

Abstract
Downregulation of brain-derived neurotrophic factor (BDNF) in the hippocampus occurs early in the progression of Alzheimer's disease (AD). Since BDNF plays a critical role in neuronal survival and dendrite growth, BDNF upregulation may contribute to rescue dendrite atrophy and cell loss in AD. Low-level laser therapy (LLLT) has been demonstrated to regulate neuronal function both in vitro and in vivo. In the present study, we found that LLLT rescued neurons loss and dendritic atrophy via upregulation of BDNF in both Aβ-treated hippocampal neurons and cultured APP/PS1 mouse hippocampal neurons. Photoactivation of transcription factor CRE-binding protein (CREB) increased both BDNF mRNA and protein expression, since knockdown CREB blocked the effects of LLLT. Furthermore, CREB-regulated transcription was in an ERK-dependent manner. Inhibition of ERK attenuated the DNA-binding efficiency of CREB to BDNF promoter. In addition, dendrite growth was improved after LLLT, characterized by upregulation of Rac1 activity and PSD-95 expression, and the increase in length, branching, and spine density of dendrites in hippocampal neurons. Together, these studies suggest that upregulation of BDNF with LLLT by activation of ERK/CREB pathway can ameliorate Aβ-induced neurons loss and dendritic atrophy, thus identifying a novel pathway by which LLLT protects against Aβ-induced neurotoxicity. Our research may provide a feasible therapeutic approach to control the progression of AD.
Introduction
Neurotrophins exert biological actions primarily on cells of the nervous system (Lewin and Barde, 1996). In addition to their classical role in supporting survival of neuronal populations, brain-derived neurotrophic factor (BDNF), in particular, is a strong candidate to modulate dendritic structure and potentiate synaptic transmission in the CNS (Katz and Shatz, 1996; Connor and Dragunow, 1998; Murer et al., 2001). In patients with AD, neurites atrophy and synaptic loss are considered the major causes of cognitive impairment (Einstein et al., 1994; Masliah et al., 2001; Selkoe, 2002). Recent evidence suggests Aβ-associated neurotoxicity and dendrite atrophy may be a consequence of BDNF deficiency. Several studies indicate that the cortex and hippocampus exhibit both extensive amyloid pathology and decreased levels of BDNF in Alzheimer's disease (AD; Hu and Russek, 2008; Zuccato and Cattaneo, 2009). Learning and memory deficits exhibited by transgenic mouse models of AD can be rescued by BDNF delivery (Nagahara et al., 2009). Increasing BDNF expression may be an important manner to attenuate dendrite atrophy in the CNS during AD pathology.
In recent times, low-level laser therapy (LLLT) constitutes a novel intervention shown to regulate neuronal function in cell cultures, animal models, and clinical conditions (Eells et al., 2003; Rojas et al., 2008). The mechanism of LLLT at the cellular level has been ascribed to the acceleration of electron transfer reactions, resulting in increase of reactive oxygen species and Ca2+ as versatile second messengers (Lavi et al., 2003; Lan et al., 2012). Previous studies have shown that the application of LLLT could have an influence on cellular process including altering DNA synthesis and protein expression (Feng et al., 2012; Yazdani et al., 2012), biomodulation in cytoskeleton organization (Ricci et al., 2009; Song et al., 2012), and stimulating cellular proliferation (Zhang et al., 2009; Feng et al., 2012). Studies have shown that Aβ-induced cell apoptosis was significantly diminished with light irradiation (Liang et al., 2012; Zhang et al., 2012). LLLT can efficiently penetrate into biological tissue including the CNS, producing noninvasive beneficial photobiomodulation effects such as promoting nerve regeneration and increasing ATP synthesis (Anders et al., 1993; Mochizuki-Oda et al., 2002). Such properties support that LLLT, or interventions with similar neurobiological effects, may have a role in the treatment of neurodegeneration, a phenomenon that underlies debilitating clinical conditions.
Dendritic growth is crucially dependent on targeted changes in the cytoskeleton. Activation of the Rho-family GTPases, including Rac and Cdc42, regulate dendritic growth and complexity in several areas of the brain (Li et al., 2000). Furthermore, in mature visual cortical neurons, BDNF can increase the overall amount of the postsynaptic density 95 (PSD-95) scaffolding protein to promote synaptic maturation (Yoshii and Constantine-Paton, 2007).
In this study, we investigated the effects of LLLT on Aβ-induced neurotoxicity in hippocampal neurons. We also found that LLLT rescues the decrease of dendrite length and branching by upregulation of BDNF, via the ERK/CRE-binding protein (CREB) pathway. In addition, we showed that these changes were paralleled by activation of Rac1 and increase in cytoskeleton protein, notably PSD-95. Our data further demonstrate that LLLT has potential therapeutic value in treating AD by targeting BDNF.
Materials and Methods
Chemicals and plasmids.
The following reagents were used: Aβ25–35 and Aβ1–42 were purchased from Sigma-Aldrich, and Aβ25–35 and Aβ1–42 stock solution of 1 mm were prepared in distilled and deionized water and stored at −20°C. Before a treatment, peptides were preincubated at 37°C for 5 d to promote aggregation and the diluted with medium to desired concentration (25 μm). PD98059 and API-2 were purchased from Santa Cruz Biotechnology. Mithramycin-A (MTA) was purchased from Enzo. Gö6983 was purchased from Merck. H89 and farnesyl thiosalicylic acid (FTS) were purchased from Calbiochem. TrkB-Fc peptide was purchased from R&D Systems. Rhodamine-phalloidin, FITC-phalloidin, actinomycin D (Act D), and xestospongin B were obtained from Sigma-Aldrich. Lipofectamine 2000 was purchased from Invitrogen.
The following antibodies were used: anti-ERK, anti-p-ERK, anti-CREB, anti-p-CREB (ser133), anti-PSD-95, and anti-Ras. All were purchased from Cell Signaling Technology. Anti-BDNF, anti-MAP2, and anti-Rac1 antibodies were purchased from Sigma-Aldrich. Anti-NGF antibody was obtained from Bioworld Technology. Anti-NT-3 antibody was purchased from R&D Systems. Anti-β-actin and anti-histone antibody were obtained from Santa Cruz Biotechnology.
Transgenic mice.
The transgenic mice (APP/PS1) used in this study were produced by coinjection with the APPswe and PS1dE9 vectors (Zhou et al., 2011; Zhang et al., 2013). All the experimental mice were of a C57BL/6 background, and Wild-Type (WT) and transgenic mice were paired from the litters and housed under the same living conditions. Male APP/PS1 transgenic mice and their WT littermates were killed at 6 month of age. Brains were removed and the hippocampus was dissected. Tissues were flash frozen in liquid nitrogen and stored at −80°C until analysis.
The present study was performed in accordance with the guidelines of the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council). It was approved by the Institutional Animal Care and Use Committee of our university (South China Normal University, Guangzhou, China).
Cell culture and transfection.
The human neuroblastoma cell line SH-SY5Y was cultured in DMEM containing 10% heat-inactivated fetal bovine serum, penicillin (100 Us/ml), and streptomycin (100 μg/ml) in 5% CO2, 95% air at 37°C in a humidified incubator. SH-SY5Y cells were transfected using Lipofectamine 2000 following the supplier's instructions. The cells were treated 24 h after transfection.
Primary neuronal culture.
Primary neurons were derived from hippocampal of C57BL/6 mice embryonic day 14 (E14) as previously described (Neumann et al., 1995). Neurons were seeded to a density 6 × 105 viable cells/35 mm culture dishes previously coated with poly-l-lysine (100 μg/ml) for at least 1 h at 37°C. Cultures were maintained at 37°C with 5% CO2, supplemented with Neurobasal medium with 2% B27 (Invitrogen), 2 mm l-glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml). APP/PS1 transgenic mice neuronal cultures were used for 14 d in vitro (DIV). The genotype of the animals was determined by PCR on DNA obtained from fibroblasts.
LLLT treatment.
The experiment was conducted as described in our previous work (Liang et al., 2012). Aβ was added to the culture medium 30 min before an LLLT treatment. The cells were irradiated with He-Ne laser (632.8 nm, 10 mW, 12.74 mW/cm2, HN-1000; Laser Technology Application Research Institute, Guangzhou, China) for 0.7, 1.25, 2.5, and 5 min in the dark, with the corresponding fluences of 0.5, 1, 2, and 4 J/cm2, respectively. Throughout each experiment, the cells were kept either in a complete dark or a very dim environment, except when subjected to the light irradiation, to minimize the ambient light interference.
Cell viability assay and cell apoptosis assay.
SH-SY5Y cells or primary hippocampal neurons were cultured in 96-well microplates at a density of 5 × 103 cells/well. Cell viability was assessed with CCK-8 (Dojindo Laboratories) after Aβ and/or LLLT. At the indicated time, CCK-8 was added and incubated for 1.5 h. OD450, the absorbance value at 450 nm, was read with a 96-well plate reader (DG5032; Huadong). The value is directly proportional to the number of viable cells in a culture medium.
Quantification of apoptosis by Annexin-V/PI staining was performed as described previously (Liang et al., 2012). Apoptosis cell death was determined using the BD ApoAlert Annexin-V-FITC Apoptosis Kit (Becton Dickinson, Biosciences) according to the manufacturer's instructions. Flow cytometry was performed on a BD FACSCanto II flow cytometer (Becton Dickinson).
Immunocytochemistry.
After being treated under different experimental conditions, cells were fixed in 4% paraformaldehyde in PBS, pH 7.4, for 15 min at room temperature and were then membrane-permeabilized with 0.5% Triton X-100 in PBS for 5 min. After blocking with 3% bovine serum albumin at 37°C for 1 h, cells were incubated with primary antibodies: anti-MAP2 antibody (1:200), anti-CREB antibody (1:200), or anti-p-CREB (Ser 133) (1:200), at 4°C overnight. Cells were then incubated with secondary antibodies conjugated to FITC (Proteintech Group) for 2 h. After five additional washes with PBS, slides were mounted and analyzed by confocal microscopy (LSM 510 META; Carl Zeiss MicroImaging) and LSM 510 META software (Carl Zeiss MicroImaging).
Phalloidin staining.
The treated neurons were rinsed with PBS before being fixed in 4% formaldehyde and washed again in PBS. The cells were then incubated at room temperature with 0.1% Triton X-100 buffer for 5 min and washed again in PBS. Rhodamine-phalloidin or FITC-phalloidin (1:50 diluted in PBS) was added to the coverslips and incubated at room temperature protected from light for 30 min. The coverslips were mounted on glass slides. The Rhodamine- or FITC-labeled phalloidin was viewed under a confocal microscope.
Western blot analysis.
Expressions of proteins were quantified by Western blot analysis. After individual incubations, cell proteins were extracted in lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Triton X-100, 100 μg/ml PMSF) supplemented with protease inhibitor cocktail set I for 60 min on ice. After centrifugation (4°C, 12,000 rpm, 20 min), the resulting lysates were resolved by SDS-PAGE Bis-Tris gels (30 mg/lane; Invitrogen, Life Technologies) and transferred to PVDF membranes (Millipore). The membranes were blocked in TBST (10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.1% Tween 20) containing 5% nonfat milk and then incubated with a designated primary antibody and a secondary antibody. The signals were detected with an ODYSSEY Infrared Imaging System (LI-COR). The intensity of the Western blot signals was quantitated using ImageJ software (National Institutes of Health (NIH), Bethesda, MD), and the densitometry analyses are presented as the ratio of protein/β-actin protein, and are compared with controls and normalized to 1.
ELISA assay for BDNF detection.
BDNF secretion in neurons was assessed using the ELISA BDNF Emax Immunoassay Kit (Promega). Supernatant collected from cells in the same conditions were used for the viability assay and were diluted 1:100 and plate on wells previously coated following the manufacturer's instructions.
RNAi-mediated gene silencing.
For RNAi-mediated gene silencing, we used CREB-specific siRNA (5′-GGUGGAAAAUGGACUGGCUtt-3′) (Tullai et al., 2007). Cells were assayed for gene silencing at 24 h post-transfection.
Semiquantitative reverse transcription-PCR.
RNeasy Lipid Tissue Mini Kit (Qiagen) was used to extract total RNA from SH-SY5Y cells and ImProm-II Reverse Transcription System (Promega) was used to synthesize cDNA. To determine the relative amount of cDNA molecules per sample, we performed real-time PCR using protocols provided by Light Cycler Fast Start DNA Master SYBR Green I (Roche Diagnostics) system. BDNF was detected with specific primer pairs (5′-CTCCGCCATGCAATTTCCACT-3′, 5′-GCCTTCATGCAACCGAAGTA-3′) (Tabuchi et al., 2002). GAPDH from each sample was also amplified to serve as an internal control. The primer pairs of GAPDH were 5′-TGACAACTTTGGCATCGTGGAAGG-3′ and 5′-CAACGGATACATTGGGGGTAGGAAC-3′. The annealing temperature was 59°C. The amplified products were confirmed by running 1.5% agarose gels.
Chromatin immunoprecipitation assay.
ChIP assay kit was used for ChIP assay (Millipore). SH-SY5Y cells following indicated treatments were cross-linked in 1% formaldehyde at 37°C for 15 min, and 0.125 m glycine was incubated for 5 min to stop the fixation. After several washes using ice-cold PBS, SDS lysis buffer containing 1 mm PMSF was used to homogenize the cells. The lysates were sonicated to shear DNA to lengths between 200 and 800 base pairs. Sonicated DNA (400 μg) taken from each sample was incubated with anti-p-CREB (Ser133) antibody at 4°C overnight and then with 60 μl of salmon sperm DNA/protein A agarose-50% slurry for 1 h to form the antibody/DNA/agarose complex. Negative control was done by using rabbit-IgG instead of anti-p-CREB (Ser133) antibody. The precipitated DNA was eluted by 250 μl of elution buffer. After adding 10 μl of 5 m NaCl, histone-DNA cross-links were reversed at 65°C overnight followed by the addition of 10 μl of 0.5 m EDTA, 20 μl of 1 m Tris-HCl, and 2 μl of 10 mg/ml proteinase K (Sigma) incubated for 1 h at 45°C. DNA fragments were recovered by using the QIAquick PCR purification kit (Qiagen) for subsequent reverse transcription (RT)-PCR. Chromatin immunoprecipitation (ChIP) data were normalized to input DNA from each sample. The amounts of p-CREB binding to respective BDNF promoters were expressed as a percentage of those in naive controls. Primers for RT-PCR were designed to amplify using BDNF promoter (Tabuchi et al., 2002): The primer sequences used were the following: promoter III forward, 5′-ATGCAATGCCCTGGAAC-3′; promoter III reverse, 5′-GTGAATGGGAAAGTGGGTG-3′. The annealing temperature of both cases was 59°C. The amplified products were confirmed by running 1.5% agarose gels.
Ras activity assay.
The activity of Ras was measured by detecting the levels of Ras-GTP using a Ras activation kit (Millipore). Briefly, cultured hippocampal neurons at 4 DIV were treated with different experimental conditions. After 24 h, cells were lysed at 4°C and Ras-GTP was immunoprecipitated using the Ras-GTP-binding domain of Raf1 before Western blot analysis.
Rac1 activity assay.
We measured Rac1 activity using Rac1 Activation Assay Kit (Millipore) according to the manufacturer's instructions. Briefly, cultured hippocampal neurons at 4 DIV were treated with different experimental conditions. After 24 h, cells were lysed at 4°C and incubated with Pak-PBD agarose with constant rocking at 4°C for 1 h. The proteins bound to the beads were washed three times with lysis buffer at 4°C, eluted in SDS sample buffer, and analyzed for bound Rac1 by Western blot using monoclonal antibody against Rac1. GTPase activity was quantified by densitometry analysis of the blots.
Analysis of spine density.
Dendritic spines of hippocampal neurons at 14 DIV under different treatments were identified as small protrusions that extended ≤3 μm from the parent dendrite, and counted off-line in maximum-intensity projections of the z-stacks using ImageJ software (NIH). Care was taken to ensure that each spine was counted only once by following its projection course through the stack of z-sections. Spines were counted only if they appeared continuous with the parent dendrite. Spine density was calculated by quantifying the number of spines per dendritic segment, and normalized to 1 μm dendrite length.
Statistical analysis.
Data are from one representative experiment among at least three independent experiments and are expressed as the mean ± SEM. Significant differences between groups were compared using the one-way ANOVA procedure followed by Student's t tests using SPSS software (SPSS) and the differences were considered statistically significant at p < 0.05.
Results
Effects of LLLT on Aβ-induced neurotoxicity
To examine the neuroprotective effect of LLLT against Aβ cytotoxicity, we used neuronal cell line SH-SY5Y, which is an in vitro model to mimic responses of neurons. As shown in Figure 1A, cell viability was increased by treatment of LLLT (2 J/cm2), and the relative cell viability (Aβ25–35 + LLLT-treated cells − Aβ25–35-treated cells) was raised by LLLT to a similar extent at both 24 and 48 h. Therefore, we used 24 h as the optimal time in the following experiments.
Figure 1.
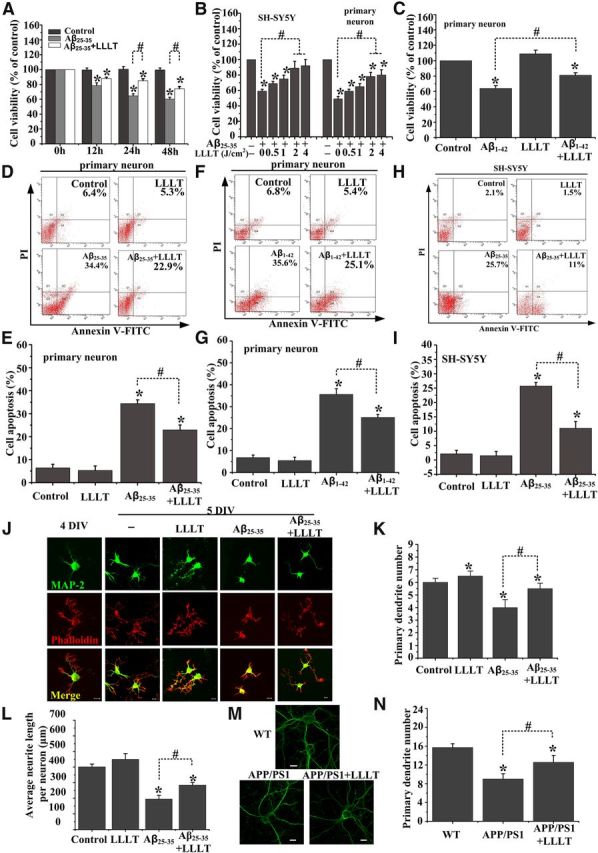
LLLT exerts neuroprotection against Aβ toxicity. A, LLLT (2 J/cm2) protects SH-SY5Y cells against Aβ25–35 (25 μm) neurotoxicity using the CCK-8 assay after being irradiated with LLLT for 12 h, 24 h, or 48 h. B, Primary hippocampal neurons or SH-SY5Y cells were exposed to Aβ25–35 followed by irradiation with LLLT at 0.5 J/cm2, 1 J/cm2, 2 J/cm2, or 4 J/cm2, respectively. Cell viability was assessed by the CCK-8 assay after 24 h. C, LLLT (2 J/cm2) protects primary hippocampal neurons against Aβ1–42 (25 μm) neurotoxicity using the CCK-8 assay after irradiation with LLLT. D–G, Primary hippocampal neurons treated with Aβ25–35 and/or LLLT or Aβ1–42 and/or LLLT were double stained with apoptosis markers Annexin V/PI using flow cytometric analysis. H–I, Apoptosis in SH-SY5Y cells was analyzed in H and I. J, Representative immunofluorescent images of 5 DIV hippocampal neurons under indicated treatments with MAP2 antibody to visualize dendrite (green). Staining with Rhodamine-labeled phalloidin to visualize F-actin (red). Scale bar, 10 μm. K, Effects of these treatments on the number of primary dendrites per neurons. For each group, >25 neurons were measured. L, Effects of these treatments on average dendritic length per neuron. For each group, >25 neurons were measured. M, Representative photomicrographs of FITC-phalloidin labeling in hippocampal neurons derived from APP/PS1 mice embryo on 10 DIV under the treatment with or without LLLT. Scale bar, 10 μm. N, Quantification of primary dendrite numbers per neurons under indicated treatments. For each group, >25 neurons were measured. All the data in these figures are presented as mean ± SEM four individual experiments. *p < 0.05 versus control group; #p < 0.05 versus indicated group.
To examine the optimal dose of LLLT, SH-SY5Y cells or primary hippocampal neurons were exposed to Aβ25–35 followed by exposure of cells to different dose of LLLT (0.5, 1, 2, or 4 J/cm2). Twenty-four hours later cell viability was measured using the CCK-8 assay. As shown in Figure 1B, cell viability was evidently increased by LLLT in a dose-dependent manner. A significant increase was observed at the dose of 2 and 4 J/cm2; meanwhile, to minimize thermal effect, 2 J/cm2 was selected as the optimum irradiation dose in our following studies. Furthermore, we also validated the protective effect of LLLT on hippocampal neurons treated with full-length peptide Aβ1–42 (Fig. 1C).
Since Aβ mediated neurotoxicity by the programmed cell death pathway, we next sought to examine if LLLT-mediated protection of neurons involved inhibition of Aβ-induced neuronal apoptosis using flow cytometric analysis. Compared with the untreated group, the apoptosis ratio significantly increased in primary hippocampal neurons within 24 h under Aβ25–35 or Aβ1–42 treatment. In contrast, the cell apoptosis rate was significantly decreased after treatment with LLLT (Fig. 1D–G). Similar results were obtained in SH-SY5Y cells (Fig. 1H,I). These results further confirmed that LLLT attenuated Aβ-induced neurotoxicity.
We next investigated the effects of LLLT on “dendrite initiation” (i.e., the number of primary dendrites per neurons) and “dendrite elongation.” Hippocampal neurons were cultured for 4 d before stimulating with Aβ25–35 and/or LLLT. At 5 DIV, immunofluorescent staining with MAP2 antibody showed that, compared with untreated group, Aβ25–35 decreased the primary dendrite number (Fig. 1J,K) and average dendrite length (Fig. 1J,L) per neurons. However, LLLT treatment attenuated Aβ-induced dendrite atrophy (Fig. 1J–L). Additionally, dendritic atrophy rescued by LLLT was verified by culturing hippocampal neurons derived from APP/PS1 transgenic mice embryos at 10 DIV (Fig. 1M,N).
LLLT increases BDNF protein levels in hippocampal neurons
BDNF promotes the survival of neurons and dendrite growth both in vitro and in vivo (Katz and Shatz, 1996; Connor and Dragunow, 1998; Murer et al., 2001). A growing body of evidence indicates that BDNF levels are decreased in brains of AD patients (Hu and Russek, 2008; Zuccato and Cattaneo, 2009) and APP transgenic mice (Peng et al., 2009; Francis et al., 2012). We first characterized changes in BDNF levels in the APP/PS1 transgenic mouse model at 6 months of age (Fig. 2A). Western blot analysis of hippocampal homogenate samples demonstrated dramatic reductions in baseline levels of mature BDNF in APP/PS1 transgenic mice. Similarly, decreases in the levels of BDNF protein had been observed in both Aβ25–35- and Aβ1–42-treated SH-SY5Y cells (Fig. 2B).
Figure 2.
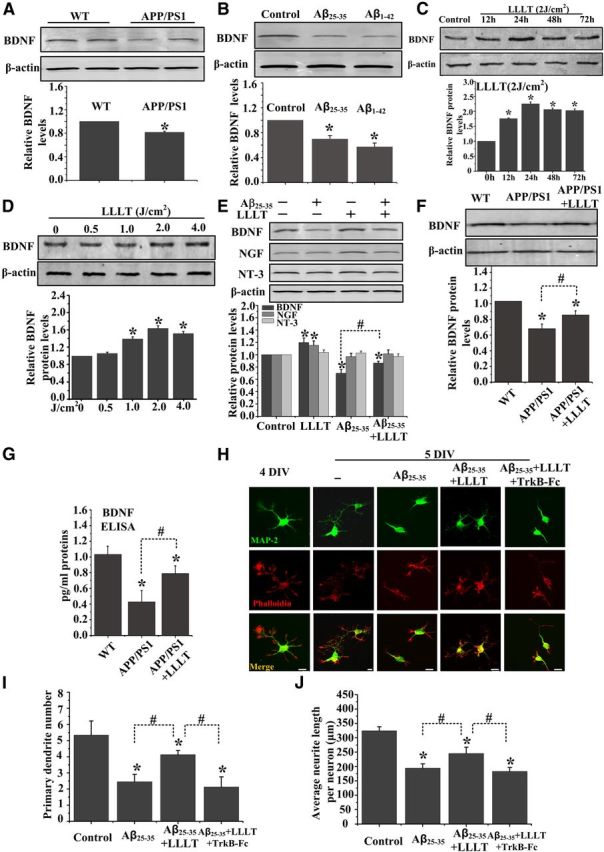
LLLT upregulates BDNF expression in cultured neurons derived from APP/PS1 transgenic mice and in Aβ-treated hippocampal neurons. A, Western blot analysis of hippocampal lysates from APP/PS1 transgenic mice and age-matched WT littermate mice at 6 months of age. B, Western blot analysis of BDNF expression in Aβ25–35-and Aβ1–42-treated primary hippocampal neurons. C, Representative Western blot assay for detecting the time-dependent effect of 2 J/cm2 LLLT on BDNF expression in SH-SY5Y cells. D, Representative Western blot assay for detecting the dose-dependent effect of LLLT on BDNF expression after 24 h. E, Western blot was performed to detect the BDNF, nerve growth factor (NGF), and NT-3 expression after Aβ25–35 treatment with or without LLLT in primary hippocampal neurons. F–G, BDNF expression was detected by Western blot (F) and ELISA (G) in hippocampal neurons derived from WT mice embryos or APP/PS1 embryos at 14 DIV with or without LLLT. H, Representative immunofluorescent images of 5 DIV hippocampal neurons under indicated treatments with MAP2 antibody to visualize dendrite (green). Staining with Rhodamine-labeled phalloidin to visualize F-actin (red). Scale bar, 10 μm. I, Effects of these treatments on the number of primary dendrites per neurons. For each group, >25 neurons were measured. J, Effects of these treatments on average dendritic length per neuron. For each group, >25 neurons were measured. All the data in these figures are presented as mean ± SEM four individual experiments. *p < 0.05 versus control group; #p < 0.05 versus indicated group.
We next examined if LLLT could regulate BDNF expression in neuronal cells. Our results showed that treatment of SH-SY5Y cells with LLLT (2 J/cm2) significantly increased BDNF protein expression (Fig. 2C). The maximal response was observed at 24 h of 2 J/cm2 LLLT treatment. In addition, there was a dose-dependent increase in BDNF protein expression, with statistically significant increases observed with 2 J/cm2 LLLT (Fig. 2D).
To further investigate if LLLT could upregulate BDNF expression in response to Aβ treatment, primary hippocampal neurons were exposed to Aβ25–25 followed by LLLT. As shown in Figure 2E, LLLT could effectively increase BDNF protein expression, but not NGF or NT-3, even in neurons exposed to Aβ25–35. The upregulation of BDNF production and secreted by LLLT was verified by culturing hippocampal neurons derived from APP/PS1 transgenic mice embryos at 14 DIV (Fig. 2F,G), which produced high levels of human Aβ40 and Aβ42 (Wu et al., 2010). These results strongly indicated that BDNF expression was enhanced during LLLT-induced neuroprotective effects.
Scavenging extracellular BDNF with TrkB-Fc allowed testing if dendritic growth caused by LLLT was mediated by increased BDNF levels. Indeed, TrkB-Fc (20 μg/ml) prevented the increase in the length and number of dendritic branches observed in Aβ25–35-treated neurons after LLLT treatment (Fig. 2H,J). These results suggest that enhanced BDNF expression is responsible for the rescue of dendrite atrophy induced by LLLT in Aβ25–35-treated neurons.
LLLT treatment increases BDNF expression by a CREB-dependent transcriptional mechanism
We next investigated if LLLT-induced increase in BDNF protein required new mRNA synthesis by pharmacologically blocking transcription with Act D. We have observed that pretreatment of SH-SY5Y cells with Act D significantly inhibits LLLT-induced increase in BDNF mRNA expression in Aβ25–35-treated SH-SY5Y cells (Fig. 3A). These data suggest a requirement for gene transcription in LLLT-induced increase in BDNF protein.
Figure 3.

LLLT-induced activation of CREB enhances BDNF transcription upon Aβ treatment. A, Act D inhibits LLLT-induced increase in BDNF mRNA expression in Aβ25–35-treated SH-S5Y cells. B, SH-SY5Y cells were transfected with CREB siRNA. Western blot analysis of BDNF expression after Aβ25–35 treatment with or without LLLT. C, Western bolt analysis of BDNF expression in SH-SY5Y cells under indicated treatments. The concentration of MTA is 200 nm. D, Western blot analysis of CREB phosphorylation at Ser133 in primary hippocampal neurons treated with Aβ25–35 and/or LLLT. E, Representative immunofluorescent images of p-CREB (Ser 133) (green) in SH-SY5Y cells under the indicated treatments. Staining with propidium iodide (PI) to visualize nucleus. F, ChIP assay was done to examine DNA-binding activity of CREB to BDNF promoter in SH-SY5Y cells and primary hippocampal neurons with different treatments. All the data in these figures are presented as mean ± SEM four individual experiments. *p < 0.05 versus control group; #p < 0.05 versus indicated group.
It is reported that CREB is a transcription factor of BDNF under activity-dependent stimulation (Aid et al., 2007). Some studies have suggested that upregulation of SP1 transcription activity protects against neurodegenerative disorders (Ryu et al., 2003). To detect which one is the transcription factor of BDNF under LLLT-treatment, we blocked the activity of CREB and SP1 by using, respectively, CREB siRNA and MTA, a drug that inhibits the activity of the transcription factors of SP1 family. Knocking down CREB antagonized the LLLT-induced increase of BDNF expression but not MTA (Fig. 3B,C).
CREB phosphorylation at serine 133 is a key event required for recruiting different transcription effectors (Shaywitz and Greenberg, 1999). To answer the question if LLLT induces CREB phosphorylation at serine 133 under Aβ25–35 treatment, Western blot assay was performed in primary neurons. As shown in Figure 3D, the phosphorylation level of CREB at ser 133 was lower at the Aβ-treated group compared with control. Treatment of these cells with 2 J/cm2 LLLT induced a significant increase in the level of CREB phosphorylation. These results were further confirmed by immunofluorescent staining with antibodies against p-CREB (ser 133) in SH-SY5Y cells (Fig. 3E).
It has been reported that there was a CREB binding site within the exon of the BDNF gene that plays an important role in neuroprotection (Aid et al., 2007). We used ChIP assay to examine of CREB bound to the promoter region of BDNF was altered after LLLT in both SH-SY5Y cells and Aβ-treated primary mouse hippocampal neuronal cultures. Figure 3F showed that the activated transcription factors p-CREB binding to promoter region of BDNF were decreased by Aβ25–35 treatment. However, there was a significant increase of p-CREB binding to BDNF promoter by the treatment with LLLT even in the Aβ-treated group. All these results suggested that LLLT potentially upregulated the expression of BDNF through a CREB-dependent transcription in Aβ-treated neurons.
Photoactivated ERK is responsible for CREB-dependent upregulation of BDNF
To investigate the kinases phosphorylating CREB upon LLLT in Aβ25–35-treated cells, we studied the effect of specific inhibitors of these pathways: Gö6983, API-2, and PD98059 (for PKCs, Akt, and MEK/ERKs, respectively) in Western bolt assays. We found that LLLT increased the phosphorylated form of CREB (serine 133) in Aβ25–35-treated SH-SY5Y cells, which was reversed by pretreatment with PD98059, but not Gö6983 and API-2 (Fig. 4A).
Figure 4.

LLLT upregulates BDNF expression and CREB Ser 133 phosphorylation in Aβ25–35-treated cells via ERK activation. A, Representative Western blot assay of CREB Ser133 phosphorylation stimulated with Aβ25–35 and/or LLLT in the presence of Gö6983 (20 μm), API-2 (2 μm), and PD98059 (1 μm) in SH-SY5Y cells. B, Representative Western blot assay of the CREB Ser133 phosphorylation stimulated with Aβ25–35 and/or LLLT in the presence of PD98059 (1 μm) and H89 (10 μm) in primary hippocampal neurons. C, Primary hippocampal neurons were treated with Aβ25–35 and/or LLLT, or pretreated with PD98059. Western blot analysis was performed to detect the levels of BDNF, total-ERK, p-ERK, total-CREB, and p-CREB. D, Representative Western blot assay for detecting the levels of ERK phosphorylation after indicated treatments in cytoplasm and nuclear lysates, respectively. E, Representative Western blot assay of the p-ERK, total-ERK, GTP-Ras, and total-Ras stimulated with Aβ25–35 and/or LLLT in the presence of PD98059 (1 μm) and FTS (20 μm) in primary hippocampal neurons. F, Representative Western blot assay of the GTP-Ras and total-Ras pretreated with Ca2+-free medium, or xestospongin B (xpB, 40 μm). All the data in these figures are presented as mean ± SEM four individual experiments. *p < 0.05 and **p < 0.05 versus control group; #p < 0.05 versus indicated group.
CREB can be phosphorylated by cAMP-dependent protein kinase A (PKA), other than ERK (Gonzalez and Montminy, 1989). It is also reported that LLLT could increase the level of cAMP via enhanced cytochrome c oxidase activity (Hu et al., 2007; de Lima et al., 2011). We next tried to detect which is the major kinase phosphorylating CREB under LLLT-treatment in Aβ-treated hippocampal neurons. We blocked the activity of ERK and PKA by using, respectively, PD98059 and H89. As shown in Figure 4B, LLLT-induced phosphorylation of CREB was markedly attenuated by PD98059, but not by H89, suggesting that MEK/ERK signaling is obligatory for LLLT-stimulated phosphorylation of CREB in Aβ-treated hippocampal neurons.
Western blot was performed to test the effect of ERK activation on the expression of BDNF in response to LLLT in Aβ25–35-treated primary neurons. As shown in Figure 4C, an increase in p-ERK (activated ERK) and p-CREB (activated CREB), as well as the expression of BDNF, was seen in the samples after LLLT treatment even in Aβ25–35-treated group. Inhibition of ERK by PD98059 before LLLT resulted in decreased phosphorylation of CREB at Ser133 (Fig. 4C) and blocked CREB binding to a BDNF promoter (Fig. 3F). Meanwhile, the phosphorylation of ERK in the nucleus was also showing a progressive level after LLLT (Fig. 4D).
LLLT-induced Ras/ERK activation depending on the calcium release from intracellular stores
Ras activation is the first step in activation of the mitogen-activated protein kinase (MAPK) cascade. It has been reported that Ras was activated by LLLT for the formation of LLLT-induced circular ruffles (Gao et al., 2009). To examine the requirement of Ras for LLLT-induced ERK activation, we measured Ras kinase activity. As shown in Figure 4E, LLLT significantly increased Ras kinase activity even in Aβ-induced neurons. Inhibition of Ras by FTS attenuated LLLT-induced ERK phosphorylation, indicating that LLLT-induced activation of ERK is Ras dependent.
A rise in free intracellular calcium concentrations can initiate the Ras/ERK cascade (Chao et al., 1992; Rosen et al., 1994). One of the suggestions to the LLLT mechanical pathway is the contribution of intracellular calcium as a versatile second messenger (Lavi et al., 2003; Lan et al., 2012). We next detected if intracellular calcium is involved in Ras/ERK cascade activation by LLLT in Aβ-treated neurons. To examine if Ras activation was dependent on calcium influx from extracellular medium, neurons were incubated for 5 min before LLLT treatment in calcium-free medium. As shown in Figure 4F, there was no significant effect on LLLT-induced Ras activation in extracellular calcium-free medium. However, after cells preincubated with inositol triphosphate (IP3) receptor inhibitor, xestospongin B, marked inhibition of LLLT-induced Ras activation was observed. These results indicated that LLLT-triggered calcium release from intracellular stores mediated photoactivation of Ras/ERK cascades.
ERK/CREB activation is necessary for LLLT to increase dendrite growth in Aβ25–35-treated hippocampal neurons
We next detected if BDNF upregulation by LLLT via ERK/CREB pathway regulates dendrite patterning in Aβ-treated neurons. As shown in Figure 5A, LLLT stimulation promoted dendrite growth of Aβ25–35-treated hippocampal neurons. However, the primary dendrite number and average dendrite length per neuron were significantly decreased by inhibition of ERK (Fig. 5A–C). Furthermore, knocking down of CREB inhibited LLLT-induced dendrite growth (Fig. 5D–F). These data support the idea that LLLT activates ERK/CREB pathway to increase the expression of BDNF and, as a result, dendrite growth.
Figure 5.
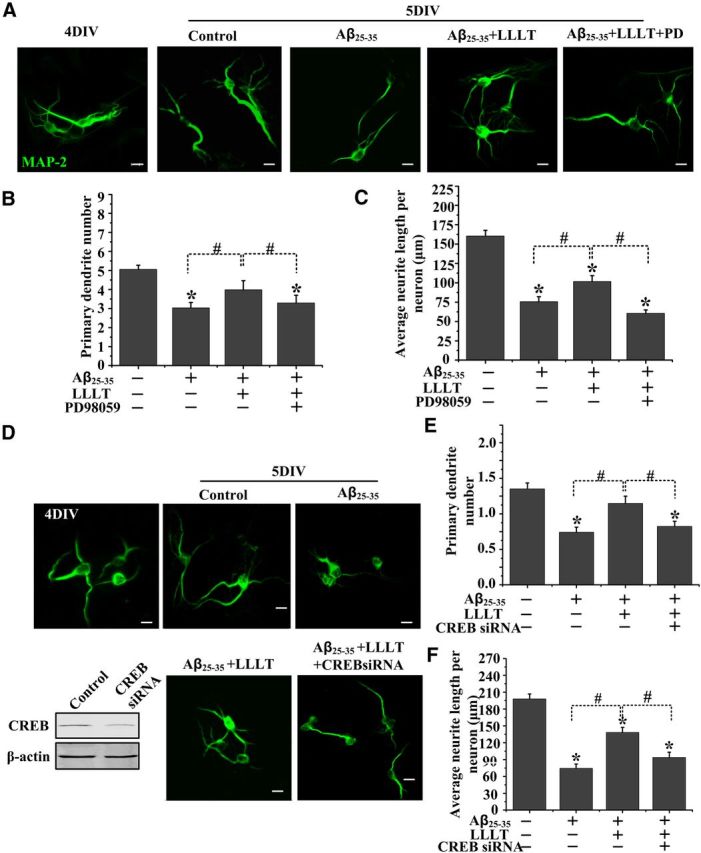
Altered dendritic morphology after LLLT in Aβ25–35-treated hippocampal neurons at 5 DIV via ERK/CREB pathway. A, Representative images of 5 DIV hippocampal neurons with indicated treatments. Scale bar, 10 μm. B, E, Quantification of primary dendrites numbers per neurons under indicated treatments. For each group, >25 neurons were measured. C, F, Quantification of average dendritic length per neurons under indicated treatments. For each group, >25 neurons were measured. D, Hippocampal neurons were transfected with CREB siRNA at 3 DIV and treated with Aβ25–35 and LLLT at 4 DIV. Representative immunofluorescent images with MAP2 antibody are shown at 5 DIV. Scale bar, 10 μm. All the data in these figures are presented as mean ± SEM four individual experiments. *p < 0.05 versus control group; #p < 0.05 versus indicated group.
The small G-protein Rac1 activation is involved in LLLT-stimulated dendritic growth
In neurons, Rac1 is involved in the regulation of dendrite morphogenesis; we were interested in delineating if Rac1 activity contribute to LLLT-stimulated dendritic growth in Aβ-treated hippocampal neurons. Cell lysates were then examined for the amount of activated, GTP-bound Rac1, using a pull-down assay performed with the p21-binding domain of Pak (Pak-PBD). Treatment of cultured hippocampal neurons with Aβ25–35 decreased Rac1 activity (Fig. 6A,B). In contrast to the inhibition of Rac1 activity in Aβ25–35-treated neurons, LLLT stimulation resulted in an increased amount of GTP-bound Rac1 (Fig. 6A,B).
Figure 6.

Dendritic cytoskeletal protein contributes to LLLT-promoted dendrite growth. A, LLLT induced Rac1 activity in Aβ25–35-treated hippocampal neurons, as indicated by an increase in GTP-bound Rac1. B, Quantitative analysis of the levels of GTP-bound Rac1 after different treatments. All the data in these figures are presented as mean ± SEM four individual experiments. *p < 0.05 versus control group; #p < 0.05 versus indicated group.
Dendritic spine loss in neurons derived from APP/PS1 transgenic mice is rescued by LLLT
It is reported that BDNF could induce elongation of dendritic spines and formation of new ones in neurons within 60 min (Adasme et al., 2011). To examine if upregulating of BDNF by LLLT could regulate the dendritic spine morphology, we therefore visualized spines using phalloidin staining in hippocampal neurons derived from E14 APP/PS1 transgenic mice on 14 DIV, which produced high levels of human Aβ40 and Aβ42 (Wu et al., 2010). Neurons treated with LLLT showed a higher density of spine-like protrusion (Fig. 7A,B).
Figure 7.

LLLT rescues dendritic spine loss in neurons derived from APP/PS1 transgenic mice. A, Representative photomicrographs of FITC-phalloidin labeling in hippocampal neurons derived from APP/PS1 mice on 14 DIV under the treatment with or without LLLT. Scale bar, 10 μm. B, Quantification of spine density under indicated treatments. For each group, we measured >25 dendrites from 13 to 15 neurons. C, Representative Western blot analysis was performed to detect the levels of PSD-95 in Aβ25–35 and/or LLLT-treated hippocampal neurons. D, Quantitative analysis of the levels of PSD-95 after indicated treatments. All the data in these figures are presented as mean ± SEM four individual experiments. *p < 0.05 versus control group; #p < 0.05 versus indicated group.
The PSD-95 scaffolding protein has been identified as marker for synaptic strength (El-Husseini et al., 2000). In visual cortical neurons, BDNF can increase the overall amount of PSD-95 in dendrites within 60 min (Yoshii and Constantine-Paton, 2007). Thus, we next detected if LLLT could increase the amount of PSD-95 in hippocampal neurons treated with Aβ25–35. As shown in Figure 7, C and D, irradiation with LLLT increased PSD-95 synthesis even in Aβ25–35-treated neurons. These results indicate that LLLT markedly rescues the dendritic spine loss in Aβ-treated neurons.
Discussion
APP and its catabolite, Aβ, play critical roles in the etiology of AD (Selkoe and Schenk, 2003). In addition to neuronal death, numerous changes in dendritic architecture have been observed, including decrease of dendrite length and branching and loss of spines in transgenic mice overexpressing APP and in brains of persons dying of AD (Einstein et al., 1994; Masliah et al., 2001). The dendritic atrophy correlates well with the decrease of neurotrophins, such as BDNF (Hu and Russek, 2008; Zuccato and Cattaneo, 2009). In the present study, we found a regulatory role of LLLT for neuroprotection and dendritic morphogenesis. We demonstrated the ability of LLLT to rescue Aβ-induced dendritic atrophy and neuronal death. In Aβ-treated neurons, LLLT attenuated the decrease of both BDNF mRNA and protein levels and p-CREB, a transcriptional regulator of BDNF. Additionally, dendrite growth was improved after LLLT treatment, characterized by upregulation of PSD-95 expression, Rac1 activity, and the increase in length, branching, and spine density of dendrites in hippocampal neurons. Moreover, we found that LLLT upregulated expression of BDNF by an ERK/CREB pathway. Understanding the mechanism and function significance of LLLT-induced neuroprotection and dendrite growth may lead to new neurotherapies.
LLLT is a nonthermal irradiation using light in visible to near infrared range, which has been used clinically to accelerate wound healing and reduce pain and inflammation in a variety of pathologies (Schindl et al., 1999; Khuman et al., 2012). Transcranial LLLT has shown good effects on treatment of stroke, traumatic brain injury, and neurodegenerative disease (Naeser and Hamblin, 2011). Moreover, LLLT could reduce amyloid plaques and attenuate behavioral deficits in APP transgenic mice (De Taboada et al., 2011). Although in the preclinical and clinical studies the 810 nm light was often used for nerve repairs (Naeser and Hamblin, 2011), the effects of laser irradiation with different wavelengths on dendrite growth remain unclear. Studies had shown that the 633 nm laser had advantages over other wavelengths in treating neurological diseases (Anders et al., 1993; Rojas et al., 2008). We found that compared with 810 nm laser treatment, irradiation with 633 nm light induced higher BDNF expression (data not shown). Previous studies from this lab, and others, have shown that cell death induced by Aβ25–35 is significantly diminished with 633 nm LLLT (Yang et al., 2010; Liang et al., 2012; Zhang et al., 2012). In this study, after confirming the prosurvival effect of 633 nm LLLT on Aβ25–35-treated primary hippocampal neurons in a dose–response manner (Fig. 1), we found similar effect of LLLT on Aβ1–42-treated hippocampal neurons as well (Fig. 1C,F,G). The dose of 2 J/cm2 was highly effective at preventing the Aβ-induced neuronal death and BDNF deficits (Figs. 1, 2). We have further found that, for the first time to our knowledge, LLLT significantly rescued the dendritic atrophy in Aβ-treated neurons (Figs. 1, 7). Therefore, LLLT using the 633 nm laser may have high clinical relevance.
BDNF is an attractive candidate for molecular signals that regulate neuronal survival and dendritic growth both in vivo and in vitro (Connor and Dragunow, 1998; Murer et al., 2001). Marked reduction in the levels of BDNF has occurred in AD patients (Hu and Russek, 2008; Zuccato and Cattaneo, 2009) and in APP transgenic mice, such as APPNLh and TgCRND8 mice (Peng et al., 2009; Francis et al., 2012). The decrease in BDNF expression was observed both in Aβ-treated hippocampal neurons and for the first time in APP/PS1 transgenic mice hippocampal tissue (Fig. 2A,B). Learning and memory deficits exhibited by transgenic mouse models of AD can be rescued by BDNF delivery (Nagahara et al., 2009). CREB-dependent transcription upregulation of BDNF produced neuroprotective signals promoting cell survival (Fang et al., 2003; Ou and Gean, 2007). Recently, it has been reported that LLLT promotes human Schwann cell proliferation via regulating neurotrophic factor gene expression (Yazdani et al., 2012). These data lead to a new hypothesis that LLLT may be capable of enhancing the generation of BDNF via CREB-mediated gene expression in Aβ-treated hippocampal neurons. Our results support this hypothesis, since pharmacological inhibition of transcription process and knockdown of CREB blocked LLLT-mediated increases in BDNF expression (Fig. 3A,B).
CREB plays a key role in mediating dendritic development in response to neuronal activity (Lonze and Ginty, 2002). Oligomeric Aβ decreases specifically p-CREB and the BDNF transcript in neurons (Tong et al., 2004; Garzon and Fahnestock, 2007). The BDNF exon III CRE site is the main target bound by CREB (Ou and Gean, 2007). We validated that LLLT markedly increases the p-CREB level and enhances DNA-binding activity of CREB to BDNF promoter even under Aβ treatment in both SH-SY5Y cells and primary hippocampal neurons (Fig. 3D–F). Although CREB can be phosphorylated by PKC, cAMP/PKA, and MAPK (Gonzalez and Montminy, 1989; Xing et al., 1998; Shaywitz and Greenberg, 1999), we found, in the present study, that the inhibition of ERK activity suppressed phosphorylation of CREB induced by LLLT in Aβ-treated hippocampal neurons (Fig. 4).
MAPK pathways are evolutionarily conserved signaling modules by which cells transduce extracellular signals into intracellular responses (Samuels et al., 2009). Once activated, ERK partly translocates from cytoplasm to nucleus (Feng et al., 2012) and phosphorylates CREB (Xing et al., 1998; Bonni et al., 1999). It has been reported that Aβ oligomer decreases active ERK and subsequently active CREB in neuroblastoma cells and in primary neurons (Ma et al., 2007). We found that LLLT triggered significant activation and nuclear translocation of ERK (Fig. 4C,D). In addition, CREB activated by LLLT was ERK-dependent. LLLT-mediated ERK/CREB activation could significantly increase BDNF expression (Fig. 4C).
How could LLLT activate MEK/ERK signaling pathway? The best characterized upstream of MEK/ERK cascade is the small GTPase, Ras (Marshall, 1996). Activated Ras promotes movement of the protein serine/threonine kinase Raf-1 to the plasma membrane where it phosphorylates another protein kinase, MEK, which in turn activates ERKs (Vojtek et al., 1993; Warne et al., 1993). We have previously observed that Ras was activated by LLLT for the formation of circular ruffles (Gao et al., 2009). LLLT induces cell proliferation via Ras/Raf/ERK signaling transduction pathways (Shefer et al., 2003). In this study, we confirmed that LLLT could activate Ras even in Aβ25–35-treated hippocampal neurons (Fig. 4E). Additionally, LLLT-induced activation of ERK was Ras dependent (Fig. 4E).
One of the suggestions to the LLLT-induced biologic effects is the contribution of intracellular calcium as a signaling molecule. Absorption of photons by intracellular photo-acceptors, such as cytochrome oxidase, leads to electronically excited states and consequently can lead to acceleration of electron transfer reactions (Yu et al., 1997; Pastore et al., 2000). More electron transport necessarily leads to increased production of ATP (Mochizuki-Oda et al., 2002; Karu, 2010), which activates P2 receptors to induce inward calcium currents and release of calcium from intracellular stores (Salter and Hicks, 1995). Increase in calcium concentration has been shown to modulate the Ras-dependent activation of MEK/ERK cascades (Chao et al., 1992; Rosen et al., 1994). We found that LLLT-induced Ras/ERK activation was slightly inhibited in Ca2+-free medium (Fig. 4F). However, LLLT-induced Ras activation was significantly blocked by xestospongin B (Fig. 4F). These results indicate that LLLT-induced Ras/ERK activation dependent on the calcium release from intracellular stores.
Activation of ERK/CREB/BDNF was found to be sufficient to increase the total dendritic length and branch point number of neurons (Finsterwald et al., 2010). Aβ-induced dendritic atrophy was rescued by LLLT in the present study (Fig. 1). We further identified the cellular mechanisms underlying this phenomenon by showing that activation of ERK/CREB is necessary to mediate the effects of LLLT on dendritic growth. Indeed, inhibition of ERK or knockdown CREB prevented the increased dendritic growth by LLLT in Aβ25–35-treated hippocampal neurons (Fig. 5).
Dendrite number and branching are regulated by the interplay between extrinsic factors and intrinsic factors. The external factors including neurotrophins (Katz and Shatz, 1996), estrogen (Sakamoto et al., 2003), and electrical activity (Vaillant et al., 2002). Intrinsic factors are the small GTPases Rho A, Rac1, and Cdc42 (Li et al., 2000), and PSD-95 (Yoshii and Constantine-Paton, 2007). To address if cytoskeleton changes were involved in LLLT-mediated dendrite growth, we investigated the Rac1 activation after LLLT. The activity of Rac1 was markedly enhanced after LLLT even in Aβ25–35-treated neurons (Fig. 6A,B). In mature visual cortical neurons, BDNF can increase the overall amount of the PSD-95 scaffolding protein to promote synaptic maturation (Yoshii and Constantine-Paton, 2007). We found that LLLT induced dendritic spine growth in APP/PS1 transgenic mice hippocampal neurons at 14 DIV (Fig. 7A,B). The expression of PSD-95 was also increased (Fig. 7C,D).
Together, the current investigation demonstrates that LLLT can inhibit Aβ-induced neurotoxicity and rescue dendrite atrophy through activation of the ERK/CREB/BDNF pathway. Although cultured mouse neurons and their treatment with Aβ may not truly resemble neurons in the brain of patients, our results suggest that upregulating BDNF may be an important step for the attenuation of dendritic atrophy by LLLT. Better understanding of the regulation mechanism of photobiomodulation may provide a therapeutic strategy to control the progression of AD.
Footnotes
This work is supported by the National Basic Research Program of China (2011CB910402, 2010CB732602), the Program for Changjiang Scholars and Innovative Research Team in University (IRT0829), and the National Natural Science Foundation of China (81101741, 31101028). We Are grateful to Dr. Zhicheng Xiao for help with APPswe/PS1dE9 mice.
The authors declare no competing financial interests.
References
- Adasme T, Haeger P, Paula-Lima AC, Espinoza I, Casas-Alarcón MM, Carrasco MA, Hidalgo C. Involvement of ryanodine receptors in neurotrophin-induced hippocampal synaptic plasticity and spatial memory formation. Proc Natl Acad Sci U S A. 2011;108:3029–3034. doi: 10.1073/pnas.1013580108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85:525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders JJ, Borke RC, Woolery SK, Van de Merwe WP. Low power laser irradiation alters the rate of regeneration of the rat facial nerve. Lasers Surg Med. 1993;13:72–82. doi: 10.1002/lsm.1900130113. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Chao TS, Byron KL, Lee KM, Villereal M, Rosner MR. Activation of MAP kinases by calcium-dependent and calcium-independent pathways. Stimulation by thapsigargin and epidermal growth factor. J Biol Chem. 1992;267:19876–19883. [PubMed] [Google Scholar]
- Connor B, Dragunow M. The role of neuronal growth factors in neurodegenerative disorders of the human brain. Brain Res Brain Res Rev. 1998;27:1–39. doi: 10.1016/S0165-0173(98)00004-6. [DOI] [PubMed] [Google Scholar]
- de Lima FM, Moreira LM, Villaverde AB, Albertini R, Castro-Faria-Neto HC, Aimbire F. Low-level laser therapy (LLLT) acts as cAMP-elevating agent in acute respiratory distress syndrome. Lasers Med Sci. 2011;26:389–400. doi: 10.1007/s10103-010-0874-x. [DOI] [PubMed] [Google Scholar]
- De Taboada L, Yu J, El-Amouri S, Gattoni-Celli S, Richieri S, McCarthy T, Streeter J, Kindy MS. Transcranial laser therapy attenuates amyloid-beta peptide neuropathology in amyloid-beta protein precursor transgenic mice. J Alzheimers Dis. 2011;23:521–535. doi: 10.3233/JAD-2010-100894. [DOI] [PubMed] [Google Scholar]
- Eells JT, Henry MM, Summerfelt P, Wong-Riley MT, Buchmann EV, Kane M, Whelan NT, Whelan HT. Therapeutic photobiomodulation for methanol-induced retinal toxicity. Proc Natl Acad Sci U S A. 2003;100:3439–3444. doi: 10.1073/pnas.0534746100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einstein G, Buranosky R, Crain BJ. Dendritic pathology of granule cells in Alzheimer's disease is unrelated to neuritic plaques. J Neurosci. 1994;14:5077–5088. doi: 10.1523/JNEUROSCI.14-08-05077.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Fang H, Chartier J, Sodja C, Desbois A, Ribecco-Lutkiewicz M, Walker PR, Sikorska M. Transcriptional activation of the human brain-derived neurotrophic factor gene promoter III by dopamine signaling in NT2/N neurons. J Biol Chem. 2003;278:26401–26409. doi: 10.1074/jbc.M211539200. [DOI] [PubMed] [Google Scholar]
- Feng J, Zhang Y, Xing D. Low-power laser irradiation (LPLI) promotes VEGF expression and vascular endothelial cell proliferation through the activation of ERK/Sp1 pathway. Cell Signal. 2012;24:1116–1125. doi: 10.1016/j.cellsig.2012.01.013. [DOI] [PubMed] [Google Scholar]
- Finsterwald C, Fiumelli H, Cardinaux JR, Martin JL. Regulation of dendritic development by BDNF requires activation of CRTC1 by glutamate. J Biol Chem. 2010;285:28587–28595. doi: 10.1074/jbc.M110.125740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis BM, Kim J, Barakat ME, Fraenkl S, Yücel YH, Peng S, Michalski B, Fahnestock M, McLaurin J, Mount HT. Object recognition memory and BDNF expression are reduced in young TgCRND8 mice. Neurobiol Aging. 2012;33:555–563. doi: 10.1016/j.neurobiolaging.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Xing D, Liu L, Tang Y. H-Ras and PI3K are required for the formation of circular dorsal ruffles induced by low-power laser irradiation. J Cell Physiol. 2009;219:535–543. doi: 10.1002/jcp.21693. [DOI] [PubMed] [Google Scholar]
- Garzon DJ, Fahnestock M. Oligomeric amyloid decreases basal levels of brain-derived neurotrophic factor (BDNF) mRNA via specific downregulation of BDNF transcripts IV and V in differentiated human neuroblastoma cells. J Neurosci. 2007;27:2628–2635. doi: 10.1523/JNEUROSCI.5053-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- Hu WP, Wang JJ, Yu CL, Lan CC, Chen GS, Yu HS. Helium-neon laser irradiation stimulates cell proliferation through photostimulatory effects in mitochondria. J Invest Dermatol. 2007;127:2048–2057. doi: 10.1038/sj.jid.5700826. [DOI] [PubMed] [Google Scholar]
- Hu Y, Russek SJ. BDNF and the diseased nervous system: a delicate balance between adaptive and pathological processes of gene regulation. J Neurochem. 2008;105:1–17. doi: 10.1111/j.1471-4159.2008.05237.x. [DOI] [PubMed] [Google Scholar]
- Karu T. Mitochondrial mechanisms of photobiomodulation in context of new data about multiple roles of ATP. Photomed Laser Surg. 2010;28:159–160. doi: 10.1089/pho.2010.2789. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Khuman J, Zhang J, Park J, Carroll JD, Donahue C, Whalen MJ. Low-level laser light therapy improves cognitive deficits and inhibits microglial activation after controlled cortical impact in mice. J Neurotrauma. 2012;29:408–417. doi: 10.1089/neu.2010.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan CC, Wu SB, Wu CS, Shen YC, Chiang TY, Wei YH, Yu HS. Induction of primitive pigment cell differentiation by visible light (helium-neon laser): a photoacceptor-specific response not replicable by UVB irradiation. J Mol Med. 2012;90:321–330. doi: 10.1007/s00109-011-0822-7. [DOI] [PubMed] [Google Scholar]
- Lavi R, Shainberg A, Friedmann H, Shneyvays V, Rickover O, Eichler M, Kaplan D, Lubart R. Low energy visible light induces reactive oxygen species generation and stimulates an increase of intracellular calcium concentration in cardiac cells. J Biol Chem. 2003;278:40917–40922. doi: 10.1074/jbc.M303034200. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Barde YA. Physiology of the neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- Liang J, Liu L, Xing D. Photobiomodulation by low-power laser irradiation attenuates Abeta-induced cell apoptosis through the Akt/GSK3beta/beta-catenin pathway. Free Radic Biol Med. 2012;53:1459–1467. doi: 10.1016/j.freeradbiomed.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Li Z, Van Aelst L, Cline HT. Rho GTPases regulate distinct aspects of dendritic arbor growth in Xenopus central neurons in vivo. Nat Neurosci. 2000;3:217–225. doi: 10.1038/72920. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/S0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Ma QL, Harris-White ME, Ubeda OJ, Simmons M, Beech W, Lim GP, Teter B, Frautschy SA, Cole GM. Evidence of Abeta- and transgene-dependent defects in ERK-CREB signaling in Alzheimer's models. J Neurochem. 2007;103:1594–1607. doi: 10.1111/j.1471-4159.2007.04869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CJ. Ras effectors. Curr Opin Cell Biol. 1996;8:197–204. doi: 10.1016/S0955-0674(96)80066-4. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr, Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology. 2001;56:127–129. doi: 10.1212/WNL.56.1.127. [DOI] [PubMed] [Google Scholar]
- Mochizuki-Oda N, Kataoka Y, Cui Y, Yamada H, Heya M, Awazu K. Effects of near-infra-red laser irradiation on adenosine triphosphate and adenosine diphosphate contents of rat brain tissue. Neurosci Lett. 2002;323:207–210. doi: 10.1016/S0304-3940(02)00159-3. [DOI] [PubMed] [Google Scholar]
- Murer MG, Yan Q, Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer's disease and Parkinson's disease. Prog Neurobiol. 2001;63:71–124. doi: 10.1016/S0301-0082(00)00014-9. [DOI] [PubMed] [Google Scholar]
- Naeser MA, Hamblin MR. Potential for transcranial laser or LED therapy to treat stroke, traumatic brain injury, and neurodegenerative disease. Photomed Laser Surg. 2011;29:443–446. doi: 10.1089/pho.2011.9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Cavalié A, Jenne DE, Wekerle H. Induction of MHC class I genes in neurons. Science. 1995;269:549–552. doi: 10.1126/science.7624779. [DOI] [PubMed] [Google Scholar]
- Ou LC, Gean PW. Transcriptional regulation of brain-derived neurotrophic factor in the amygdala during consolidation of fear memory. Mol Pharmacol. 2007;72:350–358. doi: 10.1124/mol.107.034934. [DOI] [PubMed] [Google Scholar]
- Pastore D, Greco M, Passarella S. Specific helium-neon laser sensitivity of the purified cytochrome c oxidase. Int J Radiat Biol. 2000;76:863–870. doi: 10.1080/09553000050029020. [DOI] [PubMed] [Google Scholar]
- Peng S, Garzon DJ, Marchese M, Klein W, Ginsberg SD, Francis BM, Mount HT, Mufson EJ, Salehi A, Fahnestock M. Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer's disease. J Neurosci. 2009;29:9321–9329. doi: 10.1523/JNEUROSCI.4736-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci R, Pazos MC, Borges RE, Pacheco-Soares C. Biomodulation with low-level laser radiation induces changes in endothelial cell actin filaments and cytoskeletal organization. J Photochem Photobiol B. 2009;95:6–8. doi: 10.1016/j.jphotobiol.2008.11.007. [DOI] [PubMed] [Google Scholar]
- Rojas JC, Lee J, John JM, Gonzalez-Lima F. Neuroprotective effects of near-infrared light in an in vivo model of mitochondrial optic neuropathy. J Neurosci. 2008;28:13511–13521. doi: 10.1523/JNEUROSCI.3457-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen LB, Ginty DD, Weber MJ, Greenberg ME. Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron. 1994;12:1207–1221. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- Ryu H, Lee J, Zaman K, Kubilis J, Ferrante RJ, Ross BD, Neve R, Ratan RR. Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J Neurosci. 2003;23:3597–3606. doi: 10.1523/JNEUROSCI.23-09-03597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto H, Mezaki Y, Shikimi H, Ukena K, Tsutsui K. Dendritic growth and spine formation in response to estrogen in the developing Purkinje cell. Endocrinology. 2003;144:4466–4477. doi: 10.1210/en.2003-0307. [DOI] [PubMed] [Google Scholar]
- Salter MW, Hicks JL. ATP causes release of intracellular Ca2+ via the phospholipase Cβ/IP3 pathway in astrocytes from the dorsal spinal cord. J Neurosci. 1995;15:2961–2971. doi: 10.1523/JNEUROSCI.15-04-02961.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels IS, Saitta SC, Landreth GE. MAPing CNS development and cognition: an ERKsome process. Neuron. 2009;61:160–167. doi: 10.1016/j.neuron.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindl A, Schindl M, Pernerstorfer-Schön H, Kerschan K, Knobler R, Schindl L. Diabetic neuropathic foot ulcer: successful treatment by low-intensity laser therapy. Dermatology. 1999;198:314–316. doi: 10.1159/000018140. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D. Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Shefer G, Barash I, Oron U, Halevy O. Low-energy laser irradiation enhances de novo protein synthesis via its effects on translation-regulatory proteins in skeletal muscle myoblasts. Biochim Biophys Acta. 2003;1593:131–139. doi: 10.1016/S0167-4889(02)00350-6. [DOI] [PubMed] [Google Scholar]
- Song S, Zhou F, Chen WR. Low-level laser therapy regulates microglial function through Src-mediated signaling pathways: implications for neurodegenerative diseases. J Neuroinflammation. 2012;9:219. doi: 10.1186/1742-2094-9-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi A, Sakaya H, Kisukeda T, Fushiki H, Tsuda M. Involvement of an upstream stimulatory factor as well as cAMP-responsive element-binding protein in the activation of brain-derived neurotrophic factor gene promoter I. J Biol Chem. 2002;277:35920–35931. doi: 10.1074/jbc.M204784200. [DOI] [PubMed] [Google Scholar]
- Tong L, Balazs R, Thornton PL, Cotman CW. Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J Neurosci. 2004;24:6799–6809. doi: 10.1523/JNEUROSCI.5463-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullai JW, Chen J, Schaffer ME, Kamenetsky E, Kasif S, Cooper GM. Glycogen synthase kinase-3 represses cyclic AMP response element-binding protein (CREB)-targeted immediate early genes in quiescent cells. J Biol Chem. 2007;282:9482–9491. doi: 10.1074/jbc.M700067200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant AR, Zanassi P, Walsh GS, Aumont A, Alonso A, Miller FD. Signaling mechanisms underlying reversible, activity-dependent dendrite formation. Neuron. 2002;34:985–998. doi: 10.1016/S0896-6273(02)00717-1. [DOI] [PubMed] [Google Scholar]
- Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993;74:205–214. doi: 10.1016/0092-8674(93)90307-C. [DOI] [PubMed] [Google Scholar]
- Warne PH, Viciana PR, Downward J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature. 1993;364:352–355. doi: 10.1038/364352a0. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Kornhauser JM, Xia Z, Thiele EA, Greenberg ME. Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol Cell Biol. 1998;18:1946–1955. doi: 10.1128/mcb.18.4.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Askarova S, Sheng W, Chen JK, Sun AY, Sun GY, Yao G, Lee JC. Low energy laser light (632.8 nm) suppresses amyloid-beta peptide-induced oxidative and inflammatory responses in astrocytes. Neuroscience. 2010;171:859–868. doi: 10.1016/j.neuroscience.2010.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani SO, Golestaneh AF, Shafiee A, Hafizi M, Omrani HA, Soleimani M. Effects of low level laser therapy on proliferation and neurotrophic factor gene expression of human Schwann cells in vitro. J Photochem Photobiol B. 2012;107:9–13. doi: 10.1016/j.jphotobiol.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Yoshii A, Constantine-Paton M. BDNF induces transport of PSD-95 to dendrites through PI3K-AKT signaling after NMDA receptor activation. Nat Neurosci. 2007;10:702–711. doi: 10.1038/nn1903. [DOI] [PubMed] [Google Scholar]
- Yu W, Naim JO, McGowan M, Ippolito K, Lanzafame RJ. Photomodulation of oxidative metabolism and electron chain enzymes in rat liver mitochondria. Photochem Photobiol. 1997;66:866–871. doi: 10.1111/j.1751-1097.1997.tb03239.x. [DOI] [PubMed] [Google Scholar]
- Zhang H, Wu S, Xing D. Inhibition of Abeta(25–35)-induced cell apoptosis by low-power-laser-irradiation (LPLI) through promoting Akt-dependent YAP cytoplasmic translocation. Cell Signal. 2012;24:224–232. doi: 10.1016/j.cellsig.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Zhang L, Xing D, Gao X, Wu S. Low-power laser irradiation promotes cell proliferation by activating PI3K/Akt pathway. J Cell Physiol. 2009;219:553–562. doi: 10.1002/jcp.21697. [DOI] [PubMed] [Google Scholar]
- Zhang L, Ma Q, Yang W, Qi X, Yao Z, Liu Y, Liang L, Wang X, Ma C, Huang L, Xu Y, Zhu H, Deng W, Gao Y, Ruan L, Xiao Z, Qin C. Recombinant DNA vaccine against neurite outgrowth inhibitors attenuates behavioral deficits and decreases Abeta in an Alzheimer's disease mouse model. Neuropharmacology. 2013;70:200–210. doi: 10.1016/j.neuropharm.2012.10.023. [DOI] [PubMed] [Google Scholar]
- Zhou ZD, Chan CH, Ma QH, Xu XH, Xiao ZC, Tan EK. The roles of amyloid precursor protein (APP) in neurogenesis: implications to pathogenesis and therapy of Alzheimer disease. Cell adhesion and migration. 2011;5:280–292. doi: 10.4161/cam.5.4.16986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009;5:311–322. doi: 10.1038/nrneurol.2009.54. [DOI] [PubMed] [Google Scholar]