

Abstract
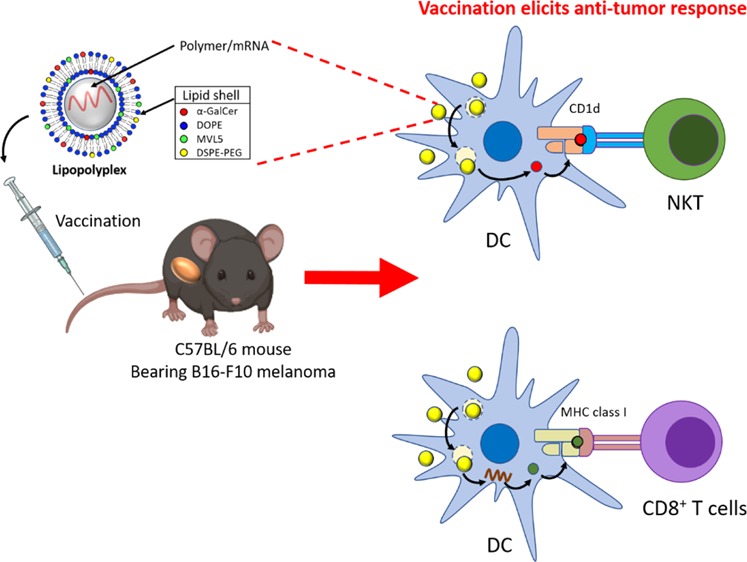
Recently, the use of mRNA-based vaccines for cancer immunotherapy has gained growing attention. Several studies have shown that mRNA delivered in a vectorized format can generate a robust and efficient immune response. In this work, a new lipopolyplex vector (multi-LP), incorporating the immune adjuvant α-galactosylceramide (α-GalCer) and a multivalent cationic lipid, was proposed for the in vivo delivery of mRNA into antigen-presenting cells. We demonstrate that dendritic cells (DCs) can be targeted in vivo by intravenous administration of a α-GalCer-/mRNA-loaded multi-LP vector, without the need for its functionalization with cell-specific antibodies or ligands. The multi-LP nanoparticles loaded with a reporter mRNA efficiently led to high expression of the enhanced green fluorescence protein in DCs both in vitro and in vivo, exhibiting an intrinsic selectivity for DCs. Finally, the TRP2-mRNA/α-GalCer-based multi-LP vaccine induced a significant therapeutic effect against a highly malignant B16-F10 melanoma tumor. This study provides the first evidence that a combination of antigen-mRNA and α-GalCer can be used as an effective antitumor vaccine, inducing strong innate and adaptive immune responses.
1. Introduction
Cancer immunotherapy is a therapeutic strategy that exploits the natural ability of the immune system to recognize and kill cancer cells.1 Several cancer immunotherapies have been proposed such as vaccines and antibody- and cell-based therapies.2−4 Vaccines, compared to other immunotherapies, offer the advantage of promoting both humoral and cell-mediated immune responses and, more importantly, they can create immunological memory, which could ensure the promotion of an immunological response of greater magnitude and faster kinetics in the case of tumor recurrence.5
Nucleic acids have been extensively investigated in the context of vaccine development for the priming of tumor-specific cytotoxic T lymphocytes (CTLs).6−8 The use of nucleic acid-based vaccines is a novel approach developed to address the issues associated with more traditional antigen forms, such as proteins and peptides. They are easily produced, provide opportunities for molecular engineering, and have the potential to promote both innate and adaptive immune responses.7,9 Moreover, unlike peptide-based vaccines, nucleic acid-based vaccines expressing complete genes are not a priori human leucocyte antigen (HLA)-restricted.7,9
Particularly, mRNA-based vaccines are attractive because they retain the same appealing characteristics of DNA-based vaccines and also offer additional benefits, such as superior immunogenicity, and, unlike DNA, mRNA only needs to gain entry to the cytoplasm, where translation occurs, in order to achieve cell transfection.8−10 Moreover, mRNA does not have any risk of integration into the genome and therefore has no oncogenic potential and can be easily synthesized.10 Finally, mRNA can act not only as a source for antigen but also as an adjuvant by providing costimulatory signals, for example, via toll-like receptor (TLR) 3, TLR7, and TLR8, to amplify the immune response.8,10 The main challenge associated with the use of mRNA for vaccine development is its sensitivity toward catalytic hydrolysis by omnipresent ribonucleases.9,10 Unprotected mRNA is highly unstable under physiological conditions and hence unsuitable for broad therapeutic applications. Because of the potential advantages of mRNA-based vaccines, intense efforts have been made toward the stabilization of mRNA in vivo.9,11 Several strategies have been proposed for mRNA delivery, including viral vectors and nonviral vectors.12−19
Nonviral vectors represent a simple, flexible, and, more importantly, safer alternative to viral vectors. Thanks to their relatively simple quantitative production and their low host immunogenicity, nonviral vectors are attractive tools in vaccine strategies. With the development of new biocompatible materials and innovative fabrication approaches, nonviral vectors are becoming the preferred vehicle to deliver mRNA. A variety of nonviral vectors have been explored as platforms for mRNA-based vaccine development, focusing mainly on the use of lipids and polymers.20−26
An additional advantage associated with the use of nonviral vectors for vaccine development is that they can facilitate codelivery of antigens with adjuvants, enhancing significantly the therapeutic efficacy of the vaccine.27−30 The codelivery of antigens with adjuvants has been extensively investigated for peptide-based vaccines, but little is known whether mRNA-based vaccines may benefit from its combination with vaccine adjuvants. In this framework, α-GalCer represents an optimal new class of vaccine adjuvant as it acts as an effective link between innate and adaptive antitumor immunity. Indeed, α-GalCer, also known as KRN7000, is an invariant natural killer T (iNKT) cell antigen presented on CD1d of antigen-presenting cells (APCs).31−33 Previous studies have reported that α-GalCer shows stronger ability to generate antigen-specific CD8+ T cells, compared to TLR ligands.29,34,35 In response to α-GalCer, invariant iNKT cells rapidly produce immunostimulatory cytokines, particularly interferon-γ (IFN-γ), and elicit the induction of several costimulatory molecules.31−33 These events promote the activation APCs, which release key Th1 cytokines (e.g., interleukin (IL)-12), and the downstream activation of CD4+ and CD8+ T lymphocytes, as well as NK cells and B cells, with important effects on the magnitude and effectiveness of the immune responses.31−33 Despite this, there is a limitation for the clinical application of α-GalCer; its poor hydrophilicity makes it essential to incorporate α-GalCer into a vector in order to improve its solubility, uptake, and immunogenicity.29,36,37 In addition, it has been demonstrated that vectorization of α-GalCer can prevent iNKT cell anergy, which usually occurs after repeated injections of α-GalCer.29,35,36,38
In this study, we have developed a lipopolyplex vector termed multi-LP, which consists of a poly-(β-amino ester) polymer (PbAE)/mRNA polyplex core entrapped into a lipid shell composed of multivalent cationic lipid (MLV5), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), DSPE-PEG, and α-GalCer. In the multi-LP platform, the cationic polymer condenses the mRNA into a polyplex nanoparticle by electrostatic interaction. A lipid shell is included in the system to improve mRNA delivery into dendritic cells (DCs) and to enhance the immune response because of the adjuvant activity mediated by α-GalCer.39 The resulting construct can not only protect mRNA molecules in the polyplex core structure from RNase attack but also be efficiently internalized by DCs, promoting the release of mRNA molecules to the cytosol for antigen translation.
Here, the nanovaccine was prepared by co-loading into the carrier system α-GalCer and antigen-mRNA coding for tyrosinase-related protein 2 (TRP2) and was tested in a poorly immunogenic and highly aggressive B16-F10 murine melanoma model, generated by injecting the tumor cells into syngeneic C57BL/6 mice.40
2. Materials and Methods
2.1. Materials
Lipids 1,2-dioleoyl-sn-glycero-3-ethylphosphocholine (EDOPC), DOPE, N1-[2-((1S)-1-[(3-aminopropyl)amino]-4-[di(3-amino-propyl)amino]butylcarboxamido)ethyl]-3,4-di[oleyloxy]-benzamide (MVL5), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG), and α-galactosyl ceramide (α-GalCer or KRN700) were purchased from Avanti Polar Lipids. Sodium acetate buffer solution (3 M, pH 5.2), nuclease-free water, and bovine serum albumin (BSA) were purchased from Sigma-Aldrich. Tyrosinase-related protein 2 (TRP-2, 180–188) peptide was purchased from GenScript. The following antibodies were purchased from BD Biosciences: anti-TCRβ (clone H57-597), anti-CD8a (clone 53–6.7), anti-CD11b (clone M1/70), anti-CD45R/B220 (clone RA3-6B2), anti-IFN-γ, and anti-CD45 (clone 30-F11); BioLegend: anti-F4/80 (clone BM8), anti-CD19 (clone B4), anti-CD11c (clone N418), anti-CD69 (clone H1.2F3), anti-MHC-II (clone M5/114.15.2), and anti-NK1.1 (clone PK136). Anti-CD8 (clone KT15) and iTAg Tetramer H-2 Kb TRP2 (SVYDFFVWL) were purchased from MBL International.
Unmodified TRP2-mRNA, enhanced green fluorescence protein (eGFP)-mRNA, Cy5-mRNA, ovalbumin (OVA)-mRNA, and luciferase (LUC)-mRNA were purchased from TriLink Biotechnologies. Six to eight week-old female C57BL/6 mice were purchased from Charles River (Canada).
2.2. Synthesis of PbAE
The PbAE was synthesized in a two-step reaction procedure as previously described.22,41 In the first step, the base polymer (B4S4) was synthesized by mixing 1,4-butanediol diacrylate (B4) (Sigma-Aldrich) with 5-amino-1-pentanol (S4) (Sigma-Aldrich) at a molar ratio of B4/S4 = 1.2:1. The reaction was maintained at 90 °C and 1000 rpm of stirring speed for the first 4 h. After 4 h, the stirring speed was slowed to 300 rpm and the reaction was maintained at 90 °C for further 12–16 h. The B4S4 polymer was dissolved in 10 mL of anhydrous tetrahydrofuran (THF) at a final concentration of 100 mg/mL. In a separate glass vial, 1-(3-aminopropyl)-4-methylpiperazine (E7) (Alfa Aesar) was dissolved in 40 mL of THF at 0.2 M final concentration. End-chain capping reaction was performed by mixing the two solutions and leaving it at room temperature, under stirring at 400 rpm for 24 h, and protected from light. The final product is termed 447.
The resulting 447 was purified by precipitation in anhydrous diethyl ether (1:3 v/v THF/diethyl ether) and dried under vacuum for 24 h. Polymers were then dissolved in acetate buffer (10 mM Tris-HCl buffer pH 7.4) and stored at −20 °C until use.
2.3. Preparation and Characterization of PbAE/mRNA Polyplex
PbAE/mRNA polyplex was prepared by mixing one volume of the PbAE polymer with one volume of mRNA molecules in acetate buffer using a PbAE/mRNA (w/w) mass ratio = 30. After a 20 min incubation at room temperature, the polyplex was analyzed for size distribution and zeta potential using a Zetasizer NanoZS dynamic light-scattering instrument (Malvern Instruments). The PbAE/mRNA polyplex was also analyzed in a gel retardation assay. Briefly, a polyplex sample containing 250 ng of mRNA was loaded into each well and separated by electrophoresis in a 0.7% agarose gel with 1× Tris–borate–EDTA buffer (BioRad, Hercules, CA). RNA bands were stained with Gelred nucleic acid gel stain (Biotium) and visualized with a GelDoc system (BioRad).
2.4. Preparation and Characterization of the Lipopolyplex Incorporating α-GalCer
All the lipids were dissolved in chloroform at a final concentration of 20 mg/mL and used to prepare thin lipid films by rotary evaporation in a Rotavapor (Buchi) under partial vacuum. The thin lipid film was rehydrated with a solution containing PbAE/mRNA polyplex to prepare the lipopolyplex. The lipid film was composed of 49% EDOPC, 49% DOPE, and 2% DSPE-PEG or 16% MVL5, 82% DOPE, and 2% DSPE-PEG for mono-LP and multi-LP, respectively. The formulations included 0.5 μg of α-GalCer. Hydrodynamic size and zeta potential of the lipopolyplexes were measured with a Zetasizer Nano ZS dynamic light-scattering instrument (Malvern). The complete construct was also analyzed by transmission electron microscopy (TEM). The incorporation of α-GalCer was confirmed as previously reported.29 Briefly, multi-LP/α-GalCer + mRNA nanoparticles loaded with different amounts of α-GalCer (50, 100, 250, and 500 ng) were added to the RPMI-1640 medium with 1 × 105 BMDCs. After 24 h of incubation, the supernatants were removed, and 1 × 105 DN32.D3 mouse iNKT hybridoma cells were added and cocultured with bone marrow-derived DCs (BMDCs). IL-2 levels were measured by ELISA after 24 h of incubation.
2.5. Preparation of BMDCs
BMDCs were prepared from C57BL/6 mice as previously described.22,42 Briefly, bone marrow cells from the femur and tibia were flushed out with 2% fetal bovine serum (FBS)-containing phosphate-buffered saline (PBS) using a syringe. Cells were centrifuged at 500g for 5 min, treated with ACK lysis buffer (Quality Biological) to remove red blood cells, and resuspended in RPMI-1640 culture medium supplemented with 10% FBS, 0.1% β-mercaptoethanol, 1% penicillin/streptomycin, 20 ng/mL of granulocyte–macrophage colony-stimulating factor (GM-CSF), and 20 ng/mL of interleukin-4 (IL-4). Cells were seeded into six-well plates at a seeding density of 1 × 106 cells/mL; the growth medium was changed every other day. The nonadherent DCs were harvested on day 5. BMDCs were stained for CD11b, CD11c, and MHC-II and analyzed by flow cytometry (BD Fortessa X-20).
2.6. MTS Test with BMDCs
To test the potential cytotoxicity from vaccine formulations, BMDCs were seeded in a 96-well plate at a seeding density of 3 × 104 cells/well and treated with 0.1 μg of mRNA loaded into mono-LP or multi-LP. Cell viability was measured 24 h later with a tetrazolium-based Cell Titer 96Aqueous One Solution Cell Proliferation (MTS) assay (Promega) following the manufacturer’s instructions.
2.7. Transfection of BMDCs
Enhanced GFP-mRNA was used as a reporter to test the transfection efficiency in BMDCs. On day 5, 3 × 105 cells/well were seeded in 24-well plates and treated in complete RPMI-1640 with mono-LP and multi-LP loaded with 0.5 μg of eGFP-mRNA. The expression of eGFP in cells was determined with a fluorescence microscope (Carl Zeiss) and the percentage of eGFP-expressing cells was measured by flow cytometry (BD Fortessa X-20).
2.8. Bioluminescence Imaging in Live Mice
C57BL/6 mice (n = 5 per group) were injected with multi-LP/LUC-mRNA (20 μg of mRNA) following an intravenous (i.v.) administration route. After 6 h, the mice were injected intraperitoneally with 200 μL of d-luciferin (15 mg/mL) (Gold Biotechnology), and bioluminescence was measured using a PerkinElmer IVIS Spectrum imaging system. The mice were monitored for body weight every 2 days for 4 weeks. Luminescence quantification was carried out using LivingImage software (PerkinElmer).
2.9. In Vivo APC Uptake and Transfection
C57BL/6 mice (n = 5 in each group from two pooled experiments) were intravenously or intradermally (i.d.) injected with lipopolyplexes loaded with Cy5-eGFP-mRNA (20 μg) for the evaluation of the cellular uptake and transfection efficiency. After 24 h, mice were sacrificed and the spleen and draining lymph nodes (dLNs) (inguinal lymph nodes, ILNs) were harvested and processed into single-cell suspensions.
Single-cell suspensions were obtained as described previously.22 Briefly, spleens or lymph nodes were pressed through a 70 μM cell strainer with the plunger of a 5 mL syringe. Erythrocytes were removed by the addition of ACK lysis buffer (Quality Biological). The cells were stained with anti-B220, anti-CD19, anti-CD11c, and anti-F4/80 in order to gate DCs (CD11c+ and F4/80–), macrophages (CD11c– and F4/80+), and B-cells (CD11c–, B220+ and CD19+) populations. The samples were analyzed by flow cytometry (BD Fortessa X-20). Animal experiments in this study were carried out in accordance with guidelines evaluated and approved by the ethics committee of the University of Ottawa to ensure the humane animal care and use.
2.10. Detection of CD69 in Spleen and Lymph Nodes by Flow Cytometry
Mice (C57BL/6, n = 5 in each group from two pooled experiments) were injected i.d. or i.v. with multi-LP vaccine loaded with 10 μg of TRP2-mRNA and with or without 1 μg of α-GalCer. After 24 h, mice were sacrificed, and lymph nodes (ILNs and axillary lymph nodes) and spleens were harvested and processed into single-cell suspensions.
Lymphocytes were stained for V450/viability dye, TCRβ, NK1.1, and CD69 to gate NK cells (TCRβ– and NK1.1+), NKT cells (TCRβ+ and NK1.1+), and T cells (TCRβ+ and NK1.1–). The samples were analyzed by flow cytometry (BD Fortessa X-20).
2.11. Measurement of Proinflammatory Cytokines
C57BL/6 mice (n = 8 for each group from three pooled experiments) were vaccinated with different vaccine formulations (multi-LP/α-GalCer + mRNA, multi-LP/mRNA, multi-LP/α-GalCer, free α-GalCer + mRNA, and free α-GalCer) loaded with 10 μg of TRP-2 mRNA and/or 0.5 μg of α-GalCer. After 6 h, blood was collected by lateral saphenous bleeding and allowed to clot overnight at 4 °C; serum was separated by centrifugation at 12 000 rpm for 15 min, 4 °C. The serum was quickly frozen at −80 °C and stored until use. Serum TNF-α, IFN-α, IFN-β, IFN-γ, IL-1β, IL-6, and IL-12 levels were measured by enzyme-linked immunosorbent assay (ELISA) kits (eBioscience).
2.12. Detection of Antigen-Specific T Cells by Intracellular and Tetramer Staining
Mice (C57BL/6, n = 5 in each group from two pooled experiments) were injected intravenously with different vaccine formulations, such as multi-LP/α-GalCer + TRP2-mRNA, multi-LP/TRP2-mRNA, multi-LP/TRP2-mRNA + free α-GalCer, free α-GalCer + TRP2-mRNA, and PBS as a negative control. The vaccine formulations were prepared using 10 μg of TRP2-mRNA and 0.5 μg of α-GalCer. Blood was collected from vaccinated mice by saphenous bleeding and erythrocytes were removed by addition of ACK lysis buffer (Quality Biological). peripheral blood mononuclear cells (PBMCs) were stimulated with 1 μg/mL of TRP2-peptide (SVYDFFVWL) for 6 h in the presence of a protein transport inhibitor, Brefeldin A (BD GolgiPlug). After incubation, cells were washed and stained with Abs for surface markers-TCRβ, CD8, and V450/viability dye. Cells were then washed, fixed, and permeabilized with a BD Fixation/Permeabilization Solution Kit and stained with anti-IFN-γ. The cells were washed, resuspended in FACS buffer (1× PBS and 0.5% BSA), and analyzed by flow cytometry (BD Fortessa X-20).
SVYDFFVWL-specific tetramer (MBL International) staining was carried out by incubation at room temperature for 10 min in FACS buffer. After washing, PBMCs were stained with anti-TCRβ, anti-CD8 clone KT15, and V450/viability for 30 min at 4 °C in FACS buffer. Then, the samples were analyzed by flow cytometry (BD Fortessa X-20).
2.13. Measurement of Antibody Titers
OVA-specific antibody titers were measured in the serum of C57BL/6 mice (n = 5 in each group from two pooled experiments) immunized on days 0, 7, and 14 by i.v. injection. Blood was collected by saphenous bleeding on day 28 after priming and allowed to clot overnight at 4 °C. The serum was separated by centrifugation at 10 000 rpm for 20 min. The serum was collected, and anti-OVA antibody titers were determined by ELISA, as previously described.22 Briefly, 96-well plates (Nunc-Immuno) were coated by overnight incubation at 4 °C with 2 μg/well of OVA in PBS. Plates were blocked with 1% BSA in PBS for 2 h, and serial two-fold dilutions of serum samples in PBS were added to the wells. After a 2 h incubation, the plates were washed with PBS containing 0.05% Tween 20 and incubated for 1 h at room temperature with HRP-conjugated goat anti-mouse immunoglobulin G (IgG), IgG1, IgG2b, and IgG2c antibodies (Southern Biotechnology Associates). The plates were incubated with tetramethylbenzidine (TMB) substrate and the reaction was stopped by the addition of 1 N HCl (Sigma). Absorbance was read at 450 nm with a microplate reader (BioRad). Antibody titers were calculated as the inverse dilution at which the absorbance equaled that of control mice plus 2 standard deviations.
2.14. Therapeutic Efficacy Using a Subcutaneous B16-F10 Tumor Model
C57BL/6 mice (n = 5 in each group) were inoculated with 2 × 105 B16-F10 melanoma tumor cells by subcutaneous injection to establish subcutaneous tumors, following a previously described protocol.43 Five days after tumor inoculation, mice were intravenously vaccinated with different formulations (10 μg of unmodified TRP2-mRNA with or without 0.5 μg of α-GalCer) and boosted at days 9 and 13 post-tumor inoculation. B16-F10 tumor bearing mice were monitored for survival, body weight, and tumor growth every 2 days. Tumor volume was calculated using the following formula: V = (L × W × W)/2, where V is the tumor volume, L is the tumor length, and W is the tumor width.
2.15. Evaluation of Tumor Infiltrating Lymphocytes and Tetramer-Positive CD8+ T Cells in s.c. B16-F10 Tumors
C57BL/6 mice (n = 5 in each group from two pooled experiments) were inoculated subcutaneously with 2 × 105 B16-F10 melanoma tumor cells. Eight days after tumor inoculation, mice were intravenously vaccinated with different formulations (10 μg of TRP2-mRNA with or without 0.5 μg of α-GalCer) and boosted 15 days post-tumor inoculation. Mice were sacrificed 17 days post-tumor inoculation and the tumors were harvested for tumor infiltrating lymphocyte (TIL) quantification.
Tumor cell suspensions were prepared from solid tumors by mechanical disaggregation as described previously.44 Erythrocytes were removed by the addition of ACK lysis buffer (Quality Biological). The resulting suspension was passed through a 40 μm cell strainer, washed once with PBS, and resuspended in FACS buffer for flow cytometry analysis. For TIL evaluation, cells were stained with a viability dye, anti-CD45, anti-TCRβ, anti-CD8, and anti-CD4 and analyzed by flow cytometry (BD Fortessa X-20). SVYDFFVWL-specific tetramer (MBL International) staining was carried out by incubation at room temperature for 10 min in FACS buffer, followed by surface staining with anti-CD45, anti-TCRβ, anti-CD4, and anti-CD8 clone KT15 for 30 min at 4 °C in FACS buffer.
2.16. Statistical Analysis
One-way analysis of variance (ANOVA) post hoc tests were used to determine the significance of differences between experimental and relevant control values within each experiment. All analyses were performed using the Graph Pad Prism 8.0 statistical software. Significant differences were reported as *p < 0.05, **p < 0.01, and ***p < 0.001. Survival curves were plotted for each condition and the log-rank (Mantel–Cox) test was used to evaluate statistical significance on GraphPad Prism 8.
3. Results and Discussion
3.1. Multi-LP Delivery System Development
We designed a novel platform for mRNA-based vaccine that consisted of a PbAE/mRNA polyplex core structure coated by a lipid bilayer shell (Figure 1a). Two different formulations were prepared using a monovalent cationic lipid (EDOPC) or a multivalent cationic lipid (MLV5) to obtain a mono-LP or a multi-LP vector, respectively. This study was aimed to assess the effect of the surface charge density on important properties of hybrid lipid-/polymer-based vaccine, such as transfection efficiency in DCs, toxicity profile, biodistribution, and immunological and therapeutic activities.
Figure 1.

(a) Schematic representation of the multi-LP vaccine platform. PbAE/mRNA polyplex is entrapped into a liposome. (b) Gel retardation assay of the naked mRNA (mRNA) and PbAE/mRNA polyplexes prepared using different N/P ratios (10, 20, 30, and 40). (c) Hydrodynamic size of PbAE/mRNA polyplex, mono-LP, and multi-LP with the respective polydispersity index. (d) Zeta potential of PbAE/mRNA polyplex, mono-LP, and multi-LP. (e) TEM image of multi-LP nanoparticle. (f) BM-DCs (1 × 105 cells/well) cocultured with DN32.D3 iNKT hybridoma cells (BMDC/iNKT ratio 1:1) for 24 h in the presence of different amounts of vectorized α-GalCer (0.05, 0.1, 0.25, and 0.5 μg/mL) and fix amount of mRNA (0.5 μg/mL). The production of IL-2 was measured by ELISA. Data are mean ± S.D. of three independent experiments.
Agarose gel retardation assay was performed to assess the mRNA-binding capacity of the cationic PbAE polymer. The gel retardation assay revealed complete mRNA incorporation into the polyplex core at the PbAE/mRNA ratio of 10–40 (Figure 1b). The multi-LP loaded with α-GalCer/mRNA was characterized for physical properties, such as size and zeta potential (Figure 1c,d, respectively), exhibiting a hydrodynamic diameter of 132.3 ± 3.78 nm, as confirmed by TEM analysis (Figure 1e), and a strongly positive zeta potential of +33.6 mV because of the cationic lipid included in the external layer, whereas the polyplex core alone, prepared using a PbAE/mRNA (w/w) ratio of 20, showed a hydrodynamic size of 113 ± 3.16 nm and a zeta potential of +35.8 mV. The lipopolyplex vector prepared using a monovalent cationic lipid resulted in a nanoparticle with a size of 150.9 ± 3.62 nm and a zeta potential of +38 mV.
Size and charge are essential parameters for efficient passive targeting of APCs. For instance, nanoparticles with a size range from ∼25 to 200 nm have been proven to traffic to the dLN where they are rapidly taken up by APCs and can, therefore, be used to elicit different immunological outcomes.45 Furthermore, it is well known that positively charged nanoparticles exhibit a faster uptake rate by phagocytic cells than negatively charged or neutral nanoparticles, and up to date, they have been revealed as the most effective carriers for gene delivery.46
The encapsulation of α-GalCer into multi-LP nanoparticles was confirmed indirectly through the quantification of IL-2 levels released by DN32.D3 iNKT hybridoma cells cocultured with BMDCs pretreated with multi-LP/α-GalCer + mRNA loaded with different amounts of α-GalCer (Figure 1f).
In line with the above, we designed a delivery platform with optimized physical–chemical properties for passive DC targeting and for the delivery of macromolecules, such as antigen-encoding mRNAs.
3.2. Multi-LP Is an Optimal Platform for mRNA-Based Transfection
The multi-LP and mono-LP platforms were prepared using two different lipid formulations, MVL5/DOPE/DSPE-PEG (16:82:2) and EDOPC/DOPE/DSPE-PEG (49:49:2), respectively. To compare the transfection efficiency of the two formulations, mono-LP and multi-LP were loaded with an mRNA encoding for eGFP and the expression of the gene reporter was quantified by flow cytometry (Figure 2a) and confirmed by fluorescence microscopy (Figure 2b). Quantification of the transfection efficiency was carried out at 24 and 48 h post-transfection and peak expression was observed at 48 h. Multi-LP loaded with eGFP-mRNA was highly efficient to transfect BMDCs in vitro, with an efficiency of ∼50% (Figure 2a), whereas mono-LP showed a lower transfection efficiency; naked mRNA failed to transfect BMDCs. These data demonstrate that the incorporation of the multivalent cationic lipid (MLV5) significantly increased the in vitro transfection efficiency of mRNA-loaded lipopolyplex platforms. It is known that efficient gene delivery requires a large molar charge ratio (cationic lipid/nucleic acid), which often results in the use of a relatively high amount of cationic lipid per cell, with a consequent increase in toxicity. In this context, the use of MVLs can improve the safety profile of the formulation compared to monovalent lipids because a smaller number of MVLs are required to achieve superior transfection efficiency compared to monovalent DOTAP and with significantly less toxicity.47−49
Figure 2.

(a) Quantification of eGFP-positive BMDCs by flow cytometry. (b) Fluorescence microscopy images of DCs transfected with eGFP-mRNA. BMDCs were seeded at a density of 3 × 105 cells/mL in a 24-well plate and transfected with 0.5 μg of eGFP-mRNA naked (mRNA) or encapsulated into mono-LP and multi-LP. Error bars represent standard error of mean with n = 3 per group from two independent experiments. (c) MTS analysis of BMDCs was performed at 24 h after exposure to mono-LP or multi-LP (0.1 μg of mRNA). Values are expressed as mean ± SD (n = 3 per group). (d) Bioluminescence imaging of mice 6 h after i.v. injection of multi-LP loaded with LUC-mRNA (20 μg). (e) Ex vivo quantitative expression of multi-LP/LUC-mRNA in the individual organs (liver, spleen, kidney, lung, heart, ILNs, and MLNs) was conducted (mean ± SD, n = 5). (f) In vivo toxicity measured as changes in the body weight of C57BL/6 mice after the treatment with multi-LP and PBS as a negative control. Data are mean ± S.D., n = 5 mice/group. (g) Representative flow cytometry plots for the expression of maturation surface markers (CD40, CD80, and CD86) in BMDCs treated with multi-LP/α-GalCer + mRNA nanoparticles.
Importantly, cytotoxicity evaluations performed employing MTS assay indicated that the multi-LP formulation was completely nontoxic in BMDCs (Figure 2c). Thus, we provide evidence that these nanoparticles are biocompatible and can be effectively used for mRNA delivery. Finally, we tested multi-LP nanoparticles loaded with mRNA encoding for LUC reporter in vivo because transfection systems that work efficiently in vitro often fail to function or have serious toxicity in vivo. Six hours after administration, a significant bioluminescence signal was detected in the abdominal region, demonstrating successful transfection by mRNA-loaded multi-LP particles (Figure 2d). Ex vivo bioluminescence of organs revealed that LUC expression was mostly localized in the liver, spleen, inguinal lymph nodes, and mesenteric lymph nodes (MLNs) (Figure 2e). Our data are in line with previous studies, which have found that nanoparticles with a size larger than 50 nm rapidly accumulated in the liver and spleen upon i.v. administration.50 Conventionally, liver accumulation of drug-loaded nanoparticles is strongly unwanted, and it is unanimously recognized as the major barrier to clinical translation of nanomaterials. However, in this study, liver accumulation could be desirable, given that our strategy involves the delivery of two distinct immune agents, α-GalCer and an mRNA coding for a selected tumor antigen, that are recognized by iNKT (most abundant in the liver) and T cells (most abundant in lymphoid organs), respectively.51 Bioluminescence distribution revealed efficient dual targeting of the liver and lymphoid organs (mostly spleen, ILNs, and MLNs) and excluded any undesired off-target signal.
Moreover, multi-LP nanoparticles did not elicit signs of toxicity in mice. No changes in the body mass or behavior of mice were detected for up to 4 weeks following a relatively high administration dose (Figure 2f).
The expression of DC maturation markers was quantified in BMDCs after treatment with multi-LP/α-GalCer + mRNA and compared with the control that was treated with the empty vector. The post-treatment DCs had a significantly increased level of CD40, CD80, and CD86 on their surface (Figure 2g). These data confirm that our nanovaccine is not only able to provide a source of a protein antigen, as demonstrated by eGFP expression after multi-LP/eGFP-mRNA treatment, but also to efficiently induce the expression of important costimulatory molecules that are necessary for efficient antigen presentation to lymphocytes.
3.3. Passive DC Targeting in Vivo for mRNA Delivery
APCs are responsible for driving the induction of CD8+ T-cell-mediated immune responses. Therefore, it was of particular interest to analyze the ability of the multi-LP vaccine to target the different APC subsets, such as DCs, macrophages, and B cells. For this study, mice were immunized once i.d. or i.v. with mono-LP and multi-LP nanoparticles loaded with fluorescently labeled mRNA (Cy5-eGFP-mRNA). Twenty four hours after vaccination, single-cell suspensions obtained from spleens and dLNs were stained with an antibody panel directed against common APC markers: DCs (CD11c+ and F4/80–), macrophages (CD11c– and F4/80+), and B cells (CD11c–, B220+ and CD19+).
At 24 h after i.v. administration, multi-LP nanoparticles were taken up predominately by DCs, while a lower uptake was also observed for B cells and macrophages (Figure 3a,b). In contrast, Cy5-eGFP-mRNA-loaded multi-LP failed to target APCs and efficiently release the mRNA into their cytoplasm after i.d. injection (Figure 3a,b).
Figure 3.

Quantification of multi-LP/Cy5-eGFP-mRNA (a,b) and mono-LP/Cy5-eGFP-mRNA (c,d) uptake by APCs in the spleen (a,c) and ILNs (b,d) 24 h after i.v. or i.d. immunization with 20 μg of Cy5-eGFP-mRNA. In vivo transfection efficiency of APCs in the spleen (e) and ILNs (f) 24 h after i.v. injection of 20 μg of eGFP-mRNA loaded into multi-LP. Values represent the mean ± SD of two separate experiments involving three to five mice per group. Values were considered significant at *p < 0.05, **p < 0.01, ***p < 0.001, n.s. = not significant. Statistical significance was evaluated using a nonparametric one-way ANOVA with a Dunn’s post-test.
On the other hand, the uptake of mono-LP nanoparticles by the different APC populations in both spleen and dLNs was significantly lower than that from multi-LP (Figure 3c,d). Surprisingly, the vector showed a certain preference for the i.v. administration route, in terms of its ability to target APC populations. The in vivo transfection efficiency study also confirmed that the expression of eGFP was predominantly observed in DCs both in spleen and dLNs following i.v. vaccination with multi-LP, whereas a minor fraction of eGFP-positive B cells and macrophages were detected in the spleen (Figure 3e,f). Oppositely, we could not detect any transfected DCs, B-cells, and macrophages upon i.d. injection of multi-LP/eGFP-mRNA (data not shown). In accordance with these results, we can affirm that the multi-LP vector has a superior capability to target passively APCs in lymphoid organs, compared to mono-LP, showing a preferential uptake by DCs after systemic administration.
3.4. Comparison of i.d. and i.v. Routes for the Capacity To Promote Immune Cell Activation
It has been demonstrated that the simultaneous activation of various immune pathways, by combining α-GalCer with TLRs adjuvants, can synergistically enhance the effectiveness and duration of antitumor immune responses; therefore, in the present study, we hypothesized that the combination of the lipid antigen α-GalCer and self-adjuvanted mRNA may potentially enhance their immunological properties.52−54 For this reason, naïve mice were injected either i.v. or i.d. with multi-LP nanoparticles loaded with antigen-mRNA with or without α-GalCer. At 24 h post-immunization, single-cell suspensions obtained from spleens and dLNs were analyzed by flow cytometry in order to measure the upregulation of the lymphocyte activation marker CD69 on NKT, NK, and T cells.
TRP2 was identified as a candidate antigen given that it is commonly overexpressed in mouse and human melanocytes, and it currently represents one of the major and better-characterized melanoma antigens.55,56 Immunization with multi-LP loaded with α-GalCer/TRP2-mRNA resulted in a significantly stronger activation of NKT, NK, and T cells in both spleen and dLNs (data not shown), suggesting that α-GalCer and antigen-mRNA together may potentiate the immune response particularly after i.v. administration, thus confirming that our formulation is well suited for systemic delivery (Figure 4).
Figure 4.

Flow cytometry analysis of CD69 in spleen NKT, NK, and T cells from mice treated with multi-LP loaded with TRP2-mRNA and with or without α-GalCer administrated i.d. or i.v. Values represent the mean ± SD of two separate experiments involving two to three mice per group. Values were considered significant at *p < 0.05, **p < 0.01, ***p < 0.001, n.s. = not significant. Statistical significance was evaluated using a nonparametric one-way ANOVA with a Dunn’s post-test.
Likewise, at 6 h after i.v. vaccination with multi-LP loaded with α-GalCer and TRP2-mRNA, we observed a high induction of cytokine secretion. Particularly, the formulation stimulated the release of high levels of type-I interferon (IFN), IL-6, IL-12, and IFN-γ (Figure 5). Although multi-LP/mRNA alone induced the release of IL-12, the addition of α-GalCer to the vaccine formulation resulted in significantly higher levels of IL-12 secretion. Notably, the induction of IFN-γ was only attributable to the treatment with α-GalCer. In fact, multi-LP vector loaded with TRP2-mRNA alone did not induce a significant release of IFN-γ while both α-GalCer/TRP2-mRNA- and α-GalCer-loaded multi-LP nanoparticles induced a comparable level of IFN-γ without a significant difference. Although the levels of type-I IFNs were higher in the group receiving multi-LP/α-GalCer + mRNA compared to the group vaccinated with multi-LP/mRNA, the difference was not statistically significant. High levels of type-I IFNs are clinically relevant, as previous studies have shown that type-I IFNs directly activate immune cells, including DCs, CD4+ T cells, and CD8+ T cells, and are required for efficient cross-priming of CD8+ T cells.57 Importantly, the nonvectorized formulation did not induce detectable levels of type-I IFNs, thus confirming the importance of mRNA encapsulation for immune activation.58,59 The introduction of α-GalCer in the vaccine formulation ensures the stimulation or enhancement of the release of key cytokines that nonadjuvanted mRNA vaccine lacks.
Figure 5.

Quantification of serum levels of IFN-α, IFN-β, IFN-γ, IL-1β, IL-6, IL-12, and TNF-α, 6 h after i.v. injection of different vaccine formulations. The data represent the mean ± SD of three separate experiments involving two to three mice per group. Values were considered significant at *p < 0.05, **p < 0.01, ***p < 0.001, n.s. = not significant. Statistical significance was evaluated using a nonparametric one-way ANOVA with a Dunn’s post-test.
3.5. Antigen-Specific T-Cell Response
To evaluate the impact of our vaccine formulation on the induction of cellular and humoral immunity, mice were intravenously immunized with multi-LP/α-GalCer + TRP2-mRNA, multi-LP/TRP2-mRNA, multi-LP/TRP2-mRNA + free α-GalCer, and multi-LP/α-GalCer on days 0 and 7. Ten days after the last immunization, the presence of TRP2-specific CD8+ T cells was evaluated by intracellular staining for IFN-γ- and TRP2-specific tetramer staining.
The number of TRP2(180–188)-specific IFN-γ-secreting CD8+ T cells was significantly higher in mice immunized with the vectorized vaccine formulation compared to the group immunized with multi-LP/TRP2-mRNA + free α-GalCer (Figure 6a).
Figure 6.

(a) Quantification of CD8+ T cells expressing IFN-γ in peripheral blood. (b) Quantification of SVYDFFVWL+ CD8+ T cells in peripheral blood. C57BL/6 mice were vaccinated two times with TRP-2-mRNA with a 1-week interval by i.v. administration. All the formulations were prepared with 10 μg of TRP-2-mRNA with or without 0.5 μg of α-GalCer. Values represent the mean ± SD of two separate experiments involving two to three mice per group. *p < 0.05, **p < 0.01, ***p < 0.001. Antibody titers of OVA-specific total IgG, IgG1, IgG2b, and IgG2c (c), and IgG2c/IgG1 ratio (d) 4 weeks after prime immunization. Values represent the mean ± SD of two separate experiments involving two to three mice per group. Values were considered significant at *p < 0.05, **p < 0.01, ***p < 0.001, n.s. = not significant. Statistical significance was evaluated using a nonparametric one-way ANOVA with a Dunn’s post-test.
In addition, the percentage of TRP2-specific IFN-γ-secreting CD8+ T cells was significantly enhanced in the mice immunized with multi-LP/α-GalCer + TRP2-mRNA compared with those immunized with multi-LP/TRP2-mRNA and multi-LP/α-GalCer, demonstrating that by combining α-GalCer with antigen-mRNA, it is possible to achieve a stronger antigen-specific CD8+ T-cell immune response (Figure 6a). We also performed tetramer staining which confirmed the same trend obtained by intracellular IFN-γ staining (Figure 6b). In particular, the frequency of TRP2 (180–188) tetramer-positive CD8+ T cells was significantly increased in mice treated with vectorized vaccine formulation including both α-GalCer and TRP2-mRNA (Figure 6b).
The induction of humoral immune responses was also evaluated in response to vaccination (Figure 6c,d). For this purpose, C57BL/6 mice were immunized with multi-LP vector loaded with 10 μg of OVA-encoding mRNA (OVA-mRNA) and/or α-GalCer (0.5 μg). The serum was collected at 4 weeks after the prime immunization from the vaccinated mice, and levels of total IgG, IgG1, IgG2b, and IgG2c antibodies were determined by ELISA (Figure 6c). The results of IgG subclass analysis show that treatment with multi-LP/α-GalCer + OVA-mRNA leads to the production of antibodies with a higher IgG2c/IgG1 ratio, compared with the other formulations that have been tested such as multi-LP/OVA-mRNA, multi-LP/α-GalCer, and multi-LP/OVA-mRNA + Free α-GalCer (Figure 6d).
Vaccination with multi-LP/α-GalCer + mRNA enhanced IgG1 and IgG2c antibody titers ∼1700- and ∼4000-fold, respectively, over multi-LP/mRNA, multi-LP/α-GalCer, and multi-LP/mRNA + free α-GalCer (Figure 6c). In contrast to other vaccine formulations, the immunization with multi-LP/mRNA failed to further enhance the secretion of IgG2b antibodies.25 Probably, the difference in IgG isotypes could be in part attributed to the different structural characteristics between the delivery platforms employed in this study and in previous works.60,61
3.6. Therapeutic Efficacy of Vectorized α-GalCer/TRP2-mRNA Vaccine in a B16-F10 Melanoma Tumor Model
Once we confirmed that the addition of α-GalCer into the mRNA-vaccine elicited a robust antigen-specific immune response, we wanted to investigate if this could translate into increased therapeutic efficacy compared to the nonadjuvanted mRNA-vaccine, in a relevant tumor model. To test this, B16-F10 melanoma-bearing mice were i.v. vaccinated using unmodified TRP2-mRNA at days 5, 9, and 13 post-tumor inoculation and the therapeutic efficacy of the combined treatments was evaluated in terms of tumor growth inhibition and survival. As shown in Figure 7a, treatment with multi-LP/mRNA and multi-LP/α-GalCer alone exhibited a modest inhibitory effect on tumor growth, while multi-LP loaded with an irrelevant mRNA (irrel. mRNA) (OVA-mRNA) had no effect, excluding any nonspecific immune response. On the other hand, mice immunized with multi-LP/α-GalCer + TRP2-mRNA exhibited a significant delay in tumor growth, demonstrating that although mRNA possesses self-adjuvant property, the addition of vaccine adjuvants can significantly potentiate its therapeutic efficacy.
Figure 7.

C57BL/6 mice were inoculated with 2 × 105 B16-F10 cells. The mice were immunized with three vaccinations at days 5, 9, and 13 post-tumor inoculation. The mice were evaluated for tumor size (a) and survival (b). Representative gross tumor size excised from PBS, multi-LP/mRNA, multi-LP/Irrel. mRNA, multi-LP/α-GalCer, and multi-LP/α-GalCer + mRNA-treated mice sacrificed 18 days post-tumor inoculation. Data represent the mean ± SD of three separate experiments involving five mice per group. Differences in tumor size were considered significant at *p < 0.05, **p < 0.01, ***p < 0.001, n.s. = not significant (one-way ANOVA with post-Turkey test). For the survival curves, the significance of the difference with the control was evaluated by log-rank (Mantel–Cox) test. Mice were treated by i.v. administration of two doses on days 8 and 15 post-tumor inoculation. Mice were sacrificed on day 17 and tumor-infiltrating CD8 and CD4 T cells were quantified by flow cytometry (c). (d) Quantification of intra-tumoral SVYDFFVWL+ CD8+ T cells after vaccination with different formulations. Data represent the mean ± SD of two independent experiments involving two to three mice per group. Values were considered significant at *p < 0.05, **p < 0.01, ***p < 0.001, n.s. = not significant. Statistical significance was evaluated using a nonparametric one-way ANOVA with a Dunn’s post-test.
Moreover, mice vaccinated with TRP2-mRNA/α-GalCer-loaded multi-LP nanoparticles (multi-LP/α-GalCer + mRNA) showed a significantly extended survival if compared with those treated with TRP2-mRNA-loaded multi-LP nanoparticles (multi-LP/mRNA), multi-LP/α-GalCer, or multi-LP/irrel. mRNA (Figure 7b). These findings clearly demonstrate that the codelivery of antigen-mRNA with α-GalCer by multi-LP vector robustly improves the therapeutic efficacy of anticancer mRNA-vaccines.
Consistent with the efficacy studies, we found that multi-LP/α-GalCer + TRP2-mRNA vaccination promotes increased infiltration of CD8+ T cells within the tumor microenvironment (Figure 7c). The combined treatment increased the CTL density and CD8/CD4 ratio (Figure 7c). Furthermore, antigen-specific CD8+ T cell analysis revealed a higher frequency of TRP2-positive tumor infiltrating CD8+ T cells in tumor-bearing mice vaccinated with multi-LP/α-GalCer + TRP2-mRNA compared with multi-LP/TRP2-mRNA, multi-LP/α-GalCer, or multi-LP/Irrel. mRNA (Figure 7d).
4. Conclusions
The incorporation of α-GalCer into the lipopolyplex vector loaded with TRP2-mRNA potentiated the antitumor immune response and its therapeutic efficacy. Our results revealed that the proposed vector for the delivery of antigen-mRNA in combination with α-GalCer possesses a natural ability to passively target DCs in vivo and to efficiently transfect them, following the i.v. administration route. Our results also showed that the use of multi-LP vector for α-GalCer/TRP2-mRNA codelivery significantly enhanced the induction of antigen-specific CD8+ T cells both systemically and intratumorally. Likewise, multi-LP/α-GalCer + TRP2-mRNA vaccination has shown to be an efficient strategy for the induction of humoral immune responses in vivo, eliciting the secretion of high levels of antigen-specific IgG1, IgG2b, and IgG2c. Mice vaccinated with multi-LP/α-GalCer + TRP2-mRNA showed a significantly higher IgG2c/IgG1 ratio compared to mice immunized with nanoparticles loaded only with antigen-mRNA, demonstrating that by combining mRNA-based vaccine with α-GalCer, it is possible to alter the Th1/Th2 balance toward an increased Th1 response.
All this resulted in marked tumor growth retardation and increased survival of B16-F10 melanoma-bearing mice. We were able to achieve the induction of an effective antitumor immune response with a lower amount of mRNA, compared to other proposed vaccination strategies based on the use of lipoplexes.21
These data may have important implications in the design of novel formulations combining mRNA with lipidic adjuvants for the development of cancer vaccines with enhanced antitumor properties.
Acknowledgments
The authors would like to thank Children’s Hospital of Eastern Ontario (CHEO) Foundation for supporting this project. This work was financially supported by the National Cancer Institute of Canada (to D.F.S.). The funders had no role in the study design, data collection, data interpretation, or decision to submit the work for publication.
Glossary
Abbreviations
- α-GalCer
α-galactosylceramide
- APC
antigen-presenting cell
- BMDC
bone marrow-derived DC
- CD
cluster of differentiation
- CTL
cytotoxic T lymphocyte
- DC
dendritic cell
- DOPE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- DSPE-PEG
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-polyethylene glycol
- EDOPC
1,2-dioleoyl-sn-glycero-3-ethylphosphocholine
- eGFP
enhanced green fluorescence protein
- HLA
human leucocyte antigen
- i.d.
intradermal
- iNKT
invariant natural killer T
- IFN-γ
interferon gamma
- IL
interleukin
- i.v.
intravenous
- LUC
luciferase
- MHC
major histocompatibility complex
- mRNA
messenger RNA
- MLV5
N1-[2-((1S)-1-[(3-aminopropyl)amino]-4-[di(3-amino-propyl)amino]butylcarboxamido)ethyl]-3,4-di[oleyloxy]-benzamide
- OVA
ovalbumin
- PbAE
poly-(β-amino ester) polymer
- TLR
toll-like receptor
- TRP2
tyrosinase-related protein 2
The authors declare no competing financial interest.
References
- Old L. J.; Boyse E. A. Immunology of experimental tumors. Annu. Rev. Med. 1964, 15, 167–186. 10.1146/annurev.me.15.020164.001123. [DOI] [PubMed] [Google Scholar]
- van der Burg S. H.; Arens R.; Ossendorp F.; van Hall T.; Melief C. J. M. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- Scott A. M.; Allison J. P.; Wolchok J. D. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012, 12, 14. [PMC free article] [PubMed] [Google Scholar]
- Jackson H. J.; Rafiq S.; Brentjens R. J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016, 13, 370–383. 10.1038/nrclinonc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melief C. J. M.; van Hall T.; Arens R.; Ossendorp F.; van der Burg S. H. Therapeutic cancer vaccines. J. Clin. Invest. 2015, 125, 3401–3412. 10.1172/jci80009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioretti D.; Iurescia S.; Fazio V. M.; Rinaldi M. DNA Vaccines: Developing New Strategies against Cancer. J. Biomed. Biotechnol. 2010, 2010, 174378. 10.1155/2010/174378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weide B.; Garbe C.; Rammensee H.-G.; Pascolo S. Plasmid DNA- and messenger RNA-based anti-cancer vaccination. Immunol. Lett. 2008, 115, 33–42. 10.1016/j.imlet.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Pardi N.; Hogan M. J.; Porter F. W.; Weissman D. mRNA vaccines - a new era in vaccinology. Nat. Rev. Drug Discovery 2018, 17, 261–279. 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U.; Karikó K.; Türeci Ö. mRNA-based therapeutics - developing a new class of drugs. Nat. Rev. Drug Discovery 2014, 13, 759–780. 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- Ulmer J. B.; Mason P. W.; Geall A.; Mandl C. W. RNA-based vaccines. Vaccine 2012, 30, 4414–4418. 10.1016/j.vaccine.2012.04.060. [DOI] [PubMed] [Google Scholar]
- Islam M. A.; Reesor E. K. G.; Xu Y.; Zope H. R.; Zetter B. R.; Shi J. Biomaterials for mRNA Delivery. Biomater. Sci. 2015, 3, 1519–1533. 10.1039/c5bm00198f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman K. J.; Webber M. J.; Anderson D. G. Materials for non-viral intracellular delivery of messenger RNA therapeutics. J. Control. Release 2016, 240, 227–234. 10.1016/j.jconrel.2015.12.032. [DOI] [PubMed] [Google Scholar]
- Schott J. W.; Morgan M.; Galla M.; Schambach A. Viral and Synthetic RNA Vector Technologies and Applications. Mol Ther 2016, 24, 1513–1527. 10.1038/mt.2016.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S.; Tonnu N.; Tachikawa K.; Limphong P.; Vega J. B.; Karmali P. P.; et al. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E1941–E1950. 10.1073/pnas.1619653114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman K. J.; Dorkin J. R.; Yang J. H.; Heartlein M. W.; DeRosa F.; Mir F. F.; et al. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. 10.1021/acs.nanolett.5b02497. [DOI] [PubMed] [Google Scholar]
- Yanez Arteta M.; Kjellman T.; Bartesaghi S.; Wallin S.; Wu X.; Kvist A. J.; et al. Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E3351–E3360. 10.1073/pnas.1720542115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinlay C. J.; Vargas J. R.; Blake T. R.; Hardy J. W.; Kanada M.; Contag C. H.; et al. Charge-altering releasable transporters (CARTs) for the delivery and release of mRNA in living animals. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E448–E456. 10.1073/pnas.1614193114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski P. S.; Rudra A.; Miao L.; Anderson D. G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. 10.1016/j.ymthe.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Zhang X.; Dong Y. Nanoscale platforms for messenger RNA delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11, e1530. 10.1002/wnan.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberli M. A.; Reichmuth A. M.; Dorkin J. R.; Mitchell M. J.; Fenton O. S.; Jaklenec A.; et al. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017, 17, 1326–1335. 10.1021/acs.nanolett.6b03329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz L. M.; Diken M.; Haas H.; Kreiter S.; Loquai C.; Reuter K. C.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- Persano S.; Guevara M. L.; Li Z.; Mai J.; Ferrari M.; Pompa P. P.; et al. Lipopolyplex potentiates anti-tumor immunity of mRNA-based vaccination. Biomaterials 2017, 125, 81–89. 10.1016/j.biomaterials.2017.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phua K. K.; Staats H. F.; Leong K. W.; Nair S. K. Intranasal mRNA nanoparticle vaccination induces prophylactic and therapeutic anti-tumor immunity. Sci. Rep. 2015, 4, 5128. 10.1038/srep05128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persano S. A Self-Assembled Non-Viral vector as Potential Platform for mRNA-Based Vaccines. Trans. Biomed. 2017, 8, 119. 10.21767/2172-0479.100119. [DOI] [Google Scholar]
- Haabeth O. A. W.; Blake T. R.; McKinlay C. J.; Waymouth R. M.; Wender P. A.; Levy R. mRNA vaccination with charge-altering releasable transporters elicits human T cell responses and cures established tumors in mice. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E9153–E9161. 10.1073/pnas.1810002115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capasso Palmiero U.; Kaczmarek J. C.; Fenton O. S.; Anderson D. G. Poly(β-amino ester)-co-poly(caprolactone) Terpolymers as Nonviral Vectors for mRNA Delivery In Vitro and In Vivo. Adv. Healthc. Mater. 2018, 7, 1800249. 10.1002/adhm.201800249. [DOI] [PubMed] [Google Scholar]
- Hess K. L.; Andorko J. I.; Tostanoski L. H.; Jewell C. M. Polyplexes assembled from self-peptides and regulatory nucleic acids blunt toll-like receptor signaling to combat autoimmunity. Biomaterials 2017, 118, 51–62. 10.1016/j.biomaterials.2016.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn G. M.; Laga R.; Darrah P. A.; Ishizuka A. S.; Balaci A. J.; Dulcey A. E.; et al. In vivo characterization of the physicochemical properties of polymer-linked TLR agonists that enhance vaccine immunogenicity. Nat. Biotechnol. 2015, 33, 1201–1210. 10.1038/nbt.3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölen Y.; Kreutz M.; Gileadi U.; Tel J.; Vasaturo A.; van Dinther E. A. W.; et al. Co-delivery of PLGA encapsulated invariant NKT cell agonist with antigenic protein induce strong T cell-mediated antitumor immune responses. Oncoimmunology 2016, 5, e1068493. 10.1080/2162402x.2015.1068493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zhang L.; Xu Z.; Miao L.; Huang L. mRNA Vaccine with Antigen-Specific Checkpoint Blockade Induces an Enhanced Immune Response against Established Melanoma. Mol. Ther. 2018, 26, 420–434. 10.1016/j.ymthe.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendelac A.; Savage P. B.; Teyton L. The biology of NKT cells. Annu. Rev. Immunol. 2007, 25, 297–336. 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- Brennan P. J.; Brigl M.; Brenner M. B. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013, 13, 101–117. 10.1038/nri3369. [DOI] [PubMed] [Google Scholar]
- Nair S.; Dhodapkar M. V. Natural Killer T Cells in Cancer Immunotherapy. Front Immunol 2017, 8, 1178. 10.3389/fimmu.2017.01178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans I. F.; Silk J. D.; Gileadi U.; Masri S. H.; Shepherd D.; Farrand K. J.; Salio M.; Cerundolo V. Dendritic cell function can be modulated through cooperative actions of TLR ligands and invariant NKT cells. J. Immunol. 2007, 178, 2721–2729. 10.4049/jimmunol.178.5.2721. [DOI] [PubMed] [Google Scholar]
- Ghinnagow R.; De Meester J.; Cruz L. J.; Aspord C.; Corgnac S.; Macho-Fernandez E.; et al. Co-delivery of the NKT agonist α-galactosylceramide and tumor antigens to cross-priming dendritic cells breaks tolerance to self-antigens and promotes antitumor responses. Oncoimmunology 2017, 6, e1339855. 10.1080/2162402x.2017.1339855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macho-Fernandez E.; Cruz L. J.; Ghinnagow R.; Fontaine J.; Bialecki E.; Frisch B.; et al. Targeted Delivery of α-Galactosylceramide to CD8α+ Dendritic Cells Optimizes Type I NKT Cell-Based Antitumor Responses. J. Immunol. 2014, 193, 961–969. 10.4049/jimmunol.1303029. [DOI] [PubMed] [Google Scholar]
- Nakamura T.; Yamazaki D.; Yamauchi J.; Harashima H. The nanoparticulation by octaarginine-modified liposome improves α-galactosylceramide-mediated antitumor therapy via systemic administration. J. Control. Release 2013, 171, 216–224. 10.1016/j.jconrel.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Thapa P.; Zhang G.; Xia C.; Gelbard A.; Overwijk W. W.; Liu C.; et al. Nanoparticle formulated alpha-galactosylceramide activates NKT cells without inducing anergy. Vaccine 2009, 27, 3484–3488. 10.1016/j.vaccine.2009.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasar H.; Biehl A.; De Rossi C.; Koch M.; Murgia X.; Loretz B. Kinetics of mRNA delivery and protein translation in dendritic cells using lipid-coated PLGA nanoparticles. J. Nanobiotechnol. 2018, 16, 72. 10.1186/s12951-018-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Saffold S.; Cao X.; Krauss J.; Chen W. Eliciting T cell immunity against poorly immunogenic tumors by immunization with dendritic cell-tumor fusion vaccines. J. Immunol. 1998, 161, 5516. [PubMed] [Google Scholar]
- Guerrero-Cázares H.; Tzeng S. Y.; Young N. P.; Abutaleb A. O.; Quiñones-Hinojosa A.; Green J. J. Biodegradable Polymeric Nanoparticles Show High Efficacy and Specificity at DNA Delivery to Human Glioblastoma in Vitro and in Vivo. ACS Nano 2014, 8, 5141–5153. 10.1021/nn501197v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X.; Mai J.; Xu R.; Perez J. E. T.; Guevara M. L.; Shen Q.; et al. Porous Silicon Microparticle Potentiates Anti-Tumor Immunity by Enhancing Cross-Presentation and Inducing Type I Interferon Response. Cell Rep. 2015, 11, 957–966. 10.1016/j.celrep.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwijk W. W.; Restifo N. P.. B16 as a mouse model for human melanoma. In Current Protocols in Immunology; John Wiley & Sons: Hoboken, NJ, 2001; Chapter 20, 10.1002/0471142735.im2001s39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachynski R. K.; Scholz A.; Monnier J.; Butcher E. C.; Zabel B. A. Evaluation of Tumor-infiltrating Leukocyte Subsets in a Subcutaneous Tumor Model. J. Visualized Exp. 2015, 98, 52657. 10.3791/52657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolova V.; Flace A.; Bauer M.; Schwarz K.; Saudan P.; Bachmann M. F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. 10.1002/eji.200737984. [DOI] [PubMed] [Google Scholar]
- Foged C.; Brodin B.; Frokjaer S.; Sundblad A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm. 2005, 298, 315–322. 10.1016/j.ijpharm.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Ahmad A.; Evans H. M.; Ewert K.; George C. X.; Samuel C. E.; Safinya C. R. New multivalent cationic lipids reveal bell curve for transfection efficiency versus membrane charge density: lipid-DNA complexes for gene delivery. J. Gene Med. 2005, 7, 739–748. 10.1002/jgm.717. [DOI] [PubMed] [Google Scholar]
- Ewert K.; Ahmad A.; Evans H. M.; Schmidt H.-W.; Safinya C. R. Efficient Synthesis and Cell-Transfection Properties of a New Multivalent Cationic Lipid for Nonviral Gene Delivery. J. Med. Chem. 2002, 45, 5023–5029. 10.1021/jm020233w. [DOI] [PubMed] [Google Scholar]
- Ewert K. K.; Zidovska A.; Ahmad A.; Bouxsein N. F.; Evans H. M.; McAllister C. S.; et al. Cationic Liposome-Nucleic Acid Complexes for Gene Delivery and Silencing: Pathways and Mechanisms for Plasmid DNA and siRNA. Top. Curr. Chem. 2010, 296, 191–226. 10.1007/128_2010_70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshyar N.; Gray S.; Han H.; Bao G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 2016, 11, 673–692. 10.2217/nnm.16.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; He W.; Shi X.; He X.; Dou L.; Gao Y. The Role of CD1d and MR1 Restricted T Cells in the Liver. Front. Immunol. 2018, 9, 2424. 10.3389/fimmu.2018.02424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando T.; Ito H.; Ohtaki H.; Seishima M. Toll-like receptor agonists and alpha-galactosylceramide synergistically enhance the production of interferon-gamma in murine splenocytes. Sci. Rep. 2013, 3, 2559. 10.1038/srep02559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong T.; Yi T.; Yang M.; Lin S.; Li W.; Xu X.; Hu J.; Jia L.; Hong X.; Niu W. Co-operation of -galactosylceramide-loaded tumour cells and TLR9 agonists induce potent anti-tumour responses in a murine colon cancer model. Biochem. J. 2016, 473, 7–19. 10.1042/bj20150129. [DOI] [PubMed] [Google Scholar]
- Sainz V.; Moura L. I. F.; Peres C.; Matos A. I.; Viana A. S.; Wagner A. M.; Vela Ramirez J. E.; Barata T. S.; Gaspar M.; et al. α-Galactosylceramide and peptide-based nano-vaccine synergistically induced a strong tumor suppressive effect in melanoma. Acta Biomater. 2018, 76, 193–207. 10.1016/j.actbio.2018.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.-F.; Appella E.; Kawakami Y.; Kang X.; Rosenberg S. A. Identification of TRP-2 as a Human Tumor Antigen Recognized by Cytotoxic T Lymphocytes. J. Exp. Med. 1996, 184, 2207–2216. 10.1084/jem.184.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschen A.; Song M.; Osen W.; Nguyen X. D.; Mueller-Berghaus J.; Fink D.; et al. Detection of Spontaneous CD4+ T-Cell Responses in Melanoma Patients against a Tyrosinase-Related Protein-2-Derived Epitope Identified in HLA-DRB1*0301 Transgenic Mice. Clin. Cancer Res. 2005, 11, 5241–5247. 10.1158/1078-0432.ccr-05-0170. [DOI] [PubMed] [Google Scholar]
- Hervas-Stubbs S.; Perez-Gracia J. L.; Rouzaut A.; Sanmamed M. F.; Le Bon A.; Melero I. Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 2011, 17, 2619–2627. 10.1158/1078-0432.ccr-10-1114. [DOI] [PubMed] [Google Scholar]
- Pollard C.; Rejman J.; De Haes W.; Verrier B.; Van Gulck E.; Naessens T.; De Smedt S.; Bogaert P.; Grooten J.; Vanham G.; De Koker S. Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol. Ther. 2013, 21, 251–259. 10.1038/mt.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel B.; Teufel R.; Probst J.; Carralot J.-P.; Geginat J.; Radsak M.; Jarrossay D.; Wagner H.; Jung G. n.; Rammensee H.-G.; Hoerr I.; Pascolo S. Toll-like receptor-dependent activation of several human blood cell types by protamine-condensed mRNA. Eur. J. Immunol. 2005, 35, 1557–1566. 10.1002/eji.200425656. [DOI] [PubMed] [Google Scholar]
- Poecheim J.; Barnier-Quer C.; Collin N.; Borchard G. Ag85A DNA Vaccine Delivery by Nanoparticles: Influence of the Formulation Characteristics on Immune Responses. Vaccines 2016, 4, 32. 10.3390/vaccines4030032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H.; Shukla S.; Wang C.; Masarapu H.; Steinmetz N. F. Heterologous Prime-Boost Enhances the Antitumor Immune Response Elicited by Plant-Virus-Based Cancer Vaccine. J. Am. Chem. Soc. 2019, 141, 6509–6518. 10.1021/jacs.9b01523. [DOI] [PMC free article] [PubMed] [Google Scholar]