

Abstract
Inflammation is often associated with the development and progression of cancer. The cells responsible for cancer-associated inflammation are genetically stable and thus are not subjected to rapid emergence of drug resistance; therefore, the targeting of inflammation represents an attractive strategy both for cancer prevention and for cancer therapy. Tumor-extrinsic inflammation is caused by many factors, including bacterial and viral infections, autoimmune diseases, obesity, tobacco smoking, asbestos exposure, and excessive alcohol consumption, all of which increase cancer risk and stimulate malignant progression. In contrast, cancer-intrinsic or cancer-elicited inflammation can be triggered by cancer-initiating mutations and can contribute to malignant progression through the recruitment and activation of inflammatory cells. Both extrinsic and intrinsic inflammations can result in immunosuppression, thereby providing a preferred background for tumor development. The current review provides a link between inflammation and cancer development.
Keywords: Cancer, cells, inflammation, Cancer, cellules, inflammation
Résumé
L’inflammation est souvent associée au développement et à la progression du cancer. Les cellules responsables de l’inflammation associée au cancer sont génétiquement stables et ne subissent donc pas l’émergence rapide d’une pharmacorésistance; par conséquent, le ciblage de l’inflammation représente une stratégie attrayante à la fois pour la prévention du cancer et pour le traitement du cancer. L’inflammation tumorale extrinsèque est causée par de nombreux facteurs, notamment: infections bactériennes et virales, maladies auto-immunes, obésité, tabagisme, exposition à l’amiante et consommation excessive d’alcool, le tout qui augmentent le risque de cancer et stimulent la progression maligne. En revanche, l’inflammation intrinsèque au cancer ou provoquée par le cancer peut être déclenchée par des mutations initiant un cancer et peuvent contribuer à la progression maligne par le recrutement et l’activation de cellules inflammatoires. Tous les deux les inflammations extrinsèques et intrinsèques peuvent entraîner une immunosuppression, fournissant ainsi un fond préféré pour le développement de la tumeur. le l’examen actuel établit un lien entre l’inflammation et le développement du cancer.
INTRODUCTION
The presence of leukocytes within tumors, observed in the 19th century by Rudolf Virchow, provided the first indication of a possible link between inflammation and cancer. Yet, it is only during the past decade that clear evidence has been obtained that inflammation plays a critical role in tumorigenesis.[1]
However, when inflammation becomes chronic or lasts too long, it can prove harmful and may lead to disease. The role of pro-inflammatory cytokines, chemokines, adhesion molecules, and inflammatory enzymes has been linked with chronic inflammation [Figure 1].[2]
Figure 1.
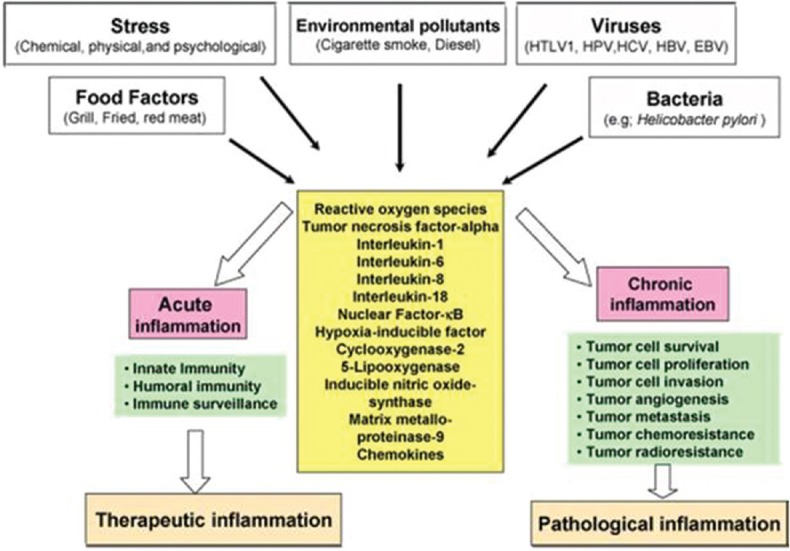
Different faces of inflammation and its role in tumorigenesis
Chronic inflammation has been found to mediate a wide variety of diseases, including cardiovascular diseases, cancer, diabetes, arthritis, Alzheimer's disease, pulmonary diseases, and autoimmune diseases.[3]
The current review, however, will be restricted to the role of chronic inflammation in cancer. Chronic inflammation has been linked to various steps involved in tumorgenesis, including cellular transformation, promotion, survival, proliferation, invasion, angiogenesis, and metastasis.[4]
Only a minority of all cancers are caused by germline mutations, whereas the vast majority (90%) are linked to somatic mutations and environmental factors. Many environmental causes of cancer and risk factors are associated with some form of chronic inflammation. Up to 20% of cancers are linked to chronic infections, 30% can be attributed to tobacco smoking and inhaled pollutants (such as silica and asbestos), and 35% can be attributed to dietary factors (20% of cancer burden is linked to obesity).[5]
Recent efforts have shed new light on molecular and cellular circuits linking inflammation and cancer. Two pathways have been schematically identified: in the intrinsic pathway, genetic events causing neoplasia initiate the expression of inflammation-related programs that guide the construction of an inflammatory microenvironment, and in the extrinsic pathway, inflammatory conditions facilitate cancer development.[6]
The triggers of chronic inflammation that increase cancer risk or progression include infections (e.g., Helicobacter pylori for gastric cancer and mucosal lymphoma; papillomavirus and hepatitis viruses for cervical and liver carcinomas, respectively), autoimmune diseases (e.g., inflammatory bowel disease for colon cancer), and inflammatory conditions of uncertain origin (e.g., prostatitis for prostate cancer). Cancer-related inflammation, the seventh hallmark of cancer, links to genetic instability.[7]
It was in 1863 that Rudolf Virchow noted leukocytes in neoplastic tissues and made a connection between inflammation and cancer. He suggested that the “lymphoreticular infiltrate” reflected the origin of cancer at sites of chronic inflammation. Over the past 10 years, our understanding of the inflammatory microenvironment of malignant tissues has supported Virchow's hypothesis, and the links between cancer and inflammation are starting to have implications for prevention and treatment.[8]
INFLAMMATION AND CAUSES
Inflammation is the body's response to tissue damage, caused by physical injury, ischemic injury (caused by an insufficient supply of blood to an organ), infection, exposure to toxins, or other types of trauma. The body's inflammatory response causes cellular changes and immune responses that result in repair of the damaged tissue and cellular proliferation (growth) at the site of the injured tissue. Inflammation can become chronic if the cause of the inflammation persists or certain control mechanisms in charge of shutting down the process fail. When these inflammatory responses become chronic, cell mutation and proliferation can result, often creating an environment that is conducive to the development of cancer. The so-called “perfect storm” is an extreme challenge that cancer patients face. This is true for the onset of cancer but also even more important for the advancement of the disease. Various signaling pathways are key contributors in creating epigenetic changes on the outside of the cell, switching on these internal mutations. Therefore, treating the inflammatory causes is always important.
Chronic inflammation has been linked to various steps involved in tumorigenesis, including cellular transformation, promotion, survival, proliferation, invasion, angiogenesis, and metastasis.
CANCER DEVELOPMENT: AN OVERVIEW
Cancer defines malignant neoplasms characterized by metastatic growth. It may occur in almost every organ and tissue relating to a variety of etiologic factors, such as genomic instability and environmental stress.[9]
However, cancer development is still accepted as a multistep process, during which genetic alterations confer specific types of growth advantages; therefore, it drives the progressive transformation from normal cells to malignant cancer cells. Malignant growth is characterized by several key changes: self-sufficiency of growth signals, insensitivity to antigrowth signals, escaping from apoptosis, unregulated proliferation potential, enhanced angiogenesis, and metastasis. Each of these shifts is complicated and accomplished by combined efforts of various signaling processes. In later discussion, we will find that inflammation may contribute to the formation of these cancer phenotypes.[10]
MECHANISMS FOR THE ASSOCIATION BETWEEN INFLAMMATION AND CANCER
Chronic inflammation is characterized by sustained tissue damage, damage-induced cellular proliferation, and tissue repair. Cell proliferation in this context is usually correlated with “metaplasia,” a reversible change in cell type. “Dysplasia,” a disorder of cellular proliferation leading to atypical cell production, follows and is regarded as the previous event of carcinoma because it was usually found adjacent to the site of neoplasm.[11]
MUTAGENIC POTENTIAL OF INFLAMMATION
The chronic inflammatory microenvironment is predominated by macrophages. Those macrophages, together with other leukocytes, generate high levels of reactive oxygen and nitrogen species to fight infection.[12] However, in a setting of continuous tissue damage and cellular proliferation, the persistence of these infection-fighting agents is deleterious. They may produce mutagenic agents, such as peroxynitrite, which react with DNA and cause mutations in proliferating epithelial and stroma cells. Macrophages and T-lymphocytes may release tumor necrosis factor-alpha (TNF-α) and macrophage migration inhibitory factor to exacerbate DNA damage.[13]
Migration inhibitory factor impairs p53-dependent protective responses, thus causing the accumulation of oncogenic mutations. Migration inhibitory factor also contributes to tumorigenesis by interfering Rb-E2F pathway.
HELICOBACTER PYLORIAND AND CANCER RISK
The bacterium H. pylori is known to colonize the human stomach and induce chronic atrophic gastritis, intestinal metaplasia, and gastric cancer. H. pylori infection is a major risk factor for gastric cancer development, which is one of the most challenging malignant diseases worldwide with limited treatments.[14]
The multistep pathogenesis of gastric cancer is the best highlighted by Correa sequence that explains the progressive pathway to gastric cancer characterized by distinct histological changes. This model predicts that infection with H. pylori triggers an inflammatory response resulting in chronic, and then, atrophic, gastritis. This is followed by intestinal metaplasia which can be further classified into complete and incomplete subtypes. At this point, some patients will then proceed to gastric cancer via the intermediate stage of dysplasia [Figure 2].[15]
Figure 2.
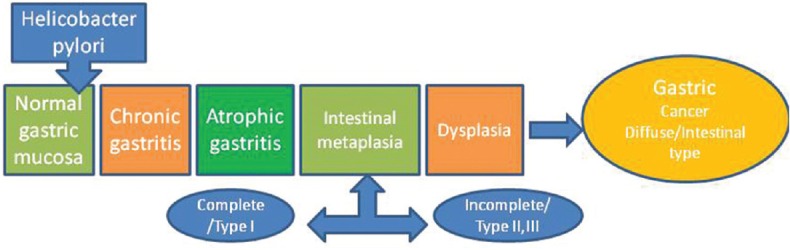
Correa sequence
The improvement or elimination of atrophy and intestinal metaplasia with H. pylori eradication could potentially inhibit gastric carcinogenesis. It is noteworthy to mention that gastric cancer can still develop even after successful eradication therapy. H. pylori eradication does not result in the regression of all precancerous lesions, which may depend on the degree and extent of preneoplastic changes at the time of eradication.[14]
INFLAMMATORY CELLS IN TUMOR MICROENVIRONMENT
The inflammatory microenvironment of tumors is characterized by the presence of host leukocytes both in the supporting stroma and in tumor areas.[16] Tumor-infiltrating lymphocytes may contribute to cancer growth and spread and to the immunosuppression associated with malignant disease.
Macrophages
Tumor-associated macrophages (TAM) are a major component of the infiltrate of most, if not all tumors. TAM derives from circulating monocytic precursors and is directed into the tumor by chemoattractant cytokines called chemokines. Many tumor cells also produce cytokines called colony-stimulating factors that prolong the survival of TAM. When appropriately activated, TAM can kill tumor cells or elicit tissue destructive reactions centered on the vascular endothelium. However, TAM also produces growth and angiogenic factors as well as protease enzymes which degrade the extracellular matrix. Hence, TAM can stimulate tumor cell proliferation, promote angiogenesis, and favor invasion and metastasis.[17]
Dendritic cells
Dendritic cells have a crucial role in both the activation of antigen-specific immunity and the maintenance of tolerance, providing a link between innate and adaptive immunity. Tumor-associated dendritic cells (TADCs) usually have an immature phenotype with defective ability to stimulate T-cells.[18]
This distribution of TADC is clearly different from that of TAM, which is evenly scattered in tumor tissue. The immaturity of TADC may reflect lack of effective maturation signals, prompt migration of mature cells to lymph nodes, or the presence of maturation inhibitors. TADC is likely to be poor inducers of effective responses to tumor antigens.
Lymphocytes
Natural killer cells are rare in the tumor microenvironment. The predominant T-cell population has a “memory” phenotype. The cytokine profile of these tumor-infiltrating T-cells has not been studied systematically, but in some tumors (e.g. Kaposi's sarcoma, Hodgkin's disease, bronchial carcinoma, and cervical carcinoma), they produce mainly interleukins (ILs) 4 and 5 and not interferon. IL-4 and 5 are cytokines associated with the T-helper type 2 (Th2) cells, whereas interferon is associated with Th1 responses.[19]
KEY MOLECULAR PLAYERS IN LINKING INFLAMMATION TO CANCER
To address the details of transition from inflammation to cancers and the further development of inflammation-associated cancers, it is necessary to investigate specific roles of key regulatory molecules involved in this process.
Pro-inflammatory cytokines
The cytokine network of several common tumors is rich in inflammatory cytokines, growth factors, and chemokines but generally lacks cytokines involved in specific and sustained immune responses.[20]
There is now evidence that inflammatory cytokines and chemokines, which can be produced by the tumor cells and/or tumor-associated leukocytes and platelets, may contribute directly to malignant progression. Many cytokines and chemokines are inducible by hypoxia, which is a major physiological difference between tumor and normal tissue. Examples are TNF, IL-1 and 6, and chemokines.
The immune response to tumors is constituted by cytokines produced by tumor cells as well as host stromal cells. Tumor-derived cytokines, such as Fas ligand, vascular endothelial growth factor (VEGF), and transforming growth factor-h, may facilitate the suppression of immune response to tumors. Moreover, inflammatory cytokines have also been reported to facilitate the spectrum of tumor development.[21]
Tumor necrosis factor
TNF is a multifunctional cytokine that plays important roles in diverse cellular events such as cell survival, proliferation, differentiation, and death. As a pro-inflammatory cytokine, TNF is secreted by inflammatory cells, which may be involved in inflammation-associated carcinogenesis. TNF exerts its biological functions through activating distinct signaling pathways such as nuclear factor-κB (NF-κB) and c-Jun N-terminal kinase (JNK). NF-κB is a major cell survival signal that is antiapoptotic while sustained JNK activation contributes to cell death. The crosstalk between the NF-κB and JNK is involved in determining cellular outcomes in response to TNF. TNF is a double-edged sword that could be either pro- or antitumorigenic. On one hand, TNF could be an endogenous tumor promoter because TNF stimulates cancer cells' growth, proliferation, invasion and metastasis, and tumor angiogenesis. On the other hand, TNF could be a cancer killer. The property of TNF in inducing cancer cell death renders it a potential cancer therapeutic.[22]
TNF can be detected in malignant and/or stromal cells in human ovarian, breast, prostate, bladder, and colorectal cancer, lymphomas, and leukemias, often in association with ILs-1 and 6 and macrophage colony-stimulating factor.[23]
Interleukins 1 and 6 in cancer regulation
IL-6 is a pleiotropic cytokine that plays important roles in immune response, inflammation, and hematopoiesis. It is produced by a variety of normal cells including monocytes and macrophages but is also expressed by multiple tumor tissue types, such as breast, prostate, colorectal, and ovarian cancer. IL-6 may also play an important role in various aspects of tumor behavior, including apoptosis, tumor growth cell proliferation, migration and invasion, angiogenesis, and metastasis.[24]
IL-10, initially termed “cytokine synthesis inhibitor” or “cytokine inhibitory factor” due to its inhibitory action on cytokine production by T helper cells, is produced by almost all leukocytes, as well as numerous human tumor cells including breast, kidney, colon, pancreas, malignant melanomas, and neuroblastomas. IL-10 is essential to suppress tumor-promoting inflammation mediators, thereby facilitating tumor growth and metastasis. Specifically, TAMs produce IL-10 and are also associated with in-tumor immunosuppression, thereby providing a suitable microenvironment for cancer growth.[25]
In mouse models of metastasis, treatment with an IL-1 receptor antagonist (which inhibits the action of IL-1) significantly decreased tumor development, suggesting that local production of this cytokine aids the development of metastasis. Moreover, mice deficient in IL-1 were resistant to the development of experimental metastasis.[26]
Chemokines
Inflammatory cytokines are major inducers of a family of chemoattractant cytokines called chemokines that play a central role in leukocyte recruitment to sites of inflammation. Most tumors produce chemokines of the two major groups α (or CXC) and β.
Typically, CXC chemokines are active on neutrophils and lymphocytes, whereas CC chemokines act on several leukocyte subsets including monocytes, eosinophils, dendritic cells, lymphocytes, and natural killer cells but not neutrophils.[27]
Human and murine tumors also frequently secrete CXC chemokines such as IL-8. These chemokines are potent neutrophil attractants, yet neutrophils are rare in tumors. However, both IL-8 and a related chemokine called “gro” induce proliferation and migration of melanoma cell.
IMPLICATIONS FOR PREVENTION AND TREATMENT
Tumor necrosis factor blockade
TNF antagonists (etanercept [Enbrel] and infliximab [Remicade]) have been licensed for a clinical trial in the treatment of rheumatoid arthritis and Crohn's disease, with over 70,000 patients now treated. Thalidomide inhibits the processing of mRNA for TNF and VEGF, and continuous low-dose thalidomide has shown activity in patients with advanced myeloma. The role of etanercept in ameliorating the adverse effects of other cancer therapies is also being evaluated. There are also ongoing and planned clinical trials with infliximab. As with other “biological” approaches to cancer treatment, anti-TNF therapy may be optimal in an adjuvant setting with minimal disease.[28]
Chemokine antagonism
Chemokine receptors belong to a family of receptors (transmembrane G-protein-coupled receptors) which is already a target of pharmacological interest. Tumors driven by chemokines and those where chemokines are implicated in metastasis (e.g. seeding to lymph nodes) may be an appropriate target for chemokine antagonists now under development.[29]
IL-6 is a major growth factor for myeloma cells. In advanced disease, there is an excess of IL-6 production, and raised serum concentrations are associated with plasmablastic proliferative activity and short survival.
Nonsteroidal anti-inflammatory agents
Nonsteroidal anti-inflammatory drugs (NSAIDs) are nonselective or selective COX-1/2 inhibitors, which are wildly prescribed for pain killing, fever reduction, and even anti-inflammation.
Patients on NSAIDs are at reduced risk of colon cancer. This may also be true for cancers of the esophagus, stomach, and rectum, and in rodents experimental bladder, breast, and colon cancer. Colon cancer is reduced when NSAIDs are administered concurrently with carcinogens. NSAIDs inhibit cyclooxygenase enzymes and angiogenesis.[30]
The mechanisms involved in the association between NSAIDs and distant metastasis inhibition remain incompletely investigated. One possible explanation is that NSAIDs inhibit COX2. Abnormally high COX2 expression is observed in multicancers. Disordered COX2/PGE pathway is involved in multicancer processes, including carcinogenesis, proliferation, and metastatic spread; in addition, inhibition of COX2/PGE pathway with NSAIDs can restrain cancer cell lines.
Mutual promotion relationship between cancer metastasis and cancer-associated thrombosis is possibly another one of the underlying mechanisms. Abnormally high constitutive level of tissue factor (TF), one key regulator of hemostasis, is expressed by metastatic cancer cells, cancer microparticles, and cancer-associated monocytes and macrophages. TF can promote thrombosis formation by activating the extrinsic pathway of coagulation cascade. Furthermore, inflammation induced by thrombosis could result in endothelial damage that results in the vascular leak, facilitating the escape of cancer cells from blood vessels. Consequently, NSAIDs may disrupt the relationship between cancer metastasis and cancer-associated thrombosis via the suppression of platelet function, which is detrimental for the disseminated cancer cells in the bloodstream.[31]
CONCLUSION
Overall, this review provides evidence for a strong link between chronic inflammation and cancer. Thus, inflammatory biomarkers as described here can be used to monitor the progression of the disease. These biomarkers can also be exploited to develop new anti-inflammatory drugs to prevent and treat cancer. These drugs can also be used as an adjuvant to the currently available chemotherapy and radiotherapy, which by themselves activate NF-κB and mediate resistance. Numerous anti-inflammatory agents including those identified from natural sources have been shown to exhibit chemopreventive activities.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: How hot is the link? Biochem Pharmacol. 2006;72:1605–21. doi: 10.1016/j.bcp.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 3.Aggarwal BB. Nuclear factor-kappaB: The enemy within. Cancer Cell. 2004;6:203–8. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 4.Mantovani A. Cancer: Inflammation by remote control. Nature. 2005;435:752–3. doi: 10.1038/435752a. [DOI] [PubMed] [Google Scholar]
- 5.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, long-term foe. Clin Cancer Res. 2009;15:425–30. doi: 10.1158/1078-0432.CCR-08-0149. [DOI] [PubMed] [Google Scholar]
- 7.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 8.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis. 2009;30:1073–81. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 9.Balkwill F, Mantovani A. Inflammation and cancer: Back to virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 10.Philip M, Rowley DA, Schreiber H. Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol. 2004;14:433–9. doi: 10.1016/j.semcancer.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Cordon-Cardo C, Prives C. At the crossroads of inflammation and tumorigenesis. J Exp Med. 1999;190:1367–70. doi: 10.1084/jem.190.10.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maeda H, Akaike T. Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochemistry (Mosc) 1998;63:854–65. [PubMed] [Google Scholar]
- 13.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 14.Lu B, Li M. Helicobacter pylori eradication for preventing gastric cancer. World J Gastroenterol. 2014;20:5660–5. doi: 10.3748/wjg.v20.i19.5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Busuttil RA, Zapparoli GV, Haupt S, Fennell C, Wong SQ, Pang JM, et al. Role of p53 in the progression of gastric cancer. Oncotarget. 2014;5:12016–26. doi: 10.18632/oncotarget.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Negus RP, Stamp GW, Hadley J, Balkwill FR. Quantitative assessment of the leukocyte infiltrate in ovarian cancer and its relationship to the expression of C-C chemokines. Am J Pathol. 1997;150:1723–34. [PMC free article] [PubMed] [Google Scholar]
- 17.Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L. The origin and function of tumor-associated macrophages. Immunol Today. 1992;13:265–70. doi: 10.1016/0167-5699(92)90008-U. [DOI] [PubMed] [Google Scholar]
- 18.Allavena P, Sica A, Vecchi A, Locati M, Sozzani S, Mantovani A. The chemokine receptor switch paradigm and dendritic cell migration: Its significance in tumor tissues. Immunol Rev. 2000;177:141–9. doi: 10.1034/j.1600-065x.2000.17714.x. [DOI] [PubMed] [Google Scholar]
- 19.Burke F, Relf M, Negus R, Balkwill F. A cytokine profile of normal and malignant ovary. Cytokine. 1996;8:578–85. doi: 10.1006/cyto.1996.0077. [DOI] [PubMed] [Google Scholar]
- 20.Smyth MJ, Cretney E, Kershaw MH, Hayakawa Y. Cytokines in cancer immunity and immunotherapy. Immunol Rev. 2004;202:275–93. doi: 10.1111/j.0105-2896.2004.00199.x. [DOI] [PubMed] [Google Scholar]
- 21.Naylor MS, Stamp GW, Foulkes WD, Eccles D, Balkwill FR. Tumor necrosis factor and its receptors in human ovarian cancer. Potential role in disease progression. J Clin Invest. 1993;91:2194–206. doi: 10.1172/JCI116446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Lin Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol Sin. 2008;29:1275–88. doi: 10.1111/j.1745-7254.2008.00889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vidal-Vanaclocha F, Fantuzzi G, Mendoza L, Fuentes AM, Anasagasti MJ, Martín J, et al. IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc Natl Acad Sci U S A. 2000;97:734–9. doi: 10.1073/pnas.97.2.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hassuneh MR, Nagarkatti M, Nagarkatti PS. Role of interleukin-10 in the regulation of tumorigenicity of a T cell lymphoma. Leuk Lymphoma. 2013;54:827–34. doi: 10.3109/10428194.2012.726721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmad N, Ammar A, Storr SJ, Green AR, Rakha E, Ellis IO, et al. IL-6 and IL-10 are associated with good prognosis in early stage invasive breast cancer patients. Cancer Immunol Immunother. 2018;67:537–49. doi: 10.1007/s00262-017-2106-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Negus RP, Stamp GW, Relf MG, Burke F, Malik ST, Bernasconi S, et al. The detection and localization of monocyte chemoattractant protein-1 (MCP-1) in human ovarian cancer. J Clin Invest. 1995;95:2391–6. doi: 10.1172/JCI117933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maini RN, Taylor PC. Anti-cytokine therapy for rheumatoid arthritis. Annu Rev Med. 2000;51:207–29. doi: 10.1146/annurev.med.51.1.207. [DOI] [PubMed] [Google Scholar]
- 28.Haghnegahdar H, Du J, Wang D, Strieter RM, Burdick MD, Nanney LB, et al. The tumorigenic and angiogenic effects of MGSA/GRO proteins in melanoma. J Leukoc Biol. 2000;67:53–62. doi: 10.1002/jlb.67.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tricot G. New insights into role of microenvironment in multiple myeloma. Lancet. 2000;355:248–50. doi: 10.1016/S0140-6736(00)00019-2. [DOI] [PubMed] [Google Scholar]
- 30.Thun MJ, Namboodiri MM, Calle EE, Flanders WD, Heath CW., Jr Aspirin use and risk of fatal cancer. Cancer Res. 1993;53:1322–7. [PubMed] [Google Scholar]
- 31.Zhao X, Xu Z, Li H. NSAIDs use and reduced metastasis in cancer patients: Results from a meta-analysis. Sci Rep. 2017;7:1875. doi: 10.1038/s41598-017-01644-0. [DOI] [PMC free article] [PubMed] [Google Scholar]