

Abstract
The coupling of the spinal postsynaptic density-95 (PSD-95) with the glutamatergic N-methyl-d-aspartate receptor NR2B subunit and the subsequent NR2B phosphorylation contribute to pain-related plasticity. Increasing evidence reveals that kalirin, a Rho-guanine nucleotide exchange factor, modulates PSD-95-NR2B-dependent neuroplasticity. Our laboratory recently demonstrated that serum-inducible and glucocorticoid-inducible kinase 1 (SGK1) participates in inflammation-associated pain hypersensitivity by modulating spinal glutamatergic neurotransmission. Because kalirin is one of the proteins in PSD that is highly phosphorylated by various kinases, we tested whether kalirin could be a downstream target of spinal SGK1 that participates in neuropathic pain development via regulation of the PSD-95–NR2B coupling-dependent phosphorylation of NR2B. We observed that spinal nerve ligation (SNL, L5) in male Sprague Dawley rats resulted in behavioral allodynia, which was associated with phosphorylated SGK1 (pSGK1), kalirin, and phosphorylated NR2B (pNR2B) expression and an increase in pSGK1–kalirin–PSD-95–pNR2B coprecipitation in the ipsilateral dorsal horn (L4–L5). SNL-enhanced kalirin immunofluorescence was coincident with pSGK1, PSD-95, and pNR2B immunoreactivity. Small-interfering RNA (siRNA) that targeted spinal kalirin mRNA expression (10 μg, 10 μl; i.t.) reduced SNL-induced allodynia, kalirin and pNR2B expression, as well as kalirin–PSD-95 and PSD-95–pNR2B coupling and costaining without affecting SGK1 phosphorylation. Daily administration of GSK-650394 (an SGK1 antagonist; 100 nm, 10 μl, i.t.) not only exhibited effects similar to the kalirin mRNA-targeting siRNA but also attenuated pSGK1–kalirin costaining and SGK1–kalirin coupling. We suggest that nerve injury could induce spinal SGK1 phosphorylation that subsequently interacts with and upregulates kalirin to participate in neuropathic pain development via PSD-95–NR2B coupling-dependent NR2B phosphorylation.
Introduction
Phosphorylation of the N-methyl-d-aspartate receptor (NMDAR) NR2B subunit is critical for the neural plasticity underlying allodynia/hyperalgesia (Kim et al., 2012) because neuropathic (Katano et al., 2011) and postinflammatory pain (Slack et al., 2008) enhances dorsal horn NR2B phosphorylation and antagonism of spinal NR2B activation prevents visceral pain-associated hyper-reflexia (Peng et al., 2008a). Neuropathic injury induces postsynaptic density-95 (PSD-95)–NR2B coupling in dorsal horn neurons (Miletic et al., 2010) and the spinal delivery of a peptide that perturbs PSD-95–NR2B interactions attenuates formalin-induced pain behavior (D'Mello et al., 2011). Moreover, painful visceral irritation (Peng et al., 2009a) and spinal nerve ligation (Peng et al., 2012a) were both demonstrated to enhance spinal PSD-95–NR2B coupling-dependent NR2B phosphorylation, indicating the interaction between PSD-95 and NR2B and the subsequent phosphorylation of NR2B at the spinal cord level are crucial for the plasticity mediating pain hypersensitivity.
The rearrangements of the actin cytoskeleton that are regulated by small GTPases activated by guanine nucleotide exchange factors (GEFs; Rossman et al., 2005) drive the synaptogenesis presumed to modify the efficacy of synaptic transmission (Tolias et al., 2011) and contribute to pain states (Griffin, 2005). Kalirin, a Rho-GEF that is the predominant splice variant of the KALRN gene in the mammalian CNS (Kiraly et al., 2011a), has repeatedly been demonstrated to have profound effects on the modulation of synaptic plasticity (Ma et al., 2003, 2008a,b). Kalirin localizes almost exclusively to the PSD and has a C-terminal PDZ-binding motif to interact with PSD-95 (Penzes et al., 2000). Electrophysiological recordings have identified kalirin as an essential modulator of NMDAR-dependent plasticity because kalirin knock-out mice exhibit structural and functional deficits in NMDAR synapses that are accompanied by impairments in NMDAR-related cognitive behavior (Lemtiri-Chlieh et al., 2011). A study investigating place preference and passive avoidance demonstrated that the interaction between NR2B and kalirin is essential for the NMDAR-actin integrity that modifies the synaptic location and function of NMDARs (Kiraly et al., 2011b). However, whether kalirin participates in neuropathic pain development by exhibiting its effects on the PSD-95–NR2B coupling-dependent NR2B activation has not been clarified.
Kalirin, one of the most highly phosphorylated targets in the PSD, is phosphorylated by kinases (Kiraly et al., 2011a) that play critical roles in plasticity mediating long-term potentiation (LTP; Katano et al., 2011) and pain pathology (Peng et al., 2009c). SGK1 is an isoform of the serum-inducible and glucocorticoid-inducible kinase (SGK) family. SGK1 phosphorylation is increased during LTP, and the transfection of dominant-negative SGK1 impairs LTP maintenance in hippocampal neurons (Ma et al., 2006). Interestingly, our laboratory has recently demonstrated that complete Freund's adjuvent (CFA)-induced paw inflammation is associated with dorsal horn SGK1-mediated glutamatergic receptor activation; antagonism of spinal SGK1 ameliorates inflammation-associated hyperalgesia, which indicates that spinal SGK1 activation participates in postinflammatory pain (Peng et al., 2012a). However, whether SGK1 has an effect on kalirin activity and thereby participates in pain pathology via kalirin-dependent cascade remains unknown. Therefore, in this study, we investigated whether spinal SGK1 contributes to neuropathic pain by modifying kalirin/PSD-95/pNR2B-dependent plasticity.
Materials and Methods
Animal preparations.
Adult male Sprague Dawley rats weighing 180–230 g were used throughout this study. All procedures for animal studies were conducted in accordance with the guidelines of the International Association for the Study of Pain (Zimmermann, 1983) and were reviewed and approved by the Institutional Review Board of National Chung-Hsing University, Taichung, Taiwan.
Spinal nerve ligation.
Ligation of the left L5 spinal nerve was performed as described by Kim and Chung (1992). Briefly, the rats were anesthetized under isoflurane anesthesia (induction 5%, maintenance 2% in oxygen). An incision was made, and the left L5 spinal nerves were carefully isolated and tightly ligated with 6-0 silk sutures 2–5 mm distal to the dorsal root ganglia (Peng et al., 2012a).
Intrathecal catheter.
Implantation of intrathecal cannulae was performed as described in our previous study (Peng et al., 2009b, 2011). Briefly, PE-10 Silastic tubing was implanted so as to reach the lumbar enlargement of the spinal cord. The outer part of the catheter was plugged and immobilized onto the skin on closure of the wound. Rats showing neurological deficits after catheter implantation were killed and excluded from statistical analyses. After implantation, animals were allowed to recover 3 d and were then used for behavioral tests, Western blotting, coprecipitation, or immunohistochemistry.
Behavioral studies.
Tactile sensitivity was assessed by measuring each rat's paw-withdrawal threshold in response to probing with von Frey monofilaments (Stoelting) according to the method of Schäfers et al. (2003). Briefly, calibrated filaments were applied to the plantar surface of the each hindpaw through wire-mesh flooring. Animals' tactile thresholds before sham operation and spinal nerve ligation were set equal to 15 g (Peng et al., 2012a). In some animals, the rota-rod test was used to assess deficits in motor function with an accelerating rota-rod apparatus (model LE8500, Ugo Basile). In a training session, the rats were placed on the rod that was set to accelerate from 3 to 30 rpm over a 180 s period, and the time that each rat was able to remain on the rota-rod was recorded. The rats were subjected to three training trials at 3–4 h intervals on 2 separate days for acclimatization purposes. In the test session, the rats were placed on the rota-rod, and their performance times were recorded up to a cutoff time of 180 s. Each animal was tested three times at intervals of 5 min, and the three measurements were averaged (Hori et al., 2010).
Western blotting.
Procedures for Western blotting were adapted from our previous work (Peng et al., 2009c). In brief, the dissected dorsal horn (L4–L5) was homogenized in 20 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 1 mm EGTA with a complete protease inhibitor mixture (Roche). After addition of Triton X-100 to a final concentration of 1%, the lysates were further incubated for 1 h on ice. The supernatant was separated on acrylamide gel and transferred to a PVDF membrane and then incubated for 1 h at room temperature in either goat anti-kalirin (1:1000, Abcam), mouse anti-PSD-95 (1:1000, Millipore), rabbit anti-NR2B (1:1000, Millipore), rabbit anti-phosphorylated NR2B (1:1000, Millipore), rabbit anti-SGK1 (1:800, Santa Cruz Biotechnology), rabbit anti-phosphorylated SGK1 (1:1000, Abcam), or mouse anti-GAPDH (1:20,000, Novus). Blots were washed and incubated in peroxidase-conjugated goat anti-rabbit IgG (1:8000, Jackson ImmunoResearch), donkey anti-goat IgG (1:8000, Jackson ImmunoResearch), or goat anti-mouse IgG (1:8000, Jackson ImmunoResearch) for 1 h at room temperature. Protein bands were visualized using an enhanced chemiluminescence detection kit (ECL Plus, Millipore), and then densitometric analysis of the Western blot membranes was performed with Science Lab 2003 (Fuji). The results were normalized against GAPDH and are presented as the mean ± SEM.
Coimmunoprecipitation.
Goat polyclonal antibody against kalirin and mouse polyclonal antibodies against SGK1, NR2B, or PSD-95 were incubated overnight at 4°C with the extraction of the lower lumbar (L4–L5) dorsal horns obtained from sham-operated or spinal nerve ligation rats. The 1:1 slurry protein agarose suspension (Millipore) was added into that immunocomplex of protein, and the mixture was incubated for 2–3 h at 4°C. Agarose beads were washed once with 1% (v/v) Triton X-100 in an immunoprecipitation buffer [50 mm Tris-Cl, pH 7.4, 5 mm EDTA, 0.02% (w/v) sodium azide], twice with 1% (v/v) Triton X-100 in an immunoprecipitation buffer plus 300 mm NaCl, and three times with an immunoprecipitation buffer only. Binding proteins were eluted with SDS-PAGE sample buffer at 95°C. Proteins were separated by SDS-PAGE, transferred to PVDF membranes electrophoretically and detected using monoclonal goat or rabbit anti-kalirin (1:1000, Millipore), rabbit anti-PSD-95 (1:1000, Cell Signaling), rabbit anti-NR2B or anti-phosphorylated NR2B, and rabbit anti-SGK1 or phosphorylated SGK1.
Immunofluorescence.
Immunofluorescence images emitted by a mercury lamp were captured and analyzed after the following steps. Rats were anesthetized and perfused with 100 ml of 0.01 m PBS, pH 7.4, followed by 300 ml of 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. The lumbar enlargement segments were then harvested, postfixed at 4°C for 4 h, and cryoprotected in 30% sucrose overnight. For double-labeling immunohistochemistry analyses, the spinal cord sections were incubated overnight at 4°C with either (1) a mixture of goat anti-kalirin (1:500, Abcam) and mouse monoclonal anti-neuronal nuclear antigen (NeuN, a neuronal marker, 1:1000, Millipore Bioscience Research Reagents), mouse anti-glial fibrillary acidic protein (GFAP, a marker of astroglial cells; 1:500, Millipore), or mouse anti-integrin αM (OX-42, a marker of microglia; 1:400, Santa Cruz Biotechnology), or (2) a mixture of goat anti-kalirin (1:500, Abcam) and mouse anti-PSD-95 (1:500, Santa Cruz Biotechnology), rabbit anti-phosphorylated SGK1 (1:500, Abcam), or rabbit anti-phosphorylated NR2B (1:200, Abcam). The sections were then incubated with Alexa Fluor 488 (1:1500) and Alexa Fluor 594 (1:1500, Invitrogen) for 1 h at 37°C. After the sections were rinsed in 0.01 m PBS, coverslips were applied.
Small-interfering RNA.
In this study, the 19 nucleotide duplexes of the siRNAs for kalirin were 5′-GCAACAAAGCCUCUGUUAA-3′ (sense) and 5′-UUAACAGAGGCUUUGUUGC-3′ (antisense), and missense nucleotides were 5′-UUCUCCGAACGUGUCACGUTT-3′ (sense) and 5′-ACGUGACACGUUCGGAGAATT-3′ (antisense).
We injected 10 μl of the missense or siRNA solution intrathecally at the L4–L5 level of the spinal cords of rats via an established intrathecal catheter once a day for 7 d.
Application of drugs.
GSK-650394 (a SGK1 inhibitor; IC50 = 62 nm for SGK1; 10, 30, and 100 nm, 10 μl; Tocris Bioscience) was administered by bolus injection at 3 or 7 d or by daily injection for 7 d (day 0–6) postspinal nerve ligation. A vehicle solution of a volume identical to that of the tested agents was dispensed to serve as a control.
Data analysis.
The data in the text and figures in this study were analyzed using SigmaPlot 10.0 (Systat Software) and are expressed as the mean ± SEM. For serial measurements over time, one-way or two-way ANOVAs were used to assess changes in values, and post hoc Tukey's tests were used to compare the means of the groups. In other cases, Student's t tests were used to compare the means of groups. Significance was set at p < 0.05.
Results
Nerve ligation induces allodynia and kalirin expression
We first examined whether neuropathic-like injury induces tactile allodynia. Spinal nerve ligation (SNL ipsilateral, 2-way ANOVA with repeated measures over time, treatment, F(3,27) = 204.300, p < 0.001; time, F(5,45) = 5.802, p < 0.001; treatment × time, F(15,135) = 16.700, p < 0.001 vs sham ipsilateral, n = 7; **p < 0.01) induced behavioral allodynia in von Frey tests, as evidenced by a significant decrease in the withdrawal threshold of the ipsilateral hindpaw (Fig. 1A, SNL IPSI) when compared with the presurgery measurement (day −1; SNL ipsilateral, 1-way ANOVA, F(5,54) = 571.089, p < 0.001 vs day −1, n = 7, ##p < 0.01). The withdrawal threshold was reduced at day 1 and reached the maximal reduction at day 7; this reduction was maintained for at least 21 d postsurgery. In contrast, SNL did not affect the withdrawal threshold of the contralateral hindpaw on days 1, 3, 7, 14, or 21 (SNL contralateral, 1-way ANOVA, F(5,54) = 0.258, p = 0.933 vs day −1, n = 7). Moreover, the sham operation had no effect on the withdrawal threshold tested in the ipsilateral and contralateral hindpaws at these time points compared with the presurgery controls (sham ipsilateral, 1-way ANOVA, F(5,54) = 0.227, p = 0.949 and sham contralateral, 1-way ANOVA, F(5,54) = 0.080, p = 0.995 vs day −1, n = 7).
Figure 1.

Nerve ligation induces behavioral allodynia and spinal kalirin expression. A, Spinal nerve ligation [SNL IPSI; **p < 0.01 vs sham operation (sham IPSI), n = 7], but not the sham operation, statistically decreased the mechanical threshold of the ipsilateral hindpaw when tested on days 1, 3, 7, 14, and 21 compared with the presurgery controls (day −1; ##p < 0.01 vs day −1, n = 7). In contrast, neither the sham operation (sham CONTRA) nor the spinal nerve ligation (SNL CONTRA) affected the mechanical threshold of the contralateral hindpaw tested at these time points (p > 0.05 vs day −1). B, Representative Western blots of kalirin expression after spinal nerve ligation. The dorsal horn lysates were blotted for kalirin, and the level of GAPDH was used as a loading control. IB, immunoblotting. C, Statistical analysis revealed that spinal nerve ligation statistically upregulated the level of kalirin in the ipsilateral dorsal horn when compared with the presurgery control [*p < 0.05, **p < 0.01 vs day −1, n = 7; ##p < 0.01 vs sham operation (sham IPSI), n = 7]. In contrast, the sham operation exerted no effect on kalirin expression quantified at identical time points (p > 0.05 vs day −1, n = 7). The data show the mean ± SEM. D, Spinal nerve ligation (II, SNL7D), but not the sham operation (I, sham 7D), notably enhanced the kalirin immunoreactivity (red) in the ipsilateral dorsal horn (IPSI) of spinal sections obtained on day 7 after surgery when compared with the contralateral dorsal horn (CONTRA). In contrast, the immunoreactivity of NeuN (III, green) was not different between the ipsilateral and the contralateral dorsal horns. Double-staining images revealed that the nerve ligation-enhanced kalirin immunoreactivity was colabeled with NeuN (IV, yellow). Amplified images of IV showed that NeuN (V, green), but not OX-42 (VI, green) or GFAP (VII, green), was colocalized with the enhanced kalirin immunoreactivity (V, yellow). Scale bar, 50 μm; thickness, 50 μm. Each of these immunofluorescence images was replicated in seven sample preparations with similar results each time.
If kalirin plays a role in the development of neuropathic pain, kalirin expression should have a temporal correlation with SNL-induced allodynia. We accordingly measured the level of spinal kalirin after an experimental nerve injury via a Western blot analysis of the dorsal horn (L4–L5). SNL significantly upregulated kalirin expression in the ipsilateral dorsal horn, corresponding to a decrease in the withdrawal threshold, when compared with the presurgery control (Fig. 1B,C, SNL IPSI; 1-way ANOVA, F(4,30) = 8.393, p < 0.001 vs day −1, n = 7, *p < 0.05, **p < 0.01; t test, p = 0.575, 0.008, <0.001, <0.001, and 0.003 in day −1, 3, 7, 14, and 21, respectively; ##p < 0.01 vs sham ipsilateral, n = 7) but did not affect the level of GAPDH (Fig. 1B, GAPDH). In contrast, the sham operation had no effect on the level of kalirin (Fig. 1B,C, sham IPSI; 1-way ANOVA, F(4,30) = 0.140, p = 0.965 vs day −1, n = 7) or GAPDH in the ipsilateral dorsal horn. Moreover, neither the SNL nor the sham operations affected the levels of kalirin or GAPDH in the contralateral dorsal horn (data not shown). These data imply that experimental neuropathic injury induced behavioral allodynia associated with spinal kalirin expression.
Nerve ligation upregulates kalirin expression in dorsal horn neurons
We next examined the cellular localization of SNL-induced kalirin expression using immunohistochemistry analysis. For these experiments, rats were killed, and their spinal cords (L4–L5) were dissected at 7 d after SNL, which was the time of maximal behavioral allodynia. Staining of spinal cord sections for kalirin immunoreactivity demonstrated that SNL (Fig. 1DII, SNL 7D), but not the sham operation (Fig. 1DI, sham 7D), increased the immunoreactivity for kalirin in the ipsilateral (IPSI) compared with the contralateral (CONTRA) dorsal horn. Immunoreactivity for NeuN (Fig. 1DIII; a neuronal marker) was not different between the ipsilateral and contralateral dorsal horns, indicating that the numbers of neurons were approximately the same on both sides. Double staining for kalirin (Fig. 1DII) and NeuN (Fig. 1DIII) revealed that the SNL-enhanced kalirin immunoreactivity colocalized with NeuN (Fig. 1DIV). To test whether kalirin expression also occurred in glial cells, we labeled the sections using antibodies for OX-42 (a microglial marker) and GFAP (an astrocyte marker). Double-staining images demonstrated that the SNL-enhanced kalirin immunoreactivity overlapped with NeuN (Fig. 1DV) but not OX-42 (Fig. 1DVI) or GFAP (Fig. 1DVII), suggesting that SNL-enhanced kalirin expression occurs in dorsal horn neurons but not in glial cells.
Kalirin mRNA-targeting siRNA prevents behavioral allodynia
To provide evidence to further support the hypothesis that SNL-enhanced kalirin expression contributes to the development of neuropathic pain, we examined whether knockdown of spinal kalirin expression could attenuate SNL-induced behavioral allodynia. We examined this possibility using daily administration of small-interfering RNA (siRNA) targeted to kalirin mRNA (Fig. 2A, siKalirin), missense siRNA (Fig. 2A, MS), or polyethylenimine (PEI), a transfection reagent (Fig. 2A, PEI), for 7 d via an intrathecal catheter that was implanted 3 d before nerve ligation. Intrathecal catheter implantation alone (Fig. 2A, it), spinal administration of missense siRNA (10 μg, 10 μl), or PEI (10 μg, 10 μl) did not affect kalirin expression in the dorsal horn of the spinal cord (Fig. 2A, SC) compared with baseline levels in naive animals (Fig. 2A, Naive). In contrast, kalirin mRNA-targeting siRNA (1, 5, and 10 μg, 10 μl) decreased the level of kalirin in a dose-dependent manner (1-way ANOVA, F(6,42) = 10.420, p < 0.001 vs naive, n = 7; **p < 0.001). Additionally, all of these treatments did not affect the level of kalirin in the dorsal root ganglion (Fig. 2B, DRG; L4–L5, 1-way ANOVA, F(6,21) = 0.094, p = 0.996 vs naive, n = 7).
Figure 2.

Kalirin mRNA-targeting siRNA prevents nerve ligation-induced behavioral allodynia. A, Representative Western blot showing kalirin expression in response to administration of siRNA that targeted kalirin mRNA. The level of kalirin was quantified from the lysate of the dorsal horn of the spinal cord (SC, L4–L5), and the level of GAPDH was used as a loading control. Statistical analysis revealed that daily administration of kalirin mRNA-targeting siRNA (siKalirin) at concentrations of 1, 5, and 10 μg (10 μl), but not intrathecal catheter implantation (it), spinal injection of PEI, or missense siRNA (MS), dose-dependently decreased the level of kalirin in the dorsal horn when compared with the naive animals (Naive; **p < 0.05 vs naive, n = 7). IB, immunoblotting. B, In contrast, the level of kalirin in the dorsal root ganglion (DRG) was not affected by any of these treatments (p > 0.05 vs naive, n = 7). C, PEI, missense siRNA (10 μg), and kalirin mRNA-targeting siRNA (10 μg) failed to produce significant differences in rota-rod performance times as measured at days 1, 3, 5, and 7 of treatment when compared with the naive animals (Naive; p > 0.05 vs naive, n = 7). D, All of the treatments, including intrathecal catheter implantation (sham+it), spinal administration of missense siRNA (sham+it+MS), or kalirin mRNA-targeting siRNA (sham+it+siKalirin), failed to produce significant differences in the withdrawal threshold of the von Frey test as measured on days 1, 3, 5, and 7 in sham-operated animals when compared with sham-operated animals without drug administration (p > 0.05 vs sham, n = 7). E, Spinal administration of kalirin mRNA-targeting siRNA (SNL+it+siKalirin), but not administration of missense siRNA (SNL+it+MS) or implantation of an intrathecal catheter (SNL+it) statistically increased the withdrawal threshold tested on days 3, 5, and 7 postsurgery compared with animals that received spinal nerve ligation only (**p < 0.01 vs SNL, n = 7). The gray bar at the bottom indicates the duration of administration. The data show the mean ± SEM.
To eliminate the possibility of an aberrant motor response caused by siRNA administration, the motor ability of the animals was assessed using the rota-rod test. We observed that animals administered either the PEI, missense siRNA, or kalirin mRNA-targeting siRNA (10 μg) did not show effects in performance time as measured 1, 3, 5, or 7 d after administration when compared with the naive group (Fig. 2C, Naive; 2-way ANOVA with repeated measures over time, treatment, F(3,27) = 0.216, p = 0.884; time, F(4,36) = 1.576, p = 0.201; treatment × time, F(12,108) = 0.327, p = 0.982, vs naive, n = 7). These results indicate that our treatments did not affect the motor function of the animals.
In the next series of experiments, we tested whether the knockdown of spinal kalirin expression could prevent SNL-induced behavioral allodynia. The spinal administration of kalirin mRNA-targeting siRNA (Fig. 2E, SNL+it+siKalirin), but not missense siRNA (Fig. 2E,, SNL+it+MS) or catheter implantation alone (Fig. 2E, SNL+it), partially prevented behavioral allodynia as evidenced by a significant increase in the withdrawal threshold at days 3, 5, and 7 postsurgery compared with the animals that received nerve ligation only (Fig. 2E, SNL; 2-way ANOVA with repeated measures over time, treatment, F(3,27) = 32.25, p < 0.001; time, F(4,36) = 324.8, p < 0.001; treatment × time, F(12,108) = 7.830, p < 0.001, vs SNL, n = 7; **p < 0.01). On the other hand, none of these treatments (Fig. 2D, sham+it+siKalirin, sham+it+MS, sham+it) affected the withdrawal threshold at any time in the sham-operated animals (2-way ANOVA with repeated measures over time, treatment, F(3,27) = 0.120, p = 0.947; time, F(4,36) = 0.488, p = 0.744; treatment × time, F(12,108) = 0.169, p = 0.999, vs sham, n = 7). These results support the hypothesis that spinal kalirin plays a critical role in SNL-associated pain hypersensitivity.
Spinal kalirin/PSD-95/NR2B coupling and subsequent NR2B activation
Our previous studies demonstrated that SNL-induced dorsal horn PSD-95–NR2B coupling and subsequent NR2B phosphorylation play a pivotal role in the development of neuropathic pain (Peng et al., 2012a). Considering that kalirin has recently been shown to regulate NMDAR-mediated neural plasticity (Lemtiri-Chlieh et al., 2011), we hypothesized that SNL-induced spinal kalirin expression would provoke downstream PSD-95–NR2B coupling-associated NR2B activation that could participate in neuropathic pain development. To test this hypothesis, we first examined the levels of total NR2B (Fig. 3A, tNR2B), phosphorylated NR2B (pNR2B), and PSD-95 in dorsal horn samples obtained 3 and 7 d postsurgery, which are time points at which our von Frey tests revealed that spinal nerve ligation provoked early and maximal reductions in the withdrawal threshold. These two time points, 3 and 7 d postsurgery, are presumed to reflect the induction (Wang et al., 2011) and maintenance (Lee et al., 2011) phases of neuropathic pain, respectively. As anticipated, immunoblotting analysis of the ipsilateral dorsal horn (L4–L5) demonstrated increased pNR2B band intensity at days 3 and 7 after SNL (Fig. 3A,B, SNL IPSI; 1-way ANOVA, F(2,18) = 9.745, p = 0.001 vs day −1, n = 7; *p < 0.05; **p < 0.01; t test, p = 0.883, 0.017, and 0.002 in day −1, 3, and 7, respectively; #p < 0.05, ##p < 0.01 vs sham ipsilateral, n = 7) but not after the sham operation (sham ipsilateral, 1-way ANOVA, F(2,18) = 0.114, p = 0.892 in pNR2B vs day −1, n = 7) when compared with the presurgery control (day −1). In contrast, SNL did not affect the levels of tNR2B (1-way ANOVA, F(2,18) = 1.120, p = 0.348 vs day −1, n = 7), PSD-95 (1-way ANOVA, F(2,18) = 0.247, p = 0.783 vs day −1, n = 7), or GAPDH at these time points. Moreover, neither SNL nor the sham operation altered the levels of assayed protein in the contralateral dorsal horn at days 3 or 7 postsurgery compared with the presurgery control (data not shown).
Figure 3.

Nerve ligation provokes dorsal horn kalirin–PSD-95 and PSD-95–NR2B coupling. A, Representative Western blot showing the expression of pNR2B, tNR2B, and PSD-95 after spinal nerve ligation (SNL IPSI) or sham operation (Sham IPSI). The lysates of the ipsilateral dorsal horn were obtained on the day before the operation and 3 and 7 d after the operation and were probed with pNR2B-specific, tNR2B-specific, and PSD-95-specific antibodies. The level of GAPDH was used as a loading control. IB, immunoblotting. B, Statistical analysis revealed that spinal nerve ligation statistically increased the band intensity of pNR2B measured on days 3 and 7 postsurgery when compared with the presurgery control (day −1; *p < 0.05, **p < 0.01 vs day −1, #p < 0.05, ##p < 0.01 vs sham operation, n = 7). In contrast, nerve ligation did not affect the levels of tNR2B (p > 0.05 vs day −1, n = 7) or PSD-95 (p > 0.05 vs day −1, n = 7) at these time points. Moreover, the sham operation (sham IPSI) had no effect on the expression of the measured proteins (all p > 0.05, vs day −1, n = 7). The data show mean ± SEM. C, Coimmunoprecipitation of kalirin, PSD-95, tNR2B, and pNR2B in the ipsilateral spinal cord. Dorsal horn samples obtained 7 d after operation were immunoprecipitated with antibodies against kalirin (IP: kalirin) or control IgG (IP: IgG) and were then probed with PSD-95, tNR2B, and pNR2B antibodies. The PSD-95-specific, tNR2B-specific, and pNR2B-specific antibodies labeled immunoreactivity in the immunoprecipitates recognized by the kalirin-selective antibody but not in those recognized by control IgG. In, input sample. D, Spinal nerve ligation notably enhanced the immunoreactivity of kalirin (I and V, red) and pNR2B (VI, green) but not PSD-95 (II, green) in the ipsilateral dorsal horn (IPSI) compared with the contralateral dorsal horn (CONTRA) of the spinal sections obtained 7 d after surgery (SNL 7D). Overlay images showing kalirin fluorescence costained with PSD-95 and pNR2B immunoreactivity (III and VII, yellow). IV, VIII, Amplified images of III and VII, respectively. Each of these immunofluorescence images was replicated in seven sample preparations with similar results each time. Scale bar, 50 μm; thickness, 50 μm. E, F, Dorsal horn samples obtained on the day before operation and 3 and 7 d after operation were immunoprecipitated with antibodies against PSD-95 (IP:PSD-95), tNR2B (IP: tNR2B) or control IgG (IP: IgG) and were then probed with PSD-95, kalirin, tNR2B, and pNR2B antibodies. Spinal nerve ligation enhanced the kalirin-labeled, tNR2B-labeled, and pNR2B-labeled immunoreactivity in the PSD-95 immunoprecipitates and the kalirin-labeled, PSD-95-labeled, and pNR2B-labeled immunoreactivity in the tNR2B immunoprecipitates when compared with the presurgery control. However, no detectable immunoreactivity was labeled by these antibodies in the precipitation recognized by the control IgG. In, input sample.
We next investigated the possibility that spinal kalirin interacts with PSD-95, tNR2B, and pNR2B. The immunoprecipitates recognized by the kalirin-specific antibody (Fig. 3C, IP: Kalirin), but not those recognized by the control IgG (Fig. 3C, IP: IgG), coprecipitated with PSD-95, tNR2B, and pNR2B in the dorsal horn sample obtained at day 7 post-SNL. This result implies that spinal kalirin directly interacts with PSD-95, tNR2B, and pNR2B in the dorsal horn. We next examined the location of the above protein–protein interactions using immunohistochemistry. We observed that SNL notably enhanced the immunoreactivity of kalirin (Fig. 3DI,DV) and pNR2B (Fig. 3DVI) in the ipsilateral (Fig. 3D, IPSI) compared with the contralateral (Fig. 3D, CONTRA) dorsal horn on day 7 after surgery, whereas no difference was observed for PSD-95 (Fig. 3DII). Moreover, double-labeled images demonstrated that the SNL-induced increase in kalirin fluorescence colocalized with PSD-95 and the SNL-enhanced pNR2B immunoreactivity (Fig. 3DIII,DVII, respectively).
Based on these results, we use coprecipitation analysis to investigate whether these protein–protein interactions are involved in the induction and maintenance of neuropathic pain. We immunoprecipitated two candidate targets, PSD-95 and tNR2B, from the dorsal horn samples obtained 3 and 7 d postsurgery. Each immunocomplex was probed with an antibody specifically targeting to PSD-95 or NR2B to serve as a loading control and was then screened for the copurification of kalirin, PSD-95, tNR2B, and pNR2B by immunoblotting. SNL increased the amount of PSD-95-bound kalirin, tNR2B, and pNR2B at days 3 and 7 after surgery (∼1.2-fold, 1.5-fold, and 1.6-fold and 1.6-fold, 1.9-fold, and 2.1-fold above day −1, respectively) compared with the presurgery control (Fig. 3E, day −1). Similarly, the amounts of tNR2B-bound kalirin, PSD-95, and pNR2B were also enhanced at the same time points (Fig. 3F; ∼1.3-fold, 1.8-fold, and 1.1-fold; and 1.5-fold, 1.9-fold, and 1.7-fold above day −1 at days 3 and 7, respectively). These results imply that SNL-enhanced kalirin may lead to PSD-95–NR2B coupling and that the subsequent NR2B activation contributes to both the induction and maintenance of neuropathic pain.
Kalirin knockdown prevents NR2B phosphorylation and protein coupling
To provide further support for the hypothesis that spinal kalirin participates in neuropathic pain development via the interaction between kalirin and PSD-95 and the downstream PSD-95–NR2B coupling-dependent NR2B phosphorylation, we knocked down kalirin expression using intrathecally administered kalirin mRNA-targeting siRNA. Western blotting of the dorsal horn samples obtained 7 d postsurgery revealed that kalirin mRNA-targeting siRNA (10 μg; Fig. 4A,B, 7D+siKalirin), but not missense siRNA (10 μg; Fig. 4A,B, 7D+MS), attenuated SNL-induced spinal NR2B phosphorylation by significantly decreasing the intensity of the pNR2B band compared with animals that did not receive siRNA (Fig. 4A,B, 7D; 1-way ANOVA, F(2,18) = 8.074, p = 0.003 vs 7D, n = 7; **p < 0.05). However, kalirin mRNA-targeting siRNA did not affect the levels of tNR2B, PSD-95, or GAPDH (1-way ANOVA, F(2,18) = 0.628, p = 0.544 in tNR2B; F(2,18) = 0.551, p = 0.585 in PSD-95, respectively, vs 7D, n = 7). Similarly, in the precipitates recognized by the PSD-95-selective antibody (Fig. 4C, IP: PSD-95), the immunoreactivity probed by the antibody against PSD-95 demonstrated a stable loading control across groups, whereas the amounts of PSD-95-bound kalirin, tNR2B, and pNR2B measured 7 d after nerve ligation were significantly reduced by the kalirin mRNA-targeting siRNA (Fig. 4C, 7D+siKalirin; ∼0.4-fold, 0.6-fold, and 0.4-fold below 7D, respectively) but not by the missense siRNA (Fig. 4C, 7D+MS; ∼1.0-fold, 1.1-fold, and 1.1-fold of 7D, respectively) when compared with animals that did not receive siRNA (Fig. 4C, 7D). In addition, the SNL-enhanced colocalizations of kalirin and PSD-95 (Fig. 4DIII), kalirin and pNR2B (Fig. 4DVII), and pNR2B and PSD-95 (Fig. 4DXI) at day 7 postoperation were all attenuated by the kalirin mRNA-targeting siRNA (Fig. 4DIV,DVIII,DXII, respectively). These results provide further support for the hypothesis that the SNL-associated kalirin–PSD-95 coupling in the dorsal horn can induce downstream PSD-95–NR2B coupling-dependent NR2B phosphorylation that participates in neuropathic pain development.
Figure 4.

Kalirin mRNA-targeting siRNA prevents NR2B phosphorylation and coupling with PSD-95. A, Representative Western blot showing the expression of pNR2B, tNR2B, and PSD-95 in response to the administration of siRNA that targeted kalirin mRNA. The lysates obtained from the ipsilateral dorsal horn 7 d after spinal nerve ligation were probed with antibodies against pNR2B, tNR2B, and PSD-95. The level of GAPDH was used as a loading control. IB, immunoblotting. B, Statistical analysis revealed that the spinal administration of kalirin-targeting siRNA (10 μg, 10 μl; 7D+siKalirin), but not missense siRNA (10 μg, 10 μl; 7D+MS), decreased the pNR2B band intensity compared with animals that received nerve ligation only (7D; **p < 0.01 vs 7D, n = 7). In contrast, neither treatment affected the tNR2B or PSD-95 band intensities (p > 0.05 vs 7D, n = 7). The data show mean ± SEM. C, Coimmunoprecipitation of PSD-95, kalirin, tNR2B, and pNR2B in the ipsilateral spinal cord. Dorsal horn samples obtained 7 d after operation were immunoprecipitated with antibodies against PSD-95 (IP: PSD-95) or control IgG (IP: IgG) and were then probed with PSD-95, kalirin, tNR2B, and pNR2B antibodies. In, input sample. The immunoreactivity probed by the kalirin-selective, tNR2B-selective, and pNR2B-selective antibodies in the PSD-95 immunoprecipitates was reduced by the kalirin mRNA-targeting siRNA, but not by the missense siRNA. No immunoreactivity was detected by PSD-95-selective, kalirin-selective, tNR2B-selective, and pNR2B-selective antibodies in the control IgG-recognized precipitates. In, input protein. D, Overlay images show the colocalized immunoreactivities of kalirin (I, red) with PSD-95 (II, green), kalirin (V, red), and pNR2B (VI, green), as well as pNR2B (IX, red) with PSD-95 (X, green) in the dorsal horn of the spinal slices obtained 7 d after spinal nerve ligation (SNL 7D; III, VII, and XI, yellow), which were remarkably reduced by kalirin mRNA-targeting siRNA (SNL 7D+siKalirin; IV, VIII, and XII, respectively). Each of these immunofluorescence images was replicated in seven sample preparations with similar results each time. Scale bar, 50 μm; thickness, 50 μm.
GSK-650394 antagonizes allodynia and protein coupling
Our laboratory has recently demonstrated that CFA-induced paw inflammation is associated with dorsal horn SGK1-mediated glutamatergic receptor activation (Peng et al., 2012b). Considering that kalirin can be phosphorylated by kinases implicated in the neural plasticity (Kiraly et al., 2011a) that regulates NR2B-dependent glutamatergic neurotransmission (Kiraly et al., 2011b), we hypothesized that nerve injury would provoke spinal SGK1 phosphorylation that would participate in the development of neuropathic pain through the kalirin/PSD-95/pNR2B cascade. To test this hypothesis, we first tested whether SNL induces spinal SGK1 phosphorylation using Western blot analysis. SNL (SNL ipsilateral, 1-way ANOVA, F(2,18) = 10.850, p < 0.001 vs day −1, n = 7; *p < 0.05, **p < 0.01; t test, p = 0.884, 0.002 and <0.001 in day −1, 3, and 7, respectively, vs sham ipsilateral, n = 7; ##p < 0.01), but not the sham operation (sham ipsilateral, 1-way ANOVA, F(2,18) = 0.220, p = 0.805 vs day −1, n = 7), upregulated the expression of pSGK1 in the ipsilateral dorsal horn without affecting the levels of total SGK1 (tSGK1) compared with the presurgery control (Fig. 5A,B, day −1; SNL ipsilateral, 1-way ANOVA, F(2,18) = 0.908, p = 0.420 vs day −1 and sham ipsilateral, F(2,18) = 0.513, p = 0.606 vs day −1) or GAPDH at days 3 and 7 postsurgery. In contrast, neither SNL nor the sham operation affected the levels of pSGK1, tSGK1, or GAPDH in the contralateral dorsal horn at these time points (data not shown). To determine the specifics of the interaction between SGK1 and the kalirin/PSD-95/pNR2B cascade, the protein–protein interactions between SGK1 and pSGK, kalirin, PSD-95, tNR2B, and pNR2B were examined via coprecipitation analyses of dorsal horn samples obtained 7 d postsurgery. We observed that pSGK1, kalirin, PSD-95, tNR2B, and pNR2B coprecipitated with tSGK1 in the SGK1 antibody-recognized immunocomplex (Fig. 5C).
Figure 5.

GSK-650394 antagonizes protein–protein interactions and behavioral allodynia. A, Representative Western blot showing the expression of pSGK1 and tSGK1 after spinal nerve ligation (SNL IPSI) or sham operation (sham IPSI). The lysates of the ipsilateral dorsal horn were probed with antibodies against pSGK1 and SGK1, and the level of GAPDH was used as a loading control. IB, immunoblotting. B, Statistical analysis revealed that spinal nerve ligation significantly increased pSGK1 band intensities measured on days 3 and 7 postsurgery (*p < 0.05, **p < 0.01 vs day −1, n = 7; ##p < 0.01 vs sham operation, n = 7) but did not affect tSGK1 band intensity when compared with the presurgery control (day −1; p > 0.05 vs day −1). The sham operation had no effect on the levels of the measured proteins at these time points. C, Coimmunoprecipitation of tSGK1, pSGK1, kalirin, PSD-95, tNR2B, and pNR2B in the ipsilateral spinal cord. Dorsal horn samples obtained 7 d after operation were immunoprecipitated with antibodies against tSGK1 (IP: tSGK1) or control IgG (IP: IgG) and were then probed with tSGK1, pSGK1, kalirin, PSD-95, tNR2B, and pNR2B antibodies. In, input sample. Antibodies selective against pSGK1-, kalirin-, PSD-95-, tNR2B-, and pNR2B-labeled immunoreactivity in the precipitates recognized by the tSGK1-specific antibody, but not in those recognized by the control IgG. In, input protein. D, Intrathecal administration of GSK-650394 at concentrations of 10, 30, and 100 nm (10 μl), but not vehicle solution (SNL 3D+Veh and SNL 7D+Veh, respectively), dose-dependently increased the withdrawal latency of the ipsilateral hindpaw at 1–3 and 1–5 h after injection at days 3 and 7 postsurgery (SNL 3D+GSK and SNL 7D+GSK, respectively) compared with animals that received nerve ligation only (SNL 3D and SNL 7D, respectively; *p < 0.05, **p < 0.01 vs SNL 3D or SNL 7D, n = 7). E, Neither vehicle solution (7D+Veh) nor GSK-650394 at a concentration of 100 nm (7D+GSK, 10 μl) affected the withdrawal threshold of the contralateral hindpaw (CONTRA) tested 2 h after injection on day 7 postsurgery (p > 0.05 vs 7D, n = 7). The data show mean ± SEM. F, In the tSGK antibody-recognized precipitates (IP: tSGK1) obtained from dorsal horn samples dissected at days 3 and 7 postsurgery (3D and 7D, respectively), the immunoreactivities probed by the pSGK1-selective, kalirin-selective, PSD-95-selective, tNR2B-selective, and pNR2B-selective antibodies were significantly reduced by intrathecal administration of GKS-650394 (100 nm, 10 μl; 3D+GSK and 7D+GSK, respectively) but not by the vehicle solution (3D+Veh and 7D+Veh, respectively).
We then pharmacologically blocked SGK1 phosphorylation to further elucidate its role in the induction and maintenance of neuropathic pain. Von Frey tests at days 3 and 7 postsurgery demonstrated that the spinal administration of GSK-650394 (an SGK1 antagonist; SNL 3D+GSK and SNL 7D+GSK; 10, 30, 100 nm, 10 μl), but not vehicle solution (SNL 3D+Veh and SNL 7D+Veh) dose-dependently ameliorated SNL-induced behavioral allodynia as evidenced by significant decreases in the withdrawal threshold of the ipsilateral hindpaw when compared with animals that received no injection (Fig. 5D, SNL 3D and SNL 7D) at 1–3 (2-way ANOVA with repeated measures over time, treatment, F(4,24) = 35.740, p < 0.001; time, F(5,30) = 8.826, p < 0.001; treatment × time, F(20,120) = 4.343, p < 0.001 vs SNL 3D; **p < 0.01, n = 7) and 1–5 h after injection (treatment, F(4,24) = 67.240, p < 0.001; time, F(5,30) = 14.110, p < 0.001; treatment × time, F(20,120) = 4.421, p < 0.001 vs SNL 7D; *p < 0.05, **p < 0.01, n = 7). Neither the vehicle solution (Fig. 5E, 7D+Veh) nor 100 nm GSK-650394 (Fig. 5E, 7D+GSK; 1-way ANOVA, F(2,18) = 0.057, p = 0.944 vs 7D, n = 7) affected the withdrawal threshold of the contralateral hindpaw 2 h after injection, which was the time point that GSK-650394 exhibited its maximal effect in the von Frey test on the ipsilateral hindpaw. Additionally, the immunoreactivity probed by the tSGK1 antibody demonstrated a stable loading control in the tSGK1 antibody-recognized precipitates obtained from dorsal horn samples dissected at days 3 and 7 postsurgery. Moreover, the increases in tSGK1-bound pSGK1, kalirin, PSD-95, tNR2B, and pNR2B immunoreactivities caused by nerve ligation (Fig. 5F IP: tSGK1; 3D and 7D, respectively) were significantly reduced by intrathecal administration of GSK-650394 (100 nm, 10 μl; ∼0.5-fold, 0.4-fold, 0.6-fold, 0.5-fold, and 0.3-fold below day −1 in 3D+GSK; and ∼0.4-fold, 0.4-fold, 0.6-fold, 0.3-fold, and 0.3-fold below day −1 in 7D+GSK, respectively) but not by administration of the vehicle solution (∼0.9-fold, 1.0-fold, 1.0-fold, 1.0-fold, and 0.9-fold compared with day −1 in 3D+Veh and ∼0.9-fold, 1.1-fold, 1.1-fold, 1.0-fold, and 1.0-fold compared with day −1 in 7D+Veh, respectively). Collectively, the results obtained from the antagonism of SGK1 activation provide pharmacological evidence that spinal SGK1 phosphorylation and the subsequent activation of the kalirin/PSD-95/pNR2B cascade participate in the induction and maintenance of neuropathic pain caused by nerve ligation.
Kalirin knockdown does not prevent SGK1 phosphorylation
The possibility that nerve injury-induced spinal SGK1 phosphorylation triggers the downstream kalirin-dependent cascade and mediates neuropathic pain development was further evaluated by intrathecal administration of siRNA targeting to kalirin mRNA. We observed that neither the kalirin mRNA-targeting siRNA (Fig. 6A,B, 7D+siKalirin; 10 μg) nor missense siRNA (Fig. 6A,B, 7D+MS; 10 μg) affected the levels of tSGK (1-way ANOVA, F(2,18) = 0.186, p = 0.831 vs 7D, n = 7), pSGK (1-way ANOVA, F(2,18) = 1.096, p = 0.355 vs 7D, n = 7), or GAPDH measured 7 d after surgery (Fig. 6A,B, 7D). Similarly, neither of these treatments affected the levels of tSGK (1-way ANOVA, F(2,9) = 0.102, p = 0.903 vs 7D, n = 7) or pSGK (1-way ANOVA, F(2,9) = 0.119, p = 0.888 vs 7D, n = 7) in the dorsal root ganglion (Fig. 6C,D, DRG). These results suggest that SNL-induced spinal SGK1 phosphorylation is upstream of kalirin in the mediation of the development of neuropathic pain.
Figure 6.
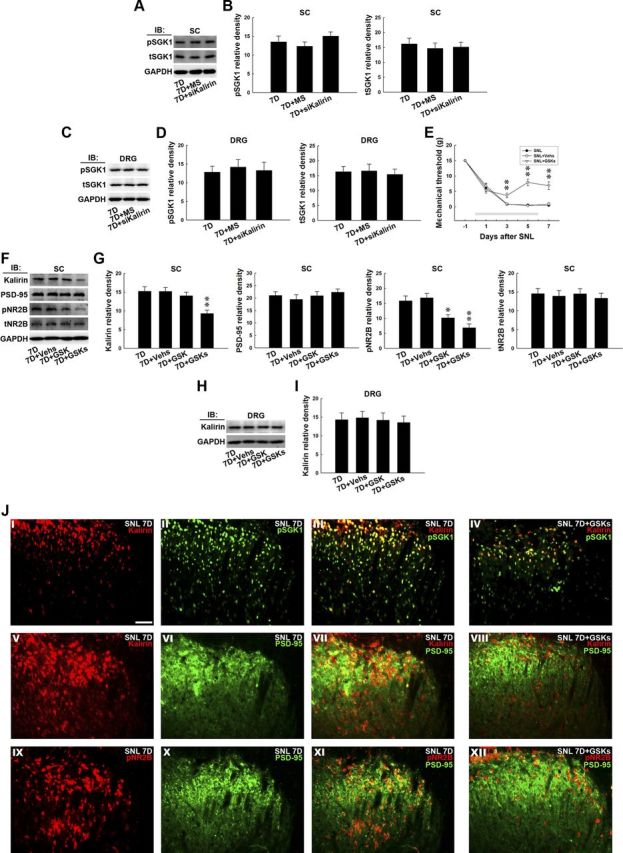
GSK-650394 prevents nerve ligation-provoked allodynia and NR2B phosphorylation. A, Representative Western blot showing the expression of pSGK1 and total tSGK1 in response to daily administration of kalirin mRNA-targeting siRNA for 7 d. The dorsal horn lysates obtained 7 d postspinal nerve ligation were probed with antibodies against pSGK1 and tSGK1. The level of GAPDH was used as a loading control. IB, immunoblotting. B, The statistical analysis revealed that spinal administration of neither the kalirin mRNA-targeting siRNA (10 μg, 10 μl; 7D+siKalirin) nor missense siRNA (10 μg, 10 μl; 7D+MS) affected the level of pSGK1 or tSGK1 in the dorsal horn of spinal cord samples (SC) when compared with animals that received nerve ligation only (7D; p > 0.05 vs 7D, n = 7). C, The lysates of the dorsal root ganglion (DRG) obtained 7 d postspinal nerve ligation were probed with antibodies against pSGK1 and tSGK1. The level of GAPDH was used as a loading control. D, Spinal administration of neither the kalirin mRNA-targeting siRNA nor missense siRNA affected the level of pSGK1 or tSGK1 in the dorsal root ganglion when compared with animals that received nerve ligation only (p > 0.05 vs 7D, n = 7). E, The decreases in the mechanical threshold of the ipsilateral hindpaw caused by spinal nerve ligation (SNL) measured on days 3, 5, and 7 postsurgery were significantly ameliorated by the daily administration of GSK-650394 (SNL+GSKs; 100 nm, 10 μl, i.t., for 7 d) but not the vehicle solution (SNL+Vehs; for 7 d, **p < 0.01 vs SNL, n = 7). The gray bar at the bottom indicates the duration of administration. F, Representative Western blot showing the expression of dorsal horn kalirin, PSD-95, tNR2B, and pNR2B in response to daily GSK-650394 administration. The dorsal horn lysates obtained 7 d after spinal nerve ligation were probed with antibodies against kalirin, PSD-95, tNR2B, and pNR2B. The level of GAPDH was used as a loading control. G, Statistical analysis revealed that the daily injection of GSK-650394 (7D+GSKs), but not the vehicle solution (7D+Vehs), decreased the band intensities of kalirin (**p < 0.01 vs 7D, n = 7) and pNR2B (*p < 0.05, **p < 0.01 vs 7D, n = 7). A single bolus of GSK-650394 on day 7 also decreased the band intensity of pNR2B but did not affect that of kalirin (t test, p = 0.038 vs 7D, n = 7; *p < 0.05). None of the treatments, including the daily administration of GSK-650394 or vehicle for 7 d or a bolus of GSK-650394 at day 7, affected the expression of PSD-95 or tNR2B. H, Representative Western blot showing the expression of dorsal root ganglion kalirin in response to daily GSK-650394 administration. The lysates of the dorsal root ganglion obtained 7 d after spinal nerve ligation were probed with antibodies against kalirin. The level of GAPDH was used as a loading control. I, Statistical analysis revealed that none of the treatments, including daily injection of GSK-650394, the vehicle solution, or a single bolus of GSK-650394 on day 7, affected the level of kalirin in the dorsal root ganglion (p > 0.05 vs 7D, n = 7). J, In the spinal slices obtained 7 d after spinal nerve ligation (SNL 7D), overlay images show the colocalized immunoreactivities of kalirin (I, red) and pSGK1 (II, green), kalirin (V, red), and PSD-95 (VI, green), as well as pNR2B (XI, red) and PSD-95 (X, green) in the dorsal horn (III, VII, and XI, yellow), which were remarkably reduced by daily administration of GSK-650394 (IV, VIII, and XII; 7D+GSKs). Each of these immunofluorescence images was replicated in seven sample preparations with similar results each time. Scale bar, 50 μm; thickness, 50 μm.
Daily GSK-650394 prevents behavioral allodynia
To further characterize the role of spinal SGK1 activation in neuropathic pain and to develop a possible therapeutic strategy, we administered GSK-650394 daily (from day 0 to 6 postsurgery; 100 nm, 10 μl, i.t.) to SNL animals and assayed the pain-related behavior, protein expression, and location in the dorsal horn. Von Frey tests demonstrated that daily GSK-650394 administration alleviated SNL-induced allodynia at days 3, 5, and 7 postsurgery (SNL+GSKs; 2-way ANOVA with repeated measures over time, treatment, F(2,18) = 50.310, p < 0.001; time, F(4,36) = 156.600, p < 0.001; treatment × time, F(8,72) = 11.510, p < 0.001, **p < 0.01, n = 7) compared with animals that received no injection (SNL). In contrast, the administration of vehicle solution exhibited no effect on the SNL-induced allodynia (Fig. 6E, SNL+Vehs). Similarly, daily administration of GSK-650394 (Fig. 6F,G, 7D+GSKs), but not the vehicle solution (Fig. 6F,G, 7D+Vehs), attenuated the SNL-enhanced kalirin and pNR2B expression (Fig. 6F,G, 7D; 1-way ANOVA, F(3,24) = 7.259, p = 0.001 vs 7D, n = 7; p < 0.01 in kalirin and 1-way ANOVA, F(3,24) = 11.520, p < 0.001 vs 7D, n = 7; *p < 0.05, **p < 0.01 in pNR2B) without affecting the levels of PSD-95 and tNR2B in the dorsal horn (1-way ANOVA, F(3,24) = 0.509, p = 0.679 in PSD-95 and F(3,24) = 0.159, p = 0.922 in tNR2B, n = 7). Additionally, a single bolus of GSK-650394 administered at day 7 postsurgery did not prevent SNL-enhanced kalirin expression but significantly decreased the expression of pNR2B (7D+GSK, 100 nm, 10 μl). In contrast, these treatments did not affect the level of kalirin found in the dorsal root ganglion 7 d after surgery (Fig. 6H,I, DRG;1-way ANOVA, F(3,12) = 0.079, p = 0.970 vs 7D). Moreover, at day 7 postsurgery, the colocalized kalirin-pSGK1, kalirin-PSD-95, and pNR2B-PSD95 immunoreactivities (Fig. 6J; III, VII, and XI, respectively) caused by nerve ligation were markedly reduced by daily GSK-650394 administration (SNL 7D+GSK). Together, these results suggest that SNL may induce spinal SGK1 phosphorylation, which participates in neuropathic pain development through the downstream kalirin/PSD-95/pNR2B cascade.
Discussion
In the present study, we found that experimental nerve injury provoked spinal SGK1 phosphorylation that possibly interacted with and enhanced the expression of kalirin, which subsequently induced the PSD-95–NR2B coupling-related NR2B subunit phosphorylation in dorsal horn neurons to underlie neuropathic pain development (Fig. 7).
Figure 7.

Schematic of the signaling caused by SGk1–kalirin interaction. Spinal nerve ligation (SNL) can promote spinal SGK1 phosphorylation that possibly interacts with and enhances the expression of kalirin, which subsequently induces PSD-95–NR2B coupling-related NR2B subunit phosphorylation in the dorsal horn.
A growing body of evidence has demonstrated Rho GTPases activated by Rho-GEFs play a key role in the development of the adult CNS (Ma et al., 2008b). Kalirin, a multifunctional Rho-GEF, is the primary product of the extensively spliced KALRN gene in the mammalian CNS (Kiraly et al., 2011a). Over the last decade, studies have identified roles for kalirin in synaptogenesis (Ma et al., 2003, 2008a). Mice lacking kalirin display, in addition to a decreased number of dendritic spines, a decrease in number of excitatory synapses, diminished LTP in hippocampal neurons (Ma et al., 2003, 2008a), and behavioral impairments in learning tasks (Ma et al., 2008b). Correlated with studies linking kalirin expression in brain areas to the plasticity underlying memory consolidation, the results of this study demonstrated that SNL induced allodynia accompanied by kalirin expression in spinal neurons, and that genetic knockdown of spinal kalirin expression protected animals from SNL-induced allodynia. These findings provide evidence extending the role of kalirin in the neural plasticity mediating neuropathic pain at the spinal cord level.
Studies investigating visceral pain demonstrated that acute irritation of pelvic organs provokes NR2B subunit phosphorylation in the lumbosacral dorsal horn (Peng et al., 2008a,b; 2009a,c). Focal knockdown of spinal NR2B subunit expression (Peng et al., 2009a; Chang et al., 2011) and spinal administration of agents selectively antagonizing NR2B phosphorylation (Peng et al., 2008a,b) both thwart irritation-induced visceral hyper-reflexia, suggesting that spinal NR2B subunit phosphorylation is involved in pain pathology (Swartjes et al., 2011). The PSD-95 is organized with the juxtamembrane intracellular domains of postsynaptic NMDARs to form signaling complexes that regulate channel function and cytoskeleton dynamics (Husi and Grant, 2001) and, therefore, PSD-95 modifies the efficacy of NMDAR-mediated neurotransmission (Elias et al., 2008). Our previous studies revealed that PSD-95–NR2B coupling in the dorsal horn contributes to visceral pain development (Peng et al., 2009a, 2010). Similarly, neuropathic injury is also demonstrated to induce spinal PSD-95–NR2B coupling (Miletic et al., 2010; Peng et al., 2012a), and the intrathecal delivery of NR2B-mimetic peptides to perturb PSD-95–NR2B interactions attenuates formalin-induced pain behavior (D'Mello et al., 2011), implying that the spinal PSD-95–NR2B association is crucial for neuropathic/postinflammatory pain development. Furthermore, studies have revealed a direct link between kalirin and PSD-95-dependent NR2B activation by showing that kalirin colocalizes with PSD-95 and NMDARs on the dendritic spines of excitatory synapses in hippocampal neurons (Ma et al., 2008a), and purified PSDs obtained from the cerebral cortex of kalirin knock-out animals contain reduced NR2B levels (Ma et al., 2008b). Additionally, electrophysiological analyses demonstrated that animals lacking kalirin have an overall decrease in the NMDAR/AMPAR current ratio caused by reducing the current conducted of NR2B subunit-containing NMDARs (Kiraly et al., 2011b). Together, these studies suggest that kalirin may participate in plasticity by regulating PSD-95–NR2B interaction-dependent NR2B activation. In the present study, we demonstrated that SNL-induced allodynia was associated with spinal kalirin expression, NR2B subunit phosphorylation, and kalirin–PSD-95 coupling at days 3 and 7 postsurgery, which are time points presumed to reflect the induction (Wang et al., 2011) and maintenance (Lee et al., 2011) phases of neuropathic pain, respectively. Both SNL-enhanced kalirin and pNR2B immunofluorescence in dorsal horn neurons were shown to colocalize with PSD-95. Moreover, the kalirin–PSD-95–pNR2B couplings were further confirmed by coprecipitation studies showing that SNL increased the immunoreactivity of PSD-95-bound kalirin and pNR2B in PSD95 precipitates as well as tNR2B-bound kalirin, PSD-95, and pNR2B in tNR2B precipitates at days 3 and 7 postsurgery. These results provide the first evidence that spinal kalirin can participate in both the induction and the maintenance of neuropathic pain via downstream PSD-95–NR2B coupling-related NMDAR NR2B subunit phosphorylation. This finding is further supported by our findings that knockdown of spinal kalirin expression decreased SNL-enhanced NR2B phosphorylation and ameliorated behavioral allodynia at days 3 and 7 postsurgery. Moreover, the physical coupling of PSD-95 with kalirin and pNR2B in the dorsal horn on days 3 and 7 postsurgery as well as the colabeling of kalirin with PSD-95 and of PSD-95 with pNR2B in the spinal slices were also blocked by siRNA targeting to kalirin mRNA.
SGK plays a crucial role in memory formation because the expression of this gene is significantly higher in the hippocampus of fast-learning compared with slow-learning rats subjected to water-maze tasks (Tsai et al., 2002). SGK phosphorylation has recently been linked to learning-related neural plasticity because high-frequency stimulation induces LTP accompanied by SGK phosphorylation, and transfection of the dominant-negative mutant SGK, which antagonizes SGK phosphorylation, impairs hippocampal LTP maintenance (Ma et al., 2006). In addition to its recognized role in the plasticity underlying memory and learning, dorsal horn SGK1 expression was shown to increase during ankle inflammation, and the intrathecal infusion of SGK1-targeting antisense delays the onset of inflammation-induced hyperalgesia (Géranton et al., 2007). Our published data demonstrated that CFA-induced paw hyperalgesia is associated with dorsal horn SGK1 phosphorylation, and that pharmacological antagonism of spinal SGK1 activation ameliorates inflammation-associated hyperalgesia; we thus proposed that spinal SGK1 activation crucially modulates pain-associated neural plasticity (Peng et al., 2012a). In the present study, SNL-induced allodynia was accompanied by spinal SGK1 phosphorylation. Neuropathic injury induced coupling of tSGK1 with pSGK1, kalirin, PSD-95, and pNR2B in the dorsal horn. Moreover, spinal pSGK1 and PSD-95 immunofluorescence colocalized with kalirin after SNL. In contrast to the kalirin knockdown, which did not affect SNL-enhanced SGK1 phosphorylation, daily administration of an agent that antagonized SGK1 activation attenuated above protein–protein colocalization/coupling and ameliorated the allodynia observed at days 3 and 7 postsurgery. Together, these findings suggest that neuropathic injury induces spinal SGK1 phosphorylation, and that the phosphorylated SGK1 may subsequently interact with kalirin to participate in both the induction and the maintenance of neuropathic pain via the kalirin/PSD-95/pNR2B cascade. Additionally, a bolus of GSK-650394 at 7 d postsurgery reversed SNL-induced behavior allodynia and spinal NR2B phosphorylation without affecting the levels of kalirin. Taking into consideration our results, which demonstrated that a bolus of GSK-650394 at 7 d postsurgery reversed the SNL-induced coupling between tSGK1 and kalirin, PSD-95, and tNR2B, as well as pNR2B, and considering that the activity of kalirin is regulated by types of kinases, we cannot exclude the possibility that SGK1 rapidly regulates signaling outside the kalirin/PSD-95/NR2B cascade to induce the NR2B activation underlying neuropathic pain. Still, we suggest that acute SGK1 administration might disrupt the protein–protein interactions of the kalirin/PSD-95/pNR2B cascade to ameliorate SNL-associated allodynia. In contrast to agents that were applied before nerve injury, the finding that a bolus of GSK-650394 at 7 d postsurgery reversed established behavioral allodynia suggests a possible therapeutic strategy for the treatment of neuropathic pain in which nerve injury occurs before a medical regimen, as in clinical scenarios.
Among the SGK1–3 isoforms (Lang et al., 2006), SGK1 and SGK3 are widely expressed in the CNS (Kobayashi et al., 1999), whereas SGK2 is present at low levels in the brain (Kobayashi et al., 1999). The forced-swim stress progressively elevates the levels SGK1 and SGK3 but not SGK2 in the prefrontal cortex, suggesting that SGK2 is not significantly involved in the functions of the CNS (Yuen et al., 2011). Because no commercial drug is currently available to antagonize SGK3 activity, we tested the role of spinal SGK1 in neuropathic pain development. However, further study is needed to elucidate the potential roles of SGK3 and SGK2 in pain pathology. Moreover, the activation of SGK1 depends on the phosphorylation of serine/threonine residues, including Thr256 (Baskin and Sayeski, 2012), Ser78 (Larsson and Broman, 2008), Ser397, Ser401 (Chen et al., 2009), and Ser422 (Chen et al., 2009; Baskin and Sayeski, 2012). As stated above, tetanus-induced LTP in the hippocampus is accompanied by SGK1 phosphorylation at Ser422, and transfection of a dominant-negative SGK1 specifically targeting SGK1 phosphorylation at Ser422 impairs LTP, suggesting that SGK1 Ser422 phosphorylation plays a crucial role in memory-related neural plasticity (Ma et al., 2006). Considering that the spinal neural plasticity underlying inflammatory/neuropathic pain shares similar mechanisms with LTP (Ji et al., 2003), we examined the participation of SGK1 in neuropathic pain development by measuring Ser422 phosphorylation in this study. However, further study is needed to elucidate the possible role of SGK1 phosphorylation at serine/threonine residues other than Ser422.
Several isoforms of kalirin are produced in the CNS through alternative splicing (McPherson et al., 2002). Our Western blot analysis demonstrated that the molecular weight of the kalirin band that was enhanced by nerve injury was ∼190 kDa; this molecular weight corresponds to rat kalirin-7 (i.e., isoform 2 of the human kalirin gene) (Tsai et al., 2012). Moreover, the small-interfering RNA probe, which was targeted to the mRNA of kalirin, knocked down the 190 kDa kalirin-7 band and prevented neuropathic injury-associated behavioral pain. Based on these observations, we suggest that spinal kalirin-7 plays a crucial role in neuropathic pain-associated plasticity. This hypothesis is corroborated by studies that link kalirin-7 with the plasticity mediating memory consolidation (Ma et al., 2003, 2008a) and learning (Ma et al., 2008b) in certain brain areas. However, clarification of the roles of isoforms other than kalirin-7 in pain pathology requires further study.
As shown by the NeuN staining in Figure 1, it is notable that the vast majority of kalirin appears to be in the soma, rather than in dendrites or terminals. These results seem to be somewhat at odds with the presumption that kalirin is important in the formation of the synaptic spine. Although the underlying cause for this discrepancy is unclear, we propose the morphology of the dorsal horn neuron in the substantia gelatinosa could be a possible cause because the dendrite extension of these neurons in the medial-lateral direction were rare and, when present, was short in the transverse sections (Hantman et al., 2004). Although rostrocaudal dendrites were observed in the sagittal sections (Hantman et al., 2004), it was difficult to observe these dendrites in the transverse slices seen in this study. Therefore, the immunofluorescence of short dendritic spines surrounding the cell body was detected and, as a result, the image appears to show the shape of a neuron (Gao et al., 2008). Even though some studies investigating spine remodeling in the spinal slice did elevate the degree of synaptogenesis of dendrites by counting the “spine-like structures,” which are morphologically identified using Golgi staining (Tan et al., 2008, 2012), this technique was not suitable for our present study, in which specific protein targets were labeled to elucidate their interactions. Moreover, the technique would have been inappropriate considering that kalirin is synthesized within the cell body and is then transcripted to the dendritic spine. The staining procedure that we used detects immunoreactivity but cannot determine which sites of neuron are labeled. We cannot exclude the possibility that we detected in the cell bodies kalirin protein upregulated by the neuropathic injury, thereby forming images with shapes like those of neurons. On the other hand, result of immunofluorescence of cultured neurons demonstrated the immunoreactivity of kalirin colocalizes with PSD-95 in the dendritic spine (Han et al., 2010), although our immunofluorescence images cannot directly show what actually occurs in the dendritic spine. Considering that PSD-95 is a postsynaptic anchoring protein presumed to locate in the dendritic spine, the PSD-95-recognized immunocomplexes could be the postsynaptic component of the neuron (i.e., very similar to the dendritic spine) (Han et al., 2010). The results of immunoprecipitation in this study demonstrated spinal nerve injury enhanced kalirin immunoreactivity in the PSD-95-recognized precipitation obtained form the dorsal horn. These data, at least in part, provide information about the role of kalirin in the dorsal horn neuron when encountering neuropathic injury. However, further study is needed to elucidate the underlying cause for this discrepancy.
Footnotes
This work was supported by National Science Council of Taiwan Grants NSC 98-2320-B-040-006-MY3 and NSC 101-2320-B-039-013-MY3 to T.-B.L. and NSC 99-2320-B-039-036 and 101-2320-B-715-001-MY3 to H.-Y.P.; and by China Medical University Grants CMU99-S-19 and CMU99-lab to T.-B.L. and DMR-100-160 to H.-Y.P.
References
- Baskin R, Sayeski PP. Angiotensin II mediates cell survival through upregulation and activation of the serum and glucocorticoid inducible kinase 1. Cell Signal. 2012;24:435–442. doi: 10.1016/j.cellsig.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Peng HY, Wu HC, Lai CY, Hsieh MC, Lin TB. Cyclophosphamide induces NR2B phosphorylation-dependent facilitation on spinal reflex potentiation. Am J Physiol Renal Physiol. 2011;300:F692–F699. doi: 10.1152/ajprenal.00531.2010. [DOI] [PubMed] [Google Scholar]
- Chen W, Chen Y, Xu BE, Juang YC, Stippec S, Zhao Y, Cobb MH. Regulation of a third conserved phosphorylation site in SGK1. J Biol Chem. 2009;284:3453–3460. doi: 10.1074/jbc.M807502200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello R, Marchand F, Pezet S, McMahon SB, Dickenson AH. Perturbing PSD-95 interactions with NR2B-subtype receptors attenuates spinal nociceptive plasticity and neuropathic pain. Mol Ther. 2011;19:1780–1792. doi: 10.1038/mt.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias GM, Elias LA, Apostolides PF, Kriegstein AR, Nicoll RA. Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc Natl Acad Sci U S A. 2008;105:20953–20958. doi: 10.1073/pnas.0811025106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géranton SM, Morenilla-Palao C, Hunt SP. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J Neurosci. 2007;27:6163–6173. doi: 10.1523/JNEUROSCI.1306-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin RS. An Epac-dependent pain pathway. J Neurosci. 2005;25:8113–8114. doi: 10.1523/JNEUROSCI.3044-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Cheng C, Zhao J, Chen M, Li X, Shi S, Niu S, Qin J, Lu M, Shen A. Developmental regulation of PSD-95 and nNOS expression in lumbar spinal cord of rats. Neurochem Int. 2008;52:495–501. doi: 10.1016/j.neuint.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Han S, Nam J, Li Y, Kim S, Cho SH, Cho YS, Choi SY, Choi J, Han K, Kim Y, Na M, Kim H, Bae YC, Choi SY, Kim E. Regulation of dendritic spines, spatial memory, and embryonic development by the TANC family of PSD-95-interacting proteins. J Neurosci. 2010;30:15102–15112. doi: 10.1523/JNEUROSCI.3128-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantman AW, van den Pol AN, Perl ER. Morphological and physiological features of spinal substantial gelatinosa neurons defined by green fluorescent protein expression. J Neurosci. 2004;24:836–842. doi: 10.1523/JNEUROSCI.4221-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori K, Ozaki N, Suzuki S, Sugiura Y. Upregulations of P2X3 and ASIC3 involve in hyperalgesia induced by cisplatin administration in rats. Pain. 2010;149:393–405. doi: 10.1016/j.pain.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Husi H, Grant SG. Isolation of 2000-kDa complexes of N-methyl-D-aspartate receptor and postsynaptic density 95 from mouse brain. J Neurochem. 2001;77:281–291. doi: 10.1046/j.1471-4159.2001.t01-1-00248.x. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory shares similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Katano T, Nakazawa T, Nakatsuka T, Watanabe M, Yamamoto T, Ito S. Involvement of spinal phosphorylation cascade of Tyr1472-NR2B, Thr286-CaMKII, and Ser831-GluR1 in neuropathic pain. Neuropharmacology. 2011;60:609–616. doi: 10.1016/j.neuropharm.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Kim Y, Cho HY, Ahn YJ, Kim J, Yoon YW. Effect of NMDA NR2B antagonist on neuropathic pain in two spinal cord injury models. Pain. 2012;153:1022–1029. doi: 10.1016/j.pain.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Kiraly DD, Stone KL, Colangelo CM, Abbott T, Wang Y, Mains RE, Eipper BA. Identification of kalirin-7 as a potential post-synaptic density signaling hub. J Proteome Res. 2011a;10:2828–2841. doi: 10.1021/pr200088w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiraly DD, Lemtiri-Chlieh F, Levine ES, Mains RE, Eipper BA. Kalirin binds the NR2B subunit of the NMDA receptor, altering its synaptic localization and function. J Neurosci. 2011b;31:12554–12565. doi: 10.1523/JNEUROSCI.3143-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Deak M, Morrice N, Cohen P. Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem J. 1999;344:189–197. [PMC free article] [PubMed] [Google Scholar]
- Lang F, Böhmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev. 2006;86:1151–1178. doi: 10.1152/physrev.00050.2005. [DOI] [PubMed] [Google Scholar]
- Larsson M, Broman J. Translocation of GluR1-containing AMPA receptors to a spinal nociceptive synapse during acute noxious stimulation. J Neurosci. 2008;28:7084–7090. doi: 10.1523/JNEUROSCI.5749-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CY, Perez FM, Wang W, Guan X, Zhao X, Fisher JL, Guan Y, Sweitzer SM, Raja SN, Tao YX. Dynamic temporal and spatial regulation of mu opioid receptor expression in primary afferent neurons following spinal nerve injury. Eur J Pain. 2011;15:669–675. doi: 10.1016/j.ejpain.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemtiri-Chlieh F, Zhao L, Kiraly DD, Eipper BA, Mains RE, Levine ES. Kalirin-7 is necessary for normal NMDA receptordependent synaptic plasticity. BMC Neurosci. 2011;12:126. doi: 10.1186/1471-2202-12-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Huang J, Wang Y, Eipper BA, Mains RE. Kalirin, a multifunctional Rho GEF, is necessary for maintenance of hippocampal pyramidal neuron dendrites and dendritic spines. J Neurosci. 2003;23:10593–10603. doi: 10.1523/JNEUROSCI.23-33-10593.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Wang Y, Ferraro F, Mains RE, Eipper BA. Kalirin-7 is an essential component of both shaft and spine excitatory synapses in hippocampal interneurons. J Neurosci. 2008a;28:711–724. doi: 10.1523/JNEUROSCI.5283-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Kiraly DD, Gaier ED, Wang Y, Kim EJ, Levine ES, Eipper BA, Mains RE. Kalirin-7 is required for synaptic structure and function. J Neurosci. 2008b;28:12368–12382. doi: 10.1523/JNEUROSCI.4269-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YL, Tsai MC, Hsu WL, Lee EH. SGK protein kinase facilitates the expression of long-term potentiation in hippocampal neurons. Learn Mem. 2006;13:114–118. doi: 10.1101/lm.179206. [DOI] [PubMed] [Google Scholar]
- McPherson CE, Eipper BA, Mains RE. Genomic organization and differential expression of kalirin isoforms. Gene. 2002;284:41–51. doi: 10.1016/s0378-1119(02)00386-4. [DOI] [PubMed] [Google Scholar]
- Miletic G, Dumitrascu CI, Honstad CE, Micic D, Miletic V. Loose ligation of the rat sciatic nerve elicits early accumulation of Shank 1 protein in the post-synaptic density of spinal dorsal horn neurons. Pain. 2010;149:152–159. doi: 10.1016/j.pain.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng HY, Chang HM, Chang SY, Tung KC, Lee SD, Chou D, Lai CY, Chiu CH, Chen GD, Lin TB. Orexin-A modulates glutamatergic NMDA-dependent spinal reflex potentiation via inhibition of NR2B subunit. Am J Physiol Endocrinol Metab. 2008a;295:E117–E129. doi: 10.1152/ajpendo.90243.2008. [DOI] [PubMed] [Google Scholar]
- Peng HY, Chang HM, Lee SD, Huang PC, Chen GD, Lai CH, Lai CY, Chiu CH, Tung KC, Lin TB. TRPV1 mediates the uterine capsaicin-induced NMDA NR2B-dependent cross-organ reflex sensitization in anesthetized rats. Am J Physiol Renal Physiol. 2008b;295:F1324–F1335. doi: 10.1152/ajprenal.00126.2008. [DOI] [PubMed] [Google Scholar]
- Peng HY, Chen GD, Tung KC, Lai CY, Hsien MC, Chiu CH, Lu HT, Liao JM, Lee SD, Lin TB. Colon mustard oil instillation induced cross-organ reflex sensitization on the pelvic-urethra reflex activity in rats. Pain. 2009a;142:75–88. doi: 10.1016/j.pain.2008.11.017. [DOI] [PubMed] [Google Scholar]
- Peng HY, Chen GD, Lee SD, Lai CY, Chiu CH, Cheng CL, Chang YS, Hsieh MC, Tung KC, Lin TB. Neuroactive steroids inhibit spinal reflex potentiation by selectively enhancing specific spinal GABA(A) receptor subtypes. Pain. 2009b;143:12–20. doi: 10.1016/j.pain.2008.12.023. [DOI] [PubMed] [Google Scholar]
- Peng HY, Chen GD, Tung KC, Chien YW, Lai CY, Hsieh MC, Chiu CH, Lai CH, Lee SD, Lin TB. Estrogen-dependent facilitation on spinal reflex potentiation involves the Cdk5/ERK1/2/NR2B cascade in anesthetized rats. Am J Physiol-Endocrinol Metab. 2009c;297:E416–E426. doi: 10.1152/ajpendo.00129.2009. [DOI] [PubMed] [Google Scholar]
- Peng HY, Chen GD, Lai CY, Hsieh MC, Hsu HH, Wu HC, Lin TB. PI3K modulates estrogen-dependent facilitation of colon-to-urethra cross-organ reflex sensitization in ovariectomized female rats. J Neurochem. 2010;113:54–66. doi: 10.1111/j.1471-4159.2010.06577.x. [DOI] [PubMed] [Google Scholar]
- Peng HY, Chang CH, Tsai SJ, Lai CY, Tung KC, Wu HC, Lin TB. Protein kinase A-dependent spinal α-amino-3-hydroxy-5-methyl-4-isoxazole proprionate-receptor trafficking mediates capsaicin-induced colon-urethra cross-organ reflex sensitization. Anesthesiology. 2011;114:70–83. doi: 10.1097/ALN.0b013e3181fe4204. [DOI] [PubMed] [Google Scholar]
- Peng HY, Chen GD, Lai CY, Hsieh MC, Lin TB. Spinal SIRPα1-SHP2 interaction regulates spinal nerve ligation-induced neuropathic pain via PSD-95-dependent NR2B activation in rats. Pain. 2012a;153:1042–1053. doi: 10.1016/j.pain.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Peng HY, Chen GD, Hsieh MC, Lai CY, Huang YP, Lin TB. Spinal SGK1/GRASP-1/Rab4 involves in complete Freund's adjuvant-induced inflammatory pain via regulating dorsal horn GluR1-containing AMPA receptor trafficking in rats. Pain. 2012b;153:2380–2392. doi: 10.1016/j.pain.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Penzes P, Johnson RC, Alam MR, Kambampati V, Mains RE, Eipper BA. An isoform of kalirin, a brain-specific GDP/GTP exchange factor, is enriched in the postsynaptic density fraction. J Biol Chem. 2000;275:6395–6403. doi: 10.1074/jbc.275.9.6395. [DOI] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Schäfers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-α induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neuron. J Neurosci. 2003;23:2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack S, Battaglia A, Cibert-Goton V, Gavazzi I. EphrinB2 induces tyrosine phosphorylation of NR2B via Src-family kinases during inflammatory hyperalgesia. Neuroscience. 2008;156:175–183. doi: 10.1016/j.neuroscience.2008.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartjes M, Morariu A, Niesters M, Aarts L, Dahan A. Nonselective and NR2B-selective N-methyl-d-aspartic acid receptor antagonists produce antinociception and long-term relief of allodynia in acute and neuropathic pain. Anesthesiology. 2011;115:165–174. doi: 10.1097/ALN.0b013e31821bdb9b. [DOI] [PubMed] [Google Scholar]
- Tan AM, Stamboulian S, Chang YW, Zhao P, Hains AB, Waxman SG, Hains BC. Neuropathic pain memory is maintained by Rac1-regulated dendritic spine remodeling after spinal cord injury. J Neurosci. 2008;28:13173–13183. doi: 10.1523/JNEUROSCI.3142-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AM, Samad OA, Fischer TZ, Zhao P, Persson AK, Waxman SG. Maladaptive dedritic spine remodeling contributes to diabetic neuropathic pain. J Neurosci. 2012;32:6795–6807. doi: 10.1523/JNEUROSCI.1017-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolias KF, Duman JG, Um K. Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog Neurobiol. 2011;94:133–148. doi: 10.1016/j.pneurobio.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KJ, Chen SK, Ma YL, Hsu WL, Lee EH. sgk, a primary glucocorticoid-induced gene, facilitates memory consolidation of spatial learning in rats. Proc Natl Acad Sci U S A. 2002;99:3990–3995. doi: 10.1073/pnas.062405399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YC, Metzger S, Riess O, Soehn AS, Nguyen HP. Genetic analysis of polymorphisms in the kalirin gene for association with age-at-onset in European Huntington disease patients. BMC Med Genet. 2012;13:48. doi: 10.1186/1471-2350-13-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Atianjoh F, Gauda EB, Yaster M, Li Y, Tao YX. Increased expression of sodium channel subunit Nav1.1 in the injured dorsal root ganglion after peripheral nerve injury. Anat Rec (Hoboken) 2011;294:1406–1411. doi: 10.1002/ar.21437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, McEwen BS, Yan Z. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol Psychiatry. 2011;16:156–170. doi: 10.1038/mp.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]