

Abstract
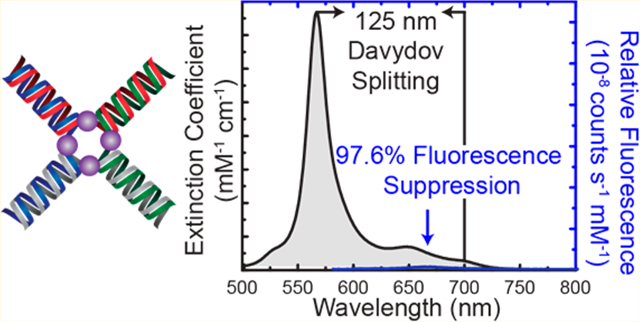
Exciton delocalization in dye aggregate systems is a phenomenon that is revealed by spectral features, such as Davydov splitting, J- and H-aggregate behavior, and fluorescence suppression. Using DNA as an architectural template to assembledye aggregates enables specific control of the aggregate size and dye type, proximal and precise positioning of the dyes within the aggregates, and a method for constructing large, modular two and three-dimensional arrays. Here, we report on dye aggregates,organized via an immobile Holliday junction DNA template, that exhibit large Davydov splitting of the absorbance spectrum (125nm, 397.5 meV), J- and H-aggregate behavior, and near-complete suppression of the fluorescence emission (~97.6% suppression).
Because of the unique optical properties of the aggregates, we have demonstrated that our dye aggregate system is a viable candidate as a sensitive absorbance and fluorescence optical reporter. DNA-templated aggregates exhibiting exciton delocalization may find application in optical detection and imaging, light-harvesting, photovoltaics, optical information processing, and quantum computing.
Graphical abstract
INTRODUCTION
Exciton delocalization, first discussed theoretically by Frenkel in the 1930s, and later by Davydov and Kasha, is an optical phenomenon that has been observed in optically active molecular (i.e., dye) aggregates.1–6 Exciton (i.e., electron− hole pair) delocalization is the spatial extension of the excited electronic eigenstates across the aggregate, with the resultant excitonic state described by a coherent linear superposition of excited states.7,8 Among the observable optical manifestations of excitonic delocalization are Davydov splitting,2,3 J- and H-aggregate behavior,4,5,7,9,10 motional narrowing,11–13 Dicke superradiance, resonance fluorescence,14 fluorescence quenching,15–19 and excitonically coupled circular dichroism (ECCD).20,21 The optical behavior of such aggregates is largely dictated by the stacking arrangement of the closely spaced dye molecules (2 nm or less apart). A single dye, termed a monomer, has a single one-exciton energy state, which is the lowest lying excited singlet state. A transition between this excited state and the ground state is allowed (Figure 1a). A two-dye aggregate system, termed a dimer, has two one-exciton states in which the degeneracy of the two excited states has been broken by the coupling between the transition dipoles, thus resulting in two energy eigenstates: E+ and E−. The exciton is delocalized between the two dyes in these eigenstates. Transitions strengths between these states and the ground state, E0, are strongly dependent on geometry. In the simplest geometric configurations, the two dyes can stack such that the transition dipoles are oriented parallel (H-dimer, Figure 1b), head-to-tail (J-dimer, Figure 1c), or at an intermediate configuration (oblique, Figure 1d). For these three possible cases, the allowed transitions are indicated by solid black arrows, while the forbidden transitions are indicated by dashed arrows in Figure 1. H-type aggregates exhibit a blue-shifted absorbance peak relative to the monomer, significantly reduced fluorescence emission intensity due to an optically forbidden energy transition from the lower energy exciton excited state (E−), and a large Stokes shift.9,15–17,22–30 Conversely, J-type aggregates, discovered by Jelley31,32 and Scheibe,33–35 exhibit a red-shift in the absorbance relative to the monomer since the only allowed optical transition is to the E− state, which consequently produce a nearly resonant fluorescence emission(i.e., small Stokes shift) with a sharp, high-intensity emission peak.7,9,12,14,27,36–42 Oblique aggregates display band (i.e., Davydov) splitting of the absorbance spectrum in which optical transitions to both E− and E+ are allowed and reflect a mix of H- and J-aggregate optical properties.2,3,27,43,44
Figure 1.

Schematics and energy level diagrams for various dye aggregate arrangements: (a) monomer, (b) H-dimer, (c) J-dimer, and(d) oblique dimer. The top schematic shows dye molecules represented as purple spheres, while the dipole orientation is depicted by black arrows. Each energy level diagram shows a ground energy state (E0), the first excited energy state (E) for the monomer, and the excited energy state’s split energy levels (higher energy, E+ and lower energy, E−) for dimers due to exciton delocalization. Allowed energy transitions are provided as solid black arrows and forbidden transitions are shown as dashed black arrows. Adapted from ref 4 with permission.
The manifestation of excitonic delocalization has been observed in both naturally occurring photosynthetic systems45–47 and in synthetic dye-solvent systems.29,39,47–51 Photosynthetic systems have evolved such that the molecular chromophores (i.e., dyes) are proximally positioned via a protein scaffold. However, using a protein scaffolding architecture in synthetic systems is difficult owing to complex folding mechanisms. The aggregation of dyes in solvents has been found to occur spontaneously and preferred dye stacking arrangements are largely driven by kinetics and thermodynamics, which is problematic for controlling the excitonic delocalization behavior. To assert greater control over the assembly of dyes into aggregates of well-defined structure, we exploit DNA as an alternative biomolecular scaffolding material. DNA assembly is governed by simple and predictable design rules due largely to Watson−Crick hybridization. In addition, DNA can be functionalized with molecular dyes through covalent attachment at precisely specified locations anywhere along its length. Thus, covalent attachment of dyes onto a DNA substrate enables (i) proximal and precise positioning of the dye molecules,27,52–58 (ii) specific and selective control of dye number and type,24,52,54,59 and (iii) the self-assembly of large two- and three-dimensional arrays.60–62
Capitalizing on the modularity enabled by DNA self-assembly allows larger, more complex arrays and networks of precisely controlled dye aggregates (i.e., dye assemblies) to be realized. Though there have been many studies that demonstrate exciton delocalization in DNA-templated dye aggregation within linear duplex structures,17,18,22–25,27,52–59,63–66 only a few studies have exploited DNA constructs of more complex geometry. Most of these have been limited to studies employing three-armed junctions for dye assembly, which have been constructed to explore light-harvesting complexes and excimer behavior.52,67 In our recent work, we were able to facilitate and control the transition of Cy5 dimers templated within a linear DNA duplex structure to a Cy5 tetramer in a mobile 4-arm junction (i.e., 4AJ) structure, thereby inducing a change in exciton delocalization from J-type to H-type behavior.68 The transition was controlled by varying salt and/or DNA concentrations. Here, we exploit an immobile 4AJ (i.e., Holliday junction)69,70 to explore exciton delocalization in multimers, including dimers, trimers, and tetramers in order to better control the effects of exciton delocalization such as Davydov splitting and J- and H-type behavior at ambient temperatures. The excitonic delocalization between the four Cy5 dyes (i.e., tetramer) located within the core of a 4AJ exhibits an unusually large Davydov splitting (125 nm, 397.5 meV) that results in a striking color change of the solution, excitonically coupled circular dichroism (EC-CD) signals, and near-complete quenching (i.e., strong suppression) of the fluorescence emission. To obtain further insight into the 4AJ-guided assembly of excitonically coupled aggregates, a variety of aggregate types were examined. It was found that increasing the number of dyes within the aggregate core of the 4AJ substantially increased the energy splitting of the system. Because of its unique optical signatures, this immobile tetramer offers two characterization modes for optical detection that have applications as optical reporters, namely a large shift in absorbance and strong suppression of the fluorescence. The construction, by DNA self-assembly, of dye aggregates exhibiting the strong optical effects reported here may pave the way for novel practical applications of exciton delocalization.
METHODS
Synthesis.
Short 26 nucleotide (nt) DNA oligomers were purchased lyophilized from Integrated DNA Technologies, Inc. Oligomers internally functionalized with Cy5 dyes were purified via high-performance liquid chromatography (HPLC), while non dye-functionalized strands were purified through standard desalting. All oligomers were rehydrated with ultrapure water (Barnstead Nanopure, Thermo Scientific) to produce a 100 μM stock solution. DNA four-armed junctions (4AJs) were prepared by combining equimolar amounts of complementary oligomers in a 1× TAE (40 mM tris-(hydroxymethyl)aminomethane, 20 mM acetic acid, 1 mM ethylenediaminetatraacetic acid) buffer solution (pH 8.0) with 15 mM added magnesium chloride (MgCl2) to obtain a final DNA concentration of 10 μM. Resulting solutions were left for 24−48 h at room temperature to allow for hybridization and self-assembly. TAE (10× stock solutions) and MgCl2 (99% purity) were purchased from Fisher Scientific and used as received. Solutions of 1.375 M MgCl2 were added to a 1× TAE buffer solution to yield final magnesium (Mg2+) concentrations of 0, 15, and 100 mM. The initial salt concentration due to the TAE buffer (<1 mM Na+ arising from the EDTA counterion) was found to be negligible compared to the subsequently added salt concentrations.
Purification.
Self-assembled DNA 4AJ structures were further purified using polyacrylamide gel-electrophoresis (PAGE). Solutions of 4AJs were prepared with various dye arrangements in a 1× TAE buffer at 10 μM double-stranded DNA (dsDNA) concentrations and 15 mM added MgCl2. A 6× New England Biolabs loading buffer was added in a 1:5 ratio with the 4AJs to give a resulting 1× concentration of the loading buffer (1× buffer: 11 mM ethylenediaminetetraacetic acid, 3.3 mM tris-hydrochloric acid, 0.017% sodium dodecyl sulfate, 0.015% bromophenol blue, 2.5% Ficoll-400). The prepared sample solutions were then injected into a 12% gel, prepared with 0.25× TAE with 3.75 mM added MgCl2 and allowed to migrate for approximately 90 min with a 150 V applied voltage. 1× TAE with 12 mM added MCl2 was used in the buffer reservoirs of the electrophoresis apparatus. Using a multiplexed gel imaging and documentation system (FluorChem Q, ProteinSimple), completed gels were imaged by brief illumination with a 254 nm ultraviolet (UV) light source while placed on a phosphor imaging plate. Well-formed 4AJ structures were identified, excised from the gels with a razor blade, and extracted. Extraction was conducted by placing select excised gel pieces into a vial of 1× TAE buffer containing 15 mM added MgCl2. The DNA 4AJs were allowed 48 h to diffuse out of the gel and into the surrounding buffer solution. The resulting buffer solution was then extracted from the vial and any remaining gel fragments removed by centrifuging the sample at 1000 relative centrifugal force (rcf) for 10 min. A further description of the gel-electrophoresis process and gel images are provided in the Supporting Information, part S5.
Optical Characterization.
All optical characterization was performed at room temperature. UV−Vis spectra were obtained using a dual-beam Cary 5000 UV−Vis−NIR spectrophotometer (Agilent Technologies). Solution samples (150 μL) were transferred into a 1 cm path length submicro, self-masking quartz spectrophotometer cell (Starna). Static absorbance spectra were monitored over a range of wavelengths and normalized by DNA concentrations. Static fluorescence spectra were obtained using a Fluorolog-3 spectrofluorometer (Horiba Scientific) and monitored as a function of wavelength. PAGE purified solution samples of each 4AJ construct (150 μL) were transferred to a 1 cm path length submicro, self-masking fluorometer cell (Starna) and were excited at or near their respective absorption maxima listed in Figure 2. The excitation and emission bandwidths used were 5.0 and 2.5 nm, respectively. Circular dichroism (CD) measurements were performed using a JACSO-J810 spectropolarimeter. Samples of purified 4AJ constructs (100 μL) were transferred to a 1 cm path length micro, self-masking quartz cuvette (Thorlabs). Spectra were monitored over a series of wavelengths and normalized by the DNA concentration.
Figure 2.

Absorbance (black traces in left column), fluorescence (blue traces), and circular dichroism (black traces in right column) spectra corresponding to the 4AJ templated (a, b) monomer, (c, d) adjacent dimer, (e, f) transverse dimer, (g, h) trimer, and (i, j) tetramer dye aggregates. The inset in part i shows the suppressed fluorescence emission peak at 664 nm that occurs due to a forbidden energy transfer mechanism. All spectra have been normalized by DNA concentration. The fluorescence studies were performed by monitoring the emission over a range of wavelengths and exciting the aggregates at their respective absorbance maxima. All samples were prepared at 10 μM DNA concentration in a 1× TAE buffer solution with 15 mM MgCl2 added, PAGE purified, and normalized by the resulting DNA concentrations (~0.5−2.5 μM).
Optical Reporter Studies.
Optical reporter studies were performed using two characterization techniques: fluorescence and absorbance. For fluorescence studies, DNA-templated 4AJs were designed with a 10 nt toehold on one of the four arms. Structures were synthesized and purified as described above. Spectra were collected using a Horiba Fluorolog-3 spectrofluorometer. Static fluorescence spectra of the samples were obtained prior to and subsequently following the reaction. Samples (250 μL) were monitored at the fluorescence peak maximum (666 nm) over 70 min while being excited at the tetramer absorbance peak maximum (565 nm). At 10 min, an invasion strand-designed to be complementary to the strand with the 10 nt toehold extension-was introduced into the system in 1:1 stoichiometry with the 4AJ and pipette mixed for ~30 s. The change in fluorescence intensity was then monitored over 70 min. The initial dsDNA concentrations of the 4AJ were ~0.4 μM, and all reactions were held at 22 °C.
Absorbance studies were conducted by initially synthesizing the 4AJ structure in two parts, described as dimers A and B, such that the two dimers are complementary and could join together to form the full 4AJ structure. Dimers A and B were purified via PAGE and diluted to ~0.1 μM dsDNA concentration. Using a Cary 5000 UV−Vis−NIR spectrophotometer, changes in absorbance at the H-tetramer peak maximum (565 nm) were monitored. Initially, a sample of dimer A was monitored. At 10 min, dimer B was introduced in stoichiometry and pipette mixed for ~30 s. Upon hybridization, the 4AJ tetramer formed, inducing an absorbance signal at 565 nm. All reactions were held at room temperature (22 °C).
Stability Studies.
Results from studies performed to monitor the stability of the 4AJs with changes in solution salt and DNA concentrations are described in Supporting Information, part S6. Samples of tetramer 4AJ constructs used for salt studies were prepared in a 1× TAE buffer solution containing 0−100 mM MgCl2 and held at a constant DNA concentration of 1.0 μM. Samples of the tetramer used for DNA concentration studies were prepared in a 1× TAE buffer solution with 0.1−3.0 μM dsDNA concentrations and held at a constant salt concentration of 15 mM MgCl2
RESULTS AND DISCUSSION
Optical Characterization of 4AJ Templated Dye Aggregates.
Optical characterization using spectroscopy techniques, including absorbance, fluorescence, and circular dichroism (CD) were performed on a variety of dye-labeled DNA 4AJ structures to identify spectral signatures of exciton delocalization. Although the majority of the data reported here were obtained from samples in a 1× TAE buffer solution containing 15 mM of MgCl2, we additionally performed absorbance measurements of samples prepared in a 1× TAE buffer solution containing a variety of salt concentrations: 0 and 100 mM MgCl2 with a constant 1 μM DNA concentration and a variety of DNA concentrations: 0.1, 1.0, and 3.0 μM with a constant 15 mM salt concentration (Supporting Information, part S6) to investigate the salt and DNA concentration dependence that might indicate higher order aggregation formation. The absorbance spectra exhibited only a weak dependence on MgCl2 and DNA concentration, indicating that the results obtained at 15 mM MgCl2 concentration are representative of a broad range of salt concentrations and that our 4AJ structures have little tendency to form higher-ordered aggregates. The minimal dependence on MgCl2 and DNA concentration to drive changes in dye aggregate formation is in contrast to what was found in our previous work with linear DNA duplex constructs that combined to form mobile 4AJ structures.68 For spectroscopic studies, all 4AJ structures were purified via polyacrylamide gel-electrophoresis (PAGE). Dye structure and strand information are provided in the Supporting Information, part S1. Fluorescence suppression is computed using the area of the emission spectrum; a general equation is given in the Supporting Information, part S2. As a control, spectral analysis of the monomer (Figure 2a) reveals single absorbance and emission peak maxima at 653 and 666 nm, respectively. Although the monomer absorbance peak maximum is slightly red-shifted from the literature value, the shift is most likely a result of the increased rigidity of the 4AJ structure, dye−DNA interactions, and surrounding base sequences.71 As expected, the dye monomer did not produce a CD signal within the visible range (Figure 2b), indicating the absence of molecular chirality and excitonic coupling. Likewise, the CD signals appearing in the UV range at 246 and 276 nm, respectively, result from the expected right-handed macro-molecular structure of the β-DNA helix.
As shown in the schematics in Figure 2, the 4AJ structure was used to organize and create four different dye aggregates in addition to the monomer: two dimer configurations, a trimer, and a tetramer. The two-dye (dimer) aggregates are distinguished by the location of the dyes, either positioned on adjacent arms (adjacent dimer, Figure 2c) or on opposing arms (transverse dimer, Figure 2e). A pronounced difference in optical properties was observed between the two dimers. The adjacent dimer shows predominantly J-type aggregate behavior, with an absorbance peak maximum at 662 nm that is slightly red-shifted relative to the monomer at 653 nm. This red-shift in absorbance relative to the fluorescence peak at 667 nm yields a small Stokes shift of 5 nm, which is near resonance fluorescence behavior. A second, smaller absorbance peak also appears at 602 nm. The appearance of the two peaks is indicative of a small Davydov splitting of 58 nm (Table 1). The adjacent dimer exhibits a split CD signal (Figure 2c) at 600 and 675 nm. The ± of the CD peaks (read from right to left), signifies right-handed chirality and is anticipated owing to the right-handed nature of the DNA helix. Because of the predominant J-type behavior and presence of Davydov splitting and CD signal, the dyes are most likely arranged head-to-tail with some break in planarity between the molecules, or oblique-like arrangement, that favors J-aggregate stacking. In contrast, the transverse dimer (Figure 2e) shows optical behavior that is roughly opposite that of the adjacent dimer. The primary absorbance peak is blue-shifted, with an absorbance maximum at 600 nm and a much smaller red-shifted absorbance peak (maximum at 636 nm) corresponding to an even smaller 36 nm Davydov splitting. A 68 nm Stokes shift is observed when comparing the primary absorbance peak to the fluorescence peak at 668 nm, which is much reduced in intensity (~97.6%). The blue-shifted primary absorbance peak, the large Stokes shift, and the reduced fluorescence intensity are all indicative of predominately H-type aggregate behavior. The small Davydov splitting and a relatively strong ∓ CD signal at 600 and 669 nm, respectively, of the transverse dimer indicates left-handed chirality and an oblique stacking arrangement that does not stack in a perfectly parallel or H-type arrangement (Figure 2f). Interestingly, the small absorbance peak at 636 nm corresponds to a split CD signal with peaks at 630 and 669 nm, which implies excitonic coupling between the dyes. The considerably decreased fluorescence emission is indicative of fluorescence suppression due to a forbidden optical transition.
Table 1.
| Optical Excitonic Delocalization Behavior per 4AJ Construct
| optical behavior | monomer | adjacent dimer | transverse dimer | trimer | tetramer |
|---|---|---|---|---|---|
| Davydov splitting (nm/meV) | 0/0 | 58/179.8 | 36/117.0 | 58/186.3 | 125/397.5 |
| abs fwhm (nm) | 34 | 30 | 24 | 49 | 16 |
| FL fwhm (nm) | 33 | 32 | 34 | 34 | 48 |
| Stokes shift (nm) | 13 | 5 | 68 | 73 | 99 |
| % FL suppression | 0.0 | 53.3 | 89.6 | 84.5 | 97.6 |
The trimer (Figure 2g) shows absorbance and fluorescence properties similar to the transverse dimer. Most notably, the trimer absorbance spectrum shows an absorbance peak at 593 nm that is blue-shifted from the monomer by 60 nm and a smaller peak at 651 nm that is slightly blue-shifted by 2 nm. The absorbance spectrum also reveals broadening of the primary peak by 15 nm compared to the monomer, as indicated by an increase in the full-width at half-maximum (fwhm) value listed in Table 1. A detailed procedure for calculating the fwhm is described in the Supporting Information, section S2. The broadening may result from an irresolvable third peak; however, because the CD spectrum (Figure 2h) does not exhibit a third peak, the broadening most likely arises from a distribution of trimer dye configurations (i.e., dye positions and orientations). In combination with the blue-shifted absorbance peak, the trimer shows ~85% suppression of the fluorescence emission intensity (relative to the monomer), indicative of net H-aggregate behavior. Similar to the other aggregate configurations, the trimer has an observed EC-CD signal that, though small, indicates a slightly imperfect stacking arrangement with minor obliqueness. Additionally, the aggregate displays right-handedness, similar to the adjacent dimer.
The tetramer dye aggregate configuration produces the most interesting optical spectra. A large Davydov splitting of 125 nm (397.5 meV), extensive enough to induce a visible color change in the solution, was observed. See the Supporting Information, Figure S2.1, for spectral data and peak fitting analysis. To our knowledge, this is the largest reported splitting for DNA-templated dye aggregates (Figure 2i). The Davydov splitting is characterized by a significantly blue-shifted intense absorbance peak at 565 nm and a red-shifted much less intense peak at 690 nm. Though difficult to resolve in the absorption spectrum, the absorbance peak at 690 nm is further substantiated by a large signal in the CD spectrum (Figure 2j) at 700 nm. Note that the difference in extinction coefficients of the various dye aggregates result from two key effects: (1) the differences in dye number and (2) exciton delocalization and exciton-vibrational interactions. The tetramer also exhibits strong fluorescence suppression behavior at 664 nm, with a 97.6% decrease in the fluorescent emission relative to the monomer, as determined by peak area. The large Davydov splitting, strong fluorescence suppression, and 99 nm Stokes shift (Table 1) provide solid evidence of a dye assembly with predominantly H-type stacking. The pronounced ∓ CD signal of the tetramer configuration indicates strong exciton coupling between the dyes and reveals that the dyes are oriented predominantly in a parallel manner with some obliqueness that is supported by the absorbance peak at 690 nm. Comparing the optical spectra of the immobile 4AJ-templated tetramer presented here with the mobile 4AJ-templated tetramer observed in our prior study, the most notable difference is in the CD spectrum. Interestingly, the immobile tetramer shows right-handedness (Figure 2J), while the mobile tetramer shows left-handedness.68 Additionally, an absorbance peak at 665 nm was observed for the mobile 4AJ-templated tetramer. In contrast, for the immobile tetramer, a very subtle peak was observed for at 690 nm that was further supported by a strong CD signal, indicating obliqueness within the immobile tetramer. These differences are most likely due to the base-pair stacking of the immobile tetramer locking the aggregate core such that the DNA junction does not undergo restacking. The mobile tetramer undergoes DNA breathing and base-pair restacking and was found to partition into two J-dimer pairs that are displaced either horizontally or vertically along the arms, which accounts for the observed 665 nm absorbance peak.68
Theoretical Spectral Modeling of 4AJ Templated Dye Aggregates.
Theoretical modeling of the absorbance and CD spectra was performed to determine the geometrical stacking configurations of the five dye aggregate configurations since specific dye positioning and orientation beyond the above simple analysis is not readily apparent from visual inspection of experimental spectral data. Following the approach used in our previous work,68 modeling of the experimental absorbance and CD spectra using a method based on the work of Kühn, Renger, and May, referred to as the Kühn−Renger−May (KRM) model, enables the extraction of the position and orientation of the dyes within each type of aggregate.72 The KRM model proceeds by constructing a system Hamiltonian that considers N dye molecules with arbitrary positions and orientations. Through judicious choice of a truncated Hilbert space, a perturbational treatment of the exciton-vibrational coupling is avoided in the diagonalization of the Hamiltonian. Information regarding the computation of the eigenvalues and eigenvectors is described in the Supporting Information, parts S3. An in-house software simulation tool that incorporates the KRM model was created to fit the absorbance and CD spectra. Smooth theoretical absorbance and CD spectra were obtained by calculating the optical transition rates for each eigenstate and convolving the transition rate line spectra with a Gaussian line shape to approximate the effect of the vibronic continuum on spectral line shape.
In Figure 3, theoretical KRM model fits of the absorbance and CD spectra are illustrated by red dashed curves, while the experimental data are shown as black open circles. To maximally constrain the theoretical modeling, the absorbance and CD data were simultaneously fit such that the two dominant peaks of the absorption and CD spectra were both matched and the mean-square difference between the experimental and theoretical absorption and CD spectra were minimized. From this fitting procedure, the relative positions and orientations of each dye are extracted (see the Supporting Information, Table S3.2).
Figure 3.

Absorbance (a, d, g, and j) and CD (b, e, h, and k) spectral fits with corresponding structures (c, f, i, and l) of the adjacent dimer, transverse dimer, trimer, and tetramer aggregates, respectively. Experimental data sets are shown as black open circles, while KRM theoretical fits are given as red curves. All samples were prepared at 10 μM DNA concentration in a 1× TAE buffer solution with 15 mM MgCl2 added, PAGE purified, and normalized by the resulting DNA concentrations (~0.5−2.5 μM). Fluorescence measurements were obtained by exciting the samples at their respective absorption maxima.
Fitting the experimental absorbance (Figure 3a) and CD (Figure 3b) spectra for the adjacent dimer revealed that the dyes are oriented with predominantly J-aggregate (or head-to-tail) stacking but in a somewhat oblique manner, which is manifested in the increased vibronic peak at 600 nm. This configuration supports the evidence provided by the red-shifted absorbance spectrum and demonstration of excitonic coupling in the CD spectrum, which only occur with an oblique stacking arrangement. Figure 3c visualizes the dimer stacking configuration within the 4AJ construct based on the fitting of the theory to the absorbance and CD data (see the Supporting Information, Table S3.2). The figure was created using UCSF Chimera visualization software.73,74 We note that only the position and orientation of the long axis of the Cy5 molecule is determined by the fitting procedure. Thus, how the molecules are oriented about the axis (i.e., rotated about the axis) and relative to the DNA is conjectural in Figure 3, parts c, f, i, and l. In contrast to the adjacent dimer, theoretical fitting of the transverse dimer absorbance (Figure 3d) and CD (Figure 3e) spectra using the KRM model determined that the dyes are indeed stacked parallel to one another (i.e., H-type) with a center-to-center distance of ~1.0 nm. The slight obliqueness of the dyes is exhibited in the EC-CD spectrum and a vibronic peak at 636 nm in the absorbance spectrum. Figure 3f illustrates the transverse dimer stacking configuration obtained from the theoretical fit of the absorbance and CD data.
Using the KRM model to fit the trimer absorbance (Figure 3g) and CD (Figure 3h) spectra substantiates our hypothesis that there are not three underlying absorbance peaks but rather that the peak at 593 nm is simply broadened. Though the theoretical fit does not provide an explanation for the peak broadening, we can conjecture the existence of a population of trimer constructs having slightly different dye configurations(i.e., dye positions and orientations within the core of the 4AJ), such that the spectrum, consisting of an average of these populations, exhibits a broadened absorption peak. The fit of the theory to the experimental absorbance and CD data indicates that the dyes of the trimer are stacked roughly parallel to each but with some twist, as depicted in Figure 3i. The configuration of the trimer dyes is predominantly H-type similar to the transverse dimer, which explains why their respective absorbance spectra appear so similar. Finally, fitting of the tetramer aggregate’s absorbance (Figure 3j) and CD (Figure 3k) spectra incorporates the red-shifted peak at 690 nm. Additionally, due to the strong EC-CD signals, obliqueness in the dye stacking arrangement was confirmed. The dye configuration obtained by the fitting procedure is illustrated in Figure 3l. Input parameters, output positions and orientations for each dye, and a quantitative characterization of the quality of the theoretical fits are provided in the Supporting Information, part S3.
Optical Reporter Functionality.
To examine the viability of the 4AJ tetramer as a highly sensitive optical reporter, we constructed and studied the reaction kinetics of two reporter systems that exploit changes in fluorescence and optical absorption, respectively. In the system that exploited fluorescence changes, strand invasion was employed to disassemble a tetramer into a one dye and three dye complex. Strand invasion was facilitated by a designed 10 nt extension (toehold) on one of the 4 arms to which the Invasion Strand can first attach by Watson−Crick base pairing. The Invasion Strand is fully complementary to the 4AJ strand with the extension and thus excises this strand from the 4AJ.75 As illustrated in Figure 4a, this 4AJ tetramer (tetramer-1), would have nearly zero fluorescence while assembled. Upon invasion, the coherent excitonic delocalization within the aggregate would cease and greatly increased fluorescence would ensue from the dyes on both the single dye (monomer) product and the three dye complex (trimer). Conversely, the system that exploited absorbance changes consisted of two halves of the 4AJ synthesized separately that can hybridize when brought together to form a full 4AJ tetramer (tetramer-2). While disassembled, the two halves of the 4AJ (dimer A and dimer B) would exhibit minimal absorption at 565 nm (the H-tetramer absorbance peak), yielding a cyan solution color. When assembled, the absorption of the complete 4AJ (tetramer-2) at 565 nm greatly increases, yielding a solution color change to violet. This color change is depicted schematically in Figure 4 by the color of the represented dye molecules (cyan and violet spheres).
Figure 4.

(a) Schematic illustration and (b) experimental demonstration of the 4AJ tetramer undergoing strand-invasion using fluorescence detection (λfl = 666 nm). (c) Schematic illustration and (d) experimental demonstration of the 4AJ tetramer formation via DNA hybridization using UV−Vis−NIR absorbance, or colorimetric, detection (λabs = 666 nm).
The operation of the fluorescence scheme (Figure 4b) yielded a 25-fold increase in fluorescence intensity upon disassembly of the 4AJ into a monomer and trimer. Similarly, the absorbance reaction (Figure 4d) gave a 10-fold increase in absorbance at 565 nm. For the DNA concentrations at which the experiments were carried out, ~ 0.4 μM for the Fluorescence scheme and ~0.1 μM for the absorbance scheme, the time to half completion of the reactions was short compared to the 30 s required to thoroughly pipette mix the reactants in the cuvette. This implies a lower bound of 8 × 105 M−1 s−1 for the rate constant of the strand invasion reaction of Figure 1a and a lower bound of 3 × 105 M−1 s−1 for the rate constant of the hybridization reaction of Figure 1c. These are consistent with the rate constants reported in the literature for these types of reactions,75,76 which are typically in the 106 M−1 s−1 range. The observed near complete (>97.6%) fluorescence suppression and large Davydov splitting (125 nm, 397.5 meV) in the 4AJ tetramer (Figure 2) signals its potential usefulness as a highly sensitive optical reporter.
CONCLUSION
We have explored a two-dimensional DNA template for proximally positioning molecular dyes to induce exciton delocalization. The four-armed junction (4AJ) DNA construct enabled the analysis of a variety of dye aggregate types (i.e., adjacent and transverse dimers, trimer, and tetramer) that revealed various exciton delocalization spectral signatures. Through static spectral characterization of the 4AJ-templated dye aggregates, it was found that the type of aggregate (i.e., H-aggregate, J-aggregate, or oblique) depended largely on the position of the dyes within the 4AJ construct. The specificity of the dye position was demonstrated explicitly by the difference in spectral properties of the two dimer constructs, such that the dimer with dyes positioned on adjacent arms (adjacent dimer) demonstrated predominantly J-aggregate behavior, whereas the transverse dimer with dyes positioned on opposite arms exhibited H-aggregate behavior. The tetramer, with four dyes positioned at each node of the 4AJ, expressed a strikingly large Davydov splitting and strong fluorescence suppression. Exploiting the distinctive optical properties of the tetramer afforded the opportunity to fabricate and operate two dynamic dye aggregate schemes for potential use as a sensitive optical reporter using either absorbance or fluorescence characterization methods. Further exploration of exciton delocalization in DNA-templated dye assemblies may yield applications in optical detection and imaging schemes, light-harvesting, photovoltaics, optical information processing, and quantum computing.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank the students and staff within the Nanoscale Materials & Device group for valuable assistance with this work, specifically Dr. Natalya Hallstrom and Allison Christy for their help with PAGE Gel electrophoresis experiments, J. Melinger and colleagues at the U.S. Naval Research Laboratory (NRL) for helpful discussions regarding the theoretical work of O. Kühn et al. and L. Li, E. Nelson, and A. Correa for modeling. The research was supported in part by the Boise State University Division of Research and Economic Development, the National Science Foundation INSPIRE No. 1648655, and the NASA Idaho Space Grant Consortium (ISGC). We acknowledge the Biomolecular Research Center for the use of their CD instrumentation supported in part by the Institutional Development Awards (IDeA) from the National Institute of General Medical Sciences No. P20GM103408 and the National Institutes of Health No P20GM109095, the National Science Foundation No. 0619793 and 0923535, the MJ Murdock Charitable Trust, the Idaho State Board of Education. B.L.C. acknowledges the Micron Technology MSE Ph.D. Fellowship. The molecular graphics and analyses were performed using the UCSF Chimera package, which is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at 10.1021/acs.jpca.7b12668
Dye specifications, strand sequences, data analysis, KRM model parameter specification, PAGE gel images, and structure stability (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Frenkel J On the Transformation of Light into Heat in Solids. II. Phys. Rev 1931, 37 (10), 1276–1294. [Google Scholar]
- (2).Davydov AS Theory of Absorption Spectra of Molecular Crystals Institute of Physics, Academy of Sciences of Ukrainian SSR: Kyiv, Ukraine, 1948; Vol. 18. [Google Scholar]
- (3).Davydov AS The Theory of Molecular Excitons. Sov. Phys 1964, 82 (3–4), 145–178. [Google Scholar]
- (4).Kasha M Energy Transfer Mechanisms and Molecular Exciton Model for Molecular Aggregates. Radiat. Res 1963, 20 (1), 55. [PubMed] [Google Scholar]
- (5).Kasha M; et al. The Exciton Model in Molecular Spectroscopy. Pure Appl. Chem 1965, 11 (3–4), 371. [Google Scholar]
- (6).Franck J; Teller E Migration and Photochemical Action of Excitation Energy in Crystals. J. Chem. Phys 1938, 6 (12), 861–872. [Google Scholar]
- (7).Eisfeld A; Briggs JS The J-Band of Organic Dyes: Lineshape and Coherence Length. Chem. Phys 2002, 281 (1), 61–70. [Google Scholar]
- (8).Fidder H; Knoester J; Wiersma DA Optical Properties of Disordered Molecular Aggregates: A Numerical Study. J. Chem. Phys 1991, 95 (11), 7880–7890. [Google Scholar]
- (9).Roden J; Eisfeld A; Briggs JS The J- and H-bands of Dye Aggregate Spectra: Analysis of the Coherent Exciton Scattering (CES) Approximation. Chem. Phys 2008, 352 (1–3), 258–266. [Google Scholar]
- (10).Eisfeld A; Briggs JS Absorption Spectra of Quantum Aggregates Interacting via Long-Range Forces. Phys. Rev. Lett 2006, 96(11), 113003. [DOI] [PubMed] [Google Scholar]
- (11).Whittaker DM; Kinsler P; Fisher TA; Skolnick MS; Armitage A; Afshar AM; Sturge MD; Roberts JS Motional Narrowing in Semiconductor Microcavities. Phys. Rev. Lett 1996, 77(23), 4792–4795. [DOI] [PubMed] [Google Scholar]
- (12).Walczak PB; Eisfeld A; Briggs JS Exchange Narrowing of the J Band of Molecular Dye Aggregates. J. Chem. Phys 2008, 128 (4), 044505. [DOI] [PubMed] [Google Scholar]
- (13).Knapp EW Lineshapes of Molecule Aggregates - Exchange Narrowing and Intersite Correlation. Chem. Phys 1984, 85 (1), 73–82. [Google Scholar]
- (14).Marcus RJ; Haugen GR Resonance Fluorescence in Chlorophyll a Solutions. Photochem. Photobiol 1965, 4, 183–192. [Google Scholar]
- (15).Ogawa M; Kosaka N; Choyke PL; Kobayashi H H-Type Dimer Formation of Fluorophores: A Mechanism for Activatable, in Vivo Optical Molecular Imaging. ACS Chem. Biol 2009, 4 (7), 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ikeda S; Kubota T; Kino K; Okamoto A Sequence Dependence of Fluorescence Emission and Quenching of Doubly Thiazole Orange Labeled DNA: Effective Design of a Hybridization-Sensitive Probe. Bioconjugate Chem 2008, 19 (8), 1719–1725. [DOI] [PubMed] [Google Scholar]
- (17).Ikeda S; Okamoto A Hybridization-Sensitive On-Off DNA Probe: Application of the Exciton Coupling Effect to Effective Fluorescence Quenching. Chem. - Asian J 2008, 3 (6), 958–968. [DOI] [PubMed] [Google Scholar]
- (18).Hara Y; Fujii T; Kashida H; Sekiguchi K; Liang XG; Niwa K; Takase T; Yoshida Y; Asanuma H Coherent Quenching of a Fluorophore for the Design of a Highly Sensitive In-Stem Molecular Beacon. Angew. Chem., Int. Ed 2010, 49 (32), 5502–5506. [DOI] [PubMed] [Google Scholar]
- (19).Bouquin N; Malinovskii VL; Haener R Highly Efficient Quenching of Excimer Fluorescence by Perylene Diimide in DNA. Chem. Commun 2008, No. 17, 1974–1976. [DOI] [PubMed] [Google Scholar]
- (20).Eisfeld A; Kniprath R; Briggs JS Theory of the Absorption and Circular Dichroism Spectra of Helical Molecular Aggregates. J. Chem. Phys 2007, 126 (10), 104904. [DOI] [PubMed] [Google Scholar]
- (21).Person RV; Monde K; Humpf HU; Berova N; Nakanishi K A New Approach in Exciton-Coupled Circular-Dichroism (ECCD)- Insertion of an Auxillary Stereogenic Center. Chirality 1995, 7 (3), 128–135. [DOI] [PubMed] [Google Scholar]
- (22).Ruedas-Rama MJ; Orte A; Martin-Domingo MC; Castello F; Talavera EM; Alvarez-Pez JM Interaction of YOYO-3 with Different DNA Templates to Form H-Aggregates. J. Phys. Chem. B 2014, 118 (23), 6098–6106. [DOI] [PubMed] [Google Scholar]
- (23).Ruedas-Rama MJ; Alvarez-Pez JM; Orte A Formation of Stable BOBO-3 H-Aggregate Complexes Hinders DNA Hybridization.J. Phys. Chem. B 2010, 114 (27), 9063–9071. [DOI] [PubMed] [Google Scholar]
- (24).Asanuma H; Fujii T; Kato T; Kashida H Coherent Interactions of Dyes Assembled on DNA. J. Photochem. Photobiol., C 2012, 13 (2), 124–135. [Google Scholar]
- (25).Asanuma H; Shirasuka K; Takarada T; Kashida H; Komiyama M DNA-Dye Conjugates for Controllable H* Aggregation. J. Am. Chem. Soc 2003, 125 (8), 2217–2223. [DOI] [PubMed] [Google Scholar]
- (26).Yang MN Influence of Energy Transfer on the Intensity Pattern of Vibronic Excitation Studied by Reduced Density-Matrix Theory. J. Mol. Spectrosc 2006, 239 (1), 108–114. [Google Scholar]
- (27).Markova LI; Malinovskii VL; Patsenker LD; Haner R J-vs. H-Type Assembly: Pentamethine Cyanine (Cy5) as a Near-IR Chiroptical Reporter. Chem. Commun 2013, 49 (46), 5298–5300. [DOI] [PubMed] [Google Scholar]
- (28).Conley NR; Pomerantz AK; Wang H; Twieg RJ; Moerner WE Bulk and Single-Molecule Characterization of an Improved Molecular Beacon Utilizing H-Dimer Excitonic Behavior. J. Phys. Chem. B 2007, 111 (28), 7929–7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lim JM; Kim P; Yoon MC; Sung J; Dehm V; Chen ZJ; Wurthner F; Kim D Exciton Delocalization and Dynamics in Helical Pi-Stacks of Self-Assembled Perylene Bisimides. Chem. Sci 2013, 4 (1), 388–397. [Google Scholar]
- (30).Ikeda S; Kubota T; Yuki M; Okamoto A Exciton-Controlled Hybridization-Sensitive Fluorescent Probes: Multicolor Detection of Nucleic Acids. Angew. Chem., Int. Ed 2009, 48 (35), 6480–6484. [DOI] [PubMed] [Google Scholar]
- (31).Jelley EE Molecular, Nematic and Crystal States of I: IDiethyl–Cyanine Chloride. Nature 1937, 139 (3519), 631–632. [Google Scholar]
- (32).Jelley EE Molecular, Nematic and Crystal States of I: IDiethyl-Cyanine Chloride. Nature 1937, 139 (3519), 631–632. [Google Scholar]
- (33).Scheibe G Über die Veranderlichkeit der Absorptionsspektren in Lösungen und die Nebenvalenzen als ihre Ursache. Angew. Chem 1937, 50 (11), 212–219. [Google Scholar]
- (34).Scheibe G; Kandler L; Ecker H Polymerisation and Polymere Adsorption as a Cause of Novel Absorption Bands of Organic Pigments. Naturwissenschaften 1937, 25, 75–75. [Google Scholar]
- (35).Scheibe G; Mareis A; Ecker H The Reversible Polymer-isation as a Cause of Unusual Absorption Bands III. Naturwissenschaften 1937, 25, 474–475. [Google Scholar]
- (36).Stiel H; Teuchner K; Becker W; Freyer W; Dahne S Flurescence Lifetime Studies of Pseudoisocyaniny J-Aggregates in the Subnanosecond Range. J. Mol. Struct 1984, 114 (MAR), 351–354. [Google Scholar]
- (37).Fidder H; Knoester J; Wiersma DA Superradiant Emission and Optical Dephasing in J-aggregates. Chem. Phys. Lett 1990, 171 (5–6), 529–536. [Google Scholar]
- (38).Eisfeld A; Briggs JS The J- and H-Bands of Organic Dye Aggregates. Chem. Phys 2006, 324 (2–3), 376–384. [Google Scholar]
- (39).Eisfeld A; Briggs JS The Shape of the J-Band of Pseudoisocyanine. Chem. Phys. Lett 2007, 446 (4–6), 354–358. [Google Scholar]
- (40).Spano FC The Spectral Signatures of Frenkel Polarons in H-and J-Aggregates. Acc. Chem. Res 2010, 43 (3), 429–439. [DOI] [PubMed] [Google Scholar]
- (41).Wuerthner F; Kaiser TE; Saha-Moeller CR J-Aggregates: From Serendipitous Discovery to Supramolecular Engineering of Functional Dye Materials. Angew. Chem., Int. Ed 2011, 50 (15), 3376–3410. [DOI] [PubMed] [Google Scholar]
- (42).Li Z. a.; Mukhopadhyay S; Jang S-H; Bredas J-L; Jen AKY Supramolecular Assembly of Complementary Cyanine Salt J-Aggregates. J. Am. Chem. Soc 2015, 137 (37), 11920–11923. [DOI] [PubMed] [Google Scholar]
- (43).Pajusalu M; Raetsep M; Trinkunas G; Freiberg A Davydov Splitting of Excitons in Cyclic Bacteriochlorophyll a Nanoaggregates of Bacterial Light-Harvesting Complexes between 4.5 and 263 K. ChemPhysChem 2011, 12 (3), 634–644. [DOI] [PubMed] [Google Scholar]
- (44).v. Berlepsch H; Bottcher C Supramolecular Structure of TTBC J-Aggregates in Solution and on Surface. Langmuir 2013, 29(16), 4948–4958. [DOI] [PubMed] [Google Scholar]
- (45).Monshouwer R; Abrahamsson M; vanMourik F; vanGrondelle R Superradiance and Exciton Delocalization in Bacterial Photosynthetic Light-Harvesting Systems. J. Phys. Chem. B 1997, 101 (37), 7241–7248. [Google Scholar]
- (46).Dahlbom M; Pullerits T; Mukamel S; Sundstrom V Exciton Delocalization in the B850 Light-Harvesting Complex: Comparison of Different Measures. J. Phys. Chem. B 2001, 105 (23), 5515–5524. [Google Scholar]
- (47).Scholes GD; Rumbles G Excitons in Nanoscale Systems. Nat. Mater 2006, 5 (9), 683–696. [DOI] [PubMed] [Google Scholar]
- (48).Blancafort L; Voityuk AA Exciton Delocalization, Charge Transfer, and Electronic Coupling for Singlet Excitation Energy Transfer Between Stacked Nucleobases in DNA: An MS-CASPT2 Study. J. Chem. Phys 2014, 140 (9), 095102. [DOI] [PubMed] [Google Scholar]
- (49).Scholes GD; Ghiggino KP; Oliver AM; Paddonrow MN Through-Space and Through-Bond Effects on Exciton Interactions in Rigidly Linked Dinaphthyl Molecules. J. Am. Chem. Soc 1993, 115(10), 4345–4349. [Google Scholar]
- (50).Saikin SK; Eisfeld A; Valleau S; Aspuru-Guzik A Photonics Meets Excitonics: Natural and Artificial Molecular Aggregates. Nanophotonics 2013, 2 (1), 21–38. [Google Scholar]
- (51).Schroter M; Ivanov SD; Schulze J; Polyutov SP; Yan Y; Pullerits T; Kühn O Exciton-Vibrational Coupling in the Dynamics and Spectroscopy of Frenkel Excitons in Molecular Aggregates. Phys.Rep 2015, 567, 1–78. [Google Scholar]
- (52).Probst M; Langenegger SM; Haner R A Modular LHC Built on the DNA Three-Way Junction. Chem. Commun 2014, 50 (2), 159–161. [DOI] [PubMed] [Google Scholar]
- (53).Markova LI; Malinovskii VL; Patsenker LD; Haner R Synthesis and Properties of Squaraine-Modified DNA. Org. Biomol. Chem 2012, 10 (45), 8944–8947. [DOI] [PubMed] [Google Scholar]
- (54).Malinovskii VL; Wenger D; Haner R Nucleic Acid-Guided Assembly of Aromatic Chromophores. Chem. Soc. Rev 2010, 39 (2), 410–422. [DOI] [PubMed] [Google Scholar]
- (55).Li SG; Langenegger SM; Haner R Control of Aggregation-Induced Emission by DNA Hybridization. Chem. Commun 2013, 49(52), 5835–5837. [DOI] [PubMed] [Google Scholar]
- (56).Haner R; Samain F; Malinovskii VL DNA-Assisted Self-Assembly of Pyrene Foldamers. Chem. - Eur. J 2009, 15 (23), 5701–5708. [DOI] [PubMed] [Google Scholar]
- (57).Garo F; Haner R A DNA-Based Light-Harvesting Antenna.Angew. Chem., Int. Ed 2012, 51 (4), 916–919. [DOI] [PubMed] [Google Scholar]
- (58).Adeyemi OO; Malinovskii VL; Biner SM; Calzaferri G; Haner R Photon Harvesting by Excimer-Forming Multichromophores. Chem. Commun 2012, 48 (77), 9589–9591. [DOI] [PubMed] [Google Scholar]
- (59).Kashida H; Asanuma H Preparation of Supramolecular Chromophoric Assemblies using a DNA Duplex. Phys. Chem. Chem. Phys 2012, 14 (20), 7196–7204. [DOI] [PubMed] [Google Scholar]
- (60).Wei B; Dai MJ; Yin P Complex Shapes Self-Assembled from Single-Stranded DNA Tiles. Nature 2012, 485 (7400), 623–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Rothemund PWK; Andersen ES Nanotechnology: The Importance of Being Modular. Nature 2012, 485 (7400), 584–585. [DOI] [PubMed] [Google Scholar]
- (62).Ke YG; Ong LL; Shih WM; Yin P Three-Dimensional Structures Self-Assembled from DNA Bricks. Science 2012, 338 (6111), 1177–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Wang MM; Silva GL; Armitage BA DNA-Templated Formation of a Helical Cyanine Dye J-Aggregate. J. Am. Chem. Soc 2000, 122 (41), 9977–9986. [Google Scholar]
- (64).Kashida H; Asanuma H; Komiyama M Alternating Hetero H Aggregation of Different Dyes by Interstrand Stacking from Two DNA-Dye Conjugates. Angew. Chem., Int. Ed 2004, 43 (47), 6522–6525. [DOI] [PubMed] [Google Scholar]
- (65).Kashida H; Tanaka M; Baba S; Sakamoto T; Kawai G; Asanuma H; Komiyama M Covalent Incorporation of Methyl Red Dyes into Double-Stranded DNA for Their Ordered Clustering. Chem.- Eur. J 2006, 12 (3), 777–784. [DOI] [PubMed] [Google Scholar]
- (66).Fujii T; Kashida H; Asanuma H Analysis of Coherent Heteroclustering of Different Dyes by Use of Threoninol Nucleotides for Comparison with the Molecular Exciton Theory. Chem. - Eur. J 2009, 15 (39), 10092–10102. [DOI] [PubMed] [Google Scholar]
- (67).Probst M; Wenger D; Biner SM; Haner R The DNA Three-Way Junction as a Mould for Tripartite Chromophore Assembly. Org. Biomol. Chem 2012, 10 (4), 755–759. [DOI] [PubMed] [Google Scholar]
- (68).Cannon BL; Kellis DL; Patten LK; Davis PH; Lee J; Graugnard E; Yurke B; Knowlton WB Coherent Exciton Delocalization in a Two-State DNA-Templated Dye Aggregate System. J. Phys. Chem. A 2017, 121 (37), 6905–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Seeman NC Nucleic Acid Junctions and Lattices. J. Theor. Biol 1982, 99, 237–247. [DOI] [PubMed] [Google Scholar]
- (70).Kallenbach NR; Ma R-I; Seeman NC An Immobile Nucleic Acid Junction Constructed from Oligonucleotides. Nature 1983, 305, 829. [Google Scholar]
- (71).Tomita G Absorption and Fluorescence Properties of Some Basic Dyes Complexing with Nucleosides or Nucleic Acids. Z. Naturforsch., B: J. Chem. Sci 1968, B23 (7), 922. [DOI] [PubMed] [Google Scholar]
- (72).Kühn O; Renger T; May V Theory of Exciton-Vibrational Dynamics in Molecular Dimers. Chem. Phys 1996, 204 (1), 99–114. [Google Scholar]
- (73).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem 2004, 25 (13), 1605–1612. [DOI] [PubMed] [Google Scholar]
- (74).Couch GS; Hendrix DK; Ferrin TE Nucleic Acid Visualization with UCSF Chimera. Nucleic Acids Res 2006, 34 (4), e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Yurke B; Turberfield AJ; Mills AP; Simmel FC; Neumann JL A DNA-Fuelled Molecular Machine Made of DNA. Nature 2000, 406 (6796), 605–608. [DOI] [PubMed] [Google Scholar]
- (76).Srinivas N; Ouldridge TE; Sulc P; Schaeffer JM; Yurke B; Louis AA; Doye JPK; Winfree E On the Biophysics and Kinetics of Toehold-Mediated DNA Strand Displacement. Nucleic Acids Res 2013, 41 (22), 10641–10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.