

Abstract
Axon formation is one of the most important events in neuronal polarization and is regulated by signaling molecules involved in cytoskeletal rearrangement and protein transport. We previously found that Partition-defective 3 (Par3) is associated with KIF3A (kinesin-2) and is transported into the nascent axon in a KIF3A-dependent fashion. Par3 interacts with the Rac-specific guanine nucleotide-exchange factors (GEFs) Tiam1/2, which activate Rac1, and participates in axon formation in cultured hippocampal neurons. However, the regulatory mechanism of the Par3-KIF3A interaction is poorly understood, and the role of Par3 in neuronal polarization in vivo remains elusive. Here, we found that extracellular signal-regulated kinase 2 (ERK2) directly interacts with Par3, that ERK2 phosphorylates Par3 at Ser-1116, and that the phosphorylated Par3 accumulates at the axonal tips in a manner dependent upon ERK2 activity. The phosphorylation of Par3 by ERK2 inhibited the interaction of Par3 with KIF3A but not with the other Par3 partners, including Par6 and aPKC. The phosphomimic mutant of Par3 (Par3-S1116D) showed less binding activity with the KIF3s and slower transport in the axons. The knockdown of Par3 by RNA interference impaired neuronal polarization, which was rescued with RNAi-resistant Par3, but not with the phosphomimic Par3 mutant, in cultured rat hippocampal neurons and mouse cortical projection neurons in vivo. These results suggest that ERK2 phosphorylates Par3 and inhibits its binding with KIF3A, thereby controlling Par3 transport and neuronal polarity.
Introduction
After neurons are born and differentiated, they lose their previous symmetry, and two structurally and functionally distinct compartments, the axons and the dendrites, are established. Axons and dendrites differ from one another in the composition of their proteins and organelles as well as in their functions. These two distinct cellular structures are fundamental for neuronal functioning because they enable neurons to receive and transmit electrical signals (Craig and Banker, 1994; Bradke and Dotti, 2000; Arimura and Kaibuchi, 2007; Barnes and Polleux, 2009; Tahirovic and Bradke, 2009).
Most of our knowledge regarding axon formation is based upon studies using dissociated neurons from the rodent brain (Dotti et al., 1988). These dissociated neurons form several immature neurites that are morphologically equivalent and undergo repeated random growth and retraction (Stage 2). One of these immature neurites stochastically initiates rapid extension. This neurite grows much longer than other neurites and finally becomes an axon (Stage 3). Thus, neuronal polarization occurs during the transition from Stage 2 to Stage 3. Many studies of this system have revealed that numerous proteins, such as kinases, small GTPases, and cytoskeleton and cytoskeleton-associated proteins, are involved in the establishment of neuronal polarity and axon formation (Bradke and Dotti, 2000; Watabe-Uchida et al., 2006; Arimura and Kaibuchi, 2007; Drees and Gertler, 2008; Barnes and Polleux, 2009; Tahirovic and Bradke, 2009).
One of the most important molecules in the regulation of neuronal polarization is Par3 (Shi et al., 2003; Nishimura et al., 2004, 2005; Schwamborn et al., 2007; Insolera et al., 2011). Par3 forms a protein complex with Par6 and aPKC and participates in several polarity events, including asymmetric cell division in Caenorhabditis elegans, the formation of tight junctions in epithelial cells, directional migration in fibroblasts, and axon formation in neurons (Kemphues et al., 1988; Izumi et al., 1998; Knoblich, 2001; Suzuki and Ohno, 2006; Dow and Humbert, 2007; Goldstein and Macara, 2007). Cdc42 plays a critical role in axon formation upstream of the Par complex (Schwamborn and Puschel, 2004). Cdc42 interacts with Par6 (Joberty et al., 2000; Johansson et al., 2000; Lin et al., 2000; Qiu et al., 2000), and Par3 mediates Cdc42-induced Rac activation through the specific Rac GEFs Tiam1 and STEF/Tiam2 (Chen and Macara, 2005; Mertens et al., 2005; Nishimura et al., 2005). This pathway promotes axon formation, presumably through the remodeling of actin filaments. Previous studies have shown that the Par complex accumulates at the distal part of growing axons and participates in axon formation (Shi et al., 2003; Nishimura et al., 2004). The transport of the Par complex into the axon is regulated by the direct interaction of Par3 and KIF3A. Thus, the kinesin-dependent transport of the Par complex is essential for axon formation in hippocampal neurons (Nishimura et al., 2004). In this context, Par3 functions as the cargo adaptor that links KIF3A to the cargo, the Par complex. To transport the Par complex to its proper destination, it is important to precisely regulate the loading and unloading of the complex from KIF3A. However, the mechanisms regulating the Par3-KIF3A interaction are not well understood.
We have recently performed comprehensive proteomic analyses of Par3-interacting proteins and identified ERK2 as a Par3-interacting candidate (Itoh et al., 2010). The Ras/Raf/MEK (mitogen-activated protein kinase/ERK kinase)/ERK pathway is required for axon formation mediated by neurotrophic factors (Markus et al., 2002; Zhou and Snider, 2006). However, the relation between ERK2 and the Par complex is poorly understood. In light of these observations, we focused on the roles of ERK2 in the Par complex. We found that ERK2 phosphorylated Par3 at Ser-1116 and inhibited its binding to KIF3A, thereby controlling Par3 transport and neuronal polarization.
Materials and Methods
Materials.
The cDNA encoding human KIF3A was obtained as previously described (Nishimura et al., 2004). The cDNA encoding rat Par3 was kindly provided by Dr. Ohno (Yokohama City University). The cDNA encoding mouse Par3 was obtained by the FANTOM Consortium (Carninci et al., 2005). The cDNA encoding Xenopus ERK2 was kindly provided by Dr. Matsumoto (Nagoya University). The cDNA encoding Xenopus MEK1-WT, constitutively active MEK1 (MEK1-CA; S218D/T222E), and dominant-negative MEK1 (MEK1-DN; S218A/T222A) were kindly provided by Dr. Nishida (Kyoto University). pTα-LPL (loxP-PolyA-loxP) and pTα-LPL-Lyn-EGFP were obtained as previously described (Nakamuta et al., 2011; Sakakibara et al., 2013). The following antibodies and materials were used: polyclonal rabbit anti-Par3 antibody raised against Par3-4N/3S-GST (Nishimura et al., 2004); polyclonal rabbit anti-Par3 and monoclonal mouse anti-Cdk5 antibodies (Millipore); polyclonal rabbit anti-c-Myc antibody (Santa Cruz Biotechnology); polyclonal rabbit anti-phospho-ERK1/2 (T202/Y204), polyclonal rabbit anti-ERK1/2, monoclonal rabbit anti-phospho-MAPK/CDK substrates (PXSP or SPXR/K) (34B2), monoclonal mouse anti-JNK1 (C26), and monoclonal mouse anti-p38α MAPK antibodies (Cell Signaling Technology); monoclonal mouse anti-ERK2, monoclonal mouse anti-KIF3A, and monoclonal mouse anti-GSK-3β antibodies (BD Biosciences); monoclonal mouse anti-KIF3A (K2.4) antibody (Abcam); monoclonal mouse anti-Myc (9E10), monoclonal mouse anti-α-tubulin (DM1A), monoclonal mouse anti-GST, and monoclonal mouse anti-Flag antibodies (Sigma); monoclonal mouse anti-Tau-1 (Millipore Bioscience Research Reagents); monoclonal mouse anti-class III β-tubulin and polyclonal rabbit anti-class III β-tubulin antibodies (Tuj1, Covance); polyclonal rabbit anti-GFP and polyclonal rabbit anti-HA antibodies (MBL); and monoclonal mouse anti-GFP antibody (Roche Diagnostics). Polyclonal rabbit maltose-binding protein (MBP) antibody was obtained as previously described (Nishimura et al., 2004). Polyclonal rabbit antibody against Par3 phosphorylated at Ser-1116 was produced against the phosphopeptide (Cys-Asn1111-Ala-Arg-Pro-Gln-phospho-Ser1116-Pro-Arg-Glu-Gly-His1121) by MBL. Alexa-488-, Alexa-555-, and Alexa-647-conjugated secondary antibodies against mouse or rabbit Ig or phalloidin were purchased from Invitrogen. Active MAP kinase 2/ERK2 was purchased from Millipore Biotechnology. GST-ERK2 was obtained from Carna Biosciences. Okadaic acid (OA) was purchased from Wako. Recombinant human neurotrophin-3 (NT-3) was purchased from PeproTech. The mitogen-activated protein kinase kinase 1/2 (MEK1/2) inhibitors PD98059 [2-(2-amino-3-methyoxy-phenyl)-4 H-1-benzopyran-4-one] and U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene] were purchased from Calbiochem. Other materials and chemicals were obtained from commercial sources.
Plasmid construct.
The mutants Par3-4N-S1116A, Par3-4N-T1145A, Par3-4N-S1316A, Par3-4N-T1328A, Par3-S1116A, Par3-4N/2-S1116A, Par3-S1116D, and Par3-4N/2-S1116D were generated with a Site-Directed Mutagenesis Kit (Stratagene) by changing Ser-1116, Thr-1145, Ser-1316, and Thr-1328 into alanine or aspartic acid. The fragment cDNAs encoding Par3 mutants were subcloned into pGEX (GE Healthcare), pMAL-c2 (New England Biolabs), and pCAGGS-Myc, pTα-LPL, and pEGFP (Clontech) vectors. All constructs were confirmed by DNA sequencing. GST- and MBP-tagged proteins were produced in BL21 (DE3) Escherichia coli cells and purified on glutathione–Sepharose 4B beads (GE Healthcare) and amylose-resin (New England Biolabs), respectively. pSico and pSicoR were generously provided by Dr. Tyler Jacks via the Addgene plasmid repository (Addgene plasmids 11578 and 11579; http://www.addgene.org). pSico-mCherry was generated by inserting the mCherry in place of the EGFP.
siRNA transfection and rescue experiments.
siRNA transfection and rescue experiments were performed as described previously (Nishimura et al., 2005). siRNA-resistant Par3 was generated by changing the targeted sequence of siPar3 #6 to the sequence 5′-GGCATGAAACGTTGGAAG-3′ (mutated nucleotides are in italics) by PCR-based site-directed mutagenesis. The mouse Par3 RNAi plasmid was generated by inserting the annealed oligonucleotides into pSico and pSicoR digested with XhoI and HpaI (Ventura et al., 2004; Wiznerowicz et al., 2006). The target sequences for Par3 RNAi #1, RNAi #2, RNAi #3, and RNAi #4 were as follows: 5′-GCGAGAAGAACAACTACTC-3′, 5′-GCAAGAGGCTCAACATCCA-3′, 5′-GCTTAAGAAAGGTACAGAA-3′ and 5′-GGCAGGAGACCGGCTAATA-3′, respectively. shRNA-resistant Par3 was generated by changing the targeted sequence of shPar3 #3 to the sequence 5′-GCTTAAAAAGGGAACAGAA-3′ (mutated nucleotides are in italics) by PCR-based site-directed mutagenesis.
Immunoprecipitation assay.
For the interaction assay of endogenous proteins, rat brains (E18) were extracted by the addition of lysis buffer (20 mm Tris-HCl, pH 7.5, 1 mm EDTA, 150 mm NaCl, 1% NP-40, 50 μg/ml PMSF, 10 μg/ml leupeptin, 2 μg/ml aprotinin, 1 μg/ml pepstatin) and then clarified by centrifugation at 100,000 × g for 30 min at 4°C. For the interaction assay of endogenous Par3 and KIF3A, brain slices were prepared from E14 mouse embryos and cultured in neurobasal medium (Invitrogen). The slices were treated with NT-3 or OA for 2 h and then extracted by the addition of lysis buffer containing the phosphatase inhibitor mixture (PhosSTOP; Roche). For the interaction assay of exogenous Par3 and KIF3A, COS7 cells were transfected with the indicated plasmids, treated with or without OA, and extracted by the addition of lysis buffer containing PhosSTOP. The soluble supernatants were incubated with the indicated antibodies for 1 h at 4°C. The immunocomplexes were then precipitated with protein A-Sepharose CL-4B (GE Healthcare). The immunocomplexes were washed three times with lysis buffer, eluted by boiling in sample buffer for SDS-PAGE, and then subjected to immunoblot analysis with the indicated antibodies.
In vitro phosphorylation assay.
The phosphorylation assay was performed as previously described (Amano et al., 1996). In brief, the kinase reactions of ERK on the MBP-Par3 mutants were performed in 100 μl of a reaction mixture (50 mm Tris/HCl, pH 7.5, 1 mm EDTA, 1 mm EGTA, 1 mm DTT, 5 mm MgCl2, 100 μm [γ-32P] ATP [1-20 GBq/mmol]), 0.05 μm active ERK2, and 0.9 μm purified MBP-Par3 mutants for 30 min at 30°C. Then, the reaction mixtures were boiled in SDS sample buffer and subjected to SDS-PAGE and silver staining. The radiolabeled proteins were visualized with an image analyzer (BAS1500; Fuji).
In vitro binding assay.
The interaction of Par3 with KIF3A was examined with an in vitro binding assay as previously described (Nishimura et al., 2004). Briefly, 90 pmol of MBP, MBP-Par3-4N, or -S1116A was incubated with glutathione-Sepharose 4B beads coated with 100 pmol of either GST or GST-KIF3A-C2 in lysis buffer for 1 h at 4°C. For the interaction of Par3 with ERK2, 50 pmol of GST-ERK2 was incubated with amylase resin coated with 50 pmol of MBP, MBP-Par3-1N, -2N, -3N, or -4N in lysis buffer for 1 h at 4°C. The beads were washed three times with lysis buffer and dissolved into SDS sample buffer. The bound proteins were analyzed by immunoblotting with the indicated antibodies.
Microtubule cosedimentation assay.
Microtubule cosedimentation assays were performed essentially as previously described (Toda et al., 2008; Yoshimura et al., 2010). Briefly, EGFP-Par3 mutants and Myc-KIF3A/B were transiently expressed in COS7 cells and lysed in PIPES buffer (100 mm PIPES, pH 6.9, 1 mm MgCl2, 1 mm EGTA, 1% Triton X-100). After centrifugation at 100,000 × g for 1 h at 4°C, the α-tubulin in the supernatant was polymerized and stabilized by the addition of 2 mm GTP, 20 μm Taxol (Sigma), and 1 mm MgCl2 for 30 min at 37°C. After incubation for 30 min at 37°C, KIF3A/B was allowed to form complexes with microtubules in response to the addition of 2 mm adenosine 5′-(β, γ-imido) triphosphate (Calbiochem) and further incubated for 30 min at 37°C. The microtubules were then sedimented through a sucrose cushion (30% sucrose, 1 mm GTP and 20 μm Taxol in assembly buffer) at 20,000 × g for 1 h at 25°C, washed once, and subjected to immunoblot analysis.
Cell culture and immunofluorescence analysis.
COS7 and Neuro2a cells were cultured in DMEM (Sigma) with 10% FBS (SAFC Biosciences). All cell lines were grown in a humidified atmosphere of 5% CO2 at 37°C. Transfection was performed using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. Hippocampal neurons were prepared from E18 rat embryos using papain as described previously (Goslin and Banker, 1989; Inagaki et al., 2001). Briefly, neurons were seeded on coverslips or dishes coated with poly-d-lysine (Sigma) and cultured for 72 h in neurobasal medium (Invitrogen) supplemented with B-27 (Invitrogen) and 1 mm GlutaMAX (Invitrogen). Neurons were transfected using a CalPhos Mammalian Transfection Kit (Clontech) according to the manufacturer's instructions before plating to analyze the morphology. Neurons were fixed at the indicated time periods with 3.7% formaldehyde in PBS for 10 min at room temperature followed by treatment with 0.05% Triton X-100 for 10 min on ice. Under extraction conditions, cells were treated with extraction buffer (100 mm PIPES, pH 6.9, 1 mm MgCl2, 1 mm EGTA, 1% Triton X-100, 2% paraformaldehyde, 10 μm Taxol) for 4 min at 25°C before fixation. After blocking with 10% donkey serum albumin in PBS, the cells were incubated with each indicated antibody overnight at 4°C. After washing, the samples were incubated with the appropriate secondary antibody. Confocal images were recorded with LSM 780 or LSM5 Pascal microscopes built around an Axio Observer Z1 or Axiovert 200M with Plan-Apochromat 20× (numerical aperture [NA] 0.75), Plan-Apochromat 20× (NA 0.8), C-Apochromat 40× (NA 1.2), or Plan Apochromat 63× (NA 1.40) lenses under the control of LSM software (Carl Zeiss). Super-Resolution images were recorded with Nikon's Structured Illumination Microscopy (N-SIM) built around ECLIPSE Ti with CFI Apo TIRF 100× H (NA 1.49) lenses under the control of NIS-elements software (Nikon). Neurons were classified as Stage 1, 2, or 3 according to their morphology as described previously (Goslin and Banker, 1989). A neuron was considered to have an axon if one process was at least twice as long as any other process and the neuron was Tau-1-positive.
Time-lapse observation.
Fluorescence recovery after photobleaching (FRAP) analysis was performed as previously described (Shinoda et al., 2007; Taya et al., 2007). In brief, hippocampal neurons were seeded on 3.5 cm glass-bottom dishes and then transfected with EGFP-Par3-WT, -S1116A, -S1116D, or -Δ4N/2 at 2 d in vitro (DIV) by Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocols. At 3 DIV, the distal parts of the axons of EGFP-positive neurons were photobleached with 30 laser irradiations and then the cells were monitored at 30 s intervals for 4 min at 37°C. In some experiments, neurons were treated with 20 μm nocodazole (Sigma) on ice for 4 min to disrupt the microtubules. Confocal images were recorded with an LSM780 built around an Axio Observer Z1 with a Plan Apochromat 63× (NA 1.40) (Carl Zeiss) lens.
In utero electroporation.
In utero electroporation was performed as described previously (Saito and Nakatsuji, 2001; Tabata and Nakajima, 2001) with some modifications. To achieve Par3 knockdown and rescue, pTα-LPL-Lyn-EGFP (2 μg/μl) was coinjected with pSico-mCherry-shPar3 #3 (3 μg/μl), pTα-Cre (0.05 μg/μl), and pTα-LPL-Par3-WT, -S1116A, or -S1116D (5 μg/μl) at a concentration ratio of 2:5:1:2. For the Par3 overexpression experiments, pTα-LPL-Lyn-EGFP (2 μg/μl) was coinjected with pTα-Cre (0.05 μg/μl) and pTα-LPL-Par3-4N/2-WT, -S1116A, or -S1116D (2 μg/μl) at a concentration ratio of 2:1:5. After plasmid injection into the lateral ventricle of the embryos, electric pulses (50 ms square pulses of 30 V with 950 ms intervals) were applied to the embryos. All electroporations were performed on E13.
Immunohistochemistry.
The brains were fixed in 4% PFA at E16 and sectioned coronally with a cryostat (Leica Microsystems) at a thickness of 60 μm. The slices were incubated with diluted primary antibodies in PBS containing 1% BSA at 4°C overnight. After several washes with PBS, the slices were treated with AlexaFluor-488- or AlexaFluor-555-conjugated secondary antibodies diluted in PBS for 1 h at room temperature. In some experiments, the nuclei were visualized by staining with Hoechst 33342 (Invitrogen). Confocal images were recorded with a Nikon A1 confocal laser scanning microscope built around an ECLIPSE Ti with CFI Plan Apo VC 20× (NA 0.75) or CFI Plan Apo VC 60×WI (NA 1.2) lenses under the control of NIS-elements software (Nikon).
Quantitative analysis of neuronal migration and morphology.
The distribution of EGFP-positive brain slices was quantified as follows. The coronal sections of cerebral cortices containing the labeled cells were classified into two regions, the cortical plate (CP) and the intermediate zone (IZ), as described previously (Kawauchi et al., 2003). The number of labeled cells in each region of at least four slices per brain was calculated. To evaluate the morphology of the migrating neurons, images of EGFP-positive neurons in the IZ were obtained as described previously (Nakamuta et al., 2011). At least three independent brains were electroporated and analyzed for each experiment. For the analysis of the neurite structure of multipolar cells, projection images of each EGFP-positive neuron were made from z-series stacks of confocal images using NIS elements (Nikon).
Animals.
All animal experiments were performed according to the guidelines of the Institute for Developmental Research. Wistar rats and ICR mice of either sex were used in the study. All animals were purchased from SLC Japan.
Statistics.
Data are expressed as the mean ± SEM. Statistical analyses were performed with GraphPad Prism version 4.01 (GraphPad Software) for Student's t test, Dunnett's, or Tukey's multiple-comparison tests and with Microsoft Excel 2011 for Scheffé's F test. p < 0.05 was considered to indicate statistical significance.
Results
Par3 interacts with ERK2
To isolate the Par3 binding partners, we previously performed Par3 affinity column chromatography and detected ERK, c-jun N-terminal kinase (JNK), and glycogen synthase kinase-3 (GSK-3) in the eluate from an affinity column coated with GST-Par3-4N among GST, GST-Par3-1N, -2N, -3N, and -4N as bait (Fig. 1A) (Itoh et al., 2010). To confirm the results obtained in the proteomic analysis, an immunoblot analysis was performed. ERK1/2 was detected in the eluate from GST-Par3-4N but not in the other eluates (Fig. 1B). JNK1 and GSK-3β were detected in the eluate from GST-Par3-3N and -4N (Fig. 1B). In contrast, p38α MAPK and cyclin-dependent kinase 5 (Cdk5) were not detected (Fig. 1B). To examine whether Par3 associates with these kinases in vivo, a coimmunoprecipitation assay was performed using rat brain lysate. ERK2 reciprocally coimmunoprecipitated with Par3 from rat brain lysate, whereas none of the other kinases coimmunoprecipitated with Par3 (Fig. 1C). Thus, we focused thereafter on ERK2. To understand the relationship between Par3 and ERK2 in neurons, we compared the subcellular distribution of Par3 and ERK2 in cultured hippocampal neurons. We found that ERK2 were partially colocalized with Par3 at the cell body and the axonal growth cone (Fig. 1D). These results suggest that Par3 specifically associates with ERK2 in vivo. To confirm the direct interaction of ERK2 and Par3, an in vitro binding assay was performed. GST-ERK2 interacted strongly with MBP-Par3-4N, weakly with MBP-Par3-2N, and not at all with MBP, MBP-Par3-1N, or -3N (Fig. 1E). Many ERK substrate proteins carry a kinase interaction motif (KIM) with the consensus sequence (V/L)-X2-(R/K)-(R/K)-X3–6-L (where X represents any amino acid) (MacKenzie et al., 2000), which facilitates the contact between ERK and its substrate. Interestingly, Par3-4N contains a putative KIM (Fig. 1F), suggesting that Par3 associates with ERK2 through the KIM of the 4N region.
Figure 1.

Identification of ERK2 as a novel Par3-interacting protein. A, Schematic representation of Par3. The numbers indicate the amino acids. CR1, Conserved region 1; PDZ, PSD-95/Dlg/ZO-1; aPKC BR, aPKC binding region. B, Validation of the results of the proteomic analysis. The eluates from affinity column chromatography were analyzed by immunoblotting using anti-ERK1/2, anti-JNK1, anti-p38α MAPK, anti-GSK-3β, and anti-Cdk5 antibodies. C, A coimmunoprecipitation assay was performed using rat brain lysate. Extracts of developing rat brain were incubated with rabbit IgG, anti-Par3, or anti-ERK1/2 antibody. The immunoprecipitates were analyzed by immunoblotting with rabbit anti-Par3 and mouse anti-ERK2, anti-JNK1, anti-p38α MAPK, anti-GSK-3β, and anti-Cdk5 antibodies. D, Super-resolution microscopy images of the cell body and the growth cone showing the colocalization of Par3 and ERK2. Hippocampal neurons were fixed at 3 DIV and then immunostained with specific antibodies against anti-ERK2 (green) and anti-Par3 (red). The peripheral regions of the growth cones were visualized by staining F-actin with Alexa-647-conjugated phalloidin (blue). Arrowheads in the enlarged images indicate the colocalization of the proteins. Scale bars, 5 μm and 0.5 μm for the enlarged images. E, GST-ERK2 was incubated with amylose-resin coated with MBP, MBP-Par3-1N, -2N, -3N, or -4N. The bound proteins were subjected to immunoblotting with anti-ERK2 antibody (top). The total amounts of MBP, MBP-Par3-1N, -2N, -3N, and -4N are shown with Coomassie Brilliant Blue (CBB) staining (bottom). Asterisks indicate intact MBP-fusion proteins. F, The putative ERK docking site KIM ((V/L)-X2-(R/K)-(R/K)-X3–6-L).
ERK2 phosphorylates Par3 in vitro and in vivo
There are at least two possible explanations for the interaction of Par3 with ERK2: Par3 may act as a scaffold protein for ERK2, or Par3 may be a substrate of ERK2. First, we examined whether Par3 regulates ERK2 activity, as the activity of JNK that is a member of MAP kinase is regulated by its interaction with scaffold proteins, such as JIP1 (Morrison and Davis, 2003; Kolch, 2005). In control cells, ERK2 was activated in response to EGF stimuli. Under this condition, ERK2 activity was not altered in Par3-depleted cells as previously described (McCaffrey et al., 2012) (data not shown), suggesting that Par3 does not regulate ERK2 activity. Next, we examined whether ERK2 phosphorylates Par3. An in vitro phosphorylation assay revealed that ERK2 effectively phosphorylated MBP-Par3-4N (Fig. 2A), suggesting that Par3 is a substrate of ERK2. Par3-4N contains four potential phosphorylation sites for ERK2, namely, P-X-S/T-P (where X represents any amino acid), including two consensus sequences. To determine the site at which Par3 is phosphorylated by ERK2, we produced Par3-4N-S1116A, -T1145A, -S1316A, and -T1328A (serine and threonine were replaced by alanine in these proteins) and examined their phosphorylation efficiency. The phosphorylation level of MBP-Par3-4N-S1116A was dramatically reduced, whereas that of the other mutants, including MBP-Par3-4N-T1145A, -S1316A, and -T1328A, was not reduced (Fig. 2B). These results suggest that ERK2's major phosphorylation site is Ser-1116. This phosphorylation site is well conserved in mammals but not in Drosophila or C. elegans (Fig. 2C), suggesting that phosphorylation may be conserved only in higher organisms. Then, we confirmed the possibility that Ser-1116 of Par3 is phosphorylated in COS7 cells. An immunoblot analysis using the anti-phospho-MAPK/CDK substrates antibody, which recognizes a phospho-serine in a PXSP or SPXR/K motif (where X represents any amino acid), revealed that the stimulation of COS7 cells with OA, an inhibitor of the serine/threonine protein phosphatase 2A, induced the phosphorylation of EGFP-Par3-WT. The mutation of Ser-1116 to Ala markedly reduced the phosphorylation of EGFP-Par3 in COS7 cells (Fig. 2D), clearly indicating that Par3 is phosphorylated at Ser-1116 in COS7 cells. To monitor the in vivo phosphorylation state of Par3 by ERK2 in detail, we prepared an antibody that specifically recognized Par3 phosphorylated at Ser-1116. The specificity of the anti-pS1116 antibody was examined with an immunoblot analysis. MBP-Par3-4N phosphorylated by ERK2 in vitro was specifically detected by the anti-pS1116 antibody in a dose-dependent manner (Fig. 3A). We also confirmed that the anti-pS1116 antibody did not recognize MBP-Par3-4N-S1116A (Fig. 3A). To examine whether ERK2 phosphorylates Par3 in COS7 cells, EGFP-Par3-WT was exogenously expressed into COS7 cells. The activation of ERK1/2 requires its phosphorylation by MEK1/2 (Errede et al., 1995). Thus, we used two independent MEK1/2-specific inhibitors, PD98059 and U0126, to inhibit ERK1/2 activation. Treatment of the cells with PD98059 or U0126 inhibited the phosphorylation of EGFP-Par3 by ERK2 (Fig. 3B). These results suggest that ERK2 can phosphorylate Par3 at Ser-1116 in COS7 cells.
Figure 2.

Identification of the phosphorylation site of Par3 by ERK2. A, The direct phosphorylation of Par3 by ERK2. Purified MBP or MBP-Par3 deletion mutants were incubated with recombinant ERK2 in the presence of [γ-32P]ATP in vitro. Samples were subjected to SDS-PAGE and silver staining (bottom) followed by autoradiography (top). Asterisks indicate intact MBP-fusion proteins. B, The phosphorylation of Par3 point mutants by ERK2. Purified MBP, MBP-Par3-4N-WT -S1116A, -T1145A, -S1316A, or -T1328A was incubated with recombinant ERK2 in the presence of [γ-32P]ATP in vitro. Samples were subjected to SDS-PAGE and silver staining (bottom) followed by autoradiography (top). Asterisks indicate intact MBP-fusion proteins. C, The alignment of ERK2 phosphorylation sites in Par3 homologs (rat, mouse, human). D, The phosphorylation of Par3-WT or -S1116A in COS7 cells. COS7 cells were transfected with EGFP-Par3-WT or -S1116A and cultured for 24 h. The cells were serum-starved for 24 h and then treated with or without 1 μm OA for 2 h. Cell lysates were incubated with anti-GFP antibody, and the immunoprecipitates were analyzed by immunoblotting with anti-phospho-MAPK/CDK substrates and anti-GFP antibodies.
Figure 3.

Phosphorylation of Par3 by ERK2 in vivo. A, Specificity of the phosphospecific anti-phospho-Par3 (pS1116) antibody. MBP-Par3-4N-WT (1 pmol) containing the indicated amounts of Par3-4N-WT or -S1116A phosphorylated by ERK2 was subjected to SDS-PAGE. Immunoblot analyses with anti-pS1116 (top) and anti-MBP antibodies (bottom) were performed. B, The phosphorylation of EGFP-Par3 in COS7 cells. COS7 cells were transfected with EGFP-Par3-WT and treated with DMSO, 50 μm PD98059, or 20 μm U0126 for 4 h. The cell lysates were analyzed by immunoblotting with anti-pS1116 and anti-GFP antibodies. The phosphorylation of ERK1/2 was also examined with anti-phospho-ERK1/2 antibody. Data represent the mean ± SEM of three independent experiments. **p < 0.01 (Dunnett's multiple-comparison test). C, D, The phosphorylation of endogenous Par3 in hippocampal neurons at 3 DIV. C, Hippocampal neurons were treated with DMSO, 50 μm PD98059, or 20 μm U0126 for 4 h and then with or without 1 μm OA for 2 h. The cell lysates were analyzed by immunoblotting with anti-pS1116 and anti-Par3 antibodies. The phosphorylation of ERK1/2 was also examined with anti-phospho-ERK1/2 antibody. Data represent the mean ± SEM of three independent experiments. Asterisks indicate statistical significance. *p < 0.05 (Tukey's multiple-comparison test). **p < 0.01 (Tukey's multiple-comparison test). ***p < 0.001 (Tukey's multiple-comparison test). D, The cells were treated with DMSO or 20 μm U0126 for 4 h and then with or without NT-3 (100 ng/ml) for 30 min. The cell lysates were analyzed by immunoblotting with anti-pS1116 and anti-Par3 antibodies. The phosphorylation of ERK1/2 was also examined with anti-phospho-ERK1/2 antibody. Data represent the mean ± SEM of four independent experiments. *p < 0.05 (Tukey's multiple-comparison test). **p < 0.01 (Tukey's multiple-comparison test). ***p < 0.001 (Tukey's multiple-comparison test). E, The spatial distribution of phosphorylated Par3 at Ser-1116 in hippocampal neurons. Hippocampal neurons were transfected with Myc-Par3-WT or -S1116A at 2 DIV. Neurons were fixed at 3 DIV and then immunostained with anti-Myc and anti-pS1116 antibodies. A merged image and the ratio (phosphorylated Par3 to total Par3) are shown. Scale bars, 20 μm. The graph plots the fluorescence intensities of Par3 phosphorylated at Ser-1116 (green) and total Par3 (red) along the line (from X to Y).
To explore Par3 phosphorylation under more realistic physiological conditions, we used hippocampal neurons. The stimulation of the hippocampal neurons with OA induced the phosphorylation not only of ERK1/2 but also of the endogenous Par3 at Ser-1116 (Fig. 3C). Treatment of the cells with PD98059 or U0126 inhibited the OA-induced phosphorylation of Par3 (Fig. 3C). It has been reported that the treatment of cultured neurons with neurotrophins, such as BDNF and NT-3, induces the activation of Ras and ERK (Iida et al., 2001) and enhances axon formation (Morfini et al., 1994; Labelle and Leclerc, 2000; Yoshimura et al., 2005; Shinoda et al., 2007). More recent studies indicate that neurotrophins derived from cultured neurons can stimulate neurite outgrowth through their tropomyosin receptor kinase (Trk) receptors in an autocrine fashion and are required for axon specification (Cheng et al., 2011; Nakamuta et al., 2011). The stimulation of hippocampal neurons with NT-3 induced the phosphorylation not only of ERK1/2 but also of the endogenous Par3 at Ser-1116 (Fig. 3D). The treatment of the cells with U0126 inhibited the NT-3-induced phosphorylation of Par3 (Fig. 3D). Together, these results suggest that OA or NT-3 provokes the phosphorylation of endogenous Par3 at Ser-1116 in an ERK-dependent fashion in cultured hippocampal neurons. Treatment with PD98059 or U0126 also lowered the basal levels of ERK1/2 and Par3 phosphorylation (Fig. 3C,D). These observations are consistent with the notion that neurotrophins derived from cultured neurons can stimulate axon formation through the ERK pathway (Nakamuta et al., 2011).
Phosphorylated Par3 localizes in the distal part of the growing axon
We investigated the spatial distribution of phosphorylated Par3 at Ser-1116. Unfortunately, the anti-pS1116 antibody could not detect the phosphorylation of endogenous Par3 during immunostaining of the hippocampal neurons because the Par3 expression levels are relatively low in differentiated neurons (Manabe et al., 2002). Accordingly, Myc-Par3 was exogenously expressed into hippocampal neurons at 2 DIV. The neurons were fixed at 3 DIV and then immunostained with anti-Myc and anti-pS1116 antibodies. Myc-Par3 (red) accumulated in the cell body and in the distal part of the growing axons in developing neurons as previously reported (Shi et al., 2003; Nishimura et al., 2004). Phosphorylated Par3 (green) was enriched in the distal part of the growing axon but not in the cell body (Fig. 3E). The merged images of Par3 and Par3 phosphorylated at Ser-1116 immunofluorescence enabled us to approximately estimate the phosphorylation levels of Par3. The merged image in the distal part of the growing axon was yellow, whereas that in the cell body was more reddish (Fig. 3E). The ratio of phosphorylated Par3 to total Par3 in the distal part of the growing axon was higher than that in the cell body (Fig. 3E). Further, clear evidence of the increased phosphorylation of the growing axon was obtained through intensity imaging of phosphorylated Par3 and total Par3 (Fig. 3E). The intensity of phosphorylated Par3 was higher than that of total Par3 in the distal part of the growing axon. ERK1/2 activity is known to be high in the neurite (Pertz et al., 2008). These results indicate that there is a Ser-1116 phosphorylated Par3 pool in the distal part of the growing axon. We confirmed that the MEK inhibitor decreased the levels of Par3 phosphorylation by immunostaining with an anti-pS1116 antibody and that the anti-pS1116 antibody did not recognize Myc-Par3-S1116A (Fig. 3E).
Phosphorylation of Par3 decreases its interaction with KIF3A
Recent studies have shown that members of the MAPK family negatively regulate the interaction of kinesin motors with their cargo (Bowman et al., 2000; Byrd et al., 2001; Verhey et al., 2001; Berman et al., 2003; Bengs et al., 2005; Burghoorn et al., 2007; Horiuchi et al., 2007; Vagnoni et al., 2011); thus, we hypothesized that ERK2 is involved in the regulation of the binding and release of Par3 from kinesins. We have previously shown that Par3 directly interacts with the C-terminal domain of KIF3A (KIF3A-C2) (Fig. 4A) through its 4N/2 domain, which contains Ser-1116 (Nishimura et al., 2004). To examine whether the ERK2-mediated phosphorylation of Par3 modulates its interaction with KIF3A, an in vitro binding assay was performed after the in vitro phosphorylation of Par3 by ERK2. MBP-Par3-4N interacted with GST-KIF3A-C2 as previously reported (Nishimura et al., 2004) (Fig. 4B). Under these conditions, the interaction of MBP-Par3-4N with GST-KIF3A-C2 was dramatically reduced in the presence of both ATP and ERK2 (Fig. 4B), but that of MBP-Par3-4N-S1116A was not (Fig. 4B, bottom). These results suggest that the phosphorylation of Par3-4N at S1116 by ERK2 suppresses its interaction with KIF3A-C2. Next, to examine whether the phosphorylation of Par3 modulates its interaction with KIF3A in COS7 cells, a coimmunoprecipitation assay was performed. When EGFP-Par3-WT was immunoprecipitated from COS7 cell lysates, Myc-KIF3A was coimmunoprecipitated (Fig. 4C). The stimulation of COS7 cells with OA inhibited the interaction of EGFP-Par3-WT with Myc-KIF3A (Fig. 4C). These results suggest that the phosphorylation of EGFP-Par3-WT suppresses its interaction with Myc-KIF3A in COS7 cells. To mimic the phosphorylation of Par3 by ERK2, aspartic acid was substituted for Ser-1116 to produce EGFP-Par3-S1116D. The amount of Myc-KIF3A that coimmunoprecipitated with EGFP-Par3-S1116D or -Δ4N/2, which lacks the KIF3A binding domain, was less than that with EGFP-Par3-WT or -S1116A (Fig. 4C). These results suggest that the substitution of aspartic acid for Ser-1116 reduces the ability of Par3 to interact with KIF3A and mimics the phosphorylated state. To examine whether the phosphorylation of Par3 affects the interaction of Par3 with KIF3A under more realistic physiological conditions, acute brain slices were treated with NT-3 or OA, and then the amounts of KIF3A that coimmunoprecipitated with Par3 were determined. When Par3 was immunoprecipitated from brain-slice lysates, KIF3A was coimmunoprecipitated. Under these conditions, the amounts of KIF3A coimmunoprecipitated with Par3 were reduced by treatment with NT-3 or OA (Fig. 4D).
Figure 4.

The effect of the phosphorylation of Par3 on the interaction of Par3 with KIF3A. A, Schematic representation of KIF3A. The numbers show the amino acids. B, The effect of ERK2 phosphorylation on the interaction of Par3 with KIF3A in vitro. Purified MBP, MBP-Par3-4N-WT, or -S1116A was incubated with or without recombinant ERK2 in the presence or absence of ATP. The reactants were incubated with glutathione beads coated with GST or GST-KIF3A-C2. The bound proteins were subjected to immunoblotting with anti-MBP antibody. The total amounts of GST and GST-KIF3A-C2 are shown with Coomassie Brilliant Blue staining. C, The interaction of KIF3A with Par3-WT, -1116A, -S1116D, or -Δ4N/2. COS7 cells were transfected with the indicated constructs, serum-starved for 24 h, and then stimulated with or without 1 μm OA for 2 h. Extracts of COS7 cells were incubated with anti-GFP antibody. The bound proteins and coimmunoprecipitates were analyzed by immunoblotting with anti-GFP and anti-Myc antibodies. The protein expression levels in cell lysates are shown on the top. D, The effect of the phosphorylation of Par3 on the interaction of Par3 with KIF3A in vivo. Mouse brain slices were cultured and treated with or without NT-3 (100 ng/ml) or 1μM OA for 2 h. Extracts of mouse brain slices were incubated with anti-Par3 antibody. The immunoprecipitates were analyzed by immunoblotting with anti-Par3, anti-KIF3A, anti-ERK1/2, and anti-phospho-ERK1/2 antibodies. E, The interaction of Par6 and aPKC with Par3-WT or -S1116D. COS7 cell lysates expressing the indicated proteins were incubated with anti-GFP antibody. The bound proteins and coimmunoprecipitates were analyzed by immunoblotting with anti-Myc, anti-Flag, and anti-GFP antibodies. Protein expression levels in the cell lysates are shown on the left. F, The interaction of LIMK2 with Par3-WT or -S1116D. COS7 cell lysates expressing the indicated proteins were incubated with anti-GFP antibody. The bound proteins and coimmunoprecipitates were analyzed by immunoblotting with anti-HA and anti-GFP antibodies. Protein expression levels in the cell lysates are shown on the left.
Several reports have shown that the phosphorylation of Par3 affects its interaction with other proteins (Nagai-Tamai et al., 2002; Hurd et al., 2003; Wang et al., 2006; Nakayama et al., 2008). RhoA/Rho-kinase inhibits Par complex formation through Par3 phosphorylation at Thr-833 (Nakayama et al., 2008). In addition, the EGF-induced phosphorylation of Par3 at Tyr-1127 reduces its association with LIMK2 (Wang et al., 2006). We examined whether the ERK2-mediated phosphorylation of Par3 modulates its interactions with Par6, aPKC, and LIMK2. A coimmunoprecipitation assay showed that the phosphorylation of Par3 by ERK2 did not influence the interaction of Par3 with Par6, aPKC, or LIMK2 (Fig. 4E,F).
The roles of Par3 and its phosphorylation in axonal transport
A previous study showed that Par3 is transported to the distal tip of the axon by KIF3A (Nishimura et al., 2004). To verify this role, we first confirmed the localization of Par3 and KIF3A in hippocampal neurons based on an immunocytochemical analysis. To examine the localization of Par3 and KIF3A in detail, we used an extraction method to focus on the high-magnification view of the axonal growth cone; this method is useful for extracting cytosolic components and visualizing the cytoskeleton- or membrane-associated components (Arimura et al., 2009a, b). Par3 and KIF3A were distributed along microtubules at the axonal shaft and the growth cone (Fig. 5A). In addition, Par3 partially colocalized with KIF3A at the axonal shaft and the growth cone (Fig. 5A), suggesting that Par3 could potentially be transported to the tip of the axon by KIF3 motor. These findings are consistent with previous observations that Par3 is transported to the tip of the axon by KIF3A (Nishimura et al., 2004).
Figure 5.
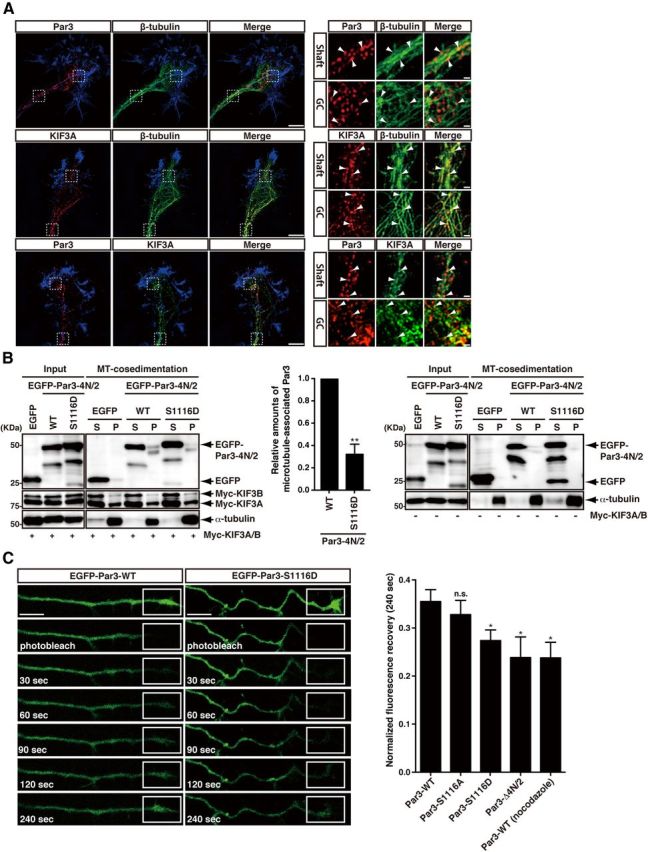
The effect of mutations of Par3 at phosphorylation sites on axonal transport of Par3. A, Super-resolution microscopy images of the axonal shaft and the growth cone (GC) showing the colocalization of Par3 and KIF3A on microtubules. Hippocampal neurons were fixed at 3 DIV and then immunostained with specific antibodies against rabbit anti-Par3 (red, top and bottom), rabbit or mouse anti-class III β-tubulin (green, top and middle), and mouse anti-KIF3A (red, middle; green, bottom). Arrowheads in the enlarged images indicate the colocalization of the proteins. The peripheral regions of the growth cones were visualized by staining F-actin with Alexa-647-conjugated phalloidin (blue). Scale bars: left, 5 μm; right, 0.5 μm. B, The interaction of Par3-WT or -S1116D with KIF3A on microtubules. COS7 cells were transfected with the indicated constructs, and the lysates were subjected to the microtubule cosedimentation assay. The resulting supernatant (S) and pellet (P) fractions were analyzed by immunoblotting with anti-GFP, anti-Myc, and anti-α-tubulin antibodies. The relative amounts of microtubule-associated Par3 were estimated by the comparison of the signal intensity of Par3 in pellets divided by that of KIF3A/B in pellets (middle). Data represent the mean ± SEM of three independent experiments. **p < 0.01 (Student's t test). C, FRAP analysis of EGFP-Par3-WT or EGFP-Par3 mutants. Hippocampal neurons were transfected with EGFP-Par3-WT, -S1116A, -S1116D, or -Δ4N/2 at 2 DIV. The distal parts of the axons in EGFP-positive neurons were photobleached in various squares and then observed at 3 DIV. Scale bars, 10 μm. The recovery of the fluorescence of EGFP-Par3-WT, -S1116A, -S1116D, or -Δ4N/2 at the photobleached sites in the distal parts of axons was quantified for 4 min. Data represent the mean ± SEM of three independent experiments (at least 10 cells in each experiment). *p < 0.05 (Student's t test). n.s., Not significant.
To examine the effect of the phosphorylation of Par3 on the interactions of KIF3A and Par3 with microtubules, a microtubule cosedimentation assay was performed. The amount of EGFP-Par3-4N/2–1116D that cosedimented with microtubules was less than that of EGFP-Par3-4N/2-WT in the presence of KIF3A/B (Fig. 5B). These results suggest that the phosphorylation of Par3 reduces the ability of Par3 to interact with KIF3A on microtubules. We further investigated the transport of Par3 in axons by FRAP analysis using EGFP-Par3-WT and EGFP-Par3 mutants as previously described (Shinoda et al., 2007; Taya et al., 2007). The fluorescent signals of EGFP-Par3-WT or EGFP-Par3 mutants were observed over entire neurons, including axon shafts and growth cones, at 3 DIV under these conditions. The distal parts of the axons of EGFP-positive neurons were photobleached, and the recovery of EGFP-Par3 fluorescence was observed. Fluorescence recovery after the photobleaching of EGFP-Par3-S1116D and EGFP-Par3-Δ4N/2, which did not bind to KIF3A, was slower than that of EGFP-Par3-WT and -S1116A (Fig. 5C). When neurons were treated with nocodazole and placed on ice for 4 min, the movement of EGFP-Par3 was prevented (Fig. 5C), whereas EGFP-GST movement was not impaired. Neither placing the neurons on ice for 4 min nor nocodazole treatment alone prevented EGFP-Par3 movement (data not shown), as previously described (Shinoda et al., 2007; Taya et al., 2007). These results suggest that the recovery of EGFP-Par3 depends on transport on the rails of microtubules and that the contribution of diffusion is negligible because transport along microtubules is inhibited by the disruption of microtubules by nocodazole (Galbraith et al., 1999). Together, these results suggest that the transport of Par3 toward the distal part of axon is regulated by the phosphorylation of Par3.
The roles of Par3 and its phosphorylation in neuronal polarization
Here, we show that the ERK-dependent phosphorylation of Par3 regulates KIF3A-mediated axonal transport. Because it has been reported that the KIF3A-dependent transport of Par3 is important for neuronal polarity (Nishimura et al., 2004), the ERK-dependent phosphorylation of Par3 may be involved in neuronal polarization. Therefore, we first examined whether the MEK/ERK pathway affected neuronal polarity. We found that the expression of MEK1-CA or MEK1-DN attenuated the length of the longest neurite and decreased the population of Stage 3 neurons (Fig. 6A–C). These results suggest that a proper balance of ERK activity is important for neuronal polarity, as described for calcium-calmodulin-dependent protein kinase kinase (Nakamuta et al., 2011). Next, to investigate whether the phosphorylation of Par3 at Ser-1116 affects neuronal polarity, knockdown and rescue experiments were performed using an RNA interference (RNAi)-resistant form of Par3 (rrPar3). We previously reported that two different siRNAs for Par3 specifically knocked down the expression of Par3 (siPar3 #4 and siPar3 #6) (Nishimura et al., 2005; Nakayama et al., 2008). Hippocampal neurons were cotransfected with siPar3 #6 and Myc-GST, Myc-rrPar3-WT, -S1116A, -S1116D, or -Δ4N/2 after plating. Neurons were fixed at 3 DIV and then immunostained with anti-Myc and anti-Tau-1 antibodies. Most of the cells expressing scramble siRNA and control GST displayed normal polarity (i.e., they had single long neurite that were Tau-1-positive and some minor processes) (Fig. 7A–C). The knockdown of Par3 attenuated the length of the longest neurite and decreased the population of Stage 3 neurons (Fig. 7A–C). These phenotypes were recovered with rrPar3-WT and -S1116A, but not with rrPar3-S1116D and -Δ4N/2, which did not bind to KIF3A (Fig. 7A–C). We also used the Par3 fragment Par3-4N/2, which functions as a dominant-negative mutant by competing with Par3 for KIF3A binding (Nishimura et al., 2004), and analyzed its effects on neuronal polarization. The expression of Par3-4N/2-WT and -S1116A decreased the number of Stage 3 neurons, as described previously, whereas Par3-4N/2-S1116D, which did not compete with Par3 for KIF3A binding, did not affect the number of Stage 3 neurons (Fig. 7D–F). Together, these results suggest that Par3 and its phosphorylation at Ser-1116 control neuronal polarity in cultured hippocampal neurons.
Figure 6.

MEK1 regulates neuronal polarity. A–C, Hippocampal neurons were transfected with Myc-GST (Control), Myc-MEK1-WT, -CA, or -DN. A, Representative images of neurons at 3 DIV. Scale bars, 20 μm. The length of the longest neurite (B) and the percentages of neurons at specific stages (C) were determined at 3 DIV. At least 30 cells per sample were measured by tracing images of immunofluorescence staining with anti-Myc (green) and anti-Tau-1 (red) antibodies. The numbers in the bars indicate the total number of neurons examined. Data represent the mean ± SEM of three independent experiments. *p < 0.05 (Dunnett's multiple-comparison test). **p < 0.01 (Dunnett's multiple-comparison test). n.s., Not significant.
Figure 7.

The effect of mutations of Par3 at phosphorylation sites on neuronal polarity. A–C, Hippocampal neurons were cotransfected with siScramble or siPar3 #6 and the indicated RNAi-resistant Myc-tagged Par3 constructs. A, Representative images of neurons at 3 DIV are shown. Scale bars, 20 μm. B, The length of the longest neurite. C, The percentages of neurons at specific stages were determined at 3 DIV. At least 50 cells per sample were measured by tracing images of immunofluorescence staining with anti-Myc (green) and anti-Tau-1 (red) antibodies. The numbers in the bars indicate the total number of neurons examined. Data represent the mean ± SEM of three independent experiments. *p < 0.05 (Tukey's multiple-comparison test). **p < 0.01 (Tukey's multiple-comparison test). n.s., Not significant. D–F, Hippocampal neurons were transfected with Myc-GST (control), Myc-Par3-4N/2-WT, -S1116A, or -S1116D. Representative images of the neurons at 3 DIV are shown (D). Scale bars represent 20 μm. The length of the longest neurite (E) and the percentages of neurons at specific stages (F) were determined at 3 DIV. At least 30 cells per sample were measured by tracing the images of immunofluorescence staining with anti-Myc (green) and anti-Tau-1 (red) antibodies. The numbers in the bars indicate the total number of neurons examined. Data represent the mean ± SEM of three independent experiments. *p < 0.05 (Dunnett's multiple-comparison test). **p < 0.01 (Dunnett's multiple-comparison test). n.s., Not significant.
The roles of Par3 and its phosphorylation in neuronal polarization in vivo
Although Par3 has been implicated in neuronal polarity in cultured neurons, its role in neuronal polarity in vivo remains elusive (Arimura and Kaibuchi, 2007; Barnes and Polleux, 2009; Insolera et al., 2011). We used cortical excitatory projection neurons to investigate the roles of Par3 in neuronal polarity in vivo because these cortical neurons exhibit processes that are similar to hippocampal neurons and are useful for dissecting neuronal polarization in vivo (Barnes and Polleux, 2009; Nakamuta et al., 2011). Cortical excitatory projection neurons are derived from neuronal progenitor cells in the ventricular zone or subventricular zone (Kriegstein and Noctor, 2004; Pinto and Gotz, 2007). The newborn neurons migrate radially and typically display a multipolar morphology with several immature neurites (multipolar neurons) in the proximal IZ. The multipolar neurons are subsequently transformed to exhibit a bipolar morphology with a leading and a trailing process (bipolar neurons) and migrate along the radial fibers of the progenitor cells toward the CP (Tabata and Nakajima, 2003; Hatanaka et al., 2004; Kriegstein and Noctor, 2004; LoTurco and Bai, 2006; Barnes and Polleux, 2009; Reiner and Sapir, 2009). The leading process becomes the apical dendrites, and the trailing process becomes the axon (Schwartz et al., 1991; Barnes and Polleux, 2009). Given that recent studies have shown that axon formation occurs during the transition from multipolar to bipolar morphologies in vivo (Nakamuta et al., 2011; Hatanaka and Yamauchi, 2012; Sakakibara et al., 2013), we focused mainly on the multipolar-to-bipolar transition in the present study.
We used an in utero electroporation system to introduce Par3 cDNA and shRNA into neuronal progenitor cells to explore the role of Par3 in axon formation in vivo. It has been reported that Par3 regulates the asymmetric division of progenitor cells in the developing neocortex (Bultje et al., 2009). To circumvent this problem, we introduced Par3 cDNA and shRNA under the control of the Cre recombinase and neuron-specific Tα promoter as described previously (Nakamuta et al., 2011). To examine the effect of Par3 and its phosphorylation on neuronal polarization in vivo, knockdown and rescue experiments were performed using rrPar3-WT, -S1116A, or -S1116D. We prepared pSico-mCherry-Par3 (shPar3), which allowed for the expression of shRNA against Par3 under the control of Cre recombinase. The transfection of four different shPar3 constructs (#1, #2, #3, and #4) with EF-Cre effectively knocked down endogenous Par3 in Neuro2a cells (Fig. 8A). The cotransfection of shPar3 #3, the most effective of these constructs, with rrPar3 restored Par3 expression (Fig. 8B). To visualize the morphologies of the neurons, Lyn-EGFP was coelectroporated with shControl, shPar3 #3, or shPar3 #3 and rrPar3-WT, -S1116A, or -S1116D into the cerebral cortices at E13, followed by fixation at E16. In control embryos, most of the EGFP-positive neurons in the IZ displayed bipolar morphologies. The knockdown of Par3 increased the number of multipolar cells. This phenotype was rescued by coelectroporation with rrPar3-WT or -S1116A but not with rrPar3-S1116D, which did not bind to KIF3A (Fig. 8C–E). Next, we examined the effect of dominant-negative Par3 fragments on neuronal polarity in vivo. Lyn-EGFP was coelectroporated with Par3-4N/2-WT, -S1116A, or -S1116D into the cerebral cortices at E13, followed by fixation at E16. The number of neurons with multipolar morphology was increased in embryos electroporated with Par3-4N/2-WT or -S1116A but not with Par3-4N/2-S1116D, which did not compete with Par3 for KIF3A binding (Fig. 8F–H). Together, these results suggest that Par3 and its phosphorylation at Ser-1116 control neuronal polarity in vivo.
Figure 8.
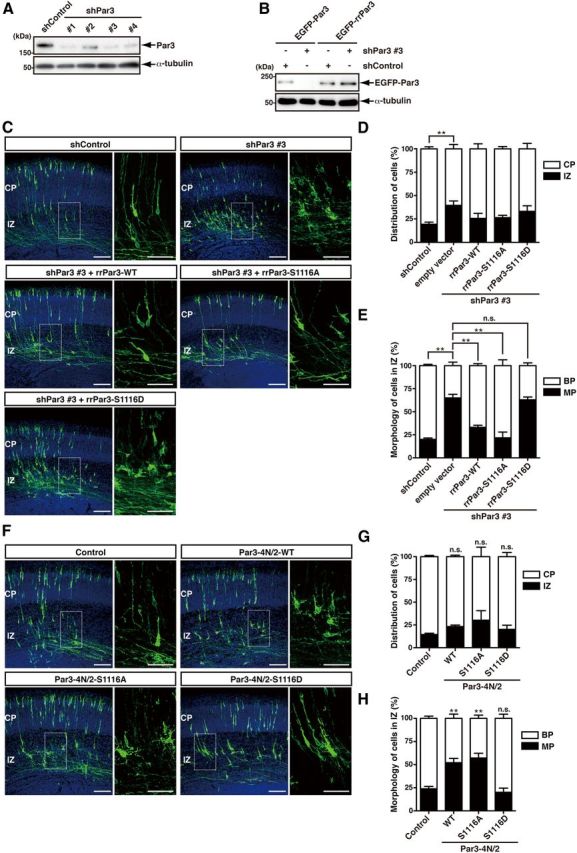
The effect of mutations of Par3 at phosphorylation sites on neuronal polarity in vivo. A, pSico-mCherry (shControl), pSico-mCherry-shPar3 #1, -shPar3 #2, -shPar3 #3, or -shPar3 #4 were cotransfected into Neuro2a cells with pEF-Cre. After 72 h, the cells were lysed and subjected to immunoblotting with anti-Par3 and anti-α-tubulin antibodies. B, pEGFP-Par3 or RNA-interference-resistant pEGFP-Par3 (pEGFP-rrPar3) were cotransfected into COS7 cells with pSico-mCherry or pSico-mCherry-shPar3 #3 with pEF-Cre. After 72 h, the cells were lysed and subjected to immunoblotting with anti-GFP and anti-α-tubulin antibodies. C, Tα-LPL-Lyn-EGFP was coelectroporated with Tα-Cre and pSico-mCherry, pSico-mCherry-shPar3 #3 or pSico-mCherry-shPar3 #3, and RNAi-resistant Par3 mutants into the cerebral cortices at E13 followed by fixation at E16. Coronal sections were prepared and immunostained with anti-GFP antibody (green). Nuclei were stained with Hoechst 33342 (blue). Scale bars: left, 100 μm; right, 50 μm. D, Quantifications of the distribution of EGFP-positive cells in distinct regions of the cerebral cortex (CP and IZ) for each experimental condition are shown on the graph. E, The percentage of EGFP-positive cells with bipolar (BP) or multipolar (MP) morphologies in the IZ of the cerebral cortex. Error bars represent the SEM. shControl + empty vector, n = 4; shPar3 #3 + empty vector, n = 4; shPar3 #3 + rrPar3-WT, n = 3; shPar3 #3 + rrPar3-S1116A, n = 3; shPar3 #3 + rrPar3-S1116D, n = 4. *p < 0.05 (Tukey's multiple-comparison test). **p < 0.01 (Tukey's multiple-comparison test). n.s., Not significant. F, pTα-LPL-Lyn-EGFP was coelectroporated with Tα-Cre and pTα-LPL (control), pTα-LPL-Par3-4N/2-WT, -S1116A, or -S1116D into the cerebral cortices at E13 followed by fixation at E16. Coronal sections were prepared and immunostained with anti-GFP antibody (green). Nuclei were stained with Hoechst 33342 (blue). Scale bars: left, 100 μm; right, 50 μm. G, Quantifications of the distribution of EGFP-positive cells in distinct regions of the cerebral cortex (CP and IZ) for each experimental condition are shown on the graph. H, The percentage of the EGFP-positive cells with bipolar (BP) or multipolar (MP) morphologies in the IZ of the cerebral cortex. Error bars represent the SEM. Control, n = 6; pTα-LPL-Par3-4N/2-WT, n = 4; pTα-LPL-Par3-4N/2-S1116A, n = 3; pTα-LPL-Par3-4N/2-S1116D, n = 5. **p < 0.01 (Dunnett's multiple-comparison test). n.s., Not significant versus control.
Discussion
The loading and unloading of cargo, such as Par3 and specific vesicles on kinesins, seem to be tightly regulated to ensure proper axon formation and neuronal function (Arimura and Kaibuchi, 2007; Hirokawa et al., 2009). It has been reported that the phosphorylation of cargo adaptors and kinesins regulates the association between cargo adaptors and kinesins and that this, in turn, regulates the loading and unloading of the kinesins' cargos (Schlager and Hoogenraad, 2009; Namba et al., 2011). The MAPKs negatively regulate the interaction of kinesin motors with their cargo (Bowman et al., 2000; Byrd et al., 2001; Verhey et al., 2001; Berman et al., 2003; Bengs et al., 2005; Burghoorn et al., 2007; Vagnoni et al., 2011). CaMKII phosphorylates the tail domain of KIF17 (kinesin-2) (Guillaud et al., 2008); this, in turn, leads to the dissociation of KIF17 from its cargo, NMDAR, and from adaptor proteins, such as LIN10/Mint1 (Guillaud et al., 2008). We previously demonstrated that Collapsin response mediator protein-2 (CRMP-2) serves as a cargo adaptor linking kinesin-1 with tubulin dimers (Kimura et al., 2005), specifically Rac-associated protein 1 (Sra-1) (Kawano et al., 2005) and TrkB (Arimura et al., 2009b) to regulate their transport in axons. We also found that GSK-3β phosphorylates CRMP-2 and inhibits its binding to kinesin-1, thereby regulating TrkB transport (Arimura et al., 2009b). Here, we found that ERK2 phosphorylated Par3 at Ser-1116 (Fig. 2A,B,D) and thereby inhibited its interaction with KIF3A (Fig. 4B–D). This phosphorylation was ERK2-dependent in vivo (Fig. 3B–D), and the phosphorylated Par3 accumulated in the growth cones (Fig. 3E). The phosphomimic mutant of Par3 (Par3-S1116D) and Par3-Δ4N/2 exhibited a reduced ability to bind KIF3s (Fig. 4C) and slower transport in axons (Fig. 5C). Together, these results suggest that ERK2 phosphorylates Par3 and inhibits its binding to KIF3A, thereby negatively controlling Par3 transport. Although the phosphorylation of Par3 by ERK2 may play a critical role in the unloading of Par3 from KIF3s, it is also possible that this phosphorylation prevents the loading of Par3 on KIF3s in the soma.
We previously showed that the Ras/Raf/MEK/ERK pathway plays a critical role in the establishment of neuronal polarity (Yoshimura et al., 2006). However, the substrates of ERK in neuronal polarization have not yet been identified. In the present study, we identified Par3 as a substrate of ERK2. Here, we found that the stimulation of the neurons with NT-3 induced the ERK-dependent phosphorylation of Par3 at Ser-1116 (Fig. 3D), that the phosphorylated Par3 accumulated in the distal part of the nascent axon, and that this phosphorylation was diminished by treatment with U0126 (Fig. 3E). Consistently, ERK1/2 activity is higher in the neurite than in the soma (Pertz et al., 2008). It has been reported that the axon formation of hippocampal neurons is enhanced by the treatment of neurotrophins, such as BDNF (Labelle and Leclerc, 2000; Shelly et al., 2007) and NT-3 (Morfini et al., 1994; Labelle and Leclerc, 2000; Yoshimura et al., 2005; Nakamuta et al., 2011). Further, the treatment of hippocampal neurons with NT-3 induces ERK1/2 phosphorylation in the distal part of the nascent axon (Shinoda et al., 2007). Together, these observations suggest that Par3 is locally phosphorylated by the activated ERK1/2 at the tip of the neurite. We also found that Par3 knockdown impaired neuronal polarization in cultured hippocampal neurons, and the inhibitory effects of Par3 knockdown were recovered with Par3-WT but not with Par3-S1116D and -Δ4N/2, which cannot bind to KIF3A (Fig. 7A–C). Interestingly, the Par3-S1116A also rescued the Par3 knockdown phenotype. A possible reason why Par3-S1116A did not affect the neuronal polarity is that Par3-S1116A at least interacted with KIF3A (Fig. 4C) and could be transported by KIF3A (Fig. 5C). FRAP analysis showed that the recovery of EGFP-Par3-S1116A fluorescence occurred at the same rate as that of EGFP-Par3-WT in axons (Fig. 5C). Once the cargo is loaded, kinesin motors have to transport them to their correct subcellular regions (e.g., the distal part of axon). Par3-S1116A may be unloaded in the distal part of axon in an unknown manner (most likely by simple dissociation). In contrast, Par3-S1116D and -Δ4N/2 lacked the ability to bind to KIF3A (Fig. 4C); therefore, these mutants were not transported to the tip of the axon in a kinesin-dependent manner (Fig. 5C). Thus, Par3-S1116A could rescue the Par3-knockdown phenotypes regarding the neuronal polarity (Fig. 7A–C). In contrast, Par3-S1116D and -Δ4N/2 could not rescue the Par3-knockdown phenotypes (Fig. 7A–C). These results suggest that the phosphorylation of Par3 by ERK2 downstream of the neurotrophins is required for the proper polarization of hippocampal neurons.
Par3 has been implicated in axon formation in cultured hippocampal neurons (Shi et al., 2003; Nishimura et al., 2004, 2005; Schwamborn et al., 2007). However, the physiological roles of Par3 in neuronal polarization in vivo remain undetermined because the removal or ectopic expression of Par3 affects the asymmetric cell division of radial glial progenitors (Bultje et al., 2009). To circumvent this problem, we introduced Par3 cDNA and shRNA under the control of Cre recombinase and the Tα promoter (Fig. 8C–H) such that Par3 was expressed shortly after the differentiation of these neurons without being expressed in progenitor cells. We found that knockdown of Par3 prevented neuronal polarization (Fig. 8C–E). This inhibitory effect was rescued by Par3-WT but not by Par3-S1116D (Fig. 8C–E). These results suggest that Par3 and its phosphorylation by ERK2 are important for neuronal polarization in cortical projection neurons in vivo.
Footnotes
This work was supported by the Japan Science and Technology Agency, Core Research for Evolutionary Science and Technology (CREST); Grant (S) 20227006 from Grants-in-Aid for Scientific Research from Japan Society for the Promotion of Science (JSPS) (K. Kaibuchi); Grant 23123507 from Grants-in-Aid for Scientific Research on Innovative Areas “Neural Diversity and Neocortical Organization” from Ministry of Education, Culture, Sports, Science and Technology (K. Kaibuchi); a Grant-in-aid for Global COE Research from MEXT; and Grant 20-4053 from Grants-in-aid for JSPS (S.N.). A part of this study is the result of “Bioinformatics of Brain Sciences” carried out under the Strategic Research Program for Brain Sciences by MEXT. We thank F. Murakami and Y. Yamasaki for in utero electroporation (Osaka University, Osaka, Japan); T. Miyata and A. Sakakibara for technical assistance and preparation of some materials (Nagoya University, Nagoya, Japan); M. Inagaki and M. Matsuyama (Aichi Cancer Center Research Institute, Nagoya, Japan) for the purification of antibodies; T. Nishimura (RIKEN Center for Developmental Biology, Kobe, Japan), N. Arimura (National Institute of Neuroscience, National Center of Neurology and Psychiatry, Tokyo, Japan), and T. Yoshimura (National Institute for Physiological Sciences, Okazaki, Japan) for helpful discussions, and the preparation of some materials; F. Ishidate for help with acquiring and analyzing images; all the members of the K. Kaibuchi laboratory for technical support; T. Ishii for secretarial assistance; M. Amano, T. Watanabe, D. Mori, D. Tsuboi, K. Kuroda, K. Sato, K. Matsuzawa, M. Matsui, and T. Hamaguchi for helpful discussions, and the preparation of some materials; the Division of Medical Research Engineering of the Nagoya University Graduate School of Medicine (I. Mizuguchi and Y. Fujita) for the use of ImageQuant LAS 4010 (GE Biohealthcare Bioscience) and Nikon-A1 microscopy; the Radioisotope Center Medical Branch, Nagoya University Graduate School of Medicine (Technical Staff, N. Hamada and Y. Nakamura); and the Nikon Imaging Laboratory of the Institute of Medical Science, University of Tokyo for the use of Nikon N-SIM (K. Tokunaga).
The authors declare no competing financial interests.
References
- Amano M, Mukai H, Ono Y, Chihara K, Matsui T, Hamajima Y, Okawa K, Iwamatsu A, Kaibuchi K. Identification of a putative target for Rho as the serine-threonine kinase protein kinase N. Science. 1996;271:648–650. doi: 10.1126/science.271.5249.648. [DOI] [PubMed] [Google Scholar]
- Arimura N, Kaibuchi K. Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat Rev Neurosci. 2007;8:194–205. doi: 10.1038/nrn2056. [DOI] [PubMed] [Google Scholar]
- Arimura N, Hattori A, Kimura T, Nakamuta S, Funahashi Y, Hirotsune S, Furuta K, Urano T, Toyoshima YY, Kaibuchi K. CRMP-2 directly binds to cytoplasmic dynein and interferes with its activity. J Neurochem. 2009a;111:380–390. doi: 10.1111/j.1471-4159.2009.06317.x. [DOI] [PubMed] [Google Scholar]
- Arimura N, Kimura T, Nakamuta S, Taya S, Funahashi Y, Hattori A, Shimada A, Ménager C, Kawabata S, Fujii K, Iwamatsu A, Segal RA, Fukuda M, Kaibuchi K. Anterograde transport of TrkB in axons is mediated by direct interaction with Slp1 and Rab27. Dev Cell. 2009b;16:675–686. doi: 10.1016/j.devcel.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Barnes AP, Polleux F. Establishment of axon-dendrite polarity in developing neurons. Annu Rev Neurosci. 2009;32:347–381. doi: 10.1146/annurev.neuro.31.060407.125536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengs F, Scholz A, Kuhn D, Wiese M. LmxMPK9, a mitogen-activated protein kinase homologue affects flagellar length in Leishmania mexicana. Mol Microbiol. 2005;55:1606–1615. doi: 10.1111/j.1365-2958.2005.04498.x. [DOI] [PubMed] [Google Scholar]
- Berman SA, Wilson NF, Haas NA, Lefebvre PA. A novel MAP kinase regulates flagellar length in Chlamydomonas. Curr Biol. 2003;13:1145–1149. doi: 10.1016/S0960-9822(03)00415-9. [DOI] [PubMed] [Google Scholar]
- Bowman AB, Kamal A, Ritchings BW, Philp AV, McGrail M, Gindhart JG, Goldstein LS. Kinesin-dependent axonal transport is mediated by the sunday driver (SYD) protein. Cell. 2000;103:583–594. doi: 10.1016/S0092-8674(00)00162-8. [DOI] [PubMed] [Google Scholar]
- Bradke F, Dotti CG. Establishment of neuronal polarity: lessons from cultured hippocampal neurons. Curr Opin Neurobiol. 2000;10:574–581. doi: 10.1016/S0959-4388(00)00124-0. [DOI] [PubMed] [Google Scholar]
- Bultje RS, Castaneda-Castellanos DR, Jan LY, Jan YN, Kriegstein AR, Shi SH. Mammalian Par3 regulates progenitor cell asymmetric division via notch signaling in the developing neocortex. Neuron. 2009;63:189–202. doi: 10.1016/j.neuron.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghoorn J, Dekkers MP, Rademakers S, de Jong T, Willemsen R, Jansen G. Mutation of the MAP kinase DYF-5 affects docking and undocking of kinesin-2 motors and reduces their speed in the cilia of Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2007;104:7157–7162. doi: 10.1073/pnas.0606974104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd DT, Kawasaki M, Walcoff M, Hisamoto N, Matsumoto K, Jin Y. UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans. Neuron. 2001;32:787–800. doi: 10.1016/S0896-6273(01)00532-3. [DOI] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, Brenner SE, Batalov S, Forrest AR, Zavolan M, Davis MJ, Wilming LG, Aidinis V, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- Chen X, Macara IG. Par-3 controls tight junction assembly through the Rac exchange factor Tiam1. Nat Cell Biol. 2005;7:262–269. doi: 10.1038/ncb1226. [DOI] [PubMed] [Google Scholar]
- Cheng PL, Song AH, Wong YH, Wang S, Zhang X, Poo MM. Self-amplifying autocrine actions of BDNF in axon development. Proc Natl Acad Sci U S A. 2011;108:18430–18435. doi: 10.1073/pnas.1115907108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AM, Banker G. Neuronal polarity. Annu Rev Neurosci. 1994;17:267–310. doi: 10.1146/annurev.ne.17.030194.001411. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow LE, Humbert PO. Polarity regulators and the control of epithelial architecture, cell migration, and tumorigenesis. Int Rev Cytol. 2007;262:253–302. doi: 10.1016/S0074-7696(07)62006-3. [DOI] [PubMed] [Google Scholar]
- Drees F, Gertler FB. Ena/VASP: proteins at the tip of the nervous system. Curr Opin Neurobiol. 2008;18:53–59. doi: 10.1016/j.conb.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errede B, Cade RM, Yashar BM, Kamada Y, Levin DE, Irie K, Matsumoto K. Dynamics and organization of MAP kinase signal pathways. Mol Reprod Dev. 1995;42:477–485. doi: 10.1002/mrd.1080420416. [DOI] [PubMed] [Google Scholar]
- Galbraith JA, Reese TS, Schlief ML, Gallant PE. Slow transport of unpolymerized tubulin and polymerized neurofilament in the squid giant axon. Proc Natl Acad Sci U S A. 1999;96:11589–11594. doi: 10.1073/pnas.96.20.11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein B, Macara IG. The PAR proteins: fundamental players in animal cell polarization. Dev Cell. 2007;13:609–622. doi: 10.1016/j.devcel.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslin K, Banker G. Experimental observations on the development of polarity by hippocampal neurons in culture. J Cell Biol. 1989;108:1507–1516. doi: 10.1083/jcb.108.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaud L, Wong R, Hirokawa N. Disruption of KIF17-Mint1 interaction by CaMKII-dependent phosphorylation: a molecular model of kinesin-cargo release. Nat Cell Biol. 2008;10:19–29. doi: 10.1038/ncb1665. [DOI] [PubMed] [Google Scholar]
- Hatanaka Y, Yamauchi K. Excitatory cortical neurons with multipolar shape establish neuronal polarity by forming a tangentially oriented axon in the intermediate zone. Cereb Cortex. 2013;23:105–113. doi: 10.1093/cercor/bhr383. [DOI] [PubMed] [Google Scholar]
- Hatanaka Y, Hisanaga S, Heizmann CW, Murakami F. Distinct migratory behavior of early- and late-born neurons derived from the cortical ventricular zone. J Comp Neurol. 2004;479:1–14. doi: 10.1002/cne.20256. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- Horiuchi D, Collins CA, Bhat P, Barkus RV, Diantonio A, Saxton WM. Control of a kinesin-cargo linkage mechanism by JNK pathway kinases. Curr Biol. 2007;17:1313–1317. doi: 10.1016/j.cub.2007.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd TW, Fan S, Liu CJ, Kweon HK, Hakansson K, Margolis B. Phosphorylation-dependent binding of 14-3-3 to the polarity protein Par3 regulates cell polarity in mammalian epithelia. Curr Biol. 2003;13:2082–2090. doi: 10.1016/j.cub.2003.11.020. [DOI] [PubMed] [Google Scholar]
- Iida N, Namikawa K, Kiyama H, Ueno H, Nakamura S, Hattori S. Requirement of Ras for the activation of mitogen-activated protein kinase by calcium influx, cAMP, and neurotrophin in hippocampal neurons. J Neurosci. 2001;21:6459–6466. doi: 10.1523/JNEUROSCI.21-17-06459.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Chihara K, Arimura N, Ménager C, Kawano Y, Matsuo N, Nishimura T, Amano M, Kaibuchi K. CRMP-2 induces axons in cultured hippocampal neurons. Nat Neurosci. 2001;4:781–782. doi: 10.1038/90476. [DOI] [PubMed] [Google Scholar]
- Insolera R, Chen S, Shi SH. Par proteins and neuronal polarity. Dev Neurobiol. 2011;71:483–494. doi: 10.1002/dneu.20867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N, Nakayama M, Nishimura T, Fujisue S, Nishioka T, Watanabe T, Kaibuchi K. Identification of focal adhesion kinase (FAK) and phosphatidylinositol 3-kinase (PI3-kinase) as Par3 partners by proteomic analysis. Cytoskeleton (Hoboken) 2010;67:297–308. doi: 10.1002/cm.20444. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Hirose T, Tamai Y, Hirai S, Nagashima Y, Fujimoto T, Tabuse Y, Kemphues KJ, Ohno S. An atypical PKC directly associates and colocalizes at the epithelial tight junction with ASIP, a mammalian homologue of Caenorhabditis elegans polarity protein PAR-3. J Cell Biol. 1998;143:95–106. doi: 10.1083/jcb.143.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joberty G, Petersen C, Gao L, Macara IG. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol. 2000;2:531–539. doi: 10.1038/35019573. [DOI] [PubMed] [Google Scholar]
- Johansson A, Driessens M, Aspenström P. The mammalian homologue of the Caenorhabditis elegans polarity protein PAR-6 is a binding partner for the Rho GTPases Cdc42 and Rac1. J Cell Sci. 2000;113:3267–3275. doi: 10.1242/jcs.113.18.3267. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Yoshimura T, Tsuboi D, Kawabata S, Kaneko-Kawano T, Shirataki H, Takenawa T, Kaibuchi K. CRMP-2 is involved in kinesin-1-dependent transport of the Sra-1/WAVE1 complex and axon formation. Mol Cell Biol. 2005;25:9920–9935. doi: 10.1128/MCB.25.22.9920-9935.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. EMBO J. 2003;22:4190–4201. doi: 10.1093/emboj/cdg413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemphues KJ, Priess JR, Morton DG, Cheng NS. Identification of genes required for cytoplasmic localization in early C. elegans embryos. Cell. 1988;52:311–320. doi: 10.1016/S0092-8674(88)80024-2. [DOI] [PubMed] [Google Scholar]
- Kimura T, Watanabe H, Iwamatsu A, Kaibuchi K. Tubulin and CRMP-2 complex is transported via kinesin-1. J Neurochem. 2005;93:1371–1382. doi: 10.1111/j.1471-4159.2005.03063.x. [DOI] [PubMed] [Google Scholar]
- Knoblich JA. Asymmetric cell division during animal development. Nat Rev Mol Cell Biol. 2001;2:11–20. doi: 10.1038/35048085. [DOI] [PubMed] [Google Scholar]
- Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27:392–399. doi: 10.1016/j.tins.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Labelle C, Leclerc N. Exogenous BDNF, NT-3 and NT-4 differentially regulate neurite outgrowth in cultured hippocampal neurons. Brain Res Dev Brain Res. 2000;123:1–11. doi: 10.1016/S0165-3806(00)00069-9. [DOI] [PubMed] [Google Scholar]
- Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T. A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat Cell Biol. 2000;2:540–547. doi: 10.1038/35019582. [DOI] [PubMed] [Google Scholar]
- LoTurco JJ, Bai J. The multipolar stage and disruptions in neuronal migration. Trends Neurosci. 2006;29:407–413. doi: 10.1016/j.tins.2006.05.006. [DOI] [PubMed] [Google Scholar]
- MacKenzie SJ, Baillie GS, McPhee I, Bolger GB, Houslay MD. ERK2 mitogen-activated protein kinase binding, phosphorylation, and regulation of the PDE4D cAMP-specific phosphodiesterases. J Biol Chem. 2000;275:16609–16617. doi: 10.1074/jbc.275.22.16609. [DOI] [PubMed] [Google Scholar]
- Manabe N, Hirai S, Imai F, Nakanishi H, Takai Y, Ohno S. Association of ASIP/mPAR-3 with adherens junctions of mouse neuroepithelial cells. Dev Dyn. 2002;225:61–69. doi: 10.1002/dvdy.10139. [DOI] [PubMed] [Google Scholar]
- Markus A, Patel TD, Snider WD. Neurotrophic factors and axonal growth. Curr Opin Neurobiol. 2002;12:523–531. doi: 10.1016/S0959-4388(02)00372-0. [DOI] [PubMed] [Google Scholar]
- McCaffrey LM, Montalbano J, Mihai C, Macara IG. Loss of the Par3 polarity protein promotes breast tumorigenesis and metastasis. Cancer Cell. 2012;22:601–614. doi: 10.1016/j.ccr.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens AE, Rygiel TP, Olivo C, van der Kammen R, Collard JG. The Rac activator Tiam1 controls tight junction biogenesis in keratinocytes through binding to and activation of the Par polarity complex. J Cell Biol. 2005;170:1029–1037. doi: 10.1083/jcb.200502129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, DiTella MC, Feiguin F, Carri N, Cáceres A. Neurotrophin-3 enhances neurite outgrowth in cultured hippocampal pyramidal neurons. J Neurosci Res. 1994;39:219–232. doi: 10.1002/jnr.490390212. [DOI] [PubMed] [Google Scholar]
- Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- Nagai-Tamai Y, Mizuno K, Hirose T, Suzuki A, Ohno S. Regulated protein-protein interaction between aPKC and PAR-3 plays an essential role in the polarization of epithelial cells. Genes Cells. 2002;7:1161–1171. doi: 10.1046/j.1365-2443.2002.00590.x. [DOI] [PubMed] [Google Scholar]
- Nakamuta S, Funahashi Y, Namba T, Arimura N, Picciotto MR, Tokumitsu H, Soderling TR, Sakakibara A, Miyata T, Kamiguchi H, Kaibuchi K. Local application of neurotrophins specifies axons through inositol 1,4,5-trisphosphate, calcium, and Ca2+/calmodulin-dependent protein kinases. Sci Signal. 2011;4:ra76. doi: 10.1126/scisignal.2002011. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Goto TM, Sugimoto M, Nishimura T, Shinagawa T, Ohno S, Amano M, Kaibuchi K. Rho-kinase phosphorylates PAR-3 and disrupts PAR complex formation. Dev Cell. 2008;14:205–215. doi: 10.1016/j.devcel.2007.11.021. [DOI] [PubMed] [Google Scholar]
- Namba T, Nakamuta S, Funahashi Y, Kaibuchi K. The role of selective transport in neuronal polarization. Dev Neurobiol. 2011;71:445–457. doi: 10.1002/dneu.20876. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Kato K, Yamaguchi T, Fukata Y, Ohno S, Kaibuchi K. Role of the PAR-3-KIF3 complex in the establishment of neuronal polarity. Nat Cell Biol. 2004;6:328–334. doi: 10.1038/ncb1118. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Yamaguchi T, Kato K, Yoshizawa M, Nabeshima Y, Ohno S, Hoshino M, Kaibuchi K. PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat Cell Biol. 2005;7:270–277. doi: 10.1038/ncb1227. [DOI] [PubMed] [Google Scholar]
- Pertz OC, Wang Y, Yang F, Wang W, Gay LJ, Gristenko MA, Clauss TR, Anderson DJ, Liu T, Auberry KJ, Camp DG, 2nd, Smith RD, Klemke RL. Spatial mapping of the neurite and soma proteomes reveals a functional Cdc42/Rac regulatory network. Proc Natl Acad Sci U S A. 2008;105:1931–1936. doi: 10.1073/pnas.0706545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto L, Götz M. Radial glial cell heterogeneity: the source of diverse progeny in the CNS. Prog Neurobiol. 2007;83:2–23. doi: 10.1016/j.pneurobio.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Qiu RG, Abo A, Steven Martin G. A human homolog of the C. elegans polarity determinant Par-6 links Rac and Cdc42 to PKCzeta signaling and cell transformation. Curr Biol. 2000;10:697–707. doi: 10.1016/S0960-9822(00)00535-2. [DOI] [PubMed] [Google Scholar]
- Reiner O, Sapir T. Polarity regulation in migrating neurons in the cortex. Mol Neurobiol. 2009;40:1–14. doi: 10.1007/s12035-009-8065-0. [DOI] [PubMed] [Google Scholar]
- Saito T, Nakatsuji N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev Biol. 2001;240:237–246. doi: 10.1006/dbio.2001.0439. [DOI] [PubMed] [Google Scholar]
- Sakakibara A, Sato T, Ando R, Noguchi N, Masaoka M, Miyata T. Dynamics of centrosome translocation and microtubule organization in neocortical neurons during distinct modes of polarization. Cereb Cortex. 2013 doi: 10.1093/cercor/bhs411. doi: 10.1093/cercor/bhs411. Advance online publication. Retrieved Jan. 10, 2013. [DOI] [PubMed] [Google Scholar]
- Schlager MA, Hoogenraad CC. Basic mechanisms for recognition and transport of synaptic cargos. Mol Brain. 2009;2:25. doi: 10.1186/1756-6606-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwamborn JC, Püschel AW. The sequential activity of the GTPases Rap1B and Cdc42 determines neuronal polarity. Nat Neurosci. 2004;7:923–929. doi: 10.1038/nn1295. [DOI] [PubMed] [Google Scholar]
- Schwamborn JC, Khazaei MR, Püschel AW. The interaction of mPar3 with the ubiquitin ligase Smurf2 is required for the establishment of neuronal polarity. J Biol Chem. 2007;282:35259–35268. doi: 10.1074/jbc.M703438200. [DOI] [PubMed] [Google Scholar]
- Schwartz ML, Rakic P, Goldman-Rakic PS. Early phenotype expression of cortical neurons: evidence that a subclass of migrating neurons have callosal axons. Proc Natl Acad Sci U S A. 1991;88:1354–1358. doi: 10.1073/pnas.88.4.1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelly M, Cancedda L, Heilshorn S, Sumbre G, Poo MM. LKB1/STRAD promotes axon initiation during neuronal polarization. Cell. 2007;129:565–577. doi: 10.1016/j.cell.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Shi SH, Jan LY, Jan YN. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell. 2003;112:63–75. doi: 10.1016/S0092-8674(02)01249-7. [DOI] [PubMed] [Google Scholar]
- Shinoda T, Taya S, Tsuboi D, Hikita T, Matsuzawa R, Kuroda S, Iwamatsu A, Kaibuchi K. DISC1 regulates neurotrophin-induced axon elongation via interaction with Grb2. J Neurosci. 2007;27:4–14. doi: 10.1523/JNEUROSCI.3825-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Ohno S. The PAR-aPKC system: lessons in polarity. J Cell Sci. 2006;119:979–987. doi: 10.1242/jcs.02898. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neuroscience. 2001;103:865–872. doi: 10.1016/S0306-4522(01)00016-1. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Multipolar migration: the third mode of radial neuronal migration in the developing cerebral cortex. J Neurosci. 2003;23:9996–10001. doi: 10.1523/JNEUROSCI.23-31-09996.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahirovic S, Bradke F. Neuronal polarity. Cold Spring Harbor Perspect Biol. 2009;1:a001644. doi: 10.1101/cshperspect.a001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taya S, Shinoda T, Tsuboi D, Asaki J, Nagai K, Hikita T, Kuroda S, Kuroda K, Shimizu M, Hirotsune S, Iwamatsu A, Kaibuchi K. DISC1 regulates the transport of the NUDEL/LIS1/14-3-3epsilon complex through kinesin-1. J Neurosci. 2007;27:15–26. doi: 10.1523/JNEUROSCI.3826-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda H, Mochizuki H, Flores R, 3rd, Josowitz R, Krasieva TB, Lamorte VJ, Suzuki E, Gindhart JG, Furukubo-Tokunaga K, Tomoda T. UNC-51/ATG1 kinase regulates axonal transport by mediating motor-cargo assembly. Genes Dev. 2008;22:3292–3307. doi: 10.1101/gad.1734608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnoni A, Rodriguez L, Manser C, De Vos KJ, Miller CC. Phosphorylation of kinesin light chain 1 at serine 460 modulates binding and trafficking of calsyntenin-1. J Cell Sci. 2011;124:1032–1042. doi: 10.1242/jcs.075168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Meissner A, Dillon CP, McManus M, Sharp PA, Van Parijs L, Jaenisch R, Jacks T. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci U S A. 2004;101:10380–10385. doi: 10.1073/pnas.0403954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey KJ, Meyer D, Deehan R, Blenis J, Schnapp BJ, Rapoport TA, Margolis B. Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J Cell Biol. 2001;152:959–970. doi: 10.1083/jcb.152.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Du D, Fang L, Yang G, Zhang C, Zeng R, Ullrich A, Lottspeich F, Chen Z. Tyrosine phosphorylated Par3 regulates epithelial tight junction assembly promoted by EGFR signaling. EMBO J. 2006;25:5058–5070. doi: 10.1038/sj.emboj.7601384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe-Uchida M, Govek EE, Van Aelst L. Regulators of Rho GTPases in neuronal development. J Neurosci. 2006;26:10633–10635. doi: 10.1523/JNEUROSCI.4084-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiznerowicz M, Szulc J, Trono D. Tuning silence: conditional systems for RNA interference. Nat Methods. 2006;3:682–688. doi: 10.1038/nmeth914. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Arimura N, Kawano Y, Kawabata S, Wang S, Kaibuchi K. Ras regulates neuronal polarity via the PI3-kinase/Akt/GSK-3beta/CRMP-2 pathway. Biochem Biophys Res Commun. 2006;340:62–68. doi: 10.1016/j.bbrc.2005.11.147. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Terabayashi T, Miki H. Par1b/MARK2 phosphorylates kinesin-like motor protein GAKIN/KIF13B to regulate axon formation. Mol Cell Biol. 2010;30:2206–2219. doi: 10.1128/MCB.01181-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FQ, Snider WD. Intracellular control of developmental and regenerative axon growth. Philos Trans R S Lond B Biol Sci. 2006;361:1575–1592. doi: 10.1098/rstb.2006.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]