

Abstract
The large isoforms of the Rab3 interacting molecule (RIM) family, RIM1α/β and RIM2α/β, have been shown to be centrally involved in mediating presynaptic active zone function. The RIM protein family contains two additional small isoforms, RIM3γ and RIM4γ, which are composed only of the RIM-specific C-terminal C2B domain and varying N-terminal sequences and whose function remains to be elucidated. Here, we report that both, RIM3γ and RIM4γ, play an essential role for the development of neuronal arborization and of dendritic spines independent of synaptic function. γ-RIM knock-down in rat primary neuronal cultures and in vivo resulted in a drastic reduction in the complexity of neuronal arborization, affecting both axonal and dendritic outgrowth, independent of the time point of γ-RIM downregulation during dendrite development. Rescue experiments revealed that the phenotype is caused by a function common to both γ-RIMs. These findings indicate that γ-RIMs are involved in cell biological functions distinct from the regulation of synaptic vesicle exocytosis and play a role in the molecular mechanisms controlling the establishment of dendritic complexity and axonal outgrowth.
Introduction
In recent years the large multidomain proteins Rab3 interacting molecules (RIM) have evolved as key regulators of presynaptic active zone function (Mittelstaedt et al., 2010). Within the cytomatrix at the active zone they occupy a central position as they interact with synaptic vesicles (SVs) via Rab3 and Synaptotagmin-1 (Wang et al., 1997; Coppola et al., 2001; Schoch et al., 2002), Ca2+ channels (Hibino et al., 2002; Kiyonaka et al., 2007; Kaeser et al., 2011), and directly or indirectly with most other active zone proteins (Betz et al., 2001; Ohtsuka et al., 2002; Schoch et al., 2002; Wang et al., 2002; Ko et al., 2003a).
The mammalian genome contains four RIM genes (RIM1–4) that through the usage of internal promoters are expressed in three principal variants α, β, and γ (Wang and Südhof, 2003; Kaeser et al., 2008): α-RIMs contain the full set of RIM domains, a N-terminal zinc-finger domain, a central PDZ domain, and two C-terminal C2A and C2B domains, which are separated by an SH3 domain binding motif; β-RIMs are identical to α-RIMs except for their lack of the N-terminal Rab3-binding sequence (RIM1β) or both the Rab3- and Munc-13 binding sequences (RIM2β); γ-RIMs, in contrast, are composed of only the C-terminal C2B domain and an isoform-specific N-terminal sequence. For RIM1 α- and β-variants have been found and RIM2 is the only gene for which all three variants have been described. In contrast, RIM3 and RIM4 only generate the shorter γ-isoform.
Genetic loss of function studies in mice have identified RIM1 and RIM2 as a key factors determining Ca2+ channel density and SV docking at the active zone (Han et al., 2011; Kaeser et al., 2011) and to be involved in the control of neurotransmitter release (Schoch et al., 2002, 2006; Calakos et al., 2004; Deng et al., 2011). RIM1 has been shown to play a role in the regulation of short- and long-term forms of presynaptic plasticity at various types of synapses (Kaeser and Südhof, 2005; Mittelstaedt et al., 2010). However, in contrast to these detailed studies on the large RIM isoforms, little is known about the physiological roles of RIM3γ and RIM4γ. In search of an analogous potential presynaptic role γ-RIMs have been suggested to modulate presynaptic Ca2+ influx via direct binding to voltage-dependent Ca2+ channel (VDCC) accessory subunits or to act in an antagonistic manner to α-RIMs in the regulation of vesicle anchoring to the VDCCs and in the regulation of transmitter release. However, γ-RIMs seem to be much less effective than α-RIMs both in binding to VDCC β-subunits and in regulating transmitter release (Uriu et al., 2010). RIM3γ was further described to be located exclusively postsynaptically (Liang et al., 2007), whereas α-RIMs are localized presynaptically (Wang et al., 1997) suggesting that the role of γ-RIMs may differ substantially from the longer isoforms.
To gain further insight into potential physiological roles of γ-RIMs we have examined their regional and subcellular localization and analyzed both in vitro and in vivo the consequences of knocking down γ-RIMs.
Materials and Methods
Antibodies.
Specific antibodies against rat RIM3γ and RIM4γ were raised against the N-terminal amino acid sequence NH2-CSKSTLQLPQPEGATK-CONH2 (RIM3γ) and NH2-CFDDEDAADSRRLKGAIQR-CONH2 (RIM4γ) after an epitope analysis (Janin, 1979) by Pineda antibody service (http://www.pineda-abservice.de). Antibodies were purified from rabbit immunosera by affinity chromatography on a column to which the synthetic peptide was linked covalently. The specificity of the antibodies was controlled for by immunoblotting of HEK-293T cells overexpressing members of the RIM protein family (see Fig. 1C) and by antigen blocking (Fig. 1F). For antigen blocking the purified RIM3γ and RIM4γ antibodies were diluted 1:200 and 1:150 in PBS containing 10% goat serum, 1% bovine serum albumin (BSA), and 0.1% Triton X-100 equally divided into two tubes. The first tube (antigen block) contained glutathione-Sepharose 4B beads bound to glutathione S-transferase (GST)-RIM3γ and RIM4γ fusion protein, at a concentration of ∼0.5 mg/ml, corresponding to the RIM3γ and RIM4γ antibody. In the second tube (control), the glutathione-Sepharose 4B beads were bound to GST alone. Both tubes were incubated at 4°C overnight, spun at 2000 rpm for 2 min and supernatants were used for immunoblot analysis (Fig. 1F) and immunocytochemistry on primary neuronal cultures (data not shown). RIM3γ and RIM4γ antibodies were applied in Western blotting, immunocytochemistry, and immunohistochemistry at a concentration of 1:200 and 1:150, respectively. The following antibodies were obtained from commercial sources: rabbit FITC-coupled anti-green fluorescent protein (GFP; Santa Cruz Biotechnology), mouse anti-Synapsin and mouse anti-Rab3 (Synaptic Systems), mouse anti-MAP2 (Millipore Bioscience Research Reagents; Millipore), mouse anti-α-Tubulin (Abcam), mouse anti-PSD-95 (NeuroMab), mouse anti-GM130 (BD Bioscience), anti-mouse or anti-rabbit FITC or Cy3 and anti-guinea pig Cy5-conjugated secondary antibodies (Jackson ImmunoResearch), horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse and biotin-conjugated anti-rabbit secondary antibodies (Vector Laboratories), and IRDye 680- and IRDye 800-conjugated anti-rabbit or anti-mouse secondary antibodies from LI-COR Odyssey. All commercial antibodies were used at the concentration indicated by the manufacturer.
Figure 1.

Overlapping but distinct expression patterns of RIM3γ and RIM4γ in adult rat brain. A, ISH micrographs showing RIM3γ and RIM4γ mRNA distribution in the whole brain. Negative controls with excess unlabeled oligonucleotides were devoid of signal (results not shown). Scale bar, 5 mm. B, Higher resolution pictures of emulsion-dipped sections of the hippocampus, the cerebellum, the olfactory bulb, and the cortex for RIM3γ (left) and RIM4γ (right). Scale bar, 300 μm. C, Homogenates of HEK-293T cells transfected with the indicated full-length RIM expression plasmids were analyzed by immunoblotting with affinity-purified antisera against RIM3γ and RIM4γ. The two antibodies were specific for the respective isoforms (peptides) they were raised against. D, Western blot analysis of adult rat tissues. Specific bands corresponding to the molecular weight of RIM3γ (32 kDa) and RIM4γ (27 kDa) were only detected in brain. E, Whole-brain homogenates from rats of the indicated ages (P0–P30) analyzed by immunoblotting with specific antibodies against RIM3γ and RIM4γ. Expression of both isoforms increased during postnatal brain development. F, Homogenates from the specified brain regions were analyzed by immunoblotting with isoform-specific antibodies and specificity of the antibody controlled by peptide blocking. Incubation of the blot with RIM3γ antibody revealed an unspecific 26 kDa band (*) in the thalamus. RIM3γ and RIM4γ proteins were ubiquitously expressed in the brain. However, the expression levels differed between the isoforms in various brain regions. GL, glomerular layer; EPL, external plexiform layer; MC, mossy cell; GC, granular cell; DG, dentate gyrus; I–VI, cortical layers.
Expression vectors and primers.
pCMV expression plasmids containing the rat cDNA of RIM1α, RIM2α, RIM2β, RIM2γ, RIM3γ, and RIM4γ were kindly provided by Dr. Thomas Südhof (Stanford University, Palo Alto, CA). The lentiviral vector for expression of short hairpin RNAs (shRNAs) under control of the U6 promoter and enhanced GFP (EGFP) under the EF1α promoter (pLenti-SHs) and the lentiviral vector for overexpression of RIM3γ and RIM4γ under an EF1α promoter (PLenti-EGFP-EF1α) were kindly provided by Philip Koch (University Bonn, Germany). Full-length and fragments of Rattus norvegicus RIM3γ and RIM4γ were cloned from brain-derived cDNA and inserted into PLenti-EGFP-EF1α between the EcoRI and the EcoRI and BamHI sites, respectively. Primers (Invitrogen) used to amplify RIM3γ full-length and RIM3γ-C2B domain were 5′-ATGTTTAACGGGGAGCCTGG-3′ (forward), 5′-GGAGCACGAGGGGCTGGTGG-3′ (C2B forward), 5′-TTAGGAGCACGAGGGGCTGG-3′ (reverse). Primers (Invitrogen) used to amplify RIM4γ full-length and RIM4γ-C2B domain were 5′-ATGGAGCGCTCGCAGAGC-3′ (forward), 5′-ACACCCATGGGGGATGTG-3′ (C2B forward), 5′-TTAAGATCGCTCGCCACAGG-3′ (reverse). PLenti-EGFP-EF1α-RIM3γ and PLenti-EGFP-EF1α–RIM4γ shRNA-resistant forms were generated using the Quick change Site-Directed Mutagenesis Kit (Stratagene). Primers used for RIM3γ mutagenesis were 5′-CTACCGATGGGAGCACCAATTCAAACAGCTCCGAGGGCACG-3′ and 5′-CGTGCCCTCGGAGCTGTTTGAATTGGTGCTCCCATCGGTAG-3′ and for RIM4γ 5′-GCAACCTGAACTATGGAGGAGTATGCTTGGCTTCGGATGCCCAGTTCA-3′ and 5′-TGAACTGGGCATCCGAAGCCAAGCATACTCCTCCATAGTTCAGGTTGC-3′. shRNA sequences were cloned into the pLenti-SH vector between MluI and ClaI sites. The target sequences for shRNA constructions were 5′-GGCAAGGTTCTACAGGTGA-3′ (RIM3γ-shRNA#1), 5′-CGCCAAGATGGTGGCTATTGT-3′ (RIM3γ-shRNA#2), 5′-GCCACCTATATCAAGGCTTAC-3′ (RIM3γ-shRNA#3), 5′-GGAGCACCAACAGTAACAGCT-3′ (RIM3γ-shRNA#4), 5′-GGATGTGGAGATCGGTTTACA-3′ (RIM4γ-shRNA#1), 5′-CGTGGGTTGGTACAAGCTCTT-3′ (RIM4γ-shRNA#2), 5′-GGAGGAGTTTGTCTAGCTTCA-3′ (RIM4γ-shRNA#3), 5′-ATCACTGGACCCGTTGTACAAC-3′ (RIM4γ-shRNA#4). The mutated shRNAs for RIM3γ and RIM4γ contained the following nucleotide exchanges (in lower case) within the target region 5′-GGAGCACtAgCcGcAACAGCT-3 (mutated shRIM3) and 5′GGAGGgGgTTGaCTgGCT-3′(mutated shRIM4).
Animals.
The studies were performed on brains of Wistar rats of either sex (Charles River). All animal use procedures were performed in accordance with the European Community Council Directive of November 24, 1986 (86/609/EEC). Rats were anesthetized by isoflurane (Baxter) inhalation before decapitation. If necessary, animals were perfused through the heart before brain removal.
In situ hybridization.
Radioactive in situ hybridization (ISH) was performed on 12 μm cryo sections as previously described (Schoch et al., 2006). Frozen rat brain sections were mounted on silane-coated glass slides, fixed with 4% (w/v) paraformaldehyde (PFA) in PBS, dried in ascending ethanol concentrations, and stored in ethanol until hybridization. The sequences of the ISH probes used were 5′-GCAATGCCCGTCTCTGTGCTCCGTCGAATGTTGCTGCGCAGCTTC-3′ (RIM3γ) and 5′-ATGGACTCATGGCTGGCTTGGCGTAACGTCCGGCTGGGCATC TCC-3′ (RIM4γ). Probes were labeled with [35S]dATP using terminal deoxyribonucleotidyl transferase (Fermentas). Each section was hybridized for 16 h at 42°C in 150 μl hybridization buffer (50% deionized formamide/10% (v/v) dextran sulfate/0.3 m NaCl/30 mm Tris/HCl, pH 8/4 mm EDTA/1× Denhardt's/0.4 mg/ml polyadenylic acid/0.5 mg/ml denatured salmon sperm DNA) containing the amount of radiolabeled probe equating to 400,000 counts per minute. Afterward, slides were washed at room temperature (RT) and 57°C for 45 min and dried in ethanol. Hybridized slices were exposed to x-ray films (KODAK BIOMAX MR; Kodak) for 2–4 weeks followed by nuclear track emulsion (NBT2; Kodak) for up to 3 months.
Subcellular fractionation.
Four rat whole brains were removed and homogenized in ice-cold homogenization buffer (0.32 m sucrose, 5 mm HEPES, pH 7.4, and 100 mm EDTA, supplemented with protease block mix from Roche) and centrifuged for 10 min at 3000 rpm (Beckman, J-20). The pellet (P1, nuclear fraction) was discarded and the supernatant (S1, crude synaptosomal fraction) centrifuged at 10,000 rpm (Beckman, J-20) for 20 min. The synaptosomal cytosol fraction (S2) was removed and the crude synaptosomal pellet fraction (P2) was resuspended in a small volume, overlaid on a sucrose density gradient and spun for 2 h and 24,600 rpm (Beckman, SW28). After centrifugation two different fractions could be distinguished: myelin in the 850/1000 mm interphase and synaptosomes in the 1000/1200 mm. The synaptosomes were carefully collected and divided in two fractions; one was used for postsynaptic density (PSD) preparation and the other for SV and synaptic plasma membrane (SPM) enrichment. For the preparation of SPM, synaptosomes were lysed by dilution into 10 volumes ice-cold water, and homogenized through three strokes at 2000 rpm (LS1). Afterward HEPES-KOH buffer, pH7.4, was added to a 1% final concentration and centrifuged for 20 min at 16,500 rpm (Beckman, SS-34). The resulting pellet contained SPM, and the supernatant was again centrifuged for 2 h at 50,000 rpm (Beckman, 50 Ti), obtaining a pellet containing the SV fraction. For the enrichment of PSDs, synaptosomes were adjusted to a final volume of 1 ml ice-cold homogenization buffer containing 0.5% (w/v) Triton X-100, rotated for 20 min, and centrifuged 20 min at 24,000 × g. The resulting pellet (TX1) was resuspended in 500 ml homogenization buffer containing 1% Triton X-100 (w/v), again rotated and centrifuged for 60 min. Supernatant of the 0.5% (w/v) or 1% (w/v) Triton X-100 soluble fraction (TX1 sup and TX2 sup, respectively) and the Triton X-100 insoluble fraction of the SPM (TX1 and TX2), together with previous aliquots, were analyzed by immunoblotting.
Immunoblotting.
Cells were washed with cold phosphate saline buffer and lysed in ice-cold buffer with 2% detergent (SDS), 10 mm EDTA in PBS, pH 7.4, containing a protease block mix (Roche). Tissue samples were sonicated and lysed in the same buffer. Protein concentration of cell and tissue extract samples was determined using a Nanodrop photometer (ND-1000). The protein extracts were adjusted to a final concentration of 5 mg/ml using 6× SDS-loading buffer and lyses buffer. Samples were heated to 95°C for 5 min and 50 μg protein was loaded and size fractionated on a 12% SDS-PAGE (PAGE) gels. Proteins were transferred to nitrocellulose membrane. Membranes were preincubated for 1 h at RT in blocking solution (5% gelatin/0.1% Tween/PBS) and subsequently incubated with the primary antibodies for 1 h at RT. Secondary antibodies were infrared (IR)-labeled (Odyssey) or HRP conjugated. The immunoblots were developed with an IR imaging system (Odyssey; LI-COR) or chemiluminescent detection reagents depending on the secondary antibody used.
Immunolabeling.
Primary neurons 13–15 d in vitro (DIV) were washed with PBS and fixed with 4% PFA in PBS for 10–15 min at room temperature. After washing, cells were permeabilized in 0.3% Triton X-100/PBS for 10 min. Directly after permeabilization a blocking step was performed by incubating the cells in blocking buffer (10% goat serum, 1% BSA, 0.1% Triton X-100 in PBS) for at least 30 min, after which a primary antibody containing solution was applied and incubated overnight at 4°C. After three washes with PBS, fluorochrome-labeled secondary antibody was applied, and incubated for 1 h at RT. Finally, cells were washed with PBS and coverslips were mounted with the Moviol medium (DAKO) and left to dry overnight at room temperature.
For paraffin brain sections, 4 μm sagittal slices were cut from paraffin-embedded brains on a Microm HM 335 E microtome. Brain slices were deparaffinized and rehydrated using xylene, a decreasing ethanol series of a 100, 95, and 70% (v/v) and PBS, for 5–10 min each. For antigen retrieval, brain slices were incubated in citrate buffer and microwaved for 20 min, and afterward cooled down for 20–30 min at RT. After a short rinse with PBS, the slices were permeabilized for 30 min in 0.5% Triton X-100 PBS and subsequently blocked in PBS 10% goat serum, 1% BSA, 0.3% Triton X-100. First the slices were incubated with antibodies in block buffer overnight at 4°C in a humidified chamber, followed by washing with PBS and by incubation with secondary antibodies for 1 h at RT. After washing off excess antibody with PBS, slices were covered using Vectashield hard set mounting medium (Vector Laboratories).
Immunohistochemical analysis of bovine retina was performed after embedding the eyes in Tissue-Tek (Sakura Finetek), and directly freezing them in liquid nitrogen. Ten μm slices were cut with a Microm HM 560 Microtom (Microm). Eye sections were heat fixated 10 min at 60°C, cooled down, blocked for 1 h in 1% BSA/PBS, and immunolabeled following the protocol for brain slices.
Primary neuron cultures.
Primary neurons were prepared from E16–E19 rat brains. Embryo heads were dissected and placed in ice-cold dissection medium (10 mm HEPES in HBSS). The cortex and hippocampus of every pup were isolated and separately collected in 5 ml dissection solution. Trypsin was added to a final concentration of 0.025–1% depending on the amount of tissue, and incubated for 10 min at 37°C. Subsequently, the enzyme solution was removed, the preparations were washed three times, and the gently dissociated cells were plated on coverslips coated with poly-l-lysine (Invitrogen). Hippocampal neurons were grown in Eagle's Basal Medium (BME; Invitrogen) supplemented with 2% B27 (Invitrogen), 1% fetal bovine serum (FBS), 0.45% glucose, and 0.2 mm l-glutamine.
In vitro transfection.
Human embryonic kidney variant HEK-293T cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin mix. Cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Transfection of primary neurons was performed using CaPO4 as described by Köhrmann et al., (1999) or using 1 μl Lipofectamine 2000 reagent and 1–2 μg DNA. Before Lipofectamine transfection, 500 μl medium (BME) was removed from each well, collected, and conserved at 37°C and 5% CO2. DNA/transfection reagent complex formation was completed in OptiMEM and added to the neurons. To increase cell survival, neurons were washed with fresh BME 2–3 h after transfection to avoid Lipofectamine toxicity effects. Finally, the previously collected culture medium was added together with fresh medium. After the growth period indicated for each experiment, the neurons were fixed with PFA, immunolabeled, mounted with Moviol mounting medium (DAKO), and analyzed using confocal microscopy (Olympus FV1000, UPLSApo 60XW UIS2, 1.2 NA objective).
Synaptic silencing.
Hippocampal neurons were transfected with a plasmid-expressing GFP to visualize axons and dendrites. At DIV 2, 1 d after transfection, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo-[f]-quinoxalin-7-sulfonamide (NBQX) was added to the culture media in a final concentration of 10 or 200 nm for 13 d. Control neurons were incubated in unsupplemented media. At DIV 14 neurons were fixed and analyzed using the Sholl Analysis plug-in for ImageJ software.
Cell viability assay.
Primary neurons were transfected on DIV 5 with plasmids expressing GFP or GFP and the shRNA against RIM3γ or RIM4γ. Cells were fixed and analyzed on DIV 14. To identify nonviable cells propidium iodide (PI) was added to cell culture medium 30 min prior to fixation. For quantification the total number of transfected neurons and PI-positive neurons were counted per micrograph image (Zeiss Axio Observer.A1, A-Plan 10× 0.25 NA Objective).
Quantification of synapse density.
Cultures were transfected at DIV 3 with a plasmid-expressing GFP alone (Control) or together with shRNAs against RIM3γ (shRIM3) and RIM4γ (shRIM4) and immunostained at DIV 14 with anti-Synapsin 1 and anti-PSD-95 antibodies. Cultured neurons were imaged with a Nikon A1/Ti-E confocal microscope using a Lambda-S CF1Apo 40× WI 1.25 NA objective. Masks representing Synapsin1/PSD-95 colocalization were created with ImageJ before counting spots on or adjacent to the transfected neuron.
Quantification of Golgi morphology.
Hippocampal neurons were transfected at DIV 3 with either shRNAs against RIM3γ (shRIM3) or RIM4γ (shRIM4), or mutated variants of both shRNAs (mutated shRIM3, mutated shRIM4), or GFP alone (control). At DIV 14 cultures were fixed and the Golgi apparatus was visualized by staining with anti-GM130 antibody. Micrograph images (Zeiss Axio Observer.A1, Plan-NEO FLUAR 40× 0.25 NA objective) of Golgi apparatus were analyzed using ImageJ. To quantify Golgi dispersion we measured the ratio of the convex area of the Golgi outline and of the Golgi fragments, and to quantify Golgi condensation the proportion of the neuron covered by the Golgi apparatus was calculated.
Axon quantification.
Hippocampal neurons were transfected at DIV 1. Axonal length was measured on DIV 5 using the ImageJ plug-in NeuronJ. Growth velocity was analyzed by life imaging of hippocampal cultures, transfected on DIV 7, between DIV 8 and DIV 11 and tracking axonal growth cones 24–48 h after transfection.
Lentiviral particle production.
Lentiviral particles containing the cloned expression constructs and helper plasmids were produced using HEK-293FT cells (Invitrogen) as previously described (Szulc et al., 2006). Lentiviral particles were produced by transfection of required plasmids in HEK-293FT cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Forty-eight hours after transfection, medium containing viral particles was harvested, filtered, and concentrated by ultracentrifugation at 34,400 rpm (Beckman Coulter, 70 Ti) for 2 h.
In vivo injection of lentiviral particles and immunofluorescence analysis.
Newborn Wistar rats were anesthetized and immobilized by covering them with ice for 3–5 min. Afterward, pups were injected with 1 μl virus suspension/HBSS in both ventricles, immediately warmed up for recovery, and returned to the mother. Postnatal day (P)7, P14, and P21 rats were anesthetized and perfused transcardially with PBS for 5 min, followed by 20 min of perfusion with 4% PFA/PBS, pH 7.4. After removal, the brain was postfixed in the same fixative (4°C, 90 min) and equilibrated in 30% sucrose for 24 h. The cerebellum was discarded and the forebrain was embedded in agarose and subsequently cut in 100 μm horizontal sections on a vibratome (Vibratome 1000; Leica Microsystems). Free-floating slices were immunolabeled with FITC-coupled anti-GFP (Abcam). GFP-positive cells were imaged with a Zeiss Axio Observer.A1 inverted microscope using the Plan-Apochromat 20×/0.8 NA air objective and images were acquired with a Jenoptik ProgRes MFcool CCD Camera. Rescue experiments were imaged with a Nikon A1/Ti-E Confocal, using a CFI Plan Apo Lambda 20× 0.75 NA objective.
Spine quantification in vivo.
PFA-fixed slices (100 μm thick) of P21 rat brains were imaged with a Nikon A1/Ti-E Confocal using a Lambda-S CF1Apo 40× WI 1.25 NA objective. Dendritic spines were counted on hippocampal and cortical neurons expressing either red fluorescent protein (RFP; control) or the shRNA against Rim3γ and RFP using Nikon NIS 4.0 software.
Statistical analysis.
Data were evaluated using Prism 4.0 software. Error bars indicate SEM. One-way ANOVA was applied to test the significance of differences among the curves. To test for differences between individual sample means t test or Tukey's multiple comparison post-test was applied.
Electrophysiology.
Miniature EPSCs (mEPSCs) were recorded in whole-cell voltage-clamp from cultured neurons 13 d after lentiviral transduction (DIV 14) with shRNAs. Cells were visualized using a Nikon Eclipse FN1 upright microscope equipped with infrared difference interference contrast optics and a water-immersion lens (×60, 1.0 NA, Nikon). Successful transduction of the neuron under investigation was verified by assessing the GFP fluorescence signal (cultures were only included in the analysis if the transduction efficiency was >90%). Somatic whole-cell voltage-clamp recordings were made with an AxoPatch 200B amplifier (Molecular Devices). Data were sampled at 10 kHz and filtered at 1 kHz with a Digidata 1322A interface controlled by pClamp software (Molecular Devices). Electrode resistance in the bath ranged from 3–5 MΩ, and series resistance ranged from 8–27 MΩ. Resting membrane potential amounted to −63.0 ± 2.3 (control), −62.0 ± 1.9 (RIM3), and −61.9 ± 0.9 (RIM4), and cell capacity was 56.3 ± 7.6, 43.3 ± 7.0, and 43.0 ± 7.5, respectively. The internal solution contained the following (in mm): 110 CH3O3SCs, 10 tetraethylammonium chloride, 11 ethylene glycol tetraacetic acid, 1 CaCl2, 2 Mg-ATP, and 10 HEPES (pH adjusted to 7.2 with CsOH, 290 mOsmol/kg). The extracellular solution contained the following (in mm): 140 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 25 d-glucose, and 300 nm tetrodotoxin, 10 HEPES (pH adjusted to 7.4 with NaOH, 310 mOsmol/kg). Holding potential was corrected off-line for a liquid junction potential of 10 mV.
Results
RIM3γ and RIM4γ exhibit a distinct distribution
To determine the expression pattern of RIM3γ and RIM4γ in the adult rat brain at high resolution we performed radioactive ISH with isoform-specific oligonucleotides that were designed based on a sequence alignment. Negative x-ray film images showed that RIM3γ and RIM4γ transcripts in adult rat brain are expressed throughout the adult rat brain, exhibiting overlapping, but distinct expression patterns (Fig. 1A). RIM4γ mRNA could be detected at high levels throughout the hippocampus being most prominent in the CA1 region (Fig. 1B). In contrast, the signal for the RIM3γ mRNA was quite weak and most strongly detected in the dentate gyrus and in scattered cells in the hilus, the stratum radiatum, and the stratum lacunosum. In the cerebellum both RIM3γ and RIM4γ were present in the granule cell layer but only RIM4γ showed a strong expression in Purkinje cells (Fig. 1B). In the olfactory bulb, expression of RIM3γ mRNA was mainly detected in the external and internal plexiform layer as well as the granule plexiform layer (Fig. 1B). In this region, RIM4γ also exhibited a divergent expression pattern, being mainly detected in the periglomerular cells and the mitral cell layer (Fig. 1B). Whereas RIM4γ was expressed at high levels throughout all cortical layers, a particularly strong labeling was observed for RIM3γ only in layer III of the cortex (Fig. 1B). In the thalamus, only RIM3γ exhibited a particularly strong expression.
To localize RIM3γ and RIM4γ protein and to confirm the mRNA analysis, we generated isoform-specific antibodies against peptides that were located in the N terminus of these proteins (Fig. 1C). Immunoblotting of homogenates from different adult rat tissues, e.g., lung, liver, and heart, revealed a highly restricted expression of both proteins only in the brain (Fig. 1D). Analysis of rat brain homogenates of different ages from P0–P30 revealed that expression of RIM3γ and RIM4γ increased during brain development, reaching a plateau around p20 for RIM3γ and p15 for RIM4γ (Fig. 1E). To confirm the previously observed regional distribution of RIM3γ and RIM4γ mRNA at the protein level, various brain regions were isolated and analyzed by immunoblotting (Fig. 1F). Both proteins could be detected in all brain regions tested. Whereas RIM4γ expression was strongest in the cerebellum, followed by cortex, hippocampus, and olfactory bulb and weak in the remaining brain regions, RIM3γ was found at high levels in both the cerebellum and the olfactory bulb and at lower levels in the other brain areas. Interestingly, at the protein level expression of RIM3γ in the thalamus was not as pronounced as observed for the mRNA (the lower molecular weight band is due to unspecific cross-reactivity that does not disappear after peptide block). Together, these results show that γ-RIMs are present in all brain areas, but exhibit divergent expression in different cell types suggesting a general role in neuronal function.
RIM3γ and RIM4γ exhibit diverging subcellular localizations
RIM1α is an integral component of the cytomatrix at the presynaptic active zone (CAZ). However, in contrast to α-RIMs that are composed of multiple protein interaction domains, the RIM γ-isoforms only contain a single C2B domain that might not be sufficient to tightly link these proteins to the scaffold of proteins composing the CAZ. To examine if γ-RIMs are associated with the presynaptic and postsynaptic densities we analyzed subcellular fractions of synaptosomes from rat forebrain by immunoblotting (Fig. 2A). Antibodies against the SV protein Rab3A and PSD-95 were used as indicators for the subcellular compartments contained within the different fractionations (Fig. 2A). Both proteins could not be detected in the myelin and SV fraction. RIM3γ and RIM4γ could be partially solubilized by Triton X-100 extraction of the synaptic membrane fraction (TX1 SUPP); however, surprisingly, the majority of the protein remained associated with the Triton X-100 insoluble fractions (TX1, TX2) resembling the pattern observed for PSD-95. This result indicates that a substantial amount of the short RIM isoforms is linked to the insoluble component of the synaptic membrane fraction, which is composed of the presynaptic and postsynaptic densities.
Figure 2.

RIM3γ and RIM4γ proteins are components of the presynaptic and postsynaptic cytomatrix. A, Rat brain homogenates were fractionated into the crude synaptosomal fraction (S1), the synaptosomal cytosol fraction (S2), the crude synaptosomal pellet fraction (P2), and the lysed synaptosomal membrane fraction (LP1, LS1), which consists of synaptosomal cytosol and SV-enriched fraction: the crude SV, the SPM, and myelin. The SPM was extracted twice with increasing Triton X-100 concentrations yielding the supernatant of the 0.5% (w/v) or 1% (w/v) Triton X-100 soluble fraction (TX1 SUPP and TX2 SUPP, respectively) and the Triton X-100 insoluble fraction of the SPM (TX1 and TX2). Fractions were analyzed using antibodies against RIM3γ and RIM4γ, as well as against Rab3A and PSD-95. Even though a fraction of RIM3γ and RIM4γ is extracted by Triton X-100, a substantial amount of the two proteins is still associated with the Triton X-100 insoluble fraction after the second extraction, resembling the pattern observed with PSD-95. B, Confocal micrographs of vertical rat retina sections labeled with antibodies against RIM3γ or RIM4γ. ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer. Scale bar, 10 μm. RIM3γ exhibits a specific labeling in both synaptic layers, whereas RIM4γ is also present at these synapses but more broadly distributed. C–E, Double immunolabelings of hippocampal neurons (DIV 14) with the presynaptic marker Synapsin (C), the dendritic marker MAP2 (D), and the postsynaptic marker PSD-95 (E). Scale bars: (in C) 30 μm; (in D, E)10 μm.
As a first step to study the differential subcellular distribution of RIM3γ and RIM4γ in a highly specialized well described synapse in native tissue, we performed immunohistochemical labeling of bovine and rodent retina sections. RIM3γ staining revealed specific labeling in both synaptic layers, the inner and the outer plexiform layer (Fig. 2B), whereas both nuclear layers were virtually devoid of signal. RIM4γ was also detected in both synaptic layers, however, with a broader distribution (Fig. 2B). These results and colabeling with the ribbon-marker protein CtBP2/Ribeye (data not shown) indicate that at the retina RIM3γ is strongly enriched at synapses and present at the presynaptic terminal.
To further investigate the subcellular localization of RIM3γ and RIM4γ we performed double immunostainings of cultured primary hippocampal neurons at DIV 14 for the γ-RIM isoforms and presynaptic and postsynaptic as well as dendritic marker proteins (Fig. 2C–E). RIM3γ revealed a mostly punctate expression pattern, showing significant colocalization with the SV protein Synapsin (Fig. 2C) and the PSD-95 (Fig. 2E). However, RIM3γ could also be detected in the soma and in dendrites (Fig. 2C). In contrast, RIM4γ was rather uniformly distributed in axons and colocalized with MAP2 in dendrites (Fig. 2C–E). These results show that in contrast to α-RIMs (Wang et al., 1997) both RIM3γ and RIM4γ are not exclusively found at presynaptic active zones.
RIM3γ and RIM4γ are required for establishing complex dendritic arbors and dendritic spines
To examine the functional role of γ-RIMs in neuronal cells we designed multiple shRNAs that specifically target either RIM3γ or RIM4γ. We first tested the knock-down efficiency as well as their isoform specificity in HEK-293T cells and cultured primary hippocampal neurons using lentiviral transduction. Immunoblotting identified one shRNA for RIM3γ, shRIM3 #4, and one for RIM4γ, shRIM4 #3, which resulted in an effective knock-down of the respective endogenous protein (Fig. 3A,B) and did not exhibit any cross-reactivity with the other γ-RIM (data not shown) and with RIM1α (data not shown).
Figure 3.
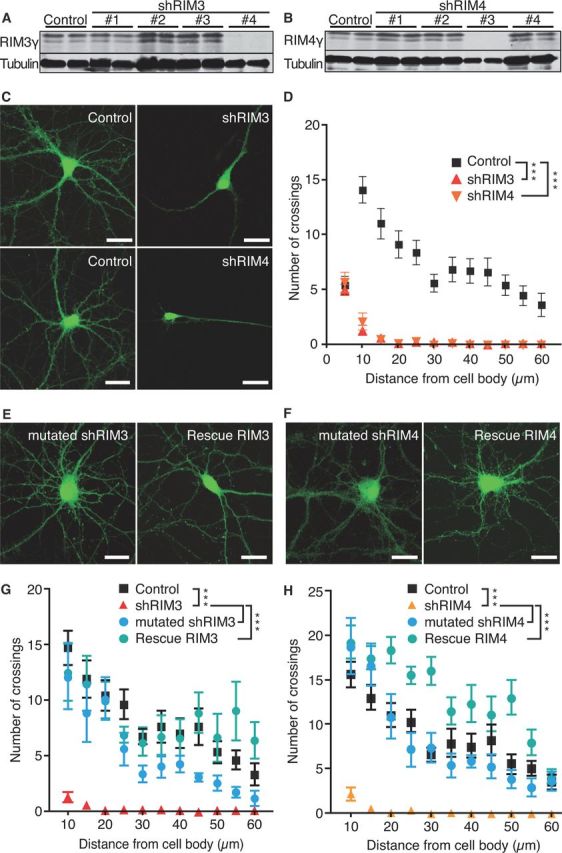
Knock-down of RIM3γ and RIM4γ results in altered neuronal morphology. A, B, Immunoblotting of cellular lysates from hippocampal primary neurons (DIV 14) transduced on DIV 1 with lentiviral particles expressing various RIM3γ-specific (A) or RIM4γ-specific (B) shRNAs (SH#1–4) and the empty vector (Control) revealed that shRIM3 results in a strong reduction of RIM3γ, and shRIM4 strongly reduces RIM4γ protein levels. Staining against tubulin was used as loading control. C, Hippocampal neurons were transduced at DIV 1 with lentiviral particles expressing GFP and either the empty vector (Control) or the RIM3γ/RIM4γ shRNA (shRIM3/4). All neurons were analyzed at DIV 14 using confocal microscopy. D, Sholl analysis revealed a loss of neuronal processes. E, F, The specificity of the observed phenotype was examined by transfecting either mutated shRNAs (mutated shRIM3/4) or functional shRNAs together with resistant RIM3γ/RIM4γ containing silent mutations (Rescue RIM3/Rescue RIM4). G, H, Quantification of the experiments in E and F by Sholl analysis for RIM3γ (G) and RIM4γ (H). Together, these control experiments show that the observed phenotype caused a reduction in the levels of RIM3γ and RIM4γ. One-way ANOVA, ***p < 0.001.
To evaluate the effects of RIM3γ and RIM4γ downregulation, we transduced hippocampal neurons at DIV 1 with lentiviral particles expressing the respective shRNAs, shRIM3, and shRIM4. These lentiviral shRNA vectors also contained an eGFP cassette for identification of neurons expressing the shRNA and allowed confocal imaging of neuronal morphology at DIV 14. Morphometric analysis revealed that the reduction of either RIM3γ or RIM4γ protein levels caused a dramatically less complex dendritic arbor, with neurons exhibiting a massive decrease in secondary and tertiary extensions (Fig. 3C, shRIM3 and shRIM4). The complexity of the dendritic tree assessed by Sholl analysis revealed that neurons transduced with the shRNA for RIM3γ or RIM4γ showed a significantly reduced number of crossings over the entire measured distance (Fig. 3D). To verify that the observed striking phenotype is not due to off-target effects of the shRNA two independent approaches were taken. First, a few nucleotides were exchanged in the respective shRNA sequences resulting in highly homologous shRNAs that should not be able to bind the target mRNA with high efficiency (mutated shRIM3 and mutated shRIM4). Second, silent mutations were introduced into the cDNA coding for RIM3γ and RIM4γ to perform rescue experiments (Rescue RIM3 and Rescue RIM4). Using immunoblotting of transfected HEK-293T cells we verified the lack of knock-down efficiency of the mutated shRNAs (data not shown). Hippocampal neurons expressing mutated shRNAs did not show any overt morphological alterations, and coexpression of resistant RIM3γ and RIM4γ with the shRNAs resulted in a rescue of the knock-down-induced phenotype (Fig. 3E–H). To rule out that the striking changes in neuronal morphology were the consequence of knock-down-induced cell death we labeled control and knock-down neurons at DIV 14 (11 d after shRNA transfection) with PI and quantified the percentage of viable neurons. The percentage of dying neurons did not differ between GFP and shRNA transfected cultures (WT 24 ± 6%, shRIM3 29 ± 2%, shRIM4 31 ± 7%). These results substantiate that the observed phenotype after knock-down of RIM3γ or RIM4γ is indeed due to the absence of these two proteins.
Next, we examined if the reduced dendritic complexity goes along with a decrease in the formation of synapses and dendritic spines. A quantitative analysis of synapse density by labeling with the presynaptic marker Synapsin 1 (SYN) and the postsynaptic marker PSD-95 revealed a significant reduction (RIM3γ ∼70% and RIM4γ ∼50%) in the number of synapses in RIM3γ- and RIM4γ-knock-down neurons (Fig. 4A,B). High-resolution confocal microscopy also revealed that the number of dendritic spines was strongly reduced after knock-down of either RIM3γ or RIM4γ (Fig. 4C; five independent cultures with more than five cells per condition each). These results establish that the presence of RIM3γ and RIM4γ is required for the generation of complex dendritic arbors, as well as for the formation of dendritic spines and synapses.
Figure 4.
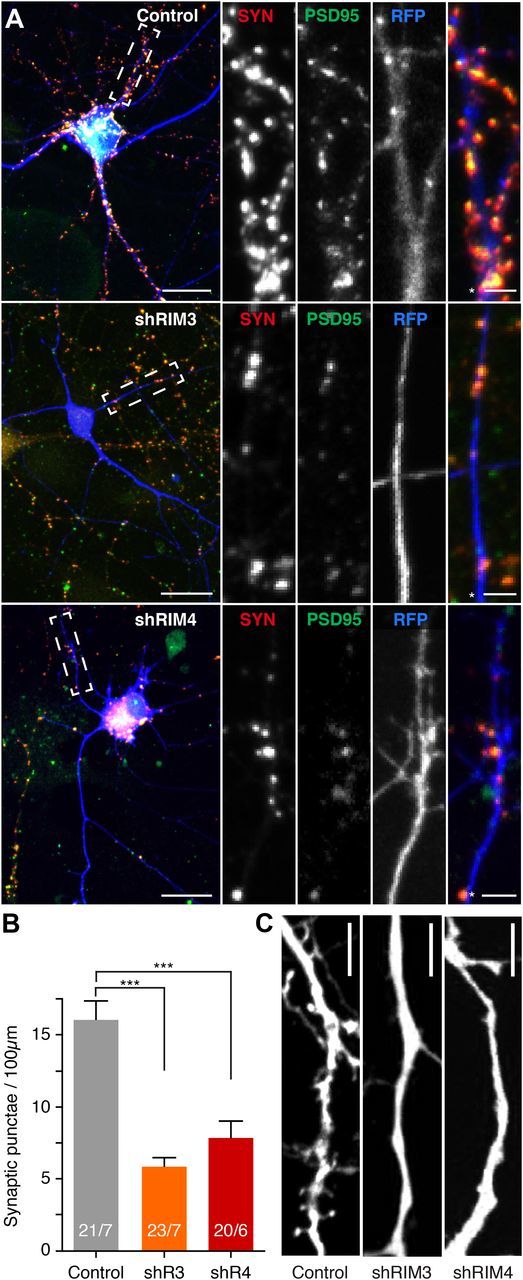
Absence of dendritic spines and reduction in synapse density in RIM3γ and RIM4γ knock-down neurons. A, Hippocampal neurons transfected at DIV 3 with either a vector-expressing GFP (Control) or GFP and the shRNA against RIM3γ (shRIM3) or RIM4γ (shRIM4). All neurons were immunostained using anti-Synapsin (SYN) and anti-PSD-95 (PSD-95) antibodies and analyzed at DIV 14 by confocal microscopy. Scale bar, 30 μm, * = 5 μm. B, Quantification of PSD-95/Synapsin colabeled synaptic punctae on RIM3γ and RIM4γ knock-down dendrites. RIM3γ-shRNA (shR3) and RIM4γ-shRNA (shR4) neurons exhibit a decreased synapse density compared with control. Quantification of Synapsin punctae density was performed using ImageJ software (n: # branches/# cells, one-way ANOVA, ***p < 0.001). C, Confocal image of a dendrite from a control and a RIM3γ-knock-down and a RIM4γ-knock-down neuron showing that substantially fewer dendritic spines can be found on knock-down dendrites (representative image of 5 independent cultures with >5 cells per condition each). Scale bar, 10 μm.
Knock-down of RIM3γ and RIM4γ alters excitatory synaptic transmission
We next examined the functional consequences of RIM3γ and RIM4γ knock-down by recording miniature postsynaptic currents from cultured hippocampal neurons transfected with either RIM3γ or RIM4γ shRNA (Fig. 5A). We hypothesized that the pronounced decrease in the complexity of the dendritic arbor and the reduction in spines would be accompanied by a loss of functional synaptic contacts. Indeed, the mEPSC frequency was markedly reduced following knock-down of either RIM3γ or RIM4γ when compared with control neurons (Fig. 5B; one-way ANOVA with Bonferroni post test, *p < 0.05, **p < 0.01, n = 7, 6, and 5 for RIM3γ and RIM4γ knock-down and control neurons, respectively). In contrast, the mEPSC amplitude following RIM4γ knock-down was unchanged and only slightly reduced after knock-down of RIM3γ (Fig. 5B,C).
Figure 5.

RIM3γ and RIM4γ knock-down decreases miniature excitatory synaptic activity. A, Representative recordings of mEPSCs from a control neuron (gray), and neurons with reduced levels of either RIM3γ (RIM3γ shRNA; black) or RIM4γ (RIM4γ shRNA; red). B, C, Analysis of mEPSC frequency and amplitude after RIM3γ or RIM4γ knock-down. B, Both RIM3γ and RIM4γ knock-down cause significant reduction in mEPSC frequency (p < 0.01 and p < 0.05, respectively; one-way ANOVA with Dunnett's post-test). C, RIM3γ knock-down causes a significant decrease of the mean amplitude (p < 0.05; one-way ANOVA with Dunnett's post-test; n = 7, 6, and 5 for RIM3γ and RIM4γ knock-down and control neurons, respectively).
Synaptic silencing has no effect on dendritic branching
To address if the observed effect on dendritic branching could be caused by a defect in synaptic transmission due to the absence of γ-RIMs we incubated primary hippocampal neurons with two different concentrations of NBQX, 10 and 200 nm, to partially and fully block AMPA-sensitive receptors (Maclean and Bowie, 2011). NBQX was applied at DIV 1 at the same time as RIM3 and RIM4 shRNA in the knock-down experiments (Fig. 6A), and dendritic branching quantified by Sholl analysis on DIV 14 (Fig. 6B). Neither concentration of NBQX significantly affected dendritic branching, indicating that the phenotype caused by the knock-down of both RIM3γ and RIM4γ cannot be the consequence of a potential alteration of synaptic input.
Figure 6.
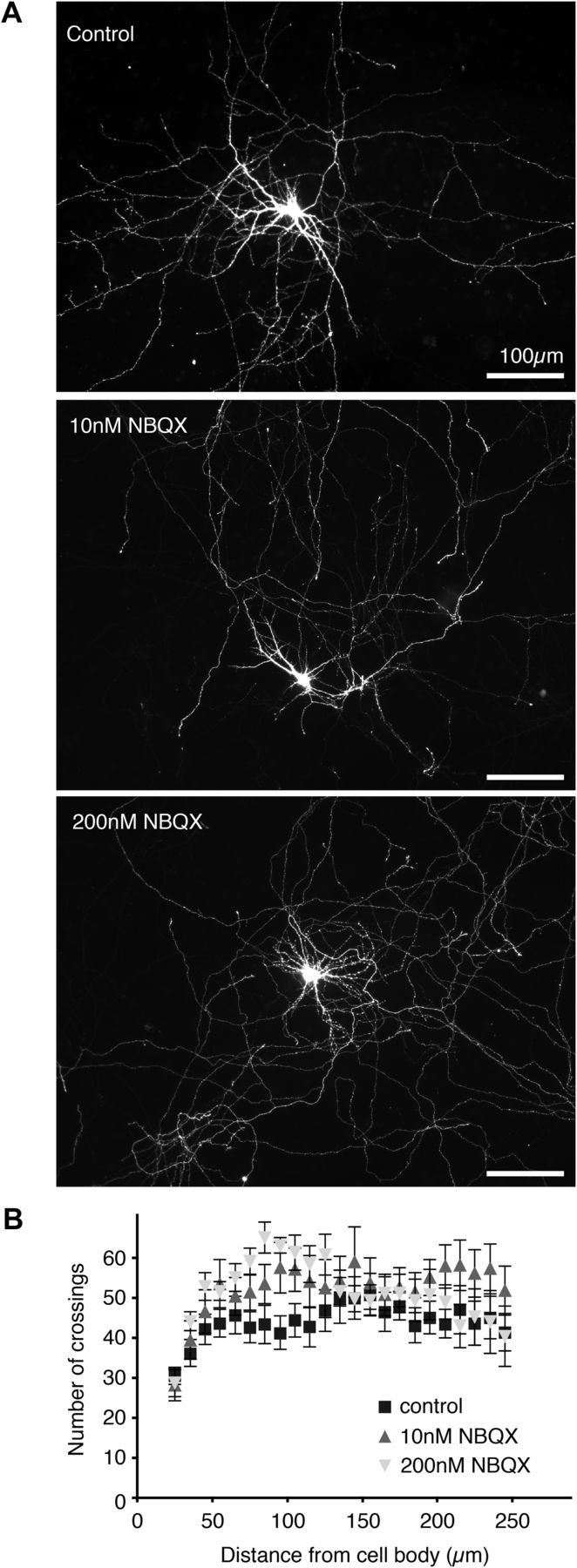
Synaptic silencing has no effect on neuronal morphology. A, Hippocampal neurons were transfected with a plasmid-expressing GFP at DIV 1 to visualize dendrites and axons. From DIV 2 to DIV 14 the cells were exposed to 10 or 200 nm NBQX. Control cells were incubated in normal media. All neurons were analyzed at DIV 14. Scale bar, 100 μm. B, Sholl analysis indicated no difference in neurite branching after synaptic silencing. To detect even small changes in distal dendrites Sholl analysis was performed up to 250 μm from the center of the neuron.
Development of neuronal processes is affected by RIM3γ and RIM4γ knock-down
To examine if RIM3γ and RIM4γ are only required at early stages of neurite development neurons were transduced at DIV 1, 3, and 7 and analyzed at DIV 14 (Fig. 7A). Viruses used for this experiment were from the same preparation to avoid variability due to different virus batches. Knock-down of RIM3γ and RIM4γ at all time points resulted in a less complex arbor compared with the respective control neurons indicating that the proteins are required for dendritic outgrowth (three independent experiments, eight neurons per condition). To approximately estimate the kinetics of RIM3γ and RIM4γ knock-down and turnover and to distinguish whether the absence of RIM3γ and RIM4γ decreases dendritic outgrowth or causes shrinkage of existing dendrites, we assessed dendritic morphology of individual cells at DIV 7 24–96 h after the transfection (Fig. 7B, sample pictures after 24, 48, 72, and 96 h). Dendrite length continually increased over the imaged period in control cells. In contrast, 50 h after RIM3γ and RIM4γ knock-down cells remained viable but outgrowth significantly stagnated (Fig. 7C).
Figure 7.

Development of neuronal processes is also affected by RIM3γ and RIM4γ knock-down at later time points. A, Hippocampal neurons transduced at DIV 1, 3, and 7, with shRNA and GFP (control) expressing lentiviral particles, and were analyzed by confocal microscopy at DIV 14. Neurons infected later exhibited a more complex dendritic tree than those in which the knock-down had been performed earlier. However, at all time points there was an obvious reduction in the number of processes in the shRNA-treated neurons as compared with control. Scale bar, 30 μm. B, Time-lapse imaging of hippocampal neurons transfected on DIV 7 with a plasmid-expressing shRIM3, shRIM4, or GFP alone (control) at 24, 48, 72, and 96 h after transfection. Already 24 h after transfection the dendritic tree of knock-down neurons appeared less complex and stopped growing after ∼50 h. Scale bar, 100 μm. C, Quantification of the length of the dendritic tree at 5 h intervals from the time-lapse experiment described in B. Dendrites were traced using ImageJ with NeuronJ plug-in; data depicted as mean± SEM were analyzed with Prism GraphPad 4 using one-way ANOVA followed by Tukey's multiple-comparison test, ***p < 0.001.
To determine whether not only dendritic growth but also axonal outgrowth was affected by RIM3γ and RIM4γ knock-down, we quantified the length of the axon on DIV 5 after transfecting the cells with shRNA-expressing plasmids on DIV 1. The total length of the axons was strongly reduced in the shRNA-expressing cells (shRIM3 547 ± 73 μm, shRIM4 693 ± 159 μm) compared with control (3119 ± 749 μm; Fig. 8A,B), and the number of axonal branches went down to one-third (Fig. 8C). Both, the reduction in total axon length and the decrease in axonal branches, were not observed if neurons were either transfected with the above described mutated shRNAs (mutated shRIM3 3108 ± 392 and mutated shRIM4 3717 ± 235.3; Fig. 8A–C) or if shRNA-resistant RIM3 and RIM4 cDNAs were coexpressed (Rescue RIM3 3389 ± 323 and Rescue RIM4 3086 ± 420; Fig. 8A–C). Furthermore, we quantified axonal growth during the first 24 h of imaging after transfection at DIV 7 (Fig. 7B). We found a significant 50% decrease in growth cone travel speed between control (17.5 ± 1.3 μm/h) and shRNA-expressing cells (shRIM3 8.5 ± 1.1 μm/h, shRIM4 9.3 ± 0.9 μm/h) (Fig. 8D). Together, these results show that RIM3γ and RIM4γ are important for dendritic as well as axonal outgrowth and arborization.
Figure 8.

RIM3γ and RIM4γ knock-down affects early axonal outgrowth. A, Hippocampal neurons transfected DIV 1 expressing GFP (Control), shRNAs against RIM3γ (shRIM3) and RIM4γ (shRIM4), shRNAs with few nucleotide exchanges (mutated shRIM3 and mutated shRIM4) and coexpressing the shRNAs with the respective resistant cDNAs (Rescue RIM3 and Rescue RIM4) were fixed and analyzed by confocal microscopy at DIV 5. B, C, Quantitative analysis of total axonal length (B) and the number of axonal branches (C) showed that axonal outgrowth and branching are strongly reduced after knock-down of RIM3γ (shRIM3) and RIM4γ (shRIM4) as compared with controls. Both parameters are unaffected after cotransfection of shRIM3 or shRIM4 and a resistant version of the respective RIM variant (Rescue RIM3 and RIM4) or using mutated shRNAs against RIM3γ (mutated shRIM3) and RIM4γ (mutated shRIM4). D, Measurements of axonal growth of hippocampal neurons at DIV 8–DIV 9 from the time-lapse experiment of Figure 7B revealed that neurons transfected with shRNAs grow with reduced velocity 24–48 h after transfection. Significance: one-way ANOVA with Tukey's multiple-comparison test, ***p < 0.001, **p < 0.01.
Decreased dendritic branching after in vivo knock-down of RIM3γ and RIM4γ
To validate the results obtained in cultured neuronal cells and to examine if knock-down of RIM3γ and RIM4γ also interferes with the acquisition of neuronal complexity within a normal functional neuronal network, we sparsely injected lentiviral particles expressing GFP only (control) or the respective shRNAs (shRIM3 and shRIM4) into the ventricle of P0 rat brains. Injected rats did not exhibit any behavioral abnormalities. Brains were collected and analyzed by immunohistochemistry at P14 and P21 (Fig. 9A; three animals per group and time point). Overall brain organization, e.g., cortical layering, and neuronal morphology of uninfected cells was not altered. Viral particles spread out after injection into different brain regions and sparsely labeled neurons in thalamus, striatum, cortex, and hippocampus. At P14 GFP-positive control neurons exhibited extended dendritic arbors, whereas neurons with reduced levels of RIM3γ or RIM4γ were almost devoid of complex dendritic structures (Fig. 9A). This effect on neuronal morphology became even more prominent at P21 (Fig. 9A,B). To substantiate the finding of decreased spinogenesis in vitro (Fig. 4C), we counted spines in a more mature and native environment, both in hippocampus and in cortex of rats, which had been injected at P0 with lentiviral particles expressing RFP only (control) or the RIM3 shRNAs (shRIM3). In accordance with our culture experiments we detected a significant decrease in spine number after knock-down of RIM3 in both brain regions (Fig. 10A,B).
Figure 9.
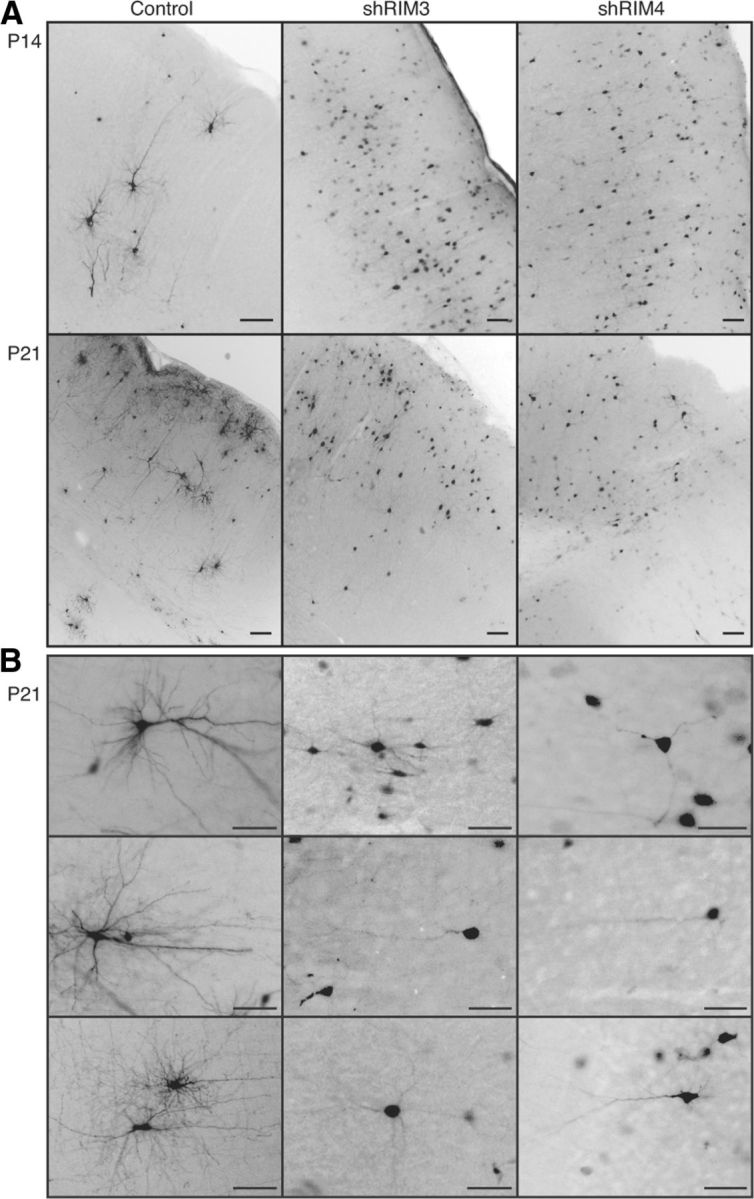
In vivo knock-down of RIM3γ and RIM4γ reproduces effect on neuronal morphology. A, Lentiviral particles expressing GFP alone (Control) or together with shRNAs against shRIM3 or shRIM4 were injected into the ventricle of P0 rat brains. Brains were analyzed at P14 (top row) and P21 (bottom row) by immunohistochemistry with an antibody against GFP. Cortical control neurons displayed a normal morphology (left), showing regular dendritic growth. In contrast, neurons transduced with the shRNA sequences exhibited a strong deficit in the number of neurites, indicating a greatly compromised neuronal branching (middle and right). Scale bar, 200 μm. B, Higher magnification images of control and knock-down cortical neurons (P21) revealed a striking loss in the dendritic arbor of neurons with decreased levels of RIM3γ and RIM4γ as compared with control (left). Scale bar, 50 μm.
Figure 10.

RIM3γ in vivo rescue of neuronal morphology, reduction in spine density after loss of RIM3γ in hippocampus and cortex. A, Hippocampal neurons in P21 rats show that neurons expressing the shRNA against RIM3γ exhibit a reduced number of spines compared with control cells expressing only RFP. B, Quantification revealed a significant loss in spine density after knock-down of RIM3γ in hippocampal and cortical neurons (t test, hippocampus ***p = 0.0003, cortex **p = 0.0042). C, Lentiviral particles expressing RFP (Control) and the shRNA against RIM3γ alone (shRIM3) or together with a green fluorescent-resistant variant of RIM3 (Rescue RIM3) were injected into P0 rat brains. At P21, RIM3 knock-down cortical neurons exhibited the expected loss in arborization, while neurons expressing both shRNA and the resistant RIM3 were undistinguishable from control neurons expressing RFP. Scale bar, 50 μm.
To verify if the observed changes in neuronal arborization in vivo could also be rescued by overexpression of the resistant protein, we injected rats at P0 with lentiviral particles expressing either RFP (control) or RFP and a shRNA against RIM3 (shRIM3) as well as with a combination of lentiviral particles expressing either the shRNA/RFP or resistant RIM3-GFP (Rescue RIM3). As in our previous experiments (Fig. 9) we observed a strong reduction in neuronal branching after knock-down of RIM3 as compared to control cells in neurons expressing RFP only. Cells that expressed both the shRNA and resistant RIM3-GFP exhibited a comparable neuronal morphology as control cells (Fig. 10C). These results demonstrate that decreasing the levels of RIM3γ and RIM4γ in the first 3 weeks after birth results in a dramatic alteration of neuronal morphology despite their contact with intact presynaptic cells and a normal microenvironment.
The conserved C2B domain is sufficient to rescue the RIM3γ and RIM4γ knock-down phenotype
To gain first insights into which steps are affected by knocking down RIM3γ and RIM4γ we performed cross-rescue experiments, in which we coexpressed a RIM3γ shRNA and RIM4γ and vice versa, and rescue experiments using only the highly conserved C2B domain (Fig. 11). These experiments revealed that the defect in neuronal growth could be almost completely rescued by the respective other isoform (Fig. 11 A–C) and even by the C2B domain alone (Fig. 11 A,D), indicating that the N-terminal isoform-specific sequences are not required for the role of RIM3γ and RIM4γ in neuronal morphogenesis.
Figure 11.

The C2B domain present in both RIM3γ and RIM4γ is sufficient to rescue the knock-down phenotype. A, Hippocampal neurons were transduced at DIV 1 with viral particles expressing GFP (Control) or shRIM3 together with either RIM3γ (nonresistant) or RIM4γ. Neurons transduced at DIV 1 with viral particles expressing GFP (Control) or shRIM4 in combination with either RIM4γ (nonresistant) or RIM3γ. B, C, Quantification shows that the phenotype caused by knock-down of either RIM3γ (B) or RIM4γ (C) can be rescued by overexpression of the respective other isoform. (A, right column) Neurons were transduced with viral particles expressing shRIM3 or shRIM4 in combination with the respective RIM3/4γ-C2B domain. D, The quantification revealed that the C2B domain is sufficient to restore dendritic complexity almost to control levels. All neurons were analyzed at DIV 14 using confocal microscopy. Sholl analysis was performed to quantify the loss of neuronal processes. One-way ANOVA; ***p < 0.001.
Knock-down of RIM3γ and RIM4γ results in structural alterations of the Golgi apparatus
A functional Golgi apparatus is essential for mediating dendritic growth and maintenance (Hanus and Ehlers, 2008). We therefore examined if knock-down of RIM3γ and RIM4γ affects the structure of the Golgi apparatus. To visualize Golgi morphology, hippocampal neurons transfected on DIV 7 with either control vector or shRNA-expressing plasmid were labeled with an antibody against the Golgi marker protein GM130 on DIV 14 (Fig. 12). Whereas in control neurons GM130 staining revealed the typical ribbon-like structure of the Golgi apparatus (Fig. 12A), it appeared fragmented into punctate structures and dispersed throughout the cytoplasm after knock-down of RIM3γ (Fig. 12A,B) and strongly condensed in RIM4γ knock-down neurons (Fig. 12A,C). Neither Golgi dispersion nor condensation was observed if shRNAs against RIM3 and RIM4 with few nucleotide exchanges were transfected (mutated shRIM3 and mutated shRIM4;, Fig. 12).
Figure 12.

Structural alteration of the Golgi apparatus in neurons lacking RIM3γ/4γ. A, Confocal images of GM130-labeled cultured hippocampal neurons, transfected at DIV 3 with either shRNAs against RIM3γ (shRIM3) or RIM4γ (shRIM4), or mutated variants of the both shRNAs (mutated shRIM3, mutated shRIM4), or GFP alone (control). Cells were fixed at DIV 14 and stained against the Golgi marker GM130 (red). Scale bar, 50 μm; * 20 μm. B, Quantification of Golgi dispersion. While Golgi dispersion in RIM4γ knock-down cells is indistinguishable from control, RIM3γ knock-down leads to increased fragmentation and dispersion. C, Quantification of Golgi size shows that knock-down of RIM4γ leads to a smaller, more condensed Golgi apparatus as compared with controls. B, C, These structural alterations were abolished using the mutated shRNAs against RIM3 or RIM4. Significance: one-way ANOVA followed by Tukey's multiple-comparison test, ***p < 0.0001.
Discussion
By using an in vitro and in vivo knock-down approach we found that both, RIM3γ or RIM4γ, in contrast to α-RIMs, play an essential role in the processes underlying the formation of the dendritic arbor, which is independent from the regulation of SV exocytosis. In particular, knock-down of RIM3γ or RIM4γ decreased dendritic and axonal growth and resulted in a reduction in the number of spines, synapses, and functional synaptic contacts. The finding that γ-RIMs are involved in basal cell biological functions is further supported by our observation that in contrast to α-RIMs, γ-RIMs exhibit a broader subcellular distribution and can also be found in the soma and dendrites.
Considering the well established roles of RIM1α/β and RIM2α/β in the regulation of SV exocytosis (Mittelstaedt et al., 2010; Han et al., 2011; Kaeser et al., 2011) and the suggested presynaptic function of γ-RIMs in (1) modulating presynaptic Ca2+ influx via direct binding to VDCC accessory subunits or (2) as agonists to α-RIMs in the regulation of vesicle anchoring to the VDCCs and in the regulation of synaptic transmission (Uriu et al., 2010), these findings were rather surprising. However, the alterations in neuronal morphology caused by the knock-down of RIM3γ and RIM4γ were not observed if corresponding shRNAs with few nucleotide exchanges were applied and could be rescued by coexpression of the according cDNAs carrying silent mutations and even by coexpression of the respective other isoform (cross-rescue), proving the phenotype is due to reduced levels of either protein. Furthermore, the phenotype was also reproduced after knock-down of RIM3γ and RIM4γ in vivo at P0. In these experiments neurons expressing the shRNAs were surrounded by unaffected neurons and thereby exposed to the native environment of signaling molecules and regulators. These results therefore firmly establish that the phenotype in neuronal arborization is cell autonomous to neurons deficient in RIM3γ and RIM4γ and independent of signaling from surrounding neurons. Accordingly, our expression analyses showed that all neurons express at least one γ-RIM isoform and that expression of both variants is already detectable at P0, the time point of virus injection in the in vivo knock-down experiments, but increases during brain development. A function for RIM3γ and RIM4γ independent of α-RIMs is also supported by their diverging subcellular distribution. In contrast to α-RIMs both γ-RIMs are not exclusively localized at presynaptic active zones, and even though they are tightly associated with the presynaptic and postsynaptic cytomatrix, they can also be found in dendrites and in the soma.
The development of a mature but plastic dendritic arbor is a complex multistep process, including outgrowth, branching, stabilization, and remodeling, highly regulated at every stage (Urbanska et al., 2008; Jan and Jan, 2010; Poulain and Sobel, 2010). The size, pattern, and stability of the dendritic tree are controlled at multiple levels by a variety of factors, e.g., transcription factors, cell-surface receptors, regulators of cytoskeletal elements, signaling cascades, and endocytic and secretory pathways. To gain a first insight into which steps are affected by knocking down RIM3γ and RIM4γ, we performed cross-rescue experiments (RIM3γ knock-down and RIM4γ overexpression and vice versa) and rescue experiments using only the C2B domain. These analyses showed that RIM3γ and RIM4γ function at the same step in dendrite development, as the knock-down phenotype of one isoform can be rescued by the respective other isoform, and that the C2B domain plays an important role in this process. Our quantitative analysis of axonal and dendritic outgrowth revealed that knock-down of either RIM3γ or RIM4γ affected the tested parameters to a similar degree, further supporting a common function for the two proteins in these processes. So far, the C2B domain of RIMs has been shown to interact with the scaffolding protein Liprin-α (Schoch et al., 2002), the β-subunits of voltage-gated calcium channels (Kiyonaka et al., 2007; Uriu et al., 2010), the synapse-localized E3 ubiquitin ligase SCRAPPER (Yao et al., 2007), and the SV protein Synaptotagmin 1 (Schoch et al., 2002). Interestingly, Liprin-α1 and three known Liprin-α1 binding proteins, GRIP, LAR-RPTP, and GIT1, have been linked to the regulation of dendrite development (Hoogenraad et al., 2005, 2007; Menon et al., 2010). Increasing Liprin-α1 levels by overexpression of degradation-resistant mutants impairs dendrite morphogenesis, whereas depletion of GRIP, LAR-RPTP, and GIT1 reduces dendritic arbor complexity. Liprin-α1 is involved in the correct targeting of GRIP, LAR-RPTP, and GIT1 (Wyszynski et al., 2002; Ko et al., 2003b; Dunah et al., 2005; Hoogenraad et al., 2007). Disruption of the Liprin-α1-LAR interaction interfered with targeting of LAR to dendrites and resulted in a concomitant decrease in dendritic arbor complexity (Hoogenraad et al., 2007). The results of these studies suggest that Liprin-α1 protein levels are closely linked to dendritic outgrowth. It could therefore be hypothesized that RIM3γ and RIM4γ by interacting with Liprin-α1 protect the protein from degradation and thereby regulate its availability. A decrease in RIM3γ and RIM4γ levels would then cause a reduction in Liprin-α1 levels and interfere with the proper targeting of Liprin-α1 binding proteins. On the other hand, RIM3γ and RIM4γ could also be involved in Liprin-α-dependent transport of different cargo complexes (Miller et al., 2005).
Interestingly, knock-down of RIM3γ or RIM4γ also caused both the absence of dendritic spines and a reduction in synapse density and in frequency of mEPSCs. This phenotype resembles the one observed after knock-down of ephrin-B3 in cultured neuronal cells. The trans-synaptic EphB2 to ephrin-B3 interaction was shown to regulate excitatory synapse density (McClelland et al., 2010) and to have the potential to rescue the loss of dendrites caused by knock-down of the Liprin-α interacting protein GRIP1 (Wyszynski et al., 2002). These observations could point to an involvement of RIM3γ and RIM4γ at some step of this signaling cascade. Knock-down of ephrin-B3 also results in a severe reduction in functional excitatory synapses as shown by the specific decrease in mEPSC but not mIPSC frequency (McClelland et al., 2010). However, we found that synaptic silencing has no effect on dendritic branching, indicating that the phenotype observed after knock-down of RIM3γ or RIM4γ is acting downstream to changes in synaptic transmission.
Our time-lapse imaging experiments showed that whereas the dendritic tree of control cells steadily increased over time, it stagnated in knock-down neurons and was significantly smaller than control after ∼48 h (Fig. 7B,C). Thereby, this experiment established that RIM3γ or RIM4γ is required for the outgrowth of both axons and dendrites but is not essential for the maintenance of dendrites. This is in contrast to the phenotype observed after knock-down of GRIP1, which affects formation and maintenance of dendrites but not axonal morphogenesis (Hoogenraad et al., 2005). However, the Liprin-α interacting protein ubiquitin ligase anaphase-promoting complex (APC) can specifically regulate axon or dendrite morphogenesis depending on whether it interacts with the coactivator fizzy-related protein homolog (CDC20) (Konishi et al., 2004; Kim et al., 2009). Therefore, differential interactions of RIM3γ or RIM4γ could be involved in the processes underlying axonal and dendritic outgrowth.
Membrane trafficking is essential for growth and maintenance of both dendritic and axonal processes and is highly regulated at multiple levels, e.g., vesicle budding, transport and fusion (Sann et al., 2009). Defects in secretory pathways, e.g., in Golgi trafficking, have been shown to affect dendritic growth (Hanus and Ehlers, 2008; Tang, 2008; Urbanska et al., 2008). Even though less is known about the membrane trafficking underlying axonal growth, similar exocytotic processes are involved in both processes (Martinez-Arca et al., 2001). RIM3γ and RIM4γ are present in the soma and are found to be associated with the Golgi apparatus. Interestingly, the structure of the Golgi apparatus is altered after knock-down of either RIM3γ or RIM4γ. While the loss of RIM3γ leads to a significant dispersion and distribution of the Golgi apparatus throughout the whole-cell, RIM4γ knock-down results in a condensation of Golgi fragments. Recently, it was reported that both Golgi fragmentation and condensation as well as dendritic complexity and axonal outgrowth depend on the expression of the microtubule plus-end tracking protein CLASP2 (Beffert et al., 2012). Therefore, our results could point toward a role for RIM3γ and RIM4γ in Golgi trafficking. Another factor equally important for neurite as well as Golgi morphogenesis is the regulation of cytoskeletal dynamics, as actin and microtubules are the major structural components underlying dendrite and axon as well as Golgi morphology (Jan and Jan, 2010; Poulain and Sobel, 2010). Signaling complexes downstream of the Rho family of GTPases are critically involved in controlling the cytoskeletal rearrangements that affect dendritic and axonal growth as well as branching and spine morphogenesis. In immunoelectron microscopy analyses RIM3γ was often detected in close proximity to microtubules (Liang et al., 2007), suggesting a potential involvement of the protein in the control of cytoskeletal dynamics downstream of Rho GTPases.
Several neurological disorders, like autism or schizophrenia, are associated with defects in dendrite growth or pruning and alterations in dendritic spine number (Kaufmann and Moser, 2000; Pardo and Eberhart, 2007; Penzes et al., 2011). Intriguingly, changes in RIM3γ expression levels have been reported in schizophrenia (Weidenhofer et al., 2006, 2009) and in lymphoblastoid cells from autism patients with either maternal duplications of 15q11q13 or fragile X syndrome (Nishimura et al., 2007). RIM3γ was further identified as a novel candidate gene for autism by a genetic approach (Kumar et al., 2010). These findings further suggest that altered levels of functional RIM3γ protein, either due to chromosomal imbalances or to rare mutations, may contribute to autism development or other neuropsychiatric disorders with shared genetic etiologies.
Footnotes
This work was supported by grants from the Deutsche Forschungsgemeinschaft EmmyNoether Program (S.S.), SFB/TR3 (H.B., D.D., S.S., A.J.B.), DI 853/3 and DI 853/4 (D.D.), and SFB894 (F.S.); the BMBF NGFNplus (H.B., S.S., A.J.B.); Unabhängige Forschergruppen in den Neurowissenschaften (S.S.); Neuron ERANET “EpiNet” (H.B.); Euroepinomics (A.J.B.); Else Kröner Fresenius Foundation (A.J.B.); German Israeli Foundation (A.J.B.); and BONFOR (S.S., H.B., D.D., AJ.B.). We thank A. Alfter, M. Gerards, G. Engels, S. Opitz, and S. Stellbogen for excellent technical assistance and J. Steinbeck for help with the viral injections.
The authors declare no competing financial interests.
References
- Beffert U, Dillon GM, Sullivan JM, Stuart CE, Gilbert JP, Kambouris JA, Ho A. Microtubule plus-end tracking protein CLASP2 regulates neuronal polarity and synaptic function. J Neurosci. 2012;32:13906–13916. doi: 10.1523/JNEUROSCI.2108-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz A, Thakur P, Junge HJ, Ashery U, Rhee JS, Scheuss V, Rosenmund C, Rettig J, Brose N. Functional interaction of the active zone proteins Munc13-1 and RIM1 in synaptic vesicle priming. Neuron. 2001;30:183–196. doi: 10.1016/s0896-6273(01)00272-0. [DOI] [PubMed] [Google Scholar]
- Calakos N, Schoch S, Südhof TC, Malenka RC. Multiple roles for the active zone protein RIM1alpha in late stages of neurotransmitter release. Neuron. 2004;42:889–896. doi: 10.1016/j.neuron.2004.05.014. [DOI] [PubMed] [Google Scholar]
- Coppola T, Magnin-Luthi S, Perret-Menoud V, Gattesco S, Schiavo G, Regazzi R. Direct interaction of the Rab3 effector RIM with Ca2+ channels, SNAP-25, and synaptotagmin. J Biol Chem. 2001;276:32756–32762. doi: 10.1074/jbc.M100929200. [DOI] [PubMed] [Google Scholar]
- Deng L, Kaeser PS, Xu W, Südhof TC. RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron. 2011;69:317–331. doi: 10.1016/j.neuron.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Hueske E, Wyszynski M, Hoogenraad CC, Jaworski J, Pak DT, Simonetta A, Liu G, Sheng M. LAR receptor protein tyrosine phosphatases in the development and maintenance of excitatory synapses. Nat Neurosci. 2005;8:458–467. doi: 10.1038/nn1416. [DOI] [PubMed] [Google Scholar]
- Han Y, Kaeser PS, Südhof TC, Schneggenburger R. RIM determines Ca(2)+ channel density and vesicle docking at the presynaptic active zone. Neuron. 2011;69:304–316. doi: 10.1016/j.neuron.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanus C, Ehlers MD. Secretory outposts for the local processing of membrane cargo in neuronal dendrites. Traffic. 2008;9:1437–1445. doi: 10.1111/j.1600-0854.2008.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H, Pironkova R, Onwumere O, Vologodskaia M, Hudspeth AJ, Lesage F. RIM binding proteins (RBPs) couple Rab3-interacting molecules (RIMs) to voltage-gated Ca(2+) channels. Neuron. 2002;34:411–423. doi: 10.1016/s0896-6273(02)00667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad CC, Milstein AD, Ethell IM, Henkemeyer M, Sheng M. GRIP1 controls dendrite morphogenesis by regulating EphB receptor trafficking. Nat Neurosci. 2005;8:906–915. doi: 10.1038/nn1487. [DOI] [PubMed] [Google Scholar]
- Hoogenraad CC, Feliu-Mojer MI, Spangler SA, Milstein AD, Dunah AW, Hung AY, Sheng M. Liprinalpha1 degradation by calcium/calmodulin-dependent protein kinase II regulates LAR receptor tyrosine phosphatase distribution and dendrite development. Dev Cell. 2007;12:587–602. doi: 10.1016/j.devcel.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Jan YN, Jan LY. Branching out: mechanisms of dendritic arborization. Nat Rev Neurosci. 2010;11:316–328. doi: 10.1038/nrn2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin J. Surface and inside volumes in globular proteins. Nature. 1979;277:491–492. doi: 10.1038/277491a0. [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Südhof TC. RIM function in short- and long-term synaptic plasticity. Biochem Soc Trans. 2005;33:1345–1349. doi: 10.1042/BST0331345. [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Kwon HB, Chiu CQ, Deng L, Castillo PE, Südhof TC. RIM1alpha and RIM1beta are synthesized from distinct promoters of the RIM1 gene to mediate differential but overlapping synaptic functions. J Neurosci. 2008;28:13435–13447. doi: 10.1523/JNEUROSCI.3235-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, Südhof TC. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell. 2011;144:282–295. doi: 10.1016/j.cell.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10:981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- Kim AH, Puram SV, Bilimoria PM, Ikeuchi Y, Keough S, Wong M, Rowitch D, Bonni A. A centrosomal Cdc20-APC pathway controls dendrite morphogenesis in postmitotic neurons. Cell. 2009;136:322–336. doi: 10.1016/j.cell.2008.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyonaka S, Wakamori M, Miki T, Uriu Y, Nonaka M, Bito H, Beedle AM, Mori E, Hara Y, De Waard M, Kanagawa M, Itakura M, Takahashi M, Campbell KP, Mori Y. RIM1 confers sustained activity and neurotransmitter vesicle anchoring to presynaptic Ca2+ channels. Nat Neurosci. 2007;10:691–701. doi: 10.1038/nn1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Na M, Kim S, Lee JR, Kim E. Interaction of the ERC family of RIM-binding proteins with the liprin-alpha family of multidomain proteins. J Biol Chem. 2003a;278:42377–42385. doi: 10.1074/jbc.M307561200. [DOI] [PubMed] [Google Scholar]
- Ko J, Kim S, Valtschanoff JG, Shin H, Lee JR, Sheng M, Premont RT, Weinberg RJ, Kim E. Interaction between liprin-alpha and GIT1 is required for AMPA receptor targeting. J Neurosci. 2003b;23:1667–1677. doi: 10.1523/JNEUROSCI.23-05-01667.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhrmann M, Haubensak W, Hemraj I, Kaether C, Lessmann VJ, Kiebler MA. Fast, convenient, and effective method to transiently transfect primary hippocampal neurons. J Neurosci Res. 1999;58:831–835. [PubMed] [Google Scholar]
- Konishi Y, Stegmüller J, Matsuda T, Bonni S, Bonni A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 2004;303:1026–1030. doi: 10.1126/science.1093712. [DOI] [PubMed] [Google Scholar]
- Kumar RA, Sudi J, Babatz TD, Brune CW, Oswald D, Yen M, Nowak NJ, Cook EH, Christian SL, Dobyns WB. A de novo 1p34.2 microdeletion identifies the synaptic vesicle gene RIMS3 as a novel candidate for autism. J Med Genet. 2010;47:81–90. doi: 10.1136/jmg.2008.065821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Zhang B, Tang J, Guo J, Li W, Ling EA, Chu H, Wu Y, Chan YG, Cao Q. RIM3gamma is a postsynaptic protein in the rat central nervous system. J Comp Neurol. 2007;503:501–510. doi: 10.1002/cne.21403. [DOI] [PubMed] [Google Scholar]
- Maclean DM, Bowie D. Transmembrane AMPA receptor regulatory protein regulation of competitive antagonism: a problem of interpretation. J Physiol. 2011;589:5383–5390. doi: 10.1113/jphysiol.2011.219485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Arca S, Coco S, Mainguy G, Schenk U, Alberts P, Bouill é P, Mezzina M, Prochiantz A, Matteoli M, Louvard D, Galli T. A common exocytotic mechanism mediates axonal and dendritic outgrowth. J Neurosci. 2001;21:3830–3838. doi: 10.1523/JNEUROSCI.21-11-03830.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland AC, Hruska M, Coenen AJ, Henkemeyer M, Dalva MB. Trans-synaptic EphB2-ephrin-B3 interaction regulates excitatory synapse density by inhibition of postsynaptic MAPK signaling. Proc Natl Acad Sci U S A. 2010;107:8830–8835. doi: 10.1073/pnas.0910644107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon P, Deane R, Sagare A, Lane SM, Zarcone TJ, O'Dell MR, Yan C, Zlokovic BV, Berk BC. Impaired spine formation and learning in GPCR kinase 2 interacting protein-1 (GIT1) knockout mice. Brain Res. 2010;1317:218–226. doi: 10.1016/j.brainres.2009.11.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, DeProto J, Kaufmann N, Patel BN, Duckworth A, Van Vactor D. Direct observation demonstrates that Liprin-alpha is required for trafficking of synaptic vesicles. Curr Biol. 2005;15:684–689. doi: 10.1016/j.cub.2005.02.061. [DOI] [PubMed] [Google Scholar]
- Mittelstaedt T, Alvaréz-Baron E, Schoch S. RIM proteins and their role in synapse function. Biol Chem. 2010;391:599–606. doi: 10.1515/BC.2010.064. [DOI] [PubMed] [Google Scholar]
- Nishimura Y, Martin CL, Vazquez-Lopez A, Spence SJ, Alvarez-Retuerto AI, Sigman M, Steindler C, Pellegrini S, Schanen NC, Warren ST, Geschwind DH. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet. 2007;16:1682–1698. doi: 10.1093/hmg/ddm116. [DOI] [PubMed] [Google Scholar]
- Ohtsuka T, Takao-Rikitsu E, Inoue E, Inoue M, Takeuchi M, Matsubara K, Deguchi-Tawarada M, Satoh K, Morimoto K, Nakanishi H, Takai Y. Cast: a novel protein of the cytomatrix at the active zone of synapses that forms a ternary complex with RIM1 and munc13-1. J Cell Biol. 2002;158:577–590. doi: 10.1083/jcb.200202083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo CA, Eberhart CG. The neurobiology of autism. Brain Pathol. 2007;17:434–447. doi: 10.1111/j.1750-3639.2007.00102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulain FE, Sobel A. The microtubule network and neuronal morphogenesis: dynamic and coordinated orchestration through multiple players. Mol Cell Neurosci. 2010;43:15–32. doi: 10.1016/j.mcn.2009.07.012. [DOI] [PubMed] [Google Scholar]
- Sann S, Wang Z, Brown H, Jin Y. Roles of endosomal trafficking in neurite outgrowth and guidance. Trends Cell Biol. 2009;19:317–324. doi: 10.1016/j.tcb.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Südhof TC. RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature. 2002;415:321–326. doi: 10.1038/415321a. [DOI] [PubMed] [Google Scholar]
- Schoch S, Mittelstaedt T, Kaeser PS, Padgett D, Feldmann N, Chevaleyre V, Castillo PE, Hammer RE, Han W, Schmitz F, Lin W, Südhof TC. Redundant functions of RIM1alpha and RIM2alpha in Ca(2+)-triggered neurotransmitter release. EMBO J. 2006;25:5852–5863. doi: 10.1038/sj.emboj.7601425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulc J, Wiznerowicz M, Sauvain MO, Trono D, Aebischer P. A versatile tool for conditional gene expression and knockdown. Nat Methods. 2006;3:109–116. doi: 10.1038/nmeth846. [DOI] [PubMed] [Google Scholar]
- Tang BL. Emerging aspects of membrane traffic in neuronal dendrite growth. Biochim Biophys Acta. 2008;1783:169–176. doi: 10.1016/j.bbamcr.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Urbanska M, Blazejczyk M, Jaworski J. Molecular basis of dendritic arborization. Acta Neurobiol Exp. 2008;68:264–288. doi: 10.55782/ane-2008-1695. [DOI] [PubMed] [Google Scholar]
- Uriu Y, Kiyonaka S, Miki T, Yagi M, Akiyama S, Mori E, Nakao A, Beedle AM, Campbell KP, Wakamori M, Mori Y. Rab3-interacting molecule gamma isoforms lacking the Rab3-binding domain induce long lasting currents but block neurotransmitter vesicle anchoring in voltage-dependent P/Q-type Ca2+ channels. J Biol Chem. 2010;285:21750–21767. doi: 10.1074/jbc.M110.101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Südhof TC. Genomic definition of RIM proteins: evolutionary amplification of a family of synaptic regulatory proteins(small star, filled) Genomics. 2003;81:126–137. doi: 10.1016/s0888-7543(02)00024-1. [DOI] [PubMed] [Google Scholar]
- Wang Y, Okamoto M, Schmitz F, Hofmann K, Südhof TC. Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature. 1997;388:593–598. doi: 10.1038/41580. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu X, Biederer T, Südhof TC. A family of RIM-binding proteins regulated by alternative splicing: implications for the genesis of synaptic active zones. Proc Natl Acad Sci U S A. 2002;99:14464–14469. doi: 10.1073/pnas.182532999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidenhofer J, Bowden NA, Scott RJ, Tooney PA. Altered gene expression in the amygdala in schizophrenia: up-regulation of genes located in the cytomatrix active zone. Mol Cell Neurosci. 2006;31:243–250. doi: 10.1016/j.mcn.2005.09.013. [DOI] [PubMed] [Google Scholar]
- Weidenhofer J, Scott RJ, Tooney PA. Investigation of the expression of genes affecting cytomatrix active zone function in the amygdala in schizophrenia: effects of antipsychotic drugs. J Psychiatr Res. 2009;43:282–290. doi: 10.1016/j.jpsychires.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Kim E, Dunah AW, Passafaro M, Valtschanoff JG, Serra-Pagès C, Streuli M, Weinberg RJ, Sheng M. Interaction between GRIP and liprin-alpha/SYD2 is required for AMPA receptor targeting. Neuron. 2002;34:39–52. doi: 10.1016/s0896-6273(02)00640-2. [DOI] [PubMed] [Google Scholar]
- Yao I, Takagi H, Ageta H, Kahyo T, Sato S, Hatanaka K, Fukuda Y, Chiba T, Morone N, Yuasa S, Inokuchi K, Ohtsuka T, Macgregor GR, Tanaka K, Setou M. SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell. 2007;130:943–957. doi: 10.1016/j.cell.2007.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]