

Abstract
FOXM1 transcription factor network is activated in over 84% of cases in high-grade serous ovarian cancer (HGSOC), and FOXM1 upregulates the expression of genes involved in the homologous recombination (HR) DNA damage and repair (DDR) pathway. However, the role of FOXM1 in PARP inhibitor response has not yet been studied. This study demonstrates that PARP inhibitor (PARPi), olaparib, induces the expression and nuclear localization of FOXM1. On the basis of ChIP-qPCR, olaparib enhances the binding of FOXM1 to genes involved in HR repair. FOXM1 knockdown by RNAi or inhibition by thiostrepton decreases FOXM1 expression, decreases the expression of HR repair genes, such as BRCA1 and RAD51, and enhances sensitivity to olaparib. Comet and PARP trapping assays revealed increases in DNA damage and PARP trapping in FOXM1-inhibited cells treated with olaparib. Finally, thiostrepton decreases the expression of BRCA1 in rucaparib-resistant cells and enhances sensitivity to rucaparib. Collectively, these results identify that FOXM1 plays an important role in the adaptive response induced by olaparib and FOXM1 inhibition by thiostrepton induces “BRCAness” and enhances sensitivity to PARP inhibitors.
Implications:
FOXM1 inhibition represents an effective strategy to overcome resistance to PARPi, and targeting FOXM1-mediated adaptive pathways may produce better therapeutic effects for PARP inhibitors.
Introduction
The primary cause of cancer-related mortality is the treatment failure resulting from intrinsic or acquired resistance to chemotherapy (1–3). In epithelial ovarian cancer, although most patients initially respond to chemotherapy, they experience recurrences and the acquired resistance to chemotherapy (4). Consequently, epithelial ovarian cancer is the most lethal gynecologic malignancies in the United States (5). Although overall survival from ovarian cancer has improved slowly over the past three decades (6), recent advances in PARP inhibitors as maintenance therapies are having positive impacts on the overall survival of patients with ovarian cancer (7). Nonetheless, acquired resistance to PARP inhibitors are being reported (8,9), and it is important to understand molecular mechanisms that contribute to acquired resistance to chemotherapeutic agents.
Intrinsic and acquired resistance to chemotherapeutic agents can be explained by the Darwinian selection of fitness-conferring genetictraits under drug treatment (10). Under this principle, cells with preexisting mutations that confer fitness under drug treatment are selected, thereby contribute to the development of resistance to treatment. Consistent with this principle, low-level revertant mutations in BRCA1 and BRCA2 are found in ovarian carcinoma samples prior to platinum-based chemotherapy, and these rare mutations become enriched in carcinoma samples from the corresponding patients during relapse (11), suggesting the selection of preexisting fitness-conferring somatic mutations by chemotherapy. Although this principle explains intrinsic resistance, it cannot fully explain the acquired resistance.
With respect to the acquired resistance in ovarian cancer, patients generally respond to platinum-based chemotherapy even after relapse from prior platinum-based chemotherapy (4). Recent evidence suggests that adaptive cellular response may provide a transitional state that allows cells to acquire fitness-conferring genetic mutations after several rounds of treatment with chemotherapeutic agents (12). A nongenetic Lamarckian mechanism of drug-induced adaptive response has been proposed as a possible mechanism for the acquisition of resistance (13). The “transient adaptive resistance” allows cells to be in “chemotherapy-tolerant state,” thereby producing “persisters” (12, 14). Extracellular matrix and tumor microenvironment have been shown to provide such transient adaptive resistance to cancer cells (15). These “persisters” subsequently acquire fitness-conferring genetic and epigenetic alterations that promote resistance to chemotherapy (16). Consistent with this concept, recent studies indicate that in vitro selection with PARP inhibitor rucaparib in MDA-MB-436 breast cancer cells resulted in resistant clones that overexpressed mutant BRCA1 at higher levels than in drug-sensitive parental MDA-MB-436 (9). In this model, epigenetic rather than genetic alterations contribute to the acquired resistance to rucaparib. In the population of cells without preexisting genetic alterations that confer fitness under drug treatment, the adaptive cellular response may represent a critical step prior to the acquisition of acquired resistance. Therefore, adaptive cellular responses may be targeted to overcome acquired resistance to chemotherapeutic agents.
A critical step in the development of effective combination therapies to extend the efficacy of existing chemotherapeutic agents is to understand the molecular mechanisms regulating the adaptive cellular responses to existing chemotherapeutic agents. According to the landmark study by the Cancer Genome Atlas (TCGA), the FOXM1 pathway is activated in approximately 84% of high-grade serous ovarian carcinomas (17). FOXM1 regulates the expression of DNA repair genes (18) as well as genes involved in adaptive response to cellular stress induced by oxidative stress and oncogenic stress (19). However, the extent to which FOXM1 pathway contributes to the adaptive cellular response to chemotherapy and the extent to which it represents an important epigenetic molecular mechanism regulating the adaptive cellular response to chemotherapy are not yet characterized.
In this study, we identified FOXM1 pathway as a component of the adaptive cellular response to PARP inhibitor olaparib. We found that olaparib induced FOXM1 expression that regulates several genes involved in homology recombination repair pathway. RNAi or FOXM1 inhibitor thiostrepton decreased FOXM1 expression and attenuated the adaptive cellular response leading to enhanced sensitivity to olaparib and carboplatin. Finally, our results showed that FOXM1 inhibitor thiostrepton decreases the expression of BRCA1 and BRCA2, produces “BRCAness,” and enhances sensitivity to olaparib. Our results support an emerging paradigm that adaptive cellular responses may be targeted to prevent acquired resistance to chemotherapeutic agents and indicate that FOXM1 pathway may be targeted to prevent acquired resistance to PARP inhibitors.
Materials and Methods
Cell lines and cell culture
ES-2, OVCAR3, and A2780 cells were maintained inMCDB105 and M199 (1:1; Sigma, USA) containing 5% FBS (Sigma), OV90 cells were maintained in MCDB105 and M199 (1:1) with 15% FBS. OVCA420* cells were cultured in DMEM (Sigma and Caisson Labs) supplemented with 10% FBS. ONCO-DG1 cells were grown in RPMI1640 (Sigma and Caisson Labs) with 10% FBS. MDA-MB-436 and its derivative rucaparib-resistant cells RR-1, RR-2, RR-3 were kind gifts from Dr. Neil Johnson laboratory at Fox Chase Cancer Center (Philadelphia, PA; ref. 9) and were maintained in RPMI1640 (Sigma and Caisson Labs) supplemented with 10% FBS. All the media were supplemented with 100 U/MI penicillin and 100 mg/mL streptomycin. ES-2, OVCAR3, OVCA420*, and OV90 cells were gifts from Dr. Viji Shridhar (Mayo Clinic, Rochester, MN). A2780 cell was from Dr. Andrew Godwin (The University of Kansas Medical Center, Kansas City, KS). ONCO-DG1 was purchased from Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures. All cell lines were subjected to cell line identity confirmation. All experiments performed on cells that were passaged less than 20 times. Mycoplasma testing was performed during the studies, and cell cultures were free of mycoplasma. Cell line identification was performed at the end of experiments. OV90 and ONCO-DG1 showed 100% short tandem repeat (STR) profiles matching to corresponding cell lines reported in ATCC or ExPASy. STR profiles for ES-2 and OVCAR3 were performed in 2014 as a supplement to our recent publication (20). STR profiles for OVCA420* does not match with any reported cell lines in ATCC, ExPASy, DSMZ, or CLIMA, and therefore we placed an asterisk to differentiate it from the original cell line.
Antibodies and compounds
Rabbit polyclonal anti-FOXM1 antibody (C-20, sc-502), rabbit polyclonal anti-BRCA1 antibody (C-20, sc-642), mouse monoclonal anti-FANCF antibody (D-2, sc-271952) were purchased from Santa Cruz Biotechnology. Rabbit polyclonal anti-Histone H3 antibody (ab1791) and rabbit monoclonal anti-b-Tubulin antibody (ab108342) were obtained from Abcam. Mouse monoclonal anti-RAD51 antibody (5B3/6, GTX23638), rabbit polyclonal anti-Histone H3 antibody (GTX122148), and rabbit polyclonal anti-BRCC3 antibody (GTX31765) were from GeneTex. Rabbit polyclonal anti-PARP antibody (9542S) and rabbit monoclonal anti-caspase-3 antibodies (9665S) were purchased from Cell Signaling Technology. Mouse monoclonal anti-Poly (ADP-ribose) antibody (PAR, 10H, ALX-804–220-R100) was obtained from Enzo Life Sciences (Alexis). Mouse monoclonal anti-β-actin antibody (A1978) was from Sigma-Aldrich. For secondary antibodies, horse anti-mouse IgG-HRP antibody (7076S) was purchased from Cell Signaling Technology, goat anti-rabbit IgG-HRP antibody (sc-2030) was from Santa Cruz Biotechnology.
Olaparib (AZD2281, Ku-0059436) was purchased from Selleckchem. Olaparib stock solutions were made with DMSO at 50 mmol/L and stored at –80°C. Thiostrepton powder was purchased from Santa Cruz Biotechnology (sc-203412A) and formulated as micelle-encapsulated thiostrepton (see Supplementary Information; Supplementary Figs. S9 and S10; Supplementary Table S1) by Dr. Laird Forrest (School of Pharmacy, University of Kansas, Lawrence, KS).
Immunoblotting
Cells were washed at least twice with PBS at the end of treatments if applicable and then lysed with an appropriate volume of 1 × electrophoresis sample buffer (Bio-Rad Laboratories) with 5% β-mercaptoethanol (Sigma-Aldrich). The cell lysates were then boiled at 95°C for 5 minutes before using. Immunoblotting procedures were performed as described previously (20). For apoptosis marker check, cells were collected at the end of treatments, and total proteins were extracted using RIPA buffer (1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS in 1 × PBS) containing protease/phosphatase inhibitor cocktail (Roche). BCA protein assay reagent kit (Pierce) was used to determine protein concentrations. Equal amounts of total proteins were loaded for SDS-PAGE and transferred onto polyvinylidene difluoride membranes (GE Healthcare). For nuclear fractionation assays, cell pellets were collected and lysed with cytoplasmic extraction buffer (CEB) in Subcellular Protein Fractionation Kit (78840, Thermo Scientific) and then centrifuged at 500 × g for 5 minutes, the supernatant was labeled as the cytoplasmic extract, while the pellets were further lysed in RIPA buffer. Protein concentration was determined by the BCA assay and equal amounts of proteins were loaded for Western blot analysis. The densitometric analysis was performed with Image J software (NIH, Bethesda, MD).
PARP trapping assay
A total of 4.5 × 105 cells were treated with the appropriate drug(s) for 4 hours before collection. The cell pellets were fractionated using Subcellular Protein Fractionation Kit according to the manufacturer’s instructions and subjected to immunoblotting.
ChIP-qPCR
Chromatin immunoprecipitation (ChIP) was carried out as described before (21). Briefly, after20 μmol/L olaparib treatment for 12 hours or 24 hours, cells were crosslinked with 1% formaldehyde (Electron Microscopy Sciences) for 10 minutes and quenched by cross-linking by glycine. The chromatin was sonicated with a BioruptorTwin (Diagenode) at maximum setting for 12 minutes. The sonicated chromatin with incubated with 1.0 μg FOXM1 antibody (C-20, sc-502, Santa Cruz Biotechnology) at 4°C for 2–4 hours before purification with 100 μL Protein A/G magnetic beads (88803, Pierce Biotechnology). The beads were washed 5 times with LiCl wash buffer [100 mmol/LTris pH 7.5, 500 mmol/L LiCl, 1% NP-40, 1% sodium deoxycholate before washing with 1 × TE buffer (10 mmol/L Tris-HCl pH7.5 and 0.1 mmol/L Na2EDTA)] and eluted with Elution Buffer (1% SDS and 0.1 mol/L NaHCO3). After reverse-crosslinking, the DNA was purified with the QIAQuick PCR Cleanup Kit (Qiagen) and used for qPCR, which was performed on a CFX384 Touch Real-Time PCR Detection System (Bio-Rad) using RT2 SYBR Green qPCR Mastermix (Qiagen).
Real-time quantitative PCR
The total RNA was extracted with the TRIzol reagent (15596–028, Invitrogen) according to the manufacturer’s manual. The cDNA was synthesized using SuperScript II reverse transcriptase (180604014, Invitrogen) with 1 μg of total RNA in a 20 μL reaction. The resulting cDNA was diluted 1:20 in nuclease-free water and 1 μL was used per qPCR reaction with triplicates. qPCR was carried out using Power SYBR Green PCR Master Mix (4367659, Thermo Fisher Scientific) on aCFX384 Real-Time PCR Detection System (Bio-Rad) including a nontemplate negative control. Amplification of GAPDH or 18S rRNA was used to normalize the level of mRNA expression. The sequences of the primer pairs were listed in Supplementary Table S2.
siRNA transfection
FOXM1 -specific siRNAs and scrambled negative control siRNAs were synthesized by Integrated DNA Technologies. A total of 3.5 × 105 cells/well were seeded in 6-well plates and incubated at 37° C overnight. Next day, 20 nmol/L of each siRNA was transfected into the cells with Oligofectamine Transfection Reagent (12252011, Invitrogen) according to the manufacturer’s instructions. Culture media was added 6–8 hours after transfection without washing cells. Forty-eight hours after transfection, the transfected cells were trypsinized and seeded on 96-well plates (for cytotoxicity assay) or 6-well plates (for colony formation assay). Drugs were added around 12 hours after seeding. For cytotoxicity assay, cells were incubated with drug for 3 days before measurement of cell viability using Sulforhodamine B assay. For colony formation assay, cells were exposed to drugs for 3 days and then changed to fresh media without drug until colonies formed and stained with Sulforhodamine B for imaging. To check the downregulation of FOXM1 expression, the transfected cells were collected to extract total RNA for qRT-PCR or proteins for Western blot analysis 72 hours after transfection. The sequences of siRNAs used are listed below:
FOXM1 siRNA#1: sense rGrUrGrCrCrArArCrCrGrCrUrArCrUr-UrGrArCrArUrUrGGA, antisense rUrCrCrArArUrGrUrCrArArGrUr-ArGrCrGrGrUrUrGrGrCrArCrUrG;
FOXM1 siRNA#2: sense rGrCrGrCrUrArUrUrArGrArUrGrUr-UrUrCrUrCrUrGrArUAA, antisense rUrUrArUrCrArGrArGrArAr-ArCrArUrCrUrArArUrArGrCrGrCrArC.
Cytotoxicity assay using Sulforhodamine B (SRB) and drug synergy studies:
SRB assays were performed as described previously (22, 23) with modifications shown below. For OVCA420*, OV90, ES-2, ONCO-DG1, and OVCAR3 cells, 3,000 cells/well were seeded in 96-well plates and treated with drugs at least 12 hours after seeding. Then, the cells were incubated for another 3 days. For MDA-MB-436 rucaparib-resistant cells RR-1, RR-2, and RR-3, 5,000 cells were seeded and incubated for 5 days after drug treatment. Dose-response curves were fitted and the IC50 for each drug was determined using GraphPad Prism 6 four parameters. All curves were constrained with 100% on top. Synergy was determined by calculating the combination index (CI) obtained from the plate reading. CI was calculated on the basis of dividing the expected effect by the observed effect.
Colony formation assay
For OVCA420*, OV90, and ES-2, 1,000 cells were seeded in 6-well plates. For MDA-MB-436 rucaparib-resistant cells RR-1 and RR-2, 2,000 or 3,000 cells were seeded per well in 6-well plates. The cells were treated with drugs at least 12 hours after seeding and further incubated for another 3 days before changing to fresh media. The medium was changed every 2–3 days to allow colonies to form. At the end of experiments, SRB assay was performed to stain the colonies which were imaged with Molecular Imager ChemiDoc MP System (Bio-Rad). The colonies were further dissolved and measured with a plate reader. Analysis of colonies was performed in GraphPad Prism 6.
Caspase-3 activity assay
Caspase-3 activity assay was performed as described previously (23). Briefly, 4 × 105 cells/well were seeded in 6-well plates and incubated overnight. The next day, cells were treated with appropriated drugs and incubated for 30 hours. Cells were then collected using a cell lifter and lysed in caspase buffer [pH 7.2, 20 mmol/L PIPES, 100 mmol/L NaCl, 1 mmol/L EDTA (pH 8.0), 0.1% (w/v) CHAPS, 10% sucrose and 10 mmol/L DTT] and quantified with BCA assay. Twenty micrograms of total protein were used to combine with 2 μL of 2 mmol/L DEVD-Afc (Millipore) in 96-well flat-bottom plates and added 200 μL/well caspase buffer. The plate was covered and incubated war 37°C for 2 hours before measuring fluorescence at excitation of 400 nm and emission of 510 nm. Measurements were analyzed using Graph-Pad Prism 6.
Alkaline comet assay
We used the CometChip Electrophoresis Starter Kit (TREVI-GEN, 4260–096-ESK) to perform alkaline comet assay according to the manufacturer’s instructions. For thiostrepton and olaparib combination experiment, cells were treated with vehicle or thiostrepton for 4 hours before seeding onto equilibrated 96-well CometChip. Then 100 μL/well of fresh culture media containing appropriate concentrations of both thiostrepton and olaparib was added to CometChip followed by another 4-hour incubation at 37°C, and alkaline comet assay was performed following the product instruction. For FOXM1 siRNA transient knockdown experiment, cells were transfected with scrambled siRNA or FOXM1 siRNA and waited 72 hours before seeding onto CometChip. Cells were then incubated with vehicle or olaparib for 4 hours at 37° C before alkaline comet assay. Comets were analyzed with Trevigen Comet Analysis Software after imaging under a 4× fluorescent microscope.
cDNA microarray analysis
cDNA microarray data from MCF7 cells treated with or without thiostrepton for 6 hours was downloaded from the Connectivity Map project and analyzed with BRB-ArrayTools (ver. 4.5.1; ref. 24). A total of 12 datasets, representing two batches of treatments, were downloaded (Supplementary Table S3). After normalization with JustRMA protocol within BRB-ArrayTools, 5813 genes passed filtering criteria, such as minimum fold change (<20% of expression values have at least a 1.5-fold change in either direction from gene’s median value), and percent missing (exceeds 50%). Subsequently, the class comparison between paired groups of arrays was performed to identify differentially expressed genes between two groups (thiostrepton vs. DMSO). A total of 716 genes were significantly changed between two groups. A cluster of 716 genes was performed using Dynamic Heatmap Viewer within BRB-ArrayTools, and select genes involved in DNA repair and apoptosis were highlighted. Functional annotation and enrichment analysis were performed using Metascape and DAVID bioinformatics tools (25, 26).
Statistical analysis
All data were analyzed using GraphPad Prism 6. Results were expressed as means ± SEM. Differences between treatment regimens were analyzed by one-way ANOVA or two-tailed Student t test. P ≤ 0.05 was considered to be statistically significant.
Results
Olaparib induces the expression of FOXM1 and HR repair genes
To establish the potential role of FOXM1 in the adaptive cellular response induced by olaparib, we treated ovarian cancer cells ES-2 and OVCA420* with olaparib and determined the expression of FOXM1, BRCA1, and RAD51 at different time points. We observed induction of FOXM1 expression by olaparib within 6 hours of treatment (Fig. 1A). In addition to the upregulation of FOXM1 expression, the increased nuclear location of FOXM1 is observed within 3 hours of olaparib treatment (Fig. 1B), indicative ofFOXM1 pathway activation. Concomitant with FOXM1 pathway activation, we observed increased expression of BRCA1 andRAD51 in these cells (Fig. 1C; Supplementary Fig. S1). Consistent with the increase in FOXM1 nuclear localization, FOXM1 binding to promoter regions of its target genes, such as BRCA1, RAD51, FANCF, RAD51D, and FANCD2, increased at 12 hours and 24 hours after olaparib treatment (Fig. 1D). These data suggest that FOXM1 plays an important role in adaptive cellular response to olaparib treatment in ovarian cancer cells.
Figure 1.
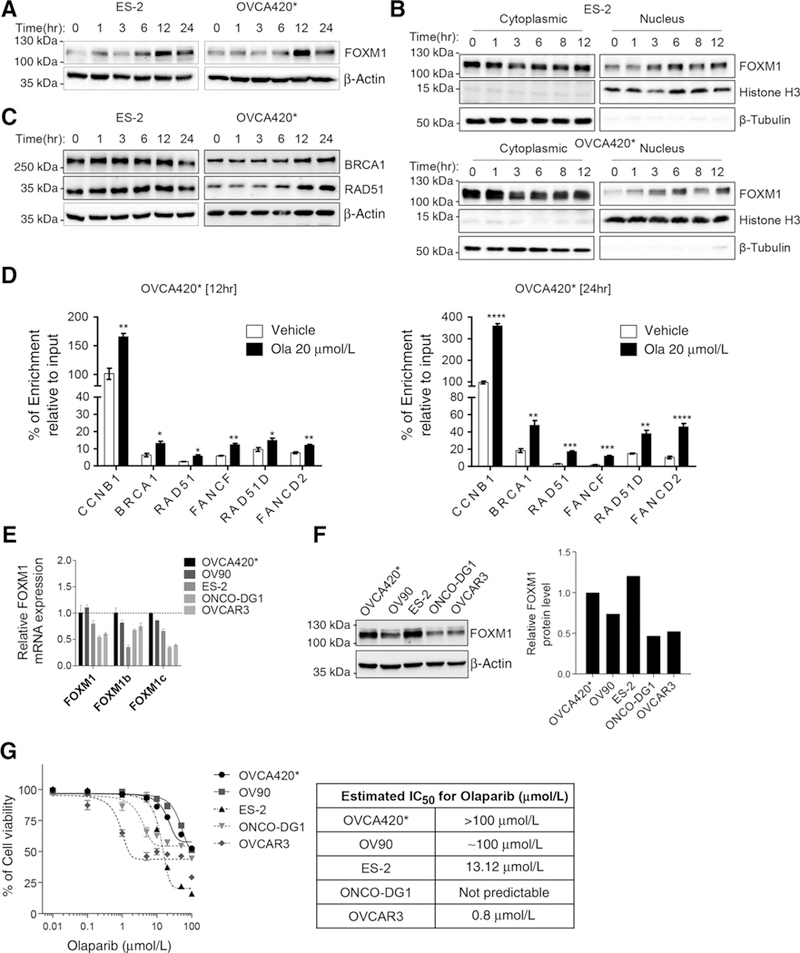
Olaparib increases FOXM1 expression and FOXM1 levels inversely correlate with olaparib sensitivity. A, Olaparib induces FOXM1 expression. The ES-2 and OVCA420* cells were treated with 10 μmol/L olaparib for 0,1,3, 6,12, or 24 hours and subjected to immunoblotting with the FOXM1 antibody. β-Actin immunoblot is used as a loading control. B, Olaparib enhances nuclear localization of FOXM1. Cells were treated with 10 μmol/L olaparib for 0,1, 3, 6, 8, or 12 hours before subcellular fractionation followed by Western blot analysis of FOXM1 in nuclear and cytoplasmic fractions. β-Tubulin and Histone H3 were used as loading controls for cytoplasmic and nuclear protein, respectively. C, Olaparib increases the expression of BRCA1 and RAD51. The whole-cell lysates were prepared from both cell lines after exposure of 10 μmol/L olaparib for 0,1, 3, 6,12, or 24 hours and blotted with BRCA1 or RAD51 antibodies. D, Olaparib increases FOXM1 occupancy at promoter regions of its target genes. OVCA420* cells were treated with 20 μmol/L olaparib for 12 hours or 24 hours before ChIP analysis using FOXM1 antibody. Results are representative of at least three experiments (A–D). E, Quantification of FOXM1 mRNA and its isoforms (isoform b and c) in multiple ovarian cancer cell lines by qRT-PCR. Data were shown as mean ± SEM. F, Measurement of FOXM1 protein levels by immunoblotting in different ovarian cancer cells. The whole-cell lysates were used for immunoblotting with the FOXM1 antibody. The Densitometry analysis was performed to quantify FOXM1 protein levels. Data was shown from a representative experiment. G, Measurement of olaparib sensitivity in ovarian cancer cells by Sulforhodamine B (SRB) cell viability assay. A total of 3,000 cells were seeded in 96-well plates and treated with increasing concentrations of olaparib for 72 hours before SRB assay. Data were shown as mean ± SEM (n = 3–4). Results were average of 3–4 independent experiments with triplicates. The table shows estimated IC50 values for olaparib in ovarian cancer cell lines. IC50 values were extrapolated from the curve using GraphPad Prism 6 software. The cells baring curves that cannot accurately extrapolate IC50 values are shown as “not predictable.” The statistics analysis was performed with two-tail Student t test: *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
FOXM1 expression is higher in cancer cells that are less responsive to olaparib
To determine the extent to which FOXM1 expression may be correlated with olaparib sensitivity, we assessed FOXM1 expression by qRT-PCR, the Western blot analysis, and olaparib sensitivity by SRB assay. Although all three forms of FOXM1 transcripts are expressed (Fig. 1E; Supplementary Fig. S2A), protein expression is reflected better by the levels of a FOXM1C transcript (Fig. 1F). Consistent with these results, analysis of the Cancer Genome Atlas ovarian cancer dataset indicates FOXM1C transcript is the most abundant among the three isoforms (Supplementary Fig. S2B), which is consistent with a report from Tassi and colleagues (18). The dose-response curves for olaparib sensitivity in these cell lines indicate that OVCA420* and OV90 with high expression of FOXM1 are less responsive to olaparib (Fig. 1G). Taken together, these results suggest a potential role of FOXM1 in olaparib sensitivity in ovarian cancer cells.
FOXM1 knockdown results in enhanced sensitivity to olaparib
To determine the extent to which FOXM1 expression contributes to olaparib sensitivity, we used two different siRNAs to downregulate FOXM1 expression. Although both siRNAs down-regulate all three isoforms of FOXM1 transcripts, FOXM1B was affected more by these siRNAs (Fig. 2A). Interestingly, FOXM1 expression was markedly decreased at protein levels, indicating that these siRNAs also affected the translation of FOXM1 protein (Fig. 2B), consistent with the translational repression of certain siRNAs (27). SRB assays indicate that cells with decreased FOXM1 expression are significantly more sensitive to olaparib (Fig. 2C; Supplementary Fig. S3A and S3B). Similarly, we observed a significant decrease in clonogenic survival in FOXM1 knocked-down cells treated with 4, 10, and 25 μmol/L olaparib compared with scrambled control siRNA (scr siRNA; Fig. 2D). Next, we used pharmacologic means to inhibit FOXM1 expression and determined the extent to which FOXM1 inhibition results in enhanced sensitivity to olaparib. Thiostrepton is a FOXM1 inhibitor, and it downregulates FOXM1 expression (20, 28). In three different ovarian cancer cell lines, we observed synergistic interactions between thiostrepton and olaparib at the concentrations tested in clonogenic assays (Fig. 2E and F) and mild synergistic activity in SRB assays (Supplementary Fig. S3C). Similarly, we observed synergistic interactions between thiostrepton and olaparib or carboplatin in other cancer cell lines (Supplementary Fig. S3D–S3F). Collectively, these results indicate FOXM1 inhibition enhances sensitivity to olaparib and carboplatin.
Figure 2.
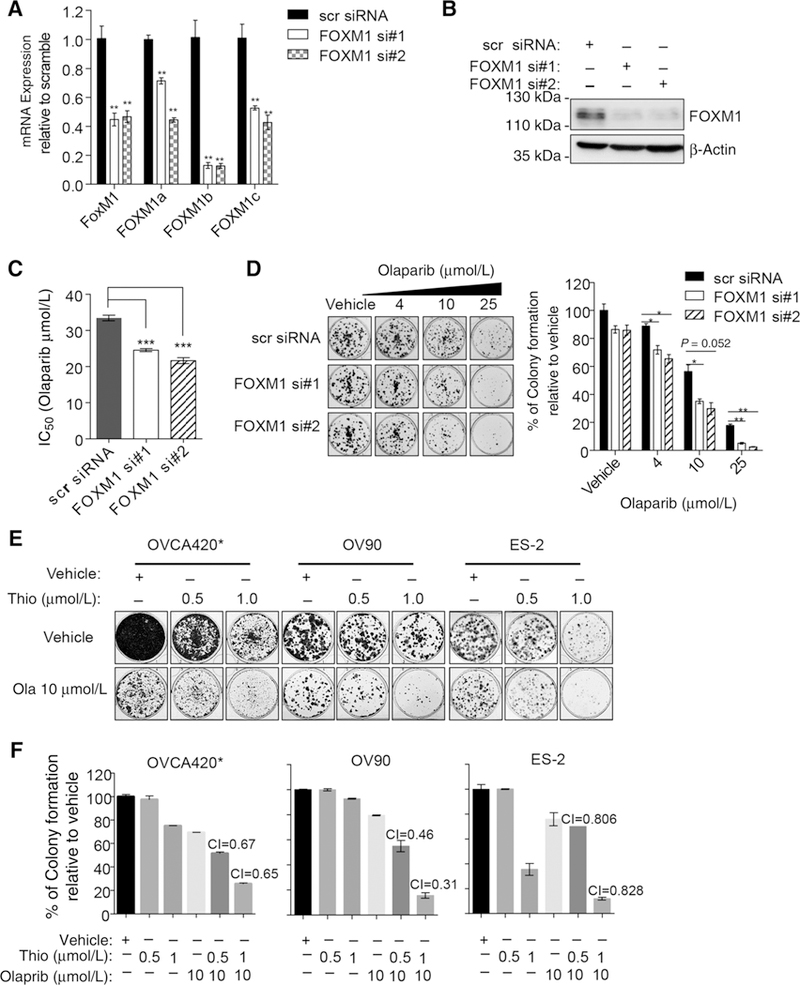
FOXM1 inhibition by either siRNA or thiostrepton sensitizes cancer cells to olaparib and decreases the number of persisting clones. A and B, Verification of FOXM1 knockdown by two different siRNAs. ES-2 cells were transfected with scrambled (scr) siRNA, FOXM1 siRNA1 (si#1), or siRNA2 (si#2) and RT-qPCR (A) and immunoblotting (B) were performed after 72 hours. **, P ≤ 0.01. Data were representatives from three independent experiments. C, Estimated IC50 values of olaparib in ES-2 cells after FOXM1 depletion. The IC50 values from SRB cell viability assays were extrapolated using Prism 6 software, and the data were shown as mean ± SEM. ***, P ≤ 0.001. Results were derived from triplicates of representative experiments. D, FOXM1 knockdown decreases persisting clones in ES-2 following olaparib treatment. Forty-eight hours after siRNA transfection, cells were seeded in 6-well plates and treated with different concentrations of olaparib for 3 days. Colonies produced by persisting cells were stained with SRB and solubilized after imaging. Percentage of colony formation relative to the vehicle was shown in a bar graph. *, P ≤0.05; **, P ≤0.01. E and F, Thiostrepton synergizes with olaparib in inhibiting the colony formation of OVCA420*, OV90, and ES-2 cells. Cells were treated with thiostrepton and olaparib alone or in combination for three days followed by colony formation around 2 weeks. Colonies were stained with SRB and imaged before solubilized in Tris buffer to measure fluorescence intensity. Relative colony formation from E was quantified, and the combination indexes (CI) were calculated. Results represent experiments performed in duplicates (D-F).
Thiostrepton decreases the expression of DNA repair genes and increases the expression of proapoptotic genes
To better characterize the effect of thiostrepton and define the molecular pathways affected by thiostrepton, we analyzed the publicly available dataset from the Connectivity Map project where cancer cells were treated with thiostrepton for 6 hours and drug-perturbed transcriptomes were profiled with array-based gene expression analysis (29). The Metascape analysis (25) of genes that are downregulated by thiostrepton at P ≤ 0.001 indicates these genes are associated with mitotic cell cycle, G1–S phase transition, and FOXM1 pathway (Supplementary Fig. S4A). On the other hand, the Metascape analysis of genes that are upregulated by thiostrepton at P ≤ 0.001 indicates these genes are associated with unfolded protein response, ER-associated degradation, and cellular redox homeostasis (Supplementary Fig. S4B). At p ≤ 0.005, we observed 716 genes that are differentially expressed between DMSO- and thiostrepton-treated cells. The classification of gene function through DAVID bioinformatics resources (26) indicates one of the pathways enriched by these genes is DNA damage and repair pathway (Supplementary Fig. S4C and S4D). Genes involved in DNA damage and repair, such as MLH3, FANCF, and BRCC3 were downregulated while proapoptotic genes such as DDIT4, DDIT3, and GADD45A were upregulated (Fig. 3A). We confirmed these observations by qRT-PCR in A2780 ovarian cancer cells treated with thiostrepton for different time points (Fig. 3B). At the same time, we also observed that thiostrepton downregulated mRNA expression of BRCA2, which is known to be regulated by FOXM1. These results indicate that select DNA repair genes were down-regulated while proapoptotic genes were upregulated by thiostrepton (Fig. 3B).
Figure 3.
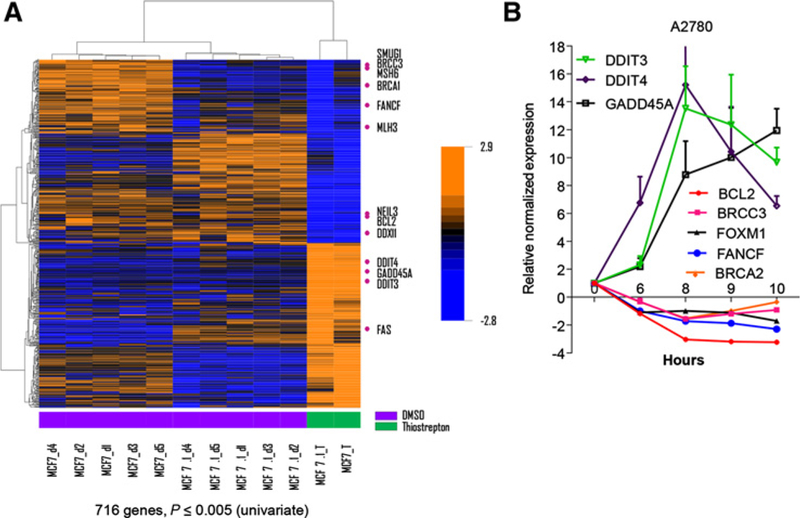
Thiostrepton decreases the expression of antiapoptotic genes and increases the expression of proapoptotic genes. A, Heatmap showing changes in gene expression profile after thiostrepton treatment. B, qRT-PCR analysis of altered genes after thiostrepton treatment in A2780 ovarian cancer cells. A2780 cells were treated with 5 μmol/L of thiostrepton for 0, 6,8, 9, and 10 hours before total RNA extraction for RT-PCR. Data are shown as mean ± SEM in log2. Results represent experiments performed in triplicates.
Thiostrepton downregulates FOXM1 target genes
To further characterize the effect of thiostrepton on the expression of FOXM1 target genes, we analyzed ENCODE dataset and focused on DNA repair genes identified from the FOXM1 ChIP-sequencing. This analysis indicates several DNA repair genes could be potentially regulated by FOXM1 (Supplementary Fig. S5). We focused on genes involved in HR repair pathway, and we analyzed the expression of FOXM1, FANCF, BRCA1, BRCC3, BRIP1, NBS1, Skp2, and Csk1. In three different cell lines, 5 μmol/L (OVCA420* and OV90) and 2.5 mmol/L (ES-2) thiostrepton downregulate the expression of FOXM1 target genes (Fig. 4A–C). Similarly, these genes were downregulated by thiostrepton in another ovarian cancer cell line A2780 (Supplementary Fig. S6A). Interestingly, thiostrepton variably upregulates FOXM1a and FOXM1b in ovarian cancer cells, while consistently down-regulating FOXM1c and its canonical target gene CCNB1 in these cells (Supplementary Fig. S6B–S6D).
Figure 4.
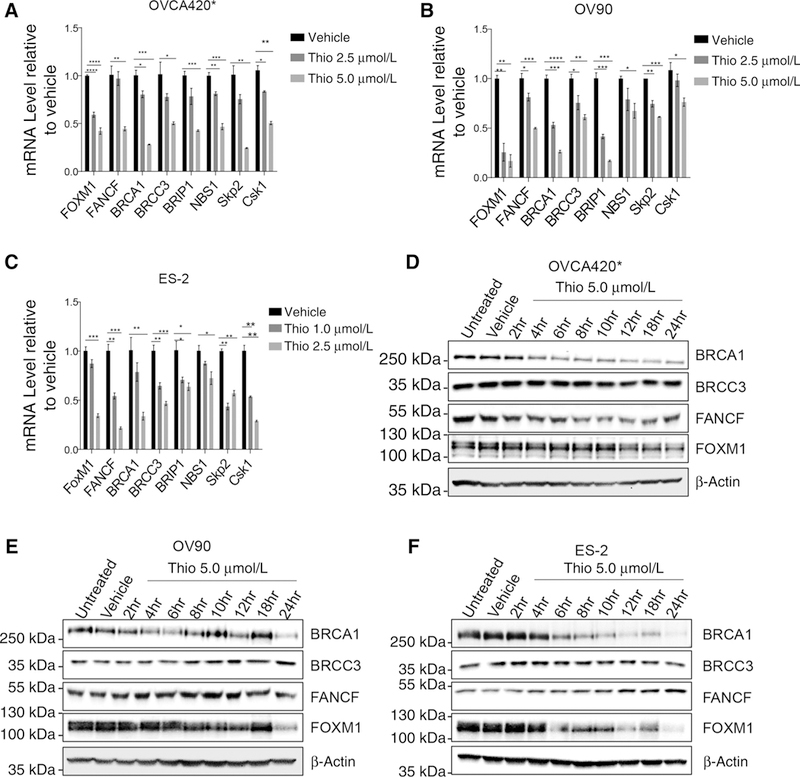
Thiostrepton downregulates FOXM1 and FOXM1 target genes involved in the homologous recombination repair. A-C, qRT-PCR of FOXM1 and its target genes involved in the homologous recombination after thiostrepton treatment. 0VCA420* (A) and OV90 (B) cells were seeded in 6-well plates and treated with vehicle, 2.5 μmol/L, or 5.0 μmol/L of thiostrepton for 24 hours before total RNA extraction. ES-2 cells (C) were treated with vehicle, 1.0 μmol/L or 2.5 μmol/L of thiostrepton for 24 hours. Statistical analysis was performed by two-tailed Student t test. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. Results represent experiments performed in triplicates. D-F, Immunoblot analysis of FOXM1 and its target genes after thiostrepton treatment. 0VCA420* (D), 0V90 (E), and ES-2 (F) cells were treated with 5.0 μmol/L thiostrepton for 0, 2, 4, 6, 8,10,12,18, or 24 hours before immunoblotting.
At protein levels, BRCA1, BRCA2, and FOXM1 were consistently downregulated by thiostrepton (Fig. 4D–F; Supplementary Fig. S6E and S6F). However, we did not observe consistent downregulation of BRCC3 and FANCF. To resolve the inconsistency between mRNA and protein expression, we analyzed the stability of these proteins in question. Following the inhibition of protein synthesis by cycloheximide, we observed a decrease in BRCA1 and FOXM1. The half-life of BRCA1 and FOXM1 was less than 60 minutes in OVCA420* (Supplementary Fig. S6G and S6H). In contrast, the half-life for BRCC3 and FANCF was longer than 24 hours (Supplementary Fig. S6G and S6H). Similarly, the half-life for BRCC3 and FANCF was longer than 24 hours in OV90 (Supplementary Fig. S6I and S6J). The longer half-life of BRCC3 and FANCF may explain why a substantial decrease in mRNA does not correspond with a decrease in protein levels. Expression of DNA repair genes including BRCA1 is known to be cell-cycle-dependent, and changes in expression of these genes may be secondary to cell-cycle changes induced by thiostrepton. However, we did not observe a marked change in cell-cycle profile at 4 hours, in two different cell lines, when BRCA1 is downregulated by thiostrepton in these cells (Supplementary Fig. S6K, Fig. 4D–F), suggesting that changes of expression of these genes including BRCA1 were due to FOXM1 inhibition. These data suggest that thiostrepton inhibits FOXM1 expression and its target HR genes, and thus induces “BRCAness” in ovarian cancer cells.
Thiostrepton induces the expression of proapoptotic genes and decreases the expression of antiapoptotic genes
In addition to genes involved in DNA repair pathway, the analysis of microarray dataset from the Connectivity Map project also indicates that thiostrepton affects the expression of genes involved in apoptosis. Therefore, we sought to confirm the expression of pro- and antiapoptotic genes affected by thiostrepton. qRT-PCR analysis indicates that thiostrepton upregulates DDIT3, GADD45A while it downregulates antiapoptotic gene BCL-2 (Fig. 5A and B). Consistent with these results, thiostrepton induces PARP1 and caspase-3 cleavages in two different cell lines (Fig. 5C and D). Finally, results from the caspase-3 activity assay indicate higher caspase-3 activity in cells treated with thiostrepton alone or in combination with olaparib (Fig. 5E and F).
Figure 5.
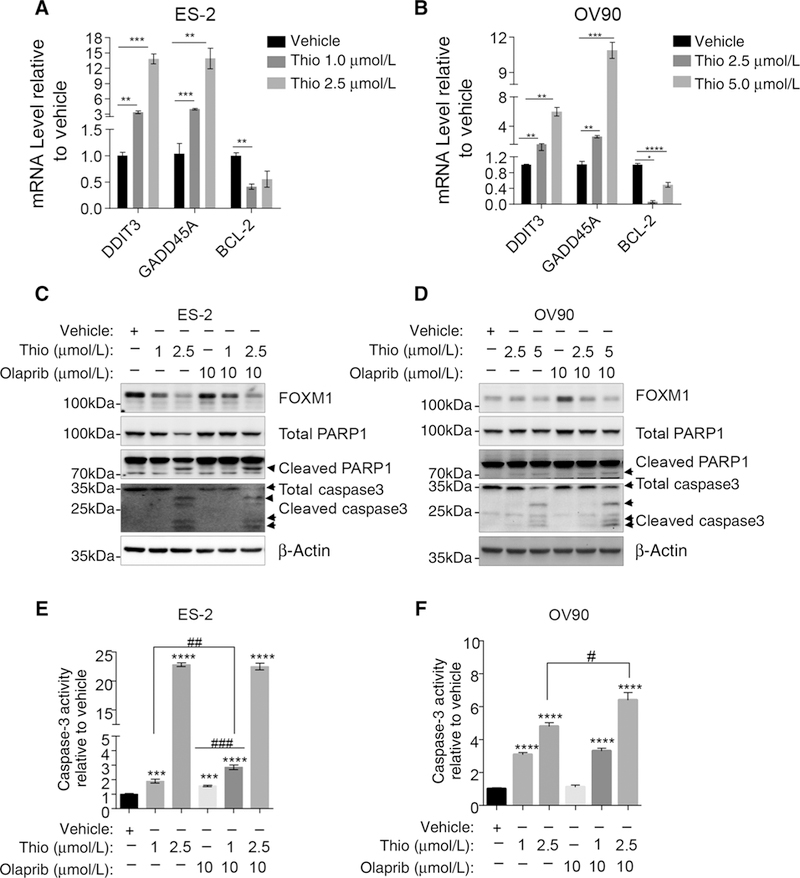
Thiostrepton induces apoptosis in ovarian cancer cells. A and B, Thiostrepton increases proapoptotic genes DDIT3 and GADD45A and decreases anti apoptotic gene BCL-2. ES-2 cells (A) and OV90 cells (B) were treated with vehicle or thiostrepton at the indicated concentration for 24 hours. C and D, Thiostrepton cooperates with olaparib in increasing PARP1 and caspase-3 cleavage. ES-2 (C) or OV90 (D) cells were treated with vehicle, thiostrepton alone, olaparib alone, or combination of thiostrepton and olaparib for 30 hours before checking for PARP1 and caspase-3 cleavage. E and F, Thiostrepton and olaparib increase caspase-3activities inovarian cancer cells. The significant analysis was performed with two-tail Student t test. *, P≤ 0.05; **, P ≤ 0.01; ***, P≤ 0.001; ****, P≤ 0.0001; #, P ≤ 0.05; ##, P < 0.01. Results represent experiments performed in duplicates.
Thiostrepton enhances DNA damage and PARP1 trapping in cells treated with olaparib
Because thiostrepton downregulates FOXM1 and its target genes involved in DNA repair pathway, we tested the extent to which thiostrepton enhances DNA damage in cells treated with olaparib. The results from comet assay indicate that thiostrepton alone increases DNA damage (Fig. 6A). In addition, higher levels of DNA in comet tail were observed in cells treated with 10 ≤ μmol/L olaparib and thiostrepton. Similar results were observed in two additional cell lines with thiostrepton and olaparib or carboplatin (Supplementary Fig. S7A and S7B). The increase in DNA damage following thiostrepton treatment may be explained in part by the downregulation of FOXM1 because FOXM1 knockdown by siRNA produces a similar increase in DNA damage (Fig. 6B). Meanwhile, increased level of phospho-H2AX (γH2AX) was seen with thiostrepton treatment and the increase was enhanced when combined with olaparib (Supplementary Fig. S7C and S7D). These results are consistent with a critical role of FOXM1 in regulating DNA repair genes, and a decreased expression of FOXM1 may compromise DNA repair function by downregulating several target genes involved in DNA repair. Consistent with an increase in DNA damage following FOXM1 inhibition, we also observed an increase in trapped PARP1 in cells treated with both 5 or 10 μmol/L thiostrepton, and 40 μmol/L olaparib (Fig. 6C). This observation provides an additional mechanism to explain the enhanced sensitivity to olaparib in ovarian cancer cells as PARP trapping onto chromatin after PARP inhibitor treatment is considered to be more cytotoxic than inhibition of enzymatic activity of PARP (30).
Figure 6.
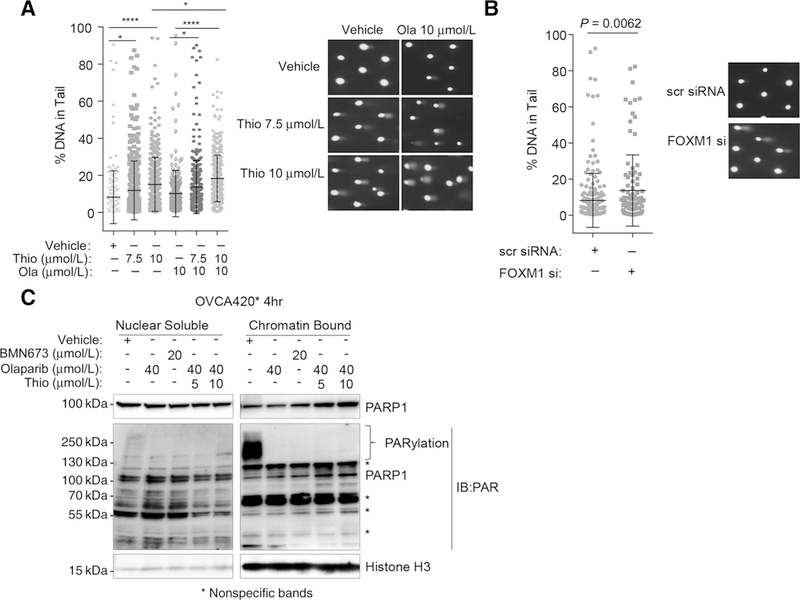
Thiostrepton enhances DNA damage and increases PARP1 trapping onto chromatin after olaparib treatment. A, Thiostrepton and olaparib increase DNA damage in OVCA420* cells. OVCA420* cells were pretreated with vehicle, 7.5 μmol/L, or 10 μmol/L thiostrepton for 4 hours in 6-well plates before trypsinizing and seeding onto CometChip. Then cells were treated with vehicle, 7.5 μmol/L, or 10 μmol/L thiostrepton and olaparib (10 μmol/L) for another 4 hours. Comets were analyzed with Trevigen Comet Analysis Software after imaging under a 4 x fluorescent microscope. Data were shown as percent of DNA in comet tail. Representative comet images were shown on the right. The significance analysis was performed with one-way ANOVA. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. Results represent experiments performed in triplicates. B, FOXM1 knockdown with siRNA increases DNA damages in Comet assay. Seventy-two hours after the FOXM1 knockdown, OVCA420* cells were trypsinized and seeded for comet assay using Trevigen CometChip Kit. Representative images were shown on the right. C, Thiostrepton increases PARP1 trapping onto chromatin. OVCA420* cells were treated with indicated concentrations of PARP inhibitors, olaparib or BMN673, alone or in combination with thiostrepton for 4 hours and fractionated as nuclear soluble and chromatin-bound fractions. The lysates were blotted first with PARP1 antibody and secondly with PARP antibody. ** shown as nonspecific bands. Results were from a representative of two independent experiments.
Thiostrepton sensitizes resistant cells to rucaparib by downregulating stabilized mutant BRCA1
Previous studies reported an enhanced expression of mutant BRCA1 as an epigenetic mechanism associated with acquired resistance to rucaparib (9). Although parental MDA-MB-436 cells are sensitive to rucaparib due to the presence of a BRCA1 mutation in these cells, the resistant clones derived from these cells expressed high levels of mutant BRCA1 that partially restore BRCA1 function and confer resistance to rucaparib. We used these resistant clones to test the extent to which thiostrepton restores sensitivity to rucaparib in these cells. Thiostrepton downregulates the expression of FOXM1 as well as mutant BRCA1 (Fig. 7A). Consistent with this downregulation, we observed an increase in sensitivity to rucaparib in thiostrepton-treated cells (Fig. 7B and C; Supplementary Fig. S8A and S8B). In addition, mild synergistic interactions between thiostrepton and rucaparib were observed at different combinations. Clonogenic assays further corroborated these synergistic interactions between the two drugs (Fig. 7D and E). This enhanced sensitivity to rucaparib was also observed in resistant cells following the FOXM1 knockdown by siRNA (Fig. 7F and G), indicating that enhanced sensitivity to rucaparib by thiostrepton could be explained in part by its effect on FOXM1 expression. These data suggest that inhibition of FOXM1 with thiostrepton can be an effective way to overcome acquired resistance to PARP inhibitors.
Figure 7.
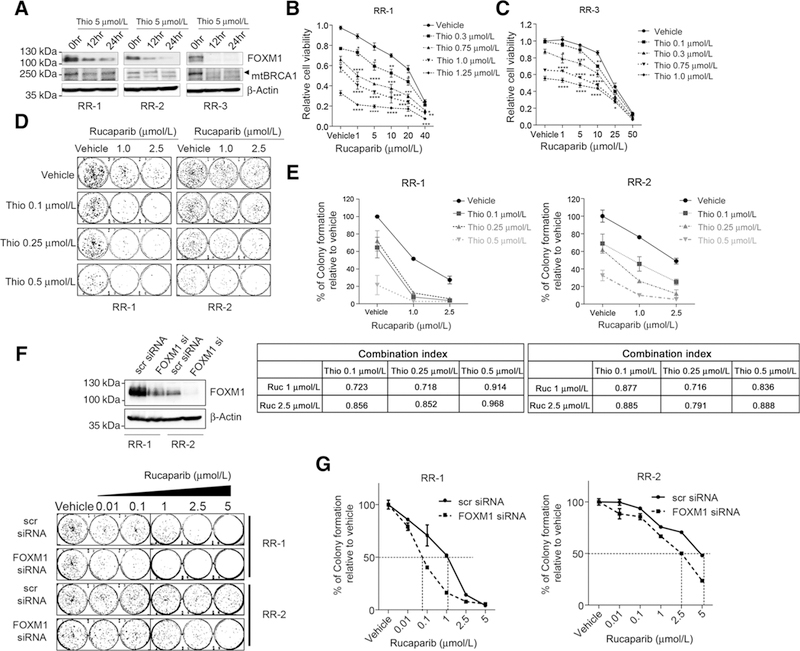
Thiostrepton sensitizes rucaparib-resistant cells by downregulating FOXM1 and mutant BRCA1. A, FOXM1 and mutant BRCA1 (mtBRCA1) decrease by thiostrepton treatment. Three rucaparib-resistant cells (RR-1, RR-2, and RR-3) were treated with 5 μmol/L thiostrepton for 0,12, or 24 hours and the FOXM1 and mutant BRCA1 protein levels were measured by immunoblotting. B and C, Synergistic effects of thiostrepton and rucaparib in rucaparib-resistant cells RR-1 (A) and RR-3 (B). Rucaparib-resistant cells were derived from MDA-MB-436 cells after long time exposure of rucaparib (9). 5000 cells were seeded in 96-well plates and treated with thiostrepton or olaparib alone or combinations of both drugs for 3 days before SRB assay. Cell survival curves were generated in the presence of vehicle (set as 100% of survival) or thiostrepton following increasing concentrations of rucaparib. Data were shown as mean ± SEM in a line graph. The statistics analysis was fulfilled with two-tailed Student t test. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. Results represent experiments with four replicates. D and E, Colony formation assay of rucaparib-resistant cells RR-1 and RR-2 with thiostrepton and rucaparib. RR-1 and RR-2 cells were seeded in 6-well plates and treated with vehicle or rucaparib with or without thiostrepton. The colony formation results were shown in (D) and quantified in E, data shown as mean ± SEM. Combination index (CI) for each combination was shown underneath. F and G, Colony formation assay after FOXM1 knockdown with siRNA. F, RR-1 and RR-2 cells were transfected with scr siRNA or FOXM1 siRNA and waited 48 hours before seeding for clonogenic assay. Cells were treated with increasing concentrations of rucaparib for 3 days and allow colonies to form for 18 days. Colonies were stained with SRB and imaged. Cell lysates were collected around 72 hours after siRNA transfection and subjected to Western blot analysis to check FOXM1 knockdown efficiency. G, Quantification of relative colony formation. Stained colonies from F were solubilized in Tris buffer and measured fluorescent intensity. Data are shown as mean ± SEM of the percentage of colony formation relative to the vehicle of scr siRNA or FOXM1 siRNA, respectively. Results represent experiments performed in duplicates (D-G).
Discussion
In this study, we identified FOXM1 as a component of the adaptive cellular pathway responsive to olaparib. Olaparib induces the expression of FOXM1 and activates the FOXM1 pathway as evidenced by the enhanced nuclear localization of FOXM1 and the increased binding of FOXM1 to its target genes. The disruption of FOXM1 pathway either by RNAi or chemical inhibition with thiostrepton decreases the levels of FOXM1 expression and sensitizes the cancer cells to olaparib and carboplatin. Furthermore, thiostrepton decreases the levels of DNA repair genes involved in HR pathway and induces “BRCAness” in ovarian cancer cells, thereby resensitizing the rucaparib-resistant breast cancer cells to rucaparib. During preparation of this article, a study from Tassi and colleagues, reported that FOXM1 expression is significantly associated with chemotherapy resistance, and FOXM1 knockdown enhances the cytotoxic effects of chemotherapeutic agents including olaparib in nonserous epithelial cancer cells (18), which is consistent with our observations described in this article.
Previous studies indicate that FOXM1 regulates the expression of BRIP1 and DNA damage repair pathway following epirubicin treatment (31). In addition, a spontaneous increase in γH2AX foci has been reported in cancer cells following the depletion of FOXM1 by RNAi (32). Consistent with these results, our studies provided additional supporting evidence that FOXM1 knockdown and thiostrepton treatment increase the γH2AX level, enhance DNA damage, and PARP1 trapping to the damaged DNA in the presence of olaparib. Olaparib has been shown to produce moderate levels of PARP trapping (30). The increase of PARP trapping in thiostrepton and olaparib treatment may be a result of increased DNA damage induced by thiostrepton, which leads to the recruitment of DNA repair machinery to the damage sites, including PARP1 which is responsible for the base excision repair (BER). A significant increase in γH2AX foci either by RNAi or thiostrepton underlies the important role FOXM1 plays in regulating DNA repair genes and maintaining DNA repair efficacy (33). Accordingly, FOXM1 is described as an “emerging master regulator of DNA damage response” (33). Therefore, our results indicating that FOXM1 is induced by olaparib and that its enhanced expression regulates the adaptive cellular response to DNA damage signal provide an important biological basis for targeting FOXM1 pathway to overcome resistance to olaparib and carboplatin.
It is important to note that previous studies have also shown that FOXM1 is stabilized by DNA-damaging agents, such as ionizing radiation, etoposide, and UV (32, 34). Tan and colleagues reported that FOXM1 is phosphorylated by Chk2 at S361, leading to the stability of FOXM1 (32). They reported that FOXM1 regulates BRCA2 and XRCC1. In addition, Monteiro and colleagues reported that FOXM1 is required for HR pathway but indispensable for NHEJ (31). Finally, Zhang and colleagues reported that FOXM1 regulates RAD51 expression (35). Consistent with these results, we observed FOXM1 occupancy to DNA repair genes, such as BRCA1, RAD51, FANCF, RAD51D, and FANCD2, in response to olaparib. In addition, inhibition of FOXM1 by thiostrepton decreases the expression of DNA repair genes, such as BRCA1 and BRCA2, thereby disrupting an adaptive response mediated by FOXM1.
Considering that adaptive responses to chemotherapy provide transient resistance to chemotherapy and may facilitate the eventual acquisition of more stable traits associated with chemotherapy resistance, our study provides an important insight into a potential role of the FOXM1 pathway in the adaptive response to olaparib. Our studies also suggest the potential use of thiostrepton as a chemotherapeutic agent to disrupt FOXM1 pathway and to enhance sensitivity to olaparib and carboplatin. Although thiostrepton was initially isolated from Streptomyces azureus and used as a topical antibiotic (36), its current use in human is limited by the low solubility and bioavailability. However, recent studies have shown that micelle-formulated thiostrepton has better solubility and pharmacodynamic effect on the tumor in xenograft models (37). Also, several studies have tested the in vivo efficacy of thiostrepton in xenograft models of other cancers, including ovarian cancer, leukemia, and laryngeal squamous cell carcinoma (20, 38, 39). In addition to thiostrepton, siomycin A is another cyclic oligopeptide isolated from Streptomyces sioyanesis and shows inhibitory effects on FOXM1 transcriptional activity and expression (40). Finally, another inhibitor FDI-6 is a small-molecule FOXM1 inhibitor that blocks FOXM1 binding to DNA (41). Although micromolar concentrations of FDI-6 is required to inhibit FOXM1, further development of this compound may result in therapeutics that can effectively target FOXM1 and enhance sensitivity to existing chemotherapeutics, such as olaparib and carboplatin.
Our studies show that FOXM1 plays an important role in an adaptive response to PARP inhibitors. FOXM1 expression levels inversely correlate with olaparib sensitivity, and knockdown of FOXM1 sensitizes ovarian cancer cells to olaparib and decreases the number of persisting cells that can produce colonies. FOXM1 inhibitor, thiostrepton, induces “BRCAness” by downregulating homologous recombination repair genes, increases apoptosis, and enhances therapeutic effects of olaparib and carboplatin in ovarian cancer cells. In addition, thiostrepton treatment overcomes resistance to rucaparib by downregulating FOXM1 and the stabilized mutant BRCA1 in resistant breast cancer cells. Inhibition of FOXM1 represents an effective strategy to overcome PARP inhibitor resistance and that disrupting FOXM1-mediated adaptive pathway may produce better therapeutic effects for PARP inhibitors.
Supplementary Material
Acknowledgments
This work was supported by grants from American Cancer Society Research Scholar(125618-RSG-14–067-01-TBE, to J. Chien), the Department of Defense Ovarian Cancer Research Program under award number (W81XWH-14–1-0116, to J. Chien), and Ann and Sol Schreiber Mentored Investigator Award (to J. Madden). The authors thank Dr. Neil Johnson (Fox Chase Cancer Center) for the breast cancer cells MDA-MB-436 and its derivative rucaparib-resistant cells. Data were generated in the UNM Shared Flow Cytometry and High Throughput Screening Resource Center supported by the University of New Mexico Health Sciences Center and the University of New Mexico Cancer Center with current funding from NCI 2P30 CA118100–11 (principal investigator: C. Willman) “UNM Cancer Center Support Grant.” The authors also thank Cristabelle De Souza for helping with flow cytometry of cell-cycle analysis.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
References
- 1.Giaccone G, Pinedo HM. Drug Resistance. Oncologist 1996;1:82–7. [PubMed] [Google Scholar]
- 2.Yardley DA. Drug resistance and the role of combination chemotherapy in improving patient outcomes. Int J Breast Cancer 2013;2013:137414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hammond WA, Swaika A, Mody K. Pharmacologic resistance in colorectal cancer: a review. Ther Adv Med Oncol 2016;8:57–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pfisterer J, Ledermann JA. Management of platinum-sensitive recurrent ovarian cancer. Semin Oncol 2006;33(2 Suppl 6):S12–S16. [DOI] [PubMed] [Google Scholar]
- 5.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29. [DOI] [PubMed] [Google Scholar]
- 6.Bast RC Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer 2009;9:415–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowtell DD, Bohm S, Ahmed AA, Aspuria PJ, Bast RC Jr, Beral V, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer 2015;15:668–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fojo T, Bates S. Mechanisms of resistance to PARP inhibitors-three and counting. Cancer Discov 2013;3:20–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci U S A 2013;110:17041–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerlinger M, Swanton C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer 2010;103:1139–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015;521:489–94. [DOI] [PubMed] [Google Scholar]
- 12.Goldman A, Majumder B, Dhawan A, Ravi S, Goldman D, Kohandel M, et al. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat Commun 2015;6:6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pisco AO, Brock A, Zhou J, Moor A, Mojtahedi M, Jackson D, et al. Non-Darwinian dynamics in therapy-induced cancer drug resistance. Nat Commun 2013;4:2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawson CC, Intapa C, Jabra-Rizk MA. “Persisters”: survival at the cellular level. PLoS Pathog 2011;7:e1002121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chien J, Kuang R, Landen C, Shridhar V. Platinum-sensitive recurrence in ovarian cancer: the role of tumor microenvironment. Front Oncol 2013; 3:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell sub-populations. Cell 2010;141:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tassi RA, Todeschini P, Siegel ER, Calza S, Cappella P, Ardighieri L, et al. FOXM1 expression is significantly associated with chemotherapy resistance and adverse prognosis in non-serous epithelial ovarian cancer patients. J Exp Clin Cancer Res 2017;36:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park HJ, Carr JR, Wang Z, Nogueira V, Hay N, Tyner AL, et al. FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO J 2009;28: 2908–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Cheng L, Minn K, Madan R, Godwin AK, Shridhar V, et al. Targeting of mutant p53-induced FoxM1 with thiostrepton induces cytotoxicity and enhances carboplatin sensitivity in cancer cells. Oncotarget 2014;5:11365–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science 2007;316:1497–502. [DOI] [PubMed] [Google Scholar]
- 22.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc 2006;1:1112–6. [DOI] [PubMed] [Google Scholar]
- 23.Bastola P, Neums L, Schoenen FJ, Chien J. VCP inhibitors induce endoplasmic reticulum stress, cause cell cycle arrest, trigger caspase-mediated cell death and synergistically kill ovarian cancer cells in combination with Salubrinal. Mol Oncol 2016;10:1559–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform 2007;3:11–7. [PMC free article] [PubMed] [Google Scholar]
- 25.Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, et al. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 2015;18:723–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J,et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol 2007;8:R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aleman LM, Doench J, Sharp PA. Comparison of siRNA-induced off-target RNA and protein effects. RNA 2007;13:385–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hegde NS, Sanders DA, Rodriguez R, BalasubramanianS.The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nat Chem 2011;3:725–31. [DOI] [PubMed] [Google Scholar]
- 29.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006;313:1929–35. [DOI] [PubMed] [Google Scholar]
- 30.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 2012;72:5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monteiro LJ, Khongkow P, Kongsema M, Morris JR, Man C, Weekes D, et al. The Forkhead Box M1 protein regulates BRIP1 expression and DNA damage repair in epirubicin treatment. Oncogene 2013;32:4634–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan Y, Raychaudhuri P, Costa RH. Chk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genes. Mol Cell Biol 2007;27:1007–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zona S, Bella L, Burton MJ, Nestal de Moraes G, Lam EW. FOXM1: an emerging master regulator of DNA damage response and genotoxic agent resistance. Biochim Biophys Acta 2014;1839:1316–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teh MT, Gemenetzidis E, Chaplin T, Young BD, Philpott MP. Upregulation of FOXM1 induces genomic instability in human epidermal keratinocytes. Mol Cancer 2010;9:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang N, Wu X, Yang L, Xiao F, Zhang H, Zhou A, et al. FoxM1 inhibition sensitizes resistant glioblastoma cells to temozolomide by downregulating the expression of DNA-repair gene Rad51. Clin Cancer Res 2012;18:5961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kutscher AH, Zegarelli EV, Rankow RM, Mercadante J, Piro JD. Clinical laboratory studies on a new topical antibiotic: thiostrepton. Oral Surg Oral Med Oral Pathol 1959;12:967–74. [DOI] [PubMed] [Google Scholar]
- 37.Wang M, Gartel AL. Micelle-encapsulated thiostrepton as an effective nanomedicine for inhibiting tumor growth and for suppressing FOXM1 in human xenografts. Mol Cancer Ther 2011;10:2287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buchner M, Park E, Geng H, Klemm L, Flach J, Passegue E, et al. Identification of FOXM1 as a therapeutic target in B-cell lineage acute lymphoblastic leukaemia. Nat Commun 2015;6:6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang L, Wu X, Wang P, Wen T, Yu C, Wei L, et al. Targeting FoxM1 by thiostrepton inhibits growth and induces apoptosis oflaryngeal squamous cell carcinoma. J Cancer Res Clin Oncol 2015;141:971–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radhakrishnan SK, Bhat UG, Hughes DE, Wang IC, Costa RH, Gartel AL. Identification of a chemical inhibitor of the oncogenic transcription factor forkhead box M1. Cancer Res 2006;66:9731–5. [DOI] [PubMed] [Google Scholar]
- 41.Gormally MV, Dexheimer TS, Marsico G, Sanders DA, Lowe C, Matak-Vinkovic D, et al. Suppression of the FOXM1 transcriptional programme via novel small molecule inhibition. Nat Commun 2014;5:5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.