

Abstract
Learning new tasks has been associated with increased growth and stabilization of new dendritic spines. We examined whether long-term potentiation (LTP), a key cellular mechanism thought to underlie learning, plays a role in selective stabilization of individual new spines during circuit plasticity. Using two-photon glutamate uncaging, we stimulated nascent spines on dendrites of rat hippocampal CA1 neurons with patterns that induce LTP and then monitored spine survival rates using time-lapse imaging. Remarkably, we found that LTP-inducing stimuli increased the long-term survivorship (>14 h) of individual new spines. Activity-induced new spine stabilization required NMDA receptor activation and was specific for stimuli that induced LTP. Moreover, abrogating CaMKII binding to the NMDA receptor abolished activity-induced new spine stabilization. Our findings demonstrate for the first time that, in addition to enhancing the efficacy of preexisting synapses, LTP-inducing stimuli promote the transition of nascent spines from a short-lived, transient state to a longer-lived, persistent state.
Introduction
Animals regularly adapt their behavior in response to sensory experience. The growth and retraction of dendritic spines and the formation and elimination of spine synapses are thought to be key substrates for the neural circuit plasticity underlying these experience-dependent processes (Holtmaat and Svoboda, 2009; Kasai et al., 2010; Bourne and Harris, 2012; Caroni et al., 2012). Indeed, manipulations that induce experience-dependent circuit plasticity in vivo also increase the rate of new spine growth and stabilization (Trachtenberg et al., 2002; Holtmaat et al., 2006; Hofer et al., 2009), and new spine stabilization is increased during learning (Xu et al., 2009; Yang et al., 2009; Roberts et al., 2010). In addition, new spines are rapidly functional (Zito et al., 2009; Kwon and Sabatini, 2011), and stabilized new spines are incorporated into anatomically mature synapses (Knott et al., 2006; Zito et al., 2009). Critically, not all new spines stabilize, suggesting that specific cues act to stabilize a subset of new spines as circuits are modified during learning.
Considerable evidence supports long-term potentiation (LTP) as a key cellular mechanism that underlies learning and memory (Sigurdsson et al., 2007; Feldman, 2009). The expression of early-phase LTP in hippocampal CA1 pyramidal neurons is proposed to depend on the activation or rapid insertion of postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) into existing synapses (Malinow and Malenka, 2002; Malenka, 2003). The mechanisms underlying expression of late-phase LTP are thought to involve protein synthesis and structural changes (Bailey et al., 1994; Yuste and Bonhoeffer, 2001), including synaptogenesis (Bourne and Harris, 2011). Yet, while time-lapse imaging studies of fluorescently labeled living neurons have demonstrated that LTP-inducing stimuli promote both the outgrowth of new spines (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999; De Roo et al., 2008b; Kwon and Sabatini, 2011) and increases in spine size (Matsuzaki et al., 2004), a direct link between LTP and the long-term stabilization of individual newly formed spine synapses thought to be critical for learning has not been demonstrated.
In this study, we use two-photon glutamate uncaging at individual nascent spines to demonstrate that LTP-inducing stimuli increase the long-term stability of newly formed dendritic spines. We show that enhanced new spine stability is not simply the result of any type of glutamatergic stimulation, but that only a stimulus strong enough to induce LTP will enhance the stability of stimulated new spines. In addition, we show that activity-induced spine stabilization requires N-methyl-d-aspartate receptor (NMDAR) activation and interaction between the NMDA receptor GluN2B subunit and CaMKII. Our results directly link LTP to the long-term stabilization of individual nascent dendritic spines in the hippocampus, and thus suggest that LTP could serve to promote the structural changes supporting the stable alteration of neural circuits during experience-dependent plasticity in the cerebral cortex in vivo.
Materials and Methods
Preparation and transfection of organotypic slice cultures.
Organotypic hippocampal slice cultures were prepared from postnatal day (P) 6 or 7 Sprague Dawley rats or C57BL/6J wild-type or GluN2B L1298A/R1300Q knock-in (KI) mice (Halt et al., 2012) of both sexes as described (Stoppini et al., 1991) in accordance with animal care and use guidelines of the University of California (Davis, CA). Pyramidal neurons were transfected using particle-mediated gene transfer (170 psi) of EGFP (Clontech) as described (Woods and Zito, 2008), except that 20 μg of DNA were coated onto 7 mg of gold particles.
Time-lapse two-photon imaging.
Image stacks (512 × 512 pixels, 1 μm z-steps) of 2–6 secondary and tertiary apical and basal dendritic segments from CA1 pyramidal neurons were acquired on a custom two-photon laser scanning microscope with a pulsed Ti::Sapphire laser (930nm, 0.5–1.5 mW at the sample; Spectra-Physics, Newport). Data acquisition was controlled by ScanImage (Pologruto et al., 2003) written in Matlab (MathWorks). The first time point was acquired in slice culture medium at room temperature (RT), after which the slice was returned to the incubator (35°C). After 1 h, the slice was placed in recirculating, oxygenated artificial cerebrospinal fluid (ACSF) (in mm: 127 NaCl, 25 NaHCO3, 1.2 NaH2PO4, 2.5 KCl, 25 d-glucose, ∼310 mOsm, pH 7.2) with 2 mm Ca2+, 0 mm Mg2+, and 1 μm tetrodotoxin at 33°C. Following new spine identification, 4-methoxy-7-nitroindolinyl-caged l-glutamate (MNI-glutamate) (2.5 – 3.5 mm), 10 mm d-serine, and, when indicated, 10 μm 3-((R)-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP) were added to the bath and allowed to permeate the slice for 5 min before stimulating the new spine. Immediately after stimulation, standard ACSF (2 mm Ca2+, 1 mm Mg2+) was washed in. For long-term time-lapse experiments, the slice was returned to slice culture medium with 100 U/ml penicillin and 0.1 mg/ml streptomycin and then placed in the incubator overnight. The following day, the final time point was imaged in standard ACSF at RT.
Image analysis and quantification.
Only new spines that grew during the initial 1 h time-lapse period were analyzed. If there was uncertainty concerning the status of a spine because of undulations in the dendrite, swellings in the z-axis, or spine movement, the spine was excluded. Spine volume was estimated as the integrated pixel intensity from an ROI positioned around the spine with local background subtracted and normalized to the mean pixel intensity from a local dendrite, as described (Woods et al., 2011). All images shown are maximum projections of 3D image stacks after applying a median filter (3 × 3) to the raw image data.
Calibration of the uncaging stimulus.
CA1 pyramidal neurons were patched (tip resistances, 4–8 MΩ) in whole-cell configuration (Vhold = −65 mV; series resistances, 14–45 MΩ) using Cs-based internal solution (in mm: 120 Cs-methanesulfonate, 10 Na2 phosphocreatine, 3 sodium l-ascorbate, 4 NaCl, 10 HEPES, 4 Mg ATP, 0.3 Na GTP, 0.6 EGTA, 20 CsCl, 0.2 Alexa Fluor 488, 290–300 mOsm, pH 7.2.) in standard ACSF (2 mm Ca2+, 1 mm Mg2+). Uncaging EPSCs (uEPSCs) were recorded from five spines per cell within 50 μm of the soma on secondary and tertiary apical and basal dendrites. Data acquisition was managed with open source Ephus software (http://openwiki.janelia.org/wiki/display/ephus/; (Suter et al., 2010). uEPSC amplitudes from individual spines were quantified as the average (5–7 test pulses at 0.1 Hz) from a 2 ms window centered on the maximum current amplitude within 50 ms following pulse delivery. Uncaging pulse power was adjusted for each individual batch of MNI-glutamate to elicit responses with an average amplitude of ∼10 pA at the soma.
LTP-inducing stimulus.
The LTP-inducing uncaging stimulus consisted of 30 pulses (720–725 nm, 8–12 mW at the sample) of 1 ms duration delivered at 0.5 Hz in the presence of 2.5–3.5 mm MNI-glutamate by parking the beam at a point ∼0.5 μm from the spine head away from the dendrite. Our stimulation protocol was based upon established protocols used by other groups to induce LTP at single spines (Matsuzaki et al., 2004; Govindarajan et al., 2006; Harvey and Svoboda, 2007). The mock stimulus was identical in parameters to the LTP stimulus, except that it was carried out in the absence of MNI-glutamate.
Electrophysiological LTP.
EGFP-expressing CA1 pyramidal neurons were patched in whole-cell configuration as described (see above paragraph, Calibration of the uncaging stimulus), except with a different internal solution (in mm: 135 Cs-methanesulfonate, 10 Na2 phosphocreatine, 3 sodium l-ascorbate, 4 NaCl, 10 HEPES, 4 MgCl, 4 Na2 ATP, 0.4 Na GTP, 300 mOsm, pH 7.2). For each cell, baseline uEPSCs were recorded from two spines (2–10 μm apart; 50–250 μm from the soma on secondary apical branches). The cell was then depolarized to 0 mV and the uncaging LTP stimulus was applied to one spine within 5 min of break-in. Following the uncaging stimulus, uEPSCs were recorded from both spines at 5 min intervals for 20 min. LTP was measured as a significant increase (>25% above baseline) in the average uEPSC amplitude that persisted at least 20 min after the LTP stimulus.
Statistical analysis.
To compare survivorship curves, the log-rank test was used. This test exhibits increased sensitivity (as compared with the rank-sum test) when a significant fraction of total observations within the dataset are right censored. To compare survivorship at individual time points, Barnard's exact test was used when the frequency of observations within any one cell was <5 and Pearson's χ2 test was used when the frequency of observations within all cells was >5. For all parametric variables, analysis was performed using a two-tailed unpaired heteroscedastic Student's t test unless otherwise noted. Error bars represent standard error of the mean (SEM).
Results
Survivorship of new spines increases in response to LTP-inducing stimuli
To directly test whether LTP-inducing stimuli increase the stability of newly formed spines, we used focal photolysis of caged glutamate to stimulate individual new spines in defined patterns, and we monitored the consequences on new spine stabilization rate. Our experimental design (Fig. 1A) consisted of three phases: new spine identification, stimulation of an individual new spine with a protocol that elicits LTP (Matsuzaki et al., 2004); Figure 1B,C), and monitoring spine stability. To identify new spines, we used time-lapse two-photon microscopy to repeatedly image dendrites of GFP-transfected hippocampal pyramidal neurons (Fig. 1D). On each cell, one new spine was stimulated with glutamate uncaging, while other new spines on the same cell served as unstimulated controls. Following the uncaging stimulation, the survivorship of all new spines was monitored using time-lapse imaging.
Figure 1.
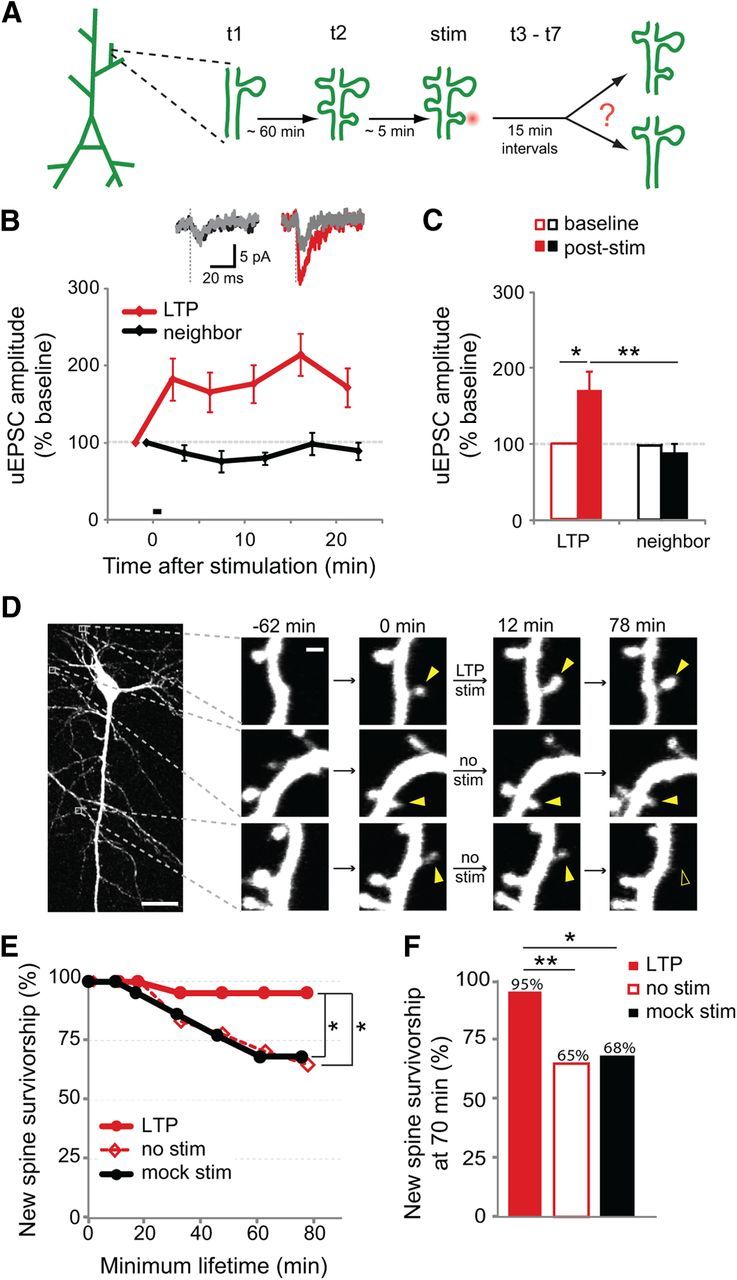
Survivorship of new spines increases in response to LTP-inducing stimuli. A, The experimental design: time-lapse imaging (t1 and t2) to identify new spines was followed by stimulation of an individual new spine using two-photon glutamate uncaging (stim) and then time-lapse imaging (t3– t7) to monitor spine stability. For display purposes, three new spines are shown here on the same dendritic segment (two unstimulated and one stimulated); however, in our data multiple new spines rarely appeared in close proximity on the same dendritic segment. B, The LTP-inducing stimulus (30 pulses at 0.5 Hz paired with postsynaptic depolarization to 0 mV) increased the uEPSC amplitude of stimulated spines (red) but not of neighboring spines (black; p < 0.05 at all poststimulus time points; 12 cells). Inset shows representative traces (mean of 5–7 trials) from individual spines during baseline (gray) and 20 min after uncaging stimulation. C, Twenty minutes after LTP induction, the uEPSC of the stimulated spine (filled red bar) was increased over baseline (open red bar; p < 0.05; paired t test) and also compared to that of the neighboring spine (filled black bar; p < 0.01; unpaired t test). D, An EGFP-transfected CA1 pyramidal neuron in organotypic slice culture (P7 + 9 DIV) and examples of three new spines (solid yellow arrowheads), one of which was selected for stimulation. One unstimulated new spine retracted (open yellow arrowhead). Scale bars, 25 μm (whole cell image); 1 μm (dendrite images). E, The LTP-inducing stimulus (30 pulses at 0.5 Hz in nominal Mg2+) increased the survivorship of stimulated new spines (solid red circles; 21 spines) as compared to unstimulated new spines on the same cells (open red diamonds; 54 spines; p < 0.05, log-rank test) or mock-stimulated new spines (solid black circles; 22 spines; p < 0.05, log-rank test). F, New spine survivorship at 70 min was increased following LTP stimulus (solid red bar) as compared to unstimulated new spines on the same cells (open red bar; p < 0.01, Barnard's exact test) or mock-stimulated new spines (solid black bar; p < 0.05, Barnard's exact test).
We found that LTP-inducing stimuli significantly increased new spine survivorship over the baseline survivorship rate of unstimulated new spines on the same cells (p < 0.05; Fig. 1E). Indeed, only 65% of unstimulated new spines were still present 70 min after initial outgrowth, whereas 95% of new spines exposed to LTP stimulation survived (p < 0.01; Fig. 1F). As a control, we examined the stabilization rate of new spines that were exposed to a mock LTP stimulus (the same laser protocol and experimental conditions in the absence of MNI-glutamate). In contrast with LTP-stimulated new spines, mock-stimulated new spines did not exhibit increased survivorship (Fig. 1E, F), demonstrating that the stabilizing effect of the LTP stimulus was due to the released glutamate and not to photo damage from the laser pulse used for uncaging. Thus, we conclude that the LTP stimulus increased the stabilization rate of new spines.
To ensure that the increased survivorship rate of stimulated new spines did not result from a selection bias in the population of new spines chosen for LTP stimulation, we compared the populations of new spines in our studies across several parameters, including initial spine volume, spine depth in the slice, distance from the soma, soma depth in slice, and location on basal or apical dendrites. Before the LTP stimulus, new spines targeted for LTP stimulation did not differ from those targeted for mock stimulation or from unstimulated new spines across any of these parameters (p > 0.26 for all comparisons; data not shown). In contrast, after the LTP stimulus we found that stimulated new spines were significantly enlarged (160 ± 17%) compared with unstimulated neighboring spines (109 ± 5%; p < 0.01) or with mock-stimulated new spines (95 ± 12%; p < 0.05; Fig. 2A,B), consistent with previous results demonstrating that LTP at individual mature dendritic spines is associated with spine volume increases (Matsuzaki et al., 2004). We conclude that although our spine populations were initially indistinguishable, the LTP stimulus induced an enlargement of stimulated new spines.
Figure 2.

New spines enlarge in response to LTP-inducing stimuli. A, New spines (solid yellow arrowheads at 0 min) and unstimulated neighboring spines (solid white arrowheads) on cells exposed to LTP stimulation (LTP stim) or mock stimulation (mock stim). Scale bar, 1 μm. B, A significant enlargement was observed for new spines stimulated with the LTP stimulus (solid red bar; 42 spines), as compared to unstimulated neighboring spines on the same cells (open black bar; 84 spines; p < 0.01, t test) or mock stimulated new spines (solid black bar, 21 spines; p < 0.05, t test).
Uncaging-induced new spine stabilization is specific to LTP-inducing stimuli
We examined whether new spine stabilization is downstream of any glutamatergic activation or whether it is induced specifically by stimuli that induce LTP. First, we reduced the strength of our uncaging stimulus (Fig. 3A) below the threshold required both for LTP (Fig. 3B) and enlargement (Fig. 3C) at single spines and tested whether this weaker stimulus would continue to support uncaging-induced new spine stabilization. Notably, we found that the weaker stimulus, which did not induce LTP, also failed to enhance new spine survivorship (Fig. 3D,E). Second, we removed the “pairing” component of our uncaging LTP protocols by eliminating the postsynaptic depolarization in our electrophysiological LTP protocol (Fig. 3F–H) and by performing our imaging only uncaging LTP protocol in the presence of physiological levels of extracellular Mg2+ (1 mm; Fig. 3I, J). In the absence of simultaneous postsynaptic depolarization, our uncaging LTP stimulus no longer induced potentiation of uEPSC amplitude (Fig. 3G) or increased spine size (Fig. 3H). Similarly, the presence of physiological levels of extracellular Mg2+ prevented uncaging-induced stabilization of new spines (Fig. 3I, J). These data strongly support that coincident glutamate binding and relief of the Mg2+ blockade of NMDARs is necessary not only for LTP, but also for the uncaging-induced increase in new spine survivorship. Thus, we conclude that uncaging-induced new spine stabilization is specific to the LTP-inducing stimuli.
Figure 3.
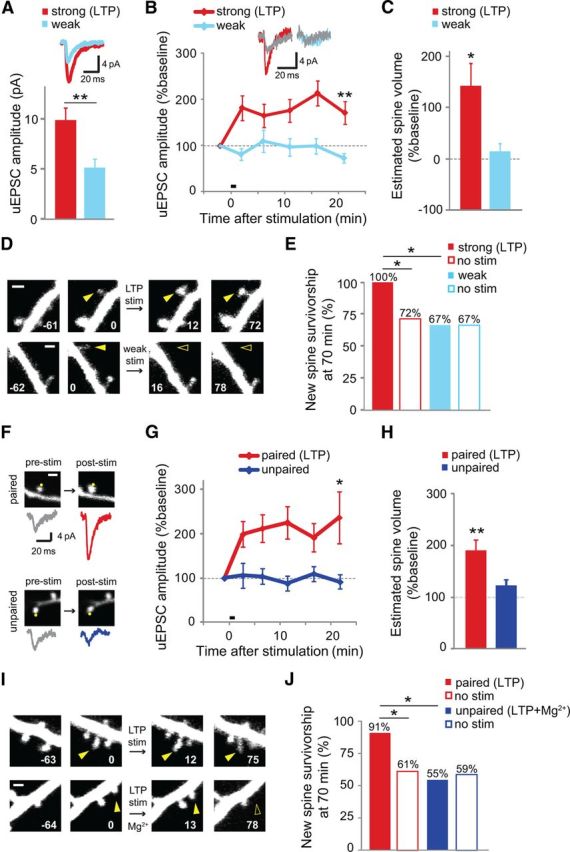
Uncaging-induced new spine stabilization is specific to LTP-inducing stimuli. A, uEPSCs evoked by the weak stimulus (blue; 2.5 mm MNI-glutamate; 5.1 ± 0.9 pA; 15 spines, 3 cells) were smaller than those evoked by the strong (LTP) stimulus (red; 3.5 mm MNI-glutamate; 9.9 ± 1.2 pA; 20 spines, 4 cells; p < 0.01). Inset shows average uEPSC trace from each condition. B, The weak stimulus did not induce LTP (blue; 13 spines, 13 cells), in contrast to the strong (LTP) stimulus, which did induce LTP (red; data from Fig. 1B). Inset shows representative traces (mean of 5–7 trials) from individual spines during baseline (gray) and 20 min after uncaging stimulation for the strong (red) and weak (blue) stimuli. C, No significant spine enlargement was observed at the first time point (∼5 min) after the weak stimulus (blue; 8 spines; p > 0.37), whereas the strong stimulus caused spine enlargement (red; 12 spines; p < 0.05, t test). D, New spines (solid yellow arrowheads at 0 min) on cells exposed to strong (LTP stim) or weak stimulation. Time stamps are in min. Scale bar = 1 μm. E, No increase in new spine survivorship was observed in response to the weak stimulus (solid blue bar; 6 spines) over unstimulated new spines (open blue bar; 18 spines; p > 0.9, Barnard's exact test). As expected, new spine survivorship increased following the strong (LTP) stimulus (solid red bar; 12 spines) over that of unstimulated new spines (open red bar; 13 spines; p < 0.05, Barnard's exact test). F, Images of spines and corresponding uEPSC traces before (pre-stim) and 20 min after (post-stim) delivery of the uncaging stimulus when paired with depolarization to 0 mV (paired) or held at −70 mV (unpaired). Scale ba, 1 μm. G, Pairing stimulation with depolarization to 0 mV resulted in robust potentiation of the uEPSC amplitude of the target spine (red; 26 spines, 26 cells), whereas potentiation was not observed in the unpaired condition (blue; 14 spines, 14 cells). H, Spines in the pairing condition (red; 26 spines, 26 cells) exhibited significant and persistent enlargement compared to baseline and compared to spines in the unpaired condition (blue; 14 spines, 14 cells) 20 min after stimulation. I, New spines (solid yellow arrowheads at 0 min) on cells exposed to the LTP stimulation in the absence or presence of Mg2+. J, New spines exposed to the LTP stimulus in the absence of Mg2+ (LTP; solid red bar; 11 spines, 11 cells) were significantly more stable than unstimulated new spines on the same cells (no stim; open red bar; 59 spines, 11 cells; p < 0.05, Barnard's exact test). In the presence of Mg2+, the uncaging stimulus had no effect on the stability of stimulated new spines (LTP + Mg2+; solid blue bar; 11 spines, 11 cells) compared with unstimulated new spines on those same cells (no stim; open blue bar; 58 spines, 11 cells; p > 0.4, Barnard's exact test).
LTP-induced new spine stabilization is long-lasting
Because we observed a significant volume increase in stimulated new spines (Fig. 2), we wondered whether the LTP stimulus might only increase new spine survivorship over a short time scale due to a transient increase in volume, while leaving long-term survivorship unaffected. To address this question, we repeated our experiment with an additional time-point at >14 h after new spine outgrowth (Fig. 4A,B), a time that lies well outside the window over which any transient influence would be expected. Remarkably, even at this long-term time point (range: 14–31 h; average: 20 h) new spine survivorship was 85% higher for stimulated new spines than for unstimulated new spines on the same cells (p < 0.05; Fig. 4C). We conclude that an LTP stimulus is capable of increasing the long-term stability of new spines.
Figure 4.

LTP-induced new spine stabilization is long lasting. A, A new spine (solid yellow arrowhead at 0 min) was stimulated with the LTP stimulus (LTP stim). Scale bars, 50 μm (whole cell image); 1 μm (dendrite images). B, After incubation overnight in culture medium, the new spine (solid yellow arrowhead) was still present. C, Even at the long-term time point (≥ 14 h), survivorship of stimulated new spines (solid red bar; 22 spines) was significantly higher than that of unstimulated new spines on the same cells (open red bar; 63 spines; p < 0.05, Pearson's χ2 test).
Larger new spines show enhanced survivorship rates
Because stimulated new spines enlarge and are also more stable, we examined whether new spine size and stabilization rate were correlated. Indeed, when we classified unstimulated new spines as “large,” “medium,” or “small” based on their relative size at the time of initial identification, both large- and medium-sized new spines were more stable than small new spines (Fig. 5A). In addition, when we divided new spines into two groups based on survivorship at 70 min (“persistent” if present and “transient” if lost), we found that spine size at the time of initial observation differed significantly between new spines that persisted and those that retracted (Fig. 5B). In contrast, no significant differences were found between the two groups for the other parameters that we examined, including distance from soma and depth in slice (Fig. 5B). We conclude that new spine size is a strong predictor of new spine survivorship.
Figure 5.

Larger new spines show enhanced survivorship rates. A, Survivorship of medium and large new spines is significantly higher than that of small new spines (p < 0.01, Pearson's χ2 < 36 a.u. = small, 41 spines, 26 cells; 36–60 a.u. = medium, 33 spines, 22 cells; >60 a.u. = large, 36 spines, 23 cells). B, Initial spine size of persistent new spines (open bars) is larger than that of transient new spines (solid bars; p < 0.01, unpaired t test; 60 persistent, 50 transient spines, 37 cells). Distance from the soma (p = 0.9) and spine depth in the slice (p = 0.5) were not different between the two groups. All values are relative to the mean. C, Survivorship of small stimulated (solid bars) new spines is higher than that of small unstimulated (open bars) new spines (p < 0.05, Barnard's exact test; bin sizes and unstimulated (no stim) spine numbers as in A; stimulated: small, 9 spines, 9 cells; medium, 20 spines, 20 cells; large, 13 spines, 13 cells). D, Volume of new spines before (pre) and after (post) stimulation to illustrate volume change of persistent (red lines; 40 spines on 40 cells) and transient (gray lines; 2 spines on 2 cells) new spines.
The observation that larger new spines are more stable than smaller new spines led us to examine whether the uncaging LTP stimulus was preferentially enhancing the stabilization rate of smaller new spines by transitioning them from a smaller, more transient bracket into a larger, more stable bracket. We therefore assessed the effect of the LTP stimulus on new spines of different volumes. We classified all LTP-stimulated new spines as either large, medium, or small based on their relative initial size and then compared the survivorship of stimulated and unstimulated new spines. For medium (p = 0.084) and large (p = 0.075) new spines, the LTP stimulus modestly but not significantly enhanced new spine survivorship. As predicted, the LTP stimulus had the largest and most significant (p < 0.05) effect on the survivorship of spines from the smallest size class (Fig. 5C), consistent with the idea that a size increase would benefit the smallest spines the most by bumping them into a larger, more stable class. In fact, the majority of stimulated new spines that stabilized became enlarged by >25% in response to the uncaging stimulus (25/40), and those stimulated new spines that did not stabilize shrank in the time point following stimulation (2/2; Fig. 5D). We conclude that LTP-induced spine size increases likely play an important role in enhancing new spine survivorship.
NMDAR activation and interaction of NMDARs with CaMKII are required for LTP-induced new spine stabilization
Which glutamatergic signaling pathways play a role in LTP-induced new spine stabilization? Because NMDAR activation contributes to LTP induction in CA1 pyramidal neurons (Grover and Teyler, 1990; Feldman, 2009), we tested whether uncaging-induced new spine stabilization also depends on NMDAR activation. In these experiments, CPP, a selective antagonist of NMDARs, was added immediately before and washed out promptly following delivery of the uncaging stimulus. New spines that were exposed to the LTP stimulus in the presence of CPP did not exhibit increased survivorship relative to unstimulated control spines on the same cells (p = 0.4; Fig. 6A,B), demonstrating that NMDAR activation is necessary for uncaging-induced new spine stabilization.
Figure 6.

NMDAR activation and interaction of NMDARs with CaMKII are required for LTP-induced new spine stabilization. A, New spines (solid yellow arrowheads at 0 min) were stimulated in the absence (LTP stim) or presence (LTP stim + CPP) of CPP. Time stamps are in minutes. Scale bar, 1 μm. B, In the presence of CPP, no significant increase in survivorship was observed for stimulated new spines (solid gray bar; 22 spines) over unstimulated new spines (open gray bar; 43 spines; p = 0.4, Barnard's exact test). As expected, in the absence of CPP, survivorship of stimulated new spines (solid red bar; 21 spines) increased as compared to that of unstimulated new spines (open red bar; 56 spines; p < 0.05, Barnard's exact test). C, New spines (solid yellow arrowheads at 0 min) on dendrites from CA1 neurons in hippocampal slice cultures from WT and GluN2B L1298/R1300Q knock-in mice (KI) were stimulated with the uncaging LTP stimulus (LTP stim). Time stamps are in minutes. D, No significant increase in survivorship was observed in GluN2B KI mice for stimulated new spines (solid orange bar; 8 spines) over unstimulated new spines (open orange bar; 30 spines; p = 0.4, Barnard's exact test). As expected, survivorship of stimulated new spines (solid red bar; 6 spines) increased in WT mice as compared to unstimulated new spines (open red bar; 30 spines; p < 0.02).
Downstream of NMDARs, CaMKII binding to the NMDAR subunit, GluN2B, has been shown to be important for induction and maintenance of LTP (Lisman et al., 2002; Barria and Malinow, 2005; Zhou et al., 2007; Sanhueza et al., 2011) and activity-dependent new spine outgrowth (Hamilton et al., 2012); interruption of this interaction causes decreased spine density (Gambrill and Barria, 2011). Moreover, CaMKII activation is important for new spine stabilization during experience-dependent plasticity (Wilbrecht et al., 2010). We therefore examined whether CaMKII interaction with GluN2B is also important for uncaging-induced spine stabilization using GluN2B-L1298A/R1300Q knock-in (GluN2B KI) mice (Halt et al., 2012). Each of the L1298A and R1300Q mutations reduce GluN2B interaction with CaMKII by over 85% in vitro (Strack et al., 2000), and together they completely block the activity-dependent increase in GluN2B-CaMKII interaction in vivo (Halt et al., 2012). In hippocampal slices from GluN2B KI mice, new spines exposed to the LTP stimulus did not exhibit increased survivorship over unstimulated new spines on the same cells (p = 0.42; Fig. 6C,D); in contrast, new spines in slices from WT mice exhibited a significant increase in survivorship in response to the LTP stimulus (p < 0.05; Fig. 6C,D). Thus, interaction between the GluN2B subunit of the NMDAR and CaMKII is necessary for LTP-induced stabilization of new dendritic spines.
Discussion
LTP and new spine stabilization
Although both LTP (Sigurdsson et al., 2007; Feldman, 2009) and new spine stabilization (Xu et al., 2009; Yang et al., 2009; Roberts et al., 2010) have been independently associated with learning, our data are the first to demonstrate a direct link between LTP-inducing stimuli and the long-term stabilization of individual nascent dendritic spines thought to underlie learning and memory. Here, we show that the long-term survivorship of individual new spines is significantly enhanced following LTP-inducing stimulation. Because LTP stabilizes nascent dendritic spines, it might be expected that LTP would be accompanied by an increase in spine density. However, several studies have found no long-term increase in spine density associated with LTP (for review, Yuste and Bonhoeffer, 2001; Bosch and Hayashi, 2012). Indeed, recent studies from the mature hippocampus (Bourne and Harris, 2011; Wosiski-Kuhn and Stranahan, 2012) show only a transient increase in spine density after LTP induction. It is likely that homeostatic mechanisms leading to the compensatory elimination of less active dendritic spines (De Roo et al., 2008b; Bourne and Harris, 2011) act to hold spine density relatively constant and could be responsible for the absence of long-term changes in spine density associated with LTP.
How does the LTP stimulus drive increased long-term survivorship of new dendritic spines? New spine survivorship is not well fit by a single exponential; instead, there is an initial rapid decline in survivorship, followed by a much slower multicomponent decay (Trachtenberg et al., 2002). One possibility is that the LTP stimulus is responsible for transitioning the new spine from an initial transient state characterized by the rapid survivorship decay into a longer-lived state. We therefore wondered how the long-term stability of our stimulated new spines would compare with that of unstimulated new spines that were present for at least1 h, and therefore likely past the initial phase of rapid decline. We found that the long-term survivorship of stimulated new spines (50%) was not significantly different from that of unstimulated new spines that had also survived for at least 1 h (42%; p = 0.4). Thus, our results support a model in which LTP-inducing stimuli promote the transition of a new spine from a shorter-lived, transient state to a longer-lived, persistent state.
We found that the LTP stimulus caused a significant volume increase at new spines, consistent with previous results at mature spines (Fifková and Van Harreveld, 1977; Lang et al., 2004; Matsuzaki et al., 2004; Okamoto et al., 2004). We also found that new spine size was correlated with stabilization rate; larger new spines stabilized at a higher rate. Because of this relationship, we predicted that the LTP stimulus might preferentially act to stabilize the smallest new spines by bumping them from a smaller, more transient state to a larger, more stable state. Indeed, our results confirmed this prediction (Fig. 5); however, it is important to note that “large” new spines are still considerably smaller than an average mature spine. How might an increase in spine volume lead to an increase in stabilization rate? One possibility is that increased volume is associated with the insertion of AMPARs or postsynaptic density (PSD) proteins, which have demonstrated roles in stabilizing the structural changes accompanying LTP in mature spines (Park et al., 2006; Kopec et al., 2007), or whose arrival has been associated with stabilization of new spines (De Roo et al., 2008a). An alternative possibility is that an increase in spine volume could further survival long enough to solidify adhesive and/or signaling interactions with the presynaptic terminal.
Cellular mechanisms of new spine stabilization
Our data suggest that LTP-inducing presynaptic activity plays an important role in the stabilization of dendritic spines. We show that new spines are capable of discriminating different patterns of glutamatergic input and therefore of identifying appropriate presynaptic partners based upon the specific pattern of presynaptic activity. However, we cannot exclude the possibility that presynaptic partners also play an activity-independent role in spine stabilization. Indeed, N-cadherin has been shown to be important for the persistence of both LTP and spine enlargement in mature spines (Okamura et al., 2004; Bozdagi et al., 2010; Mendez et al., 2010). Furthermore, axons within CA1 occupy roughly one-third of the tissue volume (Chklovskii et al., 2002), and a large number of unconnected axons pass sufficiently close to the dendritic shaft to interact with dendritic spines (Mishchenko et al., 2010). It is therefore reasonable to assume that the majority of imaged new spines in our experiments could have been engaged in adhesive interactions with nearby unlabeled axons. Adhesive interactions at the early stages of new spine development may simply be required to bring the presynaptic bouton and dendritic spine close enough to allow for synaptic communication and LTP. Alternatively, long-term stabilization in response to LTP-inducing stimuli might require adhesive interactions between the axon and dendrite. Future work combining systematic knockdown of synaptic adhesion molecules with uncaging LTP stimulation will be important for answering these questions.
We found that uncaging-induced new spine stabilization is input specific in that stimulation of an individual new spine was sufficient for stabilization of that spine. Indeed, our uncaging stimulation protocols were carried out in the presence of TTX to silence spontaneous activity, and there was no need for stimulation of multiple synapses to enhance stabilization of new spines. However, it is possible, and perhaps likely, that new spine stabilization will be influenced by activity at neighboring synapses. In fact, it has been shown that new spines preferentially grow near active synapses (De Roo et al., 2008b) and that LTP at one spine on a dendrite reduces the threshold for induction of LTP at neighboring spines (Harvey and Svoboda, 2007). Cross-talk between neighboring spines could also be at play during the stabilization of nascent spine synapses and would favor the clustering of correlated inputs within the dendritic arbor (DeBello, 2008; Govindarajan et al., 2011). Recent in vivo imaging experiments support such a clustering mechanism for stabilization of new spines during learning of repetitive tasks (Fu et al., 2012).
Molecular mechanisms of new spine stabilization
Our work demonstrates for the first time that glutamatergic stimulation alone is sufficient to enhance the stabilization rate of nascent spines. Which downstream signaling pathways are important for LTP-induced new spine stabilization? We have established a requirement for NMDAR activation and the interaction between GluN2B subunit and CaMKII. At preexisting synapses, activity induces the translocation of CaMKII to dendritic spines (Shen and Meyer, 1999; Otmakhov et al., 2004), where it interacts with the GluN2B subunit (Strack and Colbran, 1998; Leonard et al., 1999). Our data suggest that this interaction may serve as an important step in mediating the activity-dependent stabilization of nascent spines. Once in the spine, CaMKII plays diverse roles that include regulation of the PSD composition (Steiner et al., 2008), receptor trafficking (Chung et al., 2004; Correia et al., 2008), the activity or localization of other enzymes or signaling molecules (Illario et al., 2003; Oh et al., 2004; Tolias et al., 2005; Xie et al., 2007; Bingol et al., 2010), and the actin cytoskeleton (Okamoto et al., 2007). In addition, CaMKII itself can act as a structural element (Pi et al., 2010). In mature spines, LTP in hippocampal CA1 promotes translocation of polyribosomes into spines (Ostroff et al., 2002; Bourne et al., 2007) and increases the size of the PSD (Bourne and Harris, 2011), and in new spines similar processes could promote stabilization. Indeed, LTP could serve to drive spine maturation via the recruitment of specific molecules to nascent spine synapses.
Determining the relative importance of specific signaling pathways (Saneyoshi et al., 2010) will depend on defining precisely how the molecular composition and signaling mechanisms of new spines differ from those of mature spines. The implementation of new high-throughput methods, such as array tomography (Micheva and Smith, 2007), combined with careful examination of individual nascent synapses using retrospective immunoelectron microscopy (Zito et al., 1999) and live monitoring of signaling at individual nascent spines in response to LTP (Murakoshi and Yasuda, 2012) will play a key role in defining those precise molecular mechanisms that underlie new spine stabilization during experience-dependent plasticity.
Footnotes
This work was supported by National Alliance for Research on Schizophrenia and Depression, National Science Foundation CAREER Award 0845285, National Institutes of Health Training Grant T32 DC008072, and a Burroughs Wellcome Career Award in the Biomedical Sciences. We thank I. Stein and J. Hell for providing GluN2B KI mice; L. Boudewyn, J. Heiner, and S. Mikula for help with experiments and analysis; T. O'Connor for expert programming; L. Borodinsky, J. Trimmer, J. Trachtenberg, H.B. Kwon, and our colleagues at the UC Davis Center for Neuroscience for valuable discussion.
References
- Bailey CH, Alberini C, Ghirardi M, Kandel ER. Molecular and structural changes underlying long-term memory storage in Aplysia. Adv Second Messenger Phosphoprotein Res. 1994;29:529–544. doi: 10.1016/s1040-7952(06)80034-0. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Bingol B, Wang CF, Arnott D, Cheng D, Peng J, Sheng M. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell. 2010;140:567–578. doi: 10.1016/j.cell.2010.01.024. [DOI] [PubMed] [Google Scholar]
- Bosch M, Hayashi Y. Structural plasticity of dendritic spines. Curr Opin Neurobiol. 2012;22:383–388. doi: 10.1016/j.conb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Coordination of size and number of excitatory and inhibitory synapses results in a balanced structural plasticity along mature hippocampal CA1 dendrites during LTP. Hippocampus. 2011;21:354–373. doi: 10.1002/hipo.20768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Nanoscale analysis of structural synaptic plasticity. Curr Opin Neurobiol. 2012;22:372–382. doi: 10.1016/j.conb.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne JN, Sorra KE, Hurlburt J, Harris KM. Polyribosomes are increased in spines of CA1 dendrites 2 h after the induction of LTP in mature rat hippocampal slices. Hippocampus. 2007;17:1–4. doi: 10.1002/hipo.20238. [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Wang XB, Nikitczuk JS, Anderson TR, Bloss EB, Radice GL, Zhou Q, Benson DL, Huntley GW. Persistence of coordinated long-term potentiation and dendritic spine enlargement at mature hippocampal CA1 synapses requires N-cadherin. J Neurosci. 2010;30:9984–9989. doi: 10.1523/JNEUROSCI.1223-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroni P, Donato F, Muller D. Structural plasticity upon learning: regulation and functions. Nat Rev Neurosci. 2012;13:478–490. doi: 10.1038/nrn3258. [DOI] [PubMed] [Google Scholar]
- Chklovskii DB, Schikorski T, Stevens CF. Wiring optimization in cortical circuits. Neuron. 2002;34:341–347. doi: 10.1016/s0896-6273(02)00679-7. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Huang YH, Lau LF, Huganir RL. Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J Neurosci. 2004;24:10248–10259. doi: 10.1523/JNEUROSCI.0546-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia SS, Bassani S, Brown TC, Lisé MF, Backos DS, El-Husseini A, Passafaro M, Esteban JA. Motor protein-dependent transport of AMPA receptors into spines during long-term potentiation. Nat Neurosci. 2008;11:457–466. doi: 10.1038/nn2063. [DOI] [PubMed] [Google Scholar]
- DeBello WM. Micro-rewiring as a substrate for learning. Trends Neurosci. 2008;31:577–584. doi: 10.1016/j.tins.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Roo M, Klauser P, Mendez P, Poglia L, Muller D. Activity-dependent PSD formation and stabilization of newly formed spines in hippocampal slice cultures. Cereb Cortex. 2008a;18:151–161. doi: 10.1093/cercor/bhm041. [DOI] [PubMed] [Google Scholar]
- De Roo M, Klauser P, Muller D. LTP promotes a selective long-term stabilization and clustering of dendritic spines. PLoS Biol. 2008b;6:e219. doi: 10.1371/journal.pbio.0060219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Feldman DE. Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci. 2009;32:33–55. doi: 10.1146/annurev.neuro.051508.135516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fifková E, Van Harreveld A. Long-lasting morphological changes in dendritic spines of dentate granular cells following stimulation of the entorhinal area. J Neurocytol. 1977;6:211–230. doi: 10.1007/BF01261506. [DOI] [PubMed] [Google Scholar]
- Fu M, Yu X, Lu J, Zuo Y. Repetitive motor learning induces coordinated formation of clustered dendritic spines in vivo. Nature. 2012;483:92–95. doi: 10.1038/nature10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambrill AC, Barria A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc Natl Acad Sci U S A. 2011;108:5855–5860. doi: 10.1073/pnas.1012676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan A, Kelleher RJ, Tonegawa S. A clustered plasticity model of long-term memory engrams. Nat Rev Neurosci. 2006;7:575–583. doi: 10.1038/nrn1937. [DOI] [PubMed] [Google Scholar]
- Govindarajan A, Israely I, Huang SY, Tonegawa S. The dendritic branch is the preferred integrative unit for protein synthesis-dependent LTP. Neuron. 2011;69:132–146. doi: 10.1016/j.neuron.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, Hell JW. CaMKII binding to GluN2B is critical during memory consolidation. EMBO J. 2012;31:1203–1216. doi: 10.1038/emboj.2011.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton AM, Oh WC, Vega-Ramirez H, Stein IS, Hell JW, Patrick GN, Zito K. Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron. 2012;74:1023–1030. doi: 10.1016/j.neuron.2012.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450:1195–1200. doi: 10.1038/nature06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer SB, Mrsic-Flogel TD, Bonhoeffer T, Hübener M. Experience leaves a lasting structural trace in cortical circuits. Nature. 2009;457:313–317. doi: 10.1038/nature07487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Wilbrecht L, Knott GW, Welker E, Svoboda K. Experience-dependent and cell-type-specific spine growth in the neocortex. Nature. 2006;441:979–983. doi: 10.1038/nature04783. [DOI] [PubMed] [Google Scholar]
- Illario M, Cavallo AL, Bayer KU, Di Matola T, Fenzi G, Rossi G, Vitale M. Calcium/calmodulin-dependent protein kinase II binds to Raf-1 and modulates integrin-stimulated ERK activation. J Biol Chem. 2003;278:45101–45108. doi: 10.1074/jbc.M305355200. [DOI] [PubMed] [Google Scholar]
- Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010;33:121–129. doi: 10.1016/j.tins.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Knott GW, Holtmaat A, Wilbrecht L, Welker E, Svoboda K. Spine growth precedes synapse formation in the adult neocortex in vivo. Nat Neurosci. 2006;9:1117–1124. doi: 10.1038/nn1747. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Real E, Kessels HW, Malinow R. GluR1 links structural and functional plasticity at excitatory synapses. J Neurosci. 2007;27:13706–13718. doi: 10.1523/JNEUROSCI.3503-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HB, Sabatini BL. Glutamate induces de novo growth of functional spines in developing cortex. Nature. 2011;474:100–104. doi: 10.1038/nature09986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang C, Barco A, Zablow L, Kandel ER, Siegelbaum SA, Zakharenko SS. Transient expansion of synaptically connected dendritic spines upon induction of hippocampal long-term potentiation. Proc Natl Acad Sci U S A. 2004;101:16665–16670. doi: 10.1073/pnas.0407581101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-d-aspartate receptor. Proc Natl Acad Sci U S A. 1999;96:3239–3244. doi: 10.1073/pnas.96.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Malenka RC. Synaptic plasticity and AMPA receptor trafficking. Ann N Y Acad Sci. 2003;1003:1–11. doi: 10.1196/annals.1300.001. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez P, De Roo M, Poglia L, Klauser P, Muller D. N-cadherin mediates plasticity-induced long-term spine stabilization. J Cell Biol. 2010;189:589–600. doi: 10.1083/jcb.201003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Smith SJ. Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 2007;55:25–36. doi: 10.1016/j.neuron.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishchenko Y, Hu T, Spacek J, Mendenhall J, Harris KM, Chklovskii DB. Ultrastructural analysis of hippocampal neuropil from the connectomics perspective. Neuron. 2010;67:1009–1020. doi: 10.1016/j.neuron.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi H, Yasuda R. Postsynaptic signaling during plasticity of dendritic spines. Trends Neurosci. 2012;35:135–143. doi: 10.1016/j.tins.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JS, Manzerra P, Kennedy MB. Regulation of the neuron-specific Ras GTPase-activating protein, synGAP, by Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2004;279:17980–17988. doi: 10.1074/jbc.M314109200. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Narayanan R, Lee SH, Murata K, Hayashi Y. The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc Natl Acad Sci U S A. 2007;104:6418–6423. doi: 10.1073/pnas.0701656104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Tanaka H, Yagita Y, Saeki Y, Taguchi A, Hiraoka Y, Zeng LH, Colman DR, Miki N. Cadherin activity is required for activity-induced spine remodeling. J Cell Biol. 2004;167:961–972. doi: 10.1083/jcb.200406030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostroff LE, Fiala JC, Allwardt B, Harris KM. Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron. 2002;35:535–545. doi: 10.1016/s0896-6273(02)00785-7. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Tao-Cheng JH, Carpenter S, Asrican B, Dosemeci A, Reese TS, Lisman J. Persistent accumulation of calcium/calmodulin-dependent protein kinase II in dendritic spines after induction of NMDA receptor-dependent chemical long-term potentiation. J Neurosci. 2004;24:9324–9331. doi: 10.1523/JNEUROSCI.2350-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, Ehlers MD. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi HJ, Otmakhov N, El Gaamouch F, Lemelin D, De Koninck P, Lisman J. CaMKII control of spine size and synaptic strength: role of phosphorylation states and nonenzymatic action. Proc Natl Acad Sci U S A. 2010;107:14437–14442. doi: 10.1073/pnas.1009268107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K. ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online. 2003;2:13. doi: 10.1186/1475-925X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TF, Tschida KA, Klein ME, Mooney R. Rapid spine stabilization and synaptic enhancement at the onset of behavioural learning. Nature. 2010;463:948–952. doi: 10.1038/nature08759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saneyoshi T, Fortin DA, Soderling TR. Regulation of spine and synapse formation by activity-dependent intracellular signaling pathways. Curr Opin Neurobiol. 2010;20:108–115. doi: 10.1016/j.conb.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, Fernandez-Villalobos G, Stein IS, Kasumova G, Zhang P, Bayer KU, Otmakhov N, Hell JW, Lisman J. Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J Neurosci. 2011;31:9170–9178. doi: 10.1523/JNEUROSCI.1250-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Sigurdsson T, Doyère V, Cain CK, LeDoux JE. Long-term potentiation in the amygdala: a cellular mechanism of fear learning and memory. Neuropharmacology. 2007;52:215–227. doi: 10.1016/j.neuropharm.2006.06.022. [DOI] [PubMed] [Google Scholar]
- Steiner P, Higley MJ, Xu W, Czervionke BL, Malenka RC, Sabatini BL. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron. 2008;60:788–802. doi: 10.1016/j.neuron.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Strack S, Colbran RJ. Autophosphorylation-dependent targeting of calcium/ calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 1998;273:20689–20692. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 2000;275:23798–23806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
- Suter BA, O'Connor T, Iyer V, Petreanu LT, Hooks BM, Kiritani T, Svoboda K, Shepherd GM. Ephus: multipurpose data acquisition software for neuroscience experiments. Front Neural Circuits. 2010;4:100. doi: 10.3389/fncir.2010.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45:525–538. doi: 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- Wilbrecht L, Holtmaat A, Wright N, Fox K, Svoboda K. Structural plasticity underlies experience-dependent functional plasticity of cortical circuits. J Neurosci. 2010;30:4927–4932. doi: 10.1523/JNEUROSCI.6403-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods G, Zito K. Preparation of gene gun bullets and biolistic transfection of neurons in slice culture. J Vis Exp. 2008;13:pii:675. doi: 10.3791/675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods GF, Oh WC, Boudewyn LC, Mikula SK, Zito K. Loss of PSD-95 enrichment is not a prerequisite for spine retraction. J Neurosci. 2011;31:12129–12138. doi: 10.1523/JNEUROSCI.6662-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosiski-Kuhn M, Stranahan AM. Transient increases in dendritic spine density contribute to dentate gyrus long-term potentiation. Synapse. 2012;66:661–664. doi: 10.1002/syn.21545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Srivastava DP, Photowala H, Kai L, Cahill ME, Woolfrey KM, Shum CY, Surmeier DJ, Penzes P. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. 2007;56:640–656. doi: 10.1016/j.neuron.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Yu X, Perlik AJ, Tobin WF, Zweig JA, Tennant K, Jones T, Zuo Y. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature. 2009;462:915–919. doi: 10.1038/nature08389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, Gan WB. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462:920–924. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Takahashi E, Li W, Halt A, Wiltgen B, Ehninger D, Li GD, Hell JW, Kennedy MB, Silva AJ. Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. J Neurosci. 2007;27:13843–13853. doi: 10.1523/JNEUROSCI.4486-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito K, Parnas D, Fetter RD, Isacoff EY, Goodman CS. Watching a synapse grow: noninvasive confocal imaging of synaptic growth in Drosophila. Neuron. 1999;22:719–729. doi: 10.1016/s0896-6273(00)80731-x. [DOI] [PubMed] [Google Scholar]
- Zito K, Scheuss V, Knott G, Hill T, Svoboda K. Rapid functional maturation of nascent dendritic spines. Neuron. 2009;61:247–258. doi: 10.1016/j.neuron.2008.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]