

Abstract
The Neural cell adhesion molecule (NCAM) plays an important role in regulation of nervous system development. To expand our understanding of the molecular mechanisms via which NCAM influences differentiation of neurons, we used a yeast two-hybrid screening to search for new binding partners of NCAM and identified p21-activated kinase 1 (Pak1). We show that NCAM interacts with Pak1 in growth cones of neurons. The autophosphorylation and activity of Pak1 were enhanced when isolated growth cones were incubated with NCAM function triggering antibodies, which mimic the interaction between NCAM and its extracellular ligands. The association of Pak1 with cell membranes, the efficiency of Pak1 binding to its activators, and Pak1 activity were inhibited in brains of NCAM-deficient mice. NCAM-dependent Pak1 activation was abolished after lipid raft disruption, suggesting that NCAM promotes Pak1 activation in the lipid raft environment. Phosphorylation of the downstream Pak1 effectors LIMK1 and cofilin was reduced in growth cones from NCAM-deficient neurons, which was accompanied by decreased levels of filamentous actin and inhibited filopodium mobility in the growth cones. Dominant-negative Pak1 inhibited and constitutively active Pak1 enhanced the ability of neurons to increase neurite outgrowth in response to the extracellular ligands of NCAM. Our combined observations thus indicate that NCAM activates Pak1 to drive actin polymerization to promote neuronal differentiation.
Introduction
Neuronal differentiation, a process critical for the development of the complex neuronal networks in the brain, is regulated by a number of extracellular cues acting at diverse receptors expressed at the neuronal cell surface (O'Donnell et al., 2009). Among them, the neural cell adhesion molecule (NCAM), a member of the Ig superfamily of adhesion molecules, plays an important role in directing and regulating the efficiency of neurite outgrowth in the developing nervous system (Maness and Schachner, 2007; Shapiro et al., 2007; Hansen et al., 2008; Schmid and Maness, 2008). Two major transmembrane NCAM isoforms with the molecular weights of 140 kDa (NCAM140) and 180 kDa (NCAM180) are predominantly expressed at the cell surface of neurons accumulating in growth cones, while the glycophosphoinositol-anchored isoform with the molecular weight of 120 kDa (NCAM120) is enriched in glial cells (Cunningham et al., 1987; Bodrikov et al., 2005; Santuccione et al., 2005). Binding of NCAM at the cell surface of neurons to its homophilic and heterophilic extracellular multivalent ligands promotes neuronal differentiation (Hansen et al., 2008; Westphal et al., 2010; Chernyshova et al., 2011). Neurite outgrowth depends on the continuous rearrangement of the cytoskeleton along neurites and particularly at their growing tips, the growth cones (O'Donnell et al., 2009). Mechanisms linking NCAM-mediated neurite outgrowth to the cytoskeleton remodeling remain, however, poorly understood.
The p21-activated kinases (Paks) serve as key regulators of cytoskeleton dynamics in neurons (Kreis and Barnier, 2009), being the main effectors of Rac1 and cdc42 GTPases, which bind to and activate Pak (Manser et al., 1994; Bokoch, 2003). Pak activity is also regulated by binding to other partners, such as the (Pak)-interacting exchange factor (PIX) and by phosphorylation of this enzyme (Chong et al., 2001; Bokoch, 2003; Arias-Romero and Chernoff, 2008). LIM kinase (LIMK)-1 is one of the main targets of Pak (Edwards et al., 1999). Upon Pak-dependent activation, LIMK phosphorylates cofilin, an actin polymerization regulator, thereby linking cdc42 signaling to cytoskeleton dynamics and promoting neuritogenesis (Rosso et al., 2004).
By yeast two-hybrid screening, we identify Pak1, a Pak family kinase that is highly expressed in the brain (Manser et al., 1994; Ong et al., 2002), as a binding partner of NCAM. We show that activation of NCAM in growth cones results in the induction of the Pak1 signaling pathway, thereby directly linking NCAM to the cytoskeleton remodeling machinery required for NCAM-dependent neurite outgrowth.
Materials and Methods
Antibodies.
Rat monoclonal antibodies against NCAM (clone H28; Gennarini et al., 1984) were used in immunoprecipitation. Rabbit polyclonal antibodies against NCAM (Sytnyk et al., 2002; Westphal et al., 2010) were used for Western blot analysis, immunoprecipitation, immunocytochemistry, and neurite outgrowth assay. Chicken polyclonal antibodies against NCAM (Chernyshova et al., 2011) were used to stimulate isolated growth cones. Animal species of antibodies against NCAM used for immunoprecipitation or stimulation were always chosen to be different from animal species of antibodies used for consecutive Western blot analysis of NCAM immunoprecipitates or stimulated growth cones to preclude detection of antibodies used for immunoprecipitation or stimulation by secondary antibodies. Chicken and rabbit nonimmune serum was from Pineda Antibody Service. Goat polyclonal antibodies against the intracellular domain of NCAM (C-20; Santa Cruz Biotechnology) were used in the proximity ligation assay. Rabbit polyclonal antibodies against Pak1 (N-20), mouse monoclonal antibody against LIMK1 (C-10), and rabbit polyclonal antibodies against phosphatidylcholine phospholipase D2 (H-133) were from Santa Cruz Biotechnology. Rabbit polyclonal antibodies against cdc42 were from Cell Signaling Technology. Mouse monoclonal antibodies against tubulin (clone DM1A), rabbit polyclonal antibodies against actin (20–33), normal nonimmune rabbit, and rat immunoglobulins were from Sigma-Aldrich. Mouse monoclonal antibody against cofilin was from BD Biosciences. Rabbit polyclonal antibodies against tyrosine hydroxylase (TH) and Ser40-phosphorylated TH were from AbD Serotec. Rabbit polyclonal antibodies against βPIX and mouse monoclonal antibodies against GAPDH were from Millipore. Goat polyclonal antibodies against contactin-1 were from R&D Systems. Horseradish peroxidase (HRP), Cy2, Cy3, or Cy5 coupled secondary antibodies were from Jackson ImmunoResearch.
The following phospho-specific antibodies were used. Rabbit polyclonal antibodies against Thr423- and Ser199/204-phosphorylated Pak1 were from Cell Signaling Technology. The signal produced by pThr423-Pak1 antibodies has been shown to be eliminated in cells transfected with shPak1 (Siu et al., 2010) and an increase in Pak1 phosphorylation detected with pThr423-Pak1 and pSer199/204-Pak1 antibodies was shown to correlate with an increase in Pak1 enzyme activity (Parrini et al., 2009) indicating antibody specificity. Rabbit polyclonal antibodies against Thr508-phosphorylated LIMK1 were from Cell Signaling Technology and have been shown to specifically recognize overexpressed activated LIMK1 (Spratley et al., 2011), and an increase in LIMK1 phosphorylation detected with pThr508-LIMK antibodies correlated with an increase in LIMK1 enzyme activity (Gamell et al., 2008). Mouse monoclonal antibody against Thr212-phosphorylated Pak1 (clone PK-18) was from Sigma-Aldrich and has been shown to detect overexpressed wild-type Pak1, but not Pak1 with the mutated T212 residue, which does not allow phosphorylation (Rashid et al., 2001). Rabbit polyclonal antibodies against Ser3-phosphorylated cofilin (Kuz̆elov á et al., 2010; Maddala et al., 2011) were from Sigma-Aldrich, and the detection of phosphorylated cofilin by these antibodies has been found to be specifically inhibited by the phosphorylated immunizing peptide, but not by the corresponding nonphosphorylated peptide.
Mice.
One- to three-day-old wild-type (C57BL/6J, NCAM+/+) and NCAM-deficient (NCAM−/−; Cremer et al., 1994) mice of either sex were used. NCAM−/− mice had been back-crossed for at least nine generations onto the C57BL/6J background. Animals used for biochemical experiments were 1- to 3-d-old NCAM+/+ and NCAM−/− littermates obtained from heterozygous breeding. To prepare cultures of primary neurons, wild-type and NCAM−/− mice from homozygous breeding pairs were used.
DNA constructs.
Dominant-negative Pak1 (DN-Pak1) and constitutively active Pak1 (CA-Pak1) were as described previously (Hayashi et al., 2007). CA-Pak1 was generated by replacing Thr-423 in the catalytic domain of Pak1 with glutamic acid resulting in constitutively active enzyme (Sells et al., 1997). DN-Pak1 consisted of amino acid residues 67–150 (inhibitory region) of mouse Pak1 (Hayashi et al., 2002). Plasmids encoding full-length NCAM140 and NCAM140Δcys (Niethammer et al., 2002), intracellular domain of NCAM140 (Sytnyk et al., 2002; Leshchyns'ka et al., 2003), and the NCAM140ID729–750 fragment (Chernyshova et al., 2011) were as described. Intracellular domain of NCAM140 was used as a template to produce the NCAM140ID748–777 fragment, which was cloned into the pcDNA3.1/V5-His vector (Invitrogen). The NCAM miR expression vector was developed using the BLOCK-iT PolII miR RNAi expression vector kit (Invitrogen) and designed to coexpress NCAM-specific miRNA together with emerald green fluorescent protein. The following oligonucleotides were inserted into the BLOCK-iT PolII miR RNAi expression vector: top: 5′-TGC TGT GTA GTT GGA GCT TGG CAG CAG TTT TGG CCA CTG ACT GAC TGC TGC CAC TCC AAC TAC A-3′; bottom: 5′-CCT GTG TAG TTG GAG TGG CAG CAG TCA GTC AGT GGC CAA AAC TGC TGC CAA GCT CCA ACT ACA C-3′.
Negative control miR vector was from Invitrogen. Immunocytochemical labeling confirmed that transfection with the NCAM miR vector resulted in a >80% reduction in expression of NCAM in cultured hippocampal neurons at 24 h after transfection (data not shown).
Yeast two-hybrid screening.
Yeast two-hybrid screening was performed with the ProQuest Two-Hybrid System (Invitrogen) in Saccharomyces cerevisiae strain MaV203 following the manufacturer's protocol. As bait, a DNA fragment encoding the intracellular domain of mouse NCAM140 was used.
Preparation of brain homogenates.
Brain homogenates were prepared by homogenizing brain tissue with 15 strokes in a Potter homogenizer in ice-cold homogenization buffer (0.32 m sucrose, 5 mm Tris-HCl, pH 7.4, 1 mm MgCl2, 1 mm CaCl2, 1 mm NaHCO3, and 0.1 mm PMSF). The buffer was supplemented with an EDTA-free protease inhibitor mixture and a phosphatase inhibitor mixture (Roche). All experimental steps were performed on ice.
Isolation of growth cones.
Growth cones were isolated as described previously (Pfenninger et al., 1983; Chernyshova et al., 2011). Three brains of 1- to 3-d-old mice were used per preparation and all steps were performed on ice. Mouse brains were homogenized with a few strokes in 10 ml homogenization buffer (0.32 m sucrose, 1 mm MgCl2, 5 mm Tris-HCl, pH 7.4) containing a protease inhibitor mixture (Roche). The homogenates were centrifuged at 1660 g for 15 min. The supernatants were centrifuged on a discontinuous sucrose density gradient of 0.75/2.66 m sucrose at 242,000 g for 30 min. The growth cone-enriched fraction was collected at the interface between the load and 0.75 m sucrose. The growth cone-depleted fraction was collected between 0.75 m and 2.66 m sucrose and used as nongrowth cone membranes. Fractions were resuspended in the homogenization buffer and centrifuged at 100,000 g for 40 min. Pellets were resuspended in the homogenization buffer containing a protease inhibitor mixture (Roche) and used for further analysis. All centrifugation steps were performed at 4°C.
Isolation of lipid rafts.
Lipid rafts were isolated as described previously (Leshchyns'ka et al., 2003). Up to 500 μl of membrane-enriched fractions were mixed with 4× volume of ice-cold 1% Triton X-100 in Tris-buffered saline (TBS; 50 mm Tris-HCl, pH7.4, 150 mm NaCl) and incubated for 20 min on ice. The extracted membranes were mixed with an equal volume of 80% sucrose in 0.2 m Na2CO3 to a final sucrose concentration of 40%. The material was overlaid with 2 ml of 30% sucrose in TBS, 1 ml of 10% sucrose in TBS and TBS buffer, and this discontinuous gradient was centrifuged at 230,000 g for 17 h. The lipid raft fraction was collected at the top of the 10% sucrose, resuspended in TBS buffer, and pelleted down by centrifugation at 100,000 g for 1 h. All centrifugation steps were performed at 4°C. The pellets were resuspended in a minimum volume of TBS containing a protease inhibitor mixture (Roche) and used for further analysis.
Coimmunoprecipitation.
Samples containing 1 mg of total protein were lysed with ice-cold lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm Na4P2O7, 1 mm NaF, 2 mm Na3VO4, 1% (v/v) NP-40, 1 mm PMSF, EDTA-free protease inhibitor mixture; Roche) for 1 h. Lysates were centrifuged for 15 min at 20,000 g at 4°C. Supernatants were cleared with protein A/G-agarose beads (Santa Cruz Biotechnology) for 3 h, and beads were removed by centrifugation at 600 g for 5 min. The supernatant was incubated with the corresponding antibodies or nonimmune IgG overnight, followed by precipitation with protein A/G-agarose beads for 3 h. The beads were pelleted and washed four times with the lysis buffer and three times with TBS. All steps were performed at 4°C. The proteins were finally eluted from beads with 5× SDS sample buffer (310 mm Tris-HCl, pH 6.8, 25% (v/v) glycerol, 10% (w/v) SDS, 4.5% (v/v) β-mercaptoethanol, 0.015% (w/v) bromophenol blue) by boiling for 10 min and analyzed by Western blot.
Pull-down assay.
Pak1 was immunopurified from brain lysates with agarose-conjugated rabbit polyclonal antibodies (Santa Cruz Biotechnology). Recombinant intracellular domains of NCAM140 were expressed in Escherichia coli and purified as described previously (Sytnyk et al., 2002). Pak1 immobilized on agarose beads was incubated overnight at +4°C with increasing concentrations of intracellular domain of NCAM140 (NCAM140ID) in TBS containing 1% bovine serum albumin (BSA), 2 mm MgCl2, and 2 mm CaCl2. After washing with TBS containing 1% BSA, beads were subjected to Western blot analysis of the protein complexes.
In vitro protein binding assay of Pak1 to NCAM peptides.
Pak1 was immunopurified from detergent lysates of postnatal day 2 mouse brain using agarose beads conjugated to rabbit polyclonal antibodies against Pak1 (Santa Cruz Biotechnology). Pak1 was eluted from the beads with 0.2 m glycine, pH 4.0. The eluate containing Pak1 was immediately neutralized with 1 m Tris, pH 8.0. Rabbit polyclonal antibodies against Pak1 were used to immobilize Pak1 in wells of 96-well MICROLON 600 plates (Greiner). The antibodies (1 μg/ml in PBS) were adsorbed to the surface of wells overnight at 4°C. Wells were blocked with 1% BSA in PBS for 1 h at 37°C, and incubated with purified Pak1 (3.5 μm) in PBS with 0.1% Tween 20 (PBST) for 1 h at 37°C. Wells were washed with PBST and incubated for 1 h at 37°C with increasing concentrations of chemically synthesized biotinylated peptides corresponding to amino acid sequences 748–763, 756–770, and 764–777 of mouse NCAM140 (Peptide 2.0) diluted in PBST. Plates were washed five times with PBST and incubated for 1 h at 37°C with HRP-coupled NeutrAvidin diluted 1:25,000 in PBST containing 1% BSA. After washing with PBST, wells were developed with 1 mg/ml o-phenylenediamine dihydrochloride (PerbioScience). The OD was measured at 405 nm.
Treatment of growth cones.
Treatment of growth cones was performed as described previously (Westphal et al., 2010; Chernyshova et al., 2011). Growth cone fraction (1.3 ml, ice cold) was mixed with 0.5 ml of ice-cold 2× dilution buffer (44 mm HEPES buffer, pH 7.3, 100 mm sucrose, 200 mm NaCl, 2.4 mm MgCl2, 10 mm KCl, 2.4 mm NaH2PO4, 20 mm glucose), incubated for 30 min, and mixed with an additional 0.8 ml of 2× dilution buffer. Equal volumes of the growth cones were then incubated with either NCAM antibodies or pre-immune serum for 5 min, 15 min, or 30 min at 37°C with constant gentle shaking. Treatment was terminated by the addition of 5× SDS sample buffer.
Filamentous actin/globular actin assay.
The amount of filamentous actin (F-actin) versus free globular actin (G-actin) in growth cones was analyzed using an F-actin/G-actin in vivo assay kit (Cytoskeleton) in accordance with the manufacturer's protocol. Briefly, NCAM+/+ and NCAM−/− growth cones containing equal protein contents were lysed with prewarmed F-actin stabilization buffer containing the following (in mm): 50 PIPES buffer, pH 6.9, 50 NaCl, 5 MgCl2, 5 EGTA, 5% (v/v) glycerol, 0.1% (v/v) Nonidet P-40 (NP-40), 0.1% (v/v) Triton X-100, 0.1% (v/v) Tween 20, and 0.1% (v/v) β-mercaptoethanol supplemented with 1 mm ATP and 1× protease inhibitor mixture, homogenized by pipetting eight times with 200 μl fine pipette tips and incubated at 37°C for 10 min. Samples were centrifuged at 100,000 g for 1 h at 37°C. The supernatants containing G-actin were separated from the pellets and placed on ice. The pellets containing F-actin were resuspended to the same volume as the supernatants using ice-cold water containing 1 μm cytochalasin D and incubated on ice for 1 h with mixing by pipetting every 15 min to dissociate F-actin. Equal amounts of proteins from each sample were subjected to SDS-PAGE and analyzed by Western blotting with the anti-actin antibody. Tubulin was also probed for to serve as a control. The assay was performed three times in duplicates.
Pak1 kinase assay.
Pak1 kinase activity was analyzed by an HTScan Pak1 kinase assay kit (Cell Signaling Technology) with modifications. Briefly, Pak1 immunoprecipitates were mixed with the kinase buffer (25 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 5 mm β-glycerophosphate, 0.1 mm Na3VO4, 2 mm dithiothreitol) and incubated with 1.5 μm TH (Ser40) biotinylated peptide in the presence of 200 μm ATP for 30 min at 30°C with constant gentle agitation. The reaction was stopped by adding Stop Buffer (50 mm EDTA, pH 8.0). Immunoprecipitates were centrifuged and 2 μl of the supernatants were dotted onto a nitrocellulose transfer membrane and immunoblotted with antibodies against total and Ser40-phosphorylated TH. Immunoprecipitates in the pellet were boiled in 5× SDS sample buffer and subjected to the SDS-PAGE and Western blot with Pak1 antibodies to monitor the immunoprecipitation efficacy. Mock immunoprecipitates with nonimmune Ig served as a control.
Cultures of hippocampal and cortical neurons.
Cultures of hippocampal and cortical neurons were prepared from 1- to 3-d-old mice as described previously (Andreyeva et al., 2010). Neurons were maintained in Neurobasal A medium (Invitrogen) supplemented with 2% B-27 (Invitrogen), glutamine (Invitrogen), and FGF-2 (2 ng/ml; R&D Systems) on glass coverslips coated with poly-d-lysine (100 μg/ml).
Cultures and transfection of NIH 3T3 and B35 cells.
NIH 3T3 and B35 cells were maintained in DMEM/F12 with 10% donor calf serum and 2% penicillin/streptomycin at 37°C, 5% CO2, and 90% relative humidity. Cells were transfected using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions.
Immunofluorescence labeling.
Indirect immunofluorescence labeling was performed as described previously (Sytnyk et al., 2002). All steps were performed at room temperature. Neurons on glass coverslips were washed with PBS, and fixed in 4% formaldehyde in PBS for 15 min. Neurons were washed three times with PBS and blocked in 1% BSA in PBS for 20 min. Antibodies against NCAM (H28, in 0.1% BSA in PBS) were applied to fixed but nonpermeabilized cells for 1 h and detected with fluorochrome-coupled secondary antibodies applied for 45 min. The neurons were then postfixed for 5 min in 2% paraformaldehyde in PBS, washed with PBS, permeabilized with 0.25% Triton X-100 in PBS for 5 min, and blocked with 1% BSA in PBS for 20 min. Antibodies against Pak1, cdc42, and PIX were applied in 0.1% BSA in PBS for 2 h and then detected with corresponding secondary antibodies applied for 45 min. Cells were washed with PBS four times and embedded in Aqua-Poly/Mount (Polysciences). Immunofluorescence images were acquired at room temperature using a confocal laser scanning microscope LSM510, LSM510 software, (version 3), and oil Plan-Neofluar 40× objective (NA 1.3) at 3× digital zoom (all from Zeiss). Contrast and brightness of the images were further adjusted in Corel Photo-Paint 9.
Proximity ligation assay.
Cultured neurons were fixed in 4% formaldehyde in PBS, washed with PBS, permeabilized with 0.25% Triton X-100 in PBS for 5 min, and blocked with 1% BSA in PBS for 20 min. Antibodies against the intracellular domain of NCAM and Pak1, cdc42, or PIX were applied to the cells in 0.1% BSA in PBS overnight at 4°C. Further steps were performed using secondary antibodies conjugated with oligonucleotides (PLA probes; Olink Bioscience) and Duolink II fluorescence kit (Olink Bioscience) in accordance with the manufacturer's instructions. Fluorescence images were acquired at room temperature using a confocal laser scanning microscope Nikon C1si, NIS Elements software, and oil Plan Apo VC 60× objective (NA 1.4), all from Nikon.
Neurite outgrowth assay.
Cortical neurons were transfected with CA-Pak1, DN-Pak1, or the empty vector at 6 h after plating using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Fluorescent protein cherry was coexpressed in transfected neurons to visualize neuronal morphology. Rabbit polyclonal antibodies against NCAM or pre-immune serum were applied to neurons 6 h after transfection. Neurons were fixed 24 h later with 4% formaldehyde in PBS for 30 min at room temperature and washed twice with PBS. Neurons were imaged using Axiophot 2 microscope equipped with Plan-Neofluar 40× objective (NA 0.75), AxioCam HRc digital camera, and AxioVision software version 3.1 (all from Zeiss). Neurite length was measured in ImageJ (National Institutes of Health). For each experimental value at least three coverslips for each group, and at least 100 neurons from one coverslip, were measured.
Live cell imaging.
Recordings of live cultured hippocampal neurons from NCAM+/+ and NCAM−/− mice were performed with a 1 s interval using a time-lapse function of the laser scanning microscope LSM510 (Zeiss). During recordings, neurons were maintained on the microscope stage in an incubator (Zeiss) at 37°C and 5% CO2. In experiments with transfected neurons, cultures of hippocampal neurons from NCAM+/+ mice were transfected before plating by electroporation using the Neon transfection system (Invitrogen). Recordings were performed with a 1 s interval using Nikon TS100 microscope equipped with a DS-Qi1 high speed camera and Plan Fluor ELWD 40× objective (NA 0.6) and NIS Elements software (all from Nikon) with neurons maintained on the microscope stage in an incubator (OkoLab) at 37°C and 5% CO2. Numbers of filopodia and their lengths were analyzed using ImageJ software. Moving filopodia were defined as filopodia that had changed their position with respect to their position in the first frame in at least one of the consecutive frames recorded over the observation period.
Results
NCAM associates with Pak1 in growth cones
The intracellular domain of NCAM140, a major isoform of NCAM with an apparent molecular weight of 140 kDa, was used as bait in a yeast two-hybrid system screen to search for binding partners of NCAM140 within a cDNA library from mouse brain. Among >2 × 106 clones screened, 26 clones showed positive phenotypes. One clone, containing a 1.71 kb sequence, revealed an open reading frame of 164 aa encoding the C-terminal domain of Pak1 protein kinase (NCBI accession No. NM_011035.2). To confirm the interaction between NCAM and Pak1 suggested by the yeast two-hybrid screening, we first investigated the distribution of NCAM and Pak1 with respect to each other in 1-d-old cultured hippocampal neurons. Neurons were colabeled by indirect immunofluorescence with antibodies against NCAM and Pak1. The labeling showed that NCAM was accumulated in growth cones and also present at lower levels along neurites and in somata of neurons (Fig. 1A) in accordance with previous reports (Leshchyns'ka et al., 2003; Bodrikov et al., 2008). Pak1 was also accumulated in somata and present along neurites (Fig. 1A). Distributions of these two proteins partially overlapped (Fig. 1A).
Figure 1.
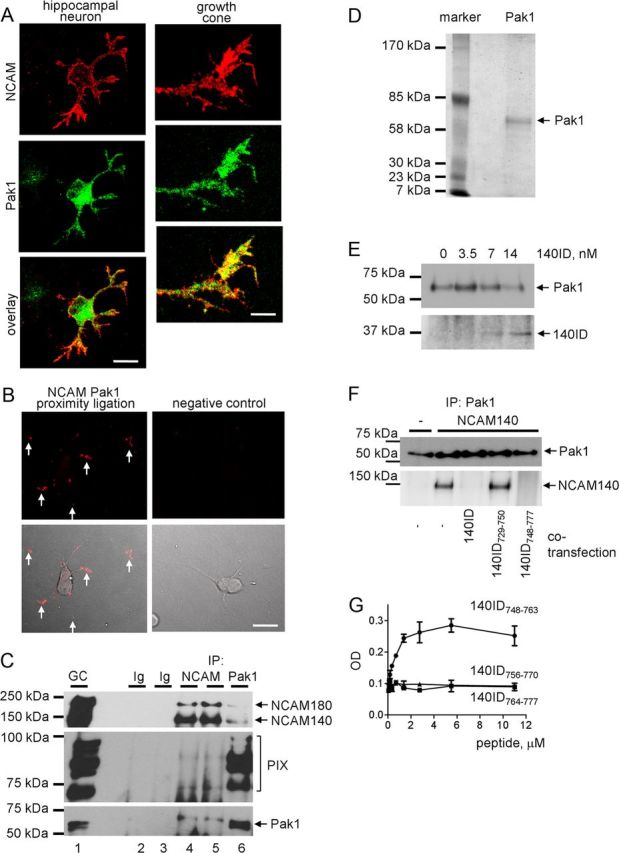
Pak1 is a novel binding partner of NCAM. A, One-day-old cultured hippocampal neuron and high magnification of a growth cone colabeled with antibodies against Pak1 and NCAM. Note clusters of NCAM overlapping with Pak1 accumulations in growth cones (yellow). Scale bars: (neuron), 20 μm; (growth cone) 5 μm. B, Proximity ligation assay with antibodies against Pak1 and the intracellular domain of NCAM (left). Arrows show examples of putative NCAM/Pak1 complexes in growth cones. Proximity ligation reaction without Pak1 antibodies served as negative control (right). Proximity ligation fluorescence signals alone (top) or in combination with differential interference contrast images (bottom) are shown. C, NCAM (lanes 4 and 5) and Pak1 (lane 6) immunoprecipitates (IP) from growth cones isolated from brains of NCAM+/+ mice and input material used for immunoprecipitation (GC, lane 1) probed by Western blot with antibodies against NCAM, PIX, and Pak1. Mock immunoprecipitation with nonimmune Ig (lanes 2 and 3) served as a control. Note that Pak1 and PIX coimmunoprecipitate with NCAM, and NCAM and PIX coimmunoprecipitate with Pak1. PIX antibody recognizes multiple splice variants of mouse βPIX isoforms. D, Silver-stained gel shows immunopurified Pak1 from mouse brain used in the in vitro protein binding assays (E, G). E, The ability of Pak1 immunopurified from mouse brain and immobilized on agarose beads to bind increasing concentrations of recombinant intracellular domain of NCAM140 (140ID) was tested in a pull-down assay. Western blots show levels of immobilized Pak1 and bound 140ID. Note that 140ID binds to Pak1 in a concentration-dependent manner. F, Analysis of Pak1 immunoprecipitates from nontransfected 3T3 cells or 3T3 cells transfected with NCAM140 alone or cotransfected with whole 140ID, or its fragments 140ID729–750 or 140ID748–777 by Western blot with antibodies against NCAM and Pak1. Note that coimmunoprecipitation of NCAM140 with Pak1 is inhibited in cells cotransfected with 140ID and 140ID748–777. G, Pak1 immunopurified from mouse brain was immobilized in wells of a 96-well plate and assayed for its ability to bind increasing concentrations of chemically synthesized peptides corresponding to amino acid sequences 748–763 (140ID748–763), 756–770 (140ID756–770), and 764–777 (140ID764–777) of the intracellular domain of NCAM140. The experiment was performed three times with the same results. Mean values (OD405) ± SEM for a representative experiment performed in triplicates are shown. Note that only 140ID748–763 binds to Pak1 in a concentration-dependent and saturable manner.
To analyze if NCAM and Pak1 form a complex, proximity ligation analysis was performed to detect sites of close proximity (<40 nm) of the two proteins indicative of complex formation. Proximity ligation with antibodies against the intracellular domain of NCAM and Pak1 produced a positive reaction (Fig. 1B) with 42.1 ± 2.4, 14.5 ± 1.6, and 43.2 ± 2.5% of NCAM/Pak1 complexes detectable in somata, along neurites, and in growth cones, respectively (n = 90 neurons were analyzed). Omission of one of the antibodies completely eliminated the signal (Fig. 1B), while proximity ligation with antibodies against NCAM and phosphatidylcholine phospholipase D2 used as a negative control produced only negligible reaction (data not shown).
To investigate if these two proteins form a complex in the brain tissue, coimmunoprecipitation experiments were performed. A high degree of colocalization of NCAM and Pak1 in growth cones (Fig. 1A,B) suggested that they interact in this subcellular compartment. To investigate this idea, NCAM was immunoprecipitated from the growth cone fraction, which was isolated from brains of 1-d-old mice using an established protocol (Pfenninger et al., 1983; Westphal et al., 2010; Chernyshova et al., 2011) (Fig. 1C). Pak1 and its activator PIX coimmunoprecipitated with NCAM (Fig. 1C). In a reverse coimmunoprecipitation experiment analyzed in parallel, NCAM140, NCAM180, and PIX coimmunoprecipitated with Pak1 (Fig. 1C). Our combined observations thus indicate that NCAM associates with Pak1 in growth cones.
The fact that the interaction between NCAM and Pak1 was identified by the yeast two-hybrid screening suggested that these two proteins can interact directly. To verify this view, binding of recombinant intracellular domains of NCAM140 (NCAM140ID) to Pak1 immunopurified from mouse brain (Fig. 1D) was analyzed in a pull-down assay. Pak1 immunobilized on beads was incubated with increasing concentrations of NCAM140ID. Western blot analysis of pull-downs showed that NCAM140ID bound to Pak1 in a concentration-dependent manner (Fig. 1E).
To identify the NCAM domains required for the interaction with Pak1, coimmunoprecipitation of NCAM with Pak1 was analyzed in lysates of 3T3 cells transfected with NCAM140 alone or cotransfected with either whole NCAM140ID or its fragments. This analysis showed that the whole NCAM140ID inhibited coimmunoprecipitation of NCAM with Pak1, most likely by competing with full-length NCAM for binding to Pak1. Coimmunoprecipitation of NCAM was also inhibited in cells cotransfected with NCAM140ID748–777 (Fig. 1F). The NCAM140729–750 fragment containing the binding site for another binding partner of NCAM, exo70 (Chernyshova et al., 2011), did not interfere with the coimmunoprecipitation of NCAM with Pak1.
To extend this analysis, Pak1 purified from mouse brain (Fig. 1D) was immobilized in the wells of a 96-well plate substrate coated with Pak1 antibody, and its ability to capture chemically synthesized peptides corresponding to amino acid sequences 748–763, 756–770, and 764–777 of the intracellular domain of NCAM140 was analyzed. Only NCAM140ID748–763 bound to Pak1 in a concentration-dependent and saturable manner (Fig. 1G), indicating that the sequence of 748–756 aa within NCAM140ID is required for this interaction. It is noteworthy that all amino acid sequences within NCAM140ID are also present in NCAM180ID. The same amino acid sequence can thus mediate the interaction between NCAM180 and Pak1.
NCAM induces activation of the Pak1 signaling pathway in growth cones
Binding of NCAM at the cell surface to its natural ligands or to function triggering NCAM antibodies induces intracellular signaling and promotes neurite outgrowth (Bodrikov et al., 2005, 2008). We investigated whether binding of NCAM to its ligands influences Pak1 activity in growth cones by treating isolated growth cones with polyclonal antibodies against the extracellular domain of NCAM. Growth cones treated with nonimmune Igs served as control. Pak1 was then immunoprecipitated from growth cones and subjected to an in vitro kinase assay. A peptide derived from TH encompassing Ser40 was used as a specific substrate for Pak1, which phosphorylates TH at this residue. Phosphorylation was analyzed by dot blot with phospho-Ser40-specific antibodies. This analysis showed that Pak1 from NCAM antibody-treated growth cones was approximately five times more efficient in phosphorylation of Ser40 than Pak1 from control Ig-treated growth cones (Fig. 2A).
Figure 2.

NCAM activation activates the Pak1 signaling pathway in isolated growth cones. A, Growth cones isolated from brains of NCAM+/+ mice were pre-incubated with NCAM antibodies or nonimmune Ig (treatment). Pak1 was then immunoprecipitated from growth cone lysates (IP: Pak1). Mock immunoprecipitates with nonimmune Ig served as control (IP: Ig). Western blot (WB) with Pak1 antibodies shows that Pak1 was immunoprecipitated with the same efficiency from nonimmune Ig-treated and NCAM antibody-treated growth cones (bottom). The precipitates were incubated with TH peptide, and total levels of TH peptide and levels of TH peptide phosphorylated at Ser40 were analyzed by dot blot (DB). Note that Pak1 from NCAM antibody-treated growth cones phosphorylates TH peptide more efficiently than Pak1 from nonimmune Ig-treated growth cones. Graph shows quantitation of dot blots from three experiments. Mean + SEM values for NCAM antibody-treated probes normalized to the values from Ig-treated probes set to 100% (dashed line) are shown. *p < 0.05, paired t test. B, Growth cones isolated from NCAM+/+ mouse brains were incubated with polyclonal antibodies against the extracellular domain of NCAM for 5, 15, or 30 min. Growth cones treated with nonimmune Ig served as control. Western blots of the growth cone lysates probed with the indicated antibodies are shown. Graphs show quantitation of Western blots from five experiments. Mean + SEM values for NCAM antibody-treated growth cones normalized to the values from nonimmune Ig-treated growth cones set to 100% (dashed lines) are shown. Note that NCAM antibodies induce phosphorylation of Pak1 at Ser199/204 and Thr423, LIMK1 at Thr508, and cofilin at Ser3, and reduce phosphorylation of Pak1 at Thr212. Labeling for GAPDH was included as a loading control. *p < 0.05, paired t test, compared with corresponding probes from nonimmune Ig-treated growth cones.
To confirm these kinase assay data, we investigated whether NCAM antibodies induce changes in Pak1 phosphorylation accompanying changes in enzyme activation status. Western blot analysis of the growth cone lysates showed that application of NCAM antibodies increased enzyme-activating autophosphorylation of Pak1 at Ser199/204 and Thr423 (Parrini et al., 2009) within 5 min after antibody application. This increase in autophosphorylation was observed for at least 30 min after NCAM antibody application, i.e., the latest time point tested (Fig. 2B). In contrast, enzyme-inhibiting phosphorylation of Pak1 at Thr212 (Nikolic et al., 1998; Rashid et al., 2001) was not strongly affected, although it tended to decrease at 15 and 30 min after NCAM antibody application (Fig. 2B).
NCAM antibody treatment also induced activation of the downstream enzymes in the Pak1-mediated signaling cascade: Pak1-mediated phosphorylation of LIMK at Thr508 was increased within 5 min after NCAM antibody application and was observed for 30 min after antibody application (Fig. 2B). LIMK-mediated phosphorylation of Ser3 in cofilin was also increased in NCAM antibody-treated growth cones (Fig. 2B). However, Ser3 phosphorylation of cofilin increased only 15 min after NCAM antibody application, indicating a slower response to NCAM antibodies when compared with PAK1 and LIMK.
NCAM deficiency results in reduced activity, Thr212 hyperphosphorylation, and impaired membrane association of Pak1
Our observations in isolated growth cones prompted us to investigate whether the kinase activity of Pak1 in the brain is affected by NCAM deficiency. Pak1 was immunoisolated from brain homogenates of wild-type (NCAM+/+) and NCAM-deficient (NCAM−/−) mice. Western blot analysis of Pak1 immunoprecipitates with Pak1 antibodies showed that similar levels of Pak1 were immunoisolated from brain homogenates of both genotypes (Fig. 3A). Pak1 was then subjected to the in vitro kinase assay. This analysis showed that the efficiency of TH Ser40 phosphorylation by Pak1 immunoisolated from NCAM−/− brains was approximately two times lower than that of Pak1 immunoisolated from NCAM+/+ brains (Fig. 3A), indicating that the kinase activity of Pak1 is inhibited in NCAM−/− brains.
Figure 3.

Kinase activity and plasma membrane association of Pak1 are reduced, and phosphorylation of Pak1 is enhanced in NCAM−/− brains. A, Pak1 was immunoprecipitated from NCAM+/+ and NCAM−/− mouse brain lysates (IP: Pak1). Mock immunoprecipitates obtained with nonimmune Ig served as control (IP: Ig). Western blot (WB) with Pak1 antibodies shows that Pak1 was immunoprecipitated with the same efficiency from NCAM+/+ and NCAM−/− probes (bottom). The precipitates were incubated with TH peptide, and total levels of TH peptide and levels of TH peptide phosphorylated by Pak1 at Ser40 were analyzed by dot blot (DB). Note that Pak1 from NCAM+/+ brains phosphorylates the TH peptide more efficiently than Pak1 from NCAM−/− brains. Graph shows quantitation of dot blots from three experiments. Mean + SEM values for NCAM−/− probes normalized to NCAM+/+ values set to 100% (dashed line) are shown. *p < 0.05, paired t test. B, Brain homogenates (BH), growth cones (GC), and nongrowth cone membranes (non-GC) from NCAM+/+ and NCAM−/− mouse brains probed by Western blot with antibodies against total Pak1 or Pak1 phosphorylated at Ser199/204, Thr212, or Thr423. Labeling for GAPDH was included as loading control. Graphs show quantitation of the blots from six experiments. Mean + SEM values for NCAM−/− probes normalized to NCAM+/+ levels set to 100% (dashed lines) are shown. *p < 0.05, paired t test, compared with corresponding NCAM+/+ values.
Next, we analyzed if reduced activity of Pak1 in NCAM−/− brains was due to abnormal phosphorylation of Pak1. Total NCAM+/+ and NCAM−/− brain homogenates were analyzed by Western blot with antibodies against different phosphorylation sites within Pak1. This analysis showed that Pak1 phosphorylation at Ser199/204 and Thr423 was not changed in NCAM−/− brain homogenates (Fig. 3B), thus ruling out a possibility that reduced activity of Pak1 was due to the overall decreased levels of its autophosphorylation. However, phosphorylation of Pak1 at Thr212 was increased in NCAM−/− brain homogenates (Fig. 3B), suggesting that hyperphosphoryla-tion of Pak1 at Thr212, which had been previously reported to inhibit Pak1 activity (Nikolic et al., 1998; Rashid et al., 2001), contributes to the downregulation of Pak1 activity in NCAM−/− brains.
Targeting of Pak1 to the membranes had also been shown to play a role in the activation of the enzyme (Daniels et al., 1998). Hence, we compared levels of Pak1 associated with plasma membranes in NCAM+/+ and NCAM−/− brains. To this aim, growth cones and nongrowth cone membranes were isolated from brains of NCAM+/+ and NCAM−/− mice by differential centrifugation in a sucrose gradient and analyzed by Western blot. Levels of Pak1 that were coisolated with NCAM−/− growth cones or nongrowth cone membranes were reduced by ∼35% when compared with NCAM+/+ samples (Fig. 3B). Similarly to total brain homogenates, levels of Pak1 phosphorylated at Thr212 were increased in NCAM−/− growth cones and nongrowth cone membranes (Fig. 3B). Surprisingly, autophosphorylation of Pak1 at Thr423 and Ser199/204 was also increased in growth cones and nongrowth cone membranes from NCAM−/− brains (Fig. 3B), indicating that it may occur independently of NCAM.
Lipid rafts are indispensable for NCAM-dependent Pak1 signaling pathway activation
NCAM-mediated signal transduction depends on cholesterol-enriched lipid microdomains, lipid rafts (Niethammer et al., 2002; Leshchyns'ka et al., 2003; Santuccione et al., 2005; Bodrikov et al., 2008). To investigate whether lipid rafts are also required for Pak1 activation in response to NCAM stimulation, NCAM-dependent Pak1 activation was analyzed under conditions where lipid rafts were acutely disrupted by treatment with methyl-β-cyclodextrin (MCD), an agent that extracts cholesterol from plasma membranes (Niethammer et al., 2002; Schneider et al., 2008). Isolated growth cones from NCAM+/+ brains where treated with function-triggering polyclonal antibodies against mouse NCAM or nonimmune control immunoglobulins either in the presence or absence of MCD. Western blot analysis of the growth cone lysates showed that NCAM antibody treatment-induced autophosphorylation of Pak1 at Ser199/204 and Thr423, accompanied by a slight but statistically significant reduction in the levels of Thr212-phosphorylated Pak1 (Figs. 2, 4A). NCAM antibody treatment also induced phosphorylation of LIMK at Thr508 and cofilin at Ser3 (Fig. 4A). NCAM antibody-induced phosphorylation of Pak1, LIMK, and cofilin was blocked in growth cones pretreated with MCD (Fig. 4A), indicating that lipid raft integrity is required for NCAM-dependent Pak1 activation. Interestingly, MCD treatment also resulted in a significant downregulation of Pak1 phosphorylation at Thr212 (Fig. 4A).
Figure 4.
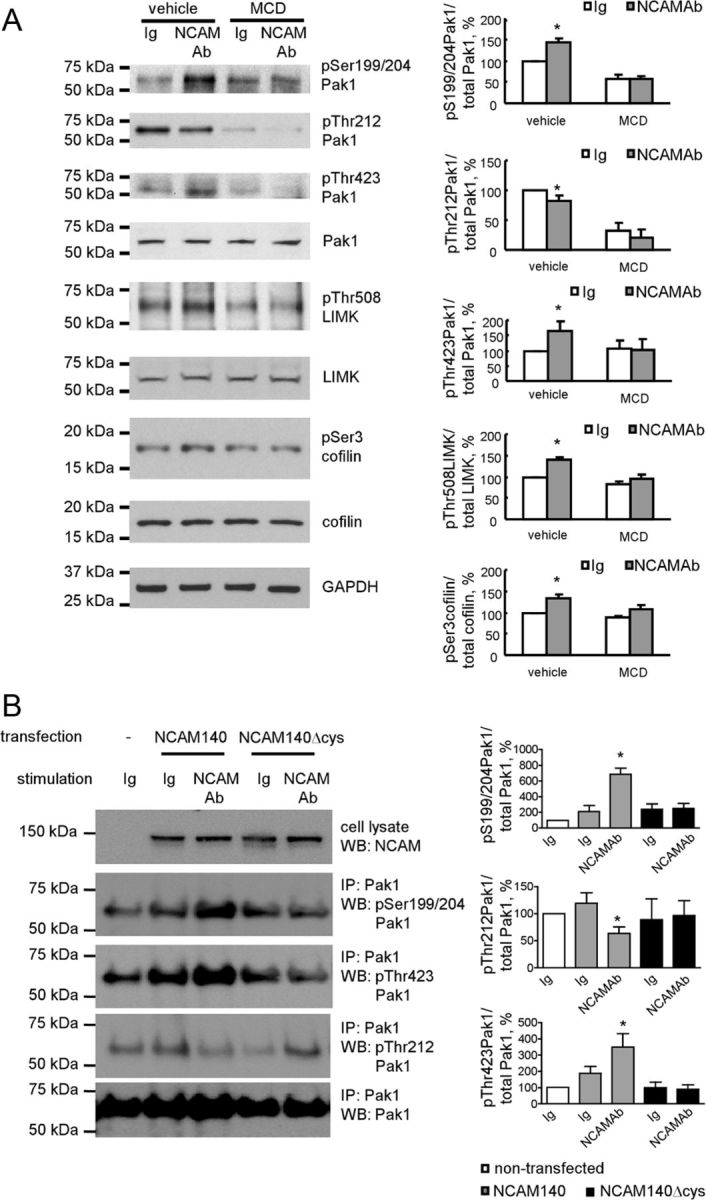
Association of NCAM with lipid rafts is required for NCAM-dependent Pak1 pathway induction. A, Western blot analysis of lysates of growth cones from NCAM+/+ mouse brains probed with the indicated antibodies. Growth cones were treated with vehicle (water used to dilute MCD) or 5 mm MCD and incubated with polyclonal antibodies against NCAM or nonimmune Ig. Graphs show quantitation of the blots from five experiments. Mean + SEM values normalized to the values for vehicle-treated and nonimmune Ig-treated growth cones set to 100% are shown. *p < 0.05, paired t test compared with the corresponding probes from nonimmune Ig-treated growth cones. Note that MCD inhibits NCAM antibody-induced phosphorylation of Pak1 at Ser199/204 and Thr423, LIMK1 at Thr508, and cofilin at Ser3. B, Western blot analysis of lysates and Pak1 immunoprecipitates (IP) from nontransfected B35 cells and B35 cells transfected with NCAM140 or NCAM140Δcys probed with the indicated antibodies. Cells were treated with nonimmune Ig or function-triggering polyclonal antibodies against NCAM. Graphs show quantitation of the blots from three experiments. Mean + SEM levels of phosphorylated Pak1 normalized to levels of total immunoprecipitated Pak1 are shown. Values for nontransfected and nonimmune Ig-treated cells were set to 100%. *p < 0.05, ANOVA with Dunnett's multiple-comparison test. Note NCAM antibody-induced phosphorylation of Pak1 at Ser199/204 and Thr423, and inhibition of Pak1 phosphorylation at Thr212 in NCAM140-transfected cells but not NCAM140Δcys-transfected cells.
Next we compared NCAM-dependent Pak1 activation in B35 neuroblastoma cells. These cells express negligible levels of endogenous NCAM (Otey et al., 2003), and were therefore transfected either with non-mutated NCAM140 or palmitoylation-deficient NCAM140Δcys, which is excluded from lipid rafts (Niethammer et al., 2002). Western blot analysis of the cell lysates confirmed that non-mutated NCAM140 and NCAM140Δcys were expressed at similar levels (Fig. 4B). Western blot analysis of Pak1 immunoprecipitates showed that application of function-triggering polyclonal antibodies against NCAM but not nonimmune control immunoglobulins increased levels of Pak1 phosphorylated at Ser199/204 and Thr423 and decreased levels of pThr212Pak1 in cells transfected with non-mutated NCAM140, but not in cells transfected with NCAM140Δcys (Fig. 4B). These observations indicate that the association of NCAM with lipid rafts is required for Pak1 activation.
Targeting of PIX to lipid rafts is reduced in NCAM−/− brains
Our observation that lipid rafts are required for NCAM-dependent Pak1 activation prompted us to analyze levels of Pak1 and its activators in lipid rafts from mouse brains. Lipid rafts were isolated either from the growth cone fraction or nongrowth cone membranes from NCAM+/+ and NCAM−/− mice. Western blot analysis of these preparations showed that they were highly enriched in the lipid raft marker contactin-1 (Fig. 5), indicating high efficiency of isolation. NCAM140 and NCAM180 were enriched in the NCAM+/+ growth cones and lipid rafts isolated from this fraction when compared with brain homogenates and nongrowth cone membranes (Fig. 5).
Figure 5.
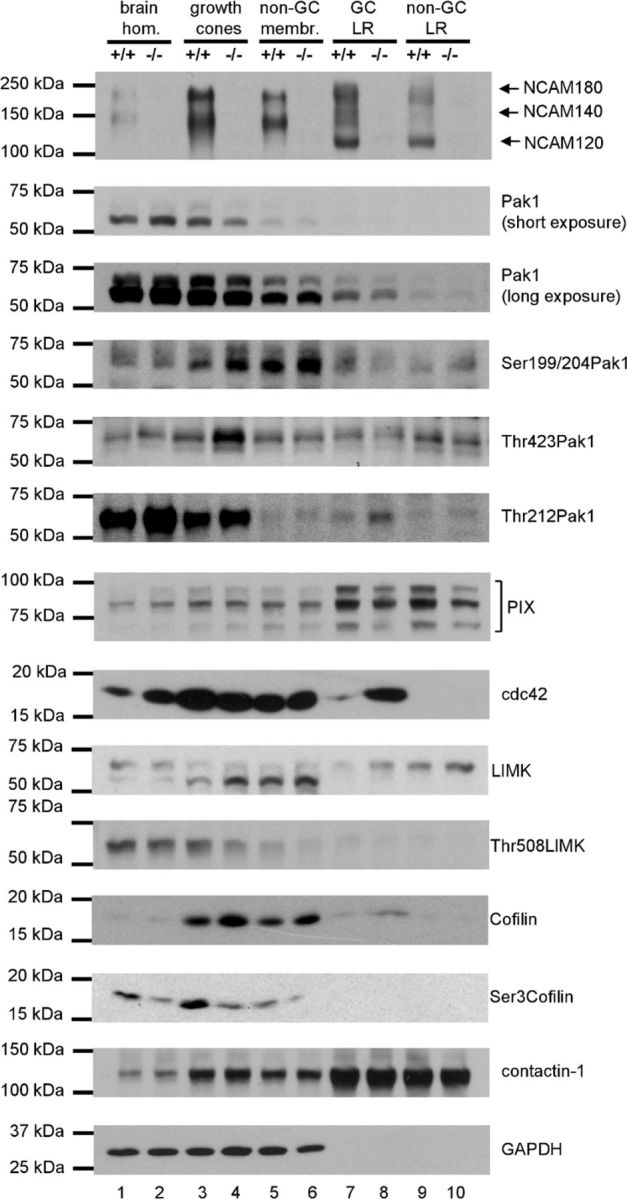
Phosphorylation of Pak1, LIMK, and cofilin as well as levels in lipid rafts of the Pak1 activators PIX and cdc42 are abnormal in NCAM−/− versus NCAM+/+ growth cones. Brain homogenates (lanes 1 and 2), growth cones (lanes 3 and 4), nongrowth cone membranes (non-GC; lanes 5 and 6), and lipid rafts from growth cones (GC LR; lanes 7 and 8) and nongrowth cone membranes (non-GC LR; lanes 9 and 10) probed by Western blot with indicated antibodies. Labeling for contactin-1 and GAPDH was included as loading control. Note that lipid raft marker contactin-1 is highly enriched in lipid rafts (lanes 7–10). Levels of Thr212-phosphorylated Pak1 and cdc42 are increased in lipid rafts from NCAM−/− versus NCAM+/+ growth cones (compare lanes 7 and 8). Levels of total LIMK and cofilin are increased in NCAM−/− versus NCAM+/+ growth cones (compare lanes 3 and 4) and growth cone lipid rafts (compare lanes 7 and 8), and levels of Thr508 phosphorylated LIMK and Ser3 phosphorylated cofilin are reduced in NCAM−/− versus NCAM+/+ growth cones (compare lanes 3 and 4). Experiments and Western blot analyses were performed three times with the same results.
Compared with brain homogenates, low levels of Pak1 were present in lipid rafts from both NCAM+/+ and NCAM−/− brains. Lipid raft levels of Pak1 tended to be increased in lipid rafts from growth cones as compared with lipid rafts from nongrowth cone membranes (Fig. 5). A similar distribution was observed for Thr212-phosphorylated Pak1, with its levels being higher in NCAM−/− versus NCAM+/+ in all fractions analyzed (Fig. 5). In contrast to total Pak1, Ser199/204- and Thr423-phosphorylated Pak1 was readily detectable in growth cone lipid rafts of both genotypes (Fig. 5) indicating enrichment of the autophosphorylated form of the enzyme relative to its total form in lipid rafts (pThr423 Pak1/total Pak1 ratio ∼0.77 in growth cone lipid rafts vs ∼0.19 in brain homogenates; pSer199/204 Pak1/total Pak1 ratio ∼1.1 in growth cone lipid rafts vs ∼0.25 in brain homogenates). Levels of Ser199/204- and Thr423-phosphorylated Pak1 were not significantly changed in NCAM−/− growth cone lipid rafts.
Since Pak1 activity is highly regulated by cdc42 and PIX, we also analyzed the distribution of these Pak1 activators. While PIX was highly enriched in lipid rafts from growth cones and nongrowth cone membranes (Fig. 5), cdc42 was detectable only at very low levels in lipid rafts, but was overall enriched in growth cones and nongrowth cone membranes (Fig. 5). These observations suggest that PIX and cdc42 are mostly segregated into distinct submembrane domains in NCAM+/+ brains. Interestingly, levels of PIX were significantly reduced by approximately 40% in lipid rafts from NCAM−/− versus NCAM+/+ growth cones (Fig. 5). In contrast, levels of cdc42 were ∼6 times higher in lipid rafts from NCAM−/− versus NCAM+/+ growth cones (Fig. 5) indicating abnormal stoichiometry of Pak1 activators specifically in lipid rafts of growth cones.
Analysis of the downstream substrates in the Pak1 signaling pathway showed that levels of LIMK and cofilin were ∼2 and 2.5 times higher, while levels of Thr508-phosphorylated LIMK and Ser3-phosphorylated cofilin were ∼30 and 50% lower, respectively, in NCAM−/− versus NCAM+/+ growth cones (Fig. 5) indicating reduced activation of the Pak1 signaling cascade in NCAM−/− growth cones.
Formation of the Pak1/PIX/cdc42 complex is reduced in NCAM−/− brains
Abnormalities in levels of Pak1 activators in lipid rafts from NCAM−/− versus NCAM+/+ growth cones suggested a possible relationship between NCAM and these Pak1 activators. Immunofluorescence labeling of cultured hippocampal neurons showed that clusters of NCAM partially colocalized with accumulations of cdc42 and PIX (Fig. 6A,C). Proximity ligation with antibodies against cdc42 and the intracellular domain of NCAM produced a positive reaction (Fig. 6B) and revealed that 35.1 ± 2.2, 16.4 ± 2.2, and 48.7 ± 2.5% of NCAM/cdc42 complexes were detectable in somata, along neurites, and in growth cones, respectively (n = 55 neurons were analyzed). Proximity ligation with antibodies against PIX and the intracellular domain of NCAM was also positive (Fig. 6D) and showed that 50.9 ± 3.2, 24.7 ± 2.2, and 24.4 ± 2.6% of NCAM/PIX complexes were localized in somata, along neurites, and in growth cones, respectively (n = 40 neurons were analyzed).
Figure 6.
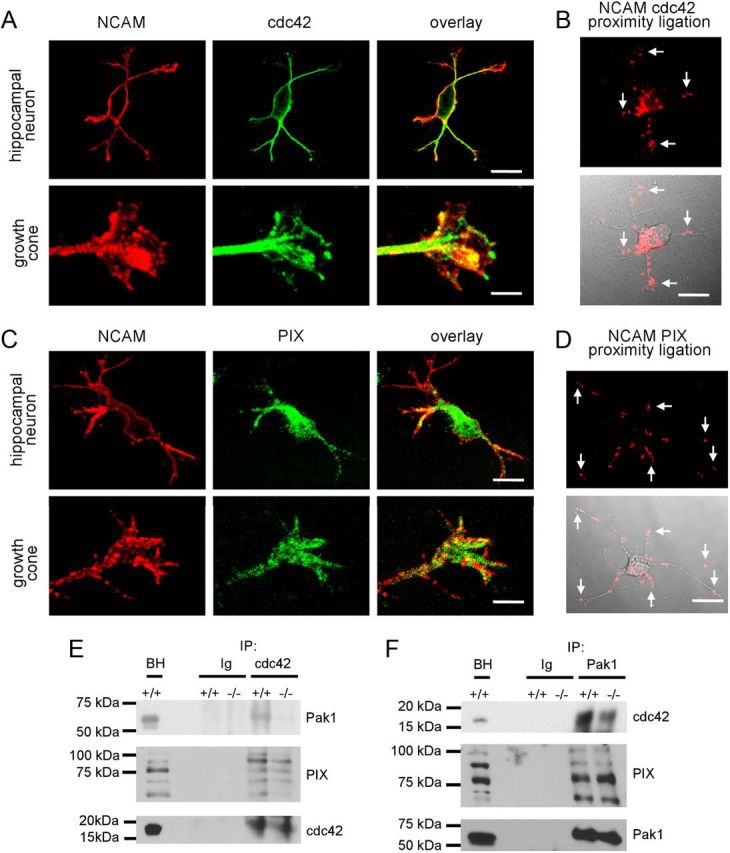
Pak1/cdc42/PIX complex formation is inhibited in NCAM−/− versus NCAM+/+ brains. A, C, Examples of hippocampal neurons and growth cones of hippocampal neurons maintained for 1 d in culture colabeled with antibodies against NCAM and cdc42 (A) or PIX (C). Note clusters of NCAM overlapping with accumulations of Cdc42 and PIX in growth cones (yellow). Scale bars: neurons, 20 μm; growth cones, 5 μm. B, D, Proximity ligation assays with antibodies against the intracellular domain of NCAM and cdc42 (B) or PIX (D). Arrows show examples of NCAM/cdc42 and NCAM/PIX complexes in growth cones. Proximity ligation fluorescence signals alone (top) or in combination with differential interference contrast images (bottom) are shown. E, F, Cdc42 and Pak1 immunoprecipitates (IP) from 1-d-old NCAM+/+ and NCAM−/− mouse brains were probed with antibodies against Pak1, cdc42, and PIX by Western blot. Mock immunoprecipitation with nonimmnune Ig served as control. Note reduced coimmunoprecipitation of Pak1 and PIX with cdc42 (E) and reduced coimmunoprecipitation of cdc42 with Pak1 (F) from NCAM−/− versus NCAM+/+ brains. BH, brain homogenates.
To analyze whether NCAM deficiency affects Pak1/PIX/cdc42 complex formation, coimmunoprecipitation experiments were performed. The efficiencies of coimmunoprecipitation of Pak1 and PIX with cdc42 from NCAM−/− brain lysates were reduced to 51.4 ± 7.9 (n = 4, p < 0.05, paired t test) and 65.7 ± 2.4% (n = 6, p < 0.01, paired t test) compared with NCAM+/+ brain lysates (Fig. 6E), respectively. Coimmunoprecipitation of cdc42 with Pak1 from NCAM−/− brain lysates was also reduced to 61.5 ± 4.8% (n = 4, p < 0.01, paired t test) compared with NCAM+/+ brain lysates (Fig. 6F). Hence, the association of Pak1 and PIX with cdc42 in NCAM−/− brains was reduced. Coimmunoprecipitation of PIX with Pak1 from NCAM−/− brain lysates was, however, not affected at 108.2 ± 2.1% (n = 5, p = 0.5, paired t test) compared with NCAM+/+ brain lysates (Fig. 6F), suggesting that Pak1/PIX complex formation is normal in NCAM−/− neurons.
Actin polymerization is reduced in NCAM−/− growth cones
LIMK-mediated phosphorylation of Ser3 within cofilin results in inhibition of its actin depolymerizing activity (Meberg et al., 1998; Bamburg et al., 1999). Western blot analysis showed that total levels of actin were not affected by NCAM deficiency in brain homogenates or in growth cones or nongrowth cone membranes (Fig. 7A). To compare the amount of F-actin versus free G-actin in NCAM+/+ and NCAM−/− growth cones, growth cones isolated from NCAM+/+ and NCAM−/− brains were lysed in F-actin stabilization buffer and centrifuged to separate the F-actin and G-actin pools. G-actin-containing supernatants and F-actin-containing pellets were then analyzed by Western blot with antibodies against actin and tubulin. This analysis showed that levels of G-actin were approximately two times higher in NCAM−/− growth cones when compared with NCAM+/+ growth cones (Fig. 7B). Proportionally, levels of F-actin were approximately two times lower in NCAM−/− growth cones when compared with NCAM+/+ growth cones (Fig. 7B). In contrast, levels of soluble and insoluble tubulin were similar in both genotypes (Fig. 7B).
Figure 7.
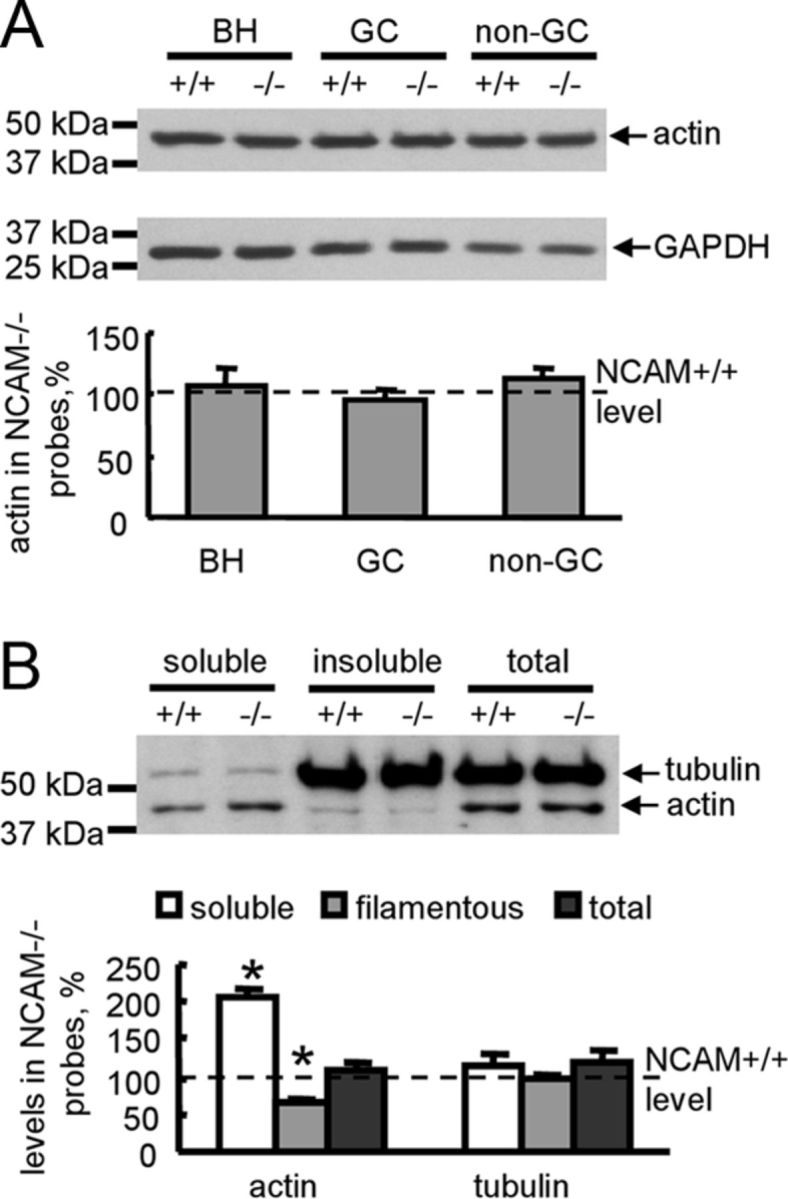
Actin polymerization is reduced in NCAM−/− growth cones. A, Brain homogenates (BH), growth cones (GC), and nongrowth cone (non-GC) membranes from NCAM+/+ and NCAM−/− mouse brains probed by Western blot with antibodies against actin. Labeling for GAPDH was included as loading control. Graph shows quantitation of the blots from six experiments. Mean + SEM values for NCAM−/− probes normalized to NCAM+/+ levels set to 100% (dashed line) are shown. B, Total growth cones from NCAM+/+ and NCAM−/− brains, and soluble and insoluble proteins from NCAM+/+ and NCAM−/− growth cones lysed in F-actin stabilization buffer probed by Western blot with antibodies against tubulin and actin. Note that the levels of soluble G-actin are higher and the levels of insoluble F-actin are lower in NCAM−/− versus NCAM+/+ growth cones. Tubulin levels are similar in both genotypes. Graph shows quantitation of the blots from six experiments. Mean + SEM values for NCAM−/− probes normalized to NCAM+/+ levels set to 100% (dashed line) are shown.
Since polymerization of actin plays a pivotal role in the formation of filopodia in growth cones (Lewis and Bridgman, 1992), we compared the behavior of growth cones in cultured hippocampal neurons from NCAM+/+ and NCAM−/− mice. Imaging of the growth cone motility of cultured hippocampal neurons over several minutes with 1 s intervals between frames showed that growth cones of NCAM+/+ neurons were more motile than growth cones of NCAM−/− neurons (Fig. 8A). This effect was associated with a reduced formation of filopodia: the number of filopodia per growth cone area was 0.08 ± 0.02 per μm2 in NCAM+/+ neurons versus 0.018 ± 0.006 per μm2 in NCAM−/− neurons (p < 0.05, t test), the number of motile filopodia per growth cone area was 0.11 ± 0.03 per μm2 in NCAM+/+ neurons versus 0.014 ± 0.006 per μm2 in NCAM−/− neurons (p < 0.05, t test), and the length of filopodia was 5.3 ± 0.3 μm in NCAM+/+ neurons versus 4.3 ± 0.3 per μm2 in NCAM−/− neurons (p < 0.05, t test).
Figure 8.

Filopodial mobility is reduced after disruption of the NCAM/Pak1 complex. A, Representative time-lapse recordings of growth cones of NCAM+/+ and NCAM−/− hippocampal neurons maintained in culture for 1 d. Scale bar, 5 μm. B, C, Diagrams show the numbers of filopodia and motile filopodia normalized to the growth cone area to account for growth cone size, and the lengths of filopodia in growth cones (GC) of NCAM+/+ neurons transfected with control miR or NCAM miR with or without constitutively active pCGC-CA-Pak1 or dominant-negative pCGC-DN-Pak1 (B), and in growth cones of NCAM+/+ neurons transfected with green fluorescent protein (GFP) alone or cotransfected with GFP and NCAM140ID729–750 or NCAM140ID748–777 (C). Mean + SEM values are shown. Note that the numbers of filopodia per growth cone area, filopodium motility, and lengths are reduced in NCAM−/− growth cones, and in growth cones of NCAM+/+ neurons transfected with NCAMmiR, pCGC-DN-Pak1, and NCAM140ID748–777. *p < 0.05, ANOVA with Dunnett's multiple-comparison test (B, C).
Filopodium formation was also reduced after knock-down of NCAM expression by transfection of neurons with miRNA engineered to target NCAM (Fig. 8B), and a similar reduction in filopodium formation was observed in neurons transfected with DN-Pak1 (Fig. 8B). A reduction in filopodia formation in response to NCAM deficiency was abolished by cotransfecting neurons with constitutively active Pak1 (Fig. 8B). Finally, transfection with NCAM140ID748–777 but not NCAM140729–750 fragment of the intracellular domain of NCAM140 inhibited filopodium formation in wild-type neurons (Fig. 8C). In combination with our biochemical data, these observations suggest that disruption of the NCAM/Pak1 complex results in abnormal remodeling of actin filaments and impaired filopodium formation in growth cones.
NCAM-dependent neurite outgrowth depends on PAK1 activity
Clustering of NCAM at the neuronal cell surface with either soluble dimeric fragments of the extracellular domain of NCAM or function-triggering NCAM antibodies led to enhanced promotion of neurite outgrowth from cultured hippocampal neurons (Bodrikov et al., 2008; Chernyshova et al., 2011; Fig. 9). To investigate if PAK1 activation is required for NCAM-dependent neurite outgrowth we analyzed the effects of DN-Pak1 versus CA-PAK1 (Hayashi et al., 2007) in cultured NCAM+/+ cortical neurons using cotransfection with cherry fluorescent protein for visualization of neurites. Neurons grown on poly-d-lysine were treated with NCAM antibodies or control nonimmune serum applied in the culture medium for 24 h, fixed, and analyzed by fluorescence microscopy for neuronal morphology. This analysis showed that the overall neurite lengths and lengths of the longest neurites considered as putative axons were increased by ∼30% in NCAM antibody-treated versus nonimmune serum-treated neurons cotransfected with the empty control vector (Fig. 9). The response to NCAM antibodies was strongly enhanced in neurons cotransfected with the constitutively active Pak1 when compared with the empty vector-transfected neurons (Fig. 9). In contrast, the response to NCAM antibodies was blocked in neurons cotransfected with the DN-Pak1 (Fig. 9). DN-Pak1 had no effect on neurite outgrowth in neurons treated with control nonimmune serum (Fig. 9). These observations indicate that Pak1 activity is required for NCAM-dependent neurite elongation.
Figure 9.

Pak1 activation is required for NCAM-dependent neurite outgrowth. A, B, Diagrams show mean + SEM lengths of the longest neurite (A) or total lengths of all neurites (B) per neuron (n = 100 neurons were analyzed in each group). Cultured cortex neurons from NCAM+/+ mice were transfected with the empty pCGC vector, constitutively active pCGC-CA-Pak1, or dominant-negative pCGC-DN-Pak1. Neurons were treated with NCAM antibodies or nonimmune serum applied 6 h after transfection for 24 h. Note increased response to NCAM antibodies in pCGC-CA-Pak-transfected neurons. Note that pCGC-DN-Pak1-transfected neurons do not respond to function-triggering NCAM antibodies. *p < 0.05, t test.
Discussion
In this study, we used a yeast two-hybrid system, pull-down, coimmunoprecipitation and colocalization analysis to show that NCAM associates with Pak1 in growth cones of differentiating neurons. We also demonstrate that activation of NCAM at the cell surface of growth cones induces Pak1 activation. Pak1 activity is highly regulated via changing the phosphorylation of the enzyme as well as its association with the cell membrane and with the Pak1-activating proteins PIX and cdc42. Our data suggest that NCAM activation influences several steps in the cascades of molecular interactions preceding Pak1 activation (Fig. 10).
Figure 10.

Proposed model of NCAM-dependent events in Pak1 kinase pathway activation. NCAM activation at the cell surface results in dephosphorylation of Pak1 at Thr212 and formation of the Pak1/PIX/cdc42 complex, followed by autophosphorylation and activation of Pak1 in a lipid raft-dependent manner. Activated Pak1 phosphorylates LIMK at Thr508, which in turn phosphorylates cofilin at Ser3, thereby reducing its actin-depolymerizing activity. An increase in actin polymerization promotes growth cone motility and generation of traction forces required for neurite outgrowth.
Pak1 activity is regulated by enzyme-inhibiting phosphorylation of Pak1 at Thr212 (Nikolic et al., 1998; Rashid et al., 2001) and enzyme-activating autophosphorylation of Pak1 at Ser199/204 and Thr423 (Parrini et al., 2009). Hyperphosphorylation of Pak1 at Thr212 in NCAM-deficient brains suggests that NCAM promotes Pak1 dephosphorylation at Thr212 (Fig. 10). In favor of this hypothesis, we show that treatment of growth cones with function-triggering NCAM antibodies induces Pak1 dephosphorylation at Thr212. While the phosphatases inducing NCAM-dependent dephosphorylation of Pak1 at Thr212 remain to be identified, PP2A is a likely candidate since it has been found in a complex with Pak1 (Westphal et al., 1999), and also associates with NCAM (Büttner et al., 2005).
The fact that the association of Pak1 and PIX with cdc42 is reduced in NCAM−/− brains suggests that NCAM also promotes formation of the Pak1/cdc42/PIX complex (Fig. 10). Interestingly, NCAM directly interacts with and recruits to the surface plasma membranes, the exocyst complex (Chernyshova et al., 2011), which directly interacts with cdc42 (Zhang et al., 2001; Wu et al., 2010) and may thus link NCAM to cdc42.
Lipid rafts play a prominent role in regulation of the signaling cascades activated by NCAM (Niethammer et al., 2002; Leshchyns'ka et al., 2003; Westphal et al., 2010). We show that lipid rafts are also important for NCAM-dependent Pak1 activation, since disruption of the association of NCAM with lipid rafts inhibited NCAM-dependent activation of the Pak1 pathway. Since PIX is enriched in lipid rafts, lipid rafts may serve as a signaling platform for Pak1/cdc42/PIX complex assembly and Pak1 activation (Fig. 10). Binding of NCAM to its extracellular ligands promotes palmitoylation of NCAM and its redistribution to lipid rafts (Niethammer et al., 2002; Ponimaskin et al., 2008). It is thus plausible that NCAM promotes Pak1/cdc42/PIX complex formation by providing nucleation sites in the lipid raft environment. Accumulation of cdc42 in NCAM−/− lipid rafts suggests that NCAM-dependent formation of Pak1/PIX/cdc42 is required for release of cdc42 from lipid rafts and redistribution of the activated Pak1/PIX/cdc42 complex to other subcellular domains accumulating downstream substrates of Pak1.
Binding to cdc42 and PIX results in autophosphorylation and full activation of Pak1 (Manser et al., 1994; Parrini et al., 2009) (Fig. 10). In agreement, we show that autophosphorylation and enzyme activity of Pak1 are increased in response to NCAM activation in growth cones. Surprisingly, the autophosphorylation of Pak1 is also increased in NCAM−/− growth cones albeit the activity of the enzyme is reduced. The latter observations suggest that autophosphorylation alone is not sufficient for Pak1 activation and can also occur independently of NCAM.
NCAM-dependent Pak1 activation results in phosphorylation of the downstream enzymes in the Pak1 pathway, including LIMK and cofilin. It is noteworthy in this context that overexpression of LIMK1 in neurons promotes accumulation of NCAM in growth cones (Rosso et al., 2004). These observations suggest an interesting scenario in which NCAM-dependent activation of Pak1 and LIMK1 kinases induces the molecular machinery targeting more NCAM to growth cones to further potentiate neurite outgrowth.
Previous reports indicate that Pak1 is required for efficient neurite formation and elongation (Hayashi et al., 2002). In this study, we demonstrate that NCAM-deficient growth cones show abnormally reduced filopodium formation correlating with reduced levels of F-actin, which plays a central role in the maintenance of neurite outgrowth induced by other adhesion molecules, such as L1 or cadherins (Nishimura et al., 2003; Bard et al., 2008). These changes in growth cone morphology observed in NCAM-deficient neurons correlate with changes in growth cones of PC12 cells overexpressing kinase-negative Pak1, which resulted in the abnormal spreading of growth cones (Obermeier et al., 1998). Actin remodeling is important for generation of the traction forces required for neurite outgrowth (Bard et al., 2008; Betz et al., 2011). NCAM functions are linked to neurite outgrowth in that neuritogenesis is inhibited in NCAM-deficient neurons (Chernyshova et al., 2011). In the present study, we show that Pak1 is the mediator of this link of NCAM to neuritogenesis in that NCAM-dependent neurite elongation is blocked in NCAM+/+ neurons overexpressing DN-Pak1 protein, indicating that Pak1 activity is required for NCAM-dependent neurite outgrowth.
Pak1 has also been reported to play a critical role in regulation of axonal guidance (Hing et al., 1999). Abnormal mobility of axonal growth cones may thus contribute to the abnormalities in axonal growth and fasciculation in the hippocampus of NCAM-deficient mice (Cremer et al., 1997; Seki and Rutishauser, 1998) and axonal path-finding errors observed in mice treated with function-inhibiting antibodies against NCAM (Monnier et al., 2001).
It remains to be investigated how the NCAM-dependent Pak1 activation pathway is interconnected with other NCAM-activated intracellular signaling cascades (Maness and Schachner, 2007; Hansen et al., 2008). It is interesting in this context that inhibitory phosphorylation of Pak1 at Thr212 is promoted by ERK2, Cdc2, and p35/Cdk5 kinases (Rashid et al., 2001; Thiel et al., 2002; Sundberg-Smith et al., 2005). Among them, ERK kinases are known downstream kinases in the NCAM signaling pathway, which are activated in response to NCAM activation (Kolkova et al., 2000; Niethammer et al., 2002). Cdk5 kinase is activated in response to glial cell line-derived neurotrophic factor (Paratcha et al., 2006), which also signals through NCAM (Paratcha et al., 2003). It is thus possible that these kinases provide a negative feedback loop by downregulating Pak1 activity via Thr212 phosphorylation. Further research will be required to fully understand and delineate a possible cross talk between Pak1 and other NCAM-activated enzymes.
Our observations on NCAM-dependent regulation of Pak1 activity are in agreement with reports showing that other adhesion molecules employ Pak1 to induce morphogenesis, cell migration, or neuronal differentiation. Among the Ig superfamily members: Down syndrome cell adhesion molecule directly binds and activates Pak1 to regulate axon guidance (Li and Guan, 2004), the cell adhesion molecule L1 activates Pak1 to induce cell migration (Schmid et al., 2004), and the close homolog of L1 (CHL1) cooperates with Pak1 in regulating the morphological development of the leading process/apical dendrite of embryonic cortical neurons (Demyanenko et al., 2010). Binding of integrins to their ligands also induces Pak1 activation required for integrin-dependent cell motility (Zhou and Kramer, 2005). It remains to be seen whether different adhesion molecules share similar pathways of Pak1 activation or use unique strategies to influence this important enzyme.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft (M.S., I.L., V.S.), National Health and Medical Research Council (V.S.), and Rebecca L. Cooper Medical Research Foundation (V.S., I.L.). We are grateful to Dr. Ohshima (Laboratory for Developmental Neurobiology, Brain Science Institute, Japan) for providing Pak1 constructs. We are also grateful to Achim Dahlmann for genotyping and Eva Kronberg for maintenance of animals. We thank Dr. Harold Cremer for NCAM−/− mice and Dr. Roger Tsien for the cherry construct.
References
- Andreyeva A, Leshchyns'ka I, Knepper M, Betzel C, Redecke L, Sytnyk V, Schachner M. CHL1 is a selective organizer of the presynaptic machinery chaperoning the SNARE complex. PLoS One. 2010;5:e12018. doi: 10.1371/journal.pone.0012018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Romero LE, Chernoff J. A tale of two Paks. Biol Cell. 2008;100:97–108. doi: 10.1042/BC20070109. [DOI] [PubMed] [Google Scholar]
- Bamburg JR, McGough A, Ono S. Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends Cell Biol. 1999;9:364–370. doi: 10.1016/s0962-8924(99)01619-0. [DOI] [PubMed] [Google Scholar]
- Bard L, Boscher C, Lambert M, Mège RM, Choquet D, Thoumine O. A molecular clutch between the actin flow and N-cadherin adhesions drives growth cone migration. J Neurosci. 2008;28:5879–5890. doi: 10.1523/JNEUROSCI.5331-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz T, Koch D, Lu YB, Franze K, Käs JA. Growth cones as soft and weak force generators. Proc Natl Acad Sci U S A. 2011;108:13420–13425. doi: 10.1073/pnas.1106145108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodrikov V, Leshchyns'ka I, Sytnyk V, Overvoorde J, den Hertog J, Schachner M. RPTPalpha is essential for NCAM-mediated p59fyn activation and neurite elongation. J Cell Biol. 2005;168:127–139. doi: 10.1083/jcb.200405073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodrikov V, Sytnyk V, Leshchyns'ka I, den Hertog J, Schachner M. NCAM induces CaMKIIalpha-mediated RPTPalpha phosphorylation to enhance its catalytic activity and neurite outgrowth. J Cell Biol. 2008;182:1185–1200. doi: 10.1083/jcb.200803045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- Büttner B, Kannicht C, Reutter W, Horstkorte R. Novel cytosolic binding partners of the neural cell adhesion molecule: mapping the binding domains of PLC gamma, LANP, TOAD-64, syndapin, PP1, and PP2A. Biochemistry. 2005;44:6938–6947. doi: 10.1021/bi050066c. [DOI] [PubMed] [Google Scholar]
- Chernyshova Y, Leshchyns'ka I, Hsu SC, Schachner M, Sytnyk V. The neural cell adhesion molecule promotes FGFR-dependent phosphorylation and membrane targeting of the exocyst complex to induce exocytosis in growth cones. J Neurosci. 2011;31:3522–3535. doi: 10.1523/JNEUROSCI.3109-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong C, Tan L, Lim L, Manser E. The mechanism of PAK activation. Autophosphorylation events in both regulatory and kinase domains control activity. J Biol Chem. 2001;276:17347–17353. doi: 10.1074/jbc.M009316200. [DOI] [PubMed] [Google Scholar]
- Cremer H, Lange R, Christoph A, Plomann M, Vopper G, Roes J, Brown R, Baldwin S, Kraemer P, Scheff S. Inactivation of the N-CAM gene in mice results in size reduction of the olfactory bulb and deficits in spatial learning. Nature. 1994;367:455–459. doi: 10.1038/367455a0. [DOI] [PubMed] [Google Scholar]
- Cremer H, Chazal G, Goridis C, Represa A. NCAM is essential for axonal growth and fasciculation in the hippocampus. Mol Cell Neurosci. 1997;8:323–335. doi: 10.1006/mcne.1996.0588. [DOI] [PubMed] [Google Scholar]
- Cunningham BA, Hemperly JJ, Murray BA, Prediger EA, Brackenbury R, Edelman GM. Neural cell adhesion molecule: structure, immunoglobulin-like domains, cell surface modulation, and alternative RNA splicing. Science. 1987;236:799–806. doi: 10.1126/science.3576199. [DOI] [PubMed] [Google Scholar]
- Daniels RH, Hall PS, Bokoch GM. Membrane targeting of p21-activated kinase 1 (PAK1) induces neurite outgrowth from PC12 cells. EMBO J. 1998;17:754–764. doi: 10.1093/emboj/17.3.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanenko GP, Halberstadt AI, Rao RS, Maness PF. CHL1 cooperates with PAK1–3 to regulate morphological differentiation of embryonic cortical neurons. Neuroscience. 2010;165:107–115. doi: 10.1016/j.neuroscience.2009.09.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- Gamell C, Osses N, Bartrons R, Rückle T, Camps M, Rosa JL, Ventura F. BMP2 induction of actin cytoskeleton reorganization and cell migration requires PI3-kinase and Cdc42 activity. J Cell Sci. 2008;121:3960–3970. doi: 10.1242/jcs.031286. [DOI] [PubMed] [Google Scholar]
- Gennarini G, Hirn M, Deagostini-Bazin H, Goridis C. Studies on the transmembrane disposition of the neural cell adhesion molecule N-CAM. The use of liposome-inserted radioiodinated N-CAM to study its transbilayer orientation. Eur J Biochem. 1984;142:65–73. doi: 10.1111/j.1432-1033.1984.tb08251.x. [DOI] [PubMed] [Google Scholar]
- Hansen SM, Berezin V, Bock E. Signaling mechanisms of neurite outgrowth induced by the cell adhesion molecules NCAM and N-cadherin. Cell Mol Life Sci. 2008;65:3809–3821. doi: 10.1007/s00018-008-8290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Ohshima T, Mikoshiba K. Pak1 is involved in dendrite initiation as a downstream effector of Rac1 in cortical neurons. Mol Cell Neurosci. 2002;20:579–594. doi: 10.1006/mcne.2002.1144. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Ohshima T, Hashimoto M, Mikoshiba K. Pak1 regulates dendritic branching and spine formation. Dev Neurobiol. 2007;67:655–669. doi: 10.1002/dneu.20363. [DOI] [PubMed] [Google Scholar]
- Hing H, Xiao J, Harden N, Lim L, Zipursky SL. Pak functions downstream of Dock to regulate photoreceptor axon guidance in Drosophila. Cell. 1999;97:853–863. doi: 10.1016/s0092-8674(00)80798-9. [DOI] [PubMed] [Google Scholar]
- Kolkova K, Novitskaya V, Pedersen N, Berezin V, Bock E. Neural cell adhesion molecule-stimulated neurite outgrowth depends on activation of protein kinase C and the Ras-mitogen-activated protein kinase pathway. J Neurosci. 2000;20:2238–2246. doi: 10.1523/JNEUROSCI.20-06-02238.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis P, Barnier JV. PAK signalling in neuronal physiology. Cell Signal. 2009;21:384–393. doi: 10.1016/j.cellsig.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Kuz̆elov á K, Pluskalov á M, Greben̆ov á D, Pavláskov á K, Halada P, Hrkal Z. Changes in cell adhesivity and cytoskeleton-related proteins during imatinib-induced apoptosis of leukemic JURL-MK1 cells. J Cell Biochem. 2010;111:1413–1425. doi: 10.1002/jcb.22868. [DOI] [PubMed] [Google Scholar]
- Leshchyns'ka I, Sytnyk V, Morrow JS, Schachner M. Neural cell adhesion molecule (NCAM) association with PKCbeta2 via betaI spectrin is implicated in NCAM-mediated neurite outgrowth. J Cell Biol. 2003;161:625–639. doi: 10.1083/jcb.200303020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AK, Bridgman PC. Nerve growth cone lamellipodia contain two populations of actin filaments that differ in organization and polarity. J Cell Biol. 1992;119:1219–1243. doi: 10.1083/jcb.119.5.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Guan KL. The Down syndrome cell adhesion molecule (DSCAM) interacts with and activates Pak. J Biol Chem. 2004;279:32824–32831. doi: 10.1074/jbc.M401878200. [DOI] [PubMed] [Google Scholar]
- Maddala R, Chauhan BK, Walker C, Zheng Y, Robinson ML, Lang RA, Rao PV. Rac1 GTPase-deficient mouse lens exhibits defects in shape, suture formation, fiber cell migration and survival. Dev Biol. 2011;360:30–43. doi: 10.1016/j.ydbio.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10:19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- Meberg PJ, Ono S, Minamide LS, Takahashi M, Bamburg JR. Actin depolymerizing factor and cofilin phosphorylation dynamics: response to signals that regulate neurite extension. Cell Motil Cytoskeleton. 1998;39:172–190. doi: 10.1002/(SICI)1097-0169(1998)39:2<172::AID-CM8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Monnier PP, Beck SG, Bolz J, Henke-Fahle S. The polysialic acid moiety of the neural cell adhesion molecule is involved in intraretinal guidance of retinal ganglion cell axons. Dev Biol. 2001;229:1–14. doi: 10.1006/dbio.2000.9970. [DOI] [PubMed] [Google Scholar]
- Niethammer P, Delling M, Sytnyk V, Dityatev A, Fukami K, Schachner M. Cosignaling of NCAM via lipid rafts and the FGF receptor is required for neuritogenesis. J Cell Biol. 2002;157:521–532. doi: 10.1083/jcb.200109059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic M, Chou MM, Lu W, Mayer BJ, Tsai LH. The p35/Cdk5 kinase is a neuron-specific Rac effector that inhibits Pak1 activity. Nature. 1998;395:194–198. doi: 10.1038/26034. [DOI] [PubMed] [Google Scholar]
- Nishimura K, Yoshihara F, Tojima T, Ooashi N, Yoon W, Mikoshiba K, Bennett V, Kamiguchi H. L1-dependent neuritogenesis involves ankyrinB that mediates L1-CAM coupling with retrograde actin flow. J Cell Biol. 2003;163:1077–1088. doi: 10.1083/jcb.200303060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermeier A, Ahmed S, Manser E, Yen SC, Hall C, Lim L. PAK promotes morphological changes by acting upstream of Rac. EMBO J. 1998;17:4328–4339. doi: 10.1093/emboj/17.15.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell M, Chance RK, Bashaw GJ. Axon growth and guidance: receptor regulation and signal transduction. Annu Rev Neurosci. 2009;32:383–412. doi: 10.1146/annurev.neuro.051508.135614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong WY, Wang XS, Manser E. Differential distribution of alpha and beta isoforms of p21-activated kinase in the monkey cerebral neocortex and hippocampus. Exp Brain Res. 2002;144:189–199. doi: 10.1007/s00221-002-1016-x. [DOI] [PubMed] [Google Scholar]
- Otey CA, Boukhelifa M, Maness P. B35 neuroblastoma cells: an easily transfected, cultured cell model of central nervous system neurons. Methods Cell Biol. 2003;71:287–304. doi: 10.1016/s0091-679x(03)01013-6. [DOI] [PubMed] [Google Scholar]
- Paratcha G, Ledda F, Ibáñez CF. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell. 2003;113:867–879. doi: 10.1016/s0092-8674(03)00435-5. [DOI] [PubMed] [Google Scholar]
- Paratcha G, Ibáñez CF, Ledda F. GDNF is a chemoattractant factor for neuronal precursor cells in the rostral migratory stream. Mol Cell Neurosci. 2006;31:505–514. doi: 10.1016/j.mcn.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Parrini MC, Camonis J, Matsuda M, de Gunzburg J. Dissecting activation of the PAK1 kinase at protrusions in living cells. J Biol Chem. 2009;284:24133–24143. doi: 10.1074/jbc.M109.015271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfenninger KH, Ellis L, Johnson MP, Friedman LB, Somlo S. Nerve growth cones isolated from fetal rat brain: subcellular fractionation and characterization. Cell. 1983;35:573–584. doi: 10.1016/0092-8674(83)90191-5. [DOI] [PubMed] [Google Scholar]
- Ponimaskin E, Dityateva G, Ruonala MO, Fukata M, Fukata Y, Kobe F, Wouters FS, Delling M, Bredt DS, Schachner M, Dityatev A. Fibroblast growth factor-regulated palmitoylation of the neural cell adhesion molecule determines neuronal morphogenesis. J Neurosci. 2008;28:8897–8907. doi: 10.1523/JNEUROSCI.2171-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid T, Banerjee M, Nikolic M. Phosphorylation of Pak1 by the p35/Cdk5 kinase affects neuronal morphology. J Biol Chem. 2001;276:49043–49052. doi: 10.1074/jbc.M105599200. [DOI] [PubMed] [Google Scholar]
- Rosso S, Bollati F, Bisbal M, Peretti D, Sumi T, Nakamura T, Quiroga S, Ferreira A, Cáceres A. LIMK1 regulates Golgi dynamics, traffic of Golgi-derived vesicles, and process extension in primary cultured neurons. Mol Biol Cell. 2004;15:3433–3449. doi: 10.1091/mbc.E03-05-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santuccione A, Sytnyk V, Leshchyns'ka I, Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol. 2005;169:341–354. doi: 10.1083/jcb.200409127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid RS, Maness PF. L1 and NCAM adhesion molecules as signaling coreceptors in neuronal migration and process outgrowth. Curr Opin Neurobiol. 2008;18:245–250. doi: 10.1016/j.conb.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid RS, Midkiff BR, Kedar VP, Maness PF. Adhesion molecule L1 stimulates neuronal migration through Vav2-Pak1 signaling. Neuroreport. 2004;15:2791–2794. [PubMed] [Google Scholar]
- Schneider A, Rajendran L, Honsho M, Gralle M, Donnert G, Wouters F, Hell SW, Simons M. Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J Neurosci. 2008;28:2874–2882. doi: 10.1523/JNEUROSCI.5345-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki T, Rutishauser U. Removal of polysialic acid-neural cell adhesion molecule induces aberrant mossy fiber innervation and ectopic synaptogenesis in the hippocampus. J Neurosci. 1998;18:3757–3766. doi: 10.1523/JNEUROSCI.18-10-03757.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol. 1997;7:202–210. doi: 10.1016/s0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- Shapiro L, Love J, Colman DR. Adhesion molecules in the nervous system: structural insights into function and diversity. Annu Rev Neurosci. 2007;30:451–474. doi: 10.1146/annurev.neuro.29.051605.113034. [DOI] [PubMed] [Google Scholar]
- Siu MK, Wong ES, Chan HY, Kong DS, Woo NW, Tam KF, Ngan HY, Chan QK, Chan DC, Chan KY, Cheung AN. Differential expression and phosphorylation of Pak1 and Pak2 in ovarian cancer: effects on prognosis and cell invasion. Int J Cancer. 2010;127:21–31. doi: 10.1002/ijc.25005. [DOI] [PubMed] [Google Scholar]
- Spratley SJ, Bastea LI, Döppler H, Mizuno K, Storz P. Protein kinase D regulates cofilin activity through p21-activated kinase 4. J Biol Chem. 2011;286:34254–34261. doi: 10.1074/jbc.M111.259424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg-Smith LJ, Doherty JT, Mack CP, Taylor JM. Adhesion stimulates direct PAK1/ERK2 association and leads to ERK-dependent PAK1 Thr212 phosphorylation. J Biol Chem. 2005;280:2055–2064. doi: 10.1074/jbc.M406013200. [DOI] [PubMed] [Google Scholar]
- Sytnyk V, Leshchyns'ka I, Delling M, Dityateva G, Dityatev A, Schachner M. Neural cell adhesion molecule promotes accumulation of TGN organelles at sites of neuron-to-neuron contacts. J Cell Biol. 2002;159:649–661. doi: 10.1083/jcb.200205098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel DA, Reeder MK, Pfaff A, Coleman TR, Sells MA, Chernoff J. Cell cycle-regulated phosphorylation of p21-activated kinase 1. Curr Biol. 2002;12:1227–1232. doi: 10.1016/s0960-9822(02)00931-4. [DOI] [PubMed] [Google Scholar]
- Westphal D, Sytnyk V, Schachner M, Leshchyns'ka I. Clustering of the neural cell adhesion molecule (NCAM) at the neuronal cell surface induces caspase-8- and -3-dependent changes of the spectrin meshwork required for NCAM-mediated neurite outgrowth. J Biol Chem. 2010;285:42046–42057. doi: 10.1074/jbc.M110.177147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal RS, Coffee RL, Jr, Marotta A, Pelech SL, Wadzinski BE. Identification of kinase-phosphatase signaling modules composed of p70 S6 kinase-protein phosphatase 2A (PP2A) and p21-activated kinase-PP2A. J Biol Chem. 1999;274:687–692. doi: 10.1074/jbc.274.2.687. [DOI] [PubMed] [Google Scholar]
- Wu H, Turner C, Gardner J, Temple B, Brennwald P. The Exo70 subunit of the exocyst is an effector for both Cdc42 and Rho3 function in polarized exocytosis. Mol Biol Cell. 2010;21:430–442. doi: 10.1091/mbc.E09-06-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bi E, Novick P, Du L, Kozminski KG, Lipschutz JH, Guo W. Cdc42 interacts with the exocyst and regulates polarized secretion. J Biol Chem. 2001;276:46745–46750. doi: 10.1074/jbc.M107464200. [DOI] [PubMed] [Google Scholar]
- Zhou H, Kramer RH. Integrin engagement differentially modulates epithelial cell motility by RhoA/ROCK and PAK1. J Biol Chem. 2005;280:10624–10635. doi: 10.1074/jbc.M411900200. [DOI] [PubMed] [Google Scholar]