

Abstract
NMNAT2 is an NAD+-synthesizing enzyme with an essential axon maintenance role in primary culture neurons. We have generated an Nmnat2 gene trap mouse to examine the role of NMNAT2 in vivo. Homozygotes die perinatally with a severe peripheral nerve/axon defect and truncated axons in the optic nerve and other CNS regions. The cause appears to be limited axon extension, rather than dying-back degeneration of existing axons, which was previously proposed for the NMNAT2-deficient Blad mutant mouse. Neurite outgrowth in both PNS and CNS neuronal cultures consistently stalls at 1–2 mm, similar to the length of truncated axons in the embryos. Crucially, this suggests an essential role for NMNAT2 during axon growth. In addition, we show that the Wallerian degeneration slow protein (WldS), a more stable, aberrant NMNAT that can substitute the axon maintenance function of NMNAT2 in primary cultures, can also correct developmental defects associated with NMNAT2 deficiency. This is dose-dependent, with extension of life span to at least 3 months by homozygous levels of WldS the most obvious manifestation. Finally, we propose that endogenous mechanisms also compensate for otherwise limiting levels of NMNAT2. This could explain our finding that conditional silencing of a single Nmnat2 allele triggers substantial degeneration of established neurites, whereas similar, or greater, reduction of NMNAT2 in constitutively depleted neurons is compatible with normal axon growth and survival. A requirement for NMNAT2 for both axon growth and maintenance suggests that reduced levels could impair axon regeneration as well as axon survival in aging and disease.
Introduction
Dying-back axon degeneration contributes to pathology in many common neurodegenerative diseases, even before loss of neuronal viability (Adalbert and Coleman, 2012). In many cases, this degeneration shares physical and biochemical features with Wallerian degeneration, so elucidation of the critical molecular changes that occur in axons in the more defined context of an acute injury should facilitate identification of suitable therapeutic targets for these disorders. Studies of WldS, an aberrant fusion protein with nicotinamide mononucleotide adenylyltransferase (NMNAT) activity that robustly delays Wallerian degeneration (Mack et al., 2001), have been very informative in this respect. They support a model in which an intrinsic axon degeneration pathway, conserved between mammals and flies, is triggered by injury and other diverse stresses (Coleman and Freeman, 2010).
NMNAT2 is one of three naturally occurring mammalian NMNAT isoforms. Like WldS, each can delay Wallerian degeneration when overexpressed (Sasaki et al., 2006; Yan et al., 2010), but depletion of NMNAT2 alone causes spontaneous neurite degeneration, which endogenous NMNAT1 and NMNAT3 cannot prevent (Gilley and Coleman, 2010). Loss of short-lived NMNAT2 has therefore been proposed as a natural trigger for Wallerian and related, Wallerian-like axon degeneration, with WldS directly substituting for NMNAT2 loss in compromised axons because it is relatively much more stable and shares critical NMNAT activity (Gilley and Coleman, 2010). Constant replenishment of NMNAT2 levels in axons, needed to prevent spontaneous degeneration, is likely achieved through rapid axonal trafficking on the surface of Golgi-derived vesicles, with palmitoylation playing a key role in vesicle attachment (Milde et al., 2013). Recent, independent studies of NMNAT2 and its Drosophila ortholog, dNmnat, have provided strong support for this model (Fang et al., 2012; Xiong et al., 2012; Babetto et al., 2013). However, the role of NMNAT2 during development of the nervous system is only just beginning to be addressed, and it is not known whether WldS can also substitute for NMNAT2 in vivo.
A severe peripheral axon defect was recently reported in Blad mutant mice (Nmnat2blad/blad) lacking NMNAT2 (Hicks et al., 2012). Dying-back axonopathy during late embryonic development was proposed as a possible cause. Here, using an independent mouse line with conditional-ready targeting of Nmnat2, we report that the underlying cause of the peripheral axon defect in NMNAT2-deficient embryos actually appears to be a failure of axons to extend beyond a threshold length from the outset, and not dying-back degeneration after relatively normal initial growth and innervation. This suggests a novel developmental role for NMNAT2 during axon growth, potentially linking axon growth and maintenance mechanisms. We also provide the first evidence that neurons in the CNS are similarly affected by NMNAT2 depletion. Finally, we not only show that WldS can substitute for NMNAT2 in vivo but find that low levels of NMNAT2 during development, which might otherwise limit axon survival, appear to trigger endogenous compensatory mechanisms. The implications of these results are discussed, with particular focus on the relationship between axon growth/regeneration and axon maintenance mechanisms, and their relevance to development, aging, and disease.
Materials and Methods
Generation of conditional-ready Nmnat2-targeted mice
ES cell clones EUCE0262a08 and EUCE0030a11 containing a conditional-ready gene trap cassette (Schnutgen et al., 2005) within intron 1 of one allele of the Nmnat2 gene were obtained from the European Conditional Mouse Mutagenesis Program (EUCOMM). Only the first 28 amino acids of murine NMNAT2 (total length 307) encoded by Nmnat2 exon 1 should be expressed from the disrupted allele as a fusion with βgeo encoded by the gene trap cassette. PCR and sequencing were used to confirm single insertion of the gene trap within intron 1 of the Nmnat2 gene in both clones. Semiquantitative endpoint RT-PCR and immunoblotting were used to assess Nmnat2 mRNA and NMNAT2 protein knock-down in the two clones, and ES cell clone EUCE0262a08 was chosen for blastocyst injection as it showed greater depletion (data not shown). EUCE0262a08 (E14TG2a) ES cells (129P2/Ola strain) were injected into C57BL/6J-Tyrc-2J blastocysts by the Babraham Institute Gene Targeting Facility to generate chimeras, which were then mated to C57BL/6J-Tyrc-2J mice. We used duplex-PCR to assess germline transmission of the Nmnat2Gt(EUCE0262a08)Hmgu allele (referred to as Nmnat2gtE in the text), and for subsequent genotyping, using the following primers (locations shown in Fig. 1A): 5′-gctggcctaggtggtgatttgc-3′ (A), 5′-actgggatgcacgagaccctgc-3′ (B), and 5′-agtcatagacactagacaatcgg-3′ (C). These primers amplified 491 bp and 552 bp products from the wild-type and trapped alleles, respectively (see Fig. 1A). Inversion of the gene trap does not alter the amplification products of the genotyping PCR, so gene trap orientation after crosses to FLPe mice was confirmed using a separate duplex-PCR using primers flanking the recombinase target sequences. Primers (3′) 5′-ACCTCGAGTACCACCACACTGG-3′, (forward) 5′-GCTAGCTTGCCAAACCTACAGG-3′, and (reverse) 5′-AACGTCGGATGGCCGCGAGACG-3′ amplify a 590-bp product from the gene trap in the forward orientation (before any FLPe or Cre-mediated recombination) and a 367 bp product from the inverted gene trap after FLPe-mediated recombination. Mice with the FLPe transgene were identified using primers 5′-GCATCATGTGCTGCTGAACTAA-3′ and 5′-GTGCCTACTAACGCTTGTCTTT-3′.
Figure 1.
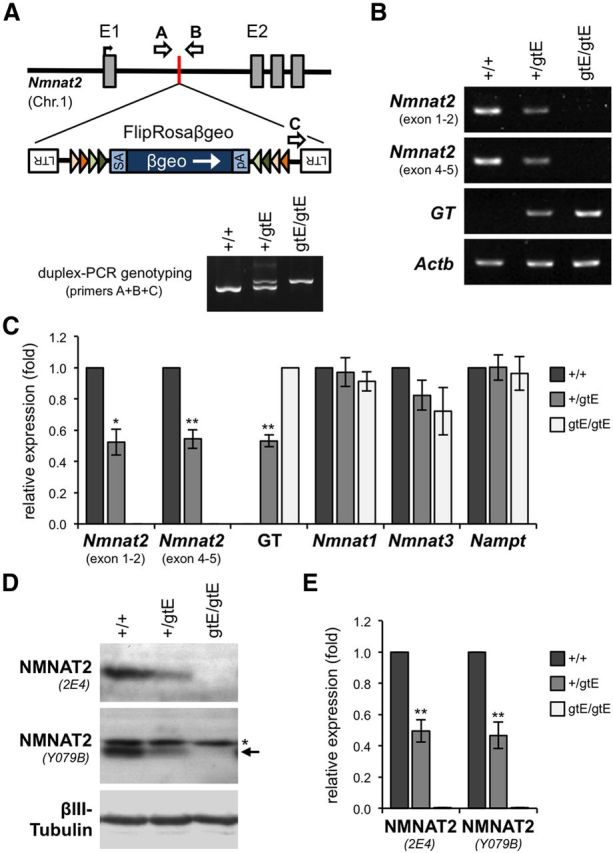
Generation and initial characterization of NMNAT2-deficient mice. A, Localization of the conditional-ready gene trap within intron 1 of the mouse Nmnat2 gene in mice derived from EUCOMM ES cell clone EUCE0262a08. Features are as described previously (Schnutgen et al., 2005). Positions of PCR genotyping primers (arrows A–C), and representative genotyping results for wild-type (+/+), Nmnat2+/gtE, and Nmnat2gtE/gtE animals, are also shown. B, Representative RT-PCR showing Nmnat2 mRNA levels (two sets of primers), levels of the mRNA derived from trapping (GT), and Actb mRNA levels (acting as a reference), in brains of wild-type (+/+), Nmnat2+/gtE, and Nmnat2gtE/gtE E18.5 embryos. C, Quantification of RT-PCR data showing relative mean levels (± SEM) of Nmnat2, GT, Nmnat1, Nmnat3, and Nampt mRNAs after normalization to Actb mRNA (n = 4 for each genotype). Data are presented relative to wild-type levels (set at 1), except for GT data, which is presented relative to Nmnat2gtE/gtE levels. *p < 0.05 (two-tailed one sample t test). **p < 0.01 (two-tailed one sample t test). D, Representative immunoblots showing Nmnat2 levels (two antibodies) in brains of wild-type (+/+), Nmnat2+/gtE, and Nmnat2gtE/gtE E18.5 embryos. βIII-Tubulin acts as the sample control. NMNAT2 migrates at ∼32 kDa. A nonspecific band (*), slightly larger than NMNAT2 (arrow), is detected by Y079B. E, Quantification of immunoblot data showing relative mean levels (± SEM) of NMNAT2 detected with each antibody after normalization to βIII-Tubulin (n = 4 for each genotype). Data are presented relative to wild-type levels (set at 1). **p < 0.01 (two-tailed one sample t test).
Other mouse strains and animal work
C57BL/6OlaHsd-Wld (WldS) mice were obtained from Harlan UK, FLPe transgenic mice (Rodríguez et al., 2000) were obtained from Martin Turner (Babraham Institute), and Nmnat2gtBay mice, derived from gene trap ES cell clone RRF238 (BayGenomics), were described previously (Mayer et al., 2010).
All animal work was performed in accordance with the Animals (Scientific Procedures) Act, 1986, under Project License PPL 80/2254. Standard timed mating procedures were followed to produce E12.5–E14.5 and E18.5 embryos. The developmental stage was confirmed using Theiler staging criteria. Pregnant females of the Nmnat2gtE strain usually littered at E19. E18.5 embryos were therefore considered essentially full-term. Mice of both sexes were studied in experiments.
RT-PCR analysis
We used semiquantitative endpoint RT-PCR to determine levels of selected mRNA species in total brain RNA extracts. RNA was extracted using TRIsure reagent (Bioline), after mechanical homogenization using an Ultra-Turrax homogenizer, and reverse transcribed by Superscript II (Invitrogen) using 1 μg RNA per reaction. DNA contamination of samples was assessed using no RT controls. Standard PCR amplification was performed using REDTaq DNA polymerase (Sigma). One-fortieth of the cDNA from the RT reaction was used in each PCR. The number of cycles needed for detection of product within the presumed exponential phase of amplification was determined empirically for each set of primers (using increments of 2 cycles). The following sets of primers were used (with reaction annealing temperatures in parentheses): Nmnat2 (exon 1–2), 5′-CAGTGCGAGAGACCTCATCCC-3′ and 5′-ACACATGATGAGACGGTGCCG-3′ (63°C); Nmnat2 (exon 4–5), 5′-GAGTGCTATCAGGACACCTGG-3′ and 5′-GTGGGCACATTGCTGTTCTGG-3′ (63°C); GT (to detect hybrid Nmnat2-gene trap mRNA), 5′-CAGTGCGAGAGACCTCATCCC-3′ and 5′-TCTTCGCTATTACGCCAGCTGG-3′ (63°C); Nmnat1, 5′-TTCAAGGCCTGACAACATCGC-3′ and 5′-GAGCACCTTCACAGTCTCCACC-3′ (63°C); Nmnat3, 5′-GGTGTGGAGGTGTGTGACAGC-3′ and 5′-GCCATGGCCACTCGGTGATGG-3′ (63°C); Nampt, 5′-GGTCATCTCCCGATTGAAGT-3′ and 5′-GAAGAGACTCCTCTGTAACC-3′ (58°C); Actb, 5′-TGTTACCAACTGGGACGACA-3′ and 5′-ATGAGGTAGTCTGTCAGGTC-3′ (58°C). Single products of the expected size were amplified for each set of primers.
Immunoblotting
Whole brains from E14.5 and E18.5 mouse embryos, early postnatal pups, and adult mice were mechanically homogenized using an Ultra-Turrax homogenizer in 1× TG lysis buffer consisting of 20 mm Tris (pH 7.5), 137 mm NaCl, 1 mm EGTA, 1% (v/v) Triton X-100, 10% (v/v) glycerol, 1.5 mm MgCl2, 1 mm Na3VO4, 50 mm NaF, and cOmplete Mini protease inhibitor cocktail (Roche). Samples were microfuged at 10,000 × g to pellet cell debris, and the supernatant was mixed with 2× Laemmli sample buffer. This step was included to facilitate detection of NMNAT2, which is a nonabundant protein (Mayer et al., 2010). There was no significant loss of NMNAT2 in the pellet. Detection of low abundance NMNAT2 required 40 μg of each extract to be loaded per lane of the gel (15 well format). To avoid signal saturation, we ran 5% of the same extracts (diluted in sample buffer) on a separate gel for detection of the far more abundant sample reference, βIII-Tubulin Extracts were separated by standard SDS-PAGE on SDS polyacrylamide gels (12% or 4–15% gradient gels) and transferred to Immobilon-P membrane (Millipore) using the Bio-Rad Mini-PROTEAN III wet transfer system. Blots were blocked and incubated with primary antibodies overnight at 4°C (in 1× TBS pH 8.3, with 0.05% Tween 20 and 5% milk powder), followed by the appropriate HRP-conjugated secondary antibody for 1–2 h at room temperature (1:2000–1:10,000), with subsequent detection using ECL or ECL plus (GE Healthcare). Multiple short washes were performed after each antibody incubation. The following primary antibodies were used: mouse monoclonal anti-NMNAT2 clone 2E4 (2.0 μg/ml, Sigma, WH0023057M1 or Abcam, ab56980), rabbit polyclonal anti-NMNAT2 Y079B (1:2000, G.Y. laboratory), mouse monoclonal anti-neuronal class βIII-Tubulin TUJ1 (1:10,000, Covance, MMS-435P), and rabbit polyclonal anti-WldS Wld18 (1:2000). Relative band densities on scanned autoradiographs were determined using ImageJ software.
Primary neuronal cultures and neurite outgrowth assays
Explant cultures.
Superior cervical ganglia (SCG) dissected from E18.5 mouse embryos, dorsal root ganglia (DRG) dissected from E14.5 or E18.5 mouse embryos, and small cortical neuron explants dissected from E18.5 mouse embryos were plated on 3.5 cm tissue culture dishes precoated with poly-l-lysine (20 μg/ml for 1–2 h; Sigma) and laminin (20 μg/ml for 1–2 h; Sigma). SCG and DRG explants were cultured in DMEM with 4500 mg/L glucose and 110 mg/L sodium pyruvate (Sigma), and supplemented with 2 mm glutamine, 1% penicillin/streptomycin, 100 ng/ml 7S NGF (all Invitrogen), and 10% FBS (Sigma) for SCG cultures or 2% B27 supplement (Invitrogen) for DRG cultures. A total of 4 μm aphidicolin (Calbiochem) was used to reduce proliferation and viability of small numbers of non-neuronal cells. Cortical neuron explants were cultured in Neurobasal Medium (Invitrogen) supplemented with 2 mm glutamine, 1% penicillin/streptomycin, and 2% B27 supplement.
Dissociated SCG cultures.
SCG ganglia from P1–P2 mouse pups were incubated in 0.025% trypsin (Sigma) in PBS (without CaCl2 and MgCl2) for 30 min followed by 0.2% collagenase type II (Invitrogen ) in PBS for 30 min. Ganglia were then gently triturated using a pipette. After a 2 h preplating stage to remove non-neuronal cells, dissociated neurons were plated on poly-l-lysine and laminin-coated ibidi μ-dishes (Thistle Scientific) and cultured as for SCG explant cultures.
Neurite outgrowth assays.
Phase-contrast images of DRG, SCG, and cortical explants and their extending neurites were captured at low magnification (4× objective) at the same time on each specified day after plating. Low magnification ensured neurite outgrowth could be captured in a single image at most time points to facilitate measurement. Radial outgrowth on each day was determined by taking the average of two measurements of representative neurite outgrowth for each explant. At later time points, measurements were made from overlapping images of the total neurite outgrowth. After 14 days in vitro (DIV), cortical neuron explants were incubated with 1 μm calcein-AM (Invitrogen) for 30 min at 37°C in the dark to label cells and neurites retaining esterase activity.
DiI labeling of peripheral nerve axons
Crystals of the lipophilic dye DiI (1,l′-dioctadecyll3,3,3′,3′tetramethylindocarbo-cyanine perchlorate) were inserted into a rostrocaudal incision along the posterior median sulcus of thoracic and subthoracic spinal cord of fixed newborn pups and embryos. Embryos were kept at 37°C in 4% paraformaldehyde for 5–8 weeks, duration being dependent on the developmental stage, to allow diffusion of the dye through neuronal membranes leading to labeling of axons in the dorsal and ventral root and spinal nerves. Tissues selected for subsequent analysis were then dissected and cleaned up before optical clearing by immersion in an increasing PBS-glycerol series (up to 100% glycerol). Lengths of DiI-labeled intercostal nerves were determined from captured images using ImageJ software.
Histology and immunostaining
Fixation and cryosectioning.
Embryos were fixed by immersion in 4% paraformaldehye and left for several days at 4°C before being cryoprotected in 0.1 m PBS, 30% sucrose, 0.01% w/v sodium azide at 4°C. Pieces of embryo were then frozen in Shandon M-1 embedding matrix (Thermo Scientific), and transverse serial sections were cut at 20 μm using a Leica CM 1850 cryostat and placed onto Superfrost plus slides (BDH). Slides were stored at −20°C.
Hematoxylin and eosin staining.
Sections warmed to room temperature were stained with Gill's III hematoxylin solution (Sigma-Aldrich) followed by a wash in tap water, differentiation in 70% ethanol, 1% HCl, and another wash in tap water. Slides were then stained with 1% (w/v) eosin (BDH), washed three times in 100% ethanol, and then cleared in xylene (twice). Sections were coverslipped with Entellan (Merck).
Nissl staining.
Sections warmed to room temperature were rinsed in PBS and then stained with 0.5% w/v cresyl violet acetate (Sigma). After washing in distilled water, sections underwent a series of ethanol washes (1× 70% 30 s, 2× 90% 2 min, 1 × 100% 30 s) and were then cleared in 1-butanol and xylene (twice). Sections were coverslipped with Entellan (Merck). Numbers of neurons in the L4 DRGs of newborn pups were counted on every fourth Nissl-stained transverse whole embryo section and multiplied by 4 to provide approximate neuron numbers for each whole ganglion. Ventral horn motor neurons were counted on multiple adjacent Nissl-stained sections from the lower lumbar spinal cord (at the level of the L3 DRG) and the average number per slide calculated.
Immunostaining of cryosections.
Sections were permeabilized with 1% Triton X-100 in PBS (10 min), blocked in 50% goat serum in PBS, 1% BSA (30 min), and incubated with rabbit polyclonal anti-NF-L (Millipore AB9568) diluted 1:250, and mouse monoclonal anti-neuronal class βIII-Tubulin TUJ1 (Covance MMS-435P) diluted 1:500 in PBS, 1% BSA for 1 h at room temperature. After three PBS washes, sections were incubated with AlexaFluor-488-cojugated anti-rabbit and AlexaFluor-568-conjugated anti-mouse secondary antibodies (Invitrogen) diluted 1:250 in PBS, 1% BSA for 1 h at room temperature. After four washes in PBS, sections were coverslipped in Vectashield containing DAPI (Vector Laboratories) for counterstaining of nuclei.
Whole-mount immunostaining.
Diaphragms dissected from E14.5 embryos that had been immersion fixed in 4% paraformaldehye were washed in PBS and then permeabilized/blocked in PTX (PBS, 0.5% Triton X-100) with 2% milk powder overnight at 4°C. Rabbit polyclonal anti-NF-H or anti-NF-L antibodies (Millipore AB1991 and AB9568) were added direct to the blocking solution (1:1000 and 1:250 dilution, respectively) and incubated for 24 h at 4°C. After multiple washes in PTX (3× 1 h at room temperature and 1× overnight at 4°C), the diaphragms were incubated with an AlexaFluor-488-cojugated anti-rabbit antibody (1:200 dilution) for 24 h at 4°C. Diaphragms were imaged after further multiple washes in PTX (3× 1 h at room temperature and 1× overnight at 4°C) and optical clearing by immersion in an increasing PBS-glycerol series (up to 100% glycerol).
Microscopy and imaging
Phase-contrast and standard fluorescence images of primary neuronal cultures and sections were acquired at room temperature using an Olympus IX81 inverted fluorescence microscope with 4×, 10×, and 20× objectives, standard DAPI, GFP, and TRITC filters, and a Soft Imaging Systems (SIS) F-View camera (analySIS imaging software). Phase-contrast images of the same field of neurites in primary cultures were captured at the indicated time points. Confocal images of immunostained and/or DiI-labeled sections were acquired at room temperature using an Olympus FV1000 confocal microscope imaging system with 40× NA 1.3 and 60× NA 1.35 objectives. Laser excitation and emission filter settings were 405 nm and 425/50 for DAPI, 488 nm and 515/30 for AlexaFluor-488, and 543 nm and 560 LP for DiI/AlexaFluor-568. All channels used the same dichroic mirror with edge wavelengths at 405, 488, and 543 nm. The different fluorescence channels were acquired sequentially. Bright-field images of Nissl-stained sections were acquired at room temperature using an Olympus BX41 upright microscope (5× objective) and MicroPublisher 3.3 camera (QIMaging). Images of hematoxylin and eosin-stained whole embryo sections were acquired using a Nikon SMZ 800 microscope at room temperature with a transillumination light source and Nikon Coolpix 990 digital camera. Images were processed using Adobe Photoshop Elements 6.
Microinjection and quantification of neurite degeneration
Microinjection.
Injections were performed on a Zeiss Axiovert 200 microscope using a FemtoJet and TransferMan NK2 microinjection system (Eppendorf). Plasmids were diluted in 0.5× PBS and passed through a Spin-X filter (Costar) before loading into Femtotips (Eppendorf) and being injected into the nuclei of SCG neurons in dissociated cultures. Plasmids pDsRed2-N1 and LV-Cre-EGFP (containing Cre-EGFP under the control of the CMV promoter) or pEGFP-C1 were injected at a final concentration of 10 ng/μl. Just 50–100 neurons were injected per dish to facilitate visualization of individual labeled neurites.
Quantification of neurite degeneration.
Numbers of continuous, healthy Ds-Red2-labeled neurites were counted in the same field of distal neurites at the same time every day after injection from the second day after injection (labeling was faint at 1 d after injection, making quantification unreliable). The percentage of healthy neurites remaining relative to the total number of labeled neurites (healthy and degenerated) at 2 d after injection was determined. Neurites were deemed unhealthy if they displayed abnormal swellings or had undergone fragmentation. Any new neurites growing into the analyzed field during the time course (mainly in wild-type cultures) were not counted.
Statistical analysis
The statistical analyses described in the text were performed using Microsoft Excel, PASWStatistics version 18.0 (SPSS), and Prism (GraphPad Software) software.
Results
Generation of a mouse line with conditional-ready disruption of Nmnat2
We generated a mouse line with targeted disruption of the Nmnat2 gene using a EUCOMM gene trap allele (Nmnat2Gt(EUCE0262a08)Hmgu) with a conditional-ready gene trap cassette inserted into intron 1 of the gene (Fig. 1A) (Schnutgen et al., 2005). Mice heterozygous and homozygous for the targeted allele will hereafter be referred to as Nmnat2+/gtE and Nmnat2gtE/gtE mice, respectively.
Using RT-PCR and immunoblotting, we found that silencing of the trapped allele appears to be robust (Fig. 1B–E). Nmnat2+/gtE animals showed an ∼50% reduction in expression of Nmnat2 mRNA and NMNAT2 protein in the brains of E18.5 embryos. No expression was detected in Nmnat2gtE/gtE embryos, although trace amounts of Nmnat2 mRNA or protein could nevertheless be present. Similar reductions were also found in E13–E14.5 brains (data not shown). Our immunoblot results, together with use of RT-PCR primers spanning exons 4–5, indicate that alternative promoter usage downstream of the gene trap is unlikely. Nmnat2gtE/gtE mice can therefore be considered to be nullizygous, or at least severely hypomorphic, for the Nmnat2 gene.
Nmnat2gtE/gtE mice show gross morphological changes that are identical to those seen in the NMNAT2-deficient Blad (Nmnat2blad/blad) mutant mouse (Hicks et al., 2012). Nmnat2gtE/gtE pups fail to initiate breathing and die at birth, they do not move in utero or at birth, and they show a hunched posture characteristic of mice with severe neuromuscular defects (Turgeon and Meloche, 2009). Nmnat2gtE/gtE embryos are also unable to micturate and develop a distended bladder by E18.5. In contrast, Nmnat2+/gtE mice are born at the expected ratios, appear normal at birth (as is the case for Nmnat2+/blad mice (Hicks et al., 2012), and show no obvious signs of abnormality up to 12 months.
We found no changes in Nmnat1 or Nmnat3 mRNA levels in the brains of either Nmnat2+/gtE or Nmnat2gtE/gtE E18.5 embryos (Fig. 1C), consistent with results for the Blad mutant mouse (Hicks et al., 2012). We additionally found no change in Nampt mRNA levels (Fig. 1C), the gene encoding the rate-limiting enzyme in the NAD+ salvage pathway. Therefore, depletion of NMNAT2 in Nmnat2+/gtE or Nmnat2gtE/gtE embryos during development does not appear to induce compensatory changes in expression of other critical enzymes in the NAD+ biosynthetic pathway.
Restricted axon extension underlies the peripheral nerve defect in Nmnat2gtE/gtE embryos
Peripheral nerve/axon defects were found to be a key feature of Blad mutant embryos, with dying-back axonopathy, beginning between E13.5 and E15.5, being proposed as a possible cause (Hicks et al., 2012). This was principally based on an immunohistological analysis of embryo sections, which revealed that axons present in the thigh region of mutant embryos at E13.5 have degenerated by E18.5 and that axons are absent in the lower leg and hindpaw at E15.5. However, the presence or absence of axons in more distal regions of the hindlimb at stages earlier than E15.5 was not reported. We therefore used a variety of techniques, including lower-magnification, whole-mount methods, to provide a more complete overview of the extent of the peripheral axon defect in NMNAT2-deficient Nmnat2gtE/gtE embryos at E13–E14.5.
DiI tracing, which can be used to anterogradely label axons (Godement et al., 1987), was first used to longitudinally label spinal nerves and their distal branches in the embryos (Fig. 2A,B). Nerve branches in the hindlimb were consistently labeled as far as the hindpaws in wild-type and Nmnat2+/gtE embryos at both E13/E13.5 and E14.5, but labeled nerves only extended a relatively short distance into the thigh region in Nmnat2gtE/gtE embryos. For intercostal nerves, DiI labeling reached a maximum of 2.39 ± 0.15 mm from the spinal cord in E14.5 Nmnat2gtE/gtE embryos (n = 7 embryos, multiple nerves), compared with 3.76 ± 0.14 mm in wild-types (n = 4) and 3.79 ± 0.16 mm in heterozygotes (n = 3). Intercostal nerves in Nmnat2gtE/gtE embryos also appeared to lack significant lateral branching. These results were indicative of truncated spinal nerve axons. Neurofilament immunostaining of cryosections additionally confirmed that axons are present in pelvic and upper thigh regions of E14.5 Nmnat2gtE/gtE embryos but are essentially absent from more distal regions of the hindlimb (data not shown). Finally, we found that the distal branches of the phrenic nerves are also absent from the diaphragms of Nmnat2gtE/gtE E14.5 embryos using whole-mount neurofilament immunostaining (Fig. 2C). Together, these findings confirmed a widespread peripheral nerve/axon defect as early as E13–E14.5 in Nmnat2gtE/gtE embryos that is characterized by axon truncation. The fact that this is already evident just a short time after axons first invade the developing limb bud at E11.5–E12.5 (Martin, 1990), together with a failure to detect any degenerated axon remnants at E14.5 using neurofilament immunostaining (Fig. 2C; and data not shown), raised the possibility that defective axon outgrowth, rather than dying-back degeneration of more established axons, could be the underlying cause.
Figure 2.
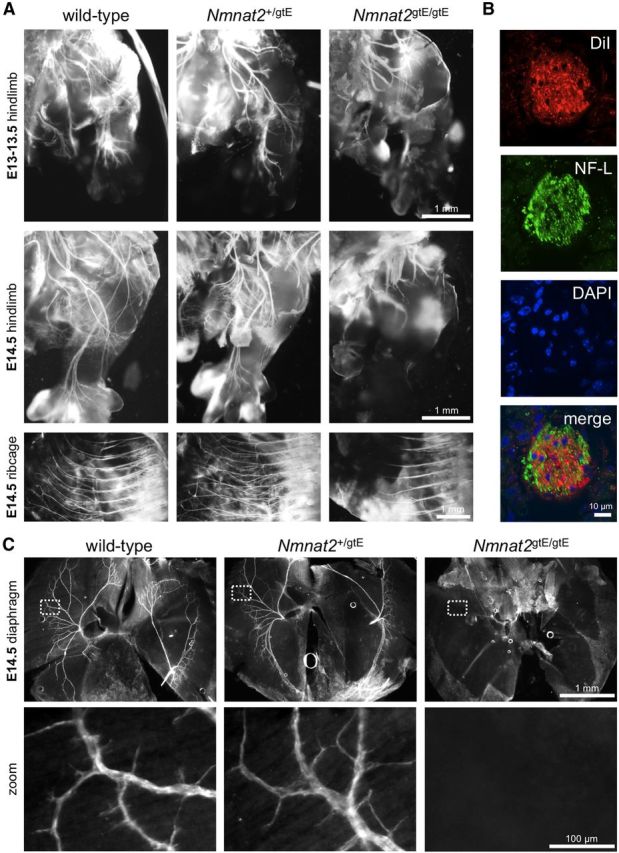
DiI labeling and neurofilament immunostaining reveal peripheral nerve defects in E13–E14.5 Nmnat2gtE/gtE embryos. A, DiI-labeled spinal nerve branches in the hindlimb and intercostal nerves in the ribcages of E13/13.5 and E14.5 wild-type, Nmnat2+/gtE, and Nmnat2gtE/gtE embryos 5 weeks after insertion of DiI crystals into the thoracic and subthoracic spinal cord (images are representative of at least n = 3 embryos of each genotype). B, Confocal optical slice from a transverse cryosection of an intercostal nerve immunostained for NF-L after diffusion of DiI from the thoracic spinal cord of a wild-type E18.5 embryo confirms labeling of structures within the nerve by DiI (DAPI counterstaining reveals nuclei). Lack of precise colocalization is consistent with DiI labeling membranes and NF-L labeling the cytoskeleton. C, Whole-mount NF-L immunostaining of diaphragms from wild-type, Nmnat2+/gtE, and Nmnat2gtE/gtE E14.5 embryos (representative of n = 3 for each genotype) to label phrenic nerve branches. No branches were detected in Nmnat2gtE/gtE diaphragms. NF-H immunostaining revealed a similar lack of phrenic nerve branches in Nmnat2gtE/gtE E14.5 embryos (n = 2 for each genotype), although staining was weaker because of low levels of NF-H expression at this stage (data not shown).
To test this, we monitored neurite outgrowth and survival over time in primary explant cultures of sensory and sympathetic PNS ganglia (Fig. 3). In E13.5–E14.5 DRG explant cultures, radial outgrowth of neurites from Nmnat2gtE/gtE ganglia initially appeared relatively normal up to 1 DIV, but subsequent extension was extremely limited. Neurite extension stalled at just >2 mm by 3 DIV, after which the majority of Nmnat2gtE/gtE neurites began to retract slightly into bundles without showing any other physical signs of deteriorating health. In contrast, neurite outgrowth in Nmnat2+/gtE cultures was comparable with that in wild-type cultures and had exceeded 5 mm by 7 DIV.
Figure 3.
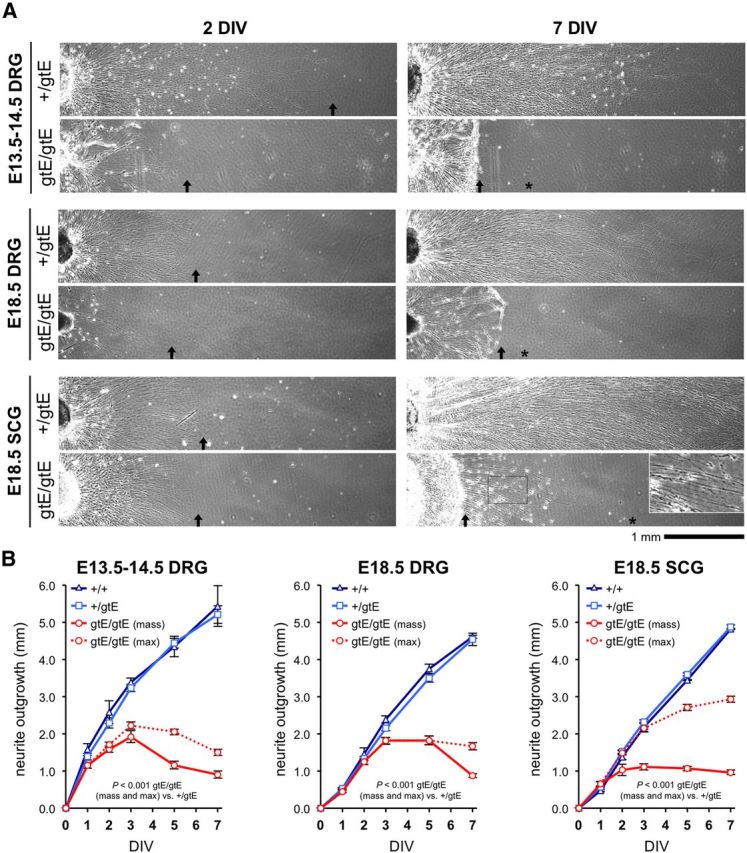
Restricted neurite outgrowth in explant cultures of Nmnat2gtE/gtE DRGs and SCGs at different embryonic stages. A, Representative images of neurite outgrowth at 2 DIV and 7 DIV from cultured E13.5–E14.5 and E18.5 DRG explants and E18.5 SCG explants from Nmnat2+/gtE and Nmnat2gtE/gtE embryos. Nmnat2+/gtE explants were indistinguishable from wild-type explants (data not shown) in all cases (see B). Nmnat2gtE/gtE DRGs taken from E18.5 embryos were noticeably smaller than wild-type and Nmnat2+/gtE DRGs, reflecting extensive DRG loss that occurs at later developmental stages in Nmnat2gtE/gtE embryos (Fig. 8C). The limit of mass outgrowth of the majority of neurites is indicated by an arrow (if located in the image shown) and maximum extension of smaller numbers of neurites in Nmnat2gtE/gtE cultures by an asterisk. Higher magnification of the boxed region in the Nmnat2gtE/gtE SCG culture at 7 DIV shows deteriorating health of the small population of longer neurites. B, Mean radial neurite outgrowth (mm ± SEM) up to 7 DIV in DRG and SCG explant cultures (as indicated) from wild-type (+/+), Nmnat2+/gtE, and Nmnat2gtE/gtE E14.5 or E18.5 embryos. Mass outgrowth (mean extension of the majority of neurites) and maximum (max) outgrowth (mean extension of smaller populations of longer neurites) are plotted separately for Nmnat2gtE/gtE cultures. Each analysis involved n = 2 wild-type, n = 7–11 Nmnat2+/gtE, and n = 6 Nmnat2gtE/gtE embryos, with average growth determined from 1 to 3 ganglia for each embryo. p values were calculated from embryo averages using two-way repeated-measures ANOVA with Bonferroni post hoc correction. Wild-type and Nmnat2+/gtE growth curves were essentially indistinguishable in all cases.
Similar patterns of neurite outgrowth were seen in DRG and SCG cultures from E18.5 embryos. Overall extension rate in E18.5 DRG cultures of all genotypes was slower than for E13.5–E14.5 DRGs, but the maximum extent of outgrowth for Nmnat2gtE/gtE neurites was nevertheless similar (∼2 mm at 3 DIV). Importantly, initial outgrowth (up to 3 DIV) from Nmnat2gtE/gtE E18.5 DRGs was relatively normal, despite the significant DRG neuron loss that has occurred by this developmental stage in embryos lacking NMNAT2 (Hicks et al., 2012) (see Fig. 8C). For Nmnat2gtE/gtE E18.5 SCGs, the length of the majority of neurites was more restricted, with a maximum of just >1 mm of radial outgrowth by 3 DIV. However, a small number of Nmnat2gtE/gtE SCG neurites showed more robust and prolonged extension, possibly reflecting previously reported SCG neuron heterogeneity (Bowers and Zigmond, 1979), but this was still relatively limited (∼3 mm compared with ∼5 mm in wild-type and Nmnat2+/gtE cultures at 7 DIV). Importantly, in contrast to the mass of more stunted Nmnat2gtE/gtE neurites, some blebbing of this small population of longer SCG neurites was evident by 7 DIV (Fig. 3A), suggestive of deteriorating health.
Figure 8.
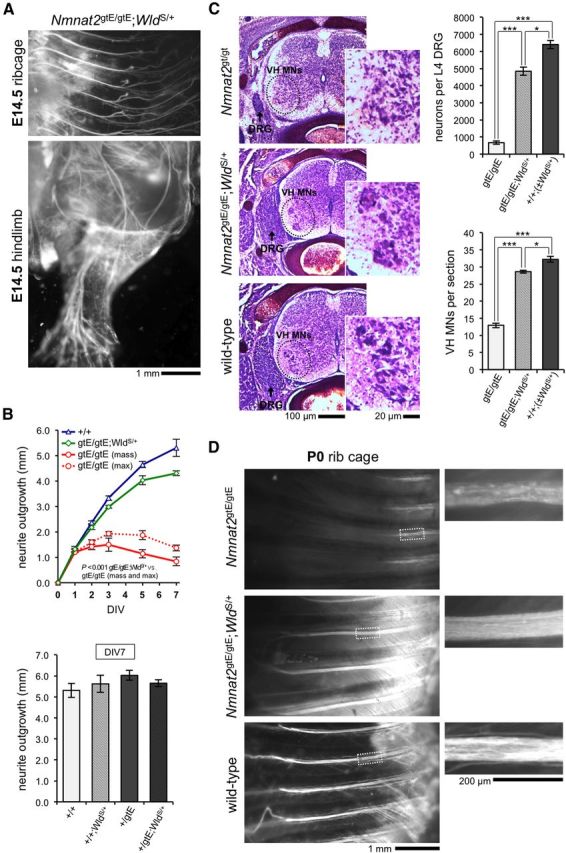
Whole nerve and neuronal defects found in newborn pups lacking NMNAT2 are rescued by WldS expression. A, DiI-labeled intercostal nerves in the ribcage and spinal nerve branches in the hindlimb of an Nmnat2gtE/gtE;WldS/+ E14.5 embryo (representative of n = 3 and matching images in Fig. 2A). B, Mean radial neurite outgrowth (mm ± SEM) up to 7 DIV in E14.5 DRG explant cultures from wild-type (+/+), Nmnat2gtE/gtE, and Nmnat2gtE/gtE;WldS/+ embryos (top: n = 2, 5, and 3 embryos, respectively, with average growth per embryo determined from 1–3 DRGs), and at 7 DIV only in cultures from Nmnat2+/gtE embryos ± WldS/+ (bottom: n = 2 or 3 embryos per genotype/3 DRGs per embryo). p values (top) were calculated from embryo averages using two-way repeated-measures ANOVA with Bonferroni post hoc correction. Data are independent of that in Figure 3B. WldS/+ does not significantly affect wild-type and Nmnat2+/gtE DRG neurite outgrowth (bottom: one-way ANOVA). C, Nissl-stained horizontal cryosections at the level of the L3 DRG (widest aspect), from newborn Nmnat2gtE/gtE, Nmnat2gtE/gtE;WldS/+, and wild-type;WldS/+ (+/+;WldS/+) pups. Reduced DRG size (arrowed) and reduced numbers of ventral horn motor neurons (VH MNs) in the spinal cord (magnified for clarity) are evident in the Nmnat2gtE/gtE pup and are rescued in the Nmnat2gtE/gtE;WldS/+ pup. Mean neuron numbers (± SEM) in L4 DRGs (top) and mean numbers (± SEM) of VH MNs per section in lower lumbar spinal cord (bottom) in each genotype. Data are from n = 3 or 4 pups for each genotype (wild-types ± WldS/+ were grouped; n = 2 each). ***p < 0.001 (one-way ANOVA with Bonferroni post hoc correction). *p < 0.05 (one-way ANOVA with Bonferroni post hoc correction). D, DiI-labeled intercostal nerves in the ribcages of newborn Nmnat2gtE/gtE, Nmnat2gtE/gtE;WldS/+, and wild-type pups (representative of n = 3 per genotype). Magnified regions highlight axon degeneration in Nmnat2gtE/gtE intercostal nerves.
These data indicate that the maximum length a neurite can extend, or can be supported, is severely limited in primary cultures of two different populations of peripheral neurons from Nmnat2gtE/gtE embryos at different developmental stages. This is an inherent defect as glia are essentially absent from these cultures. Intriguingly, the extent of radial neurite outgrowth in these cultures is broadly comparable with the consistent length of the peripheral nerve stumps in E13–E14.5 Nmnat2gtE/gtE embryos.
Axon defects in the CNS of Nmnat2gtE/gtE embryos
Gross structural changes were reported to be absent in Blad mutant brain and spinal cord, and the populations of CNS neurons that were found to be depleted in late-stage embryos (motor neurons in the ventral horn of the spinal cord and some cranial nerve nuclei) extend their axons into peripheral nerves (Hicks et al., 2012). We similarly found no obvious signs of gross structural changes in the CNS of late-stage Nmnat2gtE/gtE embryos or newborn pups using hematoxylin and eosin and Nissl staining (data not shown). However, given the profound axon defect in the periphery, we next investigated whether similar axonal defects are present in the CNS of Nmnat2gtE/gtE embryos that are not revealed using these detection methods.
We sought direct evidence for truncated axons originating from CNS neuron populations in Nmnat2gtE/gtE E18.5 embryos using a combination of NF-L and βIII-Tubulin immunostaining of brain and spinal cord sections to highlight axons and axon tracts. The clearest example was found in the visual system (Fig. 4). Both NF-L and βIII-Tubulin immunostaining revealed the presence of truncated retinal ganglion cell (RGC) axons in the optic nerve of Nmnat2gtE/gtE embryos (Fig. 4A). Axons fall short of the optic chiasma, and the optic tract is essentially absent. In addition, numerous swellings containing neurofilament-L accumulations were found toward the distal ends of the truncated axons (Fig. 4B). Importantly, the optic fiber layer in the retina, containing proximal RGC axon segments, appeared normal in Nmnat2gtE/gtE embryos, and there was no evidence for significant loss of RGC viability (Fig. 4C), indicating that this is a primary axonal defect.
Figure 4.

An RGC axon defect in Nmnat2gtE/gtE optic nerve. A, Low-magnification wide-field images of NF-L and βIII-Tubulin (TUJ1) immunostaining of horizontal cryosections of the heads of wild-type, Nmnat2gtE/gtE, and Nmnat2gtE/gtE;WldS/+ E18.5 embryos at the level of the optic nerve (ON), optic chiasma (OX), and optic tract (OT). Maximal length of NF-L and βIII-Tubulin-labeled RGC axons in the Nmnat2gtE/gtE ON (as revealed by staining of adjacent serial sections for >150 μm in each direction) is indicated by an asterisk. The RGC axon defect appears to be largely corrected in Nmnat2gtE/gtE;WldS/+ embryos. B, Confocal slices of the boxed regions in A highlight abnormal swellings containing accumulations of NF-L at the distal ends of the truncated RGC axons in Nmnat2gtE/gtE embryos. Arrows point to swellings in higher-magnification images (zoom) of the boxed regions in the panels above. C, NF-L immunostaining and DAPI counterstaining of horizontal sections through the retinas of wild-type and Nmnat2gtE/gtE E18.5 embryos revealed a normal optic fiber layer (OFL) and no evidence of significant pyknosis in the ganglion cell layer (GCL) in the mutant. Images are representative of n = 3 embryos of each genotype. Nmnat2+/gtE embryos (n = 3) were indistinguishable from wild-types (data not shown).
Reduction, or even absence, of corticospinal tract axons in the dorsal column of cervical segments of the spinal cord in E18.5 Nmnat2gtE/gtE embryos (Fig. 5A) may reflect limited axon extension in another CNS neuron population. These axons, which mostly originate from neurons in the primary motor cortex and project caudally along the spinal cord, are still in their growth phase at this developmental stage (Hsu et al., 2006). The lack of any clear signs of degenerated axon remnants at this level suggests that axon extension in the mutant is either delayed or has stalled at an earlier point along the tract.
Figure 5.
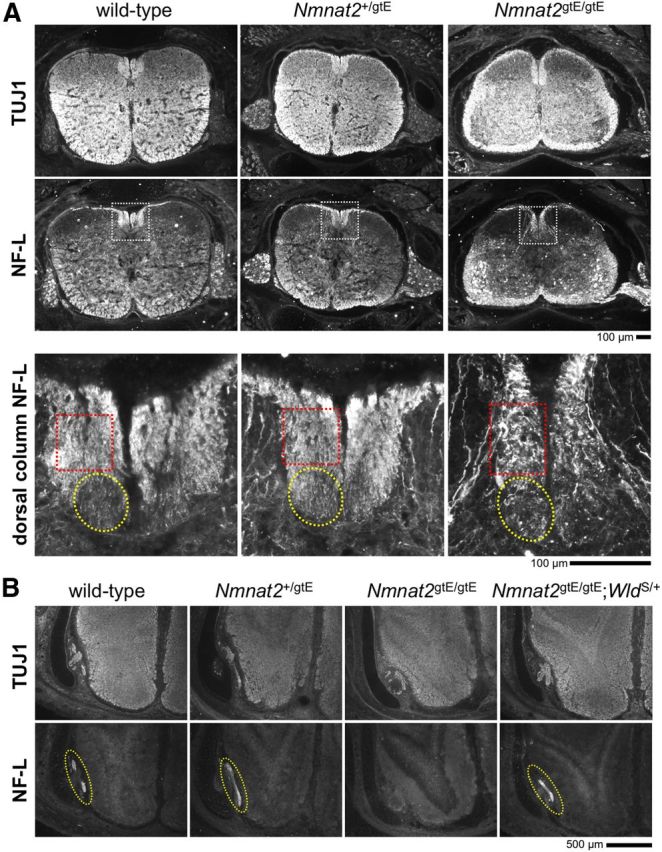
Axon defects in the spinal cord and olfactory bulb in E18.5 Nmnat2gtE/gtE embryos. Images of NF-L and βIII-Tubulin (TUJ1) immunostaining of horizontal cryosections of the lower cervical spinal cord (and surrounding tissues) (A), and the olfactory bulb (B), of wild-type, Nmnat2+/gtE, Nmnat2gtE/gtE, and Nmnat2gtE/gtE;WldS/+ E18.5 embryos (representative of serial sections from n = 3 embryos of each genotype). Higher-magnification images of NF-L immunostaining in the dorsal column of the spinal cord (white boxed region) are shown at the bottom of A. The approximate locations of the corticospinal tract (yellow circle) and gracile tract (red box) are indicated. The dorsal column appears elongated in the Nmnat2gtE/gtE embryo, but the overall area is similar to that in wild-type and Nmnat2+/gtE embryos. However, there is a clear deficiency of axons in the corticospinal tract in the Nmnat2gtE/gtE dorsal column, and the gracile tract appears to contain fewer, severely dystrophic axons. B, The olfactory nerve layer (circled in yellow) can easily be identified in wild-type, Nmnat2+/gtE, and Nmnat2gtE/gtE;WldS/+ sections but is absent from serial sections through the comparable region in Nmnat2gtE/gtE embryos.
Other axonal changes are also evident in the CNS of E18.5 Nmnat2gtE/gtE embryos, including an absence of healthy-looking gracile tract axons in the dorsal column of the spinal cord (Fig. 5A) and complete absence of the olfactory nerve fiber layer in the olfactory bulb (Fig. 5B). However, although these axon populations reside in the CNS, they are the central projections of DRG and olfactory receptor neurons located in the periphery, respectively. The extent to which early axonal defects might contribute is also not clear. Certainly, some of the axon loss and apparent axonal dystrophy in the gracile tract is likely to be the result of axon degeneration secondary to the extensive DRG cell death that is evident by E18.5/P0 in Nmnat2gtE/gtE embryos (see Fig. 8C).
These observations suggest that axonal defects may be relatively common within the CNS of Nmnat2gtE/gtE embryos, with some being evident in axon tracts derived from CNS neuron populations, and not just central projections of peripheral neurons. To establish whether this reflects defective axon extension, as appears likely in the PNS, we assessed neurite outgrowth in primary cultures of cortical explants from E18.5 embryos (Fig. 6). Cortical neurite extension was much slower than for peripheral ganglia, but outgrowth from Nmnat2gtE/gtE cortical explants was similarly severely restricted after an initial, short period of normal extension (Fig. 6A–C). Radial outgrowth reached a maximum of 1.5 mm by 7 DIV, whereas both wild-type and Nmnat2+/gtE cultures continued to extend to >2.5 mm by 14 DIV. In contrast to the majority of neurites in Nmnat2gtE/gtE DRG and SCG cultures, Nmnat2gtE/gtE cortical neurites started to show signs of significant blebbing from 7 DIV once this maximum had been reached (Fig. 6D). However, there was no subsequent dying-back of neurites (Fig. 6A), and calcein-AM staining at 14 DIV suggested that the blebbed neurites retained esterase activity and were not completely degenerated (Fig. 6B,D). Intriguingly, blebbing of the stunted Nmnat2gtE/gtE cortical neurites in these cultures mirrors the swellings seen along truncated Nmnat2gtE/gtE RGC axons in the optic nerve.
Figure 6.

Restricted neurite outgrowth in Nmnat2gtE/gtE cortical explant cultures. A, Mean radial neurite outgrowth (mm ± SEM) up to 14 DIV in cortical explant cultures from wild-type (+/+), Nmnat2+/gtE, and Nmnat2gtE/gtE E18.5 embryos (n = 4 embryos per genotype, with average growth per embryo determined from 2 separate explants). p values were calculated from embryo averages using two-way repeated-measures ANOVA with Bonferroni post hoc correction. B, Representative fluorescent images of cortical explant cultures stained with calcein-AM at 14 DIV. C, Mean radial neurite outgrowth (mm ± SEM) in cortical explant cultures as revealed by Calcein-AM staining at 14 DIV. Data are from the same cultures as in A. **p < 0.01, Nmnat2gtE/gtE versus both wild-type and Nmnat2+/gtE (calculated from embryo averages using one-way ANOVA with Bonferroni post hoc correction). Measurements based on calcein-AM staining and phase-contrast images are therefore consistent with each other. D, Representative phase-contrast images of neurites in cortical explant cultures at 7 DIV, and fluorescent images of calcein-AM stained neurites at 14 DIV. Significant neurite blebbing is visible in Nmnat2gtE/gtE cultures at both time points.
These data indicate that a lack of NMNAT2 significantly limits neurite extension in a major population of CNS neurons via a mechanism that does not appear to involve dying-back degeneration of longer neurites. This largely matches the situation in peripheral ganglia explant cultures and may underlie the RGC axon defect in the optic nerve.
Defects in Nmnat2gtE/gtE mice are rescued by WldS dose-dependently
We proposed that the WldS protein can substitute for NMNAT2 in neurites in primary SCG neuron cultures because of its shared enzyme activity and greater stability, enabling neurites to survive after axotomy or NMNAT2 knockdown (Gilley and Coleman, 2010). To address whether it can also compensate for a lack of NMNAT2 in vivo, we bred Nmnat2gtE/gtE mice that were heterozygous for the WldS mutation (Nmnat2gtE/gtE;WldS/+). Whereas Nmnat2gtE/gtE pups are hunched at birth and cannot breathe, Nmnat2gtE/gtE;WldS/+ pups were born with a normal posture and could initiate breathing in most cases (Fig. 7A,B). However, all still died within a few hours of birth.
Figure 7.
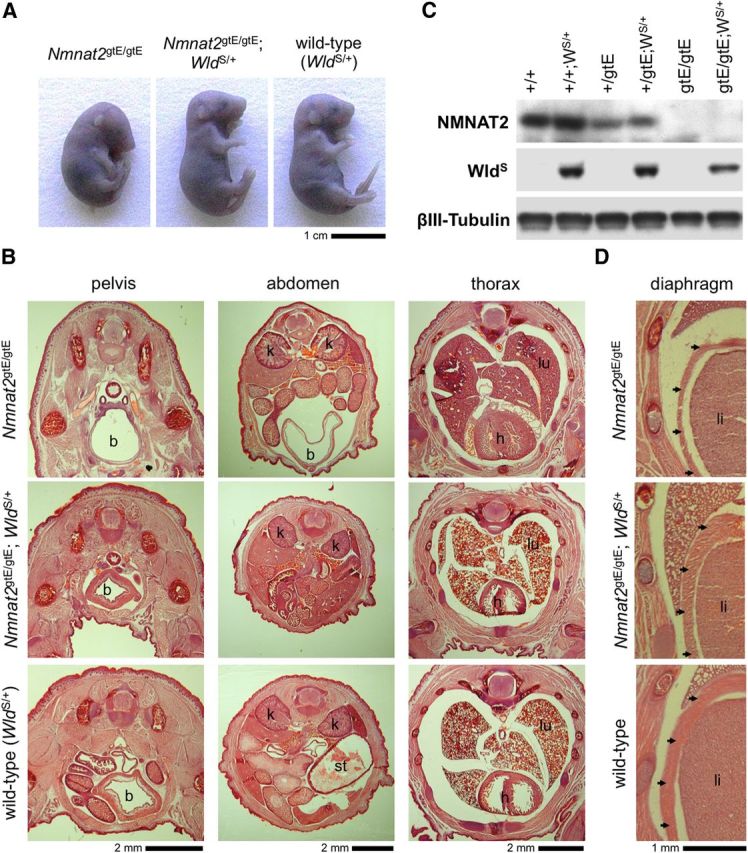
Gross morphological defects found in newborn pups lacking NMNAT2 are rescued by WldS expression. A, Posture of Nmnat2gtE/gtE, Nmnat2gtE/gtE;WldS/+, and wild-type (WldS/+) E18.5 embryos. All Nmnat2gtE/gtE;WldS/+ E18.5 embryos and newborn pups observed to date (n = 13) had a normal posture. Heterozygosity for WldS does not affect the gross appearance of wild-type or Nmnat2+/gtE embryos (which are also normal; data not shown). B, Hematoxylin and eosin-stained pelvic, abdominal, and thoracic cryosections from newborn Nmnat2gtE/gtE, Nmnat2gtE/gtE;WldS/+, and wild-type (WldS/+) pups. A distended bladder (b) can be seen to extend to the level of the kidneys (k) in the abdomen of the Nmnat2gtE/gtE pup. There is also reduced muscle mass in the thigh, pelvis, and abdominal walls, and collapsed lungs (lu) are indicative of a failure to breathe. The Nmnat2gtE/gtE;WldS/+ pup appears similar to the wild-type with a nondistended bladder, normal muscle mass, and lungs (lu) that show clear signs of inflation. A distended stomach (st) can be seen in the abdomen of the wild-type pup. C, Immunoblot showing selective expression of WldS in newborn wild-type (+/+), Nmnat2+/gtE, and Nmnat2gtE/gtE pups heterozygous for WldS (WS/+). βIII-Tubulin represents the sample control, and NMNAT2 levels confirm the Nmnat2 genotype. D, Hematoxylin and eosin-stained cryosections showing an underdeveloped diaphragm in a newborn Nmnat2gtE/gtE pup but normal diaphragms in newborn Nmnat2gtE/gtE;WldS/+ and wild-type pups. The diaphragm (arrowhead) lies between the liver (li), the lungs (top), and the ribcage (left) in these sections. B, D, Images are representative of at least n = 3 per genotype.
Despite its limited ability to extend life span, heterozygous WldS expression in these animals (confirmed by immunoblotting; Fig. 7C) was sufficient to rescue other morphological and neuronal defects associated with a lack of NMNAT2. At the gross morphological level, we found that the bladders of newborn Nmnat2gtE/gtE;WldS/+ pups and full-term embryos (E18.5) were not distended (Fig. 7B), suggesting a correction of the micturition defect (Hicks et al., 2012). Reduced muscle mass, likely because of failed innervation, is also a key feature of Nmnat2gtE/gtE and Blad embryos and newborn pups, but there was no obvious wasting of skeletal muscles in the torsos and limbs of Nmnat2gtE/gtE;WldS/+ animals (Fig. 7B). Importantly, the diaphragm, which is underdeveloped in the mutant animals, also appeared normal in the presence of WldS (Fig. 7D).
With regard to any peripheral nerve defect, we found that DiI labeling of spinal nerves in Nmnat2gtE/gtE;WldS/+ E14.5 embryos revealed a grossly normal appearance and length of intercostal nerves and nerves in the hindlimb (Fig. 8A). Consistent with this, WldS expression largely corrected the Nmnat2gtE/gtE neurite outgrowth defect in E14.5 DRG explant cultures without having any significant effect on neurite outgrowth in wild-type or Nmnat2+/gtE cultures (Fig. 8B). Rescue of the axon outgrowth defect, resulting in greatly improved innervation of target tissues, therefore likely accounts for the normal gross morphology of late-stage Nmnat2gtE/gtE;WldS/+ embryos and newborn pups. WldS expression also largely prevented the massive loss of DRG neurons and spinal cord motor neurons that occurs late in the development of NMNAT2-deficient embryos (Fig. 8C), and the degeneration of truncated axons in spinal nerve stumps (Fig. 8D), themselves possible consequences of an initial axon extension defect (see Discussion).
Interestingly, heterozygous levels of WldS also rescued CNS defects associated with a lack of NMNAT2, including the RGC axon defect in the optic nerve (Fig. 4A) and the loss of the olfactory nerve layer in the olfactory bulb (Fig. 5B). We also found that neurite outgrowth in Nmnat2gtE/gtE;WldS/+ cortical explant cultures is partially rescued, with neurites extending to 2.33 ± 0.11 mm by 14 DIV (n = 4 explants from 2 embryos, compare withFig. 6C).
Although the rescue of defects attributable to restricted axonal extension is substantial in Nmnat2gtE/gtE;WldS/+ mice, it does not significantly extend life span, presumably because it is not complete. However, as delayed Wallerian degeneration in WldS mice is dose-dependent (Mack et al., 2001), we bred Nmnat2gtE/gtE mice homozygous for WldS (Nmnat2gtE/gtE;WldS/S) and found that increased WldS expression results in more complete rescue. Nmnat2gtE/gtE;WldS/S mice survive to breeding age (2–3 months) without overt signs of abnormality, are within a healthy weight range, and are fertile. This indicates that WldS can offset an absence of NMNAT2 in vivo into adulthood, but only if levels are high enough.
Mice expressing subheterozygous levels of NMNAT2 are viable
We next investigated whether NMNAT2 expression at levels lower than those in Nmnat2+/gtE mice is compatible with survival. We crossed Nmnat2+/gtE mice to mice heterozygous for an alternative gene-trapped Nmnat2 allele (termed Nmnat2gtBay to distinguish it from the Nmnat2gtE allele), which suppresses expression from the trapped allele by only ∼50% (Mayer et al., 2010). This produced compound heterozygotes (Nmnat2gtBay/gtE) expressing Nmnat2 mRNA and NMNAT2 protein at ∼25% of wild-type levels (Fig. 9). Despite the lower expression levels, Nmnat2gtBay/gtE mice are viable and survive to at least 12 months of age without any overt abnormalities. Therefore, the threshold level of NMNAT2 required for normal development and survival appears to be ∼≤25% of wild-type levels.
Figure 9.
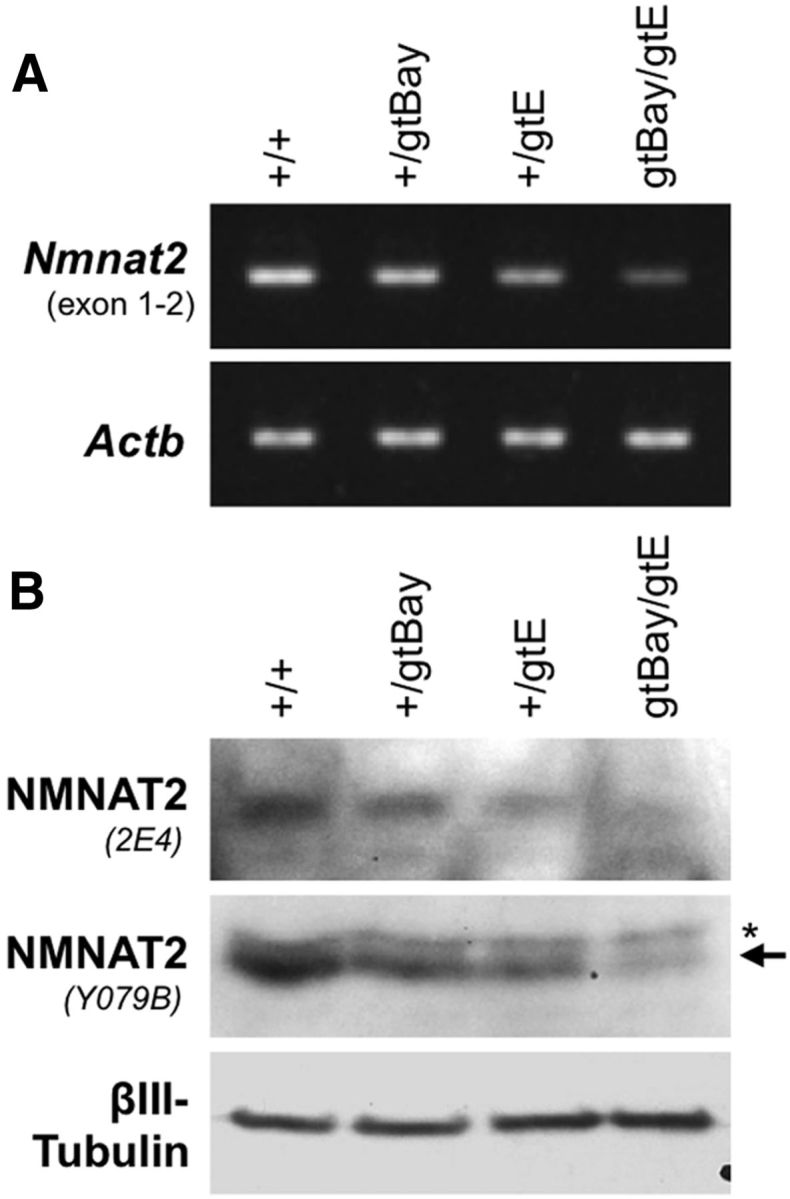
Subheterozygous levels of NMNAT2 expression in Nmnat2gtBay/gtE compound heterozygote mice. A, RT-PCR showing levels of Nmnat2 mRNA in brains of 6 month wild-type, Nmnat2+/gtBay, Nmnat2+/gtE, and Nmnat2gtBay/gtE mice. Levels of Actb mRNA act as a reference. B, Representative immunoblot showing NMNAT2 levels (two independent antibodies) in brains of 6 month wild-type, Nmnat2+/gtBay, Nmnat2+/gtE, and Nmnat2gtBay/gtE mice. βIII-Tubulin acts as the sample control.
Conditional silencing of a single Nmnat2 allele is sufficient to induce neurite degeneration
We have previously shown that siRNA-mediated depletion of NMNAT2 in primary SCG neuron cultures triggers Wallerian-like neurite degeneration (Gilley and Coleman, 2010). The gene trap cassette in the Nmnat2gtE allele is conditional-ready (Schnutgen et al., 2005) and can be inactivated and then reactivated via sequential expression of FLPe and Cre recombinases (Fig. 10A). We therefore used this functionality to test whether conditional silencing of trapped Nmnat2 alleles could replicate our siRNA results.
Figure 10.
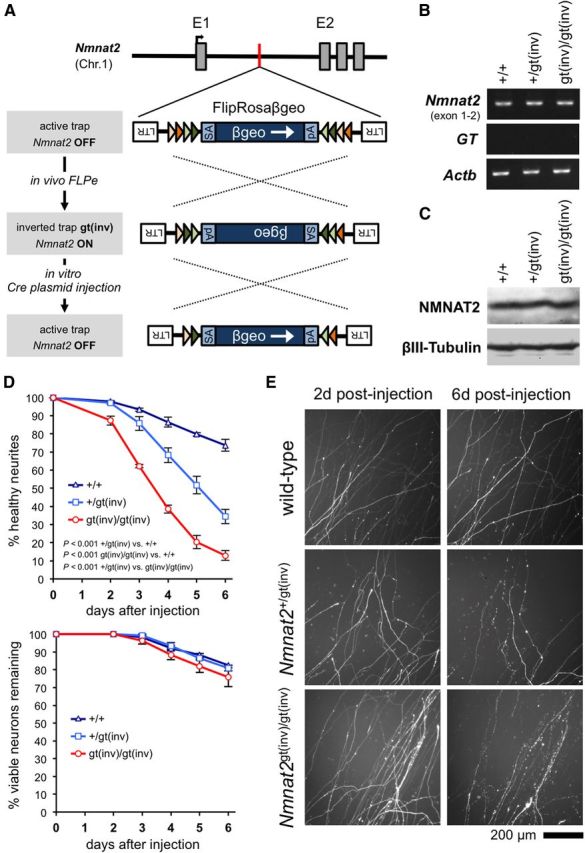
Silencing of a single Nmnat2 allele is sufficient to induce neurite degeneration in primary SCG neuron cultures. A, Schematic showing how sequential exposure to FLPe and Cre recombinases allows restoration of expression from the targeted Nmnat2 allele, and subsequent resilencing, through successive inversion of the gene trap facilitated by pairs of inversely orientated heterotypic FLPe and Cre recombinase target sequences (colored triangles) (Schnutgen et al., 2005). B, C, Representative RT-PCR (B) and representative immunoblot (C) showing Nmnat2 mRNA and NMNAT2 protein levels in brains of wild-type, Nmnat2+/gt(inv), and Nmnat2gt(inv)/gt(inv) P1 pups. Actb and βIII-Tubulin represent sample references. D, Survival of DsRed2-labeled distal neurites (top) and neuron viability (bottom) for wild-type (+/+), Nmnat2+/gt(inv), and Nmnat2gt(inv)/gt(inv) SCG neurons injected with a Cre-EGFP expression vector and pDsRed2 (both at 10 ng/μl). The mean percentage (± SEM) of healthy DsRed2-labeled neurites or viable cell bodies remaining each day after injection, of those (both healthy and abnormal) present in the same field at 2 d after injection, is plotted. Neurite data are from multiple fields in five independent injection experiments for each genotype. Injected cell bodies with normal gross morphology were classed as viable. We previously demonstrated that morphology revealed by fluorescent proteins is a reliable indicator of neuronal viability (Gilley and Coleman, 2010). p values were calculated from individual experiment averages using two-way repeated-measures ANOVA with Bonferroni post hoc correction. We also saw no increase in neurite degeneration (relative to wild-type) for Nmnat2+/gt(inv) and Nmnat2gt(inv)/gt(inv) SCG neurons injected with a control EGFP expression vector, rather than Cre-EGFP (data not shown). E, Representative fields of DsRed2-labeled distal neurites from experiments quantified in D at 2 and 6 d after injection.
Nmnat2+/gtE mice were first bred to FLPe transgenic mice to invert the gene trap cassette (Nmnat2gt(inv)) and reinstate Nmnat2 expression from the targeted allele. Mice were then bred to homozygosity. As predicted, Nmnat2 mRNA and NMNAT2 protein levels were restored to wild-type levels in both Nmnat2+/gt(inv) and Nmnat2gt(inv)/gt(inv) brains (Fig. 10B,C), and homozygotes were viable. We then prepared dissociated cultures of SCG neurons from wild-type, Nmnat2+/gt(inv), and Nmnat2gt(inv)/gt(inv) pups and injected them with an expression construct for EGFP-tagged Cre recombinase to invert the gene trap for a second time and thus resilence trapped Nmnat2 alleles. We also injected a DsRed2 expression vector to allow visualization of neurites of the injected neurons.
As predicted, we found that Cre-EGFP expression triggered robust dying-back degeneration of distal neurites in Nmnat2gt(inv)/gt(inv) cultures relative to controls (Fig. 10D,E). This occurred at a slower rate than that reported for neurite degeneration induced by Nmnat2 siRNA (Gilley and Coleman, 2010), presumably because of the different methods used to deplete NMNAT2. In the case of Cre-EGFP injection, this requires several additional steps, including expression of Cre-EGFP, recombination of the trapped allele, and natural turnover of Nmnat2 mRNA, whereas siRNA can actively target Nmnat2 mRNA for degradation as soon as injected. Notably, in contrast to siRNA-mediated NMNAT2 depletion, there was no significant reduction in neuronal viability associated with Cre-mediated Nmnat2gtE silencing relative to the control (Fig. 10D). The extent of loss of neuronal viability for all genotypes was consistent with a normal level of loss that occurs in experiments of this type and likely underlies the gradual loss of neurites seen in control cultures.
However, a much more unexpected outcome of this analysis was that distal neurites of Nmnat2+/gt(inv) neurons expressing Cre-EGFP also showed significantly increased degeneration relative to controls (Fig. 10D,E). This was delayed with respect to Nmnat2gt(inv)/gt(inv) neurons but was still extensive, with almost 70% of distal neurites having degenerated by 6 d after injection. Maintenance of established SCG neurites therefore requires NMNAT2 levels at least 50% that of wild-type levels. This directly contrasts with the normal neurite outgrowth observed in constitutive heterozygote Nmnat2+/gtE SCG cultures (Fig. 3) and the fact that mice with constitutive depletion of NMNAT2 to as low as 25% of normal levels are healthy and outwardly normal (see above). It therefore suggests that compensatory mechanisms are triggered by otherwise limiting amounts of NMNAT2, at least in primary culture and probably also in vivo, to promote normal axon extension and survival. These putative compensatory changes appear not to alter expression of other enzymes in the NAD+ salvage pathway (Fig. 1C). We have additionally found no evidence for reduced expression of SARM1, a molecule that is required for activation of Wallerian degeneration (Osterloh et al., 2012), in the brains of E14.5 or E18.5 Nmnat2+/gtE embryos or adult Nmnat2+/gtE and Nmnat2gtBay/gtE mice (data not shown).
Discussion
Nmnat2gtE/gtE embryos replicate all the initial phenotypic characteristics reported for the NMNAT2-deficient Blad mutant, providing clear confirmation that NMNAT2 has an essential developmental role. However, the extended findings presented here provide important new insight. Most significantly, neurite outgrowth assays implicate stalled extension as the origin of axonal defects in NMNAT2-deficient embryos, rather than the dying-back degeneration of established axons, which was proposed previously (Hicks et al., 2012). Critically, this suggests that NMNAT2 is required for normal axon growth, in addition to its previously reported axon maintenance function (Gilley and Coleman, 2010).
Restricted axon extension could also underlie the widespread sensory and motor neuron cell death in late-stage Nmnat2gtE/gtE and Blad embryos. Many neuron types become dependent on target-derived trophic support for survival during this stage of embryonic development (Oppenheim, 1991; Raff et al., 1993), so failure of truncated axons to innervate more distal targets from the outset would be expected to trigger programmed cell death. Consequently, the extensive degeneration of (already truncated) peripheral nerve axons seen at the same, late stages in Nmnat2gtE/gtE and Blad embryos is predicted to be secondary to neuronal death, rather than primary dying-back degeneration. The small numbers of surviving sensory and motor neurons in DRGs and ventral horn of the spinal cord of P0 Nmnat2gtE/gtE pups may be those that innervate more proximal targets within range of the limited axon length. The fact that Nmnat2gtE/gtE DRG and SCG neurons, from both mid-stage and late-stage embryos, can support secondary regrowth of neurites and maintain mostly healthy, if somewhat stunted, neurites when provided with appropriate trophic provision in primary culture, further supports this possible interpretation.
Given its central role in NAD+ biosynthesis, it seems likely that the length of axon that can be supported when NMNAT2 is lacking will be restricted by limiting levels of NAD+ itself or other related metabolites. As such, the range of influence of the other NMNATs might be a critical determinant. NMNAT3, being mitochondrial, should be present along the whole length of axons. Thus, it would be hard to explain why axon length is so restricted if this source of NMNAT activity has any meaningful capacity to promote axon extension. Indeed, this suggests that endogenous NMNAT3, which is expressed at low levels in the nervous system (Berger et al., 2005; Orsomando et al., 2012), is either too scarce or too compartmentalized to have any influence. Nuclear NMNAT1 activity and short-range diffusion of critical metabolite(s) between the cell body and axon therefore appear to be a more likely determinant of axon length in NMNAT2-deficient embryos. Alternatively, loss of chaperone activity associated with NMNAT isoforms (Zhai et al., 2008; Ljungberg et al., 2012) could perhaps be involved, with other chaperones able to partially compensate for NMNAT2 loss.
Of course, although robust reductions in Nmnat2 expression are seen in Nmnat2gtE/gtE embryos, the possibility that the initial, normal growth in Nmnat2gtE/gtE cultures is actually the result of extremely low residual levels of NMNAT2 cannot be ruled out. Gene trapping involves alternative splice acceptor usage; consequently, suppression of gene expression is sometimes incomplete (a good example being the Nmnat2gtBAY allele). Interestingly, this could also explain the less restricted outgrowth of small numbers of neurites in Nmnat2gtE/gtE SCG explant cultures if differential splicing occurs in a subpopulation of the neurons.
A requirement of NMNAT2 for normal axon extension naturally leads us to speculate about a possible role during axon regeneration. In this respect, it will be critical to clearly establish the mechanistic relationship between its axon growth and maintenance roles. Most of the stunted Nmnat2gtE/gtE neurites in SCG and DRG cultures appear healthy, and the small amount of retraction that does occur at later time points is probably the result of mechanical forces associated with neurite bundling rather than degeneration. However, the distal ends of Nmnat2gtE/gtE RGC axons in the optic nerve and cortical neurites in primary cultures, as well as some of the less stunted Nmnat2gtE/gtE neurites in SCG cultures, do develop signs of blebbing after initially appearing healthy. This suggests that they may be close to the limit of survival. Thus, it is possible that stalled extension simply reflects the point at which a threshold for degeneration is reached. Although the ability of WldS to rescue Nmnat2gtE/gtE axon extension does not differentiate these possibilities, as it ultimately fulfills the same function as NMNAT2, assessing the effects of unrelated genetic manipulations that modulate Wallerian-like degeneration, such as an absence of MORN4 or SARM1 (Bhattacharya et al., 2012; Osterloh et al., 2012), should provide important mechanistic insight. Nevertheless, the possibility that compromised function or restricted availability of NMNAT2 might influence axon regeneration is intriguing and clearly an area for future investigation.
The ability of WldS expression to correct developmental defects associated with a lack of NMNAT2 in vivo nicely complements our previous findings in primary culture (Gilley and Coleman, 2010). Furthermore, the dose dependence of this effect mirrors the dose-dependent protection of cut axons by WldS (Mack et al., 2001). Whereas WldS stability appears critical for its ability to delay Wallerian degeneration by substituting for short-lived NMNAT2, relative rates of axonal delivery are probably more critical with respect to its dose-dependent ability to rescue the extension defect in axons lacking NMNAT2. A failure to match the normal rate of supply of NMNAT2 with delivery of WldS might explain the incomplete rescue in Nmnat2gtE/gtE;WldS/+ neurons. Healthier supply when expression is higher could account for the improved survival of Nmnat2gtE/gtE;WldS/S mice. Intriguingly, relatively slower delivery and accumulation of WldS in extending axons could also explain why its capacity to delay Wallerian degeneration takes several days to become fully established in WldS neuronal cultures (Buckmaster et al., 1995).
Consistent with Nmnat2+/Blad mice (Hicks et al., 2012), Nmnat2+/gtE mice develop normally and appear healthy up to at least 12 months. Nmnat2gtBAY/gtE compound heterozygotes, with further reduced NMNAT2 levels, are also outwardly healthy up to this age. Given that NMNAT2 is a low abundance protein (Mayer et al., 2010), this sets a surprisingly low threshold of NMNAT2 for normal development and animal survival, although enzyme activity will amplify its effects. The finding that conditional silencing of a single Nmnat2 allele in primary culture is sufficient to trigger distal neurite degeneration was therefore both unexpected and intriguing. This leads us to suggest that otherwise limiting levels of NMNAT2 trigger compensatory mechanisms during development, allowing normal axon development and survival. Establishing what compensatory changes occur in these mice will likely be very revealing with regard to axon growth and/or maintenance mechanisms and could identify novel therapeutic targets for axonopathies. So far, we have ruled out altered expression of other key enzymes in the NAD+ salvage pathway or reduced expression of SARM1 as being involved.
Previously, we postulated that decreasing NMNAT2 levels in axons might precipitate dying-back axon degeneration in neurodegenerative diseases (Gilley and Coleman, 2010). Reduced Nmnat2 expression has been linked to pathogenesis in models of amyloid pathology and tauopathy (Yan et al., 2010; Ljungberg et al., 2012), but reduced delivery of NMNAT2, resulting from natural age-related or pathology-driven declines in transport, could also trigger dying-back degeneration. Thus, Nmnat2+/gtE and Nmnat2gtBAY/gtE mice should be useful resources for studying the effect of reduced NMNAT2 levels on axon loss and declining cognitive function during aging. It will be important to determine whether compensatory changes that allow normal development are also activated in this situation. The conditional functionality of the Nmnat2gtE allele should also facilitate analysis of the effect of NMNAT2 loss in axons in adult mice, as other, unrelated changes that likely occur in transected or damaged axons should be absent. These mice could also be a valuable resource for studying the influence of NMNAT2 in non-neuronal situations, such as cardiac hypertrophy (Cai et al., 2012), or other aspects of NAD+ biology.
Finally, it is intriguing that peripheral nerve and axon outgrowth defects in mice lacking the transcription factor CREB (Lonze et al., 2002) are similar to those in NMNAT2-deficient mice, given that CREB has been implicated in Nmnat2 regulation (Ljungberg et al., 2012). Of course, CREB regulates a broad range of genes in response to a variety of stimuli in neurons (Lonze and Ginty, 2002), but it would nevertheless be interesting to establish whether reduced NMNAT2 expression contributes to this phenotype in these mice.
In conclusion, we have proposed that widespread axon truncation in both the periphery and CNS of embryos lacking NMNAT2, likely a key determinant of their overall phenotype, reflects severely limited axon extension during early development, rather than later dying-back axonopathy. We have also shown that both exogenous and endogenous mechanisms can rescue the phenotype. WldS can directly substitute for NMNAT2 in vivo, both in a maintenance role similar to that in primary culture and in this previously unstudied developmental role. However, most intriguingly, our findings suggest that compensatory changes occur during development to facilitate normal axon growth and survival at limiting NMNAT2 levels. Our conditional-ready Nmnat2-targeted mouse will be a useful resource for future studies of the role of NMNAT2 in axons, as well as in a wide variety of other contexts.
Footnotes
This work was supported by United Kingdom Medical Research Council Grant G1000702. We thank Dr. Anne Segonds-Pichon for help with statistical analysis and Alexander Brown for generation of preliminary data.
The authors declare no competing financial interests.
References
- Adalbert R, Coleman MP. Axon pathology in age-related neurodegenerative disorders. Neuropathol Appl Neurobiol. 2012 doi: 10.1111/j.1365-2990.2012.01308.x. doi: 10.1111/j.1365-2990.2012.01308.x. Advance online publication. Retrieved Oct. 10, 2012. [DOI] [PubMed] [Google Scholar]
- Babetto E, Beirowski B, Russler EV, Milbrandt J, Diantonio A. The Phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep. 2013;3:1422–1429. doi: 10.1016/j.celrep.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- Bhattacharya MR, Gerdts J, Naylor SA, Royse EX, Ebstein SY, Sasaki Y, Milbrandt J, DiAntonio A. A model of toxic neuropathy in Drosophila reveals a role for MORN4 in promoting axonal degeneration. J Neurosci. 2012;32:5054–5061. doi: 10.1523/JNEUROSCI.4951-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers CW, Zigmond RE. Localization of neurons in the rat superior cervical ganglion that project into different postganglionic trunks. J Comp Neurol. 1979;185:381–391. doi: 10.1002/cne.901850211. [DOI] [PubMed] [Google Scholar]
- Buckmaster EA, Perry VH, Brown MC. The rate of Wallerian degeneration in cultured neurons from wild type and C57BL/WldS mice depends on time in culture and may be extended in the presence of elevated K+ levels. Eur J Neurosci. 1995;7:1596–1602. doi: 10.1111/j.1460-9568.1995.tb01155.x. [DOI] [PubMed] [Google Scholar]
- Cai Y, Yu SS, Chen SR, Pi RB, Gao S, Li H, Ye JT, Liu PQ. Nmnat2 protects cardiomyocytes from hypertrophy via activation of SIRT6. FEBS Lett. 2012;586:866–874. doi: 10.1016/j.febslet.2012.02.014. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Freeman MR. Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci. 2010;33:245–267. doi: 10.1146/annurev-neuro-060909-153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Soares L, Teng X, Geary M, Bonini NM. A novel Drosophila model of nerve injury reveals an essential role of Nmnat in maintaining axonal integrity. Curr Biol. 2012;22:590–595. doi: 10.1016/j.cub.2012.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godement P, Vanselow J, Thanos S, Bonhoeffer F. A study in developing visual systems with a new method of staining neurones and their processes in fixed tissue. Development. 1987;101:697–713. doi: 10.1242/dev.101.4.697. [DOI] [PubMed] [Google Scholar]
- Hicks AN, Lorenzetti D, Gilley J, Lu B, Andersson KE, Miligan C, Overbeek PA, Oppenheim R, Bishop CE. Nicotinamide mononucleotide adenylyltransferase 2 (nmnat2) regulates axon integrity in the mouse embryo. PloS One. 2012;7:e47869. doi: 10.1371/journal.pone.0047869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JY, Stein SA, Xu XM. Development of the corticospinal tract in the mouse spinal cord: a quantitative ultrastructural analysis. Brain Res. 2006;1084:16–27. doi: 10.1016/j.brainres.2006.02.036. [DOI] [PubMed] [Google Scholar]
- Ljungberg MC, Ali YO, Zhu J, Wu CS, Oka K, Zhai RG, Lu HC. CREB-activity and nmnat2 transcription are down-regulated prior to neurodegeneration, while NMNAT2 over-expression is neuroprotective, in a mouse model of human tauopathy. Hum Mol Genet. 2012;21:251–267. doi: 10.1093/hmg/ddr492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/S0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Riccio A, Cohen S, Ginty DD. Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron. 2002;34:371–385. doi: 10.1016/S0896-6273(02)00686-4. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Martin P. Tissue patterning in the developing mouse limb. Int J Dev Biol. 1990;34:323–336. [PubMed] [Google Scholar]
- Mayer PR, Huang N, Dewey CM, Dries DR, Zhang H, Yu G. Expression, localization, and biochemical characterization of nicotinamide mononucleotide adenylyltransferase 2. J Biol Chem. 2010;285:40387–40396. doi: 10.1074/jbc.M110.178913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milde S, Gilley J, Coleman MP. Subcellular localization determines the stability and axon protective capacity of axon survival factor nmnat2. PLoS Biol. 2013;11:e1001539. doi: 10.1371/journal.pbio.1001539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Orsomando G, Cialabrini L, Amici A, Mazzola F, Ruggieri S, Conforti L, Janeckova L, Coleman MP, Magni G. Simultaneous single-sample determination of NMNAT isozyme activities in mouse tissues. PloS One. 2012;7:e53271. doi: 10.1371/journal.pone.0053271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou YJ, Nathan C, Ding A, Brown RH, Jr, Conforti L, Coleman M, Tessier-Lavigne M, et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- Rodríguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, Dymecki SM. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25:139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnütgen F, De-Zolt S, Van Sloun P, Hollatz M, Floss T, Hansen J, Altschmied J, Seisenberger C, Ghyselinck NB, Ruiz P, Chambon P, Wurst W, von Melchner H. Genomewide production of multipurpose alleles for the functional analysis of the mouse genome. Proc Natl Acad Sci U S A. 2005;102:7221–7226. doi: 10.1073/pnas.0502273102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgeon B, Meloche S. Interpreting neonatal lethal phenotypes in mouse mutants: insights into gene function and human diseases. Physiol Rev. 2009;89:1–26. doi: 10.1152/physrev.00040.2007. [DOI] [PubMed] [Google Scholar]
- Xiong X, Hao Y, Sun K, Li J, Li X, Mishra B, Soppina P, Wu C, Hume RI, Collins CA. The Highwire ubiquitin ligase promotes axonal degeneration by tuning levels of Nmnat protein. PLoS Biol. 2012;10:e1001440. doi: 10.1371/journal.pbio.1001440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan T, Feng Y, Zheng J, Ge X, Zhang Y, Wu D, Zhao J, Zhai Q. Nmnat2 delays axon degeneration in superior cervical ganglia dependent on its NAD synthesis activity. Neurochem Int. 2010;56:101–106. doi: 10.1016/j.neuint.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, Bellen HJ. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452:887–891. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]