

Abstract
Several behavioral effects of nicotine are mediated by changes in serotonin (5-HT) release in brain areas that receive serotonergic afferents from the dorsal raphe nucleus (DRN). In vitro experiments have demonstrated that nicotine increases the firing activity in the majority of DRN 5-HT neurons and that DRN contains nicotinic acetylcholine receptors (nAChRs) located at both somata and presynaptic elements. One of the most common presynaptic effects of nicotine is to increase glutamate release. Although DRN receives profuse glutamatergic afferents, the effect of nicotine on glutamate release in the DRN has not been studied in detail. Using whole-cell recording techniques, we investigated the effects of nicotine on the glutamatergic input to 5-HT DRN neurons in rat midbrain slices. Low nicotine concentrations, in the presence of bicuculline and tetrodotoxin (TTX), increased the frequency but did not change the amplitude of glutamate-induced EPSCs, recorded from identified 5-HT neurons. Nicotine-induced increase of glutamatergic EPSC frequency persisted 10–20 min after drug withdrawal. This nicotinic effect was mimicked by exogenous administration of acetylcholine (ACh) or inhibition of ACh metabolism. In addition, the nicotine-induced increase in EPSC frequency was abolished by blockade of α4β2 nAChRs, voltage-gated calcium channels, or intracellular calcium signaling but not by α7 nAChR antagonists. These data suggest that both nicotine and endogenous ACh can increase glutamate release through activation of presynaptic α4β2 but not α7 nAChRs in the DRN. The effect involves long-term changes in synaptic function, and it is dependent on voltage-gated calcium channels and presynaptic calcium stores.
Introduction
Nicotinic acetylcholine receptors (nAChRs) are ligand-gated cation channels with a pentameric structure, composed of α and non-α (β) subunits. The existence of nine types of α subunits and three types of β subunits indicates a high structural and functional diversity of neuronal nAChRs, a conclusion supported by experimental studies (Wu and Lukas, 2011). However, it is known that homologous α7 and heterologous α4β2 nAChRs are more commonly expressed in the CNS (Whiting et al., 1987; Flores et al., 1992; Séguéla et al., 1993). Several studies have demonstrated that neuronal nAChRs have a preferential presynaptic location and act to increase the release of a large number of neurotransmitters (Wonnacott, 1997; Sher et al., 2004; Jensen et al., 2005).
Cholinergic nicotinic signaling is involved in a variety of processes, including anxiety, depression, arousal, memory, and attention among others (Hogg et al., 2003; Picciotto, 2003). Conversely, behavioral studies have suggested that certain effects of nicotine are mediated by increases in serotonin (5-HT) release in cortical and subcortical areas (Seth et al., 2002). Therefore, understanding the actions of nicotine on the dorsal raphe nucleus (DRN) and particularly its effects on 5-HT neurons is of broad scientific and clinical significance.
Immunocytochemical studies have demonstrated that both 5-HT and non-5-HT DRN neurons express postsynaptic nAChRs of α4β2 and α7 subtypes (Bitner et al., 2000; Bitner and Nikkel, 2002; Commons, 2008). Also, immunolabeling for the α4 receptor subunit has been detected in axon terminals in the DRN (Commons, 2008). Data obtained in our laboratory revealed that postsynaptic nAChRs of 5-HT and non-5-HT DRN neurons are functional (Galindo-Charles et al., 2008) and that nicotine increases the firing frequency of ∼80% of 5-HT DRN neurons (Mihailescu et al., 1998, 2002). Accordingly, a recent study has demonstrated the presence of functional somatic and presynaptic nAChRs in DRN neurons that project to the nucleus accumbens (Chang et al., 2011). Nicotine stimulatory effect on 5-HT DRN neurons has been explained previously through presynaptic release of noradrenaline from afferents originating in locus ceruleus (Li et al., 1998). However, DRN also receives glutamatergic afferents from cortical and subcortical areas and contains a significant number of glutamatergic interneurons (Soiza-Reilly and Commons, 2011). In addition, 5-HT DRN neurons express both ionotropic and metabotropic postsynaptic glutamatergic receptors (Celada et al., 2001; Kawashima et al., 2005). One of the most common presynaptic effects of nicotine is to increase glutamate release (McGehee et al., 1995). Taking into account these data, we hypothesized that nicotine increases glutamate release in the DRN, which explains in part the nicotinic stimulatory effects on the firing rate of 5-HT DRN neurons reported previously (Mihailescu et al., 1998, 2002; Chang et al., 2011).
The results of the present study demonstrate that both nicotine and endogenous acetylcholine (ACh) increase the release of glutamate in the DRN through activation of presynaptic α4β2 but not α7 nAChRs. This effect involves long-term changes in DRN glutamatergic synapses by a mechanism dependent on voltage-gated calcium channels (VGCCs) and Ca2+-induced Ca2+ release (CICR) from presynaptic calcium stores but independent of action potentials (APs).
Materials and Methods
Preparation of slices.
All procedures were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care Committee of the National Autonomous University of Mexico. The experiments were performed on brain slices obtained from postnatal days 21–25 male Wistar rats. Animals were deeply anesthetized with isoflurane and decapitated. Their brains were quickly removed and placed into ice-cold (4°C) bath (external) saline containing the following: 125 mm NaCl, 3 mm KCl, 25 mm NaHCO3, 1.25 mm Na2HPO4, 1 mm MgCl2, 1.2 mm CaCl2, and 25 mm glucose, 300 mOsm/L, pH 7.3 by bubbling with 95% O2 and 5% CO2. Coronal midbrain slices containing DRN (350 μm thick) were cut using a vibratome (Vibratome 1500) and stored in oxygenated bath saline at room temperature for at least 1 h before recording.
Whole-cell recordings.
Individual slices were transferred into a custom-made Plexiglas recording chamber and superfused with saline at a rate of 4–5 ml/min at 33°C. Whole-cell current- and voltage-clamp recordings were performed with a Multiclamp 700A amplifier (Molecular Devices) and monitored with a personal computer running Clampex 8 software (Molecular Devices). The micropipettes used for recordings were pulled from borosilicate glass tubes (WPI) with a Flaming-Brown puller (Sutter Instruments). The internal solution consisted of the following: 140 mm K-gluconate, 5 mm NaCl, 1 mm MgCl2, 0.02 mm EGTA, 10 mm HEPES, 2 mm Mg2-ATP, 0.5 Na2GTP, and 0.1% biocytin, pH 7.2–7.3 with Trizma base, 280–300 mOsm/L. In some experiments, we added 1–2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA; 10 mm) in the same internal solution but with 130 mm K-gluconate. The direct current impedance of the electrodes varied between 4 and 7 MΩ. Experimental data were stored in a personal computer using a Digidata 1322A analog-to-digital converter (Molecular Devices), at a sampling rate of 5 kHz. Individual neurons were visualized using an infrared video-microscopy system (BX51WI; Olympus Instruments) endowed with a 60× water-immersion objective. Neurons selected for recording were located close to the midline area of the DRN, identified as the translucent area between the medial longitudinal fasciculus and the aqueduct. Each recorded cell was identified based on its electrophysiological characteristics by generating a stimulus–response curve.
Recording of EPSCs.
Spontaneous EPSCs (sEPSCs) were recorded continuously in whole-cell voltage-clamp mode at a holding potential of −70 mV. All the experiments were made in the presence of bicuculline (10 μm) to block GABAA receptors. Nicotine or ACh were applied in the bath solution during 5 min to attain concentration equilibrium. Thereafter, the drugs were washed out, and a recording period of at least 30 min was taken for analysis. Access resistance was monitored by using depolarizing step pulses of 5 mV and 50 ms duration. When access resistance varied >15%, the experiment was discarded. In some experiments, concentric bipolar tungsten electrodes (FHC) (12 μm pencil tip) were placed just outside the boundaries of the DRN and ventral to the aqueduct. Field stimulation was made while recording from 5-HT DRN neurons. In these experiments, low stimulus intensities (subthreshold stimuli) were used to avoid the activation of action currents. Paired subthreshold stimuli were delivered at 0.1 Hz with an interstimulus interval of 50 ms. Stimuli were applied before and after nicotine and in the presence of bicuculline. An isolation unit (Digitimer) was used to adjust the stimulus parameters during the experiments.
Immunocytochemistry.
Neurons were filled with biocytin during recordings. A combination of intracellular labeling and 5-HT immunocytochemistry was used to determine the serotonergic nature of the recorded neuron. Slices containing injected neurons were fixed overnight with 4% paraformaldehyde and 1% picric acid in 0.1 m PBS, pH 7.4. The slices were then infiltrated with 30% sucrose and cut on a vibratome into 40 μm sections. The sections were incubated 4–6 h in PBS solution containing 0.2% Triton X-100 and streptavidin conjugated to Cy3 (1 mg/ml; diluted 1:100; Zymed) to label the recorded neuron. Sections were rinsed in PBS and incubated for 18–24 h at 4°C with primary rabbit anti-5-HT antisera (diluted 1:2000; ImmunoStar). After rinsing in PBS, sections were reincubated for 2 h with secondary antibodies conjugated to fluorescein (diluted 1:100; Vector Laboratories). The reacted sections were first examined with an appropriate set of filters on an epifluorescence-equipped microscope. Afterward, the sections were mounted in an anti-quenching media (Vectashield; Vector Laboratories) and examined under a confocal microscope (MRC 1024; Bio-Rad) equipped with a krypton/argon laser. A two-lines laser emitting at 550 and 500 nm wavelength was used for exciting Cy3 and fluorescein, respectively. Digitized images were transferred to a personal computer by using the image-capturing software (Confocal Assistant, T. C. Brelje, Minneapolis, MN). Omission of primary antisera resulted in no detectable signal (data not shown).
Drugs.
Drugs were dissolved into the bath saline from daily-made stock solutions and administered using a gravity-driven superfusion system. The time required for obtaining equilibrated concentrations of the drugs in the recording chamber was 3–4 min. TTX, dl-2-amino-5-phosponovaleric acid, 6-cyano-2,3-dihydroxy-7-nitroquinoxaline (CNQX), (−)bicuculline methiodide, methyllycaconitine (MLA), dihydro-β-erytroidine hydrobromide (DHβE), atropine, BAPTA, BAPTA-AM, cadmium chloride (CdCl2), and biocytin were purchased from Sigma/RBI. Thapsigargin, cyclopiazonic acid (CPA), and ryanodine were purchased from Tocris Bioscience. ω-Agatoxin-TK was obtained from Peptide Institute, and ω-conotoxin-GVIA was obtained from Alomone Labs. All reagents were added from freshly prepared stock solution to the bath saline.
Data analyses.
Offline analysis of the data was performed using Clampfit 8 (Molecular Devices), Mini Analysis (Synaptosoft), and graphing and statistical software packages (Origin version 6, Microcal; and Systat). Data are expressed as means ± SEM. Statistical comparisons between samples were made by using the Student's t test (p < 0.05 was taken as significant).
Results
5-HT neurons were identified based on their electrophysiological properties. Figure 1A shows APs elicited by increasing depolarizing current steps recorded from an identified 5-HT neuron (Fig. 1C). As previously reported (Penington et al., 1991; Bayliss et al., 1997; Galindo-Charles et al., 2008) when positive direct current is injected, 5-HT neurons exhibit repetitive firing with adaptation of the firing frequency. 5-HT neurons also exhibit prolonged afterhyperpolarizing potentials, long AP durations (∼4 ms at the base), and an inflection or “shoulder” during AP descending phase (Vandermaelen and Aghajanian, 1983; Beck et al., 2004; Marinelli et al., 2004; Galindo-Charles et al., 2008) (Fig. 1A, inset). Figure 1B illustrates the firing frequency as a function of injected current (I–F plot) calculated at the initial and final interspike intervals. At the steady state, 5-HT neurons maximal firing rate was 6.7 ± 0.7 Hz, whereas non-5HT neurons reach frequencies >25 Hz (Galindo-Charles et al., 2008). All the data were obtained from neurons identified as 5-HT positive by immunocytochemistry (Fig. 1C), bringing additional support to electrophysiological identification. This study was focused on the effects of nicotine on 5-HT DRN neurons. Therefore, when a pattern of electrophysiological activity characteristic of non-5-HT neurons was detected, the respective cell was discarded.
Figure 1.
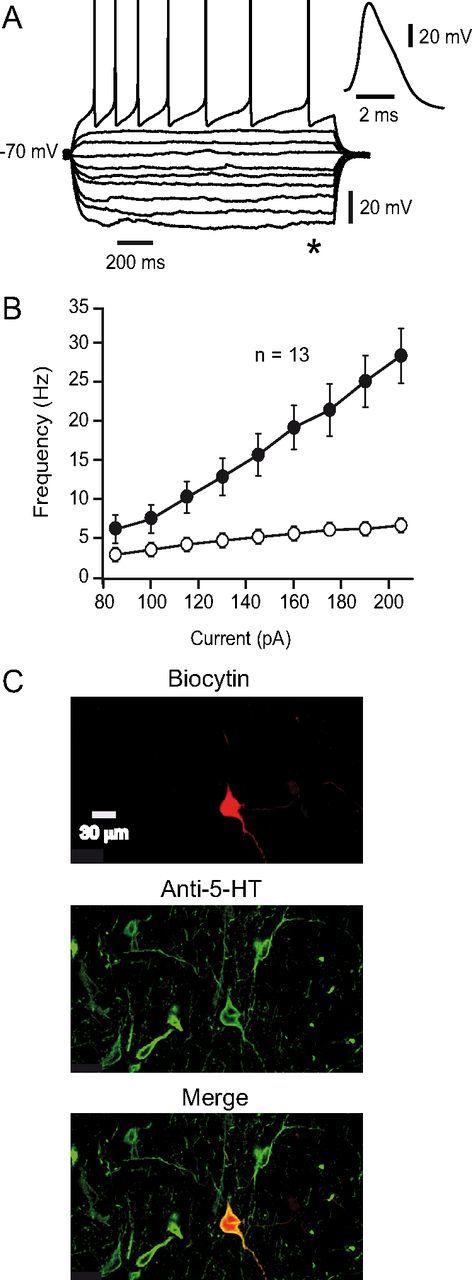
Electrophysiological characteristics of the DRN serotonergic neurons. A, Current traces in response to hyperpolarizing and depolarizing current steps recorded from a 5-HT neuron. Inset, An expanded AP from the same neuron. B, Plots of discharge frequency as a function of injected current obtained from 13 identified 5-HT neurons are shown. Filled circles represent the instantaneous frequency obtained with the reciprocal of the first interspike interval. Open circles represent the frequency obtained from the last interspike interval. C, Microphotographs illustrating the recorded cell in A, after biocytin staining (top), 5-HT immunoreactivity (middle), and merge (bottom).
Nicotine increased the frequency of sEPSCs recorded from 5-HT DRN neurons
Under voltage-clamp conditions, low concentrations of nicotine were applied to slices containing the DRN. sEPSCs were recorded from 5-HT neurons at a holding potential of −70 mV (Fig. 2A). All the experiments were made in the presence of bicuculline (10 μm). Nicotine (300 nm) increased the frequency but did not change the amplitude of sEPSCs (Fig. 2A,B). The effect of nicotine persisted for ∼15 min after drug withdrawal, and it was seen in five of seven 5-HT-positive cells tested (Fig. 2D). sEPSCs frequency increased 40 ± 8% (p < 0.05, n = 5) with respect to the baseline (Fig. 2C).
Figure 2.
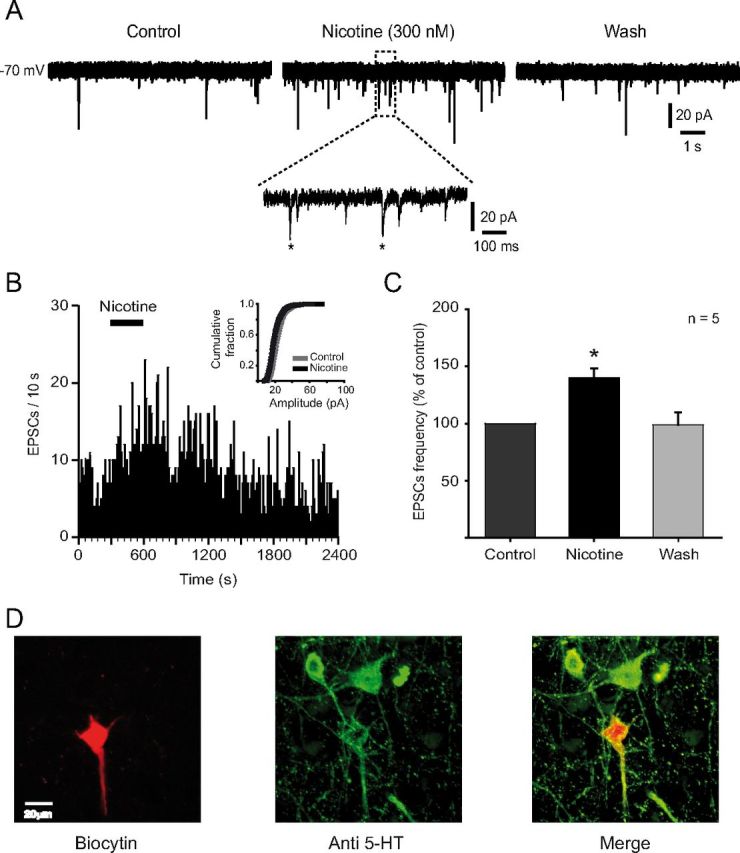
Low concentrations of nicotine enhance glutamatergic synaptic transmission. A, sEPSCs recorded by using whole-cell patch-clamp technique (voltage-clamp mode) in the presence of bicuculline (10 μm). The traces show 10 s taken in control conditions (left), after nicotine (300 nm) added to the bath solution (middle), and after 20 min wash (right). A region of the middle trace (dashed box) is shown below at a slower sweep time. Two of the synaptic events are indicated by asterisks. B, Time–frequency histogram (10 s bin) for the same cell in A, showing that nicotine increased the frequency but did not change the amplitude of sEPSCs (inset). C, Summary of the results from five neurons showing the effect on sEPSCs frequency in the presence of nicotine (*p < 0.05). D, Microphotographs illustrating the recorded cell in A after biocytin staining (left), 5-HT immunoreactivity (middle), and merge (right).
To have more consistent and robust responses, we used 1 μm nicotine in subsequent experiments. At this concentration, nicotine also increased sEPSCs frequency (Fig. 3A,B), and the effect was observed in 16 of 18 5-HT-positive neurons tested. Blocking the AMPA/kainic acid receptors with 10 μm CNQX abolished the sEPSCs completely, demonstrating their glutamatergic nature. Nicotine did not change sEPSCs amplitude (Fig. 3C), suggesting a presynaptic action mechanism. The averaged time–frequency histogram of 16 cells is shown in the Figure 3D. sEPSC frequency was increased 88 ± 20% (Fig. 3E) with respect to the baseline (p < 0.01, n = 16), and the effect extended 20 min beyond the drug administration period (Fig. 3D,E). ACh (1 mm) had similar effects to nicotine (Fig. 3E): sEPSC frequency increased 66 ± 10% by ACh in 10 of 14 5-HT-positive neurons (p < 0.01, n = 10).
Figure 3.

Nicotinic effects are mimicked by endogenous ACh. A, Traces showing sEPSCs recorded from a 5-HT-positive neuron in control, in the presence of nicotine, 10 min after nicotine washout, and 10 min after adding CNQX (10 μm). B, C, Cumulative probability distributions of frequencies and amplitudes for the same neuron in A, in control (dark gray traces), nicotine (black traces), and 10 min wash (light gray traces). D, Average normalized time–frequency histogram from 16 5-HT neurons tested with nicotine (1 μm). Inset, Normalized time–frequency histogram from five 5-HT neurons tested with eserine (10 μm). E, Summary of the results showing the effect on sEPSC frequency with nicotine alone, nicotine in the presence of TTX, ACh, and eserine. All experimental groups were compared against control (before drug application). The last three bars represents the wash time after nicotine alone. All the experiments were done in the presence of bicuculline (10 μm). ACh experiments were done in the presence of bicuculline and atropine (10 μm) (*p < 0.05, **p < 0.01).
To investigate whether endogenous ACh was able to mimic the effect of nicotine and external ACh, we applied eserine, an acetylcholinesterase inhibitor. Eserine (10 μm) also increased the sEPSCs frequency in five of seven 5-HT-positive cells (Fig. 3D, inset, E). sEPSC frequency was increased 40 ± 28% with respect to the baseline (p < 0.05). In these experiments, sEPSC frequency returned to baseline much faster than with nicotine, suggesting that acetylcholinesterase is regulating a constant cholinergic tone. Eserine effect lasted ∼8 min after drug washout (Fig. 3D, inset). The AP dependence of nicotinic effect was assessed by adding TTX (500 nm) to the perfusion solution. In the presence of TTX, nicotine (1 μm) still enhanced the frequency of sEPSCs in 11 of 12 5-HT cells tested (Fig. 3E). sEPSC frequency was increased 80 ± 10% with respect to the baseline (p < 0.01, n = 11). This increase was not statistically different from the one produced by nicotine alone.
The enhancement of glutamate-induced sEPSCs is mediated by α4β2 nAChRs
Immunocytochemical studies have revealed the presence of α7 and α4β2 nAChRs within the DRN (Bitner et al., 2000; Bitner and Nikkel, 2002). In an attempt to identify the nAChR subtype mediating the effect of nicotine, we tested the selective nAChR antagonists MLA and DHβE. Blocking the α7 nAChRs with MLA (100 nm) did not prevent the increase of glutamate sEPSC frequency induced by nicotine (1 μm) (Fig. 4A,B). Nicotine increased sEPSC frequency but did not change the amplitude (data not shown) in 10 of 11 5-HT-positive cells. sEPSC frequency was increased 82 ± 27% with respect to the baseline (p < 0.005, n = 10) in the presence of MLA. In contrast, the application of the selective α4β2 nAChR antagonist DHβE (100 nm) completely blocked the effect of nicotine in all the 5-HT-positive cells tested (Fig. 4C,D). The time–frequency histogram (Fig. 4D) illustrates the lack of effect of nicotine in the presence of DHβE. However, DHβE by itself decreased the sEPSC frequency compared with the control (2.6 ± 0.7 Hz in control; 1.4 ± 0.3 Hz in DHβE; n = 5; p < 0.05; data not shown), which supports the presence of a cholinergic tone within the DRN.
Figure 4.

Nicotinic effects are performed through β2-containing nAChRs. A, Traces showing sEPSCs recorded from a 5-HT-positive neuron in the presence of MLA (100 nm, top), MLA plus nicotine (1 μm, middle), and nicotine washout (bottom). B, Time–frequency histograms from 10 identified 5-HT neurons tested with nicotine (1 μm) in the presence of MLA. The inset shows the normalized sEPSC frequency. C, Traces showing sEPSCs recorded from a 5-HT-positive neuron in the presence of DHβE (100 nm, top), DHβE plus nicotine (1 μm, middle), and nicotine washout (bottom). D, Time–frequency histogram from 12 identified 5-HT neurons tested with nicotine (1 μm) in the presence of DHβE. The inset shows the normalized sEPSC frequency. In all the experiments, nicotine was applied after 10 min pretreatment with DHβE or MLA.
To further investigate the identity of the nAChR subtype involved in nicotine-induced glutamate release, we used α7 and α4β2 nAChR-selective agonists. The selective α4β2 nAChR agonist RJR-2403 [(E)-N-methyl-4-pyridin-3-ylbut-3-en-1-amine] (100 nm) mimicked the effect of nicotine and ACh; it increased the glutamate sEPSCs frequency in five out of six 5-HT cells. However, the effect disappeared 5 min after removing the drug from the bath. sEPSC frequency was increased 56 ± 17% with respect to the baseline (Fig. 5A,B; p < 0.05, n = 5). Conversely, the selective α7 nAChR agonist PNU-282987 (N-[(3R)-1-azabicyclo[2.2.2]octan-3-yl]-4-chlorobenzamide hydrochloride) (100 nm) did not change the glutamate sEPSC frequency in any of the five 5-HT-positive cells tested (Fig. 5C,D). These data suggest that glutamate release in the DRN is presynaptically modulated through high-affinity α4β2 nAChRs.
Figure 5.

Nicotinic effects are mimicked by the α4β2 nAChR-selective agonist RJR-2403. A, Traces showing sEPSCs recorded from a 5-HT-positive neuron in control (top), in the presence of RJR-2403 (100 nm, middle), and RJR-2403 washout (bottom). B, Time–frequency histogram from five identified 5-HT neurons tested with RJR-2403. The inset shows the normalized sEPSC frequency. C, Traces showing sEPSCs recorded from another 5-HT-positive neuron in control (top), in the presence of the selective α7 nAChR agonist PNU-282987 (100 nm, middle), and PNU-282987 washout (bottom). D, Time–frequency histograms from five identified 5-HT neurons tested with PNU-282987. The inset shows the normalized sEPSC frequency.
Buffering presynaptic intracellular calcium abolished the effect of nicotine
Recently, it has been shown that DRN neurons possess functional α7 and α4β2 nAChRs (Galindo-Charles et al., 2008). Therefore, we could not discard a possible contribution of postsynaptic nAChRs linked to the nicotinic effect observed in this study. To test this possibility, in a group of five 5-HT-positive cells, we buffered the postsynaptic Ca2+ by loading the patch pipette with an internal solution containing BAPTA (10 mm). Intracellular BAPTA had no effect on either spontaneous synaptic activity or sEPSC frequency enhancement induced by nicotine. sEPSCs frequency was increased 102 ± 2% with respect to the baseline (Fig. 6Aa; p < 0.05). On the contrary, nicotine failed to increase the sEPSC frequency when slices were perfused for 30 min with the membrane-permeable Ca2+ chelator BAPTA-AM (100 μm) in all the five cells tested (Fig. 6Ab). As expected, BAPTA-AM itself decreased the frequency of spontaneous synaptic activity. The sEPSC frequency decreased by 36 ± 0.8% after BAPTA-AM with respect to control (data not shown). A summary of these data is shown in Figure 6Ac.
Figure 6.

Nicotinic action mechanism is presynaptic. Aa, Traces showing sEPSCs recorded from a 5-HT-positive neuron with BAPTA (10 mm) in the recording pipette (top) and after adding nicotine in the bath solution (bottom). Ab, Traces showing sEPSCs recorded from a 5-HT-positive neuron with BAPTA-AM (10 mm) applied in the bath solution (top) and after adding nicotine (bottom). Ac, Bar histogram shows normalized sEPSC frequency. B, Paired-pulse protocol is illustrated in Ba. A concentric stimulation electrode (S) with a 10 μm tip diameter was placed in the proximities of the DRN, ventral to the aqueduct. Excitatory evoked currents were recorded from 5-HT neurons within the DRN in the presence of bicuculline (10 μm). Evoked current traces of a single neuron in response to paired stimuli (S1 and S2) are shown in Bb (top). Gray traces are the responses in control conditions. Black traces are the responses in the presence of nicotine (1 μm). Temporal course of the first stimulus amplitude (S1) from the cell in Bb (top) is shown in Bb (bottom). Dark gray circles represent the control, black circles represent nicotine, and light gray circles represent nicotine washout. The circles at zero current represent synaptic failures. Bc, Bar histograms show the change in the normalized S1 amplitude induced by nicotine (top; ***p < 0.001, n = 6) and the change in the paired-pulse ratio induced by nicotine (bottom; ***p < 0.001, n = 6).
In a set of experiments, we used paired pulses (50 ms interval, 100 μs duration) applied through a bipolar concentric electrode (12 μm tip diameter) placed in the boundaries of the DRN, just below the aqueduct as illustrated in Figure 6Ba. In six of eight 5-HT-positive cells, nicotine increased the amplitude of the first response (Fig. 6Bb, top, Bc, top) and decreased the number of failures to the first stimulus (Fig. 6Bb, bottom). Nicotine also decreased the paired-pulse ratio (Fig. 6Bc, bottom). Paired-pulse ratio was 0.94 ± 0.09 and 0.58 ± 0.05 in control and nicotine, respectively (p < 0.001, n = 6). These results reinforce the idea that nicotine-induced increase in glutamate sEPSC frequency depends on presynaptic nAChRs.
Blocking the VGCCs prevented nicotine-induced synaptic potentiation
It is well known that nAChRs are permeable to Ca2+ (Tsuneki et al., 2000; Fucile, 2004). Also, it has been shown that VGCCs are associated with nicotinic effects mediated through β2-subunit containing nAChRs (Tsuneki et al., 2000; Shoop et al., 2001; Dajas-Bailador et al., 2002; Dajas-Bailador and Wonnacott, 2004). Therefore, an increase of presynaptic Ca2+ via α4β2 could lead to the opening of VGCCs and facilitate glutamate release. To assess this possibility, we tested the effect of nicotine in the presence of CdCl2 (100 μm). Surprisingly, nicotine still increased the glutamate sEPSC frequency in the presence of cadmium in all six 5-HT-positive cells tested (Fig. 7A,D). Moreover, the nicotine-induced enhancement of sEPSC frequency in the presence of cadmium was 351 ± 30% (p < 0.01, n = 6) (Fig. 7D), a value that is much higher when compared with the effect of nicotine alone (see Fig. 3E). This effect could be the result of a potentiation of nAChRs produced by cadmium (Hsiao et al., 2001). Cadmium could elevate Ca2+ concentrations at glutamate terminals by enhancing Ca2+ influx through nAChRs. In these conditions, nicotine might be able to increase glutamate release even if VGCCs were blocked. Therefore, we decided to use selective Ca2+ channel blockers to see whether the effect of nicotine was dependent on VGCCs. A mixture containing ω-agatoxin-TK (400 nm), ω-conotoxin-GVIA (500 nm), and nitrendipine (10 μm) was applied during 15 min in the bath solution to block P/Q-, N-, and L-type Ca2+ channels, respectively. In these conditions, the administration of nicotine (1 μm) failed to produce significant sEPSC frequency changes (Fig. 7B,D). sEPSC frequency was 0.8 ± 0.2 and 0.68 ± 0.15 Hz in the presence of the VGCC blockers and VGCC blockers plus nicotine, respectively. These values were not statistically significant (p > 0.05, n = 6; Fig. 7D).
Figure 7.

Nicotinic effects depend on VGCCs and intracellular CICR. A, Time–frequency histogram shows the effect of nicotine on the sEPSC frequency in the presence of CdCl2 (gray bar). B, Time–frequency histogram shows the lack of effect of nicotine on the sEPSC frequency in the presence of a mixture containing the Ca2+ channel blockers ω-agatoxin-TK, ω-conotoxin-GVIA, and nitrendipine (gray bar). C, Time–frequency histogram shows the lack of effect of nicotine on the sEPSC frequency in the presence of the SERCA blocker thapsigargin (gray bar). D, Bar graph shows the effect of nicotine on the sEPSC frequency in slices pretreated with CdCl2, Ca2+ channel blockers, thapsigargin, CPA, or ryanodine (**p < 0.01). Gray bar represent the control (before nicotine application).
Depleting intracellular calcium stores abolished nicotine-induced sEPSCs
To explore whether CICR contributes to nicotine-induced synaptic potentiation, we used thapsigargin and CPA, two blockers of the sarcoplasmic/endoplasmic reticulum calcium ATPase pump (SERCA). Thapsigargin (10 μm) completely abolished the effect of nicotine in five of the 5-HT-positive cells tested. The time–frequency histogram in Figure 7C illustrates the lack of effect of nicotine in the presence of thapsigargin. sEPSC frequency was 0.7 ± 0.3 and 0.58 ± 0.2 Hz in the presence of thapsigargin alone and thapsigargin plus nicotine, respectively. These values were not statistically significant (p > 0.05, n = 5). CPA (10 μm) also blocked the nicotine-induced enhancement of sEPSC frequency. Frequency values were 1.9 ± 0.3 and 1.76 ± 0.35 Hz in the presence of CPA alone and CPA plus nicotine, respectively (p > 0.05, n = 5). Additional evidence for CICR involvement was obtained by applying nicotine on neurons pretreated (15 min) with ryanodine (100 μm) to block stored calcium. sEPSC frequency was 0.56 ± 0.3 and 0.4 ± 0.24 Hz in the presence of ryanodine alone and ryanodine plus nicotine, respectively (p > 0.05, n = 4). Figure 7D shows a summary of the effect of nicotine in the presence of CdCl2, Ca2+ channel blockers, thapsigargin, CPA, or ryanodine, on the normalized sEPSCs (p < 0.01). These results indicate that CICR is necessary for nicotine-induced glutamate release in the DRN.
Finally, Figure 8 shows a model summarizing the mechanisms by which nicotine induces a long-term increase in glutamate release in the DRN. The DRN receives cholinergic inputs originating in the pedunculopontine (PPT) and laterodorsal tegmental (LDT) nuclei (Woolf and Butcher, 1989). This model compares the physiological conditions in which only the spontaneous release of ACh is present (Fig. 8A) with the condition when nicotine is also present (Fig. 8B).
Figure 8.

Model summarizing nicotinic effects on glutamate terminals in the DRN. A, In physiological conditions, the cholinergic tone is regulating the excitatory glutamatergic input to the DRN 5-HT neurons through the activation of α4β2 nAChRs, located at glutamate terminals. Released ACh is quickly metabolized by the enzyme acetylcholinesterase. As a result, the Ca2+ influx into the glutamate terminals and glutamate release will be maintained at low levels. B, When nicotine is present (for example, in smokers), more α4β2 nAChRs will be activated, because nicotine cannot be degraded in the synaptic cleft. This will enhance Ca2+ entry, followed by depolarization of glutamate terminals and activation of VGCCs. This, in turn, will increase even more intracellular calcium and produce CICR from the endoplasmic reticulum (ER) through the activation of ryanodine receptors (RyR). This last event generates a long-term potentiation of glutamate release.
Discussion
The main finding of this study was that the activation of α4β2 nAChRs by nicotine or ACh provokes a long-lasting enhancement of the glutamatergic input to 5-HT DRN neurons. In addition, our data indicate that CICR from presynaptic stores of glutamatergic terminals is necessary for nicotine-evoked glutamate release.
According to our results, both nicotine and exogenous ACh increased the frequency of glutamate-dependent sEPSCs recorded from identified 5-HT DRN neurons. This effect was mediated by high-affinity α4β2 nAChRs because it was manifest at low nicotine concentrations (300 nm) (Zoli et al., 1998; Lambe et al., 2003), blocked by low concentrations of DHβE (100 nm) (Zoli et al., 1998), and unaffected by MLA, a selective α7 nAChR blocker. Furthermore, the α4β2 nAChR-selective agonist RJR-2403 mimicked the effect of nicotine. Although RJR-2403 exerts less desensitization on α4β2 receptors (Papke et al., 2000), our results show a more rapid washout of RJR-2403-induced effects compared with nicotine. This could be explained by the low concentrations of RJR-2403 (100 nm) used in this study. Conversely, at micromolar concentrations, RJR-2403 can lose its selectivity for neuronal α4β2 nAChRs (Yang et al., 2009).
It should be mentioned that nicotine-induced glutamate release is more frequently mediated by α7 nAChRs than other nAChRs subtypes (McGehee et al., 1995; Girod and Role, 2001; Maggi et al., 2004; Sharma et al., 2008). However, in a few brain areas, non-α7 nAChRs have also been involved in glutamate release modulation (Rousseau et al., 2005; Couey et al., 2007), and it has been reported that α4β2 nAChRs facilitate glutamate release in thalamocortical afferents (Gioanni et al., 1999; Lambe et al., 2003).
Our data indicate that, in midbrain slices, endogenous ACh also increases the glutamatergic input to 5-HT DRN neurons because the administration of eserine, an inhibitor of acetylcholinesterase, significantly increased the frequency of glutamatergic sEPSCs in 5-HT DRN neurons. In support of this finding, it has been demonstrated that DRN neurons receive a cholinergic input from the LDT and PPT nuclei (Woolf and Butcher, 1989) and that these cholinergic connections are functional in midbrain coronal slices (Galindo-Charles et al., 2008).
According to our data, the magnitude of the effect of 1 μm nicotine on glutamate release is comparable with that of 1 mm exogenous ACh (see Fig. 3 E). This could be explained by the high affinity of nicotine for α4β2 nAChRs (Alkondon and Albuquerque, 1995; Buisson et al., 1996) and also by the fast degradation of ACh by acetylcholinesterase.
In the present study, a single nicotine administration induced glutamate release effects within the DRN that persisted 10–20 min after nicotine withdrawal. In fact, nicotinic effects on glutamate transmission lasted longer and had a higher prevalence than reported previously by Chang et al. (2011). These discrepancies could be attributed to animal age or strain or the fact that they performed all physiology experiments at room temperature, whereas our experiments were done at 33°C. Recently, it was reported that the response of nAChRs to their agonists depends on temperature (Jindrichova et al., 2012).
Nicotine-evoked glutamate release in the DRN is achieved through presynaptic mechanisms because nicotine increased the frequency but not the amplitude of glutamate sEPSCs. This assertion is supported by the results obtained in this study using the paired-pulse paradigm; nicotine increased the amplitude of the response to the first stimulus and decreased the paired-pulse ratio, effects that are suggestive of a presynaptic effect (Zucker and Regehr, 2002). Furthermore, nicotine-induced glutamate release requires the increase of calcium levels in glutamatergic axon terminals because, in our experiments, buffering intracellular calcium with the membrane-permeable compound BAPTA-AM decreased both the basal and nicotine-evoked glutamate release. The possibility that a Ca2+-dependent retrograde messenger, released by the 5-HT neurons, could be responsible of the glutamate sEPSC enhancement was ruled out because intracellular postsynaptic BAPTA did not change the effect of nicotine.
Conversely, nicotine-induced increase of glutamate release persisted in the presence of TTX, which indicates that firing activity of glutamatergic neurons is not required for this effect.
Previous studies have demonstrated that nicotine excitatory actions on neurotransmitter release exerted through β2 subunit-containing nAChRs are entirely dependent on the activation of VGCCs, which induces an additional increase in intracellular calcium levels and CICR (Tsuneki et al., 2000; Shoop et al., 2001; Dajas-Bailador et al., 2002; Dickinson et al., 2008). In contrast, Ca2+ influx through presynaptic α7 nAChRs is sufficient to induce CICR and neurotransmitter release without the intervention of VGCCs (Gray et al., 1996; Sharma and Vijayaraghavan, 2003; Sharma et al., 2008). Accordingly, our data indicate that blocking N-, P/Q-, and L-type Ca2+ channels by using selective antagonists completely suppresses nicotine-induced glutamate release. Furthermore, depleting the intracellular Ca2+ stores with thapsigargin or CPA or perfusing the cells with ryanodine also abolished the effects of nicotine. These results suggest that, in the DRN, the activation of β2-containing nAChRs, located at glutamate terminals, produces changes in synaptic efficacy through a mechanism involving VGCC activation and CICR. Similar conclusions have been reached in several studies involving other brain areas (Léna and Changeux, 1997; Mansvelder et al., 2002; Lambe et al., 2003). In support of this idea, presynaptically located calcium stores have been found to regulate neurotransmitter release in different brain structures (Llano et al., 2000; Conti et al., 2004).
Unexpectedly, the VGCCs broad-spectrum blocker CdCl2 failed to block the effect of nicotine. Instead, the nicotine-induced increase of sEPSC frequency was potentiated in the presence of CdCl2. One possible explanation for this effect is that, like other divalent cations, cadmium potentiates neuronal nAChRs (Hsiao et al., 2001). If this is the case, an increased influx of Ca2+ through α4β2 nAChRs, induced by cadmium, might be sufficient to cause CICR, leading to a potentiation of nicotine-induced glutamate release without the intervention of VGCCs.
Although nicotine produces fast desensitization of β2-containing nAChRs (Wooltorton et al., 2003), it provoked a persistent increase of glutamate sEPSCs. According to our model of nicotine-induced glutamate release potentiation (Fig. 8), nicotine activates more nAChRs and is degraded much slower than ACh. As a consequence, nicotine will induce a higher Ca2+ influx, resulting in CICR, activation of Ca2+-dependent kinases (Dickinson et al., 2008; Sharma et al., 2008), and enhancement of synaptic efficacy at glutamate terminals. Once the changes in the synaptic release efficacy have occurred, they will extend beyond nAChR desensitization. Therefore, we speculate here that nicotine will induce a higher release of glutamate in smokers than endogenous ACh in non-smokers.
DRNs receive glutamatergic projections from cortical and subcortical areas, as well as from local interneurons. The results of our experiments suggest that nicotine-dependent glutamate release in this nucleus might be associated with an increased excitability of 5-HT DRN neurons. Previously, we found that all 5-HT identified neurons tested with local application of ACh exhibited either α4β2 or α7 nicotinic currents (Galindo-Charles et al., 2008). In addition, in this work, we observed that nicotine produced an increase of glutamate EPSCs in most 5-HT neurons. Therefore, we assume that presynaptic and postsynaptic effects of nicotine coexist in the majority of 5-HT neurons. Postsynaptic effects are short lasting because of nAChRs desensitization, whereas presynaptic effects are long lasting because of glutamate release potentiation. These combined presynaptic and postsynaptic effects of nicotine would increase firing frequency of DRN neurons (Mihailescu et al., 2001, 2002; Chang et al., 2011), which in turn would change the levels of 5-HT in the brain.
Contrary to our findings, systemic nicotine administration in rats transiently inhibited 5-HT DRN neurons (Engberg et al., 2000). Because 5-HT neuron firing strongly depends on the arousal state, it is possible that the inhibitory effect of nicotine in vivo was influenced by anesthesia.
Glutamate neurotransmission within the DRN has been implicated in the pathophysiology of affective disorders, such as depression (Paris and Cunningham, 1994; Grahn et al., 2000; Paul and Skolnick, 2003) and stress (Amat et al., 2005; Kirby et al., 2007). Indeed, in humans and animal models, glutamate receptor antagonists have been reported to be effective antidepressants (Trullas and Skolnick, 1990; Yilmaz et al., 2002). These effects could rely on the ability of glutamatergic neurotransmission to change the circuits regulating the serotonergic tone within the DRN and other brain regions. For example, it has been proposed that an increase of DRN serotonergic output to the nucleus accumbens could partially explain the rewarding and addictive effects of nicotine (Chang et al., 2011).
Conversely, low concentrations of nicotine applied into the DRN of rats consistently induces anxiolytic effects (Cheeta et al., 2001). Interestingly, the systemic administration of ABT-418 (3-methyl-5-[(2S)-1-methylpyrrolidin-2-yl]-1,2-oxazole), an agonist of α4β2 nAChRs, mimics the anxiolytic effects of nicotine and attenuates the anxiogenic response evoked by nicotine withdrawal (Brioni et al., 1994). Because α4β2 nAChR agonists increase glutamatergic input to 5-HT DRN neurons, we hypothesize that the effects of these agonists on mood and nicotine withdrawal symptoms are at least partly dependent on a glutamate-induced increase in activity of 5-HT DRN neurons.
Footnotes
This work was supported by Consejo Nacional de Ciencia y Tecnología Grant 50147-M and Dirección General de Asuntos del Personal Académico–UNAM Grants IN217107 and IN220112–3. We thank Lorena Alline Becerril-Melendez for technical support.
References
- Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. III. Agonist actions of the novel alkaloid epibatidine and analysis of type II current. J Pharmacol Exp Ther. 1995;274:771–782. [PubMed] [Google Scholar]
- Amat J, Baratta MV, Paul E, Bland ST, Watkins LR, Maier SF. Medial prefrontal cortex determines how stressor controllability affects behavior and dorsal raphe nucleus. Nat Neurosci. 2005;8:365–371. doi: 10.1038/nn1399. [DOI] [PubMed] [Google Scholar]
- Bayliss DA, Li YW, Talley EM. Effects of serotonin on caudal raphe neurons: inhibition of N and P/Q-type calcium channels and the afterhyperpolarization. J Neurophysiol. 1997;77:1362–1374. doi: 10.1152/jn.1997.77.3.1362. [DOI] [PubMed] [Google Scholar]
- Beck SG, Pan YZ, Akanwa AC, Kirby LG. Median and dorsal raphe neurons are not electrophysiologically identical. J Neurophysiol. 2004;91:994–1005. doi: 10.1152/jn.00744.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitner RS, Nikkel AL. Alpha-7 nicotinic receptor expression by two distinct cell types in the dorsal raphe nucleus and locus coeruleus of rat. Brain Res. 2002;938:45–54. doi: 10.1016/s0006-8993(02)02485-x. [DOI] [PubMed] [Google Scholar]
- Bitner RS, Nikkel AL, Curzon P, Donnelly-Roberts DL, Puttfarcken PS, Namovic M, Jacobs IC, Meyer MD, Decker MW. Reduced nicotinic receptor-mediated antinociception following in vivo antisense knock-down in rat. Brain Res. 2000;871:66–74. doi: 10.1016/s0006-8993(00)02442-2. [DOI] [PubMed] [Google Scholar]
- Brioni JD, O'Neill AB, Kim DJ, Buckley MJ, Decker MW, Arneric SP. Anxiolytic-like effects of the novel cholinergic channel activator ABT-418. J Pharmacol Exp Ther. 1994;271:353–361. [PubMed] [Google Scholar]
- Buisson B, Gopalakrishnan M, Arneric SP, Sullivan JP, Bertrand D. Human alpha4beta2 neuronal nicotinic acetylcholine receptor in HEK 293 cells: a patch-clamp study. J Neurosci. 1996;16:7880–7891. doi: 10.1523/JNEUROSCI.16-24-07880.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celada P, Puig MV, Casanovas JM, Guillazo G, Artigas F. Control of dorsal raphe serotonergic neurons by the medial prefrontal cortex: involvement of serotonin-1A, GABAA, and glutamate receptors. J Neurosci. 2001;21:9917–9929. doi: 10.1523/JNEUROSCI.21-24-09917.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Daniele CA, Gallagher K, Madonia M, Mitchum RD, Barrett L, Vezina P, McGehee DS. Nicotinic excitation of serotonergic projections from dorsal raphe to the nucleus accumbens. J Neurophysiol. 2011;106:801–808. doi: 10.1152/jn.00575.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeta S, Irvine EE, Kenny PJ, File SE. The dorsal raphé nucleus is a crucial structure mediating nicotine's anxiolytic effects and the development of tolerance and withdrawal responses. Psycopharmacology (Berl) 2001;155:78–85. doi: 10.1007/s002130100681. [DOI] [PubMed] [Google Scholar]
- Commons KG. Alpha4 containing nicotinic receptors are positioned to mediate postsynaptic effects on 5-HT neurons in the rat dorsal raphe nucleus. Neuroscience. 2008;153:851–859. doi: 10.1016/j.neuroscience.2008.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti R, Tan YP, Llano I. Action potential-evoked and ryanodine-sensitive spontaneous Ca2+ transients at the presynaptic terminal of a developing CNS inhibitory synapse. J Neurosci. 2004;24:6946–6957. doi: 10.1523/JNEUROSCI.1397-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couey JJ, Meredith RM, Spijker S, Poorthuis RB, Smit AB, Brussaard AB, Mansvelder HD. Distributed network actions by nicotine increase the threshold for spike-timing-dependent plasticity in prefrontal cortex. Neuron. 2007;54:73–87. doi: 10.1016/j.neuron.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador F, Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci. 2004;25:317–324. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador FA, Mogg AJ, Wonnacott S. Intracellular Ca2+ signals evoked by stimulation of nicotinic acetylcholine receptors in SH-SY5Y cells: contribution of voltage-operated Ca2+ channels and Ca2+ stores. J Neurochem. 2002;81:606–614. doi: 10.1046/j.1471-4159.2002.00846.x. [DOI] [PubMed] [Google Scholar]
- Dickinson JA, Kew JN, Wonnacott S. Presynaptic alpha 7- and beta 2-containing nicotinic acetylcholine receptors modulate excitatory amino acid release from rat prefrontal cortex nerve terminals via distinct cellular mechanisms. Mol Pharmacol. 2008;74:348–359. doi: 10.1124/mol.108.046623. [DOI] [PubMed] [Google Scholar]
- Engberg G, Erhardt S, Sharp T, Hajos M. Nicotine inhibits firing activity of dorsal raphé 5-HT neurones in vivo. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:41–45. doi: 10.1007/s002100000252. [DOI] [PubMed] [Google Scholar]
- Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of α4 and β2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol. 1992;41:31–37. [PubMed] [Google Scholar]
- Fucile S. Ca2+ permeability of nicotinic acetylcholine receptors. Cell Calcium. 2004;35:1–8. doi: 10.1016/j.ceca.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Galindo-Charles L, Hernandez-Lopez S, Galarraga E, Tapia D, Bargas J, Garduño J, Frías-Dominguez C, Drucker-Colin R, Mihailescu S. Serotoninergic dorsal raphe neurons possess functional postsynaptic nicotinic acetylcholine receptors. Synapse. 2008;62:601–615. doi: 10.1002/syn.20526. [DOI] [PubMed] [Google Scholar]
- Gioanni Y, Rougeot C, Clarke PB, Lepousé C, Thierry AM, Vidal C. Nicotinic receptors in the rat prefrontal cortex: increase in glutamate release and facilitation of mediodorsal thalamo-cortical transmission. Eur J Neurosci. 1999;11:18–30. doi: 10.1046/j.1460-9568.1999.00403.x. [DOI] [PubMed] [Google Scholar]
- Girod R, Role LW. Long-lasting enhancement of glutamatergic synaptic transmission by acetylcholine contrasts with response adaptation after exposure to low-level nicotine. J Neurosci. 2001;21:5182–5190. doi: 10.1523/JNEUROSCI.21-14-05182.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahn RE, Watkins LR, Maier SF. Impaired escape performance and enhanced conditioned fear in rats following exposure to an uncontrollable stressor are mediated by glutamate and nitric oxide in the dorsal raphe nucleus. Behav Brain Res. 2000;112:33–41. doi: 10.1016/s0166-4328(00)00161-3. [DOI] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Raggenbass M, Bertrand D. Nicotinic acetylcholine receptors: from structure to brain function. Rev Physiol Biochem Pharmacol. 2003;147:1–46. doi: 10.1007/s10254-003-0005-1. [DOI] [PubMed] [Google Scholar]
- Hsiao B, Dweck D, Luetje CW. Subunit-dependent modulation of neuronal nicotinic receptors by zinc. J Neurosci. 2001;21:1848–1856. doi: 10.1523/JNEUROSCI.21-06-01848.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen AA, Frølund B, Liljefors T, Krogsgaard-Larsen P. Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem. 2005;48:4705–4745. doi: 10.1021/jm040219e. [DOI] [PubMed] [Google Scholar]
- Jindrichova M, Lansdell SJ, Millar NS. Changes in temperature have opposing effects on current amplitude in α7 and α4β2 nicotinic acetylcholine receptors. PLoS One. 2012;7:e32073. doi: 10.1371/journal.pone.0032073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima N, Karasawa J, Shimazaki T, Chaki S, Okuyama S, Yasuhara A, Nakazato A. Neuropharmacological profiles of antagonists of group II metabotropic glutamate receptors. Neurosci Lett. 2005;378:131–134. doi: 10.1016/j.neulet.2004.12.021. [DOI] [PubMed] [Google Scholar]
- Kirby LG, Pan YZ, Freeman-Daniels E, Rani S, Nunan JD, Akanwa A, Beck SG. Cellular effects of swim stress in the dorsal raphe nucleus. Psychoneuroendocrinology. 2007;32:712–723. doi: 10.1016/j.psyneuen.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambe EK, Picciotto MR, Aghajanian GK. Nicotine induces glutamate release from thalamocortical terminals in prefrontal cortex. Neuropsychopharmacology. 2003;28:216–225. doi: 10.1038/sj.npp.1300032. [DOI] [PubMed] [Google Scholar]
- Léna C, Changeux JP. Role of Ca2+ ions in nicotinic facilitation of GABA release in mouse thalamus. J Neurosci. 1997;17:576–585. doi: 10.1523/JNEUROSCI.17-02-00576.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Rainnie DG, McCarley RW, Greene RW. Presynaptic nicotinic receptors facilitate monoaminergic transmission. J Neurosci. 1998;18:1904–1912. doi: 10.1523/JNEUROSCI.18-05-01904.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, González J, Caputo C, Lai FA, Blayney LM, Tan YP, Marty A. Presynaptic calcium stores underlie large-amplitude miniature IPSCs and spontaneous calcium transients. Nat Neurosci. 2000;3:1256–1265. doi: 10.1038/81781. [DOI] [PubMed] [Google Scholar]
- Maggi L, Sola E, Minneci F, Le Magueresse C, Changeux JP, Cherubini E. Persistent decrease in synaptic efficacy induced by nicotine at Schaffer collateral-CA1 synapses in the immature rat hippocampus. J Physiol. 2004;559:863–874. doi: 10.1113/jphysiol.2004.067041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–919. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Marinelli S, Schnell SA, Hack SP, Christie MJ, Wessendorf MW, Vaughan CW. Serotonergic and nonserotonergic dorsal raphe neurons are pharmacologically and electrophysiologically heterogeneous. J Neurophysiol. 2004;92:3532–3537. doi: 10.1152/jn.00437.2004. [DOI] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- Mihailescu S, Palomero-Rivero M, Meade-Huerta P, Maza-Flores A, Drucker-Colín R. Effects of nicotine and mecamylamine on rat dorsal raphe neurons. Eur J Pharmacol. 1998;360:31–36. doi: 10.1016/s0014-2999(98)00658-x. [DOI] [PubMed] [Google Scholar]
- Mihailescu S, Guzmán-Marín R, Drucker-Colín R. Nicotine stimulation of dorsal raphe neurons: effects on laterodorsal and pedunculopontine neurons. Eur Neuropsychopharmacol. 2001;11:359–366. doi: 10.1016/s0924-977x(01)00104-3. [DOI] [PubMed] [Google Scholar]
- Mihailescu S, Guzmán-Marín R, Domínguez Mdel C, Drucker-Colín R. Mechanisms of nicotine actions on dorsal raphe serotoninergic neurons. Eur J Pharmacol. 2002;452:77–82. doi: 10.1016/s0014-2999(02)02244-6. [DOI] [PubMed] [Google Scholar]
- Papke RL, Webster JC, Lippiello PM, Bencherif M, Francis MM. The activation and inhibition of human nicotinic acetylcholine receptor by RJR-2403 indicate a selectivity for the alpha4beta2 receptor subtype. J Neurochem. 2000;75:204–216. doi: 10.1046/j.1471-4159.2000.0750204.x. [DOI] [PubMed] [Google Scholar]
- Paris JM, Cunningham KA. Habenula lesions decrease the responsiveness of dorsal raphe serotonin neurons to cocaine. Pharmacol Biochem Behav. 1994;49:555–560. doi: 10.1016/0091-3057(94)90068-x. [DOI] [PubMed] [Google Scholar]
- Paul IA, Skolnick P. Glutamate and depression: clinical and preclinical studies. Ann NY Acad Sci. 2003;1003:250–272. doi: 10.1196/annals.1300.016. [DOI] [PubMed] [Google Scholar]
- Penington NJ, Kelly JS, Fox AP. A study of the mechanism of Ca2+ current inhibition produced by serotonin in rat dorsal raphe neurons. J Neurosci. 1991;11:3594–3609. doi: 10.1523/JNEUROSCI.11-11-03594.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR. Nicotine as a modulator of behavior: beyond the inverted U. Trends Pharmacol Sci. 2003;24:493–499. doi: 10.1016/S0165-6147(03)00230-X. [DOI] [PubMed] [Google Scholar]
- Rousseau SJ, Jones IW, Pullar IA, Wonnacott S. Presynaptic alpha7 and non-alpha7 nicotinic acetylcholine receptors modulate [3H]d-aspartate release from rat frontal cortex in vitro. Neuropharmacology. 2005;49:59–72. doi: 10.1016/j.neuropharm.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth P, Cheeta S, Tucci S, File SE. Nicotinic-serotonergic interactions in brain and behaviour. Pharmacol Biochem Behav. 2002;71:795–805. doi: 10.1016/s0091-3057(01)00715-8. [DOI] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S. Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron. 2003;38:929–939. doi: 10.1016/s0896-6273(03)00322-2. [DOI] [PubMed] [Google Scholar]
- Sharma G, Grybko M, Vijayaraghavan S. Action potential-independent and nicotinic receptor-mediated concerted release of multiple quanta at hippocampal CA3–mossy fiber synapses. J Neurosci. 2008;28:2563–2575. doi: 10.1523/JNEUROSCI.5407-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher E, Chen Y, Sharples TJ, Broad LM, Benedetti G, Zwart R, McPhie GI, Pearson KH, Baldwinson T, De Filippi G. Physiological roles of neuronal nicotinic receptor subtypes: new insights on the nicotinic modulation of neurotransmitter release, synaptic transmission and plasticity. Curr Top Med Chem. 2004;4:283–297. doi: 10.2174/1568026043451393. [DOI] [PubMed] [Google Scholar]
- Shoop RD, Chang KT, Ellisman MH, Berg DK. Synaptically driven calcium transients via nicotinic receptors on somatic spines. J Neurosci. 2001;21:771–781. doi: 10.1523/JNEUROSCI.21-03-00771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soiza-Reilly M, Commons KG. Glutamatergic drive of the dorsal raphe nucleus. J Chem Neuroanat. 2011;41:247–255. doi: 10.1016/j.jchemneu.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trullas R, Skolnick P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur J Pharmacol. 1990;185:1–10. doi: 10.1016/0014-2999(90)90204-j. [DOI] [PubMed] [Google Scholar]
- Tsuneki H, Klink R, Léna C, Korn H, Changeux JP. Calcium mobilization elicited by two types of nicotinic acetylcholine receptors in mouse substantia nigra pars compacta. Eur J Neurosci. 2000;12:2475–2485. doi: 10.1046/j.1460-9568.2000.00138.x. [DOI] [PubMed] [Google Scholar]
- Vandermaelen CP, Aghajanian GK. Electrophysiological and pharmacological characterization of serotonergic dorsal raphe neurons recorded extracellularly and intracellularly in rat brain slices. Brain Res. 1983;289:109–119. doi: 10.1016/0006-8993(83)90011-2. [DOI] [PubMed] [Google Scholar]
- Whiting PJ, Liu R, Morley BJ, Lindstrom JM. Structurally different neuronal nicotinic acetylcholine receptor subtypes purified and characterized using monoclonal antibodies. J Neurosci. 1987;7:4005–4016. doi: 10.1523/JNEUROSCI.07-12-04005.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonnacott S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- Woolf NJ, Butcher LL. Cholinergic systems in the rat brain. IV. Descending projections of the pontomesencephalic tegmentum. Brain Res Bull. 1989;23:519–540. doi: 10.1016/0361-9230(89)90197-4. [DOI] [PubMed] [Google Scholar]
- Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci. 2003;23:3176–3185. doi: 10.1523/JNEUROSCI.23-08-03176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Lukas RJ. Naturally expressed nicotinic acetylcholine receptor subtypes. Biochem Pharmacol. 2011;82:800–807. doi: 10.1016/j.bcp.2011.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Hu J, Lucero L, Liu Q, Zheng C, Zhen X, Jin G, Lukas RJ, Wu J. Distinctive nicotinic acetylcholine receptor functional phenotypes of rat ventral tegmental area dopaminergic neurons. J Physiol. 2009;587:345–361. doi: 10.1113/jphysiol.2008.162743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz A, Schulz D, Aksoy A, Canbeyli R. Prolonged effect of an anesthetic dose of ketamine on behavioral despair. Pharmacol Biochem Behav. 2002;71:341–344. doi: 10.1016/s0091-3057(01)00693-1. [DOI] [PubMed] [Google Scholar]
- Zoli M, Léna C, Picciotto MR, Changeux JP. Identification of four classes of brain nicotinic receptors using β2 mutant mice. J Neurosci. 1998;18:4461–4472. doi: 10.1523/JNEUROSCI.18-12-04461.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]