

Abstract
ATP-binding cassette transporter A7 (ABCA7) is expressed in the brain and has been detected in macrophages, microglia, and neurons. ABCA7 promotes efflux of lipids from cells to apolipoproteins and can also regulate phagocytosis and modulate processing of amyloid precursor protein (APP) to generate the Alzheimer's disease (AD) amyloid-β (Aβ) peptide. Genome-wide association studies have indicated that ABCA7 single nucleotide polymorphisms confer increased risk for late-onset AD; however, the role that ABCA7 plays in the brain in the AD context is unknown. In the present study, we crossed ABCA7-deficient (A7−/−) mice with J20 amyloidogenic mice to address this issue. We show that ABCA7 loss doubled insoluble Aβ levels and thioflavine-S–positive plaques in the brain. This was not related to changes in APP processing (assessed by analysis of full-length APP and the APP β C-terminal fragment). Apolipoprotein E regulates cerebral Aβ homeostasis and plaque load; however, the apolipoprotein E concentration was not altered by ABCA7 loss. Spatial reference memory was significantly impaired in both J20 and J20/A7−/− mice compared with wild-type mice; however, there were no cognitive differences between J20 and J20/A7−/− mice. There were also no major differences detected in hippocampal or plaque-associated microglial/macrophage markers between J20 and J20/A7−/− mice, whereas the capacity for bone marrow-derived macrophages derived from A7−/− mice to take up oligomeric Aβ was reduced by 51% compared with wild-type mice. Our results suggest that ABCA7 plays a role in the regulation of Aβ homeostasis in the brain and that this may be related to altered phagocyte function.
Introduction
Genome-wide association studies indicate that ATP-binding cassette transporter A7 (ABCA7) is a genetic risk factor for late-onset Alzheimer's disease (AD) (Harold et al., 2009; Hollingworth et al., 2011). ABCA7 is a member of the “A” subfamily of ABC transporters that predominantly transport lipids and other lipophilic molecules across membranes (Kim et al., 2008). Cell-culture studies suggest that, similar to its closest homolog, ABCA1 (54% sequence identity), ABCA7 facilitates the transport of phospholipids and, to a lesser extent, cholesterol, across cell membranes to extracellular apolipoprotein A–I (apoA–I) and apolipoprotein E (apoE) discoidal lipoprotein complexes (Wang et al., 2003; Abe-Dohmae et al., 2004; Chan et al., 2008). Overexpression of ABCA1 or ABCA7 in cell lines that constitutively express human amyloid precursor protein (APP) results in decreased extracellular levels of amyloid-β (Aβ) peptide (Sun et al., 2003; Kim et al., 2007; Chan et al., 2008). However, it is unclear whether this anti-amyloidogenic action is linked to lipid transport, because loss-of-function mutations in ABCA1 do not inhibit its capacity to downregulate Aβ levels in vitro (Kim et al., 2010).
In addition to its similarity to ABCA1, ABCA7 also shares a high degree of sequence homology (24% identity, 43% similarity) with the Caenorhabditis elegans gene CED-7 (Jehle et al., 2006). CED-7 plays a crucial role in phagocytosis and it is now recognized that ABCA7 also regulates phagocytosis in several mammalian cell types (Iwamoto et al., 2006; Jehle et al., 2006). These findings are consistent with the high expression of ABCA7 in human phagocytes, including macrophages and, in particular, microglia (Kaminski et al., 2000; Kim et al., 2006). Although ABCA7 is highly expressed in the brain (Wang et al., 2003; Kim et al., 2005; Kim et al., 2008), its function in the context of AD is unknown. Previous studies have used amyloidogenic AD mouse models to examine the impact that Abca1 deletion has on the regulation of cerebral Aβ homeostasis, amyloid plaque burden, and associated neuropathological and behavioral parameters (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005; Wahrle et al., 2005; Lefterov et al., 2009). In the present study, we have undertaken an in vivo assessment of the role that ABCA7 plays in the AD context and provide evidence that Abca7 deletion increases cerebral Aβ accumulation.
Materials and Methods
Transgenic mice.
Hemizygous hAPPSwInd J20 mice and Abca7−/− mice (all of the C57BL/6J background and backcrossed >10 generations) were crossed to generate J20/Abca7+/− breeders. These mice were mated to generate J20/Abca7+/+ and J20/Abca7−/− mice (“J20” and “J20/A7−/−,” respectively). Wild-type (WT) mice were used as controls for spatial memory testing. The generation and genotyping of the J20 and Abca7−/− mice has been described previously (Mucke et al., 2000; Kim et al., 2005). Test animals were males at ∼17 months of age (J20, n = 14, 528 ± 4 d old; J20/A7−/−, n = 11, 528 ± 12 d old; WT, n = 9, 447 ± 40 d old). Ethics approval was acquired from the University of New South Wales and the University of Wollongong animal ethics committees.
Spatial reference memory assessment using the cheeseboard paradigm.
The cheeseboard paradigm is established as a “dry-land” equivalent of the Morris water maze to assess spatial reference memory in male J20 mice (Karl et al., 2012). Testing was performed with a 9 d training period (3 2 min trials per day) during which the latency to find a food reward was measured. On the test day, a probe trial was conducted and the percentage time spent in the target zone was calculated. Male Abca7−/− mice performed the same as WT mice in the cheeseboard paradigm and were therefore not tested as an additional control group in the present study (Logge et al., 2012).
Tissue preparation.
Euthanized mice were perfused with PBS. The brains were sagittally divided; the right hemisphere was snap frozen and stored at −80°C and the left hemisphere was fixed in 4% paraformaldehyde. Cryosections were collected and stored at −20°C in antifreeze solution. Frozen brain tissue (30–50 mg) was homogenized in 8 volumes of 140 mm NaCl, 3 mm KCl, 25 mm Tris, pH 7.4, containing 1% Nonidet P-40 and Roche complete protease inhibitors (TBS/NP40 extraction buffer) using a Precellys 24 homogenizer (2 × 30 s, 6000 × g). Homogenates were centrifuged at 20,817 × g for 15 min at 4°C and the supernatant (TBS/NP40-soluble fraction) was stored at −80°C until use. The pellet was homogenized with 6 volumes of 6.25 m guanidine HCl (gHCl) in 50 mm Tris, pH 8.0, incubated for 4 h at 25°C, and centrifuged at 20,817 × g for 20 min at 4°C. The resultant supernatant was collected as the gHCl-soluble fraction and stored at −80°C until use. Where specifically indicated, hemibrains were dissected into hippocampus, cortex, and cerebellum and homogenized in TBS without Nonidet P-40 as above (Precellys 24 homogenizer, 2 × 30 s, 6000 × g), the homogenates were centrifuged at 20,817 × g for 15 min at 4°C, and the supernatant (TBS-soluble fraction) was stored at −80°C until use specifically for apoE quantification by Western blotting. Protein was measured using the bicinchoninic acid method.
Aβ ELISA.
Aβ40 and Aβ42 in TBS/NP40-soluble and gHCl-soluble fractions of brain homogenates were quantified using the Beta Amyloid x-40 and x-42 ELISA kits (Covance) as described previously (Elliott et al., 2011).
Histology and immunohistochemistry.
Sagittal sections (45 μm thick, free floating) were rinsed with TBS to remove cryoprotectants. The sections were then immersed in distilled H2O (2 min) followed by Harris hemotoxylin (2 min), distilled H2O (3 min), 1% (w/v) thioflavine S (ThS, Sigma T1892, 3 min), distilled H2O (3 min), 1% (v/v) acetic acid (20 min), distilled H2O (3 min), and mounted in buffered glycerol. Three sections per mouse were analyzed from the hippocampal region between lateral 1 mm to lateral 2 mm from the midline as defined using a mouse brain atlas (Franklin and Paxinos, 2007). Images were captured using a Nikon TE2000 microscope equipped with a SPOT digital camera (Diagnostic Instruments) and Image-Pro Plus 6.1 software (Media Cybernetics). Analysis of whole ThS-stained sagittal sections indicated that plaque deposition was predominantly within the hippocampus, with only occasional plaques present in the cortex. ThS-positive plaques in the hippocampus were therefore counted and expressed as the number of plaques per section. For the purposes of comparison, hippocampal ThS-positive plaque load was also expressed as the percentage coverage of the area of interest (measured as square micrometers) using ImageJ software.
For immunohistochemistry, sections were stained using rabbit anti-Iba1 (1:5000; Wako) or rat anti-Mac2 (1:2000; Cedarlane) for 16 h at 4°C, followed by incubation with biotinylated secondary antibodies (1:2000; Sigma or Jackson ImmunoResearch) for 2 h at 22°C, and streptavidin-HRP (1:4000; Sigma) for 1 h at 22°C. The sections were developed using DAB substrate (Vector Laboratories) and scanned at 20× magnification (Aperio Digital Pathology System, Aperio Technologies). For quantification, the percentage of pixels above background staining and the occupied area (in square micrometers) of the positive staining within the hippocampus was measured using ImageJ software. For triple-label immunofluorescence staining, the sections were incubated in primary antibodies (1:2000 Iba1, 1:1000 Mac2, and 1:5000 6E10-Biotin) for 24 h at 4°C, and incubated with corresponding fluorescently labeled secondary antibodies (Alexa Fluor 488–anti-rat, Alexa Fluor 568–streptavidin, and Alexa Fluor 647–anti-rabbit, all at 1:500) at 22°C for 2 h. The sections were mounted with antifade medium and visualized using a Leica SP5X confocal microscope.
Western blotting.
TBS/NP40-soluble (and, where specifically indicated, TBS-soluble) homogenate fractions were analyzed by SDS-PAGE (15 μg of protein per lane) and Western blotting using antibodies to: ABCA1 (1:2000; Life Research), ABCA7 (1:5000; LifeSpan Bioscience), low-density lipoprotein receptor (LDLR, 1:1000; Novus), apoE (1:5000; Calbiochem), WO2 (1:200; provided by Dr. Qiao-Xin Li and Prof. Colin Masters, University of Melbourne), synaptophysin (1:10,000; Sigma), PSD95 (1:1000; Invitrogen), APP C-terminal (1:10,000; Sigma), Iba1 (1:5000; Wako), Mac2 (1:2000; Cedarlane), and β-actin (1:10,000; Sigma). Signals were detected using species-specific HRP-conjugated secondary antibodies and enhanced chemiluminescence and quantified using ImageJ software. Integrated optical density data were normalized to β-actin levels and expressed as relative values.
Macrophage isolation and Aβ uptake.
Bone marrow-derived macrophages were obtained from five WT and five Abca7−/− mice as described previously (Elliott et al., 2008). Based on previous studies of phagocytic uptake of apoptotic cellular debris by Abca7+/− macrophages (Jehle et al., 2006), uptake of Aβ oligomers from culture media was assessed after 45 min. Aβ oligomers were prepared by sonicating human Aβ42 (100 μm in Ham's F12 cell culture medium) for 10 min, followed by 48 h of incubation at 10°C.
Statistical analysis.
For cognitive assessment, a one-way ANOVA was used to analyze effects over training days and a one-way ANOVA followed by Fisher-PLSD post hoc analysis was used to analyze memory retention in the probe trial. Biochemical and histopathological data were compared using the t test. Differences were regarded as significant if p < 0.05. All data are presented as means ± SE.
Results
Deletion of ABCA7 increases insoluble Aβ levels and ThS-positive plaque load
To assess whether ABCA7 regulates pathways involved in AD, we generated amyloidogenic J20 mice that lack ABCA7 (J20/A7−/−) and compared these with their J20 littermates. Deletion of ABCA7 resulted in significant 2.1-fold and 2.3-fold increases in Aβ42 and Aβ40 levels, respectively, in the insoluble fractions of brain homogenates (Fig. 1A). We also observed a nonsignificant trend for increased soluble Aβ42 and Aβ40 levels in the J20/A7−/− mice (Fig. 1B). In the present study, ThS-positive amyloid plaques were largely restricted to the hippocampus. Consistent with the increased insoluble Aβ detected by ELISA in the hemibrain homogenates (which include the hippocampus), the number of ThS-positive amyloid plaques detected in the hippocampus was increased by 53% in the absence of ABCA7 (Fig. 1C,D). When hippocampal ThS-positive plaque load was expressed as a percentage relative to tissue area, we also observed a significant 40% increase in the absence of ABCA7 (0.48 ± 0.04 vs 0.67 ± 0.06, mean ± SE; J20 vs J20/A7−/−, respectively, p = 0.016). Note that the Aβ42 concentration in the insoluble brain fractions was ∼200 times higher than Aβ40, whereas the levels of these two Aβ species were similar in the soluble fractions (Fig. 1A,B).
Figure 1.

Deletion of ABCA7 increases insoluble Aβ and ThS plaques in J20 mice. A, B, The gHCL-soluble “Insoluble” fraction (A) and the TBS/NP40-soluble “Soluble” fractions (B) of brain homogenates were analyzed by ELISA for Aβ42 and Aβ40. C, Brain sections (n = 6 for both the J20 and J20/A7−/− groups) were stained with ThS and images were captured using fluorescence microscopy. Scale bar, 500 μm. D, ThS staining was quantified as number of plaques per hippocampal section. Data for J20 (n = 6) and J20/A7−/− (n = 6) mice are mean ± SE (*p < 0.05; **p < 0.01). Note that the ELISA data are derived from hemibrain homogenates (see Materials and Methods) that include the hippocampus and other brain regions.
Spatial reference memory assessment
To assess whether increased amyloid deposition in the J20/A7−/− mice is correlated with changes in spatial memory, mice were tested using the cheeseboard paradigm (Karl et al., 2012). When the animals were assessed by one-way ANOVA for their ability to learn to find a food reward over a 9 d period, only the WT mice were successful in this task (p = 0.0003; Fig. 2A). Task acquisition was impaired in both the J20 and J20/A7−/− mice (p = 0.4022 and p = 0.1850, respectively; Fig. 2A). The cognitive performance of J20/A7−/− mice during training was the same as that of J20 mice (Fig. 2A). In the probe trial testing spatial memory, a significant effect (p < 0.05) of genotype was detected when the three groups were compared using ANOVA (Fig. 2B). Post hoc analysis indicated that both the J20 and J20/A7−/− mice showed impaired memory retention compared with the WT animals. The spatial memory of J20/A7−/− mice was similar to that of J20 mice (Fig. 2B).
Figure 2.
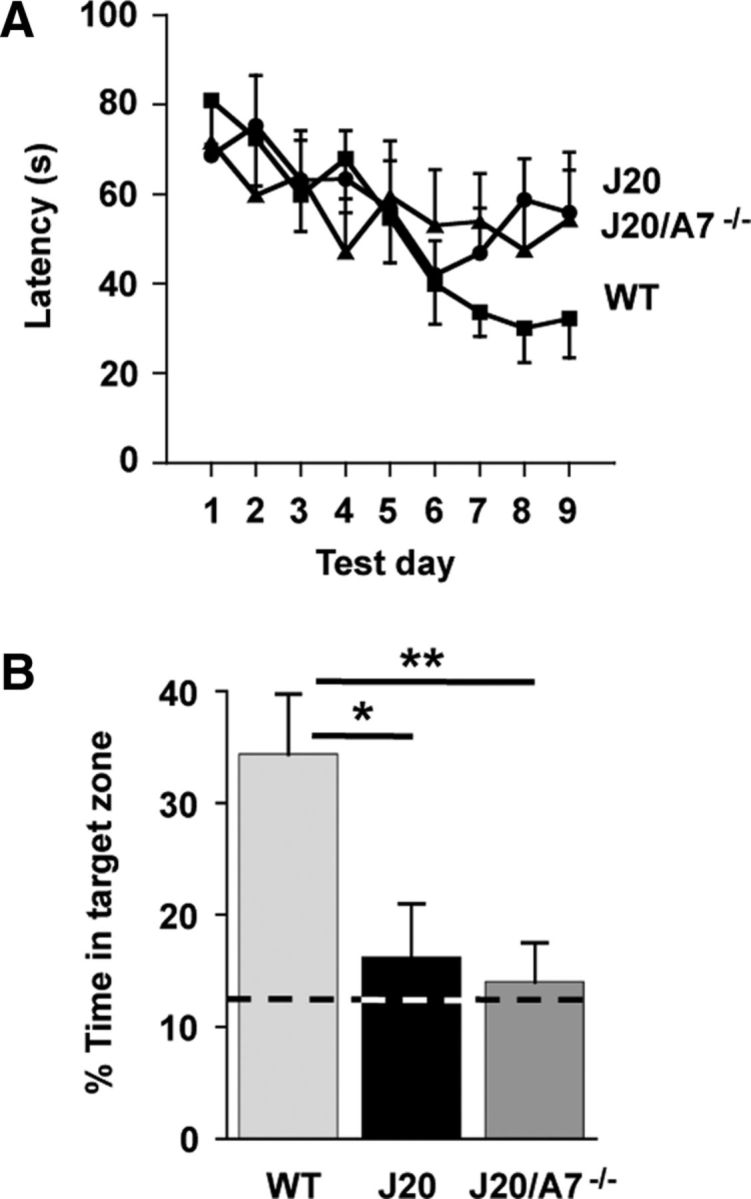
Spatial reference memory using the cheeseboard paradigm. A, Mean latency (averaged across 3 trials per day) to find the food reward. B, Percentage of time spent in the target zone during the probe trial. Time spent in the target zone due to chance (12.5%) is marked with a broken line. Data for WT control (n = 9), J20 (n = 14), and J20/A7−/− (n = 11) mice are mean ± SE. *p < 0.05 compared with WT.
We also assessed the J20 and J20/A7−/− mice in additional behavioral paradigms including a novel object recognition task, the Y-maze (short-term recognition), and a fear conditioning paradigm. We have previously validated each of these tests in the J20 mouse model (Karl et al., 2012). However, these tests did not reveal any differences between the J20 and J20/A7−/− mice (J. Low, D. Cheng, B. Garner, and T. Karl, unpublished observations).
ABCA7 deletion does not alter APP processing or apoE level
The increase in insoluble Aβ and ThS-positive plaque load associated with ABCA7 deletion (Fig. 1) may be due to altered APP processing or decreased Aβ clearance/degradation. ABCA7 overexpression inhibits processing of APP to form Aβ in vitro (Chan et al., 2008). Therefore, we assessed whether ABCA7 deletion had any impact on full-length APP and the β C-terminal fragment (βCTF) that is generated subsequent to β-secretase cleavage of APP. Neither APP nor βCTF levels were altered (Fig. 3), suggesting that amyloidogenic processing of APP is not accelerated by ABCA7 deletion.
Figure 3.
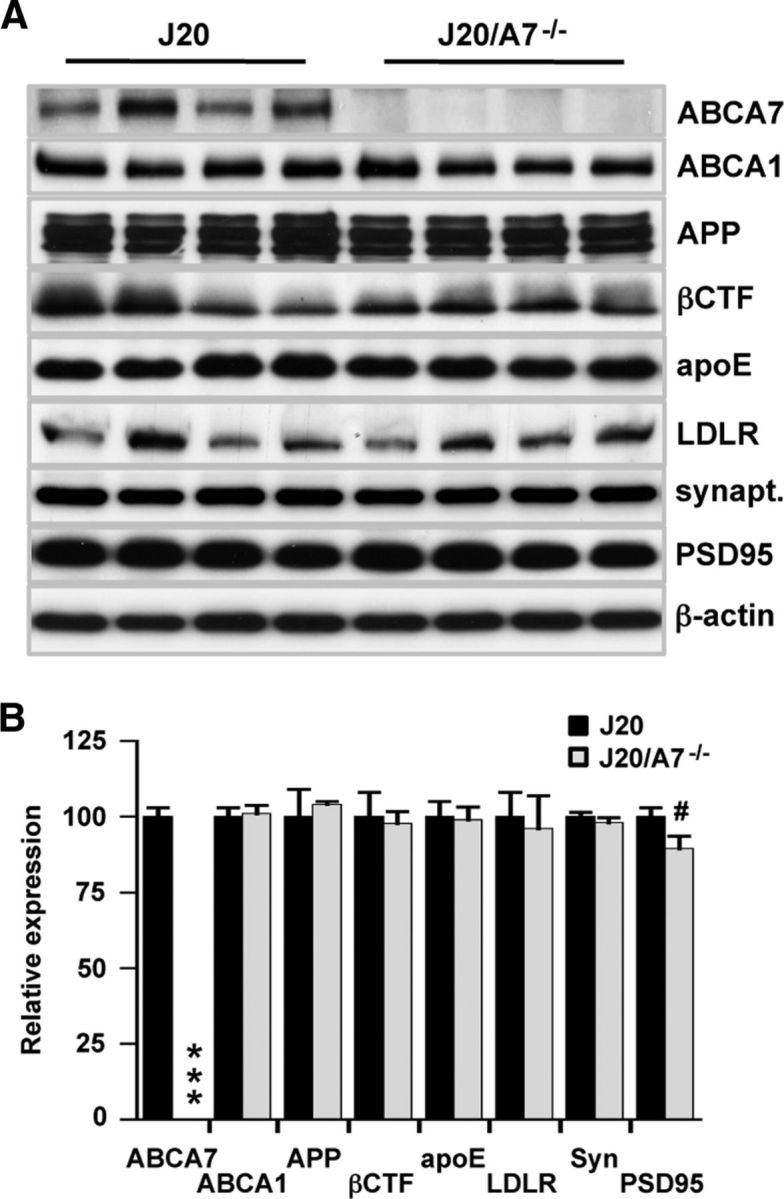
Impact of ABCA7 deletion on brain protein expression. A, Brain homogenates were probed for the proteins listed. Representative blots (n = 4 samples for each group) are shown. β-Actin is used as a loading control. B, Integrated optical density for blots of all samples (J20, n = 14; J20/A7−/−, n = 11) is shown. Levels are expressed relative to J20 mice and presented as mean ± SE. ***p < 0.0001 and #p = 0.05 compared with J20 mice.
ApoE binds to Aβ in an isoform-dependent manner and is thought to regulate Aβ degradation and clearance from the brain (LaDu et al., 1994; Jiang et al., 2008; Castellano et al., 2011; Kim et al., 2011; Bien-Ly et al., 2012; Cramer et al., 2012). ABCA1 deletion in AD mouse models is associated with significant (∼70–75%) reductions in apoE levels (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005; Wahrle et al., 2005) depending on the mouse model and age. This is thought to be due to the impaired capacity for apoE to receive its full complement of lipids in the absence of ABCA1. Because ABCA7 also promotes apoE lipidation in vitro (Chan et al., 2008), we measured apoE levels in the present study. There was no effect of ABCA7 deletion on cerebral apoE levels (Fig. 3). Because both ABCA1 and the LDLR influence apoE lipidation and recycling (Zhou et al., 2008; Kim et al., 2011), we assessed the expression of these proteins to determine their potential compensatory upregulation in J20/A7−/− mice. Neither ABCA1 nor LDLR expression was altered by ABCA7 deletion (Fig. 3). As a surrogate marker for neurodegeneration and synapse loss, the levels of synaptophysin and PSD95 were measured. Synaptophysin levels were not altered by ABCA7 deletion; however, we detected a strong trend (p = 0.05) for decreased PSD95, indicating that ABCA7 deletion has a minor impact on neurodegeneration in J20 mice (Fig. 3).
In previous studies showing that ABCA1 deletion was associated with significant reductions in cerebral apoE levels in amyloidogenic mice, the “soluble” apoE fractions were either derived from homogenates that contained detergent (0.25% SDS and 1% Triton X-100 (Koldamova et al., 2005), similar to the conditions used to generate the TBS/NP40-soluble fractions analyzed as in Figure 3, or they were derived from homogenates that contained either PBS (Hirsch-Reinshagen et al., 2005) or carbonate buffer (Wahrle et al., 2005) in the absence of detergent. In addition, in two of these studies (Hirsch-Reinshagen et al., 2005; Wahrle et al., 2005), the hippocampus and remaining cortex were analyzed separately for apoE. So that our data could be compared more directly with these previous reports, we dissected the hippocampal, cortical, and cerebellar regions from the brain and homogenized them in TBS without detergents. These TBS-soluble fractions were then analyzed by Western blotting for apoE. The data reveal that apoE levels differed significantly depending on the brain region (cortex > hippocampus > cerebellum), and we show that that these regional differences were observed clearly in both the J20 and J20/A7−/− mice (Fig. 4A,B). Moreover, when each of these brain regions was analyzed separately, we found no evidence for a change in apoE levels (Fig. 4C,D). This confirms that ABCA7 deletion has no significant impact on cerebral apoE levels.
Figure 4.
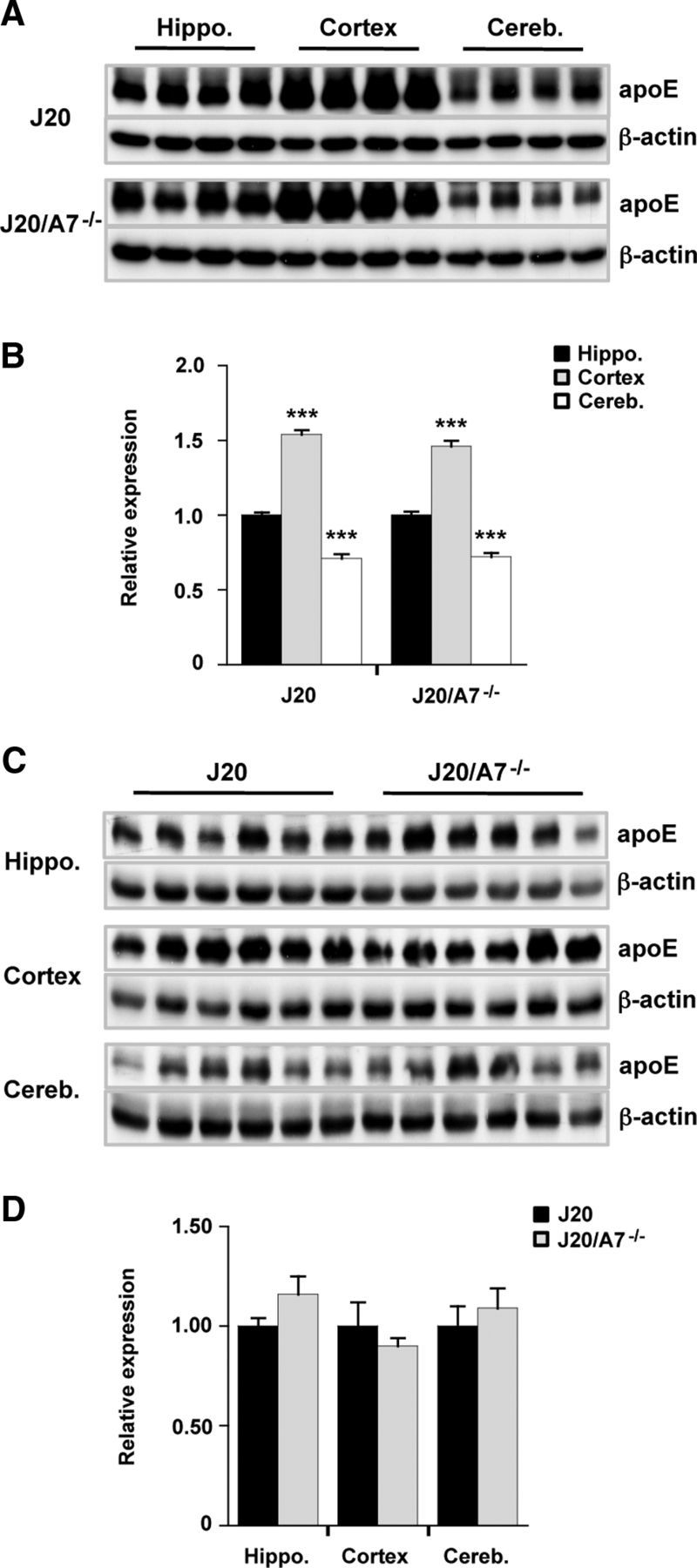
Impact of ABCA7 deletion on apoE expression in different brain regions. A, C, Brain homogenates were prepared and TBS-soluble fractions were probed for apoE. Representative blots (n = 4 or n = 6 samples for each group) are shown. β-Actin was used as a loading control. B, D, Integrated optical density for blots of all samples (J20, n = 14; J20/A7−/−, n = 11) is shown. Levels are expressed relative to hippocampus (B) or J20 mice (D) and are presented as mean ± SE. ***p < 0.0001 compared with hippocampus.
ABCA7 deletion inhibits cellular uptake of Aβ in vitro
Microglia and macrophages contribute to Aβ degradation in the brain (Bolmont et al., 2008; Thanopoulou et al., 2010; Cramer et al., 2012). There is increasing evidence that bone marrow-derived microglia (not resident microglia) play the major role in the clearance of cerebral Aβ (Malm et al., 2005; Simard et al., 2006; Mildner et al., 2011; Naert and Rivest, 2012). Based on previous studies showing that ABCA7 regulates phagocytic uptake of apoptotic debris and latex beads in a variety of phagocytic cell types (Iwamoto et al., 2006; Jehle et al., 2006), we examined the uptake of Aβ by bone marrow-derived macrophages derived from both WT and Abca7−/− mice. Aβ was sonicated and incubated to promote the formation of oligomers that resemble those detected in the human brain (Shankar et al., 2008). Macrophages accumulated Aβ predominantly as dimeric and trimeric species (Fig. 5A). ABCA7-deficient cells contained significantly less (51%) Aβ (Fig. 5A,B). These results are consistent with the proposal that ABCA7 deletion reduces the capacity of phagocytes to clear Aβ from the brain.
Figure 5.
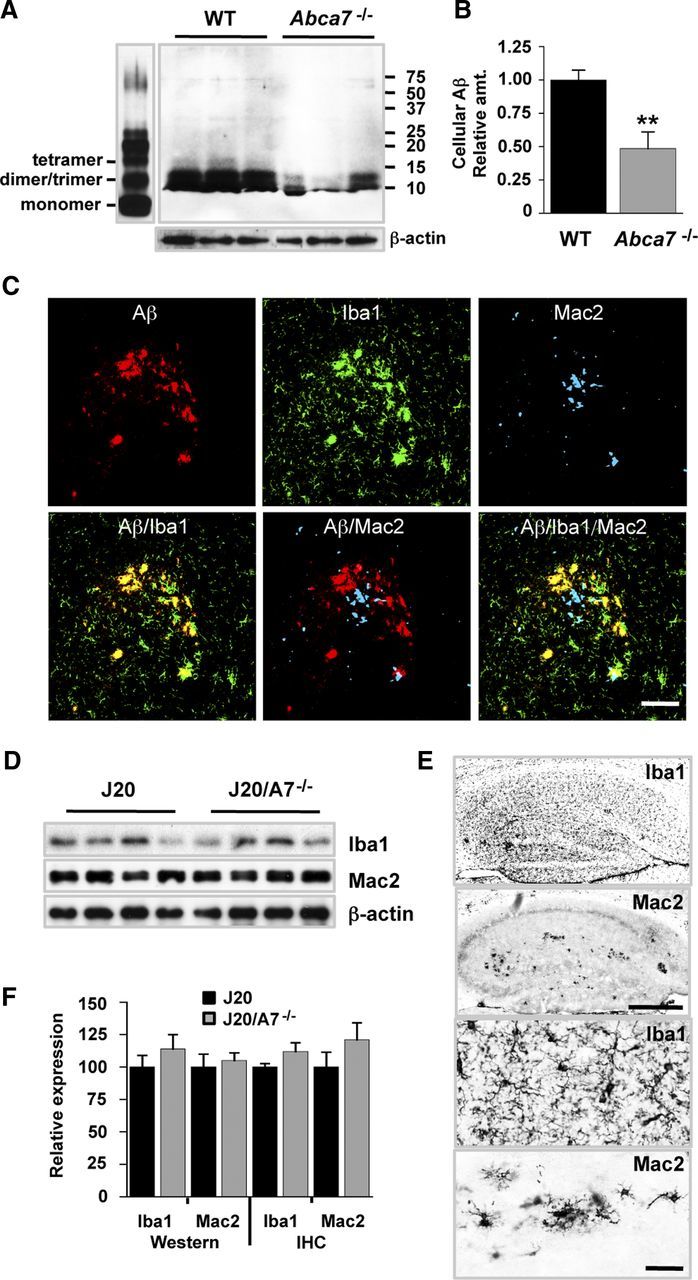
Impact of ABCA7 deletion on Aβ phagocytosis and brain macrophage/microglial markers. A, Bone marrow-derived macrophages from WT and Abca7−/− mice were incubated with 5 μm Aβ oligomers (left) for 45 min. Cells were rinsed and lysates were probed for Aβ uptake by Western blotting. β-Actin was used as a loading control. B, Integrated optical density for the Western blot is shown. Levels are expressed relative to WT mice and are mean ± SE. **p < 0.01 compared with J20. C, Hippocampal sections were stained for Aβ, Iba1, and Mac2. Images were collected using confocal microscopy and merged as indicated. Scale bar, 50 μm. D, Iba1 and Mac2 levels in brain homogenates were assessed by Western blotting. Representative blots (n = 4 samples) are shown. β-Actin was used as a loading control. E, Iba1 and Mac2 in hippocampal sections were assessed by immunohistochemistry. Scale bars: Top, 500 μm; bottom, 50 μm. F, Quantification of Iba1 and Mac2 blots (Western, J20, n = 14; J20/A7−/−, n = 11) and immunohistochemistry (IHC, J20, n = 6; J20/A7−/−, n = 6) is shown. Levels are expressed relative to J20 mice and are mean ± SE.
Assessment of microglia and macrophage markers in the brain
We next examined the levels of microglia and macrophage markers (Iba1 and Mac2, respectively) and their plaque distribution to search for obvious functional differences that may be correlated with altered phagocyte numbers or activation status in the ABCA7-deficient AD setting. Iba1 microglial staining was abundant in the brain and increased in the region of amyloid plaques (Fig. 5C). Mac2-positive cells (representing bone marrow-derived macrophages and a subset of activated microglia) were relatively uncommon throughout the parenchyma, but were clearly detected in association with amyloid plaques (Fig. 5C). Confocal analysis indicated that Iba1-postive cells colocalized with Aβ in plaques, whereas Mac2-postive cells were found in the vicinity of plaques but did not colocalize directly (Fig. 5C). Global levels of Iba1 and Mac2 assessed by Western blotting of hemibrain homogenates were not significantly altered in J20/A7−/− compared with J20 mice (Fig. 5D–F). Analysis of hippocampal Iba-positive and Mac2-positive cells also indicated no significant difference, although trends for increased levels were observed for both markers in the ABCA7-deficient mice (Fig. 5E,F). Using previously described quantitative methods (Teunissen et al., 2002), we did not detect differences in the Iba1 or Mac2 plaque association or Aβ colocalization (H. Li and B. Garner, unpublished observations). Overall, these data suggest that ABCA7 loss does not induce an obvious difference in cerebral microglia/macrophage number even though their capacity to take up Aβ in vitro may be impaired.
Discussion
Our study provides the first evidence that cerebral Aβ peptide deposition and ThS-positive plaque load are increased when ABCA7 is deleted in an AD mouse model. Even though the extent of increase in insoluble Aβ42 induced by ABCA7 deletion in J20 mice in the present study is similar to that observed when ABCA1 was deleted in APP23 and PDAPP AD mouse models (Koldamova et al., 2005; Wahrle et al., 2005), one crucial difference is that the change in Aβ level induced by ABCA7 deletion is independent of alterations in apoE concentration. This implies that ABCA7 and ABCA1 may have different functions in the brain, particularly regarding apoE function and AD. The lack of difference in TBS/NP40-soluble Aβ and βCTF in the absence of ABCA7 suggests that accelerated amyloidogenic processing of APP probably does not account for the increased Aβ detected in gHCl-soluble fractions of brain homogenates and in ThS-positive plaques. This finding is similar to the lack of impact that ABCA1 deletion has on APP processing in several AD mouse models (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005; Wahrle et al., 2005).
The finding that ABCA7 deletion did not exacerbate cognitive deficits in the J20 mice may be due to the fact that soluble Aβ levels were not significantly changed. In support of this idea, it has been shown in APP23 mice that, although ABCA1 deficiency increases insoluble Aβ levels, it is the soluble oligomeric Aβ species that are the most correlated with cognitive impairment (Lefterov et al., 2009). An alternative explanation could be due to the J20 mice reaching a “floor effect” in the cheeseboard paradigm. By chance, mice were shown to spend 12.5% of their time in the target zone (i.e., no learned preference) during a previous probe trial (Karl et al., 2012). In the present study, the J20 mice on average spent 16.1% of their time in the target zone during the probe trial; therefore it was difficult to detect any further impairment in spatial memory in the J20/A7−/− mice (which spent 14.0% of their time in the target zone).
Our data suggest that ABCA7 deletion appears to impair the uptake of Aβ in vitro (Fig. 5A). Alternative explanations for the reduced level of Aβ detected in ABCA7-deficient macrophages could include alterations to intracellular Aβ degradation or facilitation of Aβ resecretion. Regardless of the potential for these pathways to affect intracellular Aβ metabolism, it is intriguing that ABCA7 deletion was not associated with increased soluble Aβ in the J20/A7−/− mouse brain (Fig. 1B). Previous studies have highlighted the important role that microglia and macrophages play in the phagocytic clearance of Aβ deposited within amyloid plaques in AD mouse models and in Aβ42-immunized human AD patients (for review, see Perry et al., 2010). It is possible that ABCA7 deletion primarily modulates cerebral Aβ homeostasis via altered microglial/macrophage clearance of amyloid plaques rather than soluble Aβ species. Whether this is the pathway that links the recently identified ABCA7 SNPs with increased AD risk remains to be determined.
It would be worthwhile to investigate the impact that ABCA7 deletion has on the soluble Aβ dimers, trimers, and other oligomeric Aβ species in future studies because the ELISA methods we used may not detect such pathogenic species of Aβ (Mc Donald et al., 2010; Benilova et al., 2012). In preliminary studies, we probed the brain homogenates analyzed in this study for oligomeric forms of Aβ using Western blot and dot blot assays and a variety of monoclonal antibodies, including WO2 and 4G8 (which recognize Aβ) and A11 (which recognizes a generic epitope common to prefibrillar oligomers); however, we were unable to detect clearly the abovementioned oligomeric Aβ species (H. Li, B. Garner, unpublished observations). Although cerebral bone marrow-derived macrophages clearly play a role in cerebral Aβ clearance, it would also be interesting to study the phagocytic uptake of in vitro-derived oligomeric Aβ complexes by ABCA7-deficient resident microglia because there may be subtle differences in the function of these cells in the brain.
The J20 mice used in the present study express a mutant form of human APP bearing both the Swedish (K670N/M671L) and the Indiana (V717F) mutations (Mucke et al., 2000). In future studies, it will be important to also assess the modulation of Aβ homeostasis in additional amyloidogenic mouse models, including those that have a more aggressive pathology due to the transgenic expression of multiple mutant genes (e.g., the APP/PS1 and 3xTg-AD mice) (Mc Donald et al., 2010; Romberg et al., 2011). Because these mice typically display plaque pathology earlier than the J20 mice, the impact that ABCA7 deletion has on plaque development at earlier time points could also be investigated. Similarly, our present study does not exclude the possibility that ABCA7 deletion may alter cerebral apoE levels at earlier time points.
The deletion of ABCA1 in mice impairs apoE lipidation significantly, and this has been shown to play a critical role in the metabolism of Aβ and apoE in the brain (Wahrle et al., 2004; Jiang et al., 2008). The fact that ABCA7 deletion does not affect cerebral apoE levels may be interpreted as evidence that ABCA7 does not play a significant role in apoE lipidation in the brain and, furthermore, that the increased Aβ deposition detected in the absence of ABCA7 is not due to an apoE deficiency. However, the data provided herein do not prove this idea directly. Microdialysis studies have been used recently to characterize apoE in murine cerebral interstitial fluid (Fitz et al., 2012), and this approach may be useful in future studies to measure changes in apoE lipid composition and Aβ binding.
In summary, we have shown that ABCA7 deletion is associated with a significant increase in insoluble Aβ in the brain and that ABCA7-deficient macrophages have a reduced capacity to phagocytose Aβ. Although there appears to be no obvious change in cerebral microglial or macrophage markers, it remains possible that “plaque clearance” is reduced in the absence of ABCA7 and that, over the lifespan, this could lead to a higher overall Aβ load in the brain.
Footnotes
Research support was from the Australian National Health and Medical Research Council (NHMRC Project Grant 510148). T.K. is supported by an NHMRC Career Development Award (1045643) and the Motor Neuron Disease Research Institute of Australia (Mick Rodger Benalla MND Research Grant). B.G. is supported by a fellowship from the Australian Research Council (ARC Future Fellowship FT0991986). We thank Prof. Mason W. Freeman (Massachusetts General Hospital, Boston) and Prof. Lennart Mucke (Gladstone Institute of Neurological Disease, San Francisco) for providing Abca7−/− mice and J20 mice, respectively, Prof. Colin Masters and Dr. Qiao-Xin Li (University of Melbourne, Melbourne, Australia) for providing WO2 antibody and the ThS staining protocol, and Dr. Andy Liang (Neuroscience Research Australia, Sydney, Australia) for assisting with tissue scanning.
The authors declare no competing financial interests.
References
- Abe-Dohmae S, Ikeda Y, Matsuo M, Hayashi M, Okuhira K, Ueda K, Yokoyama S. Human ABCA7 supports apolipoprotein-mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J Biol Chem. 2004;279:604–611. doi: 10.1074/jbc.M309888200. [DOI] [PubMed] [Google Scholar]
- Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Bien-Ly N, Gillespie AK, Walker D, Yoon SY, Huang Y. Reducing human apolipoprotein E levels attenuates age-dependent Abeta accumulation in mutant human amyloid precursor protein transgenic mice. J Neurosci. 2012;32:4803–4811. doi: 10.1523/JNEUROSCI.0033-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SL, Kim WS, Kwok JB, Hill AF, Cappai R, Rye KA, Garner B. ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J Neurochem. 2008;106:793–804. doi: 10.1111/j.1471-4159.2008.05433.x. [DOI] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott DA, Kim WS, Jans DA, Garner B. Macrophage apolipoprotein-E knockdown modulates caspase-3 activation without altering sensitivity to apoptosis. Biochim Biophys Acta. 2008;1780:145–153. doi: 10.1016/j.bbagen.2007.10.021. [DOI] [PubMed] [Google Scholar]
- Elliott DA, Tsoi K, Holinkova S, Chan SL, Kim WS, Halliday GM, Rye KA, Garner B. Isoform-specific proteolysis of apolipoprotein-E in the brain. Neurobiol Aging. 2011;32:257–271. doi: 10.1016/j.neurobiolaging.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Fitz NF, Cronican AA, Saleem M, Fauq AH, Chapman R, Lefterov I, Koldamova R. Abca1 deficiency affects Alzheimer's disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J Neurosci. 2012;32:13125–13136. doi: 10.1523/JNEUROSCI.1937-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Ed 3. New York: Academic; 2007. [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, Maia LF, Burgess BL, Blain JF, Naus KE, McIsaac SA, Parkinson PF, Chan JY, Tansley GH, Hayden MR, Poirier J, Van Nostrand W, Wellington CL. The absence of ABCA1 decreases soluble apoE levels but does not diminish amyloid deposition in two murine models of Alzheimer's disease. J Biol Chem. 2005;280:43243–43256. doi: 10.1074/jbc.M508781200. [DOI] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto N, Abe-Dohmae S, Sato R, Yokoyama S. ABCA7 expression is regulated by cellular cholesterol through the SREBP2 pathway and associated with phagocytosis. J Lipid Res. 2006;47:1915–1927. doi: 10.1194/jlr.M600127-JLR200. [DOI] [PubMed] [Google Scholar]
- Jehle AW, Gardai SJ, Li S, Linsel-Nitschke P, Morimoto K, Janssen WJ, Vandivier RW, Wang N, Greenberg S, Dale BM, Qin C, Henson PM, Tall AR. ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J Cell Biol. 2006;174:547–556. doi: 10.1083/jcb.200601030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski WE, Orsó E, Diederich W, Klucken J, Drobnik W, Schmitz G. Identification of a novel human sterol-sensitive ATP-binding cassette transporter (ABCA7) Biochem Biophys Res Commun. 2000;273:532–538. doi: 10.1006/bbrc.2000.2954. [DOI] [PubMed] [Google Scholar]
- Karl T, Bhatia S, Cheng D, Kim WS, Garner B. Cognitive phenotyping of amyloid precursor protein transgenic J20 mice. Behav Brain Res. 2012;228:392–397. doi: 10.1016/j.bbr.2011.12.021. [DOI] [PubMed] [Google Scholar]
- Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, Basak JM, Finn MB, Holtzman DM. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J Neurosci. 2011;31:18007–18012. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WS, Fitzgerald ML, Kang K, Okuhira K, Bell SA, Manning JJ, Koehn SL, Lu N, Moore KJ, Freeman MW. Abca7 null mice retain normal macrophage phosphatidylcholine and cholesterol efflux activity despite alterations in adipose mass and serum cholesterol levels. J Biol Chem. 2005;280:3989–3995. doi: 10.1074/jbc.M412602200. [DOI] [PubMed] [Google Scholar]
- Kim WS, Guillemin GJ, Glaros EN, Lim CK, Garner B. Quantitation of ATP-binding cassette subfamily-A transporter gene expression in primary human brain cells. Neuroreport. 2006;17:891–896. doi: 10.1097/01.wnr.0000221833.41340.cd. [DOI] [PubMed] [Google Scholar]
- Kim WS, Rahmanto AS, Kamili A, Rye KA, Guillemin GJ, Gelissen IC, Jessup W, Hill AF, Garner B. Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein-E discs and suppression of amyloid-beta peptide generation. J Biol Chem. 2007;282:2851–2861. doi: 10.1074/jbc.M607831200. [DOI] [PubMed] [Google Scholar]
- Kim WS, Weickert CS, Garner B. Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J Neurochem. 2008;104:1145–1166. doi: 10.1111/j.1471-4159.2007.05099.x. [DOI] [PubMed] [Google Scholar]
- Kim WS, Bhatia S, Elliott DA, Agholme L, Kågedal K, McCann H, Halliday GM, Barnham KJ, Garner B. Increased ATP-binding cassette transporter A1 expression in Alzheimer's disease hippocampal neurons. J Alzheimers Dis. 2010;21:193–205. doi: 10.3233/JAD-2010-100324. [DOI] [PubMed] [Google Scholar]
- Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain Apoe level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005;280:43224–43235. doi: 10.1074/jbc.M504513200. [DOI] [PubMed] [Google Scholar]
- LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994;269:23403–23406. [PubMed] [Google Scholar]
- Lefterov I, Fitz NF, Cronican A, Lefterov P, Staufenbiel M, Koldamova R. Memory deficits in APP23/Abca1+/− mice correlate with the level of Abeta oligomers. ASN Neuro. 2009;1:e00006. doi: 10.1042/AN20090015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logge W, Cheng D, Chesworth R, Bhatia S, Garner B, Kim WS, Karl T. Role of Abca7 in mouse behaviours relevant to neurodegenerative diseases. PLoS One. 2012;7:e45959. doi: 10.1371/journal.pone.0045959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm TM, Koistinaho M, Pärepalo M, Vatanen T, Ooka A, Karlsson S, Koistinaho J. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–142. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Mc Donald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, Selkoe DJ, Ince PG, Walsh DM Medical Research Council Cognitive Function and Ageing Study. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain. 2010;133:1328–1341. doi: 10.1093/brain/awq065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schlevogt B, Kierdorf K, Böttcher C, Erny D, Kummer MP, Quinn M, Brück W, Bechmann I, Heneka MT, Priller J, Prinz M. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer's disease. J Neurosci. 2011;31:11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naert G, Rivest S. Hematopoietic CC-chemokine receptor 2 (CCR2) competent cells are protective for the cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer's disease. Mol Med. 2012;18:297–313. doi: 10.2119/molmed.2011.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- Romberg C, Mattson MP, Mughal MR, Bussey TJ, Saksida LM. Impaired attention in the 3xTgAD mouse model of Alzheimer's disease: rescue by donepezil (Aricept) J Neurosci. 2011;31:3500–3507. doi: 10.1523/JNEUROSCI.5242-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Sun Y, Yao J, Kim TW, Tall AR. Expression of liver X receptor target genes decreases cellular amyloid beta peptide secretion. J Biol Chem. 2003;278:27688–27694. doi: 10.1074/jbc.M300760200. [DOI] [PubMed] [Google Scholar]
- Teunissen CE, de Vente J, Steinbusch HW, De Bruijn C. Biochemical markers related to Alzheimer's dementia in serum and cerebrospinal fluid. Neurobiol Aging. 2002;23:485–508. doi: 10.1016/s0197-4580(01)00328-1. [DOI] [PubMed] [Google Scholar]
- Thanopoulou K, Fragkouli A, Stylianopoulou F, Georgopoulos S. Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc Natl Acad Sci U S A. 2010;107:20816–20821. doi: 10.1073/pnas.1005888107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, Kowalewski T, Holtzman DM. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem. 2004;279:40987–40993. doi: 10.1074/jbc.M407963200. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM, Holtzman DM. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer's disease. J Biol Chem. 2005;280:43236–43242. doi: 10.1074/jbc.M508780200. [DOI] [PubMed] [Google Scholar]
- Wang N, Lan D, Gerbod-Giannone M, Linsel-Nitschke P, Jehle AW, Chen W, Martinez LO, Tall AR. ATP-binding cassette transporter A7 (ABCA7) binds apolipoprotein A-I and mediates cellular phospholipid but not cholesterol efflux. J Biol Chem. 2003;278:42906–42912. doi: 10.1074/jbc.M307831200. [DOI] [PubMed] [Google Scholar]
- Zhou X, He W, Huang Z, Gotto AM, Jr, Hajjar DP, Han J. Genetic deletion of low density lipoprotein receptor impairs sterol-induced mouse macrophage ABCA1 expression. A new SREBP1-dependent mechanism. J Biol Chem. 2008;283:2129–2138. doi: 10.1074/jbc.M706636200. [DOI] [PubMed] [Google Scholar]