

Abstract
c-Jun N-terminal kinase (JNK) mediates cell signaling essential for axon outgrowth, but the associated substrates and underlying mechanisms are poorly understood. We identified in Xenopus laevis embryos a novel posttranscriptional mechanism whereby JNK regulates axonogenesis by phosphorylating a specific site on heterogeneous nuclear ribonucleoprotein K (hnRNP K). Both JNK inhibition and hnRNP K knockdown inhibited axon outgrowth and translation of hnRNP K-regulated cytoskeletal RNAs (tau and neurofilament medium), effects that were alleviated by expressing phosphomimetic, but not phosphodeficient, forms of hnRNP K. Immunohistochemical and biochemical analyses indicated that JNK phosphorylation of hnRNP K occurred within the cytoplasm and was necessary for the translational initiation of hnRNP K-targeted RNAs but not for hnRNP K intracellular localization or RNA binding. Thus, in addition to its known roles in transcription and cytoskeletal organization, JNK acts posttranscriptionally through hnRNP K to regulate translation of proteins crucial for axonogenesis.
Introduction
Virtually all aspects of axon development, from polarization to initiation and elongation, are directed by kinases that converge at some level on either the composition or organization and transport of cytoskeletal elements (Yoshimura et al., 2006; Polleux and Snider, 2010). c-Jun N-terminal kinases (JNKs), comprising isoforms encoded by three genes and multiple splice variants, are among those involved in these crucial functions (Gelderblom et al., 2004; Eom et al., 2005; Oliva et al., 2006; Eminel et al., 2008; Barnat et al., 2010). Although the roles of distinct JNK isoforms (Kuan et al., 1999; Haeusgen et al., 2009; Barnat et al., 2010) and upstream activators (Hammarlund et al., 2009; Itoh et al., 2009) during neural development have been studied extensively, its many substrates have complicated the identification of specific mechanisms by which JNK promotes axonogenesis (Bogoyevitch and Kobe, 2006; Waetzig et al., 2006). To date, investigators have focused mainly on transcription factors regulating cytoskeletal genes and on structural proteins controlling cytoskeletal organization, stability, and transport, while acknowledging that additional substrates likely exist as a result of the varied roles of JNK in non-neuronal cells (for review, see Zhou et al., 2004).
In addition to cytoskeletal organization and gene transcription, posttranscriptional control of cytoskeletal composition is increasingly seen as a crucial component of axonogenesis (for review, see Szaro and Strong, 2010; Jung et al., 2012). For example, in developing neurons of Xenopus laevis, heterogeneous nuclear ribonucleoprotein K (hnRNP K), an RNA-binding protein, is involved in nuclear export and translational control of multiple cytoskeletal-related transcripts needed for axonogenesis [e.g., type IV neurofilaments (NFs), actin-related protein 2 (ARP2), growth-associated protein 43 (GAP43), and microtubule-associated protein tau]; knockdown of hnRNP K by antisense morpholino oligonucleotide (MO) in Xenopus leads not only to the loss of protein as a result of the inefficient nuclear export and ineffectual loading of these hnRNP K-targeted mRNAs with polyribosomes for translation but also the failure of both embryonic axon initiation and optic axon regeneration (Liu et al., 2008, 2012; Liu and Szaro, 2011).
As a substrate for multiple kinases regulating its nucleocytoplasmic shuttling, RNA binding, and protein–protein interactions in cell lines (Schullery et al., 1999; Ostrowski et al., 2000; Habelhah et al., 2001a; Ostareck-Lederer et al., 2002; Bomsztyk et al., 2004), hnRNP K is ideally situated to serve as a focus for signaling pathways converging on the expression of cytoskeletal-related proteins during axonogenesis. Notably, Habelhah et al. (2001a) demonstrated in HEK293T cells that hnRNP K is a substrate for JNK, but the functional consequences of this phosphorylation for neuronal development have yet to be elucidated. Here, we identify a novel, posttranscriptional mode of action for JNK through the regulation of the interaction of hnRNP K with the translational machinery to control axonogenesis.
Materials and Methods
Preparation of plasmids for in vitro transcription of RNA for expression in Xenopus.
The coding region of X. laevis hnRNP K was excised from pSP6–XhnRNPK (Liu et al., 2008) and cloned into pEGFP–C3 (Clontech). Full-length EGFP–hnRNP K cDNA was isolated from this plasmid by high-fidelity PCR (Platinum Pfx DNA Polymerase; Invitrogen) and cloned into a modified pGEM–3Z vector (Lin and Szaro, 1996) to express EGFP–hnRNP K. JNK site mutations [to create S189A (phosphodeficient) and S189D (phosphomimetic)] were introduced into this plasmid using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene/Agilent) and PAGE-purified mutagenic primers. Sequences of primers (Integrated DNA Technologies) are listed in Table 1. Fidelity of the coding regions of all constructs was confirmed by sequencing (Genewiz).
Table 1.
List of oligonucleotide probe and primer sequences
| Primer name | Sequences (5′→ 3′) |
|---|---|
| Antisense MO | |
| hnRNP K MO1 | GCTGCTACCTTTCTCCTACGCCGAC |
| Primers for cloning | |
| EGFP–hnRNP K forward | AAAGCGGCCGCACCATGGTGAGCAAGGGCGAG |
| EGFP–hnRNP K reverse | CCCGCGGCCGCTCAGTTATCTAGATCCGGTGGATCCCC |
| Primers for site-directed mutagenesis | |
| S189A forward | GATCTCATCTCTGAGGCCCCTGTCAAGGGCA |
| S189A reverse | TGCCCTTGACAGGGGCCTCAGAGATGAGATC |
| S189D forward | CTTGATCTCATCTCTGAGGACCCTGTCAAGGGCAGGAC |
| S189D reverse | GTCCTGCCCTTGACAGGGTCCTCAGAGATGAGATCAAG |
| Primers for qRT-PCR | |
| GAPDH (SYBR Green) forward | TTAGCAATGCTTCCTGCACTACA |
| GAPDH (SYBR Green) reverse | TGCCAAAGTTGTCGTTGATGAC |
| Peripherin (SYBR Green and TaqMan) forward | GCTGTAAGCGACTTTGGTGCTA |
| Peripherin (SYBR Green and TaqMan) reverse | CAAGGCATGATGGGACAGAGT |
| NF-M (SYBR Green and TaqMan) forward | GTGAGGGCACTGAGCAACAG |
| NF-M (SYBR Green and TaqMan) reverse | TTGGATACAGAGATTGTCTCTTCAGAA |
| Tau (SYBR Green) forward | GCTTCTTTTATTTGCCGATGTTTCC |
| Tau (SYBR Green) reverse | CTTGAACAACAGACCGTGTGAAGCA |
| Tau (TaqMan) forward | GCTTCTTTTATTTGCCGATGTTTC |
| Tau (TaqMan) reverse | AGTATCAATGGCAGCAATGGAA |
| TaqMan probe sequences | |
| Peripherin | 6FAM-CATTCCCTGCCTCTGTGTATTGGCTGGTAMRA |
| NF-M | 6FAM-AGAGGACGAACAAGCAAAATCCCCAGATAMRA |
| Tau | 6FAM-CCGGCCCCCGTTATCTTTTTATTTCAGTATAMRA |
Embryo microinjection and dissociated neural tube/myotome cultures.
Plasmids were linearized (AatII) and transcribed in vitro (mMessage mMachine SP6 kit; Ambion) for injection into single blastomeres of two-cell stage, periodic albino X. laevis embryos of either sex, as described by Gervasi and Szaro (2004). When indicated, to test the abilities of constructs to rescue embryos from the effects of hnRNP K knockdown, 0.5 ng of RNA was coinjected with 10 ng of antisense MO (Gene Tools) targeting Xenopus hnRNP K nucleotides −54 to −30 (hnRNP K MO1; Table 1). Details concerning the efficacy and specificity of this MO in Xenopus embryos are described fully by Liu et al. (2008).
Cultures were prepared from stage 22 embryos as described previously (Tabti and Poo, 1991; Undamatla and Szaro, 2001). After dissociation, cells from one embryo per culture were plated into 35 × 10 mm Nunclon Δ culture dishes and grown at 22.5°C for 24 h before analysis. For experiments using JNK inhibitor II [SP600125 (anthra[1,9-cd]pyrazol-6(2H)-one); Calbiochem/EMD Biosciences], culture medium contained 1, 5, 10, 25, or 50 μm SP600125 with 2% dimethylsulfoxide (DMSO) or 2% DMSO alone (0 μm, vehicle control). For Western blots, cultures were rinsed with PBS to remove media and extracted with 0.3 ml of SDS sample solution [8 m urea/5% β-mercaptoethanol (βME)/2.5% SDS] added directly to the dishes. For each condition, extracts were pooled from 25 cultures and concentrated using Amicon Ultra-4 Centrifugal Filter Units 10,000 molecular weight cutoff (Millipore). For qRT-PCR, cells were lysed and extracted by adding 0.35 ml of Buffer RLT/1% βME (RNeasy Plus Mini Kit; Qiagen) directly to culture dishes. Lysates were pooled (25 cultured embryos for each replicate) and vortexed at high speed for 1 min before isolating total RNA from RNeasy spin columns in 30 μl of RNase-free water. All procedures involving animals were performed in accordance with the University at Albany Institutional Animal Care and Use Committee.
Antibodies.
Details for primary antibodies are listed in Table 2. An antibody to β-tubulin I+II was used to label neuronal cell bodies and axons. Whereas in mammals, class II β-tubulins are expressed in a variety of tissues and staining with this antibody is not restricted to the nervous system, in X. laevis, class II β-tubulins are expressed only in neurons and staining with this antibody is neuron specific (Moody et al., 1996). For the purposes of this study and to avoid confusion, we refer to the labeled antigen as neuronal-β-tubulin (N-β-tubulin), as is done routinely for studies in Xenopus (for review, see Szaro and Strong, 2011).
Table 2.
List of primary antibodies
| Antibody | Target | Origin | Working concentration | Reference |
|---|---|---|---|---|
| RMO270 (Invitrogen) | A phosphorylation-independent epitope of NF-M | Mouse monoclonal | 2 μg/ml (Western blot/immunofluorescence)a | Szaro et al., 1989 |
| 3C2 (Santa Cruz Biotechnology) | hnRNP K | Mouse monoclonal | 1:1000 (Western blot); 1:200 (immunofluorescence)b | Liu et al., 2008 |
| 6C5 (Ambion) | GAPDH | Mouse monoclonal | 5 μg/ml (Western blot) | |
| JDR0.3B8 (Sigma-Aldrich) | β-Tubulin I+II (neuron-specific in X. laevis) | Mouse monoclonal | 1:200 (immunofluorescence)a | Moody et al., 1996 |
| 14a9 supernatant (Developmental Studies Hybridoma Bank, University of Iowa) | X. laevis nuclear lamins II/III | Mouse monoclonal | 1:15 (immunofluorescence)c | |
| GFP (Rockland) | EGFP | Goat polyclonal | 1:1000 (Western blot) | |
| Tau | Tau-3′, targeting the C-terminal region conserved in all isoforms | Rabbit polyclonal | 1:2000 (Western blot) | Black et al., 1996; Liu and Szaro, 2011 |
| H3 (Active Motif) | Histone H3, C-terminal | Rabbit polyclonal | 1:15,000 (Western blot) | |
| pS189 | X. laevis hnRNP K, phosphorylated at serine 189 | Rabbit polyclonal | 1:50 (immunofluorescence)b | This study (Fig. 2) |
| XIF3 | X. laevis peripherin | Rabbit polyclonal | 1:500 (immunofluorescence)b | Liu et al., 2008; Liu and Szaro, 2011 |
| 81E11 (Cell Signaling Technology) | Phospho-JNK (Thr183/Tyr185) | Rabbit polyclonal | 1:500 (Western blot) | |
| Total JNK (Cell Signaling Technology) | SAPK/JNK | Rabbit polyclonal | 1:1000 (Western blot) | |
| D13.14.4E XP (Cell Signaling Technology) | Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Rabbit monoclonal | 1:2000 (Western blot) | |
| 137F5 (Cell Signaling Technology) | p44/42 MAPK (ERK1/2) | Rabbit polyclonal | 1:5000 (Western blot) | |
| 5G10 (Cell Signaling Technology) | S6 ribosomal protein | Rabbit monoclonal | 1:1000 |
SAPK, Stress-activated protein kinase.
aFixation in 4% paraformaldehyde in 0.1 m sodium phosphate buffer, pH 7.4.
bFixation in 100% methanol.
cFixation in dithiobis[succinimidyl propionate] (1 mg/ml; Pierce/Thermo Fisher Scientific) in phosphate buffer/40% DMSO.
An antibody (pS189) against the phosphorylated JNK site in hnRNP K was generated in rabbit, using ILDLISE(pS)PVKGRTQ as the antigenic phosphopeptide (21st Century Biochemicals). The resultant serum was immunodepleted with the corresponding non-phosphopeptide and affinity purified using the phosphopeptide. Because the antibody epitope was too scarce to be detected on Western blots, specificity was validated immunohistochemically on transverse sections of stage 37/38 methanol-fixed embryos as described in Results and in accordance with recommendations made for antibody validation for histochemical use in neuronal tissues (Saper and Sawchenko, 2003; Rhodes and Trimmer, 2006). When indicated, sections were treated with alkaline phosphatase (four units, P4252; Sigma) for 4 h at room temperature according to the instructions of the manufacturer.
Immunofluorescence.
Single-label immunofluorescence in culture was performed as described previously (Undamatla and Szaro, 2001), using TRITC-conjugated avidin D (1:1000; Vector Laboratories). Double-label immunofluorescence in culture was performed as described previously (Liu and Szaro, 2011). For all cultures, nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). Images were acquired using a Leitz conventional fluorescence microscope [25× Plan Fluotar, 0.6 numerical aperture (NA); 40× Plan Fluotar, 0.7 NA], a Nikon DS-Ri1, and NIS Elements D software version 3.10.
For single-label immunofluorescence in tissue, stage 37/38 embryos were immersion fixed (4°C, overnight); fixatives varied according to primary antibody (Table 2). Fixed embryos were immersed in 30% sucrose/0.1 m sodium phosphate, pH 7.4, for cryoprotection, embedded, and frozen in Tissue Freezing Medium (Triangle Biomedical Sciences), and cut in 20 μm transverse sections. Primary antibodies were visualized using the appropriate Alexa Fluor 546 secondary antibody (1:1000). When indicated, SYBR Green I (1:100,000; Invitrogen) was added to the primary antibody solution to label nuclei. Images were acquired with a Carl Zeiss LSM 510 confocal microscope (20× Plan ApoChromat, 0.75 NA; 63× oil Plan ApoChromat, 1.4 NA).
For both cultures and sections, coverslips were mounted with Fluoromount-G (Southern Biotechnology Associates). Images were minimally processed for pseudocoloring, brightness, and contrast using Adobe Photoshop CS5 (Adobe Systems). Intensity profiles were obtained using NIS Elements D software. Immunofluorescence was quantified, when indicated, using NIH ImageJ64. For quantification of the morpholino-rescue phenotypes, background was subtracted uniformly across the images using the background subtraction function in NIH ImageJ64 with a rolling-ball radius of 50.00 pixels before quantitation.
Nucleocytoplasmic fractionation and polysome profiling.
For subcellular fractionation and polysome profiling, 40 and 50 stage 37/38 embryos, respectively, were pooled for each sample and homogenized in 0.5 ml of Polysomal Buffer A using Pyrex 2 ml glass homogenizers, and the nuclei were pelleted by low-speed centrifugation for 3 min (Ananthakrishnan et al., 2008). For Western blots of subcellular fractions, SDS sample solution was added directly to the pellet and supernatant. For polysome profiles, 0.5 ml of cytosolic supernatant was layered onto a 10 ml, linear 15–45% sucrose gradient (Gradient Master 107; BioComp Instruments) and then spun at 37,000 rpm (4°C, 2 h) in a swinging-bucket rotor (SW41; Beckman Coulter). Fractions (0.5 ml) were collected from the top (Auto Densi-Flow Density Gradient Fractionator; Labconco) and then split into two 0.25 ml aliquots. Total RNA was recovered from one of them (phenol-chloroform extraction/ethanol precipitation), quantified (A260; ND-1000; NanoDrop/Thermo Scientific), plotted to identify premonosomal, monosomal, and polysomal fractions, and processed for qRT-PCR (see below). Remaining aliquots were pooled according to their respective fractions, and protein was prepared for SDS-PAGE by TCA precipitation and resuspension in SDS sample solution. Ten percent and 100% of protein were loaded for Western blot from the premonosomal and all other fractions, respectively.
RNA-binding protein/RNA coimmunoprecipitation.
Coimmunoprecipitation of mRNP complexes was performed as described, with minor modifications (Liu et al., 2008). Lysates from stage 37/38, RNA-injected embryos (30 pooled per sample) were incubated with protein G-agarose beads (P7700; Sigma-Aldrich) coated with anti-GFP (Rockland) for 2 h at room temperature. Ten percent of each lysate was reserved before immunoprecipitation for use as total input control for qRT-PCR normalization [ΔCT = CT (coimmunoprecipitated RNA) − CT (total input RNA)].
Western blots.
SDS-PAGE, transfer to nitrocellulose, and incubation in primary and secondary antibodies were performed either as described by Zhao and Szaro (1994) with minor modifications or exactly as recommended by the manufacturer for commercial antibodies. Details for primary antibodies are listed in Table 2. Samples were run on 7.5% (cultures, 10 μg protein) or 10% (subcellular fractions, 15 μg protein; polysome profiling) polyacrylamide minigels. Blots were visualized with horseradish peroxidase-conjugated secondary antibodies (Vector Laboratories) and SuperSignal West Dura Chemiluminescent Substrate (Pierce/Thermo Fisher Scientific), imaged with a ChemiDoc XRS system (Bio-Rad), and quantified using NIH ImageJ64.
qRT-PCR.
qRT-PCR was performed as described previously, with minor modifications (Liu et al., 2008; Liu and Szaro, 2011). For culture, cDNA was synthesized from 5 μg of total RNA using SuperScript III reverse transcriptase (Invitrogen) and oligo-dT12–18 for priming, and qRT-PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems), 1 μl of cDNA template, and 150 nm each forward and reverse primers. For polysome profiling, adjacent RNA fractions were pooled, and equal volumes were used to synthesize cDNA as above; qRT-PCR was performed using SYBR Green as above. For RNA coimmunoprecipitation experiments, cDNA was synthesized from 200 ng of RNA, and qRT-PCR was performed for NF-M and peripherin using SYBR Green as above and for tau and peripherin using TaqMan Gene Expression Master Mix (Applied Biosystems), 1 μl of cDNA template, 250 nm TaqMan probe, and 900 nm each forward and reverse primers (Table 1). Data were collected using an ABI Prism 7900HT Sequence Detection System (software version 2.3) and analyzed by the comparative CT method (Schmittgen and Livak, 2008).
Statistical analyses.
Statistical comparisons between two samples were made using t tests (one- or two-sided, as noted), as indicated in text. Comparisons among multiple samples were made using one-way ANOVA, with Tukey's post hoc analyses to identify significant differences between specific individual samples. A Kolmogorov–Smirnov nonparametric test was used to assess statistical significance between treatments to rescue neurite length and branching (see Fig. 5J,K). Post hoc power analyses were performed using G*Power 3 (Faul et al., 2007); this analysis confirmed that sufficient statistical power was reached (≥0.8) for all reported p values for t tests and ANOVAs in the study. Statistical significance was reached at p < 0.05, and the actual values are specified in the legends or text. For data involving cell counts in culture (see Figs. 1A3, 3E, 5I), a minimum of 30 cells were scored (e.g., percentage of N-β-tubulin+ cells) for each culture, and these scores were averaged over the indicated number of cultures for each condition and time point. Data from Western blots were averaged over separate blots of each pooled sample (see Figs. 1E,H, 5M,O, 6B), and qRT-PCR data (see Fig. 1F, 7B, 8D) were similarly averaged over the indicated number of biological replicates of pooled samples given in the text and legends.
Figure 5.
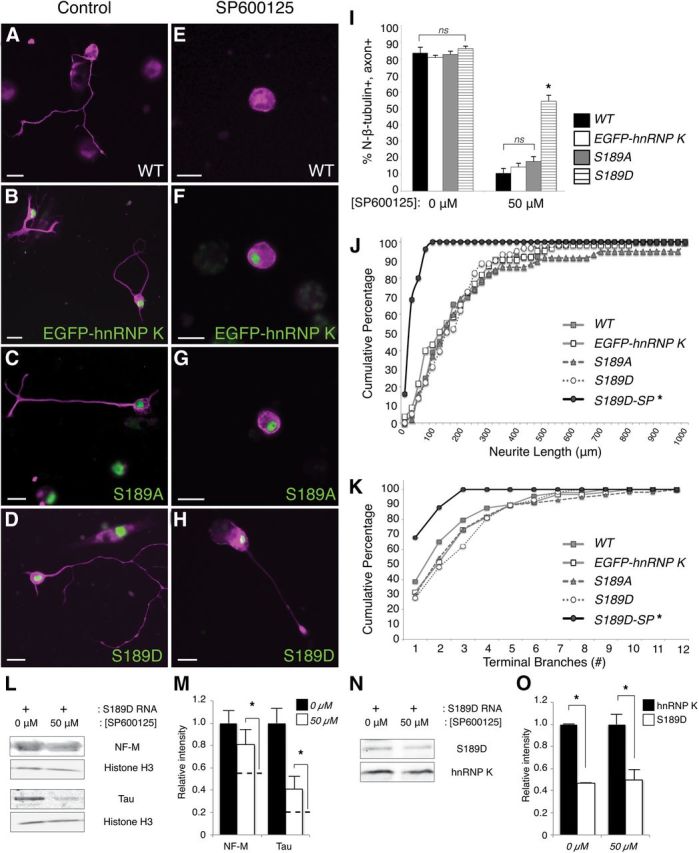
Phosphomimetic mutation of the JNK site on hnRNP K is sufficient to overcome JNK inhibition to restore axon outgrowth. A–H, Immunostaining for N-β-tubulin (magenta) visualized neuronal cell bodies and axons (if present) in representative images of control (A–D) or SP600125-treated (E–H) primary cultures. Expression of mRNA encoding EGFP–hnRNP K, S189A, and S189D is indicated by green fluorescence. Neurons isolated from uninjected WT (A, E), EGFP–hnRNP K mRNA-injected (B, F), and S189A RNA-injected (C, G) embryos successfully formed axons in the absence of pharmacological inhibition of JNK by SP600125 (A–C); the formation of axons by these neurons was severely compromised when grown in 50 μm SP600125 (E–G). However, neurons cultured from S189D mRNA-injected embryos formed axons both with and without SP600125 (D, H). Scale bars, 20 μm. I, Quantification (>30 neurons/culture, n = 5 cultures) of the percentage of N-β-tubulin+ cells that had axons in the RNA-injected cultures with SP600125 indicated that only neurons expressing the phosphomimetic mutation of the JNK site (S189D) on hnRNP K were able to rescue axon outgrowth significantly. Error bars indicate SEM. ns, Nonsignificant; *p < 0.01, one-way ANOVA with Tukey's post hoc test. J, K, Neurite lengths and terminal branch numbers were plotted as cumulative frequencies (percentages) for S189D-expressing neurons cultured in 50 μm SP600125 (S189D-SP), as well as for untreated WT, EGFP–hnRNP K-, S189A-, and S189D-expressing neurons, as indicated. Both total neurite length/neuron and terminal branch number differed significantly between S189D-SP and S189D neurons (*p < 0.001, p = 0.003, respectively, Kolmogorov–Smirnov goodness of fit test, n = 3 cultures, >25 neurons/culture), whereas they did not for untreated EGFP–hnRNP K-, S189A-, and S189D-expressing neurons compared with untreated WT (p = 0.3, p = 0.7, p = 0.4, respectively, for length; p = 0.4, p = 0.5, p = 0.2, respectively, for branching). L, M, The ability of S189D to rescue expression of tau and NF-M protein from JNK inhibition (50 μm SP600125) in culture, assayed by Western blot. L, Representative examples, with histone H3 used as a loading control. M, Average ± SEM intensities of the indicated bands (normalized to histone H3) relative to 0 μm SP600125 (n = 3 replicates of 25 pooled cultures for each condition). To show the degree of rescue, dotted lines indicate the equivalent data for WT neurons in 50 μm SP600125, which were originally presented in Figure 1. Expression of S189D significantly (although not entirely) rescued NF-M and tau protein expression from SP600125 treatment (p = 0.04, p = 0.02, respectively, one-sided t test against WT in 50 μm SP600125, n = 3 replicates of 25 pooled cultures for each condition). N, O, The level of S189D expression was compared with that of endogenous hnRNP K on the same Western blots with an hnRNP K antibody, using the same cultures as in L and M. For both control (0 μm) and SP600125-treated (50 μm) cultures, S189D was expressed at 47 ± 1 and 50 ± 9% of endogenous hnRNP K, respectively (p < 0.001, p = 0.03, respectively, one-sided t test).
Figure 1.
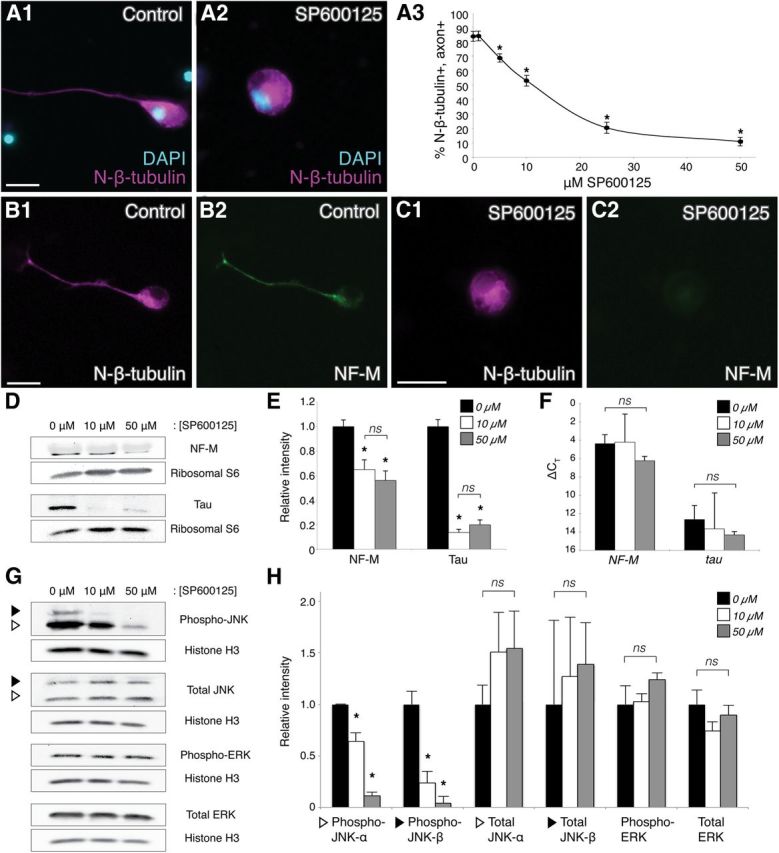
JNK inhibition compromises axon outgrowth and cytoskeletal protein expression. A1, A2, Representative images of neurons in control or SP600125-treated (50 μm) dissociated embryonic neural tube–myotome cultures, as indicated. Immunostaining for N-β-tubulin (magenta) labeled cell bodies and axons (if present), and DAPI (blue) labeled nuclei. A3, Quantification (30 neurons/culture, n = 6 cultures) of the percentage (±SEM) of N-β-tubulin+ cells that had axons in increasing concentrations of SP600125 indicated that JNK activity was required for axon initiation, reaching statistical significance at 5 μm SP600125 (p < 0.01, two-sided t-tests with control, 30 neurons/culture, n = 5 cultures). B, C, Double-label immunocytochemistry for N-β-tubulin (B1, C1, magenta) and NF-M (B2, C2, green), with or without 50 μm SP600125 (C1,C2 and B1,B2, respectively), showed a decrease in the level of NF-M expression when JNK was inhibited. Scale bars, 20 μm. D, Representative Western blots of cultures grown in 0, 10, or 50 μm SP600125. E, Intensities of NF-M and tau protein bands, after normalization against ribosomal protein S6 (loading control), were plotted relative to the intensity at 0 μm SP600125 (±SEM). JNK inhibition (10 and 50 μm) significantly reduced NF-M and tau levels compared with the 0 μm control (ns, nonsignificant; *p < 0.05 and p < 0.01 for NF-M and tau, respectively, one-way ANOVA with Tukey's post hoc test, n = 3 replicates of 25 pooled cultures for each condition); there was no significant difference in this effect between 10 and 50 μm SP600125 (p = 0.6, one-way ANOVA). F, qRT-PCR for NF-M and tau (ΔCT ± SD, relative to GAPDH) of cultures grown in 0, 10, or 50 μm SP600125 displayed no significant change in NF-M or tau RNA expression when JNK was inhibited (p = 0.5, one-way ANOVA, n = 3 replicates of 25 pooled cultures for each condition). G, H, Western blots of SP600125-treated cultures were probed with antibodies to activated (phospho-) JNK, total JNK, activated (phospho-) ERK, and total ERK to investigate effects of SP600125 on the levels of activated and total JNK versus activated and total ERK. Intensities of activated and total JNK and ERK bands, after normalization against histone H3, were plotted relative to the intensity at 0 μm SP600125 (±SEM). White and black triangles indicate the p46 (α) and p54 (β) isoforms of JNK, which in X. laevis are 44 and 48 kDa, respectively. Levels of activated JNK-α and JNK-β were significantly reduced (p < 0.01, one-way ANOVA with Tukey's post hoc test, n = 3 replicates of 25 pooled cultures for each condition) with SP600125 treatment, whereas no significant effects were observed for total JNK-α/β, activated ERK, and total ERK (p = 0.7, p = 0.9, p = 0.4, p = 0.3, respectively, one-way ANOVA), indicating that SP600125 specifically inhibited JNK activation.
Figure 3.

Phosphorylation at serine 189 on Xenopus hnRNP K is reduced by pharmacological inhibition of JNK. A–D, Representative neurons from control or SP600125-treated cultures stained for total hnRNP K and peripherin (XIF3; A, C) or pS189 and N-β-tubulin (B, D). E–H, Background intensity measurements for control (E, F) or SP600125-treated (G, H) cultures were taken from cultures stained without primary antibodies (Ab) to total hnRNP K (E, G) or pS189 (F, H). JNK inhibition caused a significant decrease in staining for pS189 but not total hnRNP K (I). Scale bars, 20 μm. ns, Nonsignificant, p = 0.5; *p = 0.02, two-sided t test, 30 neurons/culture, n = 5 cultures.
Figure 6.

Mutation of the JNK site on hnRNP K has no effect on its subcellular localization. A, B, Nucleocytoplasmic fractionation followed by Western blot analysis of lysates from uninjected WT, EGFP mRNA-, EGFP–hnRNP K mRNA-, S189A mRNA-, and S189D mRNA-injected stage 37/38 embryos. A, Chemiluminescence immunoblot analysis with antibodies to histone H3 (nuclear) and GAPDH (cytoplasmic) indicated complete separation of the fractions. EGFP fusions (EGFP–hnRNP K, S189A, and S189D) and EGFP were all visualized with anti-GFP and compared with endogenous hnRNP K localization. B, Ratios of nuclear versus cytoplasmic band intensities, normalized to histone H3 and GAPDH, respectively, indicated that all EGFP fusions localized similarly to endogenous hnRNP K. Error bars indicate SEM, n = 3 replicates of 40 embryos per group. *p < 0.01, one-way ANOVA with Tukey's post hoc test; ns, nonsignificant (p > 0.9, as before). C–H, Fluorescence immunostaining (magenta) on stage 37/38 hindbrain sections (C, E, G) and primary neuronal cultures (D, F, H). An antibody to nuclear lamins (C, E, G) and DAPI (D, F, H, blue) labeled nuclei, and N-β-tubulin labeled neurons (D, F, H). Arrows (C1–H1) indicate regions measured for intensity profiles (C3–H3). Subcellular localization of EGFP–hnRNP K (C, D), S189A (E, F), and S189D (G, H) was similar to that of endogenous hnRNP K (shown previously in Fig. 3D,E). Scale bars, 20 μm.
Figure 7.
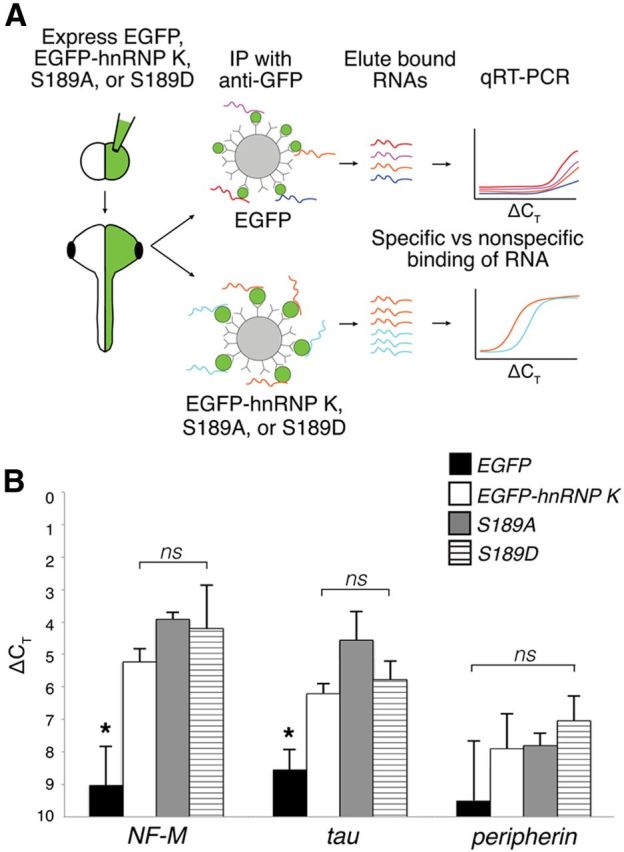
The endogenous association of EGFP–hnRNP K with targeted RNAs is unaffected by mutation of the JNK site. A, Experimental design of RNA-binding protein/RNA coimmunoprecipitation to test RNA association in vivo of EGFP fusion proteins (EGFP–hnRNP K, S189A, S189D). After expression of EGFP (control) and EGFP fusions, lysates were incubated with anti-GFP-coated beads to coimmunoprecipitate proteins with bound RNAs. In qRT-PCR, specifically bound RNAs reached threshold before RNAs that were nonspecific and therefore had smaller ΔCT values. B, Real-time qRT-PCR of RNAs eluted from anti-GFP immunoprecipitation, normalized to total input control. ΔCT values of NF-M and tau (hnRNP K targets) were significantly lower for EGFP–hnRNP K, S189A, and S189D compared with that of EGFP control (*p < 0.05, one-way ANOVA with Tukey's post hoc test) but not for peripherin (a nontarget), indicating specific binding. One-way ANOVA further showed no significant (ns) difference in binding of NF-M and tau among EGFP fusion proteins. Error bars indicate SD, n = 3 replicates, 30 embryos per group.
Figure 8.

Phosphorylation of the JNK site on EGFP–hnRNP K regulates handoff of its RNA targets to the translational machinery. Polysome profiling followed by Western blot analysis of cytosolic extracts from embryos unilaterally coinjected with hnRNP K MO and either EGFP–hnRNP K (A), S189A (B), or S189D mRNA (C). The black line indicates RNA absorbance at 260 nm (A260) across fractions, and premonosomal (Pre), monosomal (Mono), and polysomal (Poly) fractions are indicated (top). Western blots were probed with anti-GFP to visualize the EGFP fusions and anti-S6 to demonstrate proper polysomal separation (bottom). A, EGFP–hnRNP K was most abundant in the premonosomal fractions (94.6%) but was also present to a lesser extent in the monosomal (3.9%) and polysomal fractions (1.5%). B, The loss of phosphorylation at the JNK site prevented S189A from moving beyond the premonosomal fraction (99.7%) into heavier, translating fractions (0.3%). C, S189D, like EGFP–hnRNP K (A), also appeared across all fractions (premonosome, 95.4%; monosome, 3.5%; polysome, 1.1%). D, Quantitation by real-time qRT-PCR of the target (NF-M and tau) and nontarget (peripherin) RNAs of hnRNP K among non-premonosomal fractions (monosome + polysome) with real-time qRT-PCR. Relative amounts (corrected against uninjected WT; see Results) of NF-M (*p = 0.04, one-way ANOVA with Tukey's post hoc test; 3 replicates, 25 embryos per sample) and tau (*p = 0.01, as before) mRNAs in these fractions for embryos unilaterally coinjected with hnRNP K MO and S189A RNA were significantly reduced compared with those coinjected with EGFP–hnRNP K or S189D RNA, whereas relative amounts of peripherin mRNA were not significantly different among groups (p = 0.2, one-way ANOVA as above). Error bars indicate pooled SDs. ns, Nonsignificant.
Results
Pharmacological inhibition of JNK disrupted de novo axon outgrowth of Xenopus spinal neurons
To bypass JNK effects on early development (Liao et al., 2006) and exploratory neuritic outgrowth (Coffey et al., 2000; Eminel et al., 2008) and thus more specifically assess its functions during de novo axon outgrowth in Xenopus spinal neurons, a pan-JNK pharmacological inhibitor (SP600125) was added to dissociated embryonic neural tube/myotome cultures plated at stage 22 (Nieuwkoop and Faber, 1994). In these cultures, terminally specified neurons express N-β-tubulin+ before neurite outgrowth (Moody et al., 1996), and, as evidenced by expression of axonal markers (e.g., phosphorylated NF-M and Par3/Par6), all neurites that extend during the first day become axons (Lin and Szaro, 1994; Liu and Szaro, 2011). Thus, axon initiation was scored as the percentage of N-β-tubulin+ cells forming neurites by 24 h (Fig. 1A). Successively higher doses of SP600125 increasingly attenuated axon initiation, reaching statistical significance (p < 0.01, relative to untreated cultures) by 5 μm, with an IC50 of ∼12 μm. Thus, consistent with observations in hippocampal (Oliva et al., 2006) and dorsal root ganglion (Barnat et al., 2010) cultures, JNK inhibition by SP600125 reduced axon outgrowth in Xenopus spinal neurons.
JNK inhibition posttranscriptionally inhibited tau and NF-M expression
In hippocampal cultures, SP600125 suppresses tau expression in addition to axon outgrowth (Oliva et al., 2006). Because the expression of tau is regulated posttranscriptionally by hnRNP K (Liu and Szaro, 2011), we wanted to determine whether tau expression was diminished by SP600125 in Xenopus cultures and, if so, whether the inhibition occurred at the transcriptional or posttranscriptional level. Quantitative Western blot analysis of cultures treated with 0 (control), 10, or 50 μm SP600125 revealed a marked, significant reduction in expression of tau protein with JNK inhibition (Fig. 1D,E), but as assessed by real-time qRT-PCR, this suppression occurred without incurring significant reductions in tau mRNA levels. Thus, inhibition of tau expression by SP600125 occurred posttranscriptionally rather than transcriptionally (Fig. 1F). Because hnRNP K posttranscriptionally regulates multiple cytoskeletal-related transcripts during axon outgrowth (Liu and Szaro, 2011), we next determined whether JNK inhibition similarly affected expression of NF-M, an additional hnRNP K-regulated transcript. As with tau, SP600125 treatment significantly reduced NF-M protein (Fig. 1D,E) but not mRNA expression (Fig. 1F). Double-label immunocytochemistry further showed that, although 50 μm SP600125 inhibited NF-M expression, it had no effect on expression of N-β-tubulin (Fig. 1B,C), a neuronal protein not regulated by hnRNP K. These data confirmed that effects on the targeted transcripts of hnRNP K, NF-M and tau, were not caused by generalized defects in protein expression or neuronal specification.
To confirm that, under the experimental conditions used in Xenopus cultures, SP600125 targeted JNK specifically, Western blots of SP600125-treated cultures were probed for activated as well as total JNK and ERK1/2, a related mitogen-activated protein kinase that is also known to phosphorylate hnRNP K (Bomsztyk et al., 2004). Pharmacological inhibition reduced levels of activated JNK-α, the p46-homologous isoform, to 65 ± 8 and 11 ± 3% of control and activated JNK-β, the p54-homologous isoform, to 24 ± 11 and 4 ± 7% of control for 10 and 50 μm SP600125, respectively [±SEM; p < 0.01, one-way ANOVA with Tukey's post hoc, n = 3 independent replicates of 25 pooled cultures (1 embryo per culture) for each condition; Fig. 1G,H]. Conversely, treatment with SP600125 had no significant effects (p > 0.3, one-way ANOVA) on levels of total JNK, phospho-ERK, or total ERK. Thus, effects observed on axon outgrowth and translational control of the cytoskeletal-associated RNAs could be attributed specifically to JNK. Together, these data indicated that JNK similarly regulated expression of two hnRNP K-targeted neuronal transcripts posttranscriptionally.
Serine 189 of hnRNP K is a JNK target site in vivo
JNK phosphorylates human hnRNP K only on serine residues 216 and 353 in HEK293T cells (Habelhah et al., 2001a). This observation, along with the similarities observed between JNK inhibition (Fig. 1) and hnRNP K knockdown (Liu et al., 2008; Liu and Szaro, 2011), led us to hypothesize that JNK controls Xenopus embryonic axon outgrowth by acting upstream of hnRNP K. ClustalW2 alignment (Larkin et al., 2007) of hnRNP K amino acid sequences of multiple species (GenBank accession numbers: X. laevis, AAH44711; chicken, Q5ZIQ3; mouse, P61979; rat, NP_476482; human, AAB20770) indicated that sequences surrounding the serine 216 JNK target site of human hnRNP K are highly conserved among these species (Fig. 2A). In Xenopus hnRNP K, this site resides at serine residue 189. The remaining JNK target site of human hnRNP K (serine 353) lies within a nuclear shuttling domain (KNS), which Xenopus hnRNP K lacks (Siomi et al., 1993; Michael et al., 1997), leaving a single, highly conserved, prospective JNK phosphorylation site at S189 on Xenopus hnRNP K.
Figure 2.

Serine 189, the JNK target site on Xenopus hnRNP K, is phosphorylated in neurons in vivo. A, Multiple sequence alignment of the region of hnRNP K surrounding X. laevis hnRNP K S189 (indicated by *) from five species shows that the prospective JNK site is highly conserved. Black and gray shading indicates 100 and 80% conservation among the different species examined, respectively. The black line indicates the region of Xenopus hnRNP K used to generate the phospho-specific antibody (pS189). B, C, Immunohistochemistry (IHC) on transverse sections of spinal cord and somites of stage 37/38 embryos. B, Sections of embryos unilaterally injected with hnRNP K MO at the two-cell stage. Overlays indicate locations of spinal cord (SC) and somites (Som). Unilateral MO suppression of endogenous hnRNP K caused a similar reduction of immunostaining (p = 0.9, two-sided t test, 5 sections per embryo, n = 3 embryos per condition) for both total hnRNP K (55 ± 1% injected/uninjected, p = 0.03, one-sided t test as before; B2) and pS189 (56 ± 5% injected/uninjected, p = 0.01, one-sided t test as before; B4) on the injected side, indicating that pS189 is specific for hnRNP K. Phase contrast images (B1, B3) demonstrated that loss of staining in spinal cord and somites was attributable to loss of expression on the injected side and not to loss of cells. C, Sections of uninjected embryos treated with alkaline phosphatase before immunostaining for total hnRNP K (C2) or pS189 (C4), demonstrating specificity of pS189 for phosphorylated hnRNP K. C1, C3, SYBR Green staining of nuclei demonstrated that loss of staining was not attributable to cell loss. D, E, Representative images of immunostaining (magenta) of stage 37/38 hindbrain sections (D; confocal microscopy) and neurons from dissociated neural tube/myotome cultures (E; conventional microscopy) with the pS189 antibody indicated hnRNP K was phosphorylated at S189 in vivo during nervous system development. Whereas total hnRNP K shuttles to the cytoplasm but localized predominantly to the nucleus during axon outgrowth (D1, D2, SYBR Green; E1–E3, DAPI, blue), phospho-hnRNP K (pS189) was primarily cytoplasmic and perinuclear (D3, D4, E4–E6). Arrows indicate regions measured for intensity profiles (E3, E6). Scale bars, 20 μm.
To determine whether the S189 JNK site of Xenopus hnRNP K was phosphorylated in neurons in vivo, immunohistochemistry was performed using an antibody (pS189) generated against a phosphorylated Xenopus peptide (Fig. 2A, black line). Specificity of this antibody was validated for immunohistochemistry on transverse sections of stage 37/38 methanol-fixed embryos (Fig. 2B1–C4) by first showing that the intensity of pS189 and total hnRNP K immunostaining were significantly reduced on the injected relative to the uninjected side of embryos unilaterally injected with hnRNP K MO (Fig. 2B; 56 ± 5 and 55 ± 1%, respectively). Reductions in intensity of immunostaining as a result of MO knockdown were indistinguishable between the two antibodies (p = 0.9, two-sided t test), thereby demonstrating specificity of the pS189 phospho-specific antibody for hnRNP K. Second, pS189 staining but not total hnRNP K staining was effectively abolished after treating sections with alkaline phosphatase (Fig. 2C), thus demonstrating pS189 specificity for the phosphorylated protein.
In localization studies, immunostaining of transverse sections of stage 37/38 hindbrain for total hnRNP K was heaviest in neuronal nuclei, with little staining in the cytoplasm (Fig. 2D1,D2), consistent with previous studies of hnRNP K localization in embryonic Xenopus neurons (Liu et al., 2008). In the same region, pS189 immunostaining (Fig. 2D3,D4) was much less intense overall and predominantly cytoplasmic in its intracellular localization. Moreover, this cytoplasmic labeling, like that of total hnRNP K when seen in the cytoplasm of older Xenopus neurons (Liu et al., 2012), was punctate, a finding consistent with the idea that cytoplasmic RNA-binding proteins reside within mRNP granules and with observations of the distribution of total hnRNP K in mature neurons (Liu et al., 2012). To obtain better intracellular resolution, we immunostained embryonic neural tube cultures for total and pS189-hnRNP K and took intensity profiles across representative images of individual neurons. Consistent with our observations in sections of developing hindbrain and in keeping with its known behavior as a shuttling protein (Michael et al., 1997; Bomsztyk et al., 2004; Liu et al., 2008, 2012), total hnRNP K in cultured developing spinal neurons was mostly concentrated in the nucleus, with very low levels in the cytoplasmic region of the intensity profiles (Fig. 2E1–E3). As in sections, pS189 immunostaining of cultured neurons was both punctate and cytoplasmic and, interestingly, highly concentrated in perinuclear regions, especially on the side facing the axon (Fig. 2E4–E6). The staining intensity and localization observed for pS189–hnRNP K suggests that just a small subset of endogenous hnRNP K is phosphorylated at the JNK site in developing neurons and that pS189–hnRNP K resides mostly in granules within the cytoplasm.
To further validate that S189 of hnRNP K was targeted by JNK, fluorescence intensities were measured in control and SP600125-treated neural tube cultures immunostained with primary antibodies to total or pS189–hnRNP K (Fig. 3A–D). To control for nonspecific secondary antibody fluorescence, a subset of each group of cultures was immunostained without primary antibodies (Fig. 3E–H), and the intensity of staining was quantified and subtracted as background fluorescence. Whereas JNK inhibition had no significant effect on the levels of total hnRNP K (p = 0.5, two-sided t test, at least 30 neurons/culture, n = 5 cultures; Fig. 3A,C,E,G), treatment with SP600125 caused a significant reduction in pS189 staining (p = 0.02, as previous), falling to levels comparable with background staining (p = 0.9, as previous; Fig. 3B,D,F,H). Thus, S189 of Xenopus hnRNP K is primarily targeted by JNK in neurons and, based on the intracellular distribution of pS189–hnRNP K, is most likely to be involved in the cytoplasmic, as opposed to nuclear, functions of hnRNP K.
Phosphorylation of the JNK site on hnRNP K is required for its function during axon outgrowth
Consistent with previous studies (Liu et al., 2008), unilateral suppression of endogenous hnRNP K by blastomere coinjection of antisense hnRNP K MO and EGFP RNA led to the loss of both spinal motor axons and NF-M expression on the injected side at stage 37/38 (Fig. 4A,B). To estimate the efficiency of the effects of hnRNP K knockdown on axon outgrowth and translation of an hnRNP K mRNA target in the intact embryo, we compared immunofluorescence intensity of N-β-tubulin [the mRNA of which is not an hnRNP K target (Liu et al., 2008)] in somites and NF-M [the mRNA of which is an hnRNP K target (Thyagarajan and Szaro, 2004; Liu et al., 2008)] in spinal cord, respectively, between the injected and uninjected sides of embryos that were coinjected with hnRNP K MO and EGFP RNA (five sections per embryo, n = 3 embryos per condition). N-β-tubulin staining was quantified in somites to monitor the extent of peripheral axon innervation and to avoid any contribution from expression in neuronal cell bodies in the spinal cord. NF-M staining in spinal cord was quantified from sections caudal to the fifth somite to monitor protein expression in neuronal cell bodies while minimizing bias resulting from either defects in axon outgrowth or decussation of axons from the uninjected side. By these measures, hnRNP K knockdown by MO reduced somite innervation and NF-M expression to 58 ± 8% (p < 0.05, one-way ANOVA with Tukey's post hoc test; Fig. 4I) and 41 ± 7% (p < 0.01, as before; Fig. 4J), respectively, on the injected compared with the uninjected side. Immunostaining of sections from the same embryos for total hnRNP K indicated that the magnitude of these effects was comparable with that of hnRNP K knockdown itself (p = 0.2, one-way ANOVA as before; Fig. 2B; see above, Results).
Figure 4.
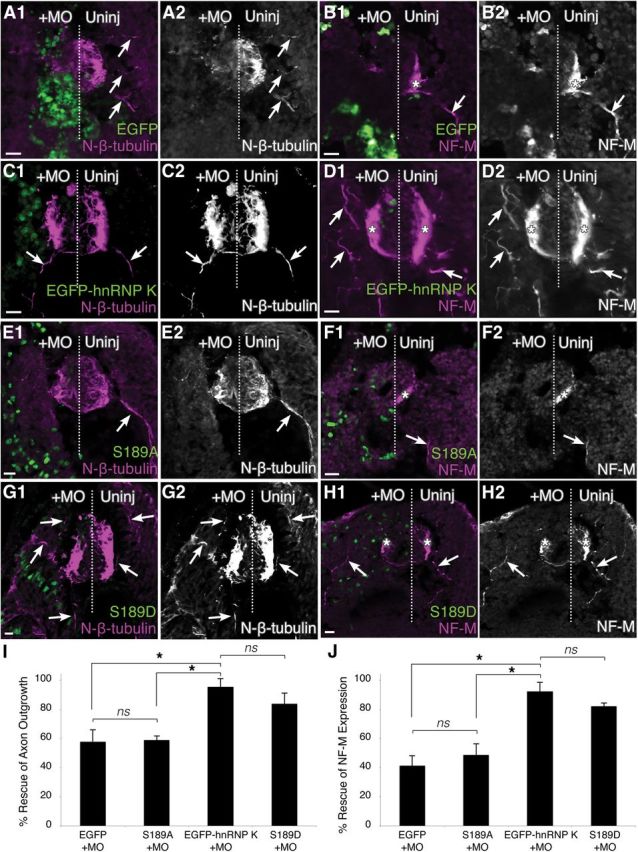
Phosphorylation of the JNK site (S189) is essential for the function of hnRNP K in axon outgrowth. Transverse sections of spinal cord through stage 37/38 embryos. Immunostaining (magenta) for N-β-tubulin (A, C, E, G) and NF-M (B, D, F, H) was used to assay rescue of hnRNP K MO phenotype. The midline is represented by a dotted line, and the injected side (+MO) was unilaterally injected at the two-cell stage with hnRNP K MO and mRNA encoding the fluorescent fusion protein indicated (green). A, B, EGFP by itself rescued neither the axonless phenotype (A1, A2) nor the loss of NF-M expression (B1, B2) caused by hnRNP K MO knockdown. NF-M was expressed (*), and motor axons (arrows) were readily visible extending from the spinal cord into somites on the uninjected side, neither of which could be seen on the injected side. C, D, Unilateral coinjection of EGFP–hnRNP K with the hnRNP K MO yielded axons and NF-M expression on both the injected and uninjected sides, indicating the unmutated hnRNP K fusion protein was fully functional in the intact organism. E, F, A serine-to-alanine mutation of the JNK site on hnRNP K (S189A) abrogated the function of hnRNP K in both axon outgrowth and NF-M expression. G, H, A serine-to-aspartic acid mutation (S189D) successfully rescued both axon outgrowth (arrows) and NF-M expression (*). Scale bars, 20 μm. I, J, Reproducibility of phenotypes was assessed using quantitative immunofluorescence to determine rescue of axon outgrowth (I; N-β-tubulin staining in somites) and translation of an mRNA target of hnRNP K (J; NF-M staining in spinal cord). Degrees of rescue and p values are described fully in Results. ns, Nonsignificant; *p < 0.05, one-way ANOVA with Tukey's post hoc test; 5 sections per embryo, n = 3 embryos per condition.
To study the function of JNK phosphorylation of hnRNP K, we first synthesized an RNA encoding an EGFP–hnRNP K fusion protein, which enabled us to distinguish the exogenous protein from endogenous hnRNP K, and tested its ability to rescue embryos from hnRNP K knockdown by antisense MO. Compared with coinjection of EGFP RNA alone with MO, coinjection of the EGFP–hnRNP K RNA successfully rescued embryos, significantly restoring both somite innervation and NF-M expression on the injected side versus uninjected side to 96 ± 6% (significantly greater than EGFP RNA coinjection, p < 0.05, one-way ANOVA with Tukey's post hoc test as before) and 92 ± 6% (p < 0.01), respectively (Fig. 4C,D,I,J), indicating that the fusion protein was functional in vivo. Next, to determine whether phosphorylation of the S189 JNK site was required for the role of hnRNP K in axonogenesis and NF-M expression, we introduced serine-to-alanine (S189A, phosphodeficient) and serine-to-aspartic acid (S189D, phosphomimetic) mutations into EGFP–hnRNP K and coinjected these RNAs with the MO, as above. Whereas the S189D (phosphomimetic) mutant form restored both somite innervation (84 ± 7% of uninjected side) and NF-M expression (82 ± 2% of uninjected side) on the injected side (Fig. 4G–J) similarly to unmutated EGFP–hnRNP K (p > 0.05, one-way ANOVA with Tukey's post hoc test), the S189A (phosphodeficient) form did not (Fig. 4E,F,I,J). With S189A, somite innervation was reduced to 59 ± 3% and NF-M expression was reduced to 49 ± 8% of the uninjected side. These values were not significantly different from the results obtained with coinjection of EGFP RNA (Fig. 4A,B,I,J; p > 0.05) but were significantly different from rescue with EGFP–hnRNP K and S189D (Fig. 4C,D,I,J; p < 0.05 and p < 0.01, respectively; one-way ANOVA with Tukey's post hoc test). Thus, phosphorylation of the JNK site was required for the functions of hnRNP K in spinal neuron axon formation and NF-M translation in vivo.
Phosphomimetic mutation of hnRNP K restored axon outgrowth during pharmacological inhibition of JNK
To confirm that JNK-dependent axon outgrowth occurred via an hnRNP K-dependent mechanism and not in a parallel pathway, 50 μm SP600125 was added to cultures expressing only endogenous hnRNP K [wild type (WT)] and cultures coexpressing WT with either EGFP–hnRNP K or mutant fusion protein. In the absence of inhibitor, the added expression of EGFP–hnRNP K, S189A, or S189D had no significant effect on axon initiation compared with WT (Fig. 5A–D,I; p = 0.4, one-way ANOVA), because >80% of neurons formed axons in all four cases. Addition of JNK inhibitor reduced this to ∼11%, and added expression of unmutated EGFP–hnRNP K (Fig. 5F,I) or S189A (Fig. 5G,I) gave no significant improvement (p = 0.3, one-way ANOVA). However, expression of S189D (phosphomimetic) significantly increased axon initiation from the effects of the inhibitor (Fig. 5H), with 54 ± 4% (±SEM) of spinal neurons forming axons (Fig. 5I; p < 0.01, one-way ANOVA with Tukey's post hoc test).
Although these data indicated that a major portion of the effects of JNK on early axon outgrowth were mediated via hnRNP K, the “rescued” (SP600125 + S189D) axons were nonetheless significantly shorter and less branched than those of untreated, S189D-expressing neurons (p < 0.001 and p = 0.003, respectively, Kolmogorov–Smirnov test; Fig. 5J,K). As with axon initiation (Fig. 5I), expression of fusion proteins (EGFP–hnRNP K, S189A, S189D) in the absence of pharmacological JNK inhibition had no significant effect on either axon elongation or branching (Fig. 5J,K), confirming that the expression of the fusion proteins alone had no effect on these parameters.
Expression of S189D also significantly rescued NF-M and tau translation from pharmacological JNK inhibition, as assayed on Western blots (Fig. 5L,M; p = 0.04 and 0.02, respectively, two-sided t test performed with Western blot data of 50 μm SP600125-treated WT neurons from Fig. 1). However, whereas rescue of NF-M expression from JNK inhibition was nearly complete (81 ± 13%) and not significantly different from S189D-expressing controls (p = 0.3, two-sided t test), that of tau was incomplete (42 ± 12%), remaining significantly lower than S189D-expressing controls (p = 0.01). Western blots confirmed that the effects of pharmacological JNK inhibition cannot be attributed to indirect effects on the levels of expression of either endogenous or S189D hnRNP K, because there were no significant differences (p > 0.8) in expression of either between SP600125-treated and control cultures. However, the residual effects of JNK inhibition after S189D rescue might be attributable to the expression of S189D being significantly less (∼50%; p < 0.03) than that of endogenous hnRNP K (Fig. 5N,O). We were unable to test this hypothesis by increasing expression of S189D because injecting greater amounts of exogenous RNA proved to be toxic to the embryos.
Mutation of the JNK site had no effect on hnRNP K subcellular localization
Studies in cell lines have implicated various kinases in regulating the nucleocytoplasmic shuttling of hnRNP K and its interactions with RNAs and other proteins (Ostrowski et al., 2000; Habelhah et al., 2001b; Ostareck-Lederer et al., 2002; Messias et al., 2006). To assess whether phosphorylation of hnRNP K at S189 regulated its nucleocytoplasmic shuttling, subcellular fractionation followed by Western blot analysis was used to quantify changes in its nuclear versus cytoplasmic localization attributable to mutation of the JNK site. Lysates from uninjected WT, EGFP mRNA-, EGFP–hnRNP K mRNA-, S189A mRNA-, or S189D mRNA-injected embryos were separated via low-speed centrifugation into nuclear and cytoplasmic fractions. Consistent with immunostaining of endogenous hnRNP K (Fig. 2), the vast majority (∼94%) of endogenous and all three hnRNP K fusion proteins were in the nuclear fraction (Fig. 6A,B), with no significant differences among them (p = 0.99, one-way ANOVA; Fig. 6B). This distribution was dependent on hnRNP K, because EGFP alone was significantly enriched in the cytoplasm (p < 0.01, one-way ANOVA with Tukey's post hoc test). Using EGFP fluorescence to localize the fusion proteins, these data were confirmed at the cellular level in sections of hindbrain at stage 37/38 (Fig. 6C,E,G; counterstained for nuclear lamins) and in primary cultures (Fig. 6D,F,H; counterstained for N-β-tubulin and with DAPI) of embryos unilaterally injected with EGFP–hnRNP K, S189A, or S189D mRNA (Fig. 6C–H). In no case did mutation of the JNK site affect the distribution of hnRNP K compared with EGFP–hnRNP K or WT, because all were predominantly nuclear. Thus, phosphorylation of the JNK site did not regulate nucleocytoplasmic shuttling activities of hnRNP K in vivo.
Mutation of the JNK site had no effect on RNA binding by hnRNP K
RNA-binding protein/RNA coimmunoprecipitation was used to test whether mutation of the JNK site affected the association of hnRNP K with RNA in vivo (Fig. 7A). After immunoprecipitation with anti-GFP from lysates of embryos expressing EGFP, EGFP–hnRNP K, S189A, or S189D, three coimmunoprecipitated RNAs were quantified by qRT-PCR: NF-M and tau, which associate endogenously with hnRNP K, and peripherin, which does not (Liu et al., 2008; Liu and Szaro, 2011). For both NF-M and tau, the average CT of coimmunoprecipitated transcript normalized to total input (ΔCT) was significantly higher for EGFP (nonspecific) than for EGFP–hnRNP K, S189A, and S189D fusion proteins (Fig. 7B; p < 0.05, one-way ANOVA with Tukey's post hoc test), indicating that both RNAs were specifically enriched in each of the three EGFP fusion protein coimmunoprecipitations over the nonspecific (EGFP) control. Moreover, none of these associations differed significantly among the three hnRNP K fusion proteins (NF-M, p = 0.55; tau, p = 0.13, one-way ANOVA), indicating comparable association with target RNAs. Conversely, these associations were specific for hnRNP K-targeted RNAs, because no significant enrichment was observed for any of the fusion proteins relative to EGFP for peripherin RNA, a nontarget (p = 0.3, one-way ANOVA). Collectively, these data indicated that phosphorylation of the JNK site did not regulate RNA binding by hnRNP K.
Phosphorylation of the JNK site on hnRNP K regulated its interaction with the translational machinery
Experiments in mammalian cell lines have led to a model for the function of hnRNP K as an RNA shuttling protein in which hnRNP K, complexed with its regulated mRNAs, enters the cytoplasm as a component of translationally repressed mRNP granules. In erythropoietic stem cells, hnRNP K in complex with its target mRNAs represses their translation by inhibiting subsequent joining of the 60S and 40S ribosomal subunits in the forming of the 80S monosome (Ostareck et al., 1997, 2001; Naarmann et al., 2008). In a mammalian proximal tubular epithelial cell line, cytoplasmic hnRNP K moves from the translationally repressed, lighter premonosomal fractions of polysome profiles into heavier monosomal and polysomal fractions as repression is released, indicating that the target mRNAs become loaded with ribosomes while remaining complexed with hnRNP K (Feliers et al., 2007).
To determine whether JNK phosphorylation of hnRNP K is required for this remodeling of premonosomal hnRNP K–mRNP complexes to associate with ribosomes, a necessary precursor for translation, polysome profiles of stage 37/38 embryos expressing each of the hnRNP K fusion proteins were assayed by Western blot and qRT-PCR to assess the association of both hnRNP K and its targeted mRNAs, respectively, with assembled ribosomes. Cytosolic extracts from embryos unilaterally coinjected with each of the three EGFP fusion RNAs and MO were fractionated by linear sucrose gradient ultracentrifugation. The resultant fractions were identified as premonosomal (Pre), 80S monosomal (Mono), and polysomal (Poly) by characteristic peaks of RNA absorbance at 260 nm (Mathews et al., 2007) and pooled accordingly (Fig. 8A–C, top). The presence of fusion protein in each pooled fraction was determined by Western blot with anti-GFP, and the efficiency of fractionation was assessed by probing blots for ribosomal S6 protein. In keeping with what was reported for hnRNP K in mammals (Feliers et al., 2007), unmutated EGFP–hnRNP K in Xenopus embryos was most abundant in the lighter, nontranslating premonosomal fractions but was also readily detectable in the monosomal and polysomal fractions, which encompass those associated with 80S ribosome-bound mRNA (Fig. 8A–C, bottom). Consistent with their differing abilities to rescue the expression of proteins derived from hnRNP K-targeted transcripts from hnRNP K knockdown, the phosphomimetic fusion protein was distributed identically to unmutated EGFP–hnRNP K (Fig. 8A,C, bottom), whereas the phosphodeficient form was detectable only in the translationally repressed, premonosomal fractions (Fig. 8B, bottom).
Using real-time qRT-PCR for two target (NF-M and tau) and one nontarget (peripherin) RNAs (Fig. 8D), we next confirmed that these heavier fractions containing ribosome-associated RNA were enriched specifically with hnRNP K-regulated mRNAs in embryos coinjected with MO (to knockdown endogenous hnRNP K) and the two hnRNP K fusion proteins that rescued protein expression and axon outgrowth (i.e., EGFP–hnRNP K and S189D) but not in those coinjected with the form that did not rescue (i.e., S189A). Because bilaterally injected embryos failed to survive to stage 37/38, we used unilaterally injected embryos and therefore corrected for the contribution of mRNA from the unperturbed, uninjected side by subtracting half the value for each mRNA that was present in the equivalent fractions of a polysomal profile of uninjected embryos of the same stage. As predicted by the model and the data from Western blots, the S189A-expressing embryos exhibited significantly less of the two hnRNP K targeted mRNAs in fractions associated with ribosomes than did either the EGFP–hnRNP K or S189D embryos (p = 0.04 for NF-M and p = 0.01 for tau, one-way ANOVA with Tukey's post hoc test). Moreover, the levels of NF-M (p > 0.9, one-way ANOVA) and tau RNA (p > 0.1) in these fractions were not significantly different between EGFP–hnRNP K- and S189D-expressing embryos, and effects were specific for the hnRNP K targeted mRNAs because there was no significant difference for peripherin among any of the groups (p = 0.2, one-way ANOVA).
Thus, in light of the model based on experiments in mammalian cell lines, wherein hnRNP K represses translation of its RNA targets at the level of initiation by inhibiting joining of the 40S and 60S ribosomal subunits to form the 80S monosome on the mRNP complex (Ostareck et al., 1997, 2001; Naarmann et al., 2008), both Western blot and qRT-PCR data support the conclusion that phosphorylation of hnRNP K at S189 by JNK is required in developing Xenopus neurons for this crucial step in the life cycle of hnRNP K-regulated RNAs during axon outgrowth.
Discussion
These data shed new light on the role of JNK in axonogenesis, demonstrating that it phosphorylates hnRNP K to posttranscriptionally regulate cytoskeletal genes that build the axon. Reminiscent of hnRNP K-knockdown studies in Xenopus embryos (Liu et al., 2008; Liu and Szaro, 2011), JNK inhibition compromised translation of NF-M and tau RNAs, but not their transcription, and severely impaired the formation of spinal motor axons. Although before our study, it was known from studies in mammalian cell lines that JNK phosphorylates hnRNP K, the function of this phosphorylation in vivo was unknown (Habelhah et al., 2001a). We demonstrated that, in Xenopus embryos, JNK phosphorylation of hnRNP K affected neither nucleocytoplasmic localization nor RNA binding of hnRNP K but rather mediated the interaction between hnRNP K and the molecular machinery for translating its targeted RNAs. Evidence for this conclusion came from demonstrating the following: (1) pharmacological inhibition of JNK specifically reduced expression of proteins whose mRNAs are hnRNP K targets (i.e., NF-M and tau) posttranscriptionally and that introducing a JNK-site phosphomimetic form of hnRNP K (S189D), but neither unmutated (EGFP–hnRNP K) nor phosphodeficient forms (S189A), into neurons ameliorated this effect; (2) these same hnRNP K-targeted transcripts efficiently entered heavier fractions of polysome profiles (i.e., monosome and polysome), which are associated with being ribosomally bound, of embryos rescued from hnRNP K knockdown by either unmutated or phosphomimetic, but not phosphodeficient, EGFP–hnRNP K; and (3) the phosphomimetic (S189D) and unmutated forms of EGFP–hnRNP K themselves entered the fractions of polysome profiles that are associated with ribosomes, whereas the phosphodeficient form (S189A) did not. In other cellular contexts, angiotensin II stimulation of translation of vascular endothelial growth factor (VEGF) mRNA, which is an hnRNP K target, leads to increased levels of hnRNP K in polysome fractions (Feliers et al., 2007), and hnRNP K bound to DICE (differentiation control) elements in 3′UTRs represses translation by blocking the ability of these RNAs to bind assembled monosomes (Naarmann et al., 2008). Taken in light of these observations from non-neuronal cells, our data support the interpretation that JNK phosphorylation of hnRNP K alleviates translational repression of specific hnRNP K-bound, cytoskeletal-related mRNAs in developing neurons through remodeling of mRNP complexes to permit their movement from premonosomal, translationally repressed mRNP granules into complexes associated with assembled ribosomes competent for translation. The predominantly perinuclear staining seen with the pS189 antibody indicates that this phosphorylation most likely begins as mRNP complexes exit the nucleus.
By demonstrating that JNK acts via hnRNP K to prime translation of transcripts crucial for building the axon, our study adds a new dimension to our understanding of its role during axonogenesis. Previous studies of the activities of JNK during neuronal development have focused primarily on either its nuclear functions as a regulator of transcription or select cytoplasmic functions, mainly within the axon itself, as a regulator of cytoskeletal organization, stability, and transport (Zhou et al., 2004; Bogoyevitch and Kobe, 2006; Waetzig et al., 2006; Oliva et al., 2006). Although our experiments demonstrate that JNK phosphorylation of hnRNP K is a major component of the role of JNK in axon outgrowth, they add to, rather than exclude, these other mechanisms. JNK inhibitor was added to neural cultures at stage 22 expressly to bypass earlier stages of neural development and target those stages known to require hnRNP K (Liu et al., 2008; Liu and Szaro, 2011). Consistent with the possibility that JNK targets additional substrates, phosphomimetic hnRNP K only partly rescued axon outgrowth from pharmacological JNK inhibition, leading to shorter and simpler neurites than normal. Interestingly, both NFs and tau, whose protein synthesis depends on JNK phosphorylation of hnRNP K, are themselves JNK substrates (Goedert et al., 1997; Brownlees et al., 2000). Linking both synthesis and phosphorylation of these proteins to JNK activation may help neurons coordinate both processes through shared extracellular signals.
Although all JNK isoforms are involved in axonogenesis (for review, see Waetzig et al., 2006), each can play distinct roles (Barnat et al., 2010; Atkinson et al., 2011). For example, in embryonic dorsal root ganglionic neurons, JNK3 is required for neurite initiation, JNK1 for sustained neurite elongation, and JNK2 contributes to both (Barnat et al., 2010). Although we made no attempt to distinguish among these isoforms, it is likely that more than one targets hnRNP K, because hnRNP K is required not only for axon initiation in embryos (Liu et al., 2008; Liu and Szaro, 2011) but also for regenerative elongation of optic axons (Liu et al., 2012), regulating NF-M and tau in both contexts. Indeed, JNK is also crucial for a successful regenerative response to axotomy (for review, see Waetzig et al., 2006), and DLK (dual leucine zipper kinase), which is a JNK-activating kinase that is required for axon regeneration (Hammarlund et al., 2009; Itoh et al., 2009), acts by increasing mRNA stability and translation (Yan et al., 2009). Thus, JNK activation of hnRNP K may be equally important in axon regeneration and in development, making it a point of convergence for the many varied JNK signaling pathways that promote axon outgrowth in different contexts.
The current study adds both support for and critical new insights into an emerging model for the role of hnRNP K in developmental and regenerative axon outgrowth through the shared, posttranscriptional control of multiple cytoskeletal-related transcripts (e.g., type IV NFs, tau, ARP2, and GAP43) that organize neuronal cytoskeletal polymers into the arrays that are both characteristic of and needed to build the axon (Szaro and Strong, 2010; Liu and Szaro, 2011; Liu et al., 2012). In this model, hnRNP K enters the nucleus to bind its targeted mRNAs, in which it accumulates until some signal stimulates export of the mRNP complexes into the cytoplasm. Once in the cytoplasm, these mRNP complexes are translationally repressed until an additional signal remodels them to promote ribosome assembly on the mRNA and relieve repression. Support for this model comes from studies of hnRNP K function in mammalian cell lines and in axon outgrowth during Xenopus development and optic axon regeneration. In mammalian cell lines, hnRNP K accumulates in the nucleus with increased transcription and shuttling of its mRNA cargoes into the cytoplasm (Michael et al., 1997). Once in the cytoplasm, it translationally represses its target mRNAs until the mRNP complexes undergo additional remodeling (Ostareck-Lederer and Ostareck, 2004; Yano et al., 2005), after which it can remain bound to the mRNAs as they are translated on polysomes (Feliers et al., 2007). In Xenopus embryos and optic axon regeneration, hnRNP K and its regulated transcripts undergo similar behaviors. Accumulation of hnRNP K in the nucleus accompanies increased transcription of hnRNP K-regulated RNAs during peak periods of axon outgrowth, and the fraction of these mRNAs shifts continually between untranslated premonosomal pools and actively translated polysomal pools (Ananthakrishnan et al., 2008; Liu et al., 2008, 2012; Ananthakrishnan and Szaro, 2009; Liu and Szaro, 2011). Increased transcription of hnRNP K-regulated mRNAs and continual shifts in the amounts of actively translated transcript could therefore explain why the vast majority of hnRNP K in developing embryos fractionated with nuclei [∼94% (Fig. 6)] and only a small fraction of the hnRNP K entering the cytoplasm associated with ribosomes on polysome profiles [6% (Fig. 8) or ∼0.4% of total cellular hnRNP K). Because phosphorylation events that regulate RNA-binding proteins are frequently transient (Allison et al., 2004; Atkins et al., 2005), it is therefore unsurprising that levels of pS189–hnRNP K were too low to detect on Western blots.
In addition to JNKs, hnRNP K is phosphorylated by other kinases that regulate its binding, nuclear export, and translation of targeted RNAs (Bomsztyk et al., 2004; Mikula et al., 2006). Several, including PKCδ, ERK1/2, and Src, have also been implicated in neuritic outgrowth (Ignelzi et al., 1994; Perron and Bixby, 1999; Waetzig and Herdegen, 2003; Zhao et al., 2003; Ling et al., 2004). ERK1/2 has been implicated in regulating nucleocytoplasmic shuttling of hnRNP K (Habelhah et al., 2001b) and may therefore act upstream of the actions of JNK on hnRNP K in regulating translation. That the fusion protein mimicking constitutive JNK phosphorylation of hnRNP K (S189D) led to unregulated accumulation of neither the fusion protein nor hnRNP K-regulated transcripts in polysomes suggests that additional controls on translation must act either in parallel with or downstream of JNK. In cell lines, Src and PKCδ phosphorylation of hnRNP K regulate translation of DICE-bearing (Ostareck-Lederer et al., 2002) and VEGF (Sataranatarajan et al., 2008) RNAs, respectively, and are therefore good candidates for such regulation. As a target of JNK and other kinases, hnRNP K is thus ideally situated for linking multiple signaling pathways with the expressions of key axonal structural components. With JNK phosphorylation as the founding example of this principle, continued progress toward understanding what signaling molecules act on hnRNP K, and in which sequence, should yield valuable insights into the mechanisms of axonogenesis.
Footnotes
This work was supported by National Science Foundation Grants IOS 951043 and IOS 1257449 (B.G.S.), an American Association of University Women Fellowship (E.J.H.), and a Sigma Xi Grant-in-Aid (E.J.H.). We thank Dr. Itzhak Fischer (Drexel University, Philadelphia, PA) for providing the tau antibody. We also thank Dr. Christine Gervasi for help with Xenopus spawnings and Drs. Min-Ho Lee and Cara Pager for providing access to critical equipment and expertise for polysomal profiling, respectively. Drs. Kurt M. Gibbs and Yuanyuan Liu provided technical assistance with pilot experiments. Chen Wang and Drs. Yuanyuan Liu, Kurt Gibbs, Amar Thyagarajan, Sohini Bose, and Cara Pager made helpful editorial comments.
References
- Allison R, Czaplinski K, Git A, Adegbenro E, Stennard F, Houliston E, Standart N. Two distinct Staufen isoforms in Xenopus are vegetally loclalized during oogenesis. RNA. 2004;10:1751–1763. doi: 10.1261/rna.7450204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthakrishnan L, Szaro BG. Transcriptional and translational dynamics of light neurofilament subunit RNAs during Xenopus laevis optic nerve regeneration. Brain Res. 2009;1250:27–40. doi: 10.1016/j.brainres.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Ananthakrishnan L, Gervasi C, Szaro BG. Dynamic regulation of middle neurofilament (NF-M) RNA pools during optic nerve regeneration. Neuroscience. 2008;153:144–153. doi: 10.1016/j.neuroscience.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Davare MA, Oh MC, Derkach V, Soderling TR. Bidirectional regulation of cytoplasmic polyadenylation element-binding protein phosphorylation by Ca2+/calmodulin-dependent protein kinase II and protein phosphatase I during hippocampal long-term potentiation. J Neurosci. 2005;25:5604–5610. doi: 10.1523/JNEUROSCI.5051-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson PJ, Cho CH, Hansen MR, Green SH. Activity of all JNK isoforms contributes to neurite growth in spiral ganglion neurons. Hear Res. 2011;278:77–85. doi: 10.1016/j.heares.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnat M, Enslen H, Propst F, Davis RJ, Soares S, Nothias F. Distinct roles of c-Jun N-terminal kinase isoforms in neurite initiation and elongation during axonal regeneration. J Neurosci. 2010;30:7804–7816. doi: 10.1523/JNEUROSCI.0372-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black MM, Slaughter T, Moshiach S, Obrocka M, Fischer I. Tau is enriched on dynamic microtubules in the distal region of growing axons. J Neurosci. 1996;16:3601–3619. doi: 10.1523/JNEUROSCI.16-11-03601.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. 2006;70:1061–1095. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomsztyk K, Denisenko O, Ostrowski J. hnRNP K: one protein multiple processes. Bioessays. 2004;26:629–638. doi: 10.1002/bies.20048. [DOI] [PubMed] [Google Scholar]
- Brownlees J, Yates A, Bajaj NP, Davis D, Anderton BH, Leigh PN, Shaw CE, Miller CC. Phosphorylation of neurofilament heavy chain side-arms by stress activated protein kinase-1b/Jun N-terminal kinase-3. J Cell Sci. 2000;113:401–407. doi: 10.1242/jcs.113.3.401. [DOI] [PubMed] [Google Scholar]
- Coffey ET, Hongisto V, Dickens M, Davis RJ, Courtney MJ. Dual roles for c-Jun N-terminal kinase in developmental and stress responses in cerebellar granule neurons. J Neurosci. 2000;20:7602–7613. doi: 10.1523/JNEUROSCI.20-20-07602.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eminel S, Roemer L, Waetzig V, Herdegen T. C-Jun N-terminal kinases trigger both degeneration and neurite outgrowth in primary hippocampal and cortical neurons. J Neurochem. 2008;104:957–969. doi: 10.1111/j.1471-4159.2007.05101.x. [DOI] [PubMed] [Google Scholar]
- Eom DS, Choi WS, Ji S, Cho JW, Oh YJ. Activation of c-Jun N-terminal kinase is required for neurite outgrowth of dopaminergic neuronal cells. NeuroReport. 2005;16:823–828. doi: 10.1097/00001756-200505310-00009. [DOI] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175–191. doi: 10.3758/BF03193146. [DOI] [PubMed] [Google Scholar]
- Feliers D, Lee MJ, Ghosh-Choudhury G, Bomsztyk K, Kasinath BS. Heterogeneous nuclear ribonucleoprotein K contributes to angiotensin II stimulation of vascular endothelial growth factor mRNA translation. Am J Physiol Renal Physiol. 2007;293:F607–F615. doi: 10.1152/ajprenal.00497.2006. [DOI] [PubMed] [Google Scholar]
- Gelderblom M, Eminel S, Herdegen T, Waetzig V. c-Jun N-terminal kinases (JNKs) and the cytoskeleton—functions beyond neurodegeneration. Int J Dev Neurosci. 2004;22:559–564. doi: 10.1016/j.ijdevneu.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Gervasi C, Szaro BG. Performing functional studies of Xenopus laevis intermediate filament proteins through injection of macromolecules into early embryos. Methods Cell Biol. 2004;78:673–701. doi: 10.1016/S0091-679X(04)78023-1. [DOI] [PubMed] [Google Scholar]
- Goedert M, Hasegawa M, Jakes R, Lawler S, Cuenda A, Cohen P. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 1997;409:57–62. doi: 10.1016/S0014-5793(97)00483-3. [DOI] [PubMed] [Google Scholar]
- Habelhah H, Shah K, Huang L, Burlingame AL, Shokat KM, Ronai Z. Identification of new JNK substrate using ATP pocket mutant JNK and a corresponding ATP analogue. J Biol Chem. 2001a;276:18090–18095. doi: 10.1074/jbc.M011396200. [DOI] [PubMed] [Google Scholar]
- Habelhah H, Shah K, Huang L, Ostareck-Lederer A, Burlingame AL, Shokat KM, Hentze MW, Ronai Z. ERK phosphorylation drives cytoplasmic accumulation of hnRNP K and inhibition of mRNA translation. Nat Cell Biol. 2001b;3:325–330. doi: 10.1038/35060131. [DOI] [PubMed] [Google Scholar]
- Haeusgen W, Boehm R, Zhao Y, Herdegen T, Waetzig V. Specific activities of individual c-Jun N-terminal kinases in the brain. Neuroscience. 2009;161:951–959. doi: 10.1016/j.neuroscience.2009.04.014. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M. Axon regeneration requires a conserved MAP kinase pathway. Science. 2009;323:802–806. doi: 10.1126/science.1165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignelzi MA, Jr, Miller DR, Soriano P, Maness PF. Impaired neurite outgrowth of src-minus cerebellar neurons on the cell adhesion molecule L1. Neuron. 1994;12:873–884. doi: 10.1016/0896-6273(94)90339-5. [DOI] [PubMed] [Google Scholar]
- Itoh A, Horiuchi M, Bannerman P, Pleasure D, Itoh T. Impaired regenerative response of primary sensory neurons in ZPK/DLK gene-trap mice. Biochem Biophys Res Comm. 2009;383:258–262. doi: 10.1016/j.bbrc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Jung H, Yoon BC, Holt CE. Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat Rev Neurosci. 2012;13:308–324. doi: 10.1038/nrn3210. [Erratum (2012) 13:597] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/S0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Liao G, Tao Q, Kofron M, Chen JS, Schloemer A, Davis RJ, Hsieh JC, Wylie C, Heasman J, Kuan CY. Jun NH2-terminal kinase (JNK) prevents nuclear beta-catenin accumulation and regulates axis formation in Xenopus embryos. Proc Natl Acad Sci U S A. 2006;103:16313–16318. doi: 10.1073/pnas.0602557103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Szaro BG. Maturation of neurites in mixed cultures of spinal cord neurons and muscle cells from Xenopus laevis embryos followed with antibodies to neurofilament proteins. J Neurobiol. 1994;25:1235–1248. doi: 10.1002/neu.480251006. [DOI] [PubMed] [Google Scholar]
- Lin W, Szaro BG. Effects of intermediate filament disruption on the early development of the peripheral nervous system of Xenopus laevis. Dev Biol. 1996;179:197–211. doi: 10.1006/dbio.1996.0251. [DOI] [PubMed] [Google Scholar]
- Ling M, Trollér U, Zeidman R, Lundberg C, Larsson C. Induction of neurites by the regulatory domains of PKCdelta and epsilon is counteracted by PKC catalytic activity and the RhoA pathway. Exp Cell Res. 2004;292:135–150. doi: 10.1016/j.yexcr.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Liu Y, Szaro BG. hnRNP K post-transcriptionally co-regulates multiple cytoskeletal genes needed for axonogenesis. Development. 2011;138:3079–3090. doi: 10.1242/dev.066993. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gervasi C, Szaro BG. A crucial role for hnRNP K in axon development in Xenopus laevis. Development. 2008;135:3125–3135. doi: 10.1242/dev.022236. [DOI] [PubMed] [Google Scholar]
- Liu Y, Yu H, Deaton SK, Szaro BG. Heterogeneous ribonucleoprotein K, an RNA-binding protein, is required for optic axon regeneration in Xenopus laevis. J Neurosci. 2012;32:3563–3574. doi: 10.1523/JNEUROSCI.5197-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews MB, Sonenberg N, Hershey JW. Origins and principles of translational control. In: Mathews MB, Sonenberg N, Hershey JW, editors. Translational control in biology and medicine. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2007. pp. 1–40. [Google Scholar]
- Messias AC, Harnisch C, Ostareck-Lederer A, Sattler M, Ostareck DH. The DICE-binding activity of KH domain 3 of hnRNP K is affected by c-Src-mediated tyrosine phosphorylation. J Mol Biol. 2006;361:470–481. doi: 10.1016/j.jmb.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Michael WM, Eder PS, Dreyfuss G. The K nuclear shuttling domain: a novel signal for nuclear import and nuclear export in the hnRNP K protein. EMBO J. 1997;16:3587–3598. doi: 10.1093/emboj/16.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikula M, Dzwonek A, Karczmarski J, Rubel T, Dadlez M, Wyrwicz LS, Bomsztyk K, Ostrowski J. Landscape of the hnRNP K protein-protein interactome. Proteomics. 2006;6:2395–2406. doi: 10.1002/pmic.200500632. [DOI] [PubMed] [Google Scholar]
- Moody SA, Miller V, Spanos A, Frankfurter A. Developmental expression of a neuron-specific beta-tubulin in frog (Xenopus laevis): a marker for growing axons during the embryonic period. J Comp Neurol. 1996;364:219–230. doi: 10.1002/(SICI)1096-9861(19960108)364:2<219::AID-CNE3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Naarmann IS, Harnisch C, Flach N, Kremmer E, Kühn H, Ostareck DH, Ostareck-Lederer A. mRNA silencing in human erythroid cell maturation: hnRNP K controls the expression of its regulator c-Src. J Biol Chem. 2008;283:18461–18472. doi: 10.1074/jbc.M710328200. [DOI] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal table of Xenopus laevis (Daudin) New York: Garland Publishing; 1994. [Google Scholar]
- Oliva AA, Jr, Atkins CM, Copenagle L, Banker GA. Activated c-Jun N-terminal kinase is required for axon formation. J Neurosci. 2006;26:9462–9470. doi: 10.1523/JNEUROSCI.2625-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostareck DH, Ostareck-Lederer A, Wilm M, Thiele BJ, Mann M, Hentze MW. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3′ end. Cell. 1997;89:597–606. doi: 10.1016/S0092-8674(00)80241-X. [DOI] [PubMed] [Google Scholar]
- Ostareck DH, Ostareck-Lederer A, Shatsky IN, Hentze MW. Lipoxygenase mRNA silencing in erythoid differentiation: the 3′UTR regulatory complex controls 60S ribosomal subunit joining. Cell. 2001;104:281–290. doi: 10.1016/S0092-8674(01)00212-4. [DOI] [PubMed] [Google Scholar]
- Ostareck-Lederer A, Ostareck DH. Control of mRNA translation and stability in haematopoietic cells: the function of hnRNPs K and E1/E2. Biol Cell. 2004;96:407–411. doi: 10.1016/j.biolcel.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Ostareck-Lederer A, Ostareck DH, Cans C, Neubauer G, Bomsztyk K, Superti-Furga G, Hentze MW. c-Src-mediated phosphorylation of hnRNP K drives translational activation of specifically silenced mRNAs. Mol Cell Biol. 2002;22:4535–4543. doi: 10.1128/MCB.22.13.4535-4543.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski J, Schullery DS, Denisenko ON, Higaki Y, Watts J, Aebersold R, Stempka L, Gschwendt M, Bomsztyk K. Role of tyrosine phosphorylation in the regulation of the interaction of heterogeneous nuclear ribonucleoprotein K protein with its protein and RNA partners. J Biol Chem. 2000;275:3619–3628. doi: 10.1074/jbc.275.5.3619. [DOI] [PubMed] [Google Scholar]
- Perron JC, Bixby JL. Distinct neurite outgrowth signaling pathways converge on ERK activation. Mol Cell Neurosci. 1999;13:362–378. doi: 10.1006/mcne.1999.0753. [DOI] [PubMed] [Google Scholar]
- Polleux F, Snider W. Initiating and growing an axon. Cold Spring Harb Perspect Biol. 2010;2:a001925. doi: 10.1101/cshperspect.a001925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes KJ, Trimmer JS. Antibodies as valuable neuroscience research tools versus reagents of mass distraction. J Neurosci. 2006;26:8017–8020. doi: 10.1523/JNEUROSCI.2728-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Sawchenko PE. Magic peptides, magic antibodies: guidelines for appropriate controls for immunohistochemistry. J Comp Neurol. 2003;465:161–163. doi: 10.1002/cne.10858. [DOI] [PubMed] [Google Scholar]
- Sataranatarajan K, Lee MJ, Mariappan MM, Feliers D. PKC-delta regulates the stimulation of vascular endothelial factor mRNA translation by angiotensin II through hnRNP K. Cell Signal. 2008;20:969–977. doi: 10.1016/j.cellsig.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Schullery DS, Ostrowski J, Denisenko ON, Stempka L, Shnyreva M, Suzuki H, Gschwendt M, Bomsztyk K. Regulated interaction of protein kinase Cδ with the heterogeneous nuclear ribonucleoprotein K protein. J Biol Chem. 1999;274:15101–15109. doi: 10.1074/jbc.274.21.15101. [DOI] [PubMed] [Google Scholar]
- Siomi H, Matunis MJ, Michael WM, Dreyfuss G. The pre-mRNA binding K protein contains a novel evolutionarily conserved motif. Nucl Acids Res. 1993;1993:1193–1198. doi: 10.1093/nar/21.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaro BG, Strong MJ. Post-transcriptional control of neurofilaments: new roles in development, regeneration and neurodegenerative disease. Trends Neurosci. 2010;33:27–37. doi: 10.1016/j.tins.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Szaro BG, Strong MJ. Regulation of cytoskeletal composition in neurons: transcriptional and post-transcriptional control in development, regeneration, and disease. In: Nixon RA, Yuan A, editors. Advances in neurobiology, Vol 3: Cytoskeleton of the nervous system. New York: Springer Science+Business Media; 2011. pp. 559–602. [Google Scholar]
- Szaro BG, Lee VMY, Gainer H. Spatial and temporal expression of phosphorylated and non-phosphorylated forms of neurofilament proteins in the developing nervous system of Xenopus laevis. Dev Brain Res. 1989;48:87–103. doi: 10.1016/0165-3806(89)90095-3. [DOI] [PubMed] [Google Scholar]
- Tabti N, Poo MM. Culturing spinal neurons and muscle cells from Xenopus embryos. In: Banker G, Goslin K, editors. Culturing nerve cells. Cambridge, MA: Massachusetts Institute of Technology; 1991. pp. 137–154. [Google Scholar]
- Thyagarajan A, Szaro BG. Phylogenetically conserved binding of specific KH domain proteins to the 3′ untranslated region of the vertebrate middle neurofilament mRNA. J Biol Chem. 2004;279:49680–49688. doi: 10.1074/jbc.M408915200. [DOI] [PubMed] [Google Scholar]
- Undamatla J, Szaro BG. Differential expression and localization of neuronal intermediate filament proteins within newly developing neurites in dissociated cultures of Xenopus laevis embryonic spinal cord. Cell Motil Cytoskel. 2001;49:16–32. doi: 10.1002/cm.1017. [DOI] [PubMed] [Google Scholar]
- Waetzig V, Herdegen T. The concerted signaling of ERK1/2 and JNKs is essential for PC12 cell neuritogenesis and converges at the level of target proteins. Mol Cell Neurosci. 2003;24:238–249. doi: 10.1016/S1044-7431(03)00126-X. [DOI] [PubMed] [Google Scholar]
- Waetzig V, Zhao Y, Herdegen T. The bright side of JNKs—multitalented mediators in neuronal sprouting, brain development and nerve fiber regeneration. Prog Neurobiol. 2006;80:84–97. doi: 10.1016/j.pneurobio.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Yan D, Wu Z, Chisholm AD, Jin Y. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell. 2009;138:1005–1018. doi: 10.1016/j.cell.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano M, Okano HJ, Okano H. Involvement of Hu and heterogeneous nuclear ribonucleoprotein K in neuronal differentiation through p21 mRNA post-transcriptional regulation. J Biol Chem. 2005;280:12690–12699. doi: 10.1074/jbc.M411119200. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Arimura N, Kaibuchi K. Signaling networks in neuronal polarization. J Neurosci. 2006;26:10626–10630. doi: 10.1523/JNEUROSCI.3824-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Szaro BG. The return of phosphorylated and nonphosphorylated epitopes of neurofilament proteins to the regenerating optic nerve of Xenopus laevis. J Comp Neurol. 1994;343:158–172. doi: 10.1002/cne.903430112. [DOI] [PubMed] [Google Scholar]
- Zhao YL, Takagawa K, Oya T, Yang HF, Gao ZY, Kawaguchi M, Ishii Y, Sasaoka T, Owada K, Furuta I, Sasahara M. Active Src expression is induced after rat peripheral nerve injury. Glia. 2003;42:184–193. doi: 10.1002/glia.10223. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Walzer MA, Snider WD. Turning on the machine: genetic control of axon regeneration by c-jun. Neuron. 2004;43:1–2. doi: 10.1016/j.neuron.2004.06.020. [DOI] [PubMed] [Google Scholar]