Mitochondrial Membrane Depolarization
Following treatment with an apoptotic stimulus as indicated, count cells and resuspend in FACS tubes to a concentration of 1 × 106 cells/mL. Wash cells once with 200 μl PBS.
Aliquot two tubes of cells for a negative (no treatment; polarized membrane) and positive (total mitochondrial membrane depolarization) control.
Optional: Add Live/Dead stain. If permeabilizing the cells, check the manufacturer’s protocol to ensure that the Live/ Dead stain is compatible with permeabilization, such as the LIVE/DEAD Fixable Far Red Stain Kit. If using this kit, dilute the Live/Dead stain 1:10,000 in PBS. Add 100 μL to the cells and incubate for 15 min on ice. After incubating, add 400 μL of PBS and spin down and flick to remove the supernatant. Wash cells once with 200 μL PBS and proceed to the next step.
Add 100 μL of PBS to each sample. For the negative control, treat with DMSO or another suitable control solution. Treat cells with 2 μL of 1 mM FCCP as a positive control. FCCP is a respiratory uncoupler that quickly depolarizes the mitochondrial membrane. Depolarization should be complete by the time the cells are done staining (see Note 10).
Stain the cells with 25 μL of450 nM rhodamine 123 for 30 min at room temperature in the dark. Do not wash prior to flow cytometric analysis.
Analyze via flow cytometry. Start with the unstained controls and adjust voltages so that there is no positive signal from the cells. The cells should be in a roughly uniform population between 0 and 102 in the unstained samples. If staining cells with multiple colors at a time, run single stain or compensation bead controls and adjust compensation accordingly.
Once the voltage and compensation has been set, run the samples. Vortex each sample before loading it onto the sample injection port. Run the samples on low if possible. Try to be consistent with the number ofevents collected for each sample; a value of 10,000 events per sample is typically sufficient.
Data analysis can be performed on FlowJo, FACSDiva, or a similar flow analysis software. Start with compensation when looking at multiple colors in one sample. For FlowJo, a compensation matrix can typically be generated automatically through the software. Start with the positive and negative control samples. The DMSO treated sample (negative control) can be used to generate a “Polarized” gate and the FCCP treated sample (positive control) can be used to generate a “Depolarized” gate. Remaining samples should either overlap with one of these two samples or fall somewhere in between if partial membrane depolarization occurred (Fig. 5).
Fig. 5.
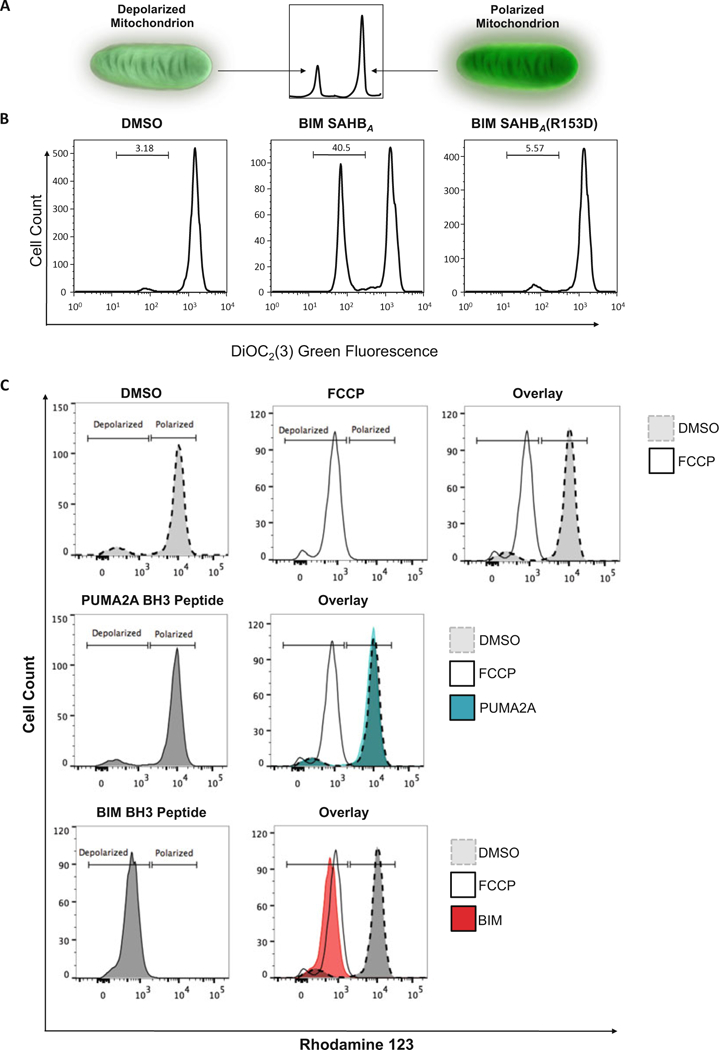
Measurement of mitochondrial outer membrane permeabilization (MOMP). (a) The general principle of how cationic dyes are used to measure MOMP is shown. As the membrane depolarizes and loses its negative charge, the cationic dyes will no longer associate with the mitochondria and the fluorescent signal will be lost. (b and c) Two examples of MOMP measurement are shown. (b) In the first example, MOMP is measured after treating cells with a stabilized alpha helix of BCL-2 domain (SAHB) modeled after the BH3-only protein BIM, which is known to induce apoptosis [30–33]. Treatment with BIM SAHBA (middle) causes partial membrane depolarization compared to DMSO (left) while treatment with a point mutant control, BIM SAHBA(R153D), results in no depolarization. (c) The second example demonstrates the use of BH3 peptides optimized to test the dependency of different cell types on specific anti-apoptotic proteins, a technique known as BH3 profiling [27, 34]. DMSO and FCCP are used as negative and positive controls respectively. These controls are overlaid and used as a reference guide in the measurement of the level of depolarization from other BH3 peptide treatments. The PUMA2A peptide serves as a negative control and does not cause membrane depolarization. BIM BH3 uniformly leads to complete depolarization in all cell types, and therefore overlaps with the FCCP treated sample
4. Notes
Sodium azide is useful to help preserve antibody–antigen interactions; however, it also affects cell viability. We have success in performing these assays in FACS buffer without sodium azide, and therefore it is listed as an optional addition.
These antibodies are to be used in conjunction with other fluorophores if the need to use additional colors arises. If already using the PE and Alexa Fluor 647 channels for other antibodies, different fluorophores can be used, or unconjugated antibodies can be used with a number of secondary antibody-conjugated fluorophores. These may need to be titrated and optimized for best results. When choosing anti-bodies for flow panels, be aware of the available lasers on the cytometer being used, and be sure that the fluorophores have an emission frequency that will be detected by the cytometer.
For primary antibodies conjugated to fluorophores, use IgG controls conjugated to the same fluorophore. If using unconjugated primary antibodies followed by incubation with a secondary antibody conjugated to a fluorophore, set aside a sample as a “no primary” control. Incubate this sample with only the secondary antibody.
Other dyes can be substituted for rhodamine 123, such as JC-1, TMRE, and DiOC family dyes. Concentration of the dyes may need to be adjusted for best results.
If there is a small amount of cells being tested, the unstained control samples do not have to be at the same density as the other samples. These samples will be used to adjust the voltages of the cytometer and ensure the fixation and permeabilization steps were effective.
If there are samples with multiple cell types of interest and there are plans to stain for cell surface markers, stain extracellular antigens as a separate step prior to fixation. A typical stain time would be 30 min on ice. Once the staining process has begun, the cells should be kept in the dark whenever possible.
Fixation and permeabilization will cause the cells to become smaller, therefore expect a shift to the left on the forward/side scatter plot during analysis.
This protocol was performed using antibodies already conjugated to fluorophores. If an unconjugated antibody is used an additional incubation step will be required for the conjugated secondary antibody.
Validation of results using Western blot analysis is a good way to confirm that the intracellular flow is working properly. If using limited number of cells, antibody validation can be performed on other cell types (e.g., immortalized cell lines). There are many cell lines in which the BCL-2 protein levels have been described. These can be useful tools when checking antibody optimization.
If there is overlap between the DMSO and FCCP treated controls, and the FCCP-treated sample is still emitting a strong positive signal, the mitochondrial membrane may not be fully depolarized. A longer incubation with the FCCP prior to staining or a higher concentration of FCCP can be used for larger peak separation.
Acknowledgment
We would like to thank Eric E. Smith for graphics in Fig. 2 and 5.
References
- 1.Cotter TG (2009) Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer 9 (7):501–507 [DOI] [PubMed] [Google Scholar]
- 2.Kerr JF, Wyllie AH, Currie AR(1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26(4):239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Czabotar PE, Lessene G, Strasser A, Adams JM (2014) Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15(1):49–63 [DOI] [PubMed] [Google Scholar]
- 4.Ludwig LM, Nassin ML, Hadji A, LaBelle JL (2016) Killing two cells with one stone: pharmacologic BCL-2 family targeting for cancer cell death and immune modulation. Front Pediatr 4:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S (1991) The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 66(2):233–243 [DOI] [PubMed] [Google Scholar]
- 6.Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116(2):205–219 [DOI] [PubMed] [Google Scholar]
- 7.Fukuhara S, Rowley JD (1978) Chromosome 14 translocations in non-Burkitt lymphomas. Int J Cancer 22(1):14–21 [DOI] [PubMed] [Google Scholar]
- 8.Tsujimoto Y, Yunis J, Onorato-Showe L, Erikson J, Nowell PC, Croce CM (1984) Molecular cloning of the chromosomal breakpoint of B-cell lymphomas and leukemias with the t(11;14) chromosome translocation. Science 224(4656):1403–1406 [DOI] [PubMed] [Google Scholar]
- 9.Vaux DL, Cory S, Adams JM (1988) Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335(6189):440–442 [DOI] [PubMed] [Google Scholar]
- 10.Delbridge AR, Grabow S, Strasser A, Vaux DL (2016) Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer 16(2):99–109 [DOI] [PubMed] [Google Scholar]
- 11.Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH (2009) Stepwise activation of bAx and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell 36(3):487–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Happo L, Strasser A, Cory S (2012) BH3-only proteins in apoptosis at a glance. J Cell Sci 125 (Pt 5):1081–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor RC, Cullen SP, Martin SJ (2008) Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 9(3):231–241 [DOI] [PubMed] [Google Scholar]
- 14.Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Castedo M, Cidlowski JA, Ciechanover A, Cohen GM, De Laurenzi V, De Maria R, Deshmukh M, Dynlacht BD, El-Deiry WS, Flavell RA, Fulda S, Garrido C, Golstein P, Gougeon ML, Green DR, Gronemeyer H, Hajnoczky G, Hardwick JM, Hengartner MO, Ichijo H, Jaattela M, Kepp O, Kimchi A, Klionsky DJ, Knight RA, Kornbluth S, Kumar S, Levine B, Lipton SA, Lugli E, Madeo F, Malomi W, Marine JC, Martin SJ, Medema jP, Mehlen P, Melino G, Moll UM, Morselli E, Nagata S, Nicholson DW, Nicotera P, Nunez G, Oren M, Penninger J, Pervaiz S, Peter ME, Piacentini M, Prehn JH, Puthalakath H, Rabinovich GA, Rizzuto R, Rodrigues CM, Rubinsztein DC, Rudel T, Scorrano L, Simon HU, Steller H, Tschopp J, Tsujimoto Y, Vandenabeele P, Vitale I, Vousden KH, Youle RJ, Yuan J, Zhivotovsky B, Kroemer G (2009) Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 16(8):1093–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G (2011) Cell death assays for drug discovery. Nat Rev Drug Discov 10(3):221–237 [DOI] [PubMed] [Google Scholar]
- 16.Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ (2017) From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov 16(4):273–284 [DOI] [PubMed] [Google Scholar]
- 17.Opferman JT (2016) Attacking cancer’s Achilles heel: antagonism of anti-apoptotic BCL-2 family members. FEBS J 283(14):2661–2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elmore SP, Nishimura Y, Qian T, Herman B, Lemasters JJ (2004) Discrimination of depolarized from polarized mitochondria by confocal fluorescence resonance energy transfer. Arch Biochem Biophys 422(2):145–152 [DOI] [PubMed] [Google Scholar]
- 19.Gottlieb E, Armour SM, Harris MH, Thompson CB (2003) Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ 10(6):709–717 [DOI] [PubMed] [Google Scholar]
- 20.Dewson G, Kluck RM (2009) Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci 122 (Pt 16):2801–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, Mac Gregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292(5517):727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rego AC, Vesce S, Nicholls DG (2001) The mechanism of mitochondrial membrane potential retention following release of cytochrome c in apoptotic GT1–7 neural cells. Cell Death Differ 8(10):995–1003 [DOI] [PubMed] [Google Scholar]
- 23.Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW, Lindsten T, Thompson CB, Crow MT, Gavathiotis E, Dorn GW 2nd, O’Rourke B, Kitsis RN (2012) Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A 109(17):6566–6571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen LB (1988) Mitochondrial membrane potential in living cells. Annu Rev Cell Biol 4:155–181 [DOI] [PubMed] [Google Scholar]
- 25.Cottet-Rousselle C, Ronot X, Leverve X, Mayol JF (2011) Cytometric assessment of mitochondria using fluorescent probes. Cytometry A 79(6):405–425 [DOI] [PubMed] [Google Scholar]
- 26.Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB (1995) Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol 260:406–417 [DOI] [PubMed] [Google Scholar]
- 27.Ryan J, Letai A (2013) BH3 profiling in whole cells by fluorimeter or FACS. Methods 61 (2):156–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engbers-Buijtenhuijs P, Kamphuis M, van der Sluijs VG, Haanen C, Poot AA, Feijen J, Vermes I (2005) A novel time resolved fluoro-metric assay of anoikis using europium-labelled Annexin V in cultured adherent cells. Apoptosis 10(2):429–437 [DOI] [PubMed] [Google Scholar]
- 29.Paoli P, Giannoni E, Chiarugi P (2013) Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta 1833 (12):3481–3498 [DOI] [PubMed] [Google Scholar]
- 30.Bird GH, Gavathiotis E, LaBelle JL, Katz SG, Walensky LD (2014) Distinct BimBH3 (Bim-SAHB) stapled peptides for structural and cellular studies. ACS Chem Biol 9(3):831–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edwards AL, Wachter F, Lammert M, Huhn AJ, Luccarelli J, Bird GH, Walensky LD (2015) Cellular uptake and Ultrastructural localization underlie the pro-apoptotic activity of a hydrocarbon-stapled BIM BH3 peptide. ACS Chem Biol 10(9):2149–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C, Fisher JK, Godes M, Pitter K, Kung AL, Walensky LD (2012) A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest 122(6):2018–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reynolds C, Roderick JE, LaBelle JL, Bird G, Mathieu R, Bodaar K, Colon D, Pyati U, Stevenson KE, Qi J, Harris M, Silverman LB, Sallan SE, Bradner JE, Neuberg DS, Look AT, Walensky LD, Kelliher MA, Gutierrez A (2014) Repression of BIM mediates survival signaling by MYC and AKT in high-risk T-cell acute lymphoblastic leukemia. Leukemia 28 (9):1819–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Certo M, Del Gaizo MV, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A (2006) Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9 (5):351–365 [DOI] [PubMed] [Google Scholar]