

Abstract
Nitric oxide (NO) is an important signaling molecule with a variety of functions in the CNS, including a potential role in modulating neuronal growth and synapse formation. In the present study, we used tractable, identified neurons in the CNS of the pond snail Lymnaea stagnalis to study the role of endogenous NO signaling in neuronal growth and synaptic remodeling after nerve injury. Axonal damage of L. stagnalis neurons B1 and B2 induces extensive central growth of neurites that is accompanied by changes in existing electrical connections, the transient formation of novel electrical connections, and the formation of a novel excitatory chemical synapse from B2 to B1 neurons. Partial chronic inhibition of endogenous NO synthesis reduces neurite growth in NO-synthase-expressing B2, but has only minor effects on NOS-negative B1 neurons. Chronic application of an NO donor while inhibiting endogenous NO synthesis rescues neurite extension in B2 neurons and boosts growth of B1 neurons. Blocking soluble guanylate cyclase activity completely suppresses neurite extension and synaptic remodeling after nerve crush, demonstrating the importance of cGMP in these processes. Interestingly, inhibition of cGMP-dependent protein kinase only suppresses chemical synapse formation without effects on neuronal growth and electrical synapse remodeling. We conclude that NO signaling via cGMP is an important modulator of both neurite growth and synaptic remodeling after nerve crush. However, differential effects of cGMP-dependent protein kinase inhibition on neurite growth and synaptic remodeling suggest that these effects are mediated by separate signaling pathways.
Introduction
The radical gas nitric oxide (NO) has attracted much attention since the first demonstration that it can act as a signaling molecule in the CNS (Garthwaite et al., 1988). NO has been implicated in many different aspects of neuronal function ranging from synaptic transmission and plasticity to neuromodulation, learning and memory formation, and nervous system development (Garthwaite, 2008; Zhou and Zhu, 2009). The majority of physiological NO functions are mediated by an evolutionarily highly conserved signaling pathway that starts with the binding of NO to soluble guanylate cyclase (sGC; Cary et al., 2006; Garthwaite, 2010). Activation of sGC leads to the production of cGMP, which in turn can act on various downstream targets, including cGMP-dependent protein kinase (PKG), a number of phosphodiesterases, cyclic-nucleotide-gated ion channels, and transcription factors (Francis et al., 2010). In addition, some NO effects have been shown to occur independently of sGC/cGMP and are probably due to direct reaction of NO with various proteins (e.g., S-nitrosylation) modifying their function (Jaffrey et al., 2001; Hess et al., 2005).
Various lines of evidence suggest that NO plays a significant role in modulating neuronal growth and synaptogenesis. In both vertebrates and invertebrates, NO synthase (NOS) expression increases progressively during embryonic and postembryonic development (Downen et al., 1999; Villani, 1999; Moreno et al., 2002; Serfozo et al., 2002; Gao et al., 2008; Stern et al., 2010). Interestingly, the increases in NOS expression during development frequently coincide with periods of synapse formation and maturation. Direct evidence for a role of NO in synapse formation and maturation and the refinement of neuronal circuits has been provided by studies demonstrating morphological changes in innervation patterns and synapse morphology after disruption of NO signaling during critical developmental periods (Cogen and Cohen-Cory, 2000; Seidel and Bicker, 2000; Gibbs et al., 2001; Gibson et al., 2001; Leamey et al., 2001; Godfrey and Schwarte, 2010). Further evidence for a role of NO in the control of neuronal growth has been provided by studies of neuronal injury and neuronal regeneration, which have shown that NO can have either beneficial or detrimental effects on recovery from nerve injury (Keilhoff et al., 2002; Lee et al., 2003; Marsala et al., 2007; Stern and Bicker, 2008; Koriyama et al., 2009).
Despite the extensive evidence, based primarily on anatomical and immunohistochemical techniques, for a role of NO in neuronal growth and synapse formation, there is currently a lack of studies that combine morphological and electrophysiological techniques to investigate the relationship between the effects of NO signaling on neuritogenesis and the formation of functional synapses. Using the large, easily identifiable neurons in the pond snail (Lymnaea stagnalis) nervous system as a model system, we demonstrate here that endogenous NO synthesis and cGMP production affect injury-induced neuronal growth, de novo synapse formation, and synaptic remodeling. We also provide evidence that these effects are at least partially independent of each other and that NO effects on synapse formation and remodeling are not simply the consequence of NO effects on neurite growth.
Materials and Methods
Animals.
The hermaphroditic snails (L. stagnalis) used in these experiments were obtained from the breeding facility at the University of Leicester. The animals were kept in 35 L plastic tanks filled with deionized water plus Instant Ocean (0.25 g/L; Aquarium Systems). Each tank was fitted with an internal foam filter and the bottom was covered with a thin layer of crushed sea shell. Animals were maintained at room temperature on a 12 h light-dark cycle and fed a diet of lettuce.
Organotypic culture.
Before dissection, snails were washed and anesthetized by washing for 10 min in 20% Listerine. All dissections were performed in sterile HEPES-buffered saline (normal saline, NS) containing the following (in mm): 50 NaCl, 1.6 KCl, 2 MgCl2, 3.5 CaCl2, and 10 HEPES, pH 7.9, in distilled water (Benjamin and Winlow, 1981). The whole CNS was isolated from dissected animals (15–30 mm shell length), including the paired buccal ganglia (BGs) plus a short stretch of esophagus. Intact pairs of BGs were separated from the rest of the nervous system by cutting the cerebral-buccal connective close to the cerebral ganglia. The intact dorsal buccal nerves (DBNs) were kept attached to a small section of esophagus and the lateral and ventral buccal nerves were cut as close to the buccal mass as possible. The isolated BGs were pinned out on a small block of black Sylgard and the outer connective tissue sheath that covers the ganglia was gently removed. Subsequently, the preparation was washed in antibiotic saline (sterile NS plus gentamicin, 150 μg/ml). To induce central neurite growth, proximal nerve crushes were performed ∼100 μm from the origin of the DBN by pressing the nerve against the Sylgard using a 0.14 mm pin with its tip bend at an angle of 90°. Similar nerve crush protocols were used in a number of studies to study neurite growth in molluscs (e.g., Wong et al., 1981; Murphy et al., 1983; Chiasson et al., 1994). Preparations were then cultured on 24-well plates for 1–4 d in individual wells filled with 1 ml of defined medium (DM; 50% v/v Leibovitz L-15; Invitrogen) containing the following (in mm): 40 NaCl, 1.7 KCl, 4.1 CaCl2, 1.5 MgCl2, and 10 HEPES, pH 7.9 in distilled water. In some experiments, DM was supplemented with one of the following as indicated in the Results: 7-nitroindazole (7NI, 0.25 mm), 7NI (0.25 mm) plus diethylenetriamine NONOate (DETA, 0.5 mm), 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, 0.4 mm), or KT5823 (10 μm). All cultures were kept in a humidified box in an incubator at 22°C.
Cell culture.
Individual B1 and B2 neurons were isolated using previously described techniques (Straub and Benjamin, 2001) and plated on either poly-l-lysine-coated glass-bottomed (MatTek) or plastic 35 mm culture dishes (Nunc) in half-strength conditioned medium (CM; 1 ml of CM plus 1 ml of DM per culture dish). CM was prepared by incubating isolated CNS preparations in DM for 3 d. The culture dishes were stored at −20°C and thawed 2–3 h before use. The NO donor DETA (final concentration: 0.5 mm) was added to cultures 1 h after plating, as indicated in the Results. Images of cultures grown at 22°C were taken at 24 and 48 h using a Nikon TE2000 inverted microscope with a 10× phase-contrast objective using a CCD camera (DP450; Deltapix) and DPXPro software (Deltapix).
Electrophysiological recordings.
Before impalement of individual B1 and B2 neurons with sharp intracellular microelectrodes, preparations were treated enzymatically to soften the inner connective tissue and to aid penetration. For this purpose, the culture medium was removed and a small amount of dry protease (protease type XIV; Sigma) was placed on top of the BG and left for 45 s. The preparation was then washed extensively with NS to remove all traces of protease. Sharp electrodes were made from 1.5 mm glass capillaries with inner filament (GC150TF-10; Harvard Apparatus) using a P-87 puller (Sutter) and filled with 3 m potassium acetate and 5 mm KCl (electrode resistance, 15–35 MΩ). Signals from these electrodes were amplified using intracellular amplifiers (Multiclamp 900A, Molecular Devices; NL102G, Digitimer), digitized with a CED 1401plus interface (Cambridge Electronics Design), and visualized and stored on a personal computer using Spike2 software (Cambridge Electronics Design). During the recording, whole nervous system preparations were superfused at a rate of ∼1.5 ml/min with NS. Each BG contains a left and right B1 and a left and right B2 neuron (Fig. 1Ai). All four neurons were impaled simultaneously and their activity was recorded in bridge mode. The incidence and properties of newly formed chemical synaptic connections were tested using 1 s, 10 Hz trains of 20 ms depolarizing current pulses (20 nA). To obtain accurate electrical coupling and input resistance measurements, neurons were impaled with a second current injection electrode to enable independent current injection and membrane potential (Vm) measurement. Electrical coupling was investigated using 1 s hyperpolarizing current pulses (−5 nA). We also recorded the resting Vm, input resistance, and action potential (AP) properties (peak-to-peak amplitude, width, and after-hyperpolarization [AHP]) of these cells. All analysis was performed in Spike2 v6.12 software aided by some custom-written scripts.
Figure 1.
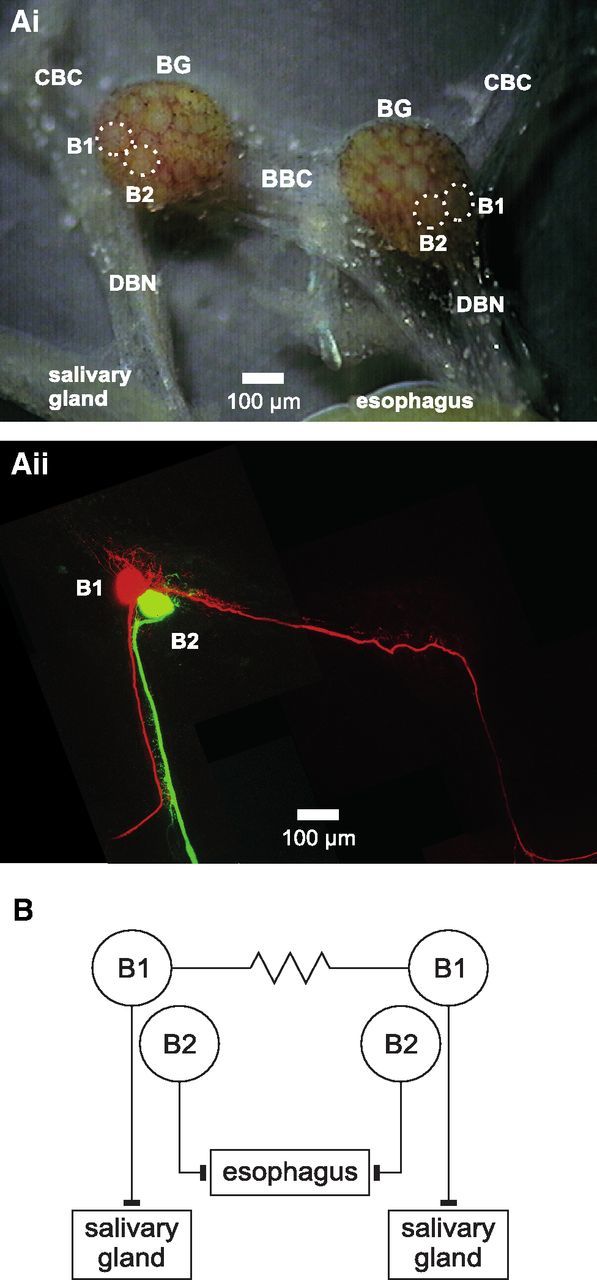
Anatomy and synaptic connectivity of L. stagnalis B1 and B2 neurons. Ai, Micrograph of isolated BGs showing soma position of B1 and B2 neurons (outline marked by dotted lines). Aii, Maximum-intensity Z projection of two-photon laser scanning image of the BGs shown in Ai after intracellular injection of a B1 neuron with Alexa Fluor 568 and a B2 neuron with Alexa Fluor 488. B, Schematic diagram showing the electrical coupling between the left and right B1 neurons and the innervation of the salivary glands by the B1 neurons and the esophagus by the B2 neurons. BBC indicates buccal-buccal commissure; CBC, cerebral-buccal connective.
Iontophoretic staining techniques.
Individual B1 and B2 cells were stained in the same preparations used for electrophysiological recordings by iontophoretic injection of either Alexa Fluor 488 fluorescent dye (Invitrogen) or Neurobiotin (Vector Laboratories). Alexa Fluor 488 (5 mm in 200 mm KCl) was injected using negative current pulses (−8 nA, 0.5 s, 1 Hz) and Neurobiotin (2% in 200 mm KCl) was injected using positive current pulses (2 nA, 0.5 s, 1 Hz) for a duration of 20 min. After injection, the electrodes were carefully removed and the preparation was kept at 4°C for a minimum of 1 h to let the dye diffuse throughout the cell's neuritic arbor. Preparations were fixed overnight in 2% formaldehyde at 4°C. After fixation, Alexa Fluor 488 fluorescence was relatively weak. Therefore, the signal was amplified using an antibody amplification technique. For this, preparations were washed extensively in 0.1 m phosphate buffer (PB; 6 × 1 h) and then incubated overnight at 4°C in an anti-Alexa Fluor 488 rabbit antibody (Invitrogen) at a dilution of 1:400 in PB and 4% Triton X-100. The incubation in the primary antibody was followed by further washes in PB (6 × 1 h) before the preparations were incubated overnight at 4°C in an Alexa Fluor 488-labeled goat anti-rabbit IgG antibody (1:1000 in PB plus 0.3% Triton X-100; Abcam). The solution also contained 0.2% streptavidin-Cy3 conjugate for the detection of the Neurobiotin label. After washing in PB (3 × 1 h), preparations were mounted in ProLong Gold (Invitrogen) and stacks of images at a z resolution of 1.5 μm were collected using a confocal microscope with a 20× objective (FV1000; Olympus).
Image analysis.
All image analysis was performed using the Fiji distribution of the ImageJ image processing package (Schindelin et al., 2012). Specific aspects of the analysis were aided by some custom macros for specific tasks.
Neuron morphology in intact BGs.
Individual image stacks were median filtered, maximum intensity projected, and assembled into single images that covered the entire neuritic arbor of the labeled neurons. Stitching of individual images was performed using either the MosaicJ (Thévenaz and Unser, 2007) or 2D stitching plug-in (Preibisch et al., 2009). The images were then thresholded manually to create a mask of all labeled structures. The automatic selection was edited manually using a set of custom macro tools to include all neuronal structures in the final mask. Analysis of neuritic growth in the DBN was restricted to the first 300 μm of the DBN that extends just beyond the original crush site. This was necessary because measurements of growth beyond the crush site were quite variable due to differences in the size of the esophageal tissue that remained at the end of the DBN. Furthermore, the crushed DBNs were very fragile and frequently broke during tissue processing after fixation at or just beyond the crush site, making it impossible to assess peripheral growth beyond the crush site. The cell soma was manually excluded from the final mask, before the mask was skeletonized and the total length of the skeleton measured using the Line8 plug-in (version 2.11; http://www.dentistry.bham.ac.uk/landinig/software/software.html).
Neuron morphology of isolated neurons in cell culture.
If necessary, multiple phase-contrast images of the total neuritic arbor of an individual neuron were assembled using the MosaicJ plug-in (Thévenaz and Unser, 2007). Images were then preprocessed (band-pass filtered, median averaged, background subtracted, and contrast enhanced) before thresholding for the automatic detection of phase-dark neuronal structures. The automatic selection was manually edited to create a mask that included all neuronal structures. The cell soma and any original axon was excluded, so that the final mask only contained neurites that had grown after isolation in cell culture. The final mask was then skeletonized and the total length of the skeleton measured using the Line8 plug-in (version 2.11; http://www.dentistry.bham.ac.uk/landinig/software/software.html).
Measurement of cGMP concentration in the whole CNS.
For the measurement of cGMP concentrations, individual whole CNS preparations were isolated from the snails and maintained in DM for 24 h at 22°C. DM was supplemented with 7NI (0.25 or 0.5 mm) or ODQ (0.4 mm), as indicated in the Results, to test for the effects of chronic inhibition of NOS and sGC, respectively. After 24 h, the preparations were removed from the culture medium, placed in 4.5 ml cryovials and quickly frozen by placing the cryovials in liquid nitrogen. The samples were then stored at −70°C before extraction and cGMP radioimmunoassay. Each whole CNS was homogenized by 3 × 20 s bursts at maximum setting in a Polytron homogenizer in 300 μl of ice-cold 0.5 m trichloroacetic acid. After centrifugation (14,000 × g, 6 min, 4°C), the supernatant was neutralized using the freon/tri-n-octylamine method. Neutral CNS extracts were diluted fivefold in 100 mm sodium acetate, pH 6.2, and acetylated by the consecutive addition of triethylamine (10 μl) and acetic anhydride (5 μl) and used in the radioimmunoassay (Brooker et al., 1979) within 60 min. cGMP standards (100 μl; 0–4 nm) were treated identically. Acetylated samples (100 μl) were mixed with 2′-O-succinyl 3-[125I]-iodotyrosine methyl ester cGMP (IM107; GE Healthcare) at 50 μl, ∼3000 disintegrations per minute made up in 50 mm sodium acetate, 0.2% bovine serum albumin (BSA), pH 6.2, and 100 μl of anti-cGMP antibody (TRK500; GE Healthcare) diluted in 50 mm sodium acetate, 0.2% BSA, pH 6.2. Samples were intermittently vortex mixed during a 4 h incubation at 4°C. Free and bound cGMP was separated by charcoal precipitation with 500 μl of a charcoal suspension (1% w/v activated charcoal in 100 mm potassium phosphate, 0.2% BSA, pH 6.2). After vortex mixing for 5 min, samples were centrifuged (13,000 × g, 4 min, 4°C) and radioactivity was determined in an aliquot of supernatant (600 μl). Unknown values were determined from the cGMP standard curve using Prism software (GraphPad).
Statistical analysis.
All statistical analysis was performed using Prism software (GraphPad). One-way ANOVA tests were performed to compare the total neurite length of B1 and B2 neurons in the isolated BGs in the nine different treatment groups (DIV0, DIV1+, DIV2+, DIV4+, DIV2−, DIV2+/7NI, DIV2+/7NI+DETA, DIV2+/ODQ, DIV2+/KT5823). Similarly, overall one-way ANOVA tests were performed for the strength of electrical and chemical synapses for all treatment groups that showed a particular connection. The one-way ANOVA tests were followed by Newman–Keuls (NK) post hoc tests for pairwise comparison of individual treatment groups. χ2 tests were used to compare the incidence of electrical and chemical synapse formation between the different treatment groups. The total growth of isolated B1 and B2 neurons in cell cultures in the presence and absence of DETA was compared using a one-way ANOVA. Other statistical analysis was performed as indicated in the text.
Results
Axonal injury induces neurite extension and specific synaptic remodeling in tissue culture
An initial objective was to characterize the regenerative response of L. stagnalis buccal B1 and B2 neurons to axonal injury. These neurons were chosen for the following reasons: first, the buccal neuron B2 is known to express NOS, whereas buccal neuron B1 does not (Moroz et al., 1994; Park et al., 1998). This enabled us to make differential comparisons between neuronal growth responses in NOS-positive and NOS-negative neurons. Second, their cell bodies are easily identifiable, which aids recording of neuronal activity and intracellular dye labeling (Fig. 1Ai). Third, both B1 and B2 neurons have axons in the DBN (Fig. 1Aii) that innervate the salivary glands (B1) and esophagus (B2; Fig. 1B). Therefore, crushing the DBN causes axonal injury to both neurons. In addition, B1 neurons possess a second axon that projects to the contralateral BG, where it forms an electrical synapse with the contralateral B1 neuron (Benjamin and Rose, 1979) and then extends into the contralateral DBN (Fig. 1Aii,B).
Intracellular injection of B1 and B2 neurons with either Neurobiotin or Alexa Fluor 488 in acutely isolated preparations (DIV0) showed the expected morphology for buccal neurons B1 and B2 (Fig. 2Ai,Bi), as has been described previously (Benjamin et al., 1979; Perry et al., 1998). Isolating and maintaining the BGs in tissue culture for 2 d without axonal injury (DIV2− group) had no apparent effect on the overall morphology of either buccal neuron B1 or B2 (Fig. 2Aii,Bii). A quantitative analysis also revealed no significant change in the total length of neurites in these preparations (B1: DIV0 7511 ± 140 μm, n = 5; DIV2− 7378 ± 1105 μm, n = 4, NK test: p > 0.05; B2: DIV0 1739 ± 125 μm, n = 7; DIV2− 2306 ± 351 μm, n = 7, NK test: p > 0.05).
Figure 2.
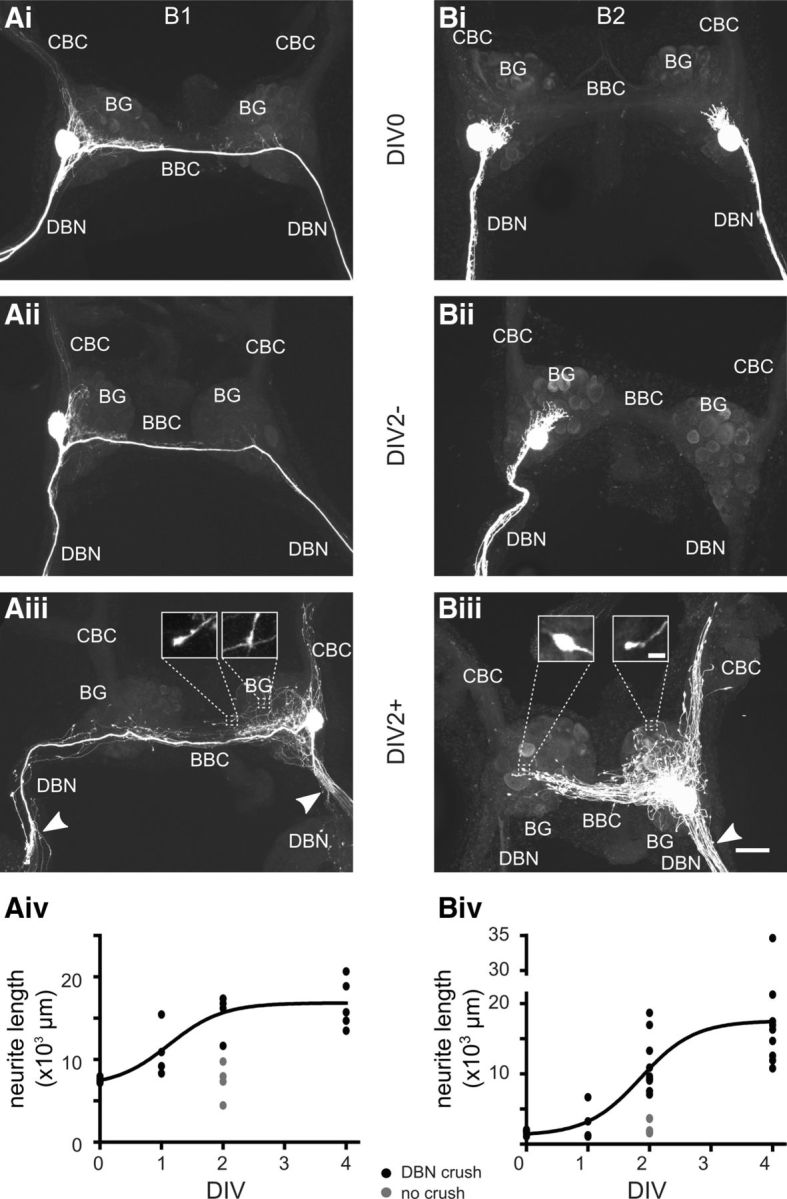
Axonal-injury-induced neurite growth in buccal neurons B1 and B2. Maximum intensity Z projections of confocal images of fluorescently labeled B1 (Ai–Aiii) and B2 neurons (Bi–Biii) are shown. Ai, Bi, B1 and B2 neurons (both the left and right B2 neuron were labeled) in acutely isolated BGs (DIV0). Aii, Bii, Single B1 and B2 neurons in BGs that were maintained in tissue culture for 2 d without crush of the DBNs (DBN; DIV2−). Aiii, Biii, Single B1 and B2 neurons in BGs in which DBNs were crushed shortly after dissection and subsequently maintained in tissue culture for 2 d (DIV2+). The sites of DBN crushes are indicated by white arrowheads. Note the extensive network of central neurites. Inserts: Higher-magnification images of the bulb-like structures at neurite tips. Aiv, Biv, Quantitative analysis of total neurite length for B1 (Aiv) and B2 neurons (Biv) at different time points. The growth data for the DIV0, DIV1+, DIV2+, and DIV4+ groups (black circles) were fitted using a sigmoid function, Y = min + , where T = 1.1 ± 0.3 d (r2 = 0.75) for B1 neurons and T = 1.9 ± 0.2 d (r2 = 0.68) for B2 neurons. Gray circles show measurements for the DIV2− group for comparison. CBC indicates cerebral-buccal connective; BBC, buccal-buccal commissure. Scale bars in Ai–Aiii, Bi–Biii, 100 μm; scale bars for inserts in Aiii, Biii, 10 μm.
However, causing damage to the main axons of the B1 and B2 neurons by crushing the DBN ∼200 μm from its exit point from the BG before culturing induced considerable neurite growth (Fig. 2Aiii, Biii). Both B1 and B2 neurons extended multiple new processes into the ipsilateral DBN toward and across the site of axonal injury that became apparent after 1 d in culture. The sprouting of multiple new processes rather than a single axon is similar to previous reports of neuronal growth after axonal damage in various molluscs and vertebrates (e.g., Wong et al., 1981; Berdan et al., 1993; Chiasson et al., 1994; Navarro et al., 2007). Neuritic growth was not restricted to the DBNs; both types of neurons also extended new neurites in the ipsilateral and contralateral BG, buccal-buccal commissure and cerebral-buccal connective covering areas well beyond the extent of their normal neuritic arborization. Novel neurites frequently terminate in a small bulbous swelling that presumably represents an active growth cone (Fig. 2Aiii, inserts, Biii, inserts). After 2 d in culture (DIV2+), the increase in total neurite length caused by axonal injury became significant and was comparable in B1 and B2 neurons. Overall, the total length of the B1 neurite arbor was significantly increased by ∼8000 μm and the total length of the B2 neurite arbor by ∼9000 μm (B1: DIV2+, 15870 ± 1077 μm, n = 5, NK test: p < 0.01; B2: DIV2+, 10845 ± 1171 μm, n = 11, NK test: p < 0.01; Fig. 2Aiv,Biv). After 2 d, the growth rate of both B1 and B2 neurons appeared to slow. Therefore, extensive central neurite growth within the BG is a specific response to axonal injury in close proximity to the cell soma, rather than just an effect of maintaining the isolated BG in tissue culture.
The extensive central growth Iof novel neurites raises the question of whetherI axonal-injury-induced neurite extensionI alters the synaptic connectivity Iof B1 and B2 neurons. ITherefore, simultaneous intracellular Irecordings from ipsilateral and contralateral Ipairs of B1 and B2 neurons were performed using sharp Imicroelectrodes to assess electrical and chemical coupling. In isolated BG preparations without crushing of the DBNs, the injection of negative current pulses I(−5 nA, 2 s) into either of the two B1 neurons after 2 d in tissue culture (DIV2−) resulted in a corresponding weaker hyperpolarization of the Vm in the contralateral B1 neuron in all cases (Fig. 3Ai,Aiii). The average coupling ratio was 9.2 ± 2.5% (n = 6). In the same preparations, no electrical coupling was found between B1 and B2 neurons and between pairs of B2 neurons (Fig. 3Ai,Aiii). This resembles the situation in acutely isolated preparations (DIV0), which also only showed significant electrical coupling between pairs of B1 neurons (Fig. 3Aiii) with an average coupling ratio of 7.4 ± 0.8% (n = 6; NK test: DIV0 vs DIV2−, p > 0.05).
Figure 3.
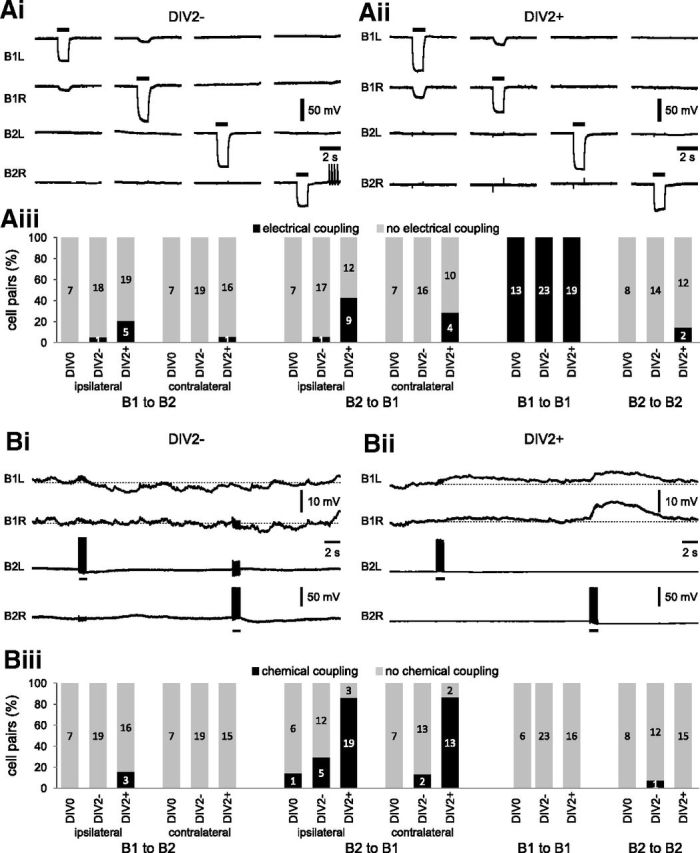
Axonal injury causes changes in synaptic coupling between B1 and B2 neurons. Electrophysiological records from B1 and B2 neurons to test for changes in electrical coupling (Ai,Aii) and chemical coupling (Bi,Bii) caused by axonal injury. Ai, Aii, Simultaneous intracellular records from the left and right B1 and B2 neurons in a preparation that was maintained in tissue culture for 2 d without injury to the DBNs (DIV2−) and in a preparation that was maintained in cell culture for 2 d after crushing the DBNs (DIV2+). The injection of negative current pulses (−5 nA, 1 s) into individual neurons to test for electrical coupling is indicated by the horizontal bars above the records. Aiii, Summary chart showing the percentage of specific cell pairs that showed electrical coupling in three experimental groups: acutely isolated BGs (DIV0), BGs maintained in tissue culture for 2 d without DBN crushing (DIV2−), and BG maintained in tissue culture for 2 d after DBN crushing (DIV2+). The numbers inside the bars indicate the n number for each observation. Bi, Bii, Simultaneous intracellular records from the left and right B1 and B2 neurons in a preparation that was maintained in tissue culture for 2 d without injury to the DBNs (DIV2−) and in a preparation that was maintained in cell culture for 2 d after DBN crushing (DIV2+). The injection of bursts of 10 positive current pulses (10 Hz, 20 nA, 20 ms) into B2 neurons to test for chemical coupling is indicated by the horizontal bars below the records. Biii, Summary chart showing the percentage of specific cell pairs that showed chemical coupling in three experimental groups (DIV0, DIV2−, DIV2+) mentioned above. The numbers inside the bars indicate n for each observation.
In contrast, crushing the DBNs followed by 2 d in tissue culture (DIV2+) resulted in a marked increase in the electrotonic coupling between the B1 neurons, significantly increasing Ithe average coupling ratio to 28.0 ± 2.2% I(n = 9; Fig. 3Aii; ANOVA: p < 0.001; post hoc NK tests: DIV0 vs DIV2+, p < 0.001, DIV2+ vs DIV2−, p < 0.001). This increase in electrical coupling persisted for at least 4 d when the average coupling ratio was 30.2 ± 1.7% (n = 5). Crushing also led to the occurrence of novel electrical connections between B1 and B2 neurons. These novel electrical synapses appeared to show some directional selectivity, with B2 neurons more likely to show coupling to B1 neurons than vice versa (Fig. 3Aiii). The coupling ratios for electrical coupling between B1 and B2 neurons were relatively weak and rarely exceeded 2%. Furthermore, these novel electrical synapses were transient and were observed most frequently 1 d after the initial DBN crush (B2 to ipsilateral B1: 64%, n = 14; B2 to contralateral B1: 64%, n = 11; B1 to ipsilateral B2: 36%, n = 14; B1 to contralateral B2: 17%, n = 12), reduced in frequency 2 d after injury (B2 to ipsilateral B1: 43%, n = 21; B2 to contralateral B1: 29%, n = 14; B1 to ipsilateral B2: 21%, n = 24; B1 to contralateral B2: 6%, n = 17), and could not be detected after 4 d in culture (B2 to ipsilateral B1: n = 14; B2 to contralateral B1: n = 15; B1 to ipsilateral B2: n = 14; B1 to contralateral B2: n = 16).
In addition to changes in electrical coupling, novel chemical synapses were also observed. Most notably, the majority of B1 neurons in the DIV2+ group responded with a slow depolarization to a burst of 10 APs in a B2 neuron (Fig. 3Bii–Biii), which was not usually seen in the DIV0 and DIV2− groups (Fig. 3Bi,Biii). These depolarizations peaked after the end of the B2 stimulation and outlasted the duration of B2 neuron stimulation considerably, indicating that they cannot be attributed to the novel electrical synapses between B1 and B2 neurons described above, but are most likely due to the formation of novel chemical synapses. Unfortunately, we were unable to provide unambiguous morphological evidence for new synaptic structures using immunohistochemical and electron microscopy approaches due to technical difficulties. Nevertheless, the electrophysiological data provide clear functional evidence for a new chemical interaction between B2 and B1 neurons, with the average peak amplitude for the ipsilateral EPSP at 3.5 ± 0.4 mV (n = 19) and 1.6 ± 0.2 mV (n = 13) for the contralateral EPSP. This novel synapse persisted for at least 4 d after injury, but its incidence declined slightly from 86% (n = 22) and 87% (n = 15) at day 2 to 69% (n = 13) and 60% (n = 15) at day 4 for chemical synapses from B2 to ipsilateral B1 and contralateral B1 neurons, respectively. In contrast, evidence for other chemical synapses between B1 and B2 neurons in all three groups (DIV0, DIV2−, DIV2+) were rare (Fig. 3Biii), which is consistent with previous results showing a lack of chemical synapses between B1 and B2 neurons (Benjamin and Rose, 1979). Therefore, we conclude that neurite extension after axonal injury leads to significant synaptic remodeling, including the formation of novel chemical synapses from B2 to B1 neurons, the formation of transient electrical synapses between B1 and B2 neurons, and a significant strengthening of electrical coupling between B1 neurons.
Inhibiting NOS activity has differential effects on neurite growth and synaptic remodeling
Using B1 and B2 neurons as a model, we studied the potential role of NO signaling in neurite extension and synaptic remodeling after axonal injury. In particular, we investigated whether endogenous NO production is important for neuronal growth. For this purpose, the NOS inhibitor 7NI was included in the tissue culture medium to block endogenous NO production during the time of neurite extension. Studies focused on 2 d after the axonal injury, because this is the period of most dynamic growth and synaptic remodeling and any effects of NO on these events should be most obvious at this time point.
Analysis of the morphology of B1 neurons cultured in the presence of 7NI (0.25 mm) for 2 d after axonal injury (DIV2+/7NI group) showed no marked effect on their ability to extend processes. B1 neurons still showed extensive growth of neurites in DBNs and in the ipsilateral and contralateral BG that extended considerably beyond the limits of the normal B1 morphology (Fig. 4Ai). The average length of the total neuritic B1 arbor was 14360 ± 1529 μm (n = 7), which is not significantly different from equivalent B1 neurons in the DIV2+ group (NK test: p > 0.05; Fig. 4Av). In contrast, injury-induced neurite growth in B2 neurons was inhibited markedly by incubation with 7NI. In the presence of 7NI, B2 neurons still displayed some neurite growth and extended neurites in the ipsilateral BG and DBN (Fig. 4Aiii); however, the total length of B2 neurites was reduced significantly, by almost 50% from 10845 ± 1171 μm (n = 11) in the DIV2+ group to 5919 ± 1013 μm (n = 10) in the DIV2+/7NI group (NK test: p < 0.05; Fig. 4Av).
Figure 4.

Endogenous NO synthesis affects neuronal growth and synaptic remodeling. Maximum-intensity Z projections of confocal images of fluorescently labeled B1 and B2 neurons 2 d after axonal injury and treatment with 7NI (Ai, B1, Aiii, B2) or treatment with 7NI + DETA (Aii, B1, Aiv, B2). Av, Average total neurite length of B1 and B2 neurons 2 d after axonal injury under control conditions (DIV2+), in the presence of 7NI (+ 7NI), and in the presence of 7NI plus DETA (+ 7NI + DETA). Individual groups were compared using NK tests. *p < 0.05. Bi, Percentage of specific cell pairs showing electrical coupling 2 d after axonal injury under the three experimental conditions outlined above. n is indicated on the bars. Bii, Average coupling ratio between pairs of B1 neurons under the three experimental conditions (DIV2+, + 7NI, + 7NI + DETA) outlined above. Groups were compared using NK tests (n.s. p > 0.05). Ci, Percentage of specific cell pairs showing excitatory chemical coupling 2 d after axonal injury under the three experimental conditions (DIV2+, + 7NI, + 7NI + DETA) outlined above. n is indicated on the bars. Cii, Average peak amplitude of excitatory chemical synapse from B2 to ipsilateral and contralateral B1 neurons under the three experimental conditions (DIV2+, + 7NI, + 7NI + DETA) outlined above. Groups were compared using NK tests (n.s. p > 0.05). Scale bars in Aii, Aiv, 100 μm.
The observation that blocking endogenous NO production resulted in a reduction in B2 neuronal growth raises the question of whether this also influences injury-induced synaptic remodeling. Simultaneous electrophysiological recordings of B1 and B2 neurons 2 d after injury showed that the incidence of formation of heterologous novel electrical synapses between B1 and B2 neurons was barely affected by incubation in 7NI, and only the incidence of electrical synapses from B2 neurons to contralateral B1 neurons showed a noticeable, but not significant, reduction from 29% (n = 14) in the DIV2+ group to 5% (n = 21) in the DIV2+/7NI group (χ2 test: p > 0.05). Similarly, B1–B1 pairs also retained their electrical coupling (Fig. 4Bi) and the small reduction in the coupling ratio was not significant (coupling ratio: DIV2+/7NI, 23.8 ± 1.2%, n = 7; DIV2+, 28.0 ± 2.2%, n = 9; NK test: p > 0.05; Fig. 4Bii).
Attenuating endogenous NO production also had comparable site-specific effects on the formation of chemical synapses. Although the formation and strength of chemical interactions between ipsilateral B2 and B1 neurons was unaffected (Fig. 4Ci,Cii), the incidence of chemical synapses from B2 to contralateral B1 neurons was reduced significantly from 87% in the DIV2+ group to 23% (n = 22) in the DIV2+/7NI group (χ2 test: p < 0.01; Fig. 4Ci). In addition, the average peak EPSP amplitude in the B1 neurons that responded to contralateral B2 activity was 0.8 ± 0.1 mV (n = 5), 50% lower than the average peak EPSP amplitude in the DIV2+ group (1.6 ± 0.2 mV, n = 13; Fig. 4Cii). The changes in synaptic coupling are unlikely to be indirect consequences of changes in the intrinsic electrophysiological properties, because the Vm, input resistance, AP amplitude, and AP AHP of B1 and B2 neurons were not significantly different in the DIV2+/7NI group compared with the DIV2+ group (Table 1). The only electrophysiological parameter affected significantly by chronic 7NI exposure was the AP width of B1, but not B2, neurons (Table 1).
Table 1.
Electrophysiological properties of B1 and B2 neurons
| Vm (mV) | Input resistance (MΩ) | AP |
|||
|---|---|---|---|---|---|
| Amplitude (mV) | Width (ms) | AHP (mV) | |||
| B1 | |||||
| DIV2+ | −68.7 ± 1.3 (25) | 18.7 ± 2.0 (11) | 75.0 ± 1.5 (24) | 6.1 ± 0.5 (24) | −14.3 ± 0.5 (24) |
| DIV2+/7NI | −67.7 ± 1.2 (26) | 23.2 ± 2.6 (9) | 75.6 ± 1.2 (18) | 7.5 ± 0.5 (18)* | −15.2 ± 0.7 (18) |
| DIV2+/7NI+DETA | −67.6 ± 1.2 (34) | 21.4 ± 2.0 (16) | 72.5 ± 1.1 (22) | 6.5 ± 0.2 (22) | −15.8 ± 0.6 (22) |
| DIV2+/ODQ | −74.8 ± 0.8 (29)* | 25.6 ± 1.4 (12) | 72.0 ± 1.8 (16) | 6.4 ± 0.4 (16) | −15.1 ± 1.4 (16) |
| DIV2+/KT5823 | −64.1 ± 0.9 (30)* | 20.2 ± 1.5 (12) | 71.3 ± 1.2 (26) | 5.2 ± 0.2 (26) | −14.5 ± 0.4 (26) |
| B2 | |||||
| DIV2+ | −61.8 ± 1.1 (24) | 18.7 ± 2.1 (7) | 76.4 ± 1.2 (24) | 9.8 ± 0.7 (24) | −17.0 ± 0.8 (24) |
| DIV2+/7NI | −65.5 ± 1.2 (27) | 22.7 ± 2.4 (12) | 78.0 ± 1.8 (26) | 12.6 ± 1.0 (26) | −16.2 ± 0.7 (26) |
| DIV2+/7NI+DETA | −66.1 ± 1.3 (30) | 24.1 ± 2.0 (15) | 77.7 ± 0.9 (28) | 13.9 ± 0.9 (28)* | −16.3 ± 0.7 (28) |
| DIV2+/ODQ | −72.6 ± 1.4 (24)* | 28.9 ± 1.5 (9)* | 70.1 ± 1.2 (20)* | 9.4 ± 1.0 (20) | −15.6 ± 0.7 (20) |
| DIV2+/KT5823 | −59.8 ± 1.2 (31) | 25.0 ± 2.6 (11) | 73.5 ± 1.0 (29) | 9.5 ± 0.4 (29) | −18.8 ± 0.5 (29) |
Show is the average Vm, input resistance, AP amplitude, AP width, and amplitude of AP AHP of B1 and B2 neurons after DBN crushing and organ culture for 2 d in the different treatment groups. The measurements were conducted in NS. AP width was measured at 50% of the peak amplitude. AHP was measured as the amplitude of the negative peak after the AP relative to the base of the AP. Data are shown as mean ± SEM (n). Statistical analysis was carried out using an ANOVA followed by Dunnett's post hoc test to compare the effects of the individual pharmacological treatments with the DIV2+ group.
*p < 0.05.
The NO donor DETA rescues injury-induced neurite growth, but not synapse remodeling after NOS inhibition
The observation that inhibiting endogenous NOS activity has specific effects on neurite growth and synapse remodeling raised the issue of whether these effects can be reversed by simultaneous application of a NO donor. In the present study, we used the very-slow-releasing NO donor DETA, which has a reported half-life of 57 h at 22–25°C (Keefer et al., 1996). Therefore, addition of DETA (0.5 mm) to the tissue culture medium provides a relatively steady exogenous source of NO. Similar to the DIV2+/7NI group, chronic coapplication of DETA with 7NI had barely any effect on the basic electrophysiological properties of B1 and B2 neurons, only resulting in a significant increase in B2 AP width (Table 1). Furthermore, acute DETA application had no significant effect on the Vm of either B1 or B2 neurons (change in Vm: B1, −1.8 ± 1.3 mV, n = 5; B2, −0.3 ± 0.5 mV, n = 4; 1-sample t test: p > 0.05 for both cases). However, DETA (DIV2+/7NI+DETA group) fully restored the regenerative potential of the B2 neuron and the morphological appearance of these neurons was not noticeable different from B2 neurons in the DIV2+ group (compare Fig. 4Aiv–Av, Fig. 2Biii; total neurite length: DIV2+/7NI+DETA 10491 ± 3098 μm, n = 6; DIV2 + 10845 ± 1171 μm, n = 11; NK test: p > 0.05). Furthermore, total B2 neurite length in the DIV2+/7NI+DETA group was significantly longer than in the DIV2+/7NI group (NK test: p < 0.05). Interestingly, despite the inhibition of endogenous NO synthesis having little effect on neurite growth in B1 neurons, the extent of B1 neurite growth in the DIV2+/7NI+DETA group was also enhanced significantly compared with B1 neurons in the DIV2+/7NI group (20337 ± 1610 μm, n = 5, vs 14360 ± 1529 μm, n = 7; NK test: p < 0.05; Fig. 4Aii,Av). The difference relative to B1 neurons in the DIV2+ group did not reach significant levels (NK test: p > 0.05), but was still very noticeable. This observation suggests that whereas neurite growth in B1 neurons appears to be less dependent on endogenous NO synthesis, NO is still able to modulate B1 neurite growth.
In contrast to the effects on neurite growth of B1 and B2 neurons, DETA application could not fully rescue the effects of inhibiting endogenous NO synthesis on synapse remodeling. Culturing BG in the presence of DETA did not reverse the reduction in the proportion of chemical interactions between B2 neurons and contralateral B1 neurons (Fig. 4Ci; χ2 test: p > 0.05). However, the peak amplitude of the B1 EPSP caused by B2 activity in the DIV2+/7NI+DETA group had an amplitude of 1.5 ± 0.2 mV (n = 8), nearly twice of the amplitude in the DIV2+/7NI group and close to the equivalent value of 1.6 ± 0.2 mV (n = 13) in the DIV2+ group (Fig. 4Cii).
Neurite extension and the formation of novel synapses depends on sGC activity
The canonical NO signaling pathway involves the activation of sGC by NO that leads to the production of cGMP. To assess whether this signaling pathway is responsible for the effects of endogenous NO synthesis on neurite growth and synapse remodeling, we included the sGC inhibitor ODQ (0.4 mm) in the tissue culture medium. Intracellular recordings from B1 and B2 neurons in these preparations after 2 d in tissue culture (DIV2+/ODQ group) showed that the neurons were healthy, with a stable Vm and the ability to generate APs both in response to injected currents and spontaneous synaptic inputs. Interestingly, in both B1 and B2 neurons, ODQ treatment caused a significant hyperpolarization of the Vm, an increase in B2 input resistance, and a decrease in B2 AP amplitude (Table 1). Intracellular labeling revealed that ODQ significantly suppressed the induction of neuronal growth and neurite extension by axonal injury compared with the DIV2+ group of both B1 and B2 neurons (compare Fig. 5Ai–Aiii, Fig. 1Aiii,Biii; NK tests: B1: p < 0.001; B2: p < 0.01). Furthermore, total neurite length in the DIV2+/ODQ group for both B1 and B2 neurons was not significantly different from the DIV0 and DIV2− groups (B1: DIV2+/ODQ vs DIV0, DIV2+/ODQ vs DIV2− and B2: DIV2+/ODQ vs DIV0, DIV2+/ODQ vs DIV2−, NK tests: p > 0.05 for all cases).
Figure 5.
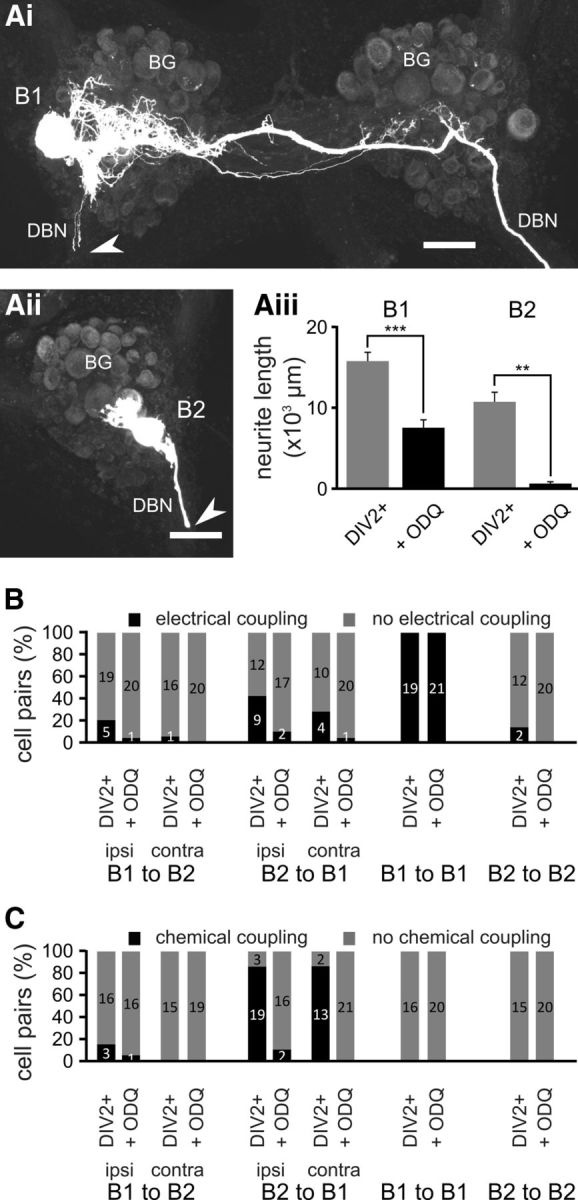
Neuronal growth and synaptic remodeling is dependent on sGC activity. Maximum-intensity Z projections of confocal images of fluorescently labeled B1 and B2 neurons 2 d after axonal injury and treatment with ODQ (Ai, B1, Aii, B2). Aiii, Average total neurite length of B1 and B2 neurons 2 d after axonal injury under control conditions (DIV2+) and in the presence of ODQ (+ ODQ). Groups were compared using NK tests: **p < 0.01; ***p < 0.001. B, Percentage of specific cell pairs showing electrical coupling 2 d after axonal injury under the two experimental conditions (DIV2+, + ODQ) outlined above. n is indicated on the bars. C, Percentage of specific cell pairs showing excitatory chemical coupling 2 d after axonal injury under the two experimental conditions (DIV2+, + ODQ) outlined above. n is indicated on the bars. Scale bars in Ai, Aii, 100 μm.
The lack of significant neurite growth in the DIV2+/ODQ was accompanied by a dramatic reduction in the cell pairs that showed the formation of novel electrical connections (Fig. 5B) and the almost complete failure to form novel chemical connections (Fig. 5C). ODQ also prevented the increase in electrical coupling between B1 neurons observed in the DIV2+ group (B1-B1 coupling ratio: DIV2+/ODQ group 10.2 ± 1.6%, n = 10, DIV2+ group 28.0 ± 2.2%, n = 9, NK test: p < 0.001). These results clearly demonstrate the importance of sGC activity for both axonal-injury-induced neurite growth and synaptic remodeling.
To understand why the effects of inhibiting sGC activity with ODQ on neurite growth and synapse formation were so much more pronounced than blocking NOS activity using 7NI, we measured directly the effects of the different treatments on cGMP concentrations in the whole CNS. The average cGMP concentration in whole CNS preparations maintained in cell culture for 1 d was 15.5 ± 1.8 pmol/mg protein (n = 6; Fig. 6A). Maintaining the isolated CNS for 1 d in the presence of 7NI (0.25 mm) reduced the average cGMP concentration to 12.4 ± 0.8 pmol/mg protein (n = 6), whereas ODQ (0.4 mm) reduced the average cGMP concentration by a greater extent, to 9.1 ± 0.7 pmol/mg protein (n = 6; Fig. 6A). The differences in cGMP concentrations between the control and the ODQ group, but not the 7NI and control group, were significant (ANOVA: p < 0.01; NK tests: control vs ODQ, p < 0.01; control vs 7NI, p > 0.05). An additional experiment using a twofold higher 7NI concentration (0.5 mm) reduced the cGMP concentration to 9.8 ± 0.8 pmol/mg protein (n = 6), which is significantly different from the control group (p < 0.01) and similar to the effect seen in the presence of ODQ (p > 0.05). Therefore, 7NI (0.25 mm) appears to produce only a partial block of NO-dependent cGMP production, which can explain why this concentration of 7NI had more subtle effects on neurite growth and synaptic remodeling compared with the treatment with ODQ.
Figure 6.

cGMP concentration in the isolated CNS. A, Average cGMP concentration measured in isolated whole CNS preparations maintained for 1 d in tissue culture under control conditions (control) or in the presence of either 7NI (0.25 mm) or ODQ (0.4 mm). Groups were compared using an ANOVA followed by NK post hoc tests. *p < 0.05. B, Plot of the average total neurite length of B1 and B2 neurons under the three different conditions (DIV2+, DIV2+/7NI, DIV2+/ODQ) against the average cGMP concentration under equivalent conditions (control, + 7NI, + ODQ).
Plotting the average cGMP concentration per CNS under the three conditions (control, 0.25 mm 7NI, 0.4 mm ODQ) against the average neurite length of B1 and B2 neurons in the DIV2+, DIV2+/7NI, and DIV2+/ODQ groups showed a good linear relationship, with regression coefficients (r2) of 0.82 for B1 neurons and 0.99 for B2 neurons (Fig. 6B), further supporting the concept that NO-dependent cGMP production is an important factor in controlling neurite growth in B1 and B2 neurons.
Novel chemical synapse formation requires PKG activity
One possible signaling pathway downstream of a NO-dependent increase in cGMP concentration is the activation of PKG. We tested the potential involvement of PKG in neurite extension and synaptic remodeling using the selective PKG antagonist KT5823. Including KT5823 (10 μm) in the culture medium for 2 d had no significant effect on B1 and B2 neurite extension after axonal crushing (Fig. 7Ai–Aiii). Neurons showed extensive growth and the total neurite length was not significantly different from the DIV2+ group (B1: 19288 ± 1993 μm, n = 6, B2: 15201 ± 956 μm, n = 5; NK tests for DIV2+ vs DIV2+/KT5823: B1, p > 0.05; B2, p > 0.5). Chronic treatment with KT5823 also had little effect on the electrophysiological properties of the neurons, causing a small but significant depolarization of the B1 Vm without altering any of the other properties of B1 and B2 neurons (Table 1). Blocking PKG activity also had only minor effects on electrical synapses between B1 and B2 neurons, with a small, nonsignificant reduction in the percentage of B2 neurons that were electrically coupled to ipsilateral and contralateral B1 neurons (Fig. 7B; χ2 tests: p > 0.05 for all cases). Similarly, the average coupling ratio between B1 neurons in the DIV2+/KT5823 group was 29.8 ± 2.1% (n = 12), very similar to the DIV2+ group (28.0 ± 2.1%, n = 9; NK test: p > 0.05).
Figure 7.
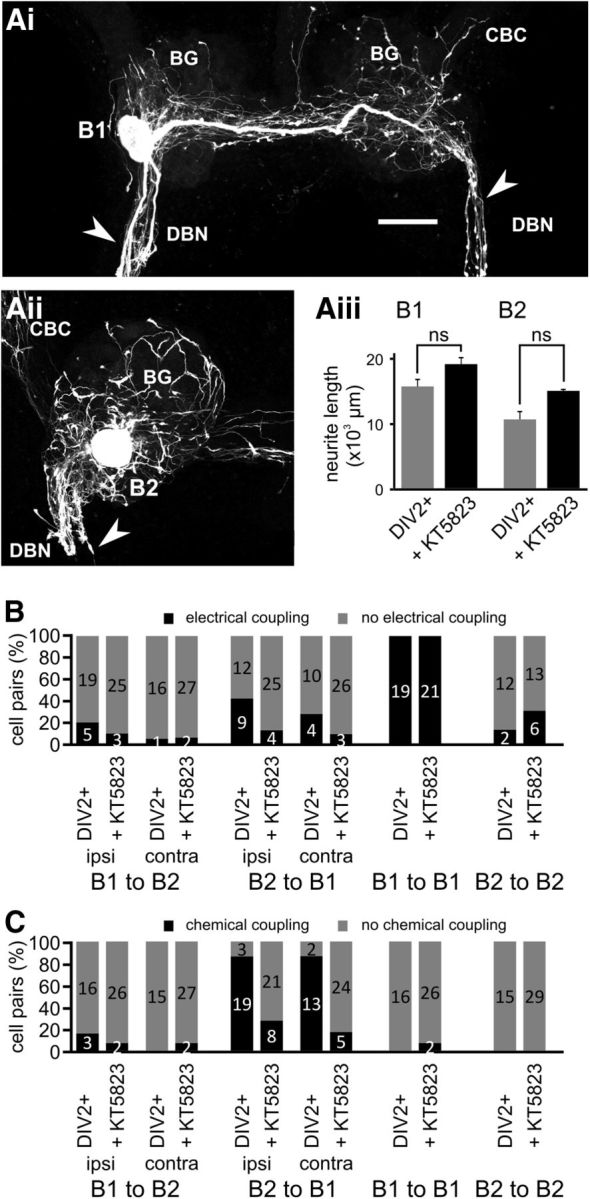
KT5823 inhibits novel chemical synapse formation without affecting neurite extension. Maximum-intensity Z projections of confocal images of fluorescently labeled B1 and B2 neurons 2 d after axonal injury and treatment with KT5823 (Ai, B1, Aii, B2). Aiii, Average total neurite length of B1 and B2 neurons 2 d after axonal injury under control conditions (DIV2+) and in the presence of KT5823 (+ KT5823). Groups were compared using NK tests (n.s. p > 0.05). B, Percentage of specific cell pairs showing electrical coupling 2 d after axonal injury under the two experimental conditions (DIV2+, + KT5823) outlined above. n is indicated on the bars. C, Percentage of specific cell pairs showing excitatory chemical coupling 2 d after axonal injury under the two experimental conditions (DIV2+, + KT5823) outlined above. n is indicated on the bars. Scale bar in Ai, 100 μm.
However, KT5823 treatment selectively reduced the occurrence of novel B2 to B1 chemical synapses, reducing it from 86% (n = 22) to 28% (n = 29) of ipsilateral and from 87% (n = 15) to 17% (n = 29) of contralateral B2-B1 pairs (χ2 test: p < 0.05 in both cases; Fig. 7C). The average amplitude of the B2 to B1 synapses (B2 to ipsilateral B1: 2.8 ± 0.4 mV, n = 8; B2 to contralateral B1: 1.4 ± 0.1 mV, n = 5) that were observed after chronic KT5823 treatment was comparable to the DIV2+ group (p > 0.05). Therefore, inhibiting KT5823 activity affects the formation of novel chemical synapses selectively without effects on neurite growth or remodeling of electrical synapses.
NO does not affect growth properties of isolated neurons in cell culture
To investigate whether NO acts directly on neuronal growth or if the observed effects in tissue culture are the indirect result of NO modulating other signaling pathways that affect neuronal growth, we performed experiments on completely isolated B1 and B2 neurons in cell culture (Fig. 8A,B). Interestingly, culturing B1 and B2 neurons in the presence of DETA (0.5 mm) had no significant effect on the total amount of growth in cell culture >2 d for individual B1 and B2 neurons. Measuring total neurite length after 1 and 2 d in cell culture showed that both B1 and B2 neurons exhibited comparable growth in the absence and presence of the NO donor DETA (Fig. 8Ci,Cii). Statistical analysis confirmed a significant increase in neurite length between days 1 and 2, but no significant effect of treatment with DETA (2-way repeated-measures ANOVA, time effect: B1, p < 0.01; B2, p < 0.01; DETA effect: B1, p > 0.05; B2, p > 0.05).
Figure 8.
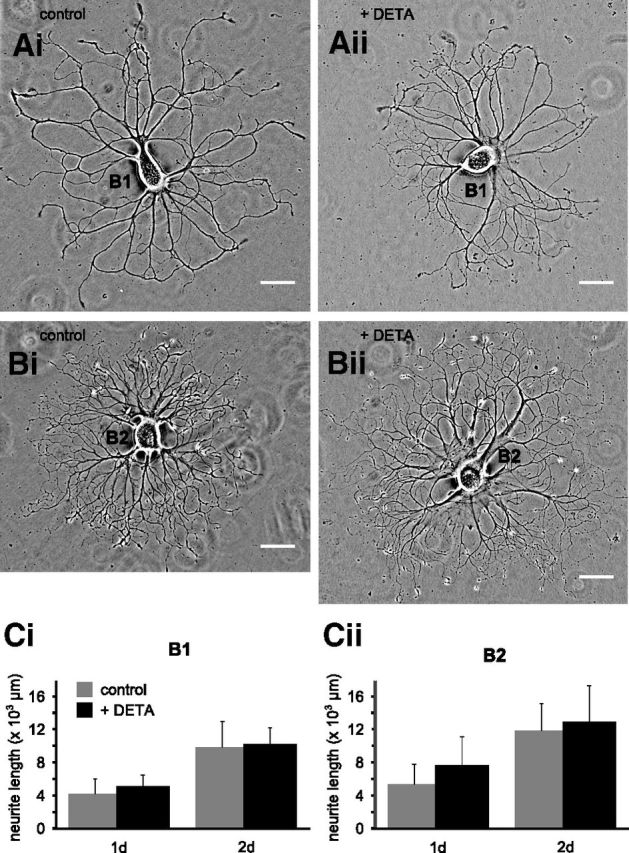
NO does not affect neuronal growth of isolated B1 and B2 neurons in cell culture. Phase-contrast images of individual isolated B1 and B2 neurons after 2 d in cell culture under control conditions (Ai, B1, Bi, B2) and in the presence of DETA (Aii, B1, Bii, B2). Ci, Cii, Average total neurite length of isolated B1 and B2 neurons after 1 and 2 d in cell culture in the absence and presence of DETA. Scale bars in Ai–Bii, 100 μm.
Discussion
The results presented here demonstrate that endogenous NO synthesis plays an important role in both axonal-injury-induced neurite extension and synapse formation/remodeling. Therefore, we have added to the growing number of studies implicating NO signaling as a modulator of neuronal growth (e.g., Seidel and Bicker, 2000; Gibbs et al., 2001; Benton et al., 2007; Bradley et al., 2010). Using easily identifiable neurons in the CNS of L. stagnalis as a model system, we were able to study not only the effects of NO signaling on the neuronal growth of individual neurons, but also the functional remodeling of synaptic connections between these neurons. This enabled us to show the apparent correlation between the effects of blocking specific aspects of the NO signaling pathway on neuronal growth and synaptic remodeling. For example, the axonal-injury-induced strengthening of electrotonic coupling between B1 neurons was only affected by inhibiting sGC, which also affected the growth of B1 neurons. This suggests that the increase in electrotonic coupling is the result of the sprouting of new collaterals and an increase in contacts between B1 neurons, similar to previous results for homologous B4 neurons in Helisoma (Murphy et al., 1983). Similarly, the reduction in the incidence of novel chemical synapses from B2 to contralateral B1 neurons, but not ipsilateral B1 neurons, as a result of partial block of endogenous NOS activity by 7NI could be considered to be the consequence of the reduced growth of B2 neurons in the presence of 7NI. However, although neurite extension of B2 neurons could be rescued by the addition of the NO donor DETA, chemical synapse formation between B2 and contralateral B1 neurons was not rescued. This suggests that neurite growth and chemical synapse formation are independently modulated by NO and have different temporal and spatial requirements for NO. The hypothesis that NO affects neurite growth and synapse formation by independent signaling pathways is further supported by our observation that the PKG inhibitor KT5823 reduced the incidence of novel chemical synapse formation between B2 and B1 neurons without any effects on axonal-injury-induced neuronal growth in either B1 or B2 neurons. Finally, the lack of effect of NO on neuronal growth of isolated B1 and B2 neurons in cell culture suggests that NO signaling modulates neuronal growth by modulating the effects of other signaling pathways that are not active in the cell culture system.
Endogenous NO synthesis, cGMP, and neurite extension
Isolated Helisoma B5 neurons, which are homologous to L. stagnalis B2 neurons, have been used previously to show that NO affects growth cone morphology and migration in cell culture (Van Wagenen and Rehder, 2001; Trimm and Rehder, 2004; Welshhans and Rehder, 2005). Our results presented here support the hypothesis that endogenous NO production is important in controlling the growth rate of extending neurites and demonstrate that NO signaling not only affects growth cone behavior transiently at a local level, but that these effects are translated into changes in global neuron morphology.
Furthermore, our results show that the growth of both NOS-positive neurons (B2) and NOS-negative neurons (B1) is positively affected by raising NO concentrations using a NO donor. This suggests that both B1 and B2 neurons possess the necessary signaling machinery to respond to NO, which is consistent with the observation that sGC expression in L. stagnalis is widespread (Ribeiro et al., 2008, 2010). However, it is noteworthy that neither B1 nor B2 neurons responded with a change in Vm to the application of the NO donor. Therefore, the effects of NO on neuronal growth in these neurons do not appear to be the consequence of changes in the activity levels of B1 and B2 neurons.
It is interesting that inhibiting endogenous NO production with 0.25 mm 7NI significantly affected neurite extension in B2 neurons, but had only very minor effects on B1 neurons. However, it is not possible to conclude on the basis of this observation that neuronal growth in B2 neurons is more dependent on endogenous NO production than in B1 neurons. Although our measurements of cGMP concentration in the whole CNS suggest that 7NI at this concentration produces an incomplete block of NO-dependent cGMP production, we have also shown previously that 0.25 mm 7NI is sufficient to produce a maximum blocking effect on the modulatory effect of endogenous NO production at an identified synapse (Straub et al., 2007). Therefore, it is possible that 7NI at this concentration may differentially affect individual NO-dependent physiological processes in different cell types. The hypothesis that endogenous NO can affect neurite growth in both B1 and B2 neurons is certainly supported by the observation that increasing NO concentrations while applying 7NI increased neurite growth in both neuron types. Furthermore, that the effects of NO on neurite growth are mediated by intracellular cGMP is strongly supported by the observation that ODQ suppressed neuronal growth in both B1 and B2 neurons. The suggestion that NO acts via the activation of sGC and cGMP production is consistent with many other studies demonstrating the effects of cGMP on neuronal growth (Hindley et al., 1997; Seidel and Bicker, 2000; Xiong et al., 2007; Murray et al., 2009; Tojima et al., 2009; Bradley et al., 2010; Shelly et al., 2010). Several of these studies on mammalian systems have also shown that the effects of cGMP on neurite extension appear to be dependent on the activation of PKG (Yamazaki et al., 2004; Xiong et al., 2007). PKG activity has also been implicated in modulating growth-cone-turning responses (Nishiyama et al., 2003) and in the reciprocal actions of cAMP and cGMP in controlling axon/dendrite formation (Shelly et al., 2010). In contrast, our data demonstrate that NO-cGMP signaling can also affect neurite growth independently of PKG activation. Therefore, our results suggest a novel cGMP-dependent, but PKG-independent, mechanism for the effect of NO on neurite growth. This could be via cGMP modulation of phosphodiesterase activity or cGMP-regulated transcription factors (Contestabile, 2008; Francis et al., 2010). However, we cannot completely rule out the possibility that ODQ's effects on neurite growth are at least in part independent of actions on GC and cGMP, because it has been reported that ODQ can affect neurite growth in an sGC-independent manner, which may result from direct interactions with unidentified redox-sensitive proteins (Lee et al., 2009).
cGMP-PKG signaling affects novel chemical synapse formation, but not neurite growth and remodeling of electrical synapses
One important finding of the present study is that blocking PKG activity selectively reduced the formation of novel chemical synapses without affecting the axonal-injury-induced neurite growth and concurrent increase in the electrical coupling between B1 neurons. This is in apparent contrast to previous studies showing that NO-cGMP-PKG signaling is necessary and sufficient for the removal of synapses after axonal injury (Sunico et al., 2005; Moreno-López et al., 2011). However, it is possible that there are differences in the response of existing and novel synapses to PKG signaling. After axonal injury, PKG signaling might lead to the removal of existing synapses while simultaneously promoting the formation of novel chemical synapses on new target sites. Our observation of an apparent role of PKG activity in the formation of novel chemical synapses is certainly consistent with previous reports on the role of NO/sGC/cGMP signaling in the development of the vertebrate neuromuscular junction, where it has been shown that this signaling pathway is required among other aspects for the aggregation of postsynaptic acetylcholine receptors (Godfrey and Schwarte, 2003, 2010; Jones and Werle, 2004). Therefore, the role of NO in the formation of neuron-neuron synapses and neuromuscular junctions is highly conserved. Furthermore, PKG has also been implicated in various forms of long-term synaptic plasticity such as LTP (Ota et al., 2010; Ping and Schafe, 2010; Luo et al., 2012), as well as learning and memory formation in both vertebrates (Izquierdo et al., 2000; Edwards et al., 2002; Chien et al., 2005; Zinn et al., 2009) and invertebrates (Kaun et al., 2007; Michel et al., 2011), processes that are associated with the formation of new synaptic sites. Because we show here that PKG inhibition reduces the formation of new chemical synapses, this provides a potential mechanism that can account for the previously reported observation that associative conditioning in L. stagnalis (Kemenes et al., 2002) and various forms of learning and memory formation in other invertebrate and vertebrate species are critically dependent on NO signaling (e.g., Holscher et al., 1996; Katzoff et al., 2002; Susswein et al., 2004; Zinn et al., 2009). The wider significance of our findings is further indicated by the fact that synaptic reorganization after injury does not only occur in invertebrates, but also in mammals (Wittenberg, 2010; e.g., Staudt, 2010; Nudo, 2011). In general, any form of mammalian CNS injury, including stroke and trauma, has been shown to result in substantial reorganization of neuronal circuits (Navarro et al., 2007; Benowitz and Carmichael, 2010). However, although axonal injury of mammalian motoneurons results in the sprouting of multiple new processes (Navarro et al., 2007) and, at least in certain cases, the expansion of the dendritic tree (Rose and Odlozinski, 1998), most of the central reorganization in the mammalian CNS is due to growth of neurons that were not injured directly, but take over some of the areas that previously mapped to the injured neurons. This can be due either to the expansion of neighboring uninjured neurons or to the growth of new contralateral projections that will occupy the space vacated by the retraction of dendrites from injured/axotomized neurons. Therefore, a better understanding of the role of NO in the processes of neurite growth and synaptic reorganization might open new treatment avenues to promote functional recovery.
Footnotes
This work was supported by the Biotechnology and Biological Sciences Research Council (Grant BB/E009972/1 to V.A.S.). We thank Dr. Jonathan McDearmid for useful comments on an early draft of the manuscript.
References
- Benjamin PR, Rose RM. Central generation of bursting in the feeding system of the snail, Lymnaea stagnalis. J Exp Biol. 1979;80:93–118. doi: 10.1242/jeb.80.1.93. [DOI] [PubMed] [Google Scholar]
- Benjamin PR, Winlow W. The distribution of 3 wide-acting synaptic inputs to identified neurons in the isolated brain of Lymnaea stagnalis (L) Comp Biochem Phys A. 1981;70:293–307. [Google Scholar]
- Benjamin PR, Rose RM, Slade CT, Lacy MG. Morphology of identified neurones in the buccal ganglia of Lymnaea stagnalis. J Exp Biol. 1979;80:119–135. [Google Scholar]
- Benowitz LI, Carmichael ST. Promoting axonal rewiring to improve outcome after stroke. Neurobiology of Disease. 2010;37:259–266. doi: 10.1016/j.nbd.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton JL, Sandeman DC, Beltz BS. Nitric oxide in the crustacean brain: regulation of neurogenesis and morphogenesis in the developing olfactory pathway. Dev Dyn. 2007;236:3047–3060. doi: 10.1002/dvdy.21340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdan RC, Easaw JC, Wang R. Alterations in membrane potential after axotomy at different distances from the soma of an identified neuron and the effect of depolarization on neurite outgrowth and calcium channel expression. J Neurophysiol. 1993;69:151–164. doi: 10.1152/jn.1993.69.1.151. [DOI] [PubMed] [Google Scholar]
- Bradley S, Tossell K, Lockley R, McDearmid JR. Nitric oxide synthase regulates morphogenesis of zebrafish spinal cord motoneurons. J Neurosci. 2010;30:16818–16831. doi: 10.1523/JNEUROSCI.4456-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooker G, Harper JF, Terasaki WL, Moylan RD. Radioimmunoassay of cyclic AMP and cyclic GMP. Adv Cyclic Nucleotide Res. 1979;10:1–33. [PubMed] [Google Scholar]
- Cary SP, Winger JA, Derbyshire ER, Marletta MA. Nitric oxide signaling: no longer simply on or off. Trends Biochem Sci. 2006;31:231–239. doi: 10.1016/j.tibs.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Chiasson BJ, Baker MW, Croll RP. Morphological changes and functional recovery following axotomy of a serotonergic cerebrobuccal neurone in the land snail Achatina fulica. J Exp Biol. 1994;192:147–167. doi: 10.1242/jeb.192.1.147. [DOI] [PubMed] [Google Scholar]
- Chien WL, Liang KC, Teng CM, Kuo SC, Lee FY, Fu WM. Enhancement of learning behaviour by a potent nitric oxide-guanylate cyclase activator YC-1. Eur J Neurosci. 2005;21:1679–1688. doi: 10.1111/j.1460-9568.2005.03993.x. [DOI] [PubMed] [Google Scholar]
- Cogen J, Cohen-Cory S. Nitric oxide modulates retinal ganglion cell axon arbor remodeling in vivo. J Neurobiol. 2000;45:120–133. doi: 10.1002/1097-4695(20001105)45:2<120::aid-neu6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Contestabile A. Regulation of transcription factors by nitric oxide in neurons and in neural-derived tumor cells. Prog Neurobiol. 2008;84:317–328. doi: 10.1016/j.pneurobio.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Downen M, Zhao ML, Lee P, Weidenheim KM, Dickson DW, Lee SC. Neuronal nitric oxide synthase expression in developing and adult human CNS. J Neuropathol Exp Neurol. 1999;58:12–21. doi: 10.1097/00005072-199901000-00002. [DOI] [PubMed] [Google Scholar]
- Edwards TM, Rickard NS, Ng KT. Inhibition of guanylate cyclase and protein kinase G impairs retention for the passive avoidance task in the day-old chick. Neurobiol Learn Mem. 2002;77:313–326. doi: 10.1006/nlme.2001.4021. [DOI] [PubMed] [Google Scholar]
- Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62:525–563. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Cheng C, Zhao J, Chen M, Li X, Shi S, Niu S, Qin J, Lu M, Shen A. Developmental regulation of PSD-95 and nNOS expression in lumbar spinal cord of rats. Neurochem Int. 2008;52:495–501. doi: 10.1016/j.neuint.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J. New insight into the functioning of nitric oxide-receptive guanylyl cyclase: physiological and pharmacological implications. Mol Cell Biochem. 2010;334:221–232. doi: 10.1007/s11010-009-0318-8. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Charles SL, Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature. 1988;336:385–388. doi: 10.1038/336385a0. [DOI] [PubMed] [Google Scholar]
- Gibbs SM, Becker A, Hardy RW, Truman JW. Soluble guanylate cyclase is required during development for visual system function in Drosophila. J Neurosci. 2001;21:7705–7714. doi: 10.1523/JNEUROSCI.21-19-07705.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson NJ, Rössler W, Nighorn AJ, Oland LA, Hildebrand JG, Tolbert LP. Neuron-glia communication via nitric oxide is essential in establishing antennal-lobe structure in Manduca sexta. Dev Biol. 2001;240:326–339. doi: 10.1006/dbio.2001.0463. [DOI] [PubMed] [Google Scholar]
- Godfrey EW, Schwarte RC. The role of nitric oxide signaling in the formation of the neuromuscular junction. J Neurocytol. 2003;32:591–602. doi: 10.1023/B:NEUR.0000020612.87729.98. [DOI] [PubMed] [Google Scholar]
- Godfrey EW, Schwarte RC. Nitric oxide and cyclic GMP regulate early events in agrin signaling in skeletal muscle cells. Exp Cell Res. 2010;316:1935–1945. doi: 10.1016/j.yexcr.2010.03.016. [DOI] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- Hindley S, Juurlink BH, Gysbers JW, Middlemiss PJ, Herman MA, Rathbone MP. Nitric oxide donors enhance neurotrophin-induced neurite outgrowth through a cGMP-dependent mechanism. J Neurosci Res. 1997;47:427–439. [PubMed] [Google Scholar]
- Hölscher C, McGlinchey L, Anwyl R, Rowan MJ. 7-Nitro indazole, a selective neuronal nitric oxide synthase inhibitor in vivo, impairs spatial learning in the rat. Learn Mem. 1996;2:267–278. doi: 10.1101/lm.2.6.267. [DOI] [PubMed] [Google Scholar]
- Izquierdo LA, Vianna M, Barros DM, Mello e Souza T, Ardenghi P, Sant'Anna MK, Rodrigues C, Medinam JH, Izquierdo I. Short- and long-term memory are differentially affected by metabolic inhibitors given into hippocampus and entorhinal cortex. Neurobiol Learn Mem. 2000;73:141–149. doi: 10.1006/nlme.1999.3925. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Jones MA, Werle MJ. Agrin-induced AChR aggregate formation requires cGMP and aggregate maturation requires activation of cGMP-dependent protein kinase. Mol Cell Neurosci. 2004;25:195–204. doi: 10.1016/j.mcn.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Katzoff A, Ben-Gedalya T, Susswein AJ. Nitric oxide is necessary for multiple memory processes after learning that a food is inedible in aplysia. J Neurosci. 2002;22:9581–9594. doi: 10.1523/JNEUROSCI.22-21-09581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaun KR, Hendel T, Gerber B, Sokolowski MB. Natural variation in Drosophila larval reward learning and memory due to a cGMP-dependent protein kinase. Learn Mem. 2007;14:342–349. doi: 10.1101/lm.505807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefer LK, Nims RW, Davies KM, Wink DA. “NONOates” (1-substituted diazen-1-ium-1,2-diolates) as nitric oxide donors: convenient nitric oxide dosage forms. Methods Enzymol. 1996;268:281–293. doi: 10.1016/s0076-6879(96)68030-6. [DOI] [PubMed] [Google Scholar]
- Keilhoff G, Fansa H, Wolf G. Differences in peripheral nerve degeneration/regeneration between wild-type and neuronal nitric oxide synthase knockout mice. J Neurosci Res. 2002;68:432–441. doi: 10.1002/jnr.10229. [DOI] [PubMed] [Google Scholar]
- Kemenes I, Kemenes G, Andrew RJ, Benjamin PR, O'Shea M. Critical time-window for NO-cGMP-dependent long-term memory formation after one-trial appetitive conditioning. J Neurosci. 2002;22:1414–1425. doi: 10.1523/JNEUROSCI.22-04-01414.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koriyama Y, Yasuda R, Homma K, Mawatari K, Nagashima M, Sugitani K, Matsukawa T, Kato S. Nitric oxide-cGMP signaling regulates axonal elongation during optic nerve regeneration in the goldfish in vitro and in vivo. J Neurochem. 2009;110:890–901. doi: 10.1111/j.1471-4159.2009.06182.x. [DOI] [PubMed] [Google Scholar]
- Leamey CA, Ho-Pao CL, Sur M. Disruption of retinogeniculate pattern formation by inhibition of soluble guanylyl cyclase. J Neurosci. 2001;21:3871–3880. doi: 10.1523/JNEUROSCI.21-11-03871.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EJ, Kim KY, Gu TH, Moon JI, Kim IB, Lee MY, Oh SJ, Chun MH. Neuronal nitric oxide synthase is expressed in the axotomized ganglion cells of the rat retina. Brain Res. 2003;986:174–180. doi: 10.1016/s0006-8993(03)03250-5. [DOI] [PubMed] [Google Scholar]
- Lee HG, Kim SY, Kim du S, Seo SR, Lee SI, Shin DM, De Smet P, Seo JT. 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one inhibits neurite outgrowth and causes neurite retraction in PC12 cells independently of soluble guanylyl cyclase. J Neurosci Res. 2009;87:269–277. doi: 10.1002/jnr.21838. [DOI] [PubMed] [Google Scholar]
- Luo C, Gangadharan V, Bali KK, Xie RG, Agarwal N, Kurejova M, Tappe-Theodor A, Tegeder I, Feil S, Lewin G, Polgar E, Todd AJ, Schlossmann J, Hofmann F, Liu DL, Hu SJ, Feil R, Kuner T, Kuner R. Presynaptically localized cyclic GMP-dependent protein kinase 1 is a key determinant of spinal synaptic potentiation and pain hypersensitivity. PLoS Biol. 2012;10:e1001283. doi: 10.1371/journal.pbio.1001283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsala J, Orendácová J, Lukácová N, Vanický I. Traumatic injury of the spinal cord and nitric oxide. Prog Brain Res. 2007;161:171–183. doi: 10.1016/S0079-6123(06)61011-X. [DOI] [PubMed] [Google Scholar]
- Michel M, Green CL, Eskin A, Lyons LC. PKG-mediated MAPK signaling is necessary for long-term operant memory in Aplysia. Learn Mem. 2011;18:108–117. doi: 10.1101/lm.2063611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno N, López JM, Sánchez-Camacho C, Crespo M, Muñoz M, González A. Early development of NADPH diaphorase-expressing neurons in the brain of the urodele amphibian Pleurodeles waltl. Brain Res Bull. 2002;57:409–412. doi: 10.1016/s0361-9230(01)00708-0. [DOI] [PubMed] [Google Scholar]
- Moreno-López B, Sunico CR, González-Forero D. NO orchestrates the loss of synaptic boutons from adult “sick” motoneurons: modeling a molecular mechanism. Mol Neurobiol. 2011;43:41–66. doi: 10.1007/s12035-010-8159-8. [DOI] [PubMed] [Google Scholar]
- Moroz LL, Winlow W, Turner RW, Bulloch AG, Lukowiak K, Syed NI. Nitric oxide synthase-immunoreactive cells in the CNS and periphery of Lymnaea. Neuroreport. 1994;5:1277–1280. doi: 10.1097/00001756-199406020-00031. [DOI] [PubMed] [Google Scholar]
- Murphy AD, Hadley RD, Kater SB. Axotomy-induced parallel increases in electrical and dye coupling between identified neurons of Helisoma. J Neurosci. 1983;3:1422–1429. doi: 10.1523/JNEUROSCI.03-07-01422.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ, Peace AG, Shewan DA. cGMP promotes neurite outgrowth and growth cone turning and improves axon regeneration on spinal cord tissue in combination with cAMP. Brain Res. 2009;1294:12–21. doi: 10.1016/j.brainres.2009.07.071. [DOI] [PubMed] [Google Scholar]
- Navarro X, Vivó M, Valero-Cabré A. Neural plasticity after peripheral nerve injury and regeneration. Prog Neurobiol. 2007;82:163–201. doi: 10.1016/j.pneurobio.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Hoshino A, Tsai L, Henley JR, Goshima Y, Tessier-Lavigne M, Poo MM, Hong K. Cyclic AMP/GMP-dependent modulation of Ca2+ channels sets the polarity of nerve growth-cone turning. Nature. 2003;423:990–995. doi: 10.1038/nature01751. [DOI] [PubMed] [Google Scholar]
- Nudo RJ. Neural bases of recovery after brain injury. J Commun Disord. 2011;44:515–520. doi: 10.1016/j.jcomdis.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota KT, Monsey MS, Wu MS, Schafe GE. Synaptic plasticity and NO-cGMP-PKG signaling regulate pre- and postsynaptic alterations at rat lateral amygdala synapses following fear conditioning. PLoS ONE. 2010;5:e11236. doi: 10.1371/journal.pone.0011236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Straub VA, O'Shea M. Anterograde signaling by nitric oxide: characterization and in vitro reconstitution of an identified nitrergic synapse. J Neurosci. 1998;18:5463–5476. doi: 10.1523/JNEUROSCI.18-14-05463.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry SJ, Straub VA, Kemenes G, Santama N, Worster BM, Burke JF, Benjamin PR. Neural modulation of gut motility by myomodulin peptides and acetylcholine in the snail Lymnaea. J Neurophysiol. 1998;79:2460–2474. doi: 10.1152/jn.1998.79.5.2460. [DOI] [PubMed] [Google Scholar]
- Ping J, Schafe GE. The NO-cGMP-PKG signaling pathway coordinately regulates ERK and ERK-driven gene expression at pre- and postsynaptic sites following LTP-inducing stimulation of thalamo-amygdala synapses. Neural Plast. 2010;2010:540940. doi: 10.1155/2010/540940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preibisch S, Saalfeld S, Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 2009;25:1463–1465. doi: 10.1093/bioinformatics/btp184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro M, Straub VA, Schofield M, Picot J, Benjamin PR, O'Shea M, Korneev SA. Characterization of NO-sensitive guanylyl cyclase: expression in an identified interneuron involved in NO-cGMP-dependent memory formation. Eur J Neurosci. 2008;28:1157–1165. doi: 10.1111/j.1460-9568.2008.06416.x. [DOI] [PubMed] [Google Scholar]
- Ribeiro M, Schofield M, Kemenes I, Benjamin PR, O'Shea M, Korneev SA. Atypical guanylyl cyclase from the pond snail Lymnaea stagnalis: cloning, sequence analysis and characterization of expression. Neuroscience. 2010;165:794–800. doi: 10.1016/j.neuroscience.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Rose PK, Odlozinski M. Expansion of the dendritic tree of motoneurons innervating neck muscles of the adult cat after permanent axotomy. J Comp Neurol. 1998;390:392–411. doi: 10.1002/(sici)1096-9861(19980119)390:3<392::aid-cne7>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel C, Bicker G. Nitric oxide and cGMP influence axonogenesis of antennal pioneer neurons. Development. 2000;127:4541–4549. doi: 10.1242/dev.127.21.4541. [DOI] [PubMed] [Google Scholar]
- Serfozo Z, Veréb Z, Roszer T, Kemenes G, Elekes K. Development of the nitric oxide/cGMP system in the embryonic and juvenile pond snail, Lymnaea stagnalis L. A comparative in situ hybridization, histochemical and immunohistochemical study. J Neurocytol. 2002;31:131–147. doi: 10.1023/a:1023945522690. [DOI] [PubMed] [Google Scholar]
- Shelly M, Lim BK, Cancedda L, Heilshorn SC, Gao H, Poo MM. Local and long-range reciprocal regulation of cAMP and cGMP in axon/dendrite formation. Science. 2010;327:547–552. doi: 10.1126/science.1179735. [DOI] [PubMed] [Google Scholar]
- Staudt M. Reorganization after pre- and perinatal brain lesions. J Anat. 2010;217:469–474. doi: 10.1111/j.1469-7580.2010.01262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern M, Bicker G. Nitric oxide regulates axonal regeneration in an insect embryonic CNS. Dev Neurobiol. 2008;68:295–308. doi: 10.1002/dneu.20585. [DOI] [PubMed] [Google Scholar]
- Stern M, Böger N, Eickhoff R, Lorbeer C, Kerssen U, Ziegler M, Martinelli GP, Holstein GR, Bicker G. Development of nitrergic neurons in the nervous system of the locust embryo. J Comp Neurol. 2010;518:1157–1175. doi: 10.1002/cne.22303. [DOI] [PubMed] [Google Scholar]
- Straub VA, Benjamin PR. Extrinsic modulation and motor pattern generation in a feeding network: a cellular study. J Neurosci. 2001;21:1767–1778. doi: 10.1523/JNEUROSCI.21-05-01767.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub VA, Grant J, O'Shea M, Benjamin PR. Modulation of serotonergic neurotransmission by nitric oxide. J Neurophysiol. 2007;97:1088–1099. doi: 10.1152/jn.01048.2006. [DOI] [PubMed] [Google Scholar]
- Sunico CR, Portillo F, González-Forero D, Moreno-López B. Nitric-oxide-directed synaptic remodeling in the adult mammal CNS. J Neurosci. 2005;25:1448–1458. doi: 10.1523/JNEUROSCI.4600-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susswein AJ, Katzoff A, Miller N, Hurwitz I. Nitric oxide and memory. Neuroscientist. 2004;10:153–162. doi: 10.1177/1073858403261226. [DOI] [PubMed] [Google Scholar]
- Thévenaz P, Unser M. User-friendly semiautomated assembly of accurate image mosaics in microscopy. Microsc Res Tech. 2007;70:135–146. doi: 10.1002/jemt.20393. [DOI] [PubMed] [Google Scholar]
- Tojima T, Itofusa R, Kamiguchi H. The nitric oxide-cGMP pathway controls the directional polarity of growth cone guidance via modulating cytosolic Ca2+ signals. J Neurosci. 2009;29:7886–7897. doi: 10.1523/JNEUROSCI.0087-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimm KR, Rehder V. Nitric oxide acts as a slow-down and search signal in developing neurites. Eur J Neurosci. 2004;19:809–818. doi: 10.1111/j.0953-816x.2004.03182.x. [DOI] [PubMed] [Google Scholar]
- Van Wagenen S, Rehder V. Regulation of neuronal growth cone filopodia by nitric oxide depends on soluble guanylyl cyclase. J Neurobiol. 2001;46:206–219. doi: 10.1002/1097-4695(20010215)46:3<206::aid-neu1003>3.3.co;2-j. [DOI] [PubMed] [Google Scholar]
- Villani L. Development of NADPH-diaphorase activity in the central nervous system of the cichlid fish, Tilapia mariae. Brain Behav Evol. 1999;54:147–158. doi: 10.1159/000006619. [DOI] [PubMed] [Google Scholar]
- Welshhans K, Rehder V. Local activation of the nitric oxide/cyclic guanosine monophosphate pathway in growth cones regulates filopodial length via protein kinase G, cyclic ADP ribose and intracellular Ca2+ release. Eur J Neurosci. 2005;22:3006–3016. doi: 10.1111/j.1460-9568.2005.04490.x. [DOI] [PubMed] [Google Scholar]
- Wittenberg GF. Experience, cortical remapping, and recovery in brain disease. Neurobiol Dis. 2010;37:252–258. doi: 10.1016/j.nbd.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RG, Hadley RD, Kater SB, Hauser GC. Neurite outgrowth in molluscan organ and cell cultures: the role of conditioning factor(s) J Neurosci. 1981;1:1008–1021. doi: 10.1523/JNEUROSCI.01-09-01008.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong G, Mojsilovic-Petrovic J, Pérez CA, Kalb RG. Embryonic motor neuron dendrite growth is stunted by inhibition of nitric oxide-dependent activation of soluble guanylyl cyclase and protein kinase G. Eur J Neurosci. 2007;25:1987–1997. doi: 10.1111/j.1460-9568.2007.05456.x. [DOI] [PubMed] [Google Scholar]
- Yamazaki M, Chiba K, Mohri T, Hatanaka H. Cyclic GMP-dependent neurite outgrowth by genipin and nerve growth factor in PC12h cells. Eur J Pharmacol. 2004;488:35–43. doi: 10.1016/j.ejphar.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Zhou L, Zhu DY. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20:223–230. doi: 10.1016/j.niox.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Zinn CG, Bevilaqua LR, Rossato JI, Medina JH, Izquierdo I, Cammarota M. On the requirement of nitric oxide signaling in the amygdala for consolidation of inhibitory avoidance memory. Neurobiol Learn Mem. 2009;91:266–272. doi: 10.1016/j.nlm.2008.09.016. [DOI] [PubMed] [Google Scholar]