

The two major cellular mechanisms responsible for clearance of proteins and organelles are the ubiquitin–proteasome system (UPS) and the autophagy–lysosomal pathway (ALP). Whereas UPS degrades most short-lived soluble proteins, the ALP is involved in degradation of long-lived protein aggregates and organelles. Of the two, autophagy is particularly important for maintaining cellular homeostasis in differentiated, postmitotic neuronal cells. However, despite their different contribution to protein degradation, both UPS and ALP have been implicated in various pathological conditions, including neurodegenerative diseases.
One disease in which dysfunctions of the UPS and ALP have been suggested to play an important role is Parkinson's disease (PD). Pathologically, PD is characterized by progressive neurodegeneration of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) and by the presence of intracellular proteinaceous inclusions (Lewy bodies) in surviving neurons. α-Synuclein (α-syn), a presynaptic protein prone to misfolding and aggregation, is the major component of Lewy bodies. However, larger aggregates and fibrils found in Lewy bodies are believed to be less toxic to neurons compared with α-syn oligomeric species, which occur earlier in α-syn aggregation (Winner et al., 2011).
Three-point mutations in the gene encoding α-syn (SNCA), as well as SNCA multiplications, are linked to dominantly inherited forms of PD. Furthermore, overexpression of wild-type human α-syn or its pathological mutants can induce features of parkinsonism in animal models. Because defects of protein degradation pathways have been observed in the presence of abnormal α-syn expression, recent studies have investigated the mechanism(s) of α-syn degradation under normal and pathological conditions (for review, see Ebrahimi-Fakhari et al., 2012). Overall, the emerging picture is that both UPS and ALP are involved in α-syn clearance, and contribution of each seems to depend on the aggregation state of the protein. More in detail, most of the in vivo studies have shown that the UPS system seems to be responsible for α-syn degradation under physiological conditions, whereas in pathological contexts, when the level of α-syn is increased, the autophagy pathway is activated.
A second protein mutated in cases of dominantly inherited PD is leucine-rich repeat kinase 2 (LRRK2), and emerging evidence suggests that LRRK2 positively regulates autophagy under normal conditions. It has been shown that LRRK2 activates a calcium-dependent protein kinase kinase β (CaMKK-β)/adenosine monophosphate (AMP)-activated protein kinase (AMPK) pathway, which is followed by an increase in formation of autophagosomes (Gómez-Suaga and Hilfiker, 2012), the double-membrane organelles that sequester cellular materials targets and then fuse with lysosomes, where degradation occurs. Additionally, the common LRRK2 mutation G2019S is associated with accumulation of autophagic vacuoles (Ramonet et al., 2011).
Based on the above observations, it seems that impaired homeostasis of α-syn, LRRK2, or both may result in autophagy defects associated with PD. However, the detailed interaction between these players in the context of PD remains unclear.
A recent article by Friedman and collaborators (2012) showed that disruption of autophagic flux in mice DA neurons may be related to PD and, in particular, to α-syn accumulation and LRRK2 regulation. The authors generated a novel Atg7-deficient mouse model (Atg7-cKOTH) in which the Atg7 gene is knocked-out specifically in tyrosine hydroxylase-positive (TH+) cells (Friedman et al., 2012, their Fig. 1A). Atg7 is essential in autophagosome formation, and therefore Atg7 gene depletion impairs autophagy, resulting in substantial accumulation of autophagy substrates, such as the ubiquitin binding protein p62 and, consequentially, ubiquitin itself (Friedman et al., 2012, their Fig. 1B). Interestingly, 9-month-old Atg7-cKOTH mice displayed a 39.8% loss of TH+ neurons (Friedman et al., 2012, their Fig. 2B), whereas younger mice (4 months) did not show significant TH+ neuronal loss, consistent with a combined effect of aging and autophagy impairment on neurodegeneration (Friedman et al., 2012, their Fig. 2A). As predicted, loss of TH+ neurons in 9-month-old transgenic mice (but not in 4-month-old mice) resulted in decreased spontaneous motor activity and coordination compared with controls (Friedman et al., 2012, their Fig. 2C,D,E).
Figure 1.
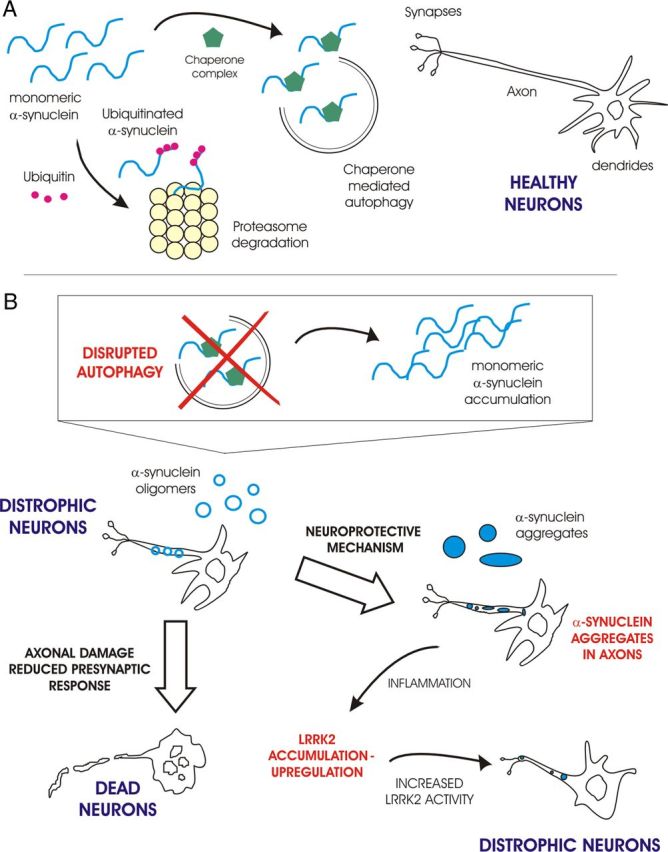
Model for toxic mechanism due to autophagy impairment in PD. A, In healthy neurons, α-syn is degraded by both the UPS and the ALP, keeping axons and dendrites functional. B, When autophagic clearance is impaired, α-syn accumulates in axons and starts forming oligomers that either damage axons and reduce presynaptic response, or further aggregate and become deposited in Lewy bodies. The aggregates slow neuronal degeneration, but cause neuroinflammation, which could induce LRRK2 expression. LRRK2 accumulation leads to increased kinase activity which further affects neurite health.
DA neurons of the SNpc provide synaptic inputs to the striatum, where efferent fibers control movement circuitry. Interestingly, Friedman and coworkers found that the surviving DA neurons in 9-month-old Atg7-cKOTH mice showed reduced TH-fiber density and dystrophic axons in the striatum (Friedman et al., 2012, their Fig. 3). They also observed axonal damage at earlier life stages (i.e., 1-month-old Atg7-cKOTHmice), suggesting that axons are more sensitive to ALP impairment than other domains of DA neurons (Friedman et al., 2012, their Fig. 3).
To better explore why disrupted autophagy affects axonal health in DA neurons, Friedman et al. (2012) examined the levels of α-syn in different cell regions. Aggregated α-syn was present in striatal TH+ axonal swellings, but not in TH+ somata of 20-month-old Atg7-cKOTH mice (Friedman et al., 2012, Fig. 5, B and A, respectively). These findings suggest that when the ALP is not functional, α-syn mainly accumulates in the axons and presynaptic terminals, leading to neuronal fiber degeneration.
Because Lewy bodies and α-syn accumulation can also occur in other neuronal cell types in PD, Friedman et al. (2012) examined mice with Atg7 depletion in all the cells of the CNS (Atg7-cKONes mice). These transgenic mice showed consistent α-syn inclusions in Purkinje cell axons terminating in the deep cerebellar nuclei at a very early stage of life (postnatal day 35) (Friedman et al., 2012, their Fig. 5C). These findings are in agreement with postmortem studies on PD brains, where it was found that Purkinje cells are also victims of α-syn pathology (Mori et al., 2003). Of note, elevated LRRK2 levels were found in Purkinje cell axons (Friedman et al., 2012, their Fig. 6A,B). Interestingly, qPCR analysis in MEF (mouse embryonic fibroblast) cells deficient in Atg7 suggests that LRRK2 accumulation results from increased mRNA content rather than impaired LRRK2 degradation. Unfortunately, the same analysis on mouse neurons is not provided by the authors.
Because Atg7-cKONes mice display a decreased survival rate, a complete analysis of these processes in older mice is not available. A conditional Atg7-cKONes mouse would allow studies of the effects of autophagy impairment at the whole-brain level at specific time points and it may also enable better assessment of the susceptibility of dopaminergic neurons compared with other neuronal types on PD.
This study by Friedman et al. (2012) shows that the depletion of the autophagy pathway in DA neurons leads to a cascade of events that begins with the formation of dystrophic axons and culminates in neuronal degeneration and death, resulting in locomotor deficits. Moreover, the authors found ubiquitinated inclusions in DA neurons, α-syn accumulation in axon terminals, and enhanced levels of LRRK2 in certain brain regions. Together, the results collected in this paper further confirm the link between disrupted autophagy and PD pathogenesis in vivo.
Despite these interesting data, it remains to be determined why α-syn specifically accumulates in the synaptic terminals and LRRK2 expression is enhanced at the transcriptional level under conditions of impaired autophagy. Considering the importance of these findings, in-depth characterization of this process in future studies would shed light on the possible interplay between autophagy, α-syn, and LRRK2. However, to our knowledge, this is the first paper that is able to form a causal link between aging and autophagy impairments in DA neurons in vivo and the PD-related proteins α-syn and LRRK2, further providing new insights in both idiopathic and genetic PD.
Taking into account the results presented by Friedman and collaborators (2012), we propose a basic model that accounts for the relationship between autophagy impairment and sporadic PD (Fig. 1). In healthy neurons (Fig. 1A), the cellular degradation pathways (UPS and ALP) are functional and are responsible for α-syn clearance, including the removal of its misfolded forms. However, if the ALP system is damaged, as in the case of Agt7-deficient mice, DA neurons start to degenerate (Fig. 1B). A possible explanation for axon and dendrite degeneration in Agt7-deficient mice is the initial formation of toxic α-syn oligomeric species (Winner et al., 2011), which occurs when the ALP is impaired and the monomeric α-syn level increases. Early on, this phenomenon is particularly apparent within axons and at presynaptic sites, where α-syn concentration is normally higher. Later, a population of more resistant neurons, still affected by axonal degeneration and showing swollen dendrites, accumulates larger α-syn aggregates (Fig. 1B). These aggregates could work as protective recruiters of α-syn toxic species, allowing these neurons to survive longer, but the accumulation of α-syn aggregates also activates microglia, releasing cytokines and triggering neuroinflammation (Roodveldt et al., 2008). It is known that the release of cytokines is related to a higher level of LRRK2 expression (Gardet et al., 2010). Increased LRRK2 expression/activity can both sustain neuroinflammation and further enhance the neuronal degeneration process, as observed for the LRRK2 G2019S mutant overexpression in cortical cultures (MacLeod et al., 2006).
The proposed model could account for a link between both sporadic and genetic PD and ALP impairment. In the case of idiopathic PD, functioning of the ALP might be impaired because of aging or environmental factors. As described for Atg7-deficient mice, the consequent α-syn accumulation and LRRK2 upregulation could seriously disrupt the homeostasis of DA neurons. This mechanism could also associate neuronal degeneration found in autosomal dominant forms of PD to ALP dysfunction. In fact, it has been shown that ALP impairment occurs when α-syn is overexpressed or mutated with any of the point mutations associated with PD (Xilouri et al., 2009), and that enhanced autophagy mediates neurite retraction induced by the disease-linked G2019S LRRK2 mutant (Ramonet et al., 2011).
Future studies are required to better understand the role of the ALP in both genetic and idiopathic PD, particularly focusing on the equilibrium among autophagic mechanism, α-syn degradation, and LRRK2 regulation. Moreover, it is still not clear why DA neurons are more sensitive to damage during PD progression. A plausible explanation is that α-syn oligomers formed in the presence of dopamine are more toxic. Further studies are needed to test this hypothesis.
Footnotes
Editor's Note: These short, critical reviews of recent papers in the Journal, written exclusively by graduate students or postdoctoral fellows, are intended to summarize the important findings of the paper and provide additional insight and commentary. For more information on the format and purpose of the Journal Club, please see http://www.jneurosci.org/misc/ifa_features.shtml.
This work was supported by the Michael J. Fox Foundation and the Italian Ministry of University and Research (MIUR). N.P. is a PhD student financed by MIUR and L.C. is a Michael J. Fox research fellow. We thank Dr. Sohail Jahid, Dr. Elisa Greggio, Dr. Marco Bisaglia and Prof. Luigi Bubacco for their helpful review of the manuscript.
The authors declare no financial conflicts of interests.
References
- Ebrahimi-Fakhari D, Wahlster L, McLean PJ. Protein degradation pathways in Parkinson's disease: curse or blessing. Acta Neuropathol. 2012;124:153–172. doi: 10.1007/s00401-012-1004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LG, Lachenmayer ML, Wang J, He L, Poulose SM, Komatsu M, Holstein GR, Yue Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of alpha-synuclein and LRRK2 in the brain. J Neurosci. 2012;32:7585–7593. doi: 10.1523/JNEUROSCI.5809-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ, Podolsky DK. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol. 2010;185:5577–5585. doi: 10.4049/jimmunol.1000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Suaga P, Hilfiker S. LRRK2 as a modulator of lysosomal calcium homeostasis with downstream effects on autophagy. Autophagy. 2012;8:692–693. doi: 10.4161/auto.19305. [DOI] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Mori F, Piao YS, Hayashi S, Fujiwara H, Hasegawa M, Yoshimoto M, Iwatsubo T, Takahashi H, Wakabayashi K. Alpha-synuclein accumulates in purkinje cells in Lewy body disease but not in multiple system atrophy. J Neuropathol Exp Neurol. 2003;62:812–819. doi: 10.1093/jnen/62.8.812. [DOI] [PubMed] [Google Scholar]
- Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L, Liu Y, Swing DA, Beal MF, Troncoso JC, McCaffery JM, Jenkins NA, Copeland NG, Galter D, Thomas B, Lee MK, et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One. 2011;6:e18568. doi: 10.1371/journal.pone.0018568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roodveldt C, Christodoulou J, Dobson CM. Immunological features of alpha-synuclein in Parkinson's disease. J Cell Mol Med. 2008;12(5B):1820–1829. doi: 10.1111/j.1582-4934.2008.00450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, Riek R. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One. 2009;4:e5515. doi: 10.1371/journal.pone.0005515. [DOI] [PMC free article] [PubMed] [Google Scholar]