

Abstract
In the present study, the mechanism of action and molecular basis for the activity of the first class of selective inhibitors of the human excitatory amino acid transporter subtype 1 (EAAT1) and its rodent ortholog GLAST are elucidated. The previously reported specificity of UCPH-101 and UCPH-102 for EAAT1 over EAAT2 and EAAT3 is demonstrated to extend to the EAAT4 and EAAT5 subtypes as well. Interestingly, brief exposure to UCPH-101 induces a long-lasting inactive state of EAAT1, whereas the inhibition exerted by closely related analogs is substantially more reversible in nature. In agreement with this, the kinetic properties of UCPH-101 unblocking of the transporter are considerably slower than those of UCPH-102. UCPH-101 exhibits noncompetitive inhibition of EAAT1, and its binding site in GLAST has been delineated in an elaborate mutagenesis study. Substitutions of several residues in TM3, TM4c, and TM7a of GLAST have detrimental effects on the inhibitory potency and/or efficacy of UCPH-101 while not affecting the pharmacological properties of (S)-glutamate or the competitive EAAT inhibitor TBOA significantly. Hence, UCPH-101 is proposed to target a predominantly hydrophobic crevice in the “trimerization domain” of the GLAST monomer, and the inhibitor is demonstrated to inhibit the uptake through the monomer that it binds to exclusively and not to affect substrate translocation through the other monomers in the GLAST trimer. The allosteric mode of UCPH-101 inhibition underlines the functional importance of the trimerization domain of the EAAT and demonstrates the feasibility of modulating transporter function through ligand binding to regions distant from its “transport domain.”
Introduction
(S)-Glutamate (l-glutamate, Glu) is the major excitatory neurotransmitter in the mammalian CNS, where it plays numerous physiological functions. Glu is also a key mediator of the neurotoxicity underlying stroke and ischemia, and glutamatergic dysfunction constitutes a major component in several neurodegenerative disorders (Corona et al., 2007; Johnson et al., 2009). Furthermore, modulation of glutamatergic neurotransmission is a highly pursued strategy in the search for new treatments of epilepsy, pain, and psychiatric disorders (Yogeeswari et al., 2009; Field et al., 2011; Gitto et al., 2012).
Glu mediates its effects through numerous receptors and is taken up into neurons and glial cells by sodium-dependent high-affinity Glu transporters (Danbolt, 2001). The five excitatory amino acid transporters (EAATs) belong to the solute carrier 1 transporter family, and the human EAAT1-EAAT5 subtypes correspond to the rodent orthologs GLAST, GLT-1, EAAC1, EAAT4, and EAAT5, respectively (Bunch et al., 2009). The subtypes are differentially expressed in the CNS and at the cellular level and thus play different roles in glutamatergic neurotransmission (Maragakis and Rothstein, 2004; Bunch et al., 2009).
The insights into EAAT structure–function aspects have in recent years been greatly advanced from crystal structures of a prokaryotic homolog of the transporters, the GltPh transporter from Pyrococcus horikoshii (Yernool et al., 2004; Boudker et al., 2007; Reyes et al., 2009; Verdon and Boudker, 2012). The EAAT (and GltPh) exists as a trimeric complex in which each monomer constitutes an independent functional unit (Gendreau et al., 2004; Grewer et al., 2005; Koch and Larsson, 2005; Leary et al., 2007). The monomer consists of eight primarily α-helical transmembrane regions (TM1-TM8) and two reentrant helical hairpin loops (HP1, HP2), and intracellular N- and C-termini. The N-terminal TM1-TM6 segment is arranged as a distorted cylinder forming the outer surface of the complex to its surroundings and with the two other monomers in the trimer. Although intermonomeric contacts in the trimer are mediated by TM2, TM4, and TM5 exclusively, the entire TM1-TM6 segment has been termed the “trimerization domain” (Reyes et al., 2009; Verdon and Boudker, 2012). The C-terminal segment (HP1/TM7/HP2/TM8) is folded into a compact core contained within this cylinder and constitutes the “transport domain” of the monomer (Kanner and Zomot, 2008; Boudker and Verdon, 2010; Jiang and Amara, 2011).
Investigations into the physiological functions of the respective EAATs have long been hampered by the lack of truly selective pharmacological tools (Bunch et al., 2009). Recently, we have discovered the first class of specific EAAT1 inhibitors (Jensen et al., 2009), and UCPH-101 from this series has subsequently been applied in several studies of EAATs in native tissues (Lane and Lawen, 2012; Lee et al., 2012; Okabe et al., 2012; Salvatore et al., 2012; Uwechue et al., 2012). Comprehensive structure–activity relationship studies have identified several key pharmacophore elements and in this way elucidated the structural determinants for EAAT1 activity of this compound scaffold (Jensen et al., 2009; Erichsen et al., 2010; Huynh et al., 2012a; b). In the present study, we have elucidated the mechanism of action and molecular basis for the EAAT1 activity of UCPH-101 and its analogs from the perspective of the transporter.
Materials and Methods
Materials.
Culture media, serum, antibiotics, and buffers for cell culture were obtained from Invitrogen. Glu was purchased from Sigma, dl-threo-β-benzyloxyaspartate (TBOA) from Tocris Bioscience Cookson, [3H]-d-aspartate ([3H]-d-Asp) from PerkinElmer, and the FLIPR Membrane Potential Blue (FMP) assay dye from Molecular Devices. The UCPH-101, UCPH-102, UCPH-100, 1, 2, and 3 analogs (Fig. 1) were synthesized as previously described (Jensen et al., 2009; Erichsen et al., 2010) (analog 1; M.N. Erichsen, J. Hansen, B. Abrahamsen, T.H.V. Huynh, C.S. Demmer, A.A. Jensen, and L. Bunch, unpublished observations). Human EAAT1 cDNA was a kind gift from Dr. S.G. Amara (University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania), and Dr. T. Rauen (University of Osnabrück, Osnabrück, Germany) kindly provided the rat GLAST and GLT-1 cDNAs. The rat EAAT4 and mouse EAAT5 cDNAs were generous gifts from Dr. J. Rothstein (Johns Hopkins University, Baltimore, Maryland) and Dr. Cordeiro (Christian-Albrechts-University, Kiel, Germany), respectively. The 5-HT3A and 5-HT3B cDNAs were kind gifts from Drs. J. Egebjerg and E.F. Kirkness, respectively, and the construction of the HA-5-HT3B-pCIneo plasmid has been described previously (Krzywkowski et al., 2008).
Figure 1.

Chemical structures of UCPH-101 and the five additional analogs used in this study. The parental chromene skeleton with its R1 and R2 substituents is given for reference.
Molecular biology.
For the electrophysiological recordings, human EAAT1, rat EAAT4, and mouse EAAT5 were expressed in tsA201 cells as fusion proteins containing an amino-terminal fluorescent protein. The construct encoding EAAT1 was formed by linking the coding region of EAAT1 via an AgeI site with a yellow fluorescent protein (YFP) into an open reading frame and subcloning into pcDNA3.1(+) was performed by using EcoRI and XbaI restriction sites. Comparable expression constructs were generated for rat EAAT4 and mouse EAAT5, which were linked via BsrGI to a monomeric YFP (mYFP). The BsrGI site was created by introduction of a silent mutation with PCR. The rEAAT4 cDNA was subcloned into pcDNA3.1(−) using BamHI and BsgI, and mEAAT5 cDNA into pcDNA3.1(+) using BspEI and HindIII restriction sites.
The rat GLAST and GLT-1 cDNAs were subcloned from their original vectors into pcDNA3.1+/hygro using the restriction enzymes NheI and XhoI. Chimeric GLAST/GLT1 constructs were constructed by use of splicing overlap extension PCR (Horton et al., 1989). This method was also used to insert a DNA sequence encoding for the HA epitope into GLAST-pcDNA3.1+/hygro and EAAT1-pcDNA3. Mutations were introduced into the GLAST- and GLT1-pcDNA3.1+/hygro plasmids by the QuikChange mutagenesis kit (Stratagene). The absence of unwanted mutations in all cDNAs created by PCR was verified by DNA sequencing (Eurofins MWG Operon).
Cell culture and transfections.
HEK293, tsA201, and stable EAAT1-HEK293 cells (Jensen and Bräuner-Osborne, 2004) were cultured in GlutaMAX-I DMEM supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml), and 5% dialyzed FBS (the medium for the EAAT1-HEK293 cell line was supplemented with 1 mg/ml G-418) in a humidified atmosphere of 5% CO2 at 37°C. For the transfection of tsA201 cells, 8 × 105 cells were split into a 6 cm tissue culture plate and transfected the following day with a total of 4 μg cDNA using Polyfect according to the manufacturer's instructions (QIAGEN), and the cells were used for the various assays 40–48 h after the transfection. Analogously to the procedure used in a previous study (Jensen et al., 2007), polyclonal HEK293 cell lines stably expressing wild-type (WT) GLAST, WT GLT-1, chimeric GLAST/GLT-1 transporters, and GLAST and GLT-1 mutants were generated by culturing the transfected cells in medium containing 300 μg/ml hygromycin B for 3–4 weeks before use. The tsA201 cells used for the electrophysiological recordings were transfected using the Ca3(PO4)2 technique as previously described (Melzer et al., 2003). In these recordings, two independent recombinants were tested and shown to exhibit indistinguishable functional properties.
Electrophysiological recordings.
Standard whole-cell patch-clamp recordings were performed using an Axopatch 200B amplifier (Molecular Devices) as described previously (Melzer et al., 2003; Winter et al., 2012). Borosilicate pipettes were pulled with resistances between 1.0 and 2.0 MΩ. Cells were clamped to 0 mV for 1 s or 2 s between test sweeps. Voltage errors were reduced by compensating at least 80% of series resistance by an analog procedure. Cells with current amplitudes >10 nA were excluded from the analysis. Currents were low-pass filtered at 5 kHz and digitized with a sampling rate of 20 kHz using a Digidata AD/DA converter (Molecular Devices).
Bath solutions contained (in mm) 140 NaNO3, 4 KCl, 2 CaCl2, 1 MgCl2, 20 HEPES, 5 tetraethylammonium chloride, and 0.5 or 1 Glu, respectively. Pipettes were filled with 115 KNO3, 2 MgCl2, 5 EGTA, and 10 HEPES. Glu- and UCPH-101-dependences were determined by application of indicated concentrations of Glu or/and the inhibitor. In bath or internal solutions, pH was adjusted to 7.4 with NaOH or KOH, respectively. External and internal salt agar bridges were made from a plastic tube filled with 140 mm KCl in 3% agar and used for electrical connection with the Ag/AgCl electrode.
[3H]-d-Asp uptake assay.
The [3H]-d-Asp uptake assay was performed essentially as previously described (Jensen and Bräuner-Osborne, 2004). Briefly, cells were split into poly-d-lysine-coated white 96-well plates (PerkinElmer). At 16–24 h later, the culture medium was aspirated, and cells were washed once with 100 μl assay buffer (HBSS supplemented with 20 mm HEPES, 1 mm CaCl2, and 1 mm MgCl2, pH 7.4). Then 50 μl assay buffer supplemented with 100 nm [3H]-d-Asp and various concentrations of test compound were added to the wells, and the plate was incubated at 37°C for 6 min. Nonspecific [3H]-d-Asp uptake was determined in wells with 3 mm Glu. The assay mixtures was quickly removed from the wells, which were then washed with 2 × 100 μl ice-cold assay buffer, and 150 μl Microscint20 scintillation fluid (PerkinElmer) was added to each well. The plate was shaken for 1 h and counted in a Wallac 1450 MicroBeta Trilux scintillation counter (GMI). The experiments were performed in duplicate at least 3 times for each ligand at the various transporters. Specific details concerning the experiments where preincubation and washing steps were performed before the execution of the [3H]-d-Asp uptake are given in the legend to Figure 3.
Figure 3.

UCPH-101 induces a long-lasting inactive state of EAAT1. A, The inhibitory potency of UCPH-101 at EAAT1-HEK293 cells increases with the length of the incubation period in the [3H]-d-Asp uptake assay, whereas those of Glu and TBOA do not. The IC50 (pIC50 ± SEM) values of UCPH-101 using incubation periods of 1.5, 3, 6, and 12 min were 1.9 μm (5.71 ± 0.05), 1.1 μm (5.97 ± 0.07), 0.82 μm (6.09 ± 0.05), and 0.44 μm (6.36 ± 0.04), respectively (n = 4). B, Preincubation of EAAT1 with UCPH-101 induces long-lasting inhibition of transporter function. EAAT1-HEK293 cells were incubated with buffer or buffer supplemented with various concentrations of UCPH-101 or TBOA for 15 min and washed for 3 × 15, 3 × 30, or 3 × 45 min before determination of specific [3H]-d-Asp uptake (n = 4). C, The degree of EAAT1 inhibition induced by preincubation with UCPH-101 increases with the duration of the preincubation. EAAT1-HEK293 cells were incubated with buffer or with buffer supplemented with UCPH-101 or TBOA for various time periods, and washed for 3 × 15 min before determination of specific [3H]-d-Asp uptake (n = 3). D, E, Glu and TBOA do not alleviate the long-lasting effects of UCPH-101 preincubation on EAAT1 function. D, EAAT1-HEK293 cells were incubated for 1 min with buffer (■) or with buffer supplemented with 3 mm Glu (□) or 300 μm TBOA (●), then incubated for 2 min with various concentrations of UCPH-101 in buffer (■) or in buffer supplemented with 3 mm Glu (□) or 300 μm TBOA (●), and then washed for 3 × 15 min before determination of specific [3H]-d-Asp uptake (n = 3). E, EAAT1-HEK293 cells were incubated for 1 min with various concentrations of UCPH-101 in buffer, then incubated for 2 min with buffer (■) or with buffer supplemented with 3 mm Glu (□) or 300 μm TBOA (●), and then washed for 3 × 15 min before determination of specific [3H]-d-Asp uptake (n = 4). F, Preincubation with UCPH-101 does not change the cell surface expression levels of EAAT1 and GLAST. HA-EAAT1- or HA-GLAST-expressing tsA201 cells were incubated with buffer or buffer supplemented with 100 μm UCPH-101 for 15 min, and washed for 3 × 15 min before the ELISA was performed (n = 4). The cell surface expression of neither HA-EAAT1 nor HA-GLAST in buffer- and UCPH-101-pretreated cells differed significantly. Preexposure of cells to the inhibitor resulted in a small but significant decrease in total expression levels (ANOVA: HA-EAAT1: p = 0.048, HA-GLAST: p = 0.049). G, Effects on EAAT1 function of preincubation with UCPH-101 and five analogs. Left, EAAT1-HEK293 cells were preincubated with buffer supplemented with various concentrations of the inhibitors for 15 min, and washed for 3 × 15 min before determination of specific [3H]-d-Asp uptake (n = 4). Right, Relationship between pIC50 values for the UCPH-101, UCPH-102, UCPH-100, and 1 in the [3H]-d-Asp uptake assay and the pIC50 values obtained for the compounds in the preincubation experiment. The R2 values for the fittings of the data for UCPH-100, 1, and UCPH-102 (black line) and the data for UCPH-100, 1, UCPH-102, and UCPH-101 (gray line) are given.
FMP assay.
The functional characterization of UCPH-101 and TBOA at stable EAAT1-HEK293 cells in the FMP assay was performed essentially as previously described (Jensen and Bräuner-Osborne, 2004). Cells were split into poly-d-lysine-coated black 96-well plates with clear bottom (BD Biosciences). At 16–24 h later, the medium was aspirated, and the cells were washed with 100 μl Krebs buffer [140 mm NaCl/4.7 mm KCl/2.5 mm CaCl2/1.2 mm MgCl2/11 mm HEPES/10 mm d -glucose, pH 7.4]; 50 μl Krebs buffer supplemented with various concentrations of UCPH-101 or TBOA was added to the wells, after which an additional 50 μl Krebs buffer supplemented with the FMP assay dye (1 mg/ml) was added to each well. The plate was incubated at 37°C in a humidified 5% CO2 incubator for 30 min and assayed in a NOVOstar plate reader (BMG Laboratory Technologies) measuring emission [in fluorescence units (FU)] at 560 nm caused by excitation at 530 nm before and up to 1 min after addition of 33 μl Glu solution. The experiments were performed in duplicate at least three times for each compound.
Quantification of transporter expression by ELISA.
The ELISA was performed essentially as previously described (Krzywkowski et al., 2008). Briefly, tsA201 cells were transfected with EAAT1-pcDNA3, GLAST-pcDNA3.1+/hygro, HA-EAAT1-pcDNA3, or HA-GLAST1-pcDNA3.1+/hygro, or cotransfected with 5-HT3A-pCIneo and HA-5-HT3B-pCIneo (ratio 1:1). At 16–24 h after the transfection, the cells were plated into poly-d-lysine-coated 48-well plates (1.5 × 105 cells/well). The following day, the cells were washed in ice-cold assay buffer (PBS supplemented with 1 mm CaCl2), after which the cells were fixed by treatment of 4% paraformaldehyde (in PBS) on ice. The remaining steps were performed at room temperature. The cells were washed three times in assay buffer and incubated with blocking solution (3% dry milk in 50 mm Tris-HCl, 1 mm CaCl2, pH 7.5) for 20 min. After blocking, the cells were incubated with mouse anti-HA antibody (Nordic Biosite, diluted 1:1000 in blocking solution) for 45 min. The cells were then washed three times with assay buffer and incubated with blocking solution for 20 min before incubation with horseradish peroxidase-conjugated secondary antibody (Invitrogen, diluted 1:400 in blocking solution) for 45 min. After three washes with assay buffer, the subunit expression was quantified using the 3,3′,5,5′-tetramethylbenzidine liquid substrate system (Sigma). The reaction was quenched with 1N H2SO4 after which the absorbance of the supernatants was determined at 450 nm. Total expression levels were determined by adding 0.1% Triton X-100 to the blocking solution used for the first blocking incubation and the incubation with anti-HA antibody. All ELISA experiments were performed in quadruplicate. Specific details concerning the preincubation and washing steps performed before the execution of the ELISA in the determination of the effects of UCPH-101 preincubation on transporter expression levels are given in the legend for Figure 3F.
Data analysis.
Data from the electrophysiological recordings were analyzed with a combination of pClamp10 (Molecular Devices) and SigmaPlot11 (Jandel Scientific) programs. Current amplitudes were used without any subtraction procedure. Data are given as mean ± SE. In experiments where UCPH-101 or UCPH-102 concentration dependences were determined, we obtained relative errors for apparent dissociation constants as SEs of fit estimates from fitting a Hill equation to the concentration dependence of EAAT1 anion currents. To evaluate time-constants for UCPH-binding and -unbinding events on EAAT1 transporters, we fitted monoexponential functions to the time courses of changes in EAAT1 mediated anion currents.
Concentration-inhibition curves for Glu and the inhibitors at the transporters in the [3H]-d-Asp uptake assay were constructed based on the radioactivity level (CPM) measured in the respective wells, after the nonspecific uptake measured in the presence of 3 mm Glu had been subtracted. Concentration–response curves for Glu and concentration–inhibition curves for UCPH-101 and TBOA at EAAT1-HEK293 cells in the FMP assay were constructed based on the difference in the fluorescence (ΔFU) between the maximal fluorescence recorded before and after addition of Glu obtained for the different ligand concentrations. All curves were generated by nonweighted least-squares fits using the program KaleidaGraph 3.6 (Synergy Software). As for the ELISA, the basal absorbance measured in wells containing tsA201 cells transfected with (untagged) WT EAAT1 or WT GLAST were subtracted from the absorbance measured from cells transfected with HA-EAAT1 cDNA or with cDNAs encoding for HA-GLAST and various HA-GLAST mutants, respectively.
Computational chemistry.
The in silico studies were performed using the comprehensive software suite MOE (Version 2012.05, Chemical Computing Group) installed on a PC-Windows 7 professional, 32-bit platform. Homology modeling: A homology model of rat GLAST was built based on an aligment of its amino acid sequence (NP_062098) with that of the GltPh transporter. The residues Phe190-Val241 segment (both residues included) between TM4b and TM4c in GLAST is absent in GltPh (Yernool et al., 2004), and analogously to a previously published homology model of EAAT1 this segment was deleted from the GLAST sequence before construction of the homology model (Huang and Vandenberg, 2007). The homology model of GLAST was built from the 3D coordinates of the crystal structure of GltPh in complex with l-aspartate and Na+ [PDB code 2NWX (Boudker et al., 2007)] using standard setup parameters. Binding site: Exploration of the binding site was done using the build-in function “Site Finder” (standard setup).
The logP and total polar surface area (tPSA) values for UCPH-101, UCPH-102, UCPH-100, 1, 2, and 3 were calculated using the standard parameters in the setup box.
Results
UCPH-101 and UCPH-102 are highly selective inhibitors of EAAT1 over other EAATs
UCPH-101 and UCPH-102 have previously been shown to be specific inhibitors of human EAAT1 and its rat ortholog GLAST displaying high nanomolar IC50 values at these transporters and negligible inhibitory activity at human EAAT2, human EAAT3, rat GLT-1, and rat EAAC1 at concentrations up to 100 μm (Jensen et al., 2009; Erichsen et al., 2010). To investigate whether this selectivity extends to the entire EAAT family, the functional properties of UCPH-101 and UCPH-102 were characterized by patch-clamp electrophysiology on tsA201 cells expressing human EAAT1, rat EAAT4, and mouse EAAT5 (Fig. 2).
Figure 2.
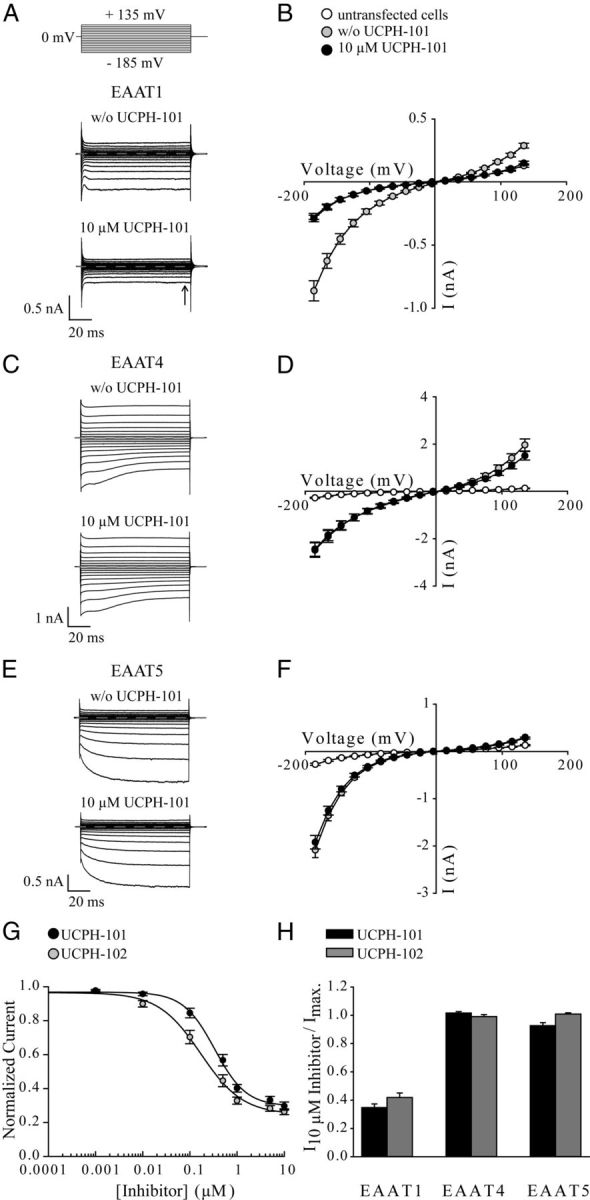
Effects of UCPH-101 and UCPH-102 on whole-cell currents of EAAT1, EAAT4, and EAAT5. A, C, E, Representative whole-cell currents from tsA201 cells expressing EAAT1 (A), EAAT4 (C), and EAAT5 (E) in the absence (top) and presence (bottom) of 10 μm UCPH-101. The compound was added to bath solution containing 140 mm NaNO3 and 0.5 mm Glu, and cells were dialyzed with 115 mm KNO3. Dotted lines represent 0 nA. B, D, F, Current–voltage relationship of EAAT1 (n = 14) (B), EAAT4 (n = 16) (D), and EAAT5 (n = 11) (F) in the absence ( ) or presence (●) of 10 μm UCPH-101. Untransfected tsA201 cells perfused with external solution containing 0.5 mm Glu were used as control (○) (n = 9). G, Application of different concentrations of UCPH-101 or UCPH-102 to cells expressing EAAT1 at a voltage step of −185 mV (solutions as in A–F). Data were normalized to the current in absence of compounds. Fitting concentration dependences of EAAT1 steady-state currents (arrow in Fig. 2A) with a Hill equation provided dissociation constants of 0.34 ± 0.03 μm (Hill coefficient = 1.3 ± 0.13, n ≥ 9) for UCPH-101 and 0.17 ± 0.02 μm (Hill coefficient = 0.97 ± 0.11, n ≥ 7) for UCPH-102. H, Effect of 10 μm UCPH-101 or 10 μm UCPH-102 on anion currents of EAAT1, EAAT4, and EAAT5 at a voltage step of −185 mV (solutions as in A–F). To determine the inhibitory effect of UCPH-101 (black bar) and UCPH-102 (gray bar) on EAAT-mediated anion currents, we divided steady-state currents in the presence of UCPH-101/UCPH-102 by currents in the absence of compounds. Data are given as mean ± SE.
) or presence (●) of 10 μm UCPH-101. Untransfected tsA201 cells perfused with external solution containing 0.5 mm Glu were used as control (○) (n = 9). G, Application of different concentrations of UCPH-101 or UCPH-102 to cells expressing EAAT1 at a voltage step of −185 mV (solutions as in A–F). Data were normalized to the current in absence of compounds. Fitting concentration dependences of EAAT1 steady-state currents (arrow in Fig. 2A) with a Hill equation provided dissociation constants of 0.34 ± 0.03 μm (Hill coefficient = 1.3 ± 0.13, n ≥ 9) for UCPH-101 and 0.17 ± 0.02 μm (Hill coefficient = 0.97 ± 0.11, n ≥ 7) for UCPH-102. H, Effect of 10 μm UCPH-101 or 10 μm UCPH-102 on anion currents of EAAT1, EAAT4, and EAAT5 at a voltage step of −185 mV (solutions as in A–F). To determine the inhibitory effect of UCPH-101 (black bar) and UCPH-102 (gray bar) on EAAT-mediated anion currents, we divided steady-state currents in the presence of UCPH-101/UCPH-102 by currents in the absence of compounds. Data are given as mean ± SE.
EAAT anion currents were measured with internal KNO3 and NaNO3-based external solution at a saturating Glu concentration. The use of NO3− increases anion currents and permits measurements of anion currents in isolation. Representative current recordings of EAAT1, EAAT4, and EAAT5 from individual cells before and after application of saturating concentrations of UCPH-101 are given in Figure 2A, C, and E, respectively. Current–voltage relationships for EAAT1 (Fig. 2B), EAAT4 (Fig. 2D), and EAAT5 (Fig. 2F) in the absence (gray circles) and presence of 10 μm UCPH-101 (black circles) illustrate the effects of the inhibitor on the different EAAT-isoforms.
UCPH-101 and UCPH-102 inhibited EAAT1 anion currents in a concentration-dependent manner, with KD values of 0.34 ± 0.03 μm (Hill = 1.3 ± 0.13, n ≥ 9) for UCPH-101 and 0.17 ± 0.02 μm (Hill = 0.97 ± 0.11, n ≥ 7) for UCPH-102 (Fig. 2G). At saturating UCPH-101 or UCPH-102 concentrations, EAAT1 currents were not different from anion currents in untransfected cells (Fig. 2B, ○), indicating complete block of EAAT1 by both compounds. Thus, the block of EAAT1 exerted by UCPH-101 and UCPH-102 can be described accurately by whole-cell recordings.
In contrast to their observed activity at EAAT1, neither UCPH-101 nor UCPH-102 caused significant inhibition of whole-cell currents in EAAT4-expressing (Fig. 2D,H) or EAAT5-expressing cells (Fig. 2F, H) at final concentrations of 10 μm. We conclude that neither EAAT4 nor EAAT5 is blocked by UCPH-101 or UCPH-102 in the low concentration range detected for EAAT1; thus, these data further support the notion of these compounds being highly selective inhibitors of EAAT1.
UCPH-101 induces long-lasting inhibition of EAAT1
In preliminary studies of various assay parameters in the [3H]-d-Asp uptake assay, the duration of the incubation time was found to be important for the activity of UCPH-101 at EAAT1. A significant, albeit modest, increase in the inhibitory potency of UCPH-101 at the transporter was observed with increasing durations of the incubation period, whereas the IC50 values for Glu and TBOA did not change significantly (Fig. 3A). Strikingly, preincubation of EAAT1-HEK293 cells with UCPH-101 followed by extensive washing resulted in a dramatic reduction in the specific [3H]-d-Asp uptake mediated by the cells (Fig. 3B). In contrast, preexposure of the cells to TBOA in concentrations up to 100-fold higher than its IC50 value followed by the same washing regimen did not impair transporter function significantly (Fig. 3B). The impact of UCPH-101 preincubation on EAAT1 function was concentration-dependent, and the degree of inhibition observed after preincubation was not altered significantly with different washing regimens (Fig. 3B). Even brief preincubation (∼6 s) with 100 μm UCPH-101 induced long-lasting inhibition of EAAT1, although the potency of the compound in its induction of this long-lasting inhibition increased with longer preincubation periods (Fig. 3C). Incubation of the EAAT1-HEK293 cells with 3 mm Glu or 300 μm TBOA before and concomitant presence of 3 mm Glu or 300 μm TBOA during the UCPH-101 preincubation did not alleviate the impairment of EAAT1 function brought on by UCPH-101 (Fig. 3D), nor did an incubation with 3 mm Glu or 300 μm TBOA in between the UCPH-101 preincubation and the execution of the [3H]-d-Asp uptake assay (Fig. 3E).
Exposure of EAAT1 and GLAST to UCPH-101 does not internalize the transporters
Sustained exposure of the serotonin transporter (from the SLC6 transporter family) to inhibitors has been reported to internalize the transporter in mammalian cell lines and neurons (Lau et al., 2008; Kittler et al., 2010), and antagonist-induced internalization has also been demonstrated for several membrane-bound receptors (Roettger et al., 1997; Gray and Roth, 2001; Pheng et al., 2003; Rojas et al., 2010). To investigate whether the observed long-lasting effects of UCPH-101 preincubation on EAAT1 function could arise from an analogous ligand-induced internalization of the transporter, the effects of UCPH-101 on the expression levels of EAAT1 and GLAST at the cell surface were investigated in an ELISA using HA-tagged transporters transiently expressed in tsA201 cells. The validity of this assay was verified in control experiments performed in parallel, where expression of an HA-tagged 5-HT3B receptor subunit in tsA201 was found not to result in significant cell surface expression, whereas of coexpression of HA-5-HT3B with 5-HT3A gave rise to the significant levels of cell surface expression of the tagged subunit (B. Abrahamsen and A.A. Jensen, unpublished observations) (Krzywkowski et al., 2008).
The HA epitope (YPYDVPDYA) was inserted between residues Pro179 and Pro180 in both EAAT1 and GLAST. In a previous study, mouse GLT-1 tagged with HA in the corresponding position has been shown to exhibit pharmacological properties not significantly different from those of the WT transporter (Peacey et al., 2009). Analogously, the IC50 values displayed by Glu, TBOA, and UCPH-101 at HA-EAAT1 and HA-GLAST in the [3H]-d-Asp uptake assay were not significantly different from those displayed by the ligands at the two WT transporters (B. Abrahamsen and A.A. Jensen, unpublished observations). As can be seen from Figure 3F, preincubation of tsA201 cells transiently expressing HA-EAAT1 and HA-GLAST with 100 μm UCPH-101 did not result in significant reduction in the numbers of cell surface-expressed transporters compared with control cells. A small but significant decrease in the total expression levels of both HA-EAAT1 and HA-GLAST was observed in cells preincubated with 100 μm UCPH-101 (p = 0.048 and p = 0.049, respectively; Fig. 3F). However, these differences are not considered pertinent from a biological perspective.
UCPH-101 and closely related analogs induce substantially different time-dependent inhibition of EAAT1
To shed light on the structural elements in the UCPH-101 molecule important for its long-lasting impairment of EAAT1-mediated uptake, the effects of preincubation of EAAT1 with five closely related UCPH-101 analogs on transporter function were investigated. The five analogs have the parental chromene skeleton in common with UCPH-101, but their 4- and 7-substituents differ (Fig. 1). Interestingly, the pronounced impairment of EAAT1 uptake observed after preincubation with UCPH-101 was contrasted by much more moderate effects on transporter function resulting from preexposure to the five analogs (Fig. 3G; Table 1). UCPH-101, UCPH-102, and 1 display similar inhibitory potencies at EAAT1 in the [3H]-d-Asp assay (Table 1) and, in the case of UCPH-101 and UCPH-102, in the electrophysiological recordings (Fig. 2). Nevertheless, UCPH-101 was found to be ∼100-fold more potent than both analogs when it came to the induction of the inactive EAAT1 state in the preincubation experiments (Fig. 3G; Table 1). Preincubation of EAAT1 with the UCPH-100, 2, and 3 analogs had moderate or negligible effects on transporter function (Fig. 3G; Table 1). The divergence of UCPH-101 from the other analogs is illustrated by the fact that the correlation between the inhibitory potencies for UCPH-102, UCPH-100, and 1 in the [3H]-d-Asp uptake assay and in the preincubation experiment is characterized by an R2 value of 0.96, which is reduced to 0.31 when the UCPH-101 data are included (Fig. 3G).
Table 1.
Physicochemical characteristics of UCPH-101 and five analogs and their functional properties at EAAT1-HEK293 cells in the [3H]-d-Asp uptake assay and in the preincubation experiments. The logP values (octanol/water) and total polar surface area (tPSA) were calculated in MOE. IC50 values obtained for the inhibitors in the [3H]-d-Asp uptake assay and in the preincubation experiments are given in μm with pIC50 ± SEM values in parentheses
| Compound | logP | tPSA | [3H]-d-Asp uptakea IC50 (μm) (pIC50 ± SEM) | Preincubation IC50 (μm) (pIC50 ± SEM) |
|---|---|---|---|---|
| UCPH-101 | 3.94 | 151 | 0.66 (6.18 ± 0.08) | 0.23 (6.64 ± 0.15) |
| UCPH-102 | 2.71 | 156 | 0.42 (6.47 ± 0.21) | 20 (4.71 ± 0.20) |
| UCPH-100 | 2.76 | 150 | 3.9 (5.48 ± 0.20) | ∼300 (∼3.5) |
| 1 | 3.45 | 155 | 0.87 (6.11 ± 0.15) | ∼30 (∼4.5) |
| 2 | 2.42 | 262 | 11 (4.97 ± 0.05) | >300 (<3.5) |
| 3 | 1.49 | 156 | 9.2 (5.04 ± 0.05) | >300 (<3.5) |
Results from the preincubation experiments are based on 3–4 individual experiments performed in duplicate as described in Materials and Methods and in the legend to Figure 3G.
a The IC50 values for the ligands in the [3H]-d-Asp uptake assay have been reported previously: UCPH-101, 2 and 3 (Jensen et al., 2009), UCPH-102 and UCPH-100 (Erichsen et al., 2010) and 1 (M.N. Erichsen, J. Hansen, B. Abrahamsen, T.H.V. Huynh, C.S. Demmer, A.A. Jensen, and L. Bunch, unpublished observations).
The observed different duration of the EAAT1 inhibition exerted by the six analogs could potentially arise from differences in the physicochemical properties of the compounds. However, the calculated tPSA values for UCPH-101, UCPH-102, UCPH-101, 1, and 3 are essentially identical, and although UCPH-101 is the most lipophilic compound of the series, the calculated logP value of analog 1 is comparable with that of UCPH-101 (3.45 and 3.94, respectively; Table 1). Furthermore, it is not possible to pinpoint a single structural feature in the UCPH-101 molecule accountable for its long-lasting effects on EAAT1: Comparisons of the IC50 value displayed by UCPH-101 to those of UCPH-100 and 2 in the uptake assay and of the IC50 values of UCPH-102 and 1 to that of 3 identify the 1-naphthalene or 1-o-biphenyl groups in these inhibitors as key for their higher inhibitory potencies at EAAT1 (Fig. 1; Table 1). However, the presence of a 1-naphthalene group in both UCPH-101 and UCPH-102 clearly demonstrates that the lipophilic 7-substituent is not the sole determinant of the sustained inhibition associated with UCPH-101. The 4-substituent in the chromene scaffold is the obvious other candidate, as UCPH-101 and UCPH-102 only differ when it comes to their respective 4-methoxyphenyl and methyl groups in this position (Fig. 1). However, because the IC50 values of 2 and UCPH-100 in the preincubation experiment correlate with their respective potencies in the [3H]-d-Asp uptake assay, the presence of a 4-methoxyphenyl or a phenyl group in this position is clearly not sufficient to induce the long-lasting inhibition characteristic in these ligands (Fig. 3G). Hence, this unique property of UCPH-101 appears to arise from system dynamics induced by small-molecule-protein interactions.
UCPH-101 and UCPH-102 display significantly different blocking and unblocking kinetics at EAAT1
To determine the binding kinetics of UCPH-101 and UCPH-102 at EAAT1, the time course of EAAT1 current amplitudes was recorded at a fixed voltage step of −145 mV upon application (wash in) or removal (wash out) of 1 μm UCPH-101 (Fig. 4A) or 1 μm UCPH-102 (Fig. 4B). The experiments were performed in the presence of saturating concentrations of Glu. Application of 1 μm UCPH-101 or 1 μm UCPH-102 resulted in monoexponential decreases of the current amplitude with time constants of 10.5 ± 1.4 s (UCPH-101, n = 11) and 4.0 ± 1.2 s (UCPH-102, n = 6) (Fig. 4A left and 4B left, respectively). After reaching steady state, the perfusion was changed to inhibitor-free solution to evaluate the time course of the unbinding of UCPH-101 and UCPH-102 from EAAT1 (Fig. 4A right and 4B right, respectively). UCPH-101 unblocking occurred on a very slow time scale (τ = 740 ± 100 s, n = 9), whereas the block induced by UCPH-102 was relieved much faster (τ = 55.7 ± 4.8 s, n = 6). Because full recovery of the EAAT current amplitude occurred only rarely after UCPH-101 block, the unblocking time constant for this compound might be underestimated.
Figure 4.

Unbinding of UCPH-101 and UCPH-102 from EAAT1 occurs on different time scales. A, B, Left, EAAT1 current-responses to application of 1 μm UCPH-101 (A) or 1 μm UCPH-102 (B) at repetitive voltage steps of −145 mV recorded with a time interval of 1 s between successive steps. Anion current peak amplitudes decayed with time constants of 10.5 ± 1.4 s (n = 11) after application of UCPH-101 and 4.0 ± 1.2 s (n = 6) after application of UCPH-102. Right, Unbinding of UCPH-101 (A) and UCPH-102 (B) from EAAT1 induced by application of inhibitor-free solution to cells expressing the transporter. Peak current amplitudes increased with time constants of 740.4 ± 100 s for UCPH-101 (n = 9) and 55.7 ± 4.8 s for UCPH-102 (n = 6). Currents were fitted with an exponential function. Data are given as mean ± SE. Insets, Representative current recordings.
UCPH-101 exhibits noncompetitive inhibition of EAAT1 function
The nature of the inhibition exerted by UCPH-101 at EAAT1 was investigated in the FMP assay and compared with that of TBOA. The pharmacological characteristics of the EAAT1-HEK293 cell line in the FMP assay have previously been shown to be in good agreement with those observed for the transporter in conventional uptake assays and electrophysiological setups (Jensen and Bräuner-Osborne, 2004). In concordance with a previous study (Shimamoto et al., 1998) and as reported for TBOA at EAAT2 (Jensen and Bräuner-Osborne, 2004), the functional profile of TBOA at EAAT1 in the FMP assay was characteristic of that of a competitive inhibitor. Glu exhibited increasing Km (EC50) values at the transporter in the presence of increasing TBOA concentrations, whereas the maximal response (Rmax) elicited by the substrate in the assay was not significantly changed (Fig. 5A). Conversely, the IC50 values determined for TBOA at EAAT1 were very dependent on the Glu concentrations used in the assay (Fig. 5B). In accordance with the principle of competitive inhibition, Ki values calculated for TBOA based on IC50 values obtained using different Glu concentrations were very similar (Fig. 5B).
Figure 5.

The nature of inhibition exerted by TBOA and UCPH-101 at EAAT1-HEK293 cells in the FLIPR Membrane Potential Blue assay. A, Concentration–response curves for Glu in the absence or the presence of five different concentrations of TBOA or UCPH-101. B, Concentration–inhibition curves for TBOA and UCPH-101 using six different Glu concentrations. Ki values for TBOA were calculated using the Cheng-Prusoff equation [Ki = IC50/(1 + [Glu]/Km)] (Craig, 1993). The figures show data from single representative experiments of a total of 3 or 4 experiments, and data are given as mean ± SD of duplicate determinations.
The EAAT1 inhibition exerted by UCPH-101 was different from that of TBOA. The presence of UCPH-101 in the FMP assay did not change the Km (EC50) values of Glu at the transporter substantially, whereas the maximal response elicited by the substrate was decreased with increasing concentrations of the inhibitor (Fig. 5A). Furthermore, the IC50 values determined for UCPH-101 at EAAT1 using six different (S)-Glu concentrations were similar (Fig. 5B).
The profile exhibited by UCPH-101 in the FMP assay was supported by patch-clamp experiments, where similar concentration dependences of block by UCPH-101 were observed in the presence of different Glu concentrations. Figure 6 shows representative whole-cell recordings of EAAT1 with external solution containing 10 mm Glu (Fig. 6A) or 0 mm Glu (Fig. 6B) in the absence (top) or presence (bottom) of 10 μm UCPH-101. The concentration dependence of block by UCPH-101 in presence of various Glu concentrations is shown in Figure 6C. KD values ∼0.3 μm were obtained for UCPH-101 in these experiments (with 0.1 mm Glu: KD UCPH-101 = 0.29 ± 0.03 μm, Hill = 1.18 ± 0.11, n ≥ 5; with 0.5 mm Glu: KD UCPH-101 = 0.34 ± 0.03 μm, Hill = 1.3 ± 0.13, n ≥ 9; with 10 mm Glu: KD UCPH-101 = 0.35 ± 0.03 μm, Hill = 1.29 ± 0.14, n = 6). EAAT1 anion currents were also blocked in the absence of Glu (KD UCPH-101 = 0.28 ± 0.09 μm; Hill = 0.78 ± 0.17, n ≥ 6), which suggests that UCPH-101 associates with EAAT1 in its outward-facing conformation. Without Glu present, EAAT1 anion currents are much smaller, resulting in larger contributions of background currents and apparently less complete block by UCPH-101. In conclusion, UCPH-101 displayed all the characteristics of a noncompetitive inhibitor at EAAT1.
Figure 6.

UCPH-101 dependence of EAAT1 in the presence of various Glu concentrations. A, B, EAAT1 whole-cell currents in the presence of 10 mm external Glu (A) or in the absence of external Glu (B) without UCPH-101 (top) or with 10 μm UCPH-101 (bottom). C, UCPH-101 concentration dependence of EAAT1 currents at a voltage step of −185 mV. Data were normalized to the current in absence of external UCPH-101, and mean current amplitudes from different cells were fitted with a Hill equation.
Identification of the allosteric site targeted by UCPH-101 in GLAST
The noncompetitive inhibition exhibited by UCPH-101 at EAAT1 prompted us to search for its putative allosteric binding site in the transporter. This was done in a multistep iterative process, where functional properties of Glu, TBOA, and UCPH-101 were determined at chimeras of GLAST and GLT-1 and at GLAST and GLT-1 mutants in the [3H]-d-Asp uptake assay. In the early stages of the project, the constructed chimeras and mutants were transiently expressed in tsA201 cells and screened for function. Major findings from the experiments were subsequently confirmed at transporters stably expressed in polyclonal HEK293 cell lines, and later steps in the project were performed exclusively at these stable polyclonal cells.
In the first phase of the project, Glu, TBOA, and UCPH-101 were characterized functionally at a considerable number of GLAST/GLT-1 chimeras in [3H]-d-Asp uptake assay. Unfortunately, most of these chimeras turned out to be nonfunctional, as no significant [3H]-d-Asp uptake could be measured in tsA201 cells expressing them (Fig. 7A). This was very much in line with previous observations made by Vandenberg and colleagues in a study of a series of EAAT1/EAAT2 chimeras (Mitrovic et al., 1998). The reasons for the observed nonfunctionality of these chimeras were not investigated further. Important for this study, the chimeras N76, N354, N403, and N461 were functional, and Glu and TBOA displayed similar IC50 values at these chimeras as those exhibited at WT GLAST and WT GLT-1 (Fig. 7B; Table 2, section I). Whereas UCPH-101 was inactive at N76, it inhibited [3H]-d-Asp uptake through the other three chimeras with IC50 values not significantly different from that at WT GLAST (Table 2, section I). The WT GLAST-like activity displayed by UCPH-101 at the N354 chimera was particularly informative, as it demonstrated that all molecular determinants for the subtype selectivity of the inhibitor reside within the Met1-Gln354 region of GLAST (Fig. 7B, C). To further narrow down the GLAST region containing these determinants, several additional GLAST/GLT-1 chimeras were constructed (Fig. 7A), but all of these were nonfunctional. Thus, we took on another approach in our search for the UCPH-101 binding site.
Figure 7.

The selectivity determinant for UCPH-101 resides within the Met1-Gln354 region of GLAST. A, Selected nonfunctional GLAST/GLT-1 chimeras. GLAST and GLT-1 regions in the chimeras are given in green and red, respectively. The prefixes N and C in the chimera names refer to the presence of GLAST parts in the N- and C-terminal, respectively, and the numbers refer to the first or last GLAST residue at the fusion point of C- and N-chimeras, respectively. B, Topology of WT GLAST and WT GLT-1 and the functional GLAST/GLT-1 chimeras N76, N354, N403, and N461. C, Concentration–inhibition curves for Glu, TBOA, and UCPH-101 at WT GLAST, WT GLT-1, and chimera N354 stably expressed in polyclonal HEK293 cells in the [3H]-d-Asp uptake assay (n = 4).
Table 2.
Functional characteristics of Glu, TBOA, and UCPH-101 at WT GLAST, WT GLT-1, GLAST/GLT-1 chimeras, and GLAST and GLT-1 mutants stably expressed in polyclonal HEK293 cells in the [3H]-d-Asp uptake assay (using 100 nm [3H]-d-Asp as tracer concentration). The position(s) of the mutation(s) introduced in the respective mutant transporters are indicated together with the corresponding residue(s) in GltPh. The IC50 values for Glu, TBOA, and UCPH-101 are given in μm with pIC50 ± SEM values in parentheses. The maximal inhibition degrees by UCPH-101 at the respective transporters are given as % ± SEM of the maximal inhibition exerted by Glu at the same transporter. Maximal inhibition percentages in the 90–100% range are stated as “Complete inhibition,” and maximal inhibition percentages below 20% as “No inhibition.”
| Transporter | Position of Mutation (Residue in GltPh) | Glu IC50 (μm) (pIC50 ± SEM) | TBOA IC50 (μm) (pIC50 ± SEM) | UCPH-101 IC50 (μm) (pIC50 ± SEM) | UCPH-101 Maximal inhibition (% ± SEM) |
|---|---|---|---|---|---|
| WT GLAST | — | 18 (4.74 ± 0.01) | 2.5 (5.60 ± 0.03) | 0.49 (6.31 ± 0.05) | Complete inhibition |
| WT GLT-1 | — | 31 (4.51 ± 0.02) | 1.4 (5.86 ± 0.03) | >100 (<4.0) | No inhibition |
| I. GLAST/GLT-1 CHIMERAS | |||||
| Chimera N76 | GLASTM1-R76/GLT-1V74-I562 | 35 (4.46 ± 0.05) | 1.9 (5.72 ± 0.09) | >100 (<4.0) | No inhibition |
| Chimera N354 | GLASTM1-Q354/GLT-1A353-I562 | 23 (4.64 ± 0.06) | 2.1 (5.68 ± 0.10) | 0.51 (6.29 ± 0.07) | Complete inhibition |
| Chimera N403 | GLASTM1-A403/GLT-1L402-I562 | 33 (4.48 ± 0.10) | 3.1 (5.51 ± 0.11) | 0.86 (6.07 ± 0.13) | Complete inhibition |
| Chimera N461 | GLASTM1-P461/GLT-1T460-I562 | 24 (4.62 ± 0.08) | 3.5 (5.46 ± 0.16) | 1.1 (5.97 ± 0.08) | Complete inhibition |
| II. GLAST MUTANTS (TM4c) | |||||
| V247I/V248G | TM4c (I255/F156) | 21 (4.69 ± 0.11) | 3.4 (5.48 ± 0.03) | 0.51 (6.29 ± 0.14) | Complete inhibition |
| V248F | TM4c (F156) | 17 (4.74 ± 0.07) | 4.6 (5.34 ± 0.08) | 0.51 (6.30 ± 0.07) | Complete inhibition |
| S250F/M251I/C252A | TM4c (A158/I159/I160) | 14 (4.87 ± 0.10) | 1.8 (5.74 ± 0.06) | 0.82 (6.07 ± 0.12) | Complete inhibition |
| M251G | TM4c (I159) | 12 (4.91 ± 0.04) | 6.5 (5.19 ± 0.03) | ∼30 (∼4.5) | ND |
| M251L | TM4c (I159) | 29 (4.53 ± 0.09) | 2.9 (5.54 ± 0.07) | 0.81 (6.09 ± 0.03) | Complete inhibition |
| M251I | TM4c (I159) | 24 (4.60 ± 0.03) | 2.7 (5.57 ± 0.05) | 0.51 (6.29 ± 0.02) | Complete inhibition |
| M251F | TM4c(I159) | 25 (4.61 ± 0.03) | 3.2 (5.50 ± 0.11) | 0.62 (6.21 ± 0.08) | Complete inhibition |
| C252G | TM4c(I160) | 12 (4.92 ± 0.07) | 2.3 (5.64 ± 0.12) | 0.89 (6.05 ± 0.12) | Complete inhibition |
| C252I | TM4c (I160) | 21 (4.68 ± 0.05) | 4.5 (5.34 ± 0.03) | 1.9 (6.42 ± 0.09) | Complete inhibition |
| F255A | TM4c (I163) | 9.9 (5.01 ± 0.13) | 2.6 (5.58 ± 0.08) | 0.95 (6.02 ± 0.13)a | 87 ± 2.8 |
| F255V | TM4c(I163) | 12 (4.92 ± 0.13) | 2.0 (5.71 ± 0.13) | 0.62 (6.32 ± 0.09)a | 79 ± 1.6 |
| F255L | TM4c (I163) | 12 (4.93 ± 0.12) | 4.8 (5.32 ± 0.12) | 1.6 (5.80 ± 0.14)a | 78 ± 4.2 |
| F255I | TM4c (I163) | 23 (4.68 ± 0.08) | 4.3 (5.37 ± 0.12) | >100 (<4.0) | No inhibition |
| F255N | TM4c (I163) | 14 (4.85 ± 0.07) | 3.6 (5.45 ± 0.14) | 0.48 (6.31 ± 0.01) | Complete inhibition |
| F255W | TM4c (I163) | 15 (4.82 ± 0.07) | 3.5 (5.45 ± 0.15) | 0.58 (6.25 ± 0.09) | Complete inhibition |
| F255R | TM4c (I163) | 20 (4.70 ± 0.07) | 2.0 (5.70 ± 0.04) | 0.53 (6.27 ± 0.06) | Complete inhibition |
| V256A/I257M/N259K | TM4c (A164/I165/Y167) | 14 (4.86 ± 0.15) | 2.9 (5.53 ± 0.06) | 0.92 (6.04 ± 0.14)a | 78 ± 4.9 |
| III. GLAST MUTANTS (TM3) | |||||
| S116F | TM3 (L78) | 28 (4.55 ± 0.04) | 3.2 (5.49 ± 0.03) | 0.80 (6.10 ± 0.01)a | 53 ± 4.1 |
| G117F | TM3 (G79) | 24 (4.62 ± 0.11) | 2.2 (5.66 ± 0.08) | 0.45 (6.34 ± 0.09) | Complete inhibition |
| G117L/K118A/M119A | TM3 (G79/R80/V81) | 18 (4.74 ± 0.10) | 4.3 (5.37 ± 0.04) | 1.4 (5.85 ± 0.05) | Complete inhibition |
| G120L/M121A/R122A | TM3 (G82/V83/K84) | 11 (4.94 ± 0.03) | 3.2 (5.49 ± 0.15) | >100 (<4.0) | No inhibition |
| G120A | TM3 (G82) | 7.5 (5.12 ± 0.16) | 2.9 (5.54 ± 0.09) | >100 (<4.0)a | 22 ± 3.1 |
| G120V | TM3 (G82) | 8.6 (5.06 ± 0.07) | 4.1 (5.39 ± 0.13) | >100 (<4.0) | No inhibition |
| G120L | TM3 (G82) | 14 (4.89 ± 0.08) | 2.8 (5.56 ± 0.14) | >100 (<4.0) | No inhibition |
| M121A | TM3 (V83) | 18 (4.74 ± 0.03) | 3.1 (5.51 ± 0.11) | 1.8 (6.16 ± 0.20) | Complete inhibition |
| R122A | TM3 (K84) | 18 (4.74 ± 0.06) | 4.3 (5.37 ± 0.07) | 0.69 (5.76 ± 0.18)a | 87 ± 3.1 |
| A123L/V124G/V125G | TM3 (I85/V86/V87) | 12 (4.91 ± 0.06) | 2.2 (5.66 ± 0.13) | ∼1 (∼6.0)a | 47 ± 11 |
| A123G | TM3 (I85) | 16 (4.79 ± 0.08) | 4.8 (5.31 ± 0.02) | 1.3 (5.89 ± 0.09)a | 72 ± 5.2 |
| A123I | TM3 (I85) | 24 (4.60 ± 0.09) | 8.7 (5.06 ± 0.03) | 1.2 (5.93 ± 0.08) | Complete inhibition |
| A123F | TM3 (I85) | 23 (4.63 ± 0.03) | 8.7 (5.06 ± 0.09) | >100 (<4.0) | No inhibition |
| V124G | TM3 (V86) | 11 (4.96 ± 0.11) | 6.5 (5.18 ± 0.15) | 0.91 (6.04 ± 0.12)a | 80 ± 4.1 |
| V124F | TM3 (V86) | 21 (4.69 ± 0.04) | 6.6 (5.18 ± 0.03) | 1.7 (5.76 ± 0.10) | Complete inhibition |
| Y126A/Y127A | TM3 (Y88/Y89) | 6.4 (5.19 ± 0.05) | 2.7 (5.56 ± 0.18) | 0.44 (6.36 ± 0.04)a | 85 ± 3.8 |
| Y126G | TM3 (Y88) | 18 (4.75 ± 0.04) | 3.2 (5.49 ± 0.02) | 0.27 (6.57 ± 0.11) | Complete inhibition |
| Y127G | TM3 (Y89) | 13 (4.89 ± 0.08) | 14 (4.84 ± 0.07) | 0.62 (6.21 ± 0.04)a | 23 ± 2.8 |
| Y127L | TM3 (Y89) | 8.2 (5.09 ± 0.13) | 5.1 (5.30 ± 0.15) | 1.5 (5.82 ± 0.17)a | 54 ± 5.4 |
| Y127I | TM3 (Y89) | 24 (4.69 ± 0.04) | 1.6 (5.85 ± 0.03) | 2.9 (5.54 ± 0.08)a | 45 ± 4.7 |
| Y127F | TM3 (Y89) | 9.5 (5.02 ± 0.04) | 2.8 (5.56 ± 0.03) | 1.1 (5.96 ± 0.13) | Complete inhibition |
| Y127R | TM3 (Y89) | 14 (4.84 ± 0.12) | 2.3 (5.63 ± 0.16) | 2.2 (5.65 ± 0.09)a | 65 ± 5.7 |
| IV. GLAST MUTANTS (TM7a) | |||||
| R385F | TM7a (G297) | 12 (4.91 ± 0.06) | 3.4 (5.47 ± 0.01) | 0.32 (6.50 ± 0.09) | Complete inhibition |
| I386G | TM7a (I298) | 16 (4.79 ± 0.03) | 2.4 (5.62 ± 0.04) | 0.22 (6.66 ± 0.07) | Complete inhibition |
| I386F | TM7a (I298) | 19 (4.73 ± 0.11) | 3.0 (5.52 ± 0.03) | 0.39 (6.41 ± 0.09) | Complete inhibition |
| F389A | TM7a (F301) | 9.2 (5.04 ± 0.07) | 9.2 (5.04 ± 0.11) | 0.21 (6.69 ± 0.18)a | 35 ± 4.3 |
| F389L | TM7a (F301) | 15 (4.83 ± 0.06) | 2.0 (5.60 ± 0.19) | 0.28 (6.55 ± 0.11)a | 27 ± 2.9 |
| F389Q | TM7a (F301) | 12 (4.92 ± 0.04) | 13 (4.89 ± 0.03) | 0.37 (6.43 ± 0.09)a | 37 ± 4.9 |
| F389T | TM7a (F301) | 17 (4.77 ± 0.03) | 7.4 (5.13 ± 0.04) | >100 (<4.0) | No inhibition |
| F389W | TM7a (F301) | 15 (4.82 ± 0.05) | 4.2 (5.38 ± 0.08) | >100 (<4.0) | No inhibition |
| V390G | TM7a (T302) | 24 (4.62 ± 0.07) | 5.1 (5.29 ± 0.11) | 0.55 (6.26 ± 0.14) | Complete inhibition |
| V393G | TM7a (L305) | 15 (4.82 ± 0.01) | 4.2 (5.38 ± 0.08) | 0.41 (6.39 ± 0.02) | Complete inhibition |
| V393I | TM7a (L305) | 19 (4.71 ± 0.05) | 6.8 (5.17 ± 0.03) | 1.9 (5.72 ± 0.01)a | 85 ± 4.9 |
| V393T | TM7a (L305) | 21 (4.67 ± 0.02) | 4.7 (5.33 ± 0.03) | >100 (<4.0) | No inhibition |
| V393Q | TM7a (L305) | 12 (4.91 ± 0.04) | 5.9 (5.23 ± 0.09) | 0.21 (6.67 ± 0.08)a | 54 ± 4.1 |
| V393F | TM7a (L305) | 13 (4.89 ± 0.06) | 5.4 (5.27 ± 0.05) | ∼0.3 (∼6.5)a | 41 ± 2.7 |
| T396G | TM7a (T308) | 27 (4.57 ± 0.07) | 6.2 (5.21 ± 0.14) | 0.47 (6.33 ± 0.10) | Complete inhibition |
| I397G | TM7a (I309) | 13 (4.90 ± 0.06) | 4.8 (5.32 ± 0.00.9) | 0.34 (6.46 ± 0.04)a | 87 ± 4.3 |
| I397F | TM7a (I309) | 10 (5.00 ± 0.04) | 3.8 (5.42 ± 0.02) | 0.56 (6.38 ± 0.08) | Complete inhibition |
| V. GLAST MUTANTS (TM2, TM2/3 LOOP, TM5 and TM8) | |||||
| L104A/V105A/T106A | TM2 (L66/V67/V68) | 11 (4.98 ± 0.13) | 5.4 (5.27 ± 0.05) | 0.33 (6.48 ± 0.19) | Complete inhibition |
| M108L/A109S/A110G/S113A | TM2/3 Loop (A70/A71/S72/A76) | 16 (4.79 ± 0.03) | 3.2 (5.59 ± 0.08) | 0.93 (6.03 ± 0.15) | Complete inhibition |
| G107V/M108A | TM2/3 Loop (G69/A70) | 8.4 (5.08 ± 0.04) | 5.9 (5.22 ± 0.12) | 2.3 (5.64 ± 0.17)a | 81 ± 5.3 |
| A109I | TM2/3 Loop (A71) | 16 (4.81 ± 0.04) | 9.4 (5.03 ± 0.09) | 2.6 (5.58 ± 0.09)a | 83 ± 2.3 |
| A109L | TM2/3 Loop (A71) | 10 (5.01 ± 0.13) | 4.7 (4.32 ± 0.09) | 1.2 (5.92 ± 0.16)a | 69 ± 4.1 |
| A109F | TM2/3 Loop (A71) | 13 (4.88 ± 0.06) | 13 (4.88 ± 0.06) | 1.6 (5.79 ± 0.12)a | 87 ± 3.4 |
| A110L | TM2/3 Loop (S72) | 9.8 (5.01 ± 0.15) | 3.6 (5.44 ± 0.10) | 0.91 (6.04 ± 0.11) | Complete inhibition |
| L111A | TM2/3 Loop (I73) | 8.7 (5.06 ± 0.09) | 2.2 (5.66 ± 0.14) | 1.5 (5.83 ± 0.10) | Complete inhibition |
| D112A | TM2/3 Loop (P75) | 8.8 (5.06 ± 0.10) | 5.1 (5.29 ± 0.10) | ∼1 (∼6.0)a | 55 ± 7.1 |
| S113G | TM2/3 Loop (A76) | 15 (4.83 ± 0.06) | 4.6 (5.34 ± 0.16) | 1.1 (6.34 ± 0.18)a | 85 ± 3.1 |
| S113L | TM2/3 Loop (A76) | 11 (4.97 ± 0.18) | 3.3 (5.48 ± 0.07) | 0.46 (6.34 ± 0.18) | Complete inhibition |
| K114A/A115G | TM2/3 Loop (R77) | 13 (4.89 ± 0.04) | 2.8 (5.56 ± 0.07) | 0.77 (6.12 ± 0.12) | Complete inhibition |
| G264A/Q265K/A266L/L267M/R268V | TM5 (A180/E181/T182/L183/L184L) | 9.1 (5.04 ± 0.06) | 2.7 (5.57 ± 0.17) | 0.54 (6.27 ± 0.10) | Complete inhibition |
| D272N/S273I | TM5 (N188/G189) | 16 (4.79 ± 0.04) | 4.3 (5.37 ± 0.15) | 1.2 (5.95 ± 0.18) | Complete inhibition |
| A277I/I278V/R280K/A283I/V284M | TM5 (A193/M194/K196/N199/G200) | 7.8 (5.11 ± 0.06) | 9.7 (5.01 ± 0.18) | 0.65 (6.19 ± 0.17)a | 88 ± 1.9 |
| R280A/L281A | TM5 (K196/I197) | 15 (4.83 ± 0.10) | 6.6 (5.18 ± 0.10) | 0.33 (6.48 ± 0.15) | Complete inhibition |
| G490L | TM8 (G408) | 19 (4.72 ± 0.05) | 2.6 (5.58 ± 0.10) | 0.31 (6.51 ± 0.09) | Complete inhibition |
| G490F | TM8 (G408) | 14 (4.86 ± 0.06) | 2.1 (5.67 ± 0.13) | 0.22 (6.66 ± 0.10)a | 81 ± 2.6 |
| V494F | TM8 (V412) | 12 (4.92 ± 0.06) | 3.1 (5.51 ± 0.07) | 1.0 (6.05 ± 0.16) | Complete inhibition |
| VI. GLAST MUTANTS (TM2 and TM5 IN NEIGHBORING MONOMER) | |||||
| F83G/P84G/G85L | TM2 (P45/F46/G47) | 13 (4.87 ± 0.08) | 2.2 (5.65 ± 0.03) | 0.35 (6.45 ± 0.13) | Complete inhibition |
| E86G/L87G/L88G | TM2 (D48/L49/F50) | 15 (4.84 ± 0.04) | 2.2 (5.27 ± 0.17) | 0.46 (6.34 ± 0.15) | Complete inhibition |
| M79G/R80G/M81G | TM2 (V51/R52/L53) | 23 (4.64 ± 0.08) | 1.9 (5.72 ± 0.12) | 0.91 (6.04 ± 0.17) | Complete inhibition |
| L92G/Q93G | TM2 (L54/K55) | 13 (4.88 ± 0.03) | 6.0 (5.21 ± 0.03) | 0.77 (6.12 ± 0.07) | Complete inhibition |
| M94G/L95G | TM2 (M56/L57) | 14 (4.85 ± 0.01) | 14 (4.86 ± 0.04) | 1.0 (5.99 ± 0.08)a | 87 ± 3.4 |
| A277L/I278G/M279G | TM5 (A193/M194/Y195) | 16 (4.80 ± 0.02) | 3.4 (5.46 ± 0.09) | 0.78 (6.11 ± 0.20) | Complete inhibition |
| R280G/L281G/V282G | TM5 (K196/I197/V198) | 20 (4.71 ± 0.05) | 3.2 (5.49 ± 0.08) | 0.69 (6.16 ± 0.14) | Complete inhibition |
| A283L/V284G/I285G | TM5 (N199/G200/V201) | 26 (4.58 ± 0.05) | 25 (4.60 ± 0.07) | 2.5 (5.60 ± 0.15)a | 85 ± 5.1 |
| M286G/W287G/Y288G | TM5 (M202/Q203/Y204) | 21 (4.68 ± 0.06) | 7.0 (5.15 ± 0.06) | 1.4 (5.86 ± 0.20) | Complete inhibition |
| A289G/P290G | TM5 (A205/P206) | 17 (4.78 ± 0.04) | 2.1 (5.68 ± 0.09) | 0.50 (6.30 ± 0.16) | Complete inhibition |
| L291G/G292L | TM5 (I207/G208) | 18 (4.75 ± 0.04) | 8.6 (5.07 ± 0.12) | 3.0 (5.53 ± 0.03)a | 81 ± 2.9 |
| VII. GLT-1 MUTANTS | |||||
| I249M | TM4c (I159) | 44 (4.36 ± 0.07) | 3.5 (5.46 ± 0.18) | >100 (<4.0) | No inhibition |
| I253F | TM4c (I163) | 38 (4.43 ± 0.06) | 3.1 (5.50 ± 0.11) | >100 (<4.0) | No inhibition |
| I249M/I253F | TM4c(I159/I163) | 33 (4.48 ± 0.09) | 1.9 (5.71 ± 0.12) | >100 (<4.0) | No inhibition |
The polyclonal cell lines were generated and the [3H]-d-Asp uptake assay was performed as described in Materials and Methods. Data are the means of 3–20 individual experiments performed in duplicate. —, Not applicable; ND, not determinable.
a UCHP-101 did not inhibit specific [3H]-d-Asp uptake in cells expressing through this mutant completely, and thus the IC50 value for UCPH-101 at this mutant represents the concentration yielding 50% of the observed inhibition.
Alignment of the amino acid sequences of human EAAT1, EAAT2, and EAAT3 and rat GLAST, GLT-1, and EAAC1 identifies a total of 104 residues in the Phe50-Gln354 region spanning the TM1 to the first half of HP1a of GLAST, which are identical in EAAT1 but different in the four other transporters. In the second phase of the project, 75 of these residues in GLAST (located in the TM1-TM4b and TM4c-HP1a segments) were mutated to the corresponding GLT-1 residues (Fig. 8), and UCPH-101 was tested at the mutants transiently expressed in tsA201 cells. The most notable observation made in this screening was the pronounced impairment of the inhibitory activity of UCPH-101 brought on by the F255I mutation in GLAST, since Glu and TBOA exhibited IC50 values at the mutant not significantly different from those at WT GLAST (data not shown). This finding was subsequently reproduced in polyclonal HEK293 cells expressing the mutant (Fig. 9; Table 2, section II).
Figure 8.
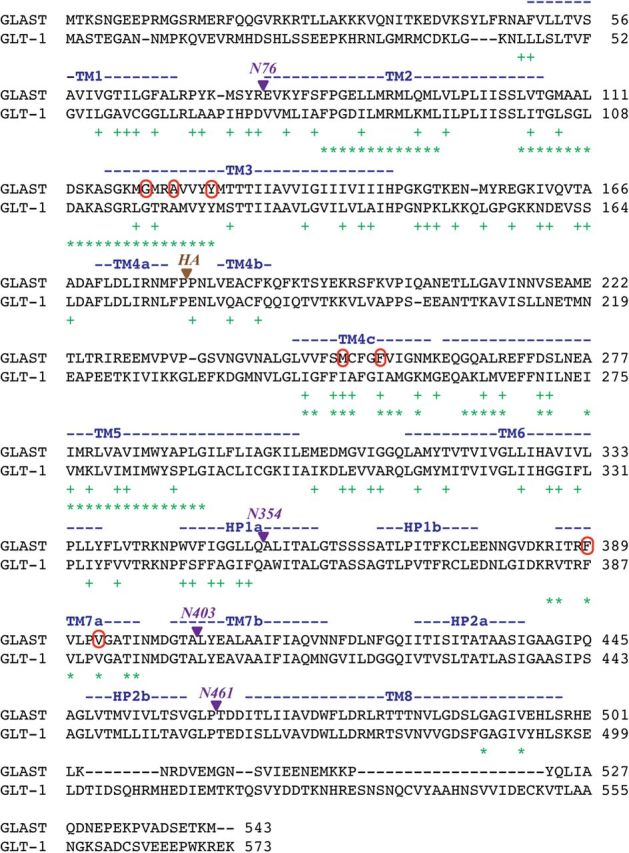
Alignment of amino acid sequences of rat GLAST and GLT-1 (extracted from an alignment of human EAAT1, EAAT2, and EAAT3 and rat GLAST, GLT-1, and EAAC1). The TM1–8 and HP1–HP2 regions are specified above the sequences. Residues in the Phe50-Gln354 segment of GLAST mutated in the preliminary mutagenesis study (+) and residues mutated in mutants stably expressed in polyclonal HEK293 in later stages of the project (*) are indicated below the sequences. The fusion points of GLAST/GLT-1 chimeras N76, N354, N403, and N461 and the site of HA-tag insertion in GLAST are given. The Gly120, Ala123, Tyr127, Met251, Phe255, Phe389, and Val393 residues in GLAST where selected mutations were found to affect UCPH-101 activity are circled.
Figure 9.

Functional properties of UCPH-101 at GLAST mutants containing mutations of the Gly120, Ala123, Tyr127, Met251, Phe255, Phe389, and Val393 residues. Concentration–inhibition curves for UCPH-101 at WT GLAST and the GLAST mutants stably expressed in polyclonal HEK293 cells in the [3H]-d-Asp uptake assay (n = 3–5).
Mapping of the UCPH-101 binding site in GLAST
The detrimental effects of the F255I mutation on UCPH-101 inhibition of GLAST prompted us to search for additional residues important for the activity of the inhibitor in the vicinity of Phe255. The residue is located in the TM4c helix of GLAST and corresponds to the Ile163 residue in the GltPh transporter (Yernool et al., 2004). In the third phase of the search for interaction partners for UCPH-101, we applied the crystal structure of GltPh in its unbound, outward-facing state (Boudker et al., 2007) to identify residues located in the vicinity of this residue. In some mutants, stretches of residues were mutated to the corresponding residues in GLT-1 but in most mutants single residues were substituted with amino acids expected to significantly alter their respective putative interactions with UCPH-101. Residues identified as important for UCPH-101 activity were subsequently subjected to additional mutations to probe the nature of and the spatial requirements for the putative interactions.
The functional properties of Glu, TBOA, and UCPH-101 were determined at polyclonal HEK293 cells stably expressing 86 different GLAST mutants in the [3H]-d-Asp uptake assay. The functional properties of the mutants are given in Table 2, sections II–VI, concentration-inhibition curves for UCPH-101 at selected mutants are given in Figure 9, and IC50 values and degrees of maximal inhibition exerted by UCPH-101 at selected mutants are summarized in Figure 10. The IC50 values displayed by Glu at the 86 mutants ranged from 7.5 to 28 μm and thus did not differ substantially from that exhibited at WT GLAST (IC50 = 18 μm; Table 2). The same was true for TBOA for the majority of mutants, as the inhibitor at 77 of the 86 GLAST mutants displayed IC50 values less than threefold different from its IC50 at WT GLAST (Table 2).
Figure 10.

Summary of the effects of mutations of residues in the TM3 (A), TM4c (B), and TM7a (C) α-helices on UCPH-101-mediated inhibition of GLAST function. Right, IC50mutant/IC50WT ratios for Glu, TBOA, and UCPH-101 at the GLAST mutants. #IC50mutant/IC50WT ratio: >200-fold. Left, The maximal inhibition degrees exhibited by UCPH-101 at WT and mutant GLAST mutants (in percentages of the maximal inhibition exerted by Glu at the respective transporters) (n = 3–5). The cutoff points for “Complete Inhibition” (>90% inhibition) and “No Inhibition” (<20% inhibition) defined in Table 2 are given as “CI” and “NI,” respectively. n.d., Not determinable.
In contrast to the inconsiderable effects of the mutations on the properties of Glu and TBOA at GLAST, UCPH-101-mediated inhibition of uptake through GLAST was dramatically impaired by mutations of seven specific residues localized in the proximity of Phe255, more specifically in the TM3, TM4c, and TM7a helices (Figs. 9 and 10). Interestingly, mutations of the seven residues gave rise to highly differential effects on the inhibitory potency and/or efficacy of UCPH-101 at GLAST. Several mutations resulted in reduced inhibitory potencies of UCPH-101 or completely eliminated the activity of the inhibitor at the transporter (Figs. 9 and 10; Table 2). However, IC50 values exhibited by UCPH-101 at some of the mutants containing substitutions of the residues Tyr127, Phe389, or Val393 were similar or only slightly higher than that at WT GLAST, whereas the efficacies displayed by the inhibitor (i.e., the maximal degrees of inhibition of [3H]-d-Asp uptake) were dramatically reduced (Figs. 9 and 10; Table 2). The potential reasons for and implications of these differential effects of the mutations on UCPH-101 potency and efficacy will be addressed in the Discussion.
TM4c
In contrast to the dramatic effect of the F255I mutation, introduction of several other amino acid residues (Ala, Val, Leu, Asn, Arg, and Trp) in this position did not result in substantial impairments of UCPH-101 activity at GLAST (Fig. 9; Table 2, section II). Removal of the side chain of the Met251 residue situated one α-helix turn (4 residues) above Phe255 (M251G) resulted in a significant increase in the UCPH-101 IC50, whereas introduction of Leu, Ile, or Phe residues in this position had negligible effects on inhibitor activity (Table 2, section II). Importantly, UCPH-101 displayed WT-like IC50 values at mutants containing mutations of the neighboring Cys252 residue and at mutants with substitutions of residues yet another helix turn below Phe255 (V247I/V248G and V248F) or residues located above the residue (V256A/I257M/N259K) (Table 2, section II). Thus, the distribution of residues found to be important for UCPH-101 activity in TM4c (Met251 and Phe255) is in agreement with the α-helical structure of the domain.
TM3
Conservative mutations of the Gly120 residue in the cytoplasmic part of TM3 were found to abolish the activity of UCPH-101 at GLAST completely (G120V, G120L) or almost completely (G120A) (Fig. 9; Table 2, section III). Mutations of Ser116 and Gly117 residues located one helix turn below Gly120 did not change UCPH-101 activity at GLAST substantially. In contrast, some mutations of Ala123 and Tyr127 situated one and two helix turns above Gly120, respectively, impaired UCPH-101 inhibition significantly (A123F, Y127G, Y127L), whereas others did not (A123G, A123I, Y123I, Y123F, Y127R). Mutations of other residues in the Gly120-Tyr127 segment (Met121, Arg122, Val124, and Tyr126) did not impact the UCPH-101 activity at the transporter (Table 2, section III). Thus, analogously to the pattern observed for TM4c, the distribution of residues of importance for UCPH-101 activity in TM3 was consistent with the α-helical structure of TM3.
TM7a
In the TM7a helix, the Phe389 residue turned out to be essential for UCPH-101 activity, as introduction of Ala, Leu, Thr, Gln, and Trp residues in this position all had detrimental effects on the inhibition mediated by the ligand (Fig. 9; Table 2, section IV). Whereas removal of the side chain and a conservative mutation of the Val393 residue situated one helix turn above Phe389 had little impact on UCPH-101 activity (V393G and V393I), introduction of hydrophilic or aromatic residues in this position resulted in significantly reduced the inhibitory potency and/or efficacy of the ligand (V393Q, V393T, V393F). Mutations introduced one helix turn below Phe389 (R385F, I386G, I386F) and one helix turn above Val393 (T396G, I397G, I397F) resulted in mutants displaying IC50 values for UCPH-101 not significantly different from WT GLAST (Table 2, section IV).
TM2, TM2–3 loop, TM5, and TM8
Substitutions of residues in TM2, the loop connecting TM2 and TM3, TM5, and TM8, were found to have little or no impact on UCPH-101-mediated inhibition of the transporter (Table 2, section V). The only exception from this general trend was the significant reduction in maximal inhibition by UCPH-101 observed upon an Ala substitution of Asp112 in GLAST, a conserved Asp residue previously shown to be important for EAAT function (Table 2, section V) (Ryan et al., 2004; Hotzy et al., 2012). We will refrain from speculating about a potential involvement in this residue in UCPH-101 binding.
The neighboring monomer
The residues in TM3, TM4c, and TM7a identified as important for UCPH-101 activity define a crevice in each monomer in the trimeric GLAST complex (Fig. 11). However, the distances between these residues in one monomer and stretches of residues in the TM2 and TM5 α-helices in the neighboring monomer (Pro45-Leu57 and Ala193-Gly208 in GltPh corresponding to Phe83-Leu95 and Ala277-Gly292 in GLAST) are of dimensions, where UCPH-101 could be envisioned to project out of this putative intramonomeric pocket and form interactions with these residues in the adjacent monomer. To investigate for this, the residues in TM2 and TM5 were subjected to mutagenesis. UCPH-101 did not display significantly different IC50 values at any of the mutants containing stretches of mutations of these residues than at the WT GLAST (Table 2, section VI).
Figure 11.

Homology model of the GLAST monomer. A, The trimeric GLAST complex viewed parallel to the membrane (left) and from the extracellular side of the membrane (right) with the three monomers given in pink, gold, and silver. In the pink monomer, the TM3, TM4c, and TM7a α-helices are highlighted in red, and the seven residues observed to be important for UCPH-101 activity in this study are given as stick representations in green. The localization of the substrate binding site in the pink monomer is indicated by the presence of l-aspartate (in purple, highlighted with a purple circle). B, The chimera N354 monomer. As in Figure 7B, the N354 domains composed of GLAST and GLT-1 regions are given in green and red, respectively. C, The residues in the GLAST monomer subjected to mutagenesis (the specific mutants are given in Table 2). Residues where none of the introduced substitutions affected UCPH-101 activity are given in yellow, whereas the seven residues where selected mutations impaired UCPH-101 potency and/or efficacy are given as stick representations in green. D, The seven residues demonstrated to be important for UCPH-101 activity at GLAST. Left, Detail of the homology model of the GLAST monomer. Spatial orientations of TM3, TM4c, and TM7a and the seven residues. Right, The distances between the seven residues (Cα to Cα distances, in Å).
GLT-1 mutants
Of the seven residues in TM3, TM4c, and TM7a where specific mutations were found to impact the inhibitory potency and/or efficacy of UCPH-101 at GLAST, only Met251 and Phe255 are not conserved in GLT-1 (Fig. 8). To investigate whether these two residues constitute all of the molecular determinants of the GLAST selectivity of UCPH-101, the corresponding two residues in GLT-1, Ile249 and Ile253, were mutated to the Met and Phe, respectively. None of the GLT-1 mutants I249M, I253F, and I249M/I253F displayed measurable sensitivity toward UCPH-101 at concentrations up to 100 μm (Table 2, section VII).
Construction of a homology model of GLAST and the composition of the proposed UCPH-101 binding site
A homology model of GLAST was constructed using the crystal structure of the bacterial homolog GltPh (PDB code: 2NWX) as template (Fig. 11) (Boudker et al., 2007). As described in the Computational chemistry section in Materials and Methods, the Phe190-Val241 segment located between TM4b and TM4c in GLAST was not included in the model, since the bacterial transporter does not contain a corresponding region. Consequently, the obtained GLAST model overlays nicely with the GltPh structure used as a template (data not shown). The spatial arrangement of and the distances between the seven residues found to be important for UCPH-101 activity in the GLAST model are given in Figure 11D. The N-terminal part of TM3 and the TM7a align and seem to be the key domains for putative binding site with, whereas TM4c based on the results from the mutagenesis study may be situated in the periphery of the binding pocket.
In TM3, Gly120 is essential for the activity of UCPH-101 at GLAST (Fig. 9). As the residue composes no side chain, it may contribute to UCPH-101 binding via hydrogen bonding to the backbone amide and/or be essential in the defining of the size of the binding pocket. In the latter scenario, any mutation of the residue would be expected to decrease binding pocket size, which seems to be consistent with the detrimental effects of the conservative G120A, G120L, and G120V mutations on UCPH-101 activity (Fig. 9). The impaired inhibitory efficacies displayed by UCPH-101 at the Y127G, Y127L, Y127I, and Y127R mutants and its WT-like profile at Y127F underline the importance of an aromatic residue in this position for UCPH-101 activity (Fig. 9). The tyrosine may not necessarily function as a binding partner to the inhibitor but could be a structural component shaping the binding pocket. Finally, judging from the WT GLAST-like properties exhibited by UCPH-101 at the A123G and A123I mutants, Ala123 is unlikely to coordinate directly to UCPH-101. However, the residue does seem to line the binding pocket because the introduction of a bulky phenylalanine in this position completely eliminates UCPH-101 activity at the transporter (Fig. 9; Table 2).
The aromatic side chain of the Phe389 residue in helix 7a is another key determinant for UCPH-101 activity at GLAST. Introduction of aliphatic or hydrophilic residues (Ala, Leu, Thr, and Gln) in this position results in dramatically decreased inhibitor activity, and the elimination of UCPH-101 activity in F389W could arise from the increased bulk of the aromatic side chain (Table 2). Substitutions of Val393 situated one α-helix turn above Phe389 with the aliphatic Gly and Ile residues are well tolerated, whereas introduction of hydrophilic or aromatic amino acids in this position clearly is not (Fig. 9). Thus, this residue is proposed to coordinate to UCPH-101, in view of its hydrophobic nature most likely via van der Waals interactions.
The role of Phe255 in TM4c for UCPH-101 binding is somewhat of a conundrum because the impaired UCPH-101 activity at F255I is contrasted by its WT-like properties at the F255A, F255L, F255V, F255N, F255R, and F255W mutants (Fig. 9, Table 2). Thus, it seems unlikely that Phe255 participates directly in UCPH-101 binding, and instead the bulky branched side chain introduced in F255I could be speculated to clash with the inhibitor by occupying a region in the binding pocket that the side chains of the residues introduced in the other mutants do not project into. Although the removal of the side chain of Met251 in M251G results in a 60-fold increased IC50 value of UCPH-101, introduction of aliphatic and aromatic side residues in this position does not impair inhibitor activity (M251L, M251I, and M251F) (Fig. 9; Table 2). The residue in position 251 may not be directly involved in UCPH-101 but play a structural role for composition of the UCPH-101 binding site. Alternatively, the impaired UCPH-101 activity at the M251G and F255I mutants may arise from allosterically induced changes in the transporter domains directly mediating the inhibitor binding, such as TM3 or TM7a.
The GLAST inhibition exerted by UCPH-101 is an intramonomeric event
The crystal structures of GltPh and molecular pharmacology studies of the EAATs strongly suggest that the three monomers in the trimeric complex function as independent functional units, also when it comes to the substrate translocation process (Gendreau et al., 2004; Grewer et al., 2005; Koch and Larsson, 2005; Leary et al., 2007; Reyes et al., 2009; Verdon and Boudker, 2012). Accordingly, binding of UCPH-101 to a site composed in each monomer in the GLAST trimer could be speculated to inhibit the transport through the targeted monomer exclusively and not to influence the translocation through the two other monomers. On the other hand, considering the close proximity of the intramonomeric binding site of UCPH-101 to the neighboring monomer in the trimer (Fig. 11), the inhibitor could also be capable of cross-inhibition (i.e., be able to inhibit the translocation process in one monomer through the binding to another).
The mode of inhibition exerted by UCPH-101 at GLAST was investigated in two sets of experiments, where the functional properties of the inhibitor were determined at tsA201 cells coexpressing WT and mutant transporters in different combinations (Figs. 12 and 13). In the first line of experiments, the inhibitory properties of UCPH-101 were determined at tsA201 cells cotransfected with cDNAs encoding for WT GLAST and the UCPH-101-insensitive mutant GLAST-G120L in different ratios (Fig. 12). The four possible compositions of GLAST trimers assembled in cells coexpressing WT GLAST and GLAST-G120L are given in Figure 12A. Because the Gly120 residue is located at some distance from the monomer/monomer interface in the GLAST trimer (Fig. 11A), it is reasonable to assume that the formation of any these four trimers will not be favored over others and that the cDNA transfection ratio thus is the predominant determinant of the transporter populations expressed. Judging from the quantification of the expression of the HA-tagged versions of the two transporters in the ELISA, total and cell surface expression levels of GLAST-G120L in tsA201 cells appear to be substantially higher than those of WT GLAST (Fig. 12B). However, the fact that the ratios of cell surface expressed WT and mutant transporters thus may not be equivalent to the transfection ratios used has no impact on the conclusions from the experiment. Glu, TBOA, and UCPH-101 exhibited similar IC50 values at cells transfected with the different ratios of WT GLAST and GLAST-G120L cDNAs (Fig. 12C, D). However, whereas Glu and TBOA completely inhibited the [3H]-d-Asp uptake in all transfected cells, a clear correlation was observed between the WT GLAST:GLAST-G120L transfection ratio and the degree of inhibition exerted by UCPH-101 at the respective cells (Fig. 12C, D).
Figure 12.
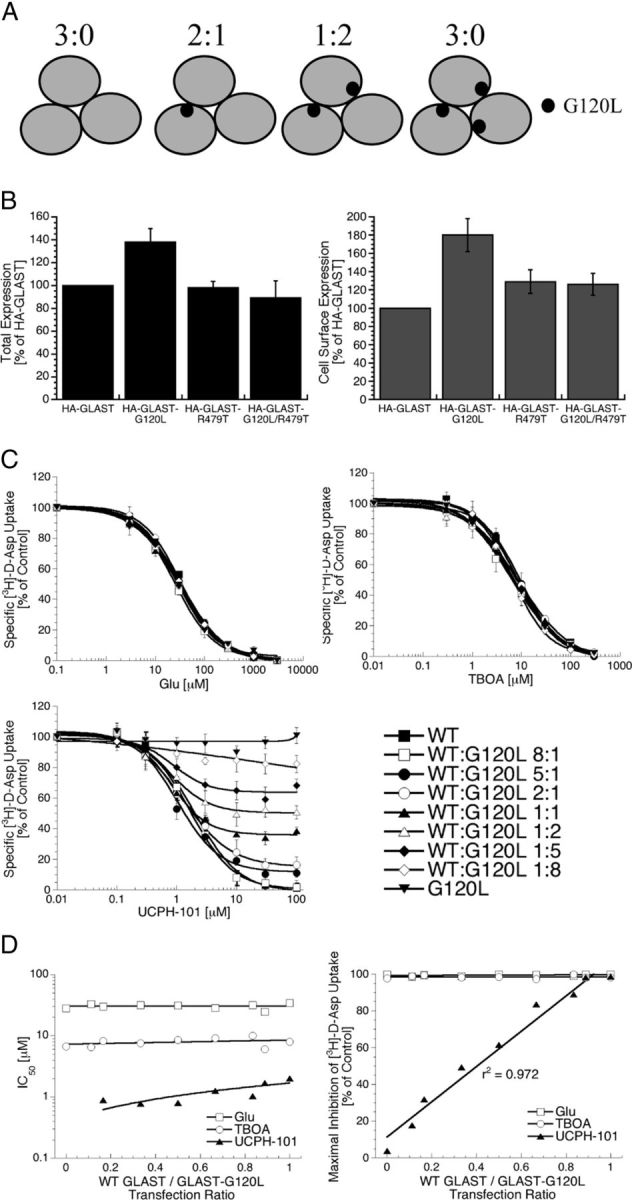
The GLAST inhibition exerted by UCPH-101 is an intramolecular event. A, The possible compositions of trimeric complexes formed in tsA201 cells co-expressing WT GLAST and GLAST-G120L. B, Total and cell surface expression levels of HA-GLAST, HA-GLAST-G120L, HA-GLAST-R497T, and HA-GLAST-G120L/R497T transiently expressed in tsA201 cells in the ELISA (n = 3). C, Concentration–inhibition curves for Glu, TBOA, and UCPH-101 at tsA201 cells transfected with WT GLAST cDNA, GLAST-G120L cDNA, or the two cDNAs in different ratios in the [3H]-d-Asp uptake assay (n = 3). D, Relationships between the transfection ratio of WT GLAST and GLAST-G120L cDNAs and the IC50 value (left) and the maximal inhibition (right) displayed by Glu, TBOA, and UCPH-101 in the [3H]-d-Asp uptake assay. IC50 values of UCPH-101 could not be determined in cells transfected with GLAST-G120L cDNA or cotransfected with WT GLAST and GLAST-G120L in a 1:8 cDNA ratio.
Figure 13.

The GLAST inhibition exerted by UCPH-101 is an intramolecular event. A, The potential GLAST trimer combinations formed in cell populations transfected with four different pairwise combinations of WT GLAST, GLAST-G120L, GLAST-R479T, and GLAST-G120L/R479T cDNAs. B, Concentration-inhibition curves for Glu, TBOA, and UCPH-101 at tsA201 cells transfected with different combinations of WT GLAST, GLAST-G120L, GLAST-R479T, and GLAST-G120L/R479T cDNAs (1:1 transfection ratios) in the [3H]-d-Asp uptake assay (n = 4).
In the second line of experiments, we took advantage of the knowledge from previous studies that mutations of Arg479 in GLAST and the corresponding Arg residue in EAAC1 completely eliminate Glu transport through the transporters (Conradt and Stoffel, 1995; Bendahan et al., 2000). This Arg residue is conserved throughout the EAATs, where its side chain is believed to form an interaction with the distal carboxylate group of Glu in the substrate binding site. In concordance with the previous studies, no significant [3H]-d-Asp uptake could be measured in tsA201 cells expressing the GLAST-R479T or GLAST-G120L/R479T mutants (B. Abrahamsen and A.A. Jensen, unpublished observations). This could not be ascribed to impaired expression of these mutants compared with the WT transporter because cell surface expression levels of HA-tagged GLAST-R479T and GLAST-G120L/R479T mutants in tsA201 cells were comparable with those of the HA-GLAST (Fig. 12B).
To investigate the effects of UCPH-101 at GLAST trimers assembled from monomers with very different functionalities in terms of substrate transport capability and UCPH-101 activity, we coexpressed all four pairwise combinations of WT GLAST, a mutant incapable of substrate transport (GLAST-R479T), a mutant insensitive to UCPH-101 (GLAST-G120L), and a mutant characterized by both of these properties (GLAST-G120L/R479T) in tsA201 cells. The possible compositions of GLAST trimers formed in each of the four combinations are outlined in Figure 13A. Glu, TBOA, and UCPH-101 inhibited [3H]-d-Asp uptake in cells coexpressing WT GLAST and GLAST-G120L/R479T, with IC50 values similar to those at WT GLAST-expressing cells (Fig. 13B). In contrast, [3H]-d-Asp uptake in cells coexpressing GLAST-R479T and GLAST-G120L was not significantly inhibited by UCPH-101, whereas Glu and TBOA displayed similar inhibitory potencies at these cells compared with WT GLAST (Fig. 13B). As expected, UCPH-101 displayed WT GLAST-like inhibitory potency at cells coexpressing WT GLAST and GLAST-R479T, and it was inactive at cells coexpressing the GLAST-G102L and GLAST-G120L/R479T mutants (Fig. 13B).
The Asp476 residue in GLAST, corresponding to Asp444 in EAAC1, is another residue thought to participate in substrate binding (Teichman and Kanner, 2007). Introduction of a D476S mutation in GLAST eliminated the [3H]-d-Asp transport capability of the transporter (B. Abrahamsen and A.A. Jensen, unpublished observations). The patterns of functionalities displayed by Glu, TBOA, and UCPH-101 at tsA201 cells coexpressing combinations of WT GLAST, GLAST-G120L, GLAST-D476S, and GLAST-G120L/D476S were very similar to those observed in the analogous studies using R479T-containing mutants described above (B. Abrahamsen and A.A. Jensen, unpublished observations).
In conclusion, the observations made in the experiments outlined in this section strongly suggest that UCPH-101 exclusively inhibits the uptake through the GLAST monomer that it binds to and does not exert cross-inhibition. The very similar IC50 values exhibited by UCPH-101 at GLAST populations formed in cells expressing different WT:G120L monomer ratios combined with the pronounced correlation between the transfection ratio and the relative efficacy of the inhibitor are certainly most reconcilable with such a model (Fig. 12D). Moreover, the complete inability of UCPH-101 to inhibit [3H]-d-Asp uptake in cells coexpressing GLAST-G120L and GLAST-R479T and its efficacious activity at WT GLAST:GLAST-G120L/R479T-transfected cells further substantiate the notion of an intramonomeric mode of action, with the inhibitor having no apparent effect at the substrate translocation through the neighboring monomers in the trimer (Fig. 13B).
Discussion
We have recently reported the discovery of the first class of subtype-selective inhibitors of the human EAAT1 and its rodent ortholog GLAST (Jensen et al., 2009). In the present study, we have investigated the mechanism of action of these inhibitors and the molecular basis for their activity at the transporter.
The functional properties of UCPH-101 and UCPH-102 at EAAT1 in the patch-clamp recordings in this study verify the potent inhibition of the transporter displayed by the inhibitors in other assays (Fig. 2) (Jensen et al., 2009; Erichsen et al., 2010). Furthermore, UCPH-101 is shown to exhibit equipotent inhibition of the substrate-activated and the uncoupled anion conductance through the transporter (Fig. 6) (Fairman et al., 1995; Wadiche et al., 1995; Melzer et al., 2003; Ryan et al., 2004; Winter et al., 2012). Finally, the previously reported specificity of the inhibitors for EAAT1/GLAST over EAAT2/GLT-1 and EAAT3/EAAC1 is demonstrated to extent to the entire EAAT family, as UCPH-101 and UCPH-102 are inactive at EAAT4 and EAAT5 as well (Fig. 2). These are important findings considering the emerging roles of UCPH-101 and UCPH-102 as pharmacological tool compounds.
Another interesting trait of UCPH-101 is its slow binding off-rate, which manifests itself in the long-lasting inactive state of EAAT1 induced by even brief exposure of the transporter to the inhibitor and in its very slow unblocking kinetics (Figs. 3C and 4A). UCPH-101 clearly does not bind irreversibly to EAAT1 because the transporter does seem able to recover from UCPH-101 exposure when subjected to a continuous wash flow (Fig. 4A). However, the rapid clearance of the extracellular solution from the transporter in the patch-clamp setup is not representative of the in vivo situation. Hence, we propose that the slow unbinding characteristics could give rise to a much more sustained inhibition of EAAT1/GLAST by UCPH-101 in vivo compared with that induced by more reversible inhibitors. Albeit equipotent at EAAT1/GLAST in an in vitro setting, the different unbinding kinetics of UCPH-101 and UCPH-102 could thus translate into different ex vivo/in vivo effects.
The differential recovery of EAAT1 function from exposure to UCPH-101 and five of its analogs conveys an intriguing tale about just how different binding kinetics of structurally similar ligands can be. The pronounced correlation between the inhibitory potencies of analogs UCPH-102, UCPH-100, 1, 2 and 3 in the [3H]-d-Asp uptake assay and in the preincubation experiment is in concordance with what would be expected for reversible inhibitors with similar binding modes, and neither the physicochemical characteristics nor the different 4- and 7-substituents of UCPH-101 seem to explain its dramatically slower unbinding from EAAT1 (Fig. 3G; Table 1). We hypothesize that the divergence arises from UCPH-101 being more embedded in the binding site than the other analogs, and these striking effects brought on by seemingly subtle structural changes in the compound scaffold are currently under further investigation in our laboratory.
In agreement with its different structure compared with substrates and nonsubstrate competitive inhibitors of the EAATs (Bunch et al., 2009), the noncompetitive inhibition of EAAT1 exerted by UCPH-101 (Figs. 5 and 6) and the WT GLAST-like activity displayed by it at chimera N354 (Figs. 7B, C and 11B) unequivocally identified it as an allosteric inhibitor (or negative allosteric modulator, NAM) of EAAT1/GLAST. The observed effect of the F255I mutation on the UCPH-101-mediated inhibition of GLAST provided a handle for the subsequent delineation of its binding site, where specific mutations of seven residues in TM3, TM4c, and TM7a were found to impair the activity of UCPH-101 significantly. As outlined in Results, the seven residues may not all contribute directly to UCPH-101 binding but several observations strongly suggest that they line the binding site for the inhibitor. Most importantly, the functional properties displayed by Glu and TBOA at mutants containing substitutions of these seven residues did not differ substantially from those at WT GLAST, which demonstrates that basic transporter function has not been compromised and that the effect of a given mutation thus is largely attributable to its impact on the GLAST-UCPH-101 interaction. Second, UCPH-101 displayed WT GLAST-like properties at numerous mutants containing mutations of other residues in TM3, TM4c, and TM7a and at mutants with substitutions in other transporter domains, which again is indicative of specific contributions of the seven residues to UCPH-101 binding and/or close proximity to the binding site (Table 2). Finally, the distribution patterns of Gly120, Ala123, and Tyr127 in TM3, Met251 and Phe255 in TM4c, and Phe389 and Val393 in TM7a imply that the three α-helices present the respective residues in the same spatial orientation, which obviously would be a prerequisite for them to contribute to or line the same binding site (Fig. 11D). It is important to stress that additional GLAST residues most likely contribute to UCPH-101 binding as well. For example, the inactivity of UCPH-101 at GLT-1-I249M/I253F bears witness to the fact that we clearly have not identified all molecular determinants in EAAT1/GLAST for the subtype selectivity of the inhibitor.
UCPH-101 is proposed to target a predominantly hydrophobic crevice situated in the trimerization domain of the GLAST monomer, and the spatial orientations and distances between the seven identified residues in the homology model of GLAST support the notion of these lining the crevice (Fig. 11). Although the overall size of the crevice in the model is too small to accommodate the UCPH-101 molecule (MOE function: Site-Finder), the EAAT1/GLAST protein is of a flexible nature and the inhibitor can be envisioned to wedge itself into the crevice during its binding pushing away side chains (induced fit binding mechanism).
The diverse functional consequences for the GLAST inhibition exerted by UCPH-101 arising from different mutations of the seven key residues are interesting. The decreased inhibitory potency or complete inactivity displayed by UCPH-101 at several of these mutants most likely reflects varying degrees of reduced binding affinity to the transporter. In contrast, we propose that the partial inhibition exerted by UCPH-101 at mutants containing specific substitutions of Tyr127, Phe389, and Val393 arise from mutation-induced changes in the binding mode of the inhibitor and/or in the translation of this binding into effects on the substrate translocation process (Figs. 9 and 10; Table 2). Here an interesting parallel can be drawn to the metabotropic Glu receptor (mGluR) field, where mutations introduced into allosteric sites in the transmembrane domains of the receptors have been shown to convert positive allosteric modulators (PAMs) and “full” NAMs into partial antagonists (Malherbe et al., 2003; Mühlemann et al., 2006; Lundström et al., 2011). In further support of the highly modifiable efficacy of ligands acting through these sites, several modulators exhibit dual NAM and PAM activity at different mGluRs (Sheffler et al., 2011), and subtle structural modifications of two series of allosteric modulators of mGluR5 have yielded everything from ago-PAMs and PAMs over full and partial NAMs to “neutral ligands” (O'Brien et al., 2003; Rodriguez et al., 2005; Noetzel et al., 2012). Whether a similar degree of functional diversity apply for ligands targeting the UCPH-101 binding site in EAAT1/GLAST depends on the inherent nature of the coupling between the allosteric site and the substrate translocation process as well as on the specific ligand. In structure–activity relationship studies of this compound scaffold, we have not identified partial inhibitors or PAMs of EAAT1, and none of the analogs developed so far have exhibited activity at EAAT2 or EAAT3 (Jensen et al., 2009; Erichsen et al., 2010; Huynh et al., 2012a; b). However, that does not rule out the possibility that future UCPH-101 analogs or other ligands targeting this site could exhibit functionalities other than full NAM activity at EAAT1 and even possess activities at other EAAT subtypes.
In conclusion, this study represents one the most elaborate explorations of the molecular basis for an allosteric modulator of a neurotransmitter transporter reported to date (Vandenberg et al., 2004; Kristensen et al., 2011). The mechanism of action of UCPH-101 and its analogs underlines the functional importance of the trimerization domain of the EAAT. Whatever molecular constraint binding of this inhibitor class to EAAT1/GLAST introduces in the transporter protein this domain is clearly more than a rigid hinge enabling the dramatic movements of the transport domain during substrate translocation (Boudker and Verdon, 2010; Jiang and Amara, 2011). Equally interesting is the feasibility of modulating EAAT function by ligands targeting regions other than its transport domain. Because of the conserved nature of the substrate binding sites in the five EAATs, precious few subtype-selective orthosteric ligands have been identified to date (Bunch et al., 2009). Another inherent flaw of the orthosteric ligand, be it a substrate or an inhibitor, is that it inevitably will inhibit Glu uptake through the EAAT, which limits the therapeutic potential in these ligands to disorders characterized by glutamatergic hypofunction (Field et al., 2011) or by reversal of EAAT function (Rossi et al., 2000). For a ligand to augment the Glu transport capacity of the EAAT, it would have to act through an allosteric site in the transporter. In this regard, the UCPH-101 binding site and other allosteric sites in the EAAT trimer constitute putative hot spots for novel ligands characterized by unique subtype selectivity and functional characteristics at the transporters.
Footnotes
This work was supported by the Lundbeck Foundation, the Deutsche Forschungsgemeinschaft, the Novo Nordisk Foundation, the Augustinus Foundation, the A. P. Møller Foundation for the Advancement of Medical Sciences, and the Danish Medical Research Council. We thank Drs. S.G. Amara, T. Rauen, J. Rothstein, and S. Cordeiro for their generous gifts of human, rat, and mouse EAAT cDNAs. Parts of this work have been previously presented in the form of a poster at the Society of Neuroscience conference in San Diego, November 13–17, 2010.
The authors declare no competing financial interests.
References
- Bendahan A, Armon A, Madani N, Kavanaugh MP, Kanner BI. Arginine 447 plays a pivotal role in substrate interactions in a neuronal glutamate transporter. J Biol Chem. 2000;275:37436–37442. doi: 10.1074/jbc.M006536200. [DOI] [PubMed] [Google Scholar]
- Boudker O, Verdon G. Structural perspectives on secondary active transporters. Trends Pharmacol Sci. 2010;31:418–426. doi: 10.1016/j.tips.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature. 2007;445:387–393. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- Bunch L, Erichsen MN, Jensen AA. Excitatory amino acid transporters as potential drug targets. Expert Opin Ther Targets. 2009;13:719–731. doi: 10.1517/14728220902926127. [DOI] [PubMed] [Google Scholar]
- Conradt M, Stoffel W. Functional analysis of the high affinity, Na+-dependent glutamate transporter GLAST-1 by site-directed mutagenesis. J Biol Chem. 1995;270:25207–25212. doi: 10.1074/jbc.270.42.25207. [DOI] [PubMed] [Google Scholar]
- Corona JC, Tovar-y-Romo LB, Tapia R. Glutamate excitotoxicity and therapeutic targets for amyotrophic lateral sclerosis. Expert Opin Ther Targets. 2007;11:1415–1428. doi: 10.1517/14728222.11.11.1415. [DOI] [PubMed] [Google Scholar]
- Craig DA. The Cheng-Prusoff relationship: something lost in the translation. Trends Pharmacol Sci. 1993;14:89–91. doi: 10.1016/0165-6147(93)90070-z. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Erichsen MN, Huynh TH, Abrahamsen B, Bastlund JF, Bundgaard C, Monrad O, Bekker-Jensen A, Nielsen CW, Frydenvang K, Jensen AA, Bunch L. Structure–activity relationship study of first selective inhibitor of excitatory amino acid transporter subtype 1: 2-amino-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (UCPH-101) J Med Chem. 2010;53:7180–7191. doi: 10.1021/jm1009154. [DOI] [PubMed] [Google Scholar]
- Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino acid-acid transporter with properties of a ligand-gated chloride channel. Nature. 1995;375:599–603. doi: 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- Field JR, Walker AG, Conn PJ. Targeting glutamate synapses in schizophrenia. Trends Mol Med. 2011;17:689–698. doi: 10.1016/j.molmed.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendreau S, Voswinkel S, Torres-Salazar D, Lang N, Heidtmann H, Detro-Dassen S, Schmalzing G, Hidalgo P, Fahlke Ch. A trimeric quaternary structure is conserved in bacterial and human glutamate transporters. J Biol Chem. 2004;279:39505–39512. doi: 10.1074/jbc.M408038200. [DOI] [PubMed] [Google Scholar]
- Gitto R, De Luca L, De Grazia S, Chimirri A. Glutamatergic neurotransmission as molecular target of new anticonvulsants. Curr Top Med Chem. 2012;12:971–993. doi: 10.2174/156802612800229242. [DOI] [PubMed] [Google Scholar]
- Gray JA, Roth BL. Paradoxical trafficking and regulation of 5-HT2A receptors by agonists and antagonists. Brain Res Bull. 2001;56:441–451. doi: 10.1016/s0361-9230(01)00623-2. [DOI] [PubMed] [Google Scholar]
- Grewer C, Balani P, Weidenfeller C, Bartusel T, Tao Z, Rauen T. Individual subunits of the glutamate transporter EAAC1 homotrimer function independently of each other. Biochemistry. 2005;44:11913–11923. doi: 10.1021/bi050987n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Hotzy J, Machtens JP, Fahlke Ch. Neutralizing aspartate 83 modifies substrate translocation of excitatory amino acid transporter 3 (EAAT3) glutamate transporters. J Biol Chem. 2012;287:20016–20026. doi: 10.1074/jbc.M112.344077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Vandenberg RJ. Mutations in transmembrane domains 5 and 7 of the human excitatory amino acid transporter 1 affect the substrate-activated anion channel. Biochemistry. 2007;46:9685–9692. doi: 10.1021/bi700647f. [DOI] [PubMed] [Google Scholar]
- Huynh TH, Abrahamsen B, Madsen KK, Gonzalez-Franquesa A, Jensen AA, Bunch L. Design, synthesis and pharmacological characterization of coumarin-based fluorescent analogs of excitatory amino acid transporter subtype 1 selective inhibitors, UCPH-101 and UCPH-102. Bioorg Med Chem. 2012a;20:6831–6839. doi: 10.1016/j.bmc.2012.09.049. [DOI] [PubMed] [Google Scholar]
- Huynh TH, Shim I, Bohr H, Abrahamsen B, Nielsen B, Jensen AA, Bunch L. Structure–activity relationship study of selective EAAT1 inhibitor 2-amino-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (UCPH-101) and absolute configurational assignment using infrared (IR) and vibrational circular dichroism (VCD) spectroscopy in combination with ab initio Hartree-Fock calculations. J Med Chem. 2012b;55:5403–5412. doi: 10.1021/jm300345z. [DOI] [PubMed] [Google Scholar]
- Jensen AA, Bräuner-Osborne H. Pharmacological characterization of human excitatory amino acid transporters EAAT1, EAAT2 and EAAT3 in a fluorescence-based membrane potential assay. Biochem Pharmacol. 2004;67:2115–2127. doi: 10.1016/j.bcp.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Jensen AA, Christesen T, Bølcho U, Greenwood JR, Postorino G, Vogensen SB, Johansen TN, Egebjerg J, Bräuner-Osborne H, Clausen RP. Functional characterization of Tet-AMPA [tetrazolyl-2-amino-3-(3-hydroxy-5-methyl-4-isoxazolyl)propionic acid] analogues at ionotropic glutamate receptors GluR1-GluR4: the molecular basis for the functional selectivity profile of 2-Bn-Tet-AMPA. J Med Chem. 2007;50:4177–4185. doi: 10.1021/jm070532r. [DOI] [PubMed] [Google Scholar]
- Jensen AA, Erichsen MN, Nielsen CW, Stensbøl TB, Kehler J, Bunch L. Discovery of the first selective inhibitor of excitatory amino acid transporter subtype 1. J Med Chem. 2009;52:912–915. doi: 10.1021/jm8013458. [DOI] [PubMed] [Google Scholar]
- Jiang J, Amara SG. New views of glutamate transporter structure and function: advances and challenges. Neuropharmacology. 2011;60:172–181. doi: 10.1016/j.neuropharm.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Conn PJ, Niswender CM. Glutamate receptors as therapeutic targets for Parkinson's disease. CNS Neurol Disord Drug Targets. 2009;8:475–491. doi: 10.2174/187152709789824606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanner BI, Zomot E. Sodium-coupled neurotransmitter transporters. Chem Rev. 2008;108:1654–1668. doi: 10.1021/cr078246a. [DOI] [PubMed] [Google Scholar]
- Kittler K, Lau T, Schloss P. Antagonists and substrates differentially regulate serotonin transporter cell surface expression in serotonergic neurons. Eur J Pharmacol. 2010;629:63–67. doi: 10.1016/j.ejphar.2009.12.010. [DOI] [PubMed] [Google Scholar]
- Koch HP, Larsson HP. Small-scale molecular motions accomplish glutamate uptake in human glutamate transporters. J Neurosci. 2005;25:1730–1736. doi: 10.1523/JNEUROSCI.4138-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AS, Andersen J, Jørgensen TN, Sørensen L, Eriksen J, Loland CJ, Strømgaard K, Gether U. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol Rev. 2011;63:585–640. doi: 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- Krzywkowski K, Davies PA, Feinberg-Zadek PL, Bräuner-Osborne H, Jensen AA. A high-frequency HTR3B variant associated with major depression dramatically augments the signaling of the human 5-HT3AB receptor. Proc Natl Acad Sci U S A. 2008;105:722–727. doi: 10.1073/pnas.0708454105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DJ, Lawen A. The glutamate aspartate transporter (GLAST) mediates L-glutamate-stimulated ascorbate-release via swelling-activated anion channels in cultured neonatal rodent astrocytes. Cell Biochem Biophys. 2012 doi: 10.1007/s12013-012-9404-8. Advance online publication. [DOI] [PubMed] [Google Scholar]
- Lau T, Horschitz S, Berger S, Bartsch D, Schloss P. Antidepressant-induced internalization of the serotonin transporter in serotonergic neurons. FASEB J. 2008;22:1702–1714. doi: 10.1096/fj.07-095471. [DOI] [PubMed] [Google Scholar]
- Leary GP, Stone EF, Holley DC, Kavanaugh MP. The glutamate and chloride permeation pathways are colocalized in individual neuronal glutamate transporter subunits. J Neurosci. 2007;27:2938–2942. doi: 10.1523/JNEUROSCI.4851-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Sidoryk-Wêgrzynowicz M, Wang N, Webb A, Son DS, Lee K, Aschner M. GPR30 regulates glutamate transporter GLT-1 expression in rat primary astrocytes. J Biol Chem. 2012;287:26817–26828. doi: 10.1074/jbc.M112.341867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundström L, Bissantz C, Beck J, Wettstein JG, Woltering TJ, Wichmann J, Gatti S. Structural determinants of allosteric antagonism at metabotropic glutamate receptor 2: mechanistic studies with new potent negative allosteric modulators. Br J Pharmacol. 2011;164:521–537. doi: 10.1111/j.1476-5381.2011.01409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malherbe P, Kratochwil N, Zenner MT, Piussi J, Diener C, Kratzeisen C, Fischer C, Porter RH. Mutational analysis and molecular modeling of the binding pocket of the metabotropic glutamate 5 receptor negative modulator 2-methyl-6-(phenylethynyl)-pyridine. Mol Pharmacol. 2003;64:823–832. doi: 10.1124/mol.64.4.823. [DOI] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Glutamate transporters: animal models to neurologic disease. Neurobiol Dis. 2004;15:461–473. doi: 10.1016/j.nbd.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Melzer N, Biela A, Fahlke Ch. Glutamate modifies ion conduction and voltage-dependent gating of excitatory amino acid transporter-associated anion channels. J Biol Chem. 2003;278:50112–50119. doi: 10.1074/jbc.M307990200. [DOI] [PubMed] [Google Scholar]
- Mitrovic AD, Amara SG, Johnston GA, Vandenberg RJ. Identification of functional domains of the human glutamate transporters EAAT1 and EAAT2. J Biol Chem. 1998;273:14698–14706. doi: 10.1074/jbc.273.24.14698. [DOI] [PubMed] [Google Scholar]
- Mühlemann A, Ward NA, Kratochwil N, Diener C, Fischer C, Stucki A, Jaeschke G, Malherbe P, Porter RH. Determination of key amino acids implicated in the actions of allosteric modulation by 3,3′-difluorobenzaldazine on rat mGlu5 receptors. Eur J Pharmacol. 2006;529:95–104. doi: 10.1016/j.ejphar.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Noetzel MJ, Rook JM, Vinson PN, Cho HP, Days E, Zhou Y, Rodriguez AL, Lavreysen H, Stauffer SR, Niswender CM, Xiang Z, Daniels JS, Jones CK, Lindsley CW, Weaver CD, Conn PJ. Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol Pharmacol. 2012;81:120–133. doi: 10.1124/mol.111.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien JA, Lemaire W, Chen TB, Chang RS, Jacobson MA, Ha SN, Lindsley CW, Schaffhauser HJ, Sur C, Pettibone DJ, Conn PJ, Williams DL., Jr A family of highly selective allosteric modulators of the metabotropic glutamate receptor subtype 5 (mGluR5) Mol Pharmacol. 2003;64:731–740. doi: 10.1124/mol.64.3.731. [DOI] [PubMed] [Google Scholar]
- Okabe Y, Takahashi T, Mitsumasu C, Kosai K, Tanaka E, Matsuishi T. Alterations of gene expression and glutamate clearance in astrocytes derived from an MeCP2-null mouse model of Rett syndrome. PLoS One. 2012;7:e35354. doi: 10.1371/journal.pone.0035354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacey E, Miller CC, Dunlop J, Rattray M. The four major N- and C-terminal splice variants of the excitatory amino acid transporter GLT-1 form cell surface homomeric and heteromeric assemblies. Mol Pharmacol. 2009;75:1062–1073. doi: 10.1124/mol.108.052829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pheng LH, Dumont Y, Fournier A, Chabot JG, Beaudet A, Quirion R. Agonist- and antagonist-induced sequestration/internalization of neuropeptide Y Y1 receptors in HEK293 cells. Br J Pharmacol. 2003;139:695–704. doi: 10.1038/sj.bjp.0705306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes N, Ginter C, Boudker O. Transport mechanism of a bacterial homologue of glutamate transporters. Nature. 2009;462:880–885. doi: 10.1038/nature08616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AL, Nong Y, Sekaran NK, Alagille D, Tamagnan GD, Conn PJ. A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol Pharmacol. 2005;68:1793–1802. doi: 10.1124/mol.105.016139. [DOI] [PubMed] [Google Scholar]
- Roettger BF, Ghanekar D, Rao R, Toledo C, Yingling J, Pinon D, Miller LJ. Antagonist-stimulated internalization of the G protein-coupled cholecystokinin receptor. Mol Pharmacol. 1997;51:357–362. [PubMed] [Google Scholar]
- Rojas C, Thomas AG, Alt J, Stathis M, Zhang J, Rubenstein EB, Sebastiani S, Cantoreggi S, Slusher BS. Palonosetron triggers 5-HT3 receptor internalization and causes prolonged inhibition of receptor function. Eur J Pharmacol. 2010;626:193–199. doi: 10.1016/j.ejphar.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- Ryan RM, Mitrovic AD, Vandenberg RJ. The chloride permeation pathway of a glutamate transporter and its proximity to the glutamate translocation pathway. J Biol Chem. 2004;279:20742–20751. doi: 10.1074/jbc.M304433200. [DOI] [PubMed] [Google Scholar]
- Salvatore MF, Davis RW, Arnold JC, Chotibut T. Transient striatal GLT-1 blockade increases EAAC1 expression, glutamate reuptake, and decreases tyrosine hydroxylase phosphorylation at ser(19) Exp Neurol. 2012;234:428–436. doi: 10.1016/j.expneurol.2012.01.012. [DOI] [PubMed] [Google Scholar]
- Sheffler DJ, Gregory KJ, Rook JM, Conn PJ. Allosteric modulation of metabotropic glutamate receptors. Adv Pharmacol. 2011;62:37–77. doi: 10.1016/B978-0-12-385952-5.00010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T. DL-threo-β-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol. 1998;53:195–201. doi: 10.1124/mol.53.2.195. [DOI] [PubMed] [Google Scholar]
- Teichman S, Kanner BI. Aspartate-444 is essential for productive substrate interactions in a neuronal glutamate transporter. J Gen Physiol. 2007;129:527–539. doi: 10.1085/jgp.200609707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uwechue NM, Marx MC, Chevy Q, Billups B. Activation of glutamate transport evokes rapid glutamine release from perisynaptic astrocytes. J Physiol. 2012;590:2317–2331. doi: 10.1113/jphysiol.2011.226605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg RJ, Ju P, Aubrey KR, Ryan RM, Mitrovic AD. Allosteric modulation of neurotransmitter transporters at excitatory synapses. Eur J Pharm Sci. 2004;23:1–11. doi: 10.1016/j.ejps.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Verdon G, Boudker O. Crystal structure of an asymmetric trimer of a bacterial glutamate transporter homolog. Nat Struct Mol Biol. 2012;19:355–357. doi: 10.1038/nsmb.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche JI, Amara SG, Kavanaugh MP. Ion fluxes associated with excitatory amino acid transport. Neuron. 1995;15:721–728. doi: 10.1016/0896-6273(95)90159-0. [DOI] [PubMed] [Google Scholar]
- Winter N, Kovermann P, Fahlke Ch. A point mutation associated with episodic ataxia 6 increases glutamate transporter anion currents. Brain. 2012;135:3416–3425. doi: 10.1093/brain/aws255. [DOI] [PubMed] [Google Scholar]
- Yernool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–818. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- Yogeeswari P, Semwal A, Mishra R, Sriram D. Current approaches with the glutamatergic system as targets in the treatment of neuropathic pain. Expert Opin Ther Targets. 2009;13:925–943. doi: 10.1517/14728220903029210. [DOI] [PubMed] [Google Scholar]