

Abstract
The cytoplasmic regulatory protein p62 (Sequestosome 1/A170) is known to modulate various receptor-mediated intracellular signaling pathways. p62 deficiency was shown to result in mature-onset obesity in mice, but the mechanisms underlying this abnormality remained unclear. Here we report that hyperphagia due to central leptin resistance is the cause of obesity in p62−/− mice. We found that these mice show hyperphagia. Restriction of food to the amount eaten by wild-type mice prevented excess body weight gain and fat accumulation, suggesting that overfeeding is the primary cause of obesity in p62−/− mice. Brain-specific p62 deficiency caused mature-onset obesity to the same extent as in p62−/− mice, further supporting a neuronal mechanism as the major cause of obesity in these mice. Immunohistochemical analysis revealed that p62 is highly expressed in hypothalamic neurons, including POMC neurons in the arcuate nucleus. Central leptin resistance was observed even in young preobese p62−/− mice. We found a defect in intracellular distribution of the transcription factor Stat3, which is essential for the action of leptin, in p62−/− mice. These results indicate that brain p62 plays an important role in bodyweight control by modulating the central leptin-signaling pathway and that lack of p62 in the brain causes leptin resistance, leading to hyperphagia. Thus, p62 could be a clinical target for treating obesity and metabolic syndrome.
Introduction
The protein p62, also termed sequestosome 1 (Sqstm1), A170, and ZIP, is a cytoplasmic endosome-associated protein known as an adaptor and scaffold for atypical protein kinase C (aPKC; Ishii et al., 1996; Joung et al., 1996; Puls et al., 1997; Moscat et al., 2007). p62 is a ubiquitin-binding protein (Vadlamudi et al., 1996) that facilitates the formation of inclusion bodies via its interaction with ubiquitinated proteins (Shin, 1998) and the formation of autophagosomes via its interaction with LC3/Atg8 (Komatsu et al., 2007). Recent studies showed that p62−/− mice exhibit various abnormalities such as mature-onset neurodegeneration accompanied by accumulation of ubiquitinated tau protein in the brain at ≥6 months of age, inducing an Alzheimer-like phenotype (Ramesh Babu et al., 2008). Reportedly, p62 also plays a regulatory role in parathyroid hormone-induced osteoclastogenesis, although basal osteoclastogenesis is not significantly affected by the loss of p62 and no apparent defects in bone morphology were observed (Durán et al., 2004). Of note, mutations in human SQSTM1 cause 5q35-linked Paget's disease of the bone (Laurin et al., 2002); osteoclasts are hyperactivated in this disease. We previously showed that p62−/− mice exhibit enhanced arterial neointimal hyperplasia after ligation, which was partly due to enhanced proliferation and migration of vascular smooth muscle cells on arterial injury or mitogenic stimulation (Sugimoto et al., 2010). These studies suggested that p62 regulates homeostasis in the brain, bone, and vasculature; however, the precise mechanisms remain to be elucidated.
p62 regulates various intracellular pathways, and its absence in p62−/− mice causes mature-onset obesity. Rodriguez et al. (2006) showed that p62−/− mice also exhibit insulin resistance when fed a standard diet. Based on the observation of enhanced basal MAPK/ERK1/2 activity and differentiation of p62-deficient adipocytes, these authors suggested that obesity in p62−/− mice is due to enhanced differentiation of preadipocytes to adipocytes (Rodriguez et al., 2006).
We previously cloned p62 (A170) from murine macrophages as an oxidative stress-inducible protein (Ishii et al., 1996) and independently generated p62−/− mice (Komatsu et al., 2007). We confirmed that p62−/− mice accumulate body fat and that long-term administration of the α-glucosidase inhibitor acarbose effectively prevents obesity and simple steatosis in these animals. These results suggest that mechanisms other than enhanced adipocyte differentiation may be involved in the onset of obesity in p62−/− mice.
In this study, we found that p62−/− mice exhibit hyperphagia and leptin resistance during their preobese youth. Moreover, we found that brain-specific p62 deficiency caused mature-onset obesity as observed in p62−/− mice, suggesting that p62 in the brain is important for body weight homeostasis. We also observed high levels of p62 expression in hypothalamic leptin responsive-neurons, and a defect in distribution of Stat3 in p62−/− mice. From these observations, we hypothesize that p62 acts as a positive regulator of leptin signaling in neurons that play critical roles in the anorectic effect of leptin. Our study implicates p62 as a novel clinical target for treating obesity and metabolic syndrome.
Materials and Methods
Chemicals.
Leptin was purchased from PeproTech EC, MT II from Bachem, and neuropeptide Y (NPY) from Sigma-Aldrich. Rat anti-GFP was purchased from Nacalai Tesque. Rabbit anti-phospho-STAT3 (Tyr705) and anti-STAT3 antibodies were purchased from Cell Signaling Technology. The rabbit anti-p62/A170 antibody was raised as described previously (Ishii et al., 2000). All other chemicals were purchased from Nacalai Tesque.
Animal care.
p62 knock-out (KO; p62−/−) mice were generated as described previously (Komatsu et al., 2007). Before analysis, these mice were crossed with C57BL/6J mice (Charles River) for >10 generations to produce fertile offspring that grew normally. POMC-EGFP and NPY-EGFP transgenic mice were described previously (Pinto et al., 2004). To generate p62flox/flox mice, we constructed a targeting vector from 11.8 kb of p62 DNA (from the HindIII site in 5′UTR to the EcoRI site in intron 6). The p62 locus was targeted by insertion of a loxP site in the 5′ upstream sequence of exon 1 and intron 1. Thus, the exon containing the start codon was flanked with loxP sites and could be deleted by Cre recombinase. Target vector DNA was electroporated into C57BL/6J mouse embryonic stem (ES) cells. Positive clones were selected with G418 and verified by PCR and Southern blotting. Recombinant ES cells were injected into mouse blastocysts to produce chimeras. These chimeras were crossed with C57BL/6J mice to produce heterozygous p62 floxed offspring. Homozygotes were obtained by intercrossing heterozygotes. To delete p62 in neuronal cells, p62flox/flox mice were crossed with Nestin-Cre transgenic mice, generating p62flox/flox; Nestin-Cre+ mice. Deletion of the loxP-flanked fragment was confirmed by PCR performed using the following primers: primer 1, 5′-GGCAATGGCTGGTCTACTTT-3′; primer 2, 5′-GGACTGAGCCTCTGAGCAAC-3′; Nestin F, 5′-TGCAACGAGTGATGAGGTTC-3′; and Nestin R, 5′-GAACCTGGTCGAAATCAGTG-3′. A schematic of the p62 floxed construct and the positions of Primers 1 and 2 is presented in Figure 2A. All mice were kept under specific pathogen-free conditions in an environmentally controlled clean room at the Laboratory Animal Resource Center, University of Tsukuba. They were housed at an ambient temperature of 24°C with a daily 12 h light/dark cycle (08:00–20:00). The mice had free access to drinking water and standard chow containing 0.9% calcium and 0.7% phosphorus (MF: 5.1% fat, 23.1% protein, 360 kcal/100 g; Oriental Yeast). Male mice were used in all experiments. The University of Tsukuba and/or the Kanazawa University Animal Research Committee approved all the animal experiments.
Figure 2.
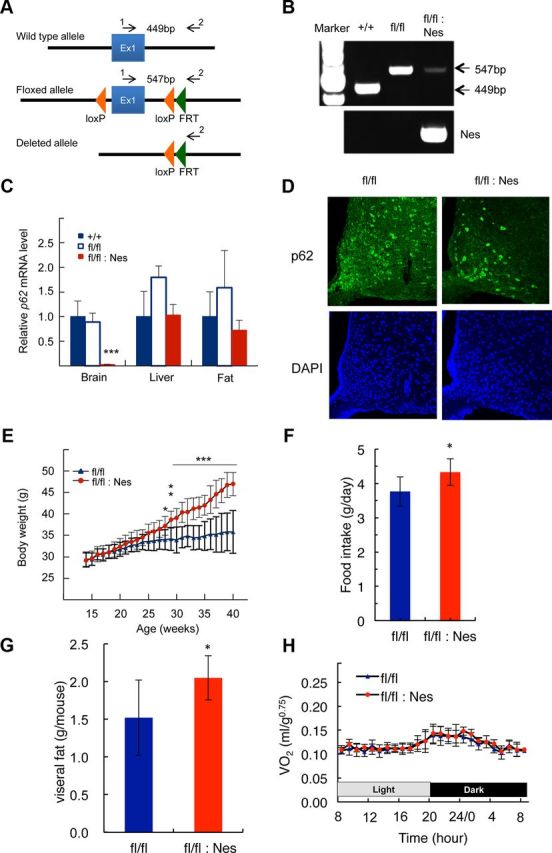
Construction of brain-specific p62−/− mice and their characterization. A, Schematic representation of the wild-type, floxed, and deleted allele of p62. Arrows represent the primers used to determine genotype. B, PCR analysis for genotyping using hypothalamic genomic DNA. C, p62 mRNA expression levels in the whole brain, liver, and epididymal fat tissue measured by quantitative real-time PCR (n = 5). D, Immunofluorescence staining of p62 in the arcuate nucleus of the hypothalamus. E, Weight gain curve of male p62flox/flox (n = 10) and p62flox/flox;Nes-Cre (n = 9) mice. F, G, The amount food intake and visceral fat in >30 weeks old mice (n = 9). H, Daily oxygen consumption rates of body weight-matched p62flox/flox (blue, 26.7 ± 1.6 g) and p62flox/flox;Nes-Cre (red, 27.7 ± 2.0 g) mice (12–18-weeks-old; n = 5). All data represent the mean ± SD. Asterisks represent statistical significance; ***p < 0.001, **p < 0.01, *p < 0.05.
CT analysis.
For Computed tomography (CT) analysis of body fat composition, mice were anesthetized with intraperitoneal injections of sodium pentobarbital and then scanned using a Latheta (LCT-100 M) experimental animal CT system (Aloka). Continuous 5 mm slice images between the liver and tail were used for quantitative assessments using Latheta software (version 1.00). Visceral and subcutaneous fat were distinguished and evaluated quantitatively.
Pair-feeding and oxygen consumption.
Each mouse (10 weeks old; n = 5) was caged separately and given one portion of a standard diet (3.0 ± 0.1 g) every day at 18:00. The body weight of each mouse was measured at 10:00 A.M. two times a week. Energy expenditure in body weight-matched wild-type (15–16-weeks-old; n = 6) and p62−/− (13–14-weeks-old; n = 6) mice was evaluated by measuring oxygen consumption using an O2/CO2 metabolism measuring system (Muromachikikai) and following a standard protocol at 24°C under a 12 h light/dark cycle. To allow acclimatization, each mouse was placed in the apparatus for >3 h before performing the measurements. The mice had free access to drinking water and standard chow during this procedure.
Glucose tolerance test.
Mice were fasted for 13 h before intraperitoneal injection of glucose (1 g/kg body weight). A blood drop was taken from the tail after 0.25, 1, and 2 h. The tail tip was cut 2 mm from the end, and the tail was pressed to collect a drop of blood at the indicated time. Control blood (time 0) was collected before glucose injection. Glucose levels were measured using a portable glucose meter (Ascensia; Bayer Health Care).
Intracerebroventricular injection.
Mice (10–15-weeks-old; body weight matched) were anesthetized with intraperitoneal injection of sodium pentobarbital (0.5 mg/kg). The mice were then positioned in a stereotaxic frame (David Kopf Instruments) and a guide cannula was implanted into the lateral ventricle under sterile conditions. The coordinates used to map the position of the implant were 0.2 mm posterior to the bregma, 1.0 mm lateral from the midline, and 2.5 mm ventral from the skull surface. The mice were then housed individually for a recovery period of at least 7 d and handled daily between 10:00 and 11:00 A.M. Leptin (0.3 or 3 μg), MT II (1 or 5 nmol), and NPY (0.3 nmol) were dissolved in physiological saline. Between 10:00 and 11:00 A.M., all injections were administered in a volume of 3 μl over 60 s. The injector was left in position for a further 60 s to ensure complete dispersal.
Slice preparation and electrophysiological recording of POMC neurons.
Under deep anesthesia with forane (Abbott), POMC-EGFP mice and POMC-EGFP p62−/− mice (3–4-weeks-old) were decapitated. Brains were isolated in an ice-cold cutting solution consisting of 280 mm sucrose, 2 mm KCl, 10 mm HEPES, 0.5 mm CaCl2, 10 mm MgCl2, 10 mm glucose, which was adjusted to pH 7.4 with NaOH, and bubbled with 100% O2. The brains were then cut coronally into 350 μm slices using a vibratome (VTA-1000S; Leica). Slices containing arcuate nucleus (ARH) were transferred for 1 h to an incubation chamber at room temperature and filled with physiological solution containing 140 mm NaCl, 2 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, 10 mm glucose, which was adjusted to pH 7.4 with KOH. The slices were then transferred to a recording chamber (RC-27L, Warner Instrument) at room temperature on a fluorescence microscope stage (BX51WI, Olympus). Neurons that showed EGFP fluorescence were subjected to patch-clamp electrophysiological measurements. The fluorescence microscope was equipped with an infrared camera (C-3077 78; Hamamatsu Photonics) for infrared differential interference contrast (IR-DIC) imaging and a CCD camera (JK-TU53H; Olympus) for fluorescence imaging. Each image was displayed separately on a monitor (Gawin; EIZO).
Recordings were performed using an Axopatch 200B amplifier (Molecular Devices) and a borosilicate pipette (GC150-10, Harvard Apparatus), which was prepared by a micropipette puller (P-97, Sutter Instruments) filled with intracellular solution (4–10 MΩ) consisting of 145 mm KCl, 10 mm HEPES, 1.1 mm EGTA-Na3, 1 mm MgCl2, 0.5 mm Na2GTP, 2 mm MgATP, which was adjusted to pH 7.3 with KOH. The osmolarity of the solution was determined using a vapor pressure osmometer (model 5520; Wescor). The osmolarity of the internal and external solutions was 280–290 and 320–330 mOsm/l, respectively. The liquid junction potential of the patch pipette and perfused extracellular solution was estimated as −16.2 mV; this value was applied to the data. The recording pipette was maintained under positive pressure while being advanced toward individual cells in the slice. Tight seals of 0.5–1.0 GΩ were made by negative pressure. The membrane patch was then ruptured by suction. The series resistance during recording was 10–25 MΩ and was compensated. The reference electrode was an Ag-AgCl pellet immersed in the bath solution. During recording, the cell was superfused with extracellular solution at a rate of 1.0–2.0 ml/min using a peristaltic pump (Miniplus3; Gilson) at room temperature. The trace was processed for presentation using Origin 6.1 software (Origin Lab).
Immunohistochemistry.
Mice deeply anesthetized with sodium pentobarbital were killed by perfusion with PBS and then perfused with 10–20 ml of 4% paraformaldehyde in 0.1 m sodium phosphate buffer, pH 7.4. The whole brain was isolated, postfixed in paraformaldehyde solution overnight at 4°C, equilibrated in 30% sucrose in PBS for 1–2 d at 4°C, embedded in O.C.T. Compound (Sakura Finetek), and frozen on crushed dry ice. The brain was cut into 30-μm-thick sections and stored in 0.1 m sodium phosphate buffer at 4°C. The sections were washed three times for 10 min each in blocking buffer (0.1 m sodium phosphate buffer, 1% BSA, 0.25% Triton X-100) at room temperature and incubated overnight with anti-A170 antiserum (1:400), anti-STAT3 antibody (1:400, Cell Signaling Technology) or anti-GFP (1:1000, Nacalai Tesque) at 4°C. The sections were again washed three times and incubated for 1 h at room temperature with the secondary antibody, AlexaFluor 564-conjugated goat anti-rabbit IgG, AlexaFluor 633-conjugated goat anti-rabbit IgG, or AlexaFluor 488-conjugated goat anti-rat IgG (1:800; Invitrogen). After three washes, the sections were mounted on APS-coated glass slides and covered with 50% glycerol in PBS or ProLong Gold antifade reagent containing DAPI (Invitrogen). Fluorescence was observed using a confocal laser-scanning microscope (TCS SP2; Leica and LSM 5 Pascal; Carl Zeiss). To determine p62 distribution in the brain, wild-type mice brain sections that include the ARH, ventromedial hypothalamus (VMH), dorsomedial hypothalamic nucleus (DMH), lateral hypothalamic area (LHA), nucleus tractus solitarius (NTS), paraventricular nucleus (PVH), or ventral tegmental area (VTA) were selected by anatomical landmarks (the shape of lateral or third/fourth ventricles) based upon the Rat Brain in Stereotaxic Coordinates (Paxinos and Watson, 2005). For Stat3 staining, the ARH including sections from wild-type or p62−/− mice were used. For counting the number of POMC- or NPY-neurons in Figure 7, POMC-EGFP:p62+/+, NPY-EGFP:p62+/+, POMC-EGFP:p62−/−, and NPY-EGFP:p62−/− mice were generated, and only sections that included the entire ARH area were used. We analyzed three or four animals for each condition and the numbers of section per condition were five to eight. The staining images were analyzed by two independent observers.
Figure 7.
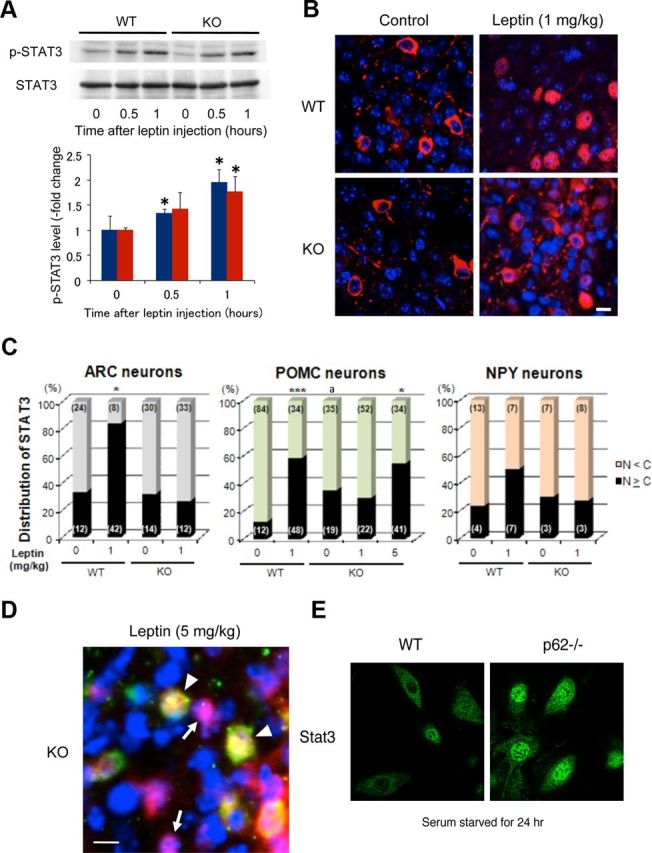
Defect in intracellular distribution of the transcription factor Stat3 in ARH neurons of p62−/− mice and MEFs. A, Immunoblotting of Stat3 and phosphorylated Stat3 in the hypothalamus (top). Animals (8–12-weeks-old; body weight-matched: wild-type, 26.0 ± 1.2 g; p62−/−, 25.5 ± 0.5 g) were fasted for 16 h and killed 0.5 and 1 h after intraperitoneal injection of leptin (1 mg/kg body weight). The levels of phosphorylated Stat3 were quantified relative to the levels of Stat3 in three independent animals (bottom). Results were shown mean ± SEM; *p < 0.05. B, Merged images of anti-Stat3 (red) and DAPI (blue, nucleus) staining in ARH neurons. Body weight-matched wild-type and p62−/− mice (8–12-weeks-old) were fasted for 16 h and killed 1 h after intraperitoneal injection of leptin (1 mg/kg body weight) or an equal amount of vehicle (control). Stat3 signals were concentrated into the nucleus after leptin stimulation in wild-type mice but were less in p62−/− mice. Similar results were observed in at least three independent animals. Scale bar, 10 μm. C, Distribution of Stat3 1 h after leptin or vehicle injection in ARH, POMC, or NPY neurons was scored from three to four independent animals. In addition to wild-type and p62−/− mice, POMC-EGFP and NPY-EGFP transgenic mice with (+/+) or without (−/−) p62 were used (8–12-weeks-old). Triple immunostaining was performed using anti-Stat3 (red), anti-GFP (green), and DAPI (blue, nucleus). Antibody stained POMC neurons are shown in Figure 3. Black columns represent the percentage of neurons in which anti-Stat3 staining of the nucleus was greater than or equal to that of the cytoplasm (N ≥ C). Total numbers of neurons counted in each fraction from 3 to 4 different animals are shown in parentheses. N, Nucleus; C, cytosol. “a” represents significantly different from wild-type control; p < 0.05. ***p < 0.001, *p < 0.05 compared with their controls (n = 3–4). D, Merged image of anti-Stat3 (red), POMC-EGFP (green), and nuclear (blue) staining of ARH neurons in p62−/− mice (8–12-weeks-old), 1 h after intraperitoneal injection of a high-dose leptin (5 mg/kg body weight). Stat3 signals were concentrated in the nucleus after leptin stimulation both in POMC (arrowhead) and other ARH neurons (arrow). Similar results were observed in three independent animals. Scale bar, 10 μm. E, MEFs from both types of mice were cultured and serum starved for 24 h, then Stat3 was visualized by immunofluorescence.
Leptin measurements.
Leptin was measured in serum using a mouse leptin ELISA kit (Morinaga Biochemical).
Quantitative real-time PCR.
Total RNA was isolated from the hypothalamus using Sepasol-RNA I Super (Nacalai Tesque) and purified using a High Pure RNA Tissue Kit (Roche Diagnostics). The PCR thermocycling parameters were 95°C for 10 s, 40 cycles of 95°C for 5 s, and 60°C for 31 s. Reverse transcription and real-time PCR were performed using Perfect Real Time (Takara) and a 7000 sequence detector (Applied Biosystems). Primers were designed using Primer Express (Applied Biosystems). The primers for ObRb: forward, 5′-TGAGCAGGCGTGCvCATC-3′; reverse, 5′-GTACCCGTCAGTTTCACATGATATA-3′. Each sample was assayed in triplicate and normalized by cyclophilin B.
Preparation of MEFs.
Mouse embryonic fibroblasts (MEFs) were isolated from 13.5 postcoitum mouse embryos. The embryos were dissociated and then trypsinized to produce single-cell suspensions. MEFs were only used for experiments after stable proliferation was observed over several passages.
Statistics.
Results are expressed as the mean ± SEM or SD. Statistical analysis was performed using a Student's t test for two-group comparisons or one-way ANOVA followed by a Tukey's multiple-comparison post hoc test; p < 0.05 was considered as a significant.
Results
Hyperphagia causes obesity in p62-deficient mice
Similar body weights were observed in p62−/− mice and their wild-type littermates at birth. Thereafter, p62−/− mice gained weight much faster than wild-type and heterozygous knock-out (p62+/−) mice (F(2,13) = 20.7, p = 0.0001; Fig. 1A). The food intake of p62−/− mice increased linearly from 3.3 g/d/mouse at age 5 weeks to 5.0 g/d/mouse at 50 weeks. In contrast, wild-type and p62+/− mice increased their food intake only slightly with age, with the average intake at 5 weeks being 3.0 g/d/mouse (Fig. 1B).
Figure 1.
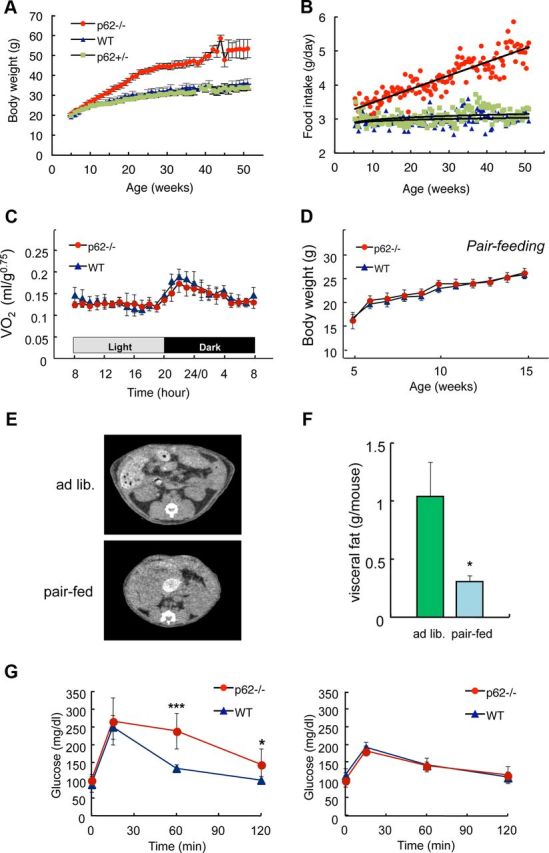
Hyperphagia causes mature-onset obesity in p62−/− mice. A, Weight change (mean ± SD; n = 5) and (B) food intake (mean ± SD; n = 5) of p62−/− (red), p62+/− (green), and control p62+/+ (wild-type, blue) male mice fed a standard diet ad libitum. C, Daily oxygen consumption rates of body weight-matched wild-type (blue, 30.3 ± 2.8 g) and p62−/− (red, 29.8 ± 1.6 g) mice (13–16-weeks-old; mean ± SD; n = 6). D, Pair-feeding experiment. Pellets were provided at 3.0 ± 0.1 g/mouse/d. A 3.0 g portion of food was given to each mouse in a separate cage at 18:00, and the body weight (mean ± SD; n = 5) was measured every week. E, F, Computed tomography (CT) analysis of the visceral fat content. Restricted daily food intake (3.0 g/d/mouse) for 10 weeks from the age of 5–15 weeks (Fig. 1D) suppressed visceral fat accumulation compared with that under ad libitum-feeding conditions in the p62−/− mice. The amount of visceral fat (F) was calculated from CT data. G, Glucose tolerance tests in ad libitum fed (left, 25–28-weeks-old) or diet restricted for 10 weeks (right, 20-weeks-old) wild-type and p62−/− mice. Blood glucose levels after intraperitoneal injection of glucose (1 g/kg body weight) are shown. All data in F and G represent the mean (n = 5–6) ± SD. Asterisks represent statistical significance; ***p < 0.001, *p < 0.05.
The rates and daily rhythm of O2 consumption with free access to drinking water and standard chow were not significantly different between p62−/− and wild-type mice, suggesting that the basal metabolic rates of the 2 genotypes were comparable (Fig. 1C). Pair-feeding experiments, in which the amount of standard chow was restricted to 3.0 ± 0.1 g/d/mouse, showed a similar body weight change in p62−/− and wild-type mice (Fig. 1D), resulting in reduced visceral fat weight and restored insulin sensitivity as judged by a glucose tolerance test in p62−/− mice, whereas ad libitum fed p62−/− mice showed impaired glucose tolerance (F(1,10) = 4.1, p = 0.015; Fig. 1E–G). These results show that the rate of basal energy expenditure was not significantly different between p62−/− and wild-type mice, indicating that increased food intake drives the obesity observed in p62−/− mice.
Neuron-specific p62-deficient mice develop obesity
Because the phenotype of p62−/− mice suggests a possibility that p62 plays an important role in leptin signaling in the CNS, we examined neuronal cell-specific p62-deficient mice in which p62 was abrogated only in the CNS. We generated p62flox/flox mice in which exon 1 of p62 is flanked by loxP sequences (Fig. 2A). We crossed these mice with Nestin-Cre mice in which Cre recombinase is expressed under the control of the rat nestin promoter and enhancer and obtained p62flox/flox; Nestin-Cre mice. Deletion of the loxP-flanked region was confirmed by PCR (Fig. 2B). Compared with wild-type mice, the p62 mRNA expression level in the hypothalamus of p62flox/flox; Nestin-Cre mice was strongly decreased, whereas that in the liver and adipose tissue was unchanged (Fig. 2C). Immunohistochemical analysis of the hypothalamus clearly showed abrogated p62 protein expression in p62flox/flox; Nestin-Cre mice (Fig. 2D).
The body weight of p62flox/flox;Nestin-Cre mice gradually increased after 20 weeks compared with that of p62flox/flox mice, and this difference became statistically significant after 27 weeks (F(1,17) = 35.9, p = 0.0001). The body weight gains of p62flox/flox;Nestin-Cre and control mice over 25 weeks (15–40 weeks of age) were 17 ± 2.7 g and 3.9 ± 3.5 g, respectively (Fig. 2E). These values were comparable to those calculated for p62−/− and wild-type mice (Fig. 1A; 18 ± 5.3 g and 6.9 ± 2.8 g, respectively). The amount of food intake over 45 weeks and the visceral fat weight and were also significantly higher than control mice (Fig. 2F,G). In addition, daily oxygen consumption was not altered as in p62−/− mice (Fig. 2H). These results indicate that the neuron-specific p62 deficiency causes similar mature-onset obesity as in p62−/− mice (Fig. 1A), suggesting that neuronal p62 plays a primary role in body weight control.
p62 distribution in the brain
Given that p62−/− mice showed abnormal regulation of food intake, we examined the expression pattern of p62 in the brain. We found that p62 was expressed in ARH, PVH, and VMH, where leptin receptors are localized (Elmquist et al., 1998). We also observed the expression of p62 in the DMH, LHA, NTS, and VTA (Fig. 3A). These areas play important roles in leptin-mediated regulation of food intake and energy homeostasis (Elmquist et al., 1999; Schwartz et al., 2000; Friedman, 2002; Myers et al., 2008).
Figure 3.
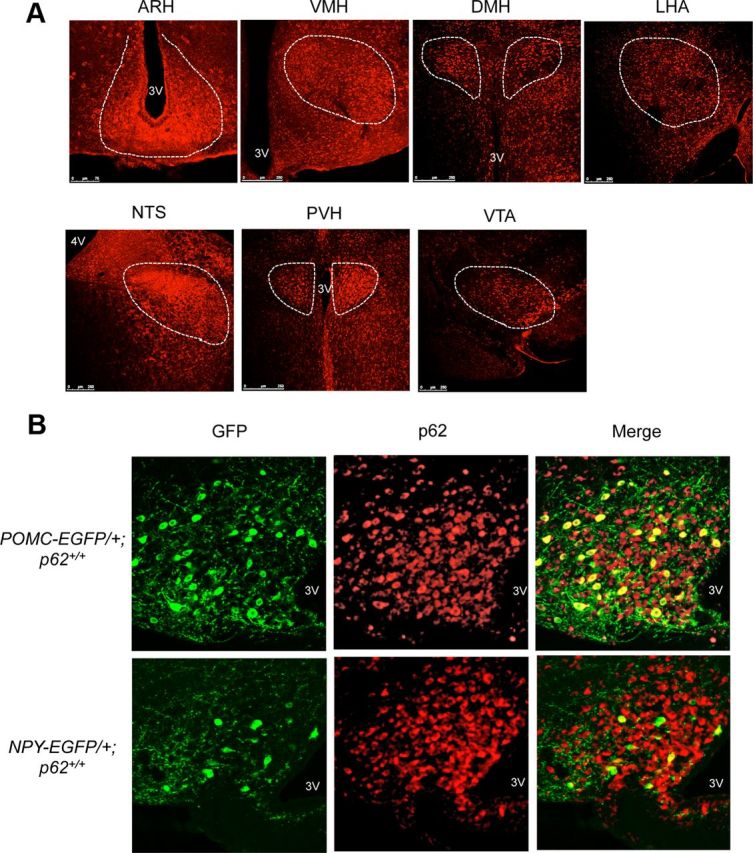
p62 expression in the hypothalamus. A, Immunofluorescence analysis showing that p62 is expressed in the brain regions implicated in the feeding behavior. p62 staining was abundantly expressed in the arcuate nucleus (ARH), ventromedial hypothalamus (VMH), dorsomedial hypothalamic nucleus (DMH), lateral hypothalamic area (LHA), nucleus tractus solitarius (NTS), paraventricular nucleus (PVH), or ventral tegmental area (VTA). 3V, Third ventricle; 4V, fourth ventricle. B, Double-staining with anti-p62 and anti-GFP antibodies in POMC (top) and NPY (bottom) neurons in POMC-EGFP and NPY-EGFP mice, respectively.
We further examined the chemical identity of these neurons using POMC-EGFP and NPY-EGFP transgenic mice. Double-staining of brain slices with antibodies against p62 and GFP clearly showed that most POMC-expressing neurons and some NPY-expressing neurons expressed p62 in ARH of wild-type mice (Fig. 3B).
Impaired effect of leptin on food intake in p62−/− mice
Serum leptin levels in p62−/− mice were much higher than those in wild-type mice, even when body weights were comparable at the young age of 3 weeks, and continued to increase with age (F(5,34) = 80.5, p = 0.0001; Fig. 4A). Furthermore, leptin levels in p62−/− mice were significantly higher than those in wild-type mice of about the same body weight (F(7,52) = 191.5, p = 0.0001; Fig. 4B). Conversely, adiponectin levels were comparable between wild-type and p62−/− mice at young age (18.2 ± 4.0 vs 16.6 ± 2.8 μg/ml; n = 10–11; 8–9-weeks-old, p = 0.311).
Figure 4.
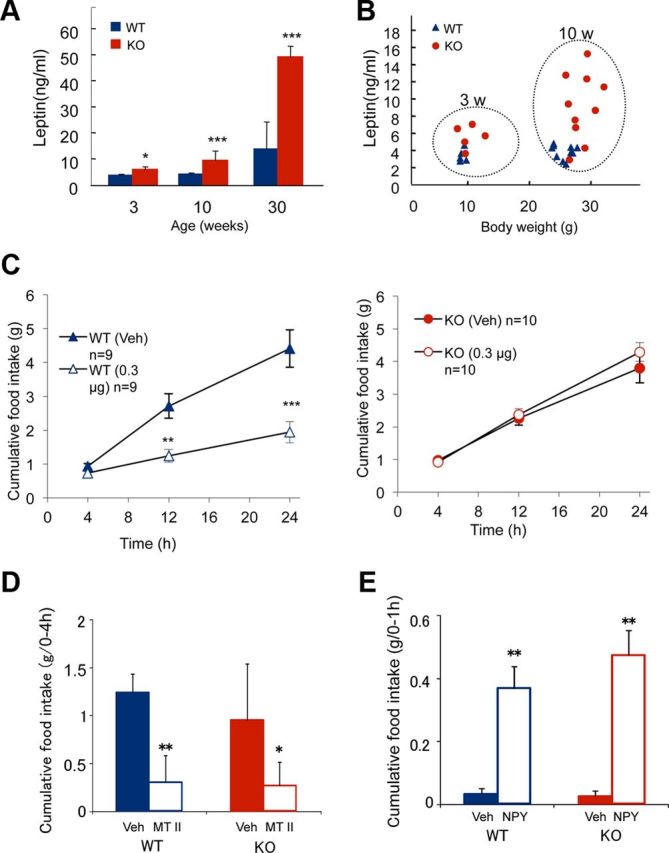
Serum leptin levels and effects of ICV injection of leptin, MTII, and NPY on eating behavior. A, Serum leptin levels in 3-week-old p62−/− (n = 11) and wild-type (n = 12); 10-week-old p62−/− (n = 10) and wild-type (n = 10); and 30-week-old p62−/− (n = 5) and wild-type (n = 5) mice. Means ± SDs are shown. Asterisks represent statistical significance; ***p < 0.001 and *p < 0.05 between the two groups. B, Serum leptin levels versus body weight of 3- and 10-week-old wild-type and p62−/− mice. C, Either leptin (0.3 μg/mouse) or physiological saline (vehicle) was ICV injected into body weight-matched wild-type (25.2 ± 1.7 g) and p62−/− (25.1 ± 4.6 g) mice after 16 h fasting. Food intake was measured for 24 h after ICV injection (10–15-weeks-old; mean ± SEM; n = 9–11). D, Effect of injecting MTII (5 nmol) on food intake 4 h after a 16 h fasting period (15–17-weeks-old; mean ± SEM; n = 5–6). E, Rapid effect of NPY (0.3 nmol/mouse) injection on food intake under ad libitum-feeding conditions was measured at 1 h after injection (10–17-weeks-old; mean ± SEM; n = 6–7). Asterisks represent statistical significance compared with vehicle injection; ***p < 0.001, **p < 0.01, *p < 0.05.
We examined the molecular mechanisms underlying hyperphagia in p62−/− mice. Feeding behavior is regulated by the hypothalamus, which acquires metabolic information from the activity of afferent neurons and from circulating factors such as blood glucose and leptin. Hyperleptinemia in p62−/− mice suggested impaired leptin action. To decipher the mechanism underlying this apparent leptin resistance, leptin was administered by intracerebroventricular (ICV) injection into the lateral ventricle because impaired transport across the blood–brain barrier is a suspected cause of leptin resistance. Wild-type and preobese p62−/− mice were fasted for 16 h and then leptin (0.3 μg/mouse) was administered. Cumulative food intake was measured for 24 h, revealing that 0.3 μg/mouse of leptin significantly inhibited food intake in wild-type mice but had little effect on p62−/− mice (Fig. 4C). These results support the hypothesis that the brain leptin response is impaired in p62−/− mice.
We next examined the anorectic effect of melanotan II (MTII), a melanocortin 4 receptor agonist, and the orexigenic effect of NPY. Injection of MTII inhibited food intake in p62−/− mice, whereas injection of NPY enhanced food intake with similar potency and efficacy as in wild-type mice (Fig. 4D,E). These results suggest that α-MSH and NPY signaling pathways, which act downstream of ARH neurons, remained intact in p62−/− mice. Collectively, these observations suggest that the impairment of leptin function in p62−/−mice was caused by an insufficient response of first-order leptin-responsive neurons in the ARH.
Expression of leptin-regulated genes, ObRb, and Socs3 in the hypothalamus
To examine the function of leptin-regulated neurons in p62−/−mice, we analyzed the expression levels of known leptin-regulated genes and leptin signal transduction-related genes in the hypothalamus. Although the blood–leptin level was significantly increased in p62−/− mice, the mean expression level of hypothalamic Pomc mRNA was lower (∼20%) than that in wild-type mice, although this was not statistically significant under ad libitum-feeding conditions (Fig. 5). The expression levels of Cart, Npy, and Agrp mRNAs were also not significantly different between wild-type and p62−/− mice (Fig. 5). After fasting for 48 h, leptin levels decreased from 3.87 ± 0.81 to 1.50 ± 0.64 ng/ml in wild-type mice and from 9.20 ± 3.90 to 0.90 ± 0.43 ng/ml in p62−/− mice. The increase in Npy and Agrp mRNA levels in the fasted hypothalamus were much lower in p62−/− mice than in wild-type mice (Fig. 5). In addition, leptin receptor (ObRb) and Socs3, a known negative regulator of JAK/STAT pathway, expression were similar in wild-type and p62−/− mice (Fig. 5). Collectively, when the p62−/− mice were fed ad libitum, mRNA expression of Pomc, Cart, Npy, and Agrp were similar to WT mice, although serum leptin levels were much higher in p62−/− mice. Moreover, upregulation of Npy and Agrp mRNA expression caused by fasting decreased leptin levels; however, these changes were marginal in p62−/− compared with WT mice, suggesting that p62−/− mice may have a decreased sensitivity to leptin.
Figure 5.
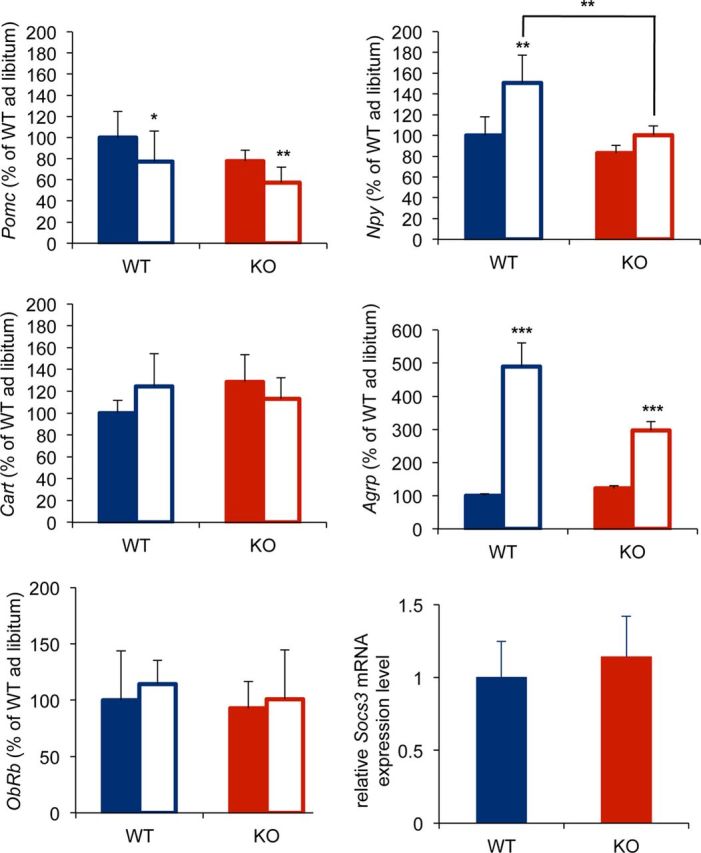
Expression levels of genes in the hypothalamus of p62−/− mice compared with wild-type controls. The relative expression levels of Pomc, Cart, Npy, Agrp, ObRb, and Socs3 mRNAs in the hypothalamus under ad libitum feeding conditions or after fasting for 48 h in body weight-matched wild-type (blue, 27.6 ± 0.9 g) and p62−/− (red, 28.4 ± 1.2 g) mice were measured by quantitative real-time PCR. Each sample was assayed in triplicate and normalized to cyclophilin B (9–11-weeks-old; mean ± SD; n = 5–7). Asterisks represent statistical significance compared with each ad libitum-fed control; ***p < 0.001, **p < 0.01, *p < 0.05.
Numbers of POMC neurons in ARH are not altered in p62−/− mice
It is reported that an absence of p62 promotes mature-onset neurodegeneration (Ramesh Babu et al., 2008). We monitored the number of POMC and NPY neurons using tissue slices through the ARH area of POMC-EGFP and NPY-EGFP transgenic wild-type and p62−/− mice. Interestingly, the number of POMC neurons did not significantly differ between p62−/− and wild-type mice and did not change with age (F(5,13) = 2.7, p = 1.00; Table 1). The number of NPY neurons, however, was higher in young p62−/− mice than in young wild-type mice and gradually decreased with age in p62−/− mice (F(5,14) = 3.9, p = 0.008; Table 1).
Table 1.
Number of POMC-EGFP and NPY-EGFP neurons in the hypothalamus ARH in wild-type and p62−/− mice
| Neuron type | Age (weeks) | No. of neurons in ARH |
|
|---|---|---|---|
| (wild-type) | (p62−/−) | ||
| POMC | 6–8 | 3952 ± 584 (n = 4) | 4441 ± 720 (n = 3) |
| 13–17 | 3792 ± 192 (n = 3) | 3412 ± 352 (n = 3) | |
| 21–25 | 3912 ± 744 (n = 3) | 4888 ± 448 (n = 3) | |
| NPY | 6–8 | 1040 ± 176 (n = 3) | 1559 ± 368 (n = 3) |
| 13–17 | 1007 ± 48 (n = 3) | 944 ± 128 (n = 3)* | |
| 21–25 | 1000 ± 320 (n = 4) | 768 ± 56 (n = 4)* | |
*p < 0.01 compared with 6- to 8-week-old p62−/− mice.
Values represent mean ± SD.
Electrophysiological responses of POMC neurons to leptin
Leptin increases firing frequency in anorexigenic POMC neurons (Cowley et al., 2001). We therefore evaluated the activation of these neurons by leptin using whole-cell patch clamp recording of visually identified POMC neurons in slice preparations from POMC-EGFP transgenic mice. In wild-type mice, the electrophysiological characteristics of POMC neurons were consistent with those presented in a previous report (Cowley et al., 2001). Bath application of leptin (50 nm for 240–300 s) caused depolarization to 8.7 ± 1.9 mV from the resting level (n = 4) and increased spontaneous firing of POMC neurons (Fig. 6). Brain slices prepared from p62−/− mice yielded almost the same results (7.4 ± 0.9 mV) as those prepared from wild-type mice (Fig. 6). Therefore, p62 deficiency did not affect the acute leptin-mediated electrophysiological effect on POMC neurons.
Figure 6.

Leptin-mediated electrophysiological activation of POMC neurons. A, The spontaneous firing of POMC neurons was increased by leptin (50 nm) exposure in both wild-type and p62−/− neurons. Brain slices containing the hypothalamus from POMC-EGFP mice (3–4-weeks-old) were used for whole-cell patch-clamp recording. The recordings show the duration before and after leptin application. Representative traces are shown. B, Changes in membrane voltage (left) and frequency (right) determined from the recordings were not significantly different between wild-type and p62−/− mice. Values represent the mean ± SEM (wild-type, n = 3; KO, n = 8).
Defect in intracellular distribution of the transcription factor Stat3
Leptin activates the transcription factor Stat3 (Vaisse et al., 1996). Subsequent tyrosine phosphorylation and nuclear translocation of Stat3 in hypothalamic neurons leads to important leptin-induced satiety signals. Tyrosine phosphorylation of Stat3 in the hypothalamus, as monitored by immunoblotting, showed a normal response to intraperitoneal injection of leptin (Fig. 7A), and immunostaining of ARH neurons with a Stat3 antibody confirmed Stat3 nuclear translocation after leptin administration in these wild-type mice (Fig. 7B, top). In p62−/− mice, however, Stat3 remained in the cytoplasm after leptin administration (Fig. 7B, bottom). Figure 7C summarizes the scoring of total ARH, POMC, and NPY neurons, showing Stat3 localization in either the cytoplasm or nucleus. p62−/− mice had a defect in Stat3 nuclear translocation in ARH neurons after leptin administration, whereas Stat3 tyrosine phosphorylation was normal. Nuclear accumulation of Stat3 was significantly greater in POMC nucleus neurons in p62−/− mice compared with wild-type controls (Fig. 7C). We found similar phenomenon in cultured MEFs (Fig. 7E). MEFs from p62−/− mice showed much higher accumulation of Stat3 in nucleus after serum starvation for 24 h. Of note, a high-dose leptin injection (5 mg/kg) increased Stat3 nuclear localization in POMC neurons of p62−/− mice (Fig. 7C,D).
Discussion
In this study, we report for the first time that p62 is important for the central regulation of feeding behavior. The p62-deficient mouse exhibits severe leptin resistance that is at least partly due to a defect in the activation of Stat3, an essential factor for leptin (Myers et al., 2008).
A previous study showed that p62 deficiency in mice enhances the differentiation of preadipocytes to mature adipocytes, resulting in increased fat accumulation in adipose tissue (Rodriguez et al., 2006). Our study therefore questions the relative contributions of adipose and brain p62 to body weight control. To examine the role of p62 in the brain, we generated p62flox/flox;Nes-Cre mice. We found that p62flox/flox;Nes-Cre mice gained body weight at a similar rate to p62−/− mice, and both genotypes developed mature-onset obesity and had similar body weights at 40 weeks of age (p62flox/flox;Nes-Cre: 47 ± 2.7 g vs p62−/− mice: 50 ± 4.8 g). However, the onset of obesity in p62flox/flox;Nes-Cre mice was apparently slower than that in null mice (Figs. 2E and 1A). This difference may due to incomplete deletion of p62 in p62flox/flox;Nes-Cre mice. We found very abundant expression of p62-immunoreactivity in the hypothalamus of control mice (Fig. 3A), although previous reports showed very low levels of endogenous p62 in the hypothalamus of wild-type mice. This discrepancy might be due to difference of specificity and affinities of antibodies. We raised an antibody against p62 that has a very high specificity and affinity for p62, and have already published data demonstrating that this antibody specifically detects endogenous p62/A170 (Ishii et al., 1996, 2000; Nakaso et al., 1999, 2000; Sugimoto et al., 2010).
The actions of leptin in the hypothalamus are mediated by a large number of orexigenic and anorectic neuropeptides (Elmquist et al., 1999; Schwartz et al., 2000; Sahu, 2004; Valassi et al., 2008). Recent studies show that leptin triggers signal transduction through its membrane receptors, which leads to specific nuclear changes that include increased Pomc and decreased Npy expression in POMC- and NPY-producing neurons, respectively. In p62−/− mice, expression of Pomc and Npy mRNAs was comparable to that in wild-type mice (Fig. 5), despite their significantly higher blood leptin levels (Fig. 4A,B). However, changes in gene expression induced by fasting were less pronounced in p62−/−mice than in wild-type mice, despite their markedly reduced leptin levels (Fig. 5). p62−/− mice showed decreased sensitivity to the anorectic effect of leptin when administered intracerebroventricularly (Fig. 4C). In addition, ICV injection of MTII or NPY into p62−/− mice had the same negative and positive effects on food intake as that observed in wild-type mice (Fig. 4D,E), suggesting that the downstream signaling pathways in POMC and NPY neurons are normal in p62-deficient mice. From these results, we concluded that the most significant defect of leptin signaling in p62−/− mice occurred in the ARH neurons, which are the primary targets sites of leptin.
The transcription factor Stat3 plays an important role in mediating the transcriptional response that leads to the anorectic effect of leptin (Vaisse et al., 1996). Stat3 in the CNS plays an essential role in the regulation of energy homeostasis and reproduction (Gao et al., 2004). Leptin induces phosphorylation and nuclear translocation of Stat3 to regulate gene expression, and this activation is defective under the leptin-resistant state for a number of reasons (Myers et al., 2008). A defect in Stat3 tyrosine phosphorylation in the hypothalamus after leptin administration was reported in animals with high-fat diet-induced obesity (Münzberg et al., 2004). Moreover, recent studies showed that leptin resistance is partly caused by ER stress in the hypothalamus following a high-fat diet (Hosoi et al., 2008; Zhang et al., 2008; Ozcan et al., 2009). In contrast, the defect in Stat3 activation that we found in young p62-deficient mice was different from that described previously because intraperitoneal injection of leptin into these mice led to normal Stat3 phosphorylation in the hypothalamus (Fig. 7A). However, translocation of Stat3 from the cytoplasm to the nuclei after leptin administration was much lower in the whole ARH neurons of p62−/− mice than of wild-type mice (Fig. 7C). This trend was also observed in POMC and NPY neurons (Fig. 7B,C). Interestingly, POMC neurons in p62−/− mice already have a higher amount of nuclear accumulation of Stat3 compared with wild-type controls (Fig. 7C). In addition, we observed Stat3 accumulation in the nucleus in serum starved MEFs from p62−/− mice (Fig. 7E). Our data also suggest that a high dose of leptin increases Stat3 translocation into nucleus in p62−/− POMC neurons (Fig. 7C,D). These results suggest that p62 is involved in intracellular distribution of Stat3. We speculate that the accumulated Stat3 in the nucleus in p62−/− POMC neurons might prevent the additional nuclear import of cytosolic Stat3 triggered by leptin, potentially accounting for the decrease the sensitivity to leptin.
It is worth noting that a previous study showed decreased Stat3 tyrosine phosphorylation in aged (5 months) obese p62-deficient mice. However, consistent with our results, Stat3 tyrosine phosphorylation was normal in young (3 months) nonobese p62−/− mice (Rodriguez et al., 2006). These results suggest that reduced Stat3 phosphorylation in aged obese p62−/− mice is secondary to obesity.
Although the precise mechanisms underlying regulation of intracellular distribution of Stat3 by p62 remain unknown, Stat3 translocation by cytokines has been extensively studied using cultured cells (Reich and Liu, 2006; Sehgal, 2008). It is proposed that a “signaling endosome” compartment mediates vesicular trafficking from the plasma membrane to the nucleus before transcriptional activation (Howe, 2005). Recent studies indicate that endocytosis of the receptor is required as a primary step for transport of Stat3 from the plasma membrane to the perinuclear region (Bild et al., 2002). Stat3 colocalizes with these receptors in transiting early endosomes (Bild et al., 2002; Shah et al., 2006; Chandra et al., 2008; Kermorgant and Parker, 2008). The transport of activated Stat3 to the nucleus requires importin-αs (Liu et al., 2005; Ushijima et al., 2005) and is mediated by a nuclear transport complex containing MgcRacGAP and GTP-bound Rac1, a member of the Rho family of GTPases (Kawashima et al., 2006, 2009). Interestingly, there are no reports of direct interaction between p62 and Stat3. We also failed to coimmunoprecipitate Stat3 with a p62-specific antibody using extracts from the brain and embryonic fibroblasts of wild-type mice, corroborating the absence of direct interactions of p62 and Stat3.
Previous studies have established that p62 plays important roles in neuronal receptor trafficking and axonal transport of signaling endosomes. Moreover, through direct interactions, p62 facilitates transport of the NGF receptor TrkA (Geetha and Wooten, 2003) and D2-type dopamine receptors (Kim et al., 2008) from the plasma membrane to the endosomes on stimulation by their ligands. Inside the cell, p62 also interacts with AMPA receptor subunits to influence their trafficking and phosphorylation (Jiang et al., 2009) and with GABAc receptor subunit ρ 1–3, forming a ternary postsynaptic complex with PKC-ζ (Croci et al., 2003). The multiple functions of p62 in trafficking and sorting of membrane vesicles may partly be mediated through its interactions with aPKCs and LC3, and both of these proteins localize to endosomes and mediate microtubule and Rab protein binding to dynein motors (Munafó and Colombo, 2002; Gutierrez et al., 2004; Fader et al., 2008; Tisdale et al., 2009). Leptin receptors are internalized in a clathrin-dependent manner after monoubiquitination (Belouzard and Rouille, 2006) and are transported through the endocytic pathway to lysosomes, where they are terminally degraded with their ligands (Barr et al., 1999; Uotani et al., 1999).
As p62 is involved in vesicle trafficking and in the autophagic degradation of ubiquitinated protein aggregates (Kirkin et al., 2009), its deficiency may affect leptin-induced Stat3 nuclear translocation in ARH neurons. Recent reports suggest that loss of hypothalamic autophagy leads to mild obesity in mice due to metabolic dysregulation (Meng and Cai, 2011; Coupe et al., 2012; Kaushik et al., 2012; Quan et al., 2012). It is not clear whether loss of p62 is linked with impaired autophagy, because p62 is not an essential factor for autophagy. Nevertheless, it is possible that selective autophagy may be decreased in the absence of p62 and this might lead to the obese phenotype observed in p62−/− mice. However, it may be possible that other mechanisms contribute to obesity in p62−/− mice because POMC specific autophagy deficient mice only develop mild obesity compared with the p62−/− mice phenotype. Further studies are required to clarify the molecular mechanism underlying p62-mediated regulation of intracellular distribution of Stat3.
Our study has established that p62 is a novel player in appetite regulation and that p62 deficiency in the brain leads to the onset of obesity. p62−/− mice provide a useful model for studying leptin resistance observed in human obesity as well as the leptin-mediated signaling pathway in neurons. If an obesity-related impairment of p62 expression or function can be identified in humans, this would provide a novel therapeutic drug target to combat obesity.
Footnotes
This study was supported by grants from the Uehara Memorial Foundation (to E.W.), the Japan Science and Technology Agency (to T.Y.), the Cabinet Office, Government of Japan through its Funding Program for World-Leading Innovative R&D in Science and Technology (FIRST Program; to T.M.), and The Japan Ministry of Education, Culture, Sports, Science, and Technology (MEXT; to T.Y., 21659459 and T.I., 21500386), Japan Society for the Promotion of Science (JSPS; to E.W., 24790232 and H.H., 21526). We thank Professor Jeffrey M. Friedman for providing POMC-EGFP and NPY-EGFP transgenic mice. We thank Drs Akihiro Yamanaka, Michihiro Mieda, Rika Sugimoto, and Satoshi Sakai for valuable discussions and continuous support throughout this study. We also thank Ms Keiko Yamatsu and Ms. Miki Kiuchi for technical assistance.
References
- Barr VA, Lane K, Taylor SI. Subcellular localization and internalization of the four human leptin receptor isoforms. J Biol Chem. 1999;274:21416–21424. doi: 10.1074/jbc.274.30.21416. [DOI] [PubMed] [Google Scholar]
- Belouzard S, Rouillé Y. Ubiquitylation of leptin receptor OB-ra regulates its clathrin-mediated endocytosis. EMBO J. 2006;25:932–942. doi: 10.1038/sj.emboj.7600989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bild AH, Turkson J, Jove R. Cytoplasmic transport of Stat3 by receptor-mediated endocytosis. EMBO J. 2002;21:3255–3263. doi: 10.1093/emboj/cdf351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra V, Kar-Roy A, Kumari S, Mayor S, Jameel S. The hepatitis E virus ORF3 protein modulates epidermal growth factor receptor trafficking, STAT3 translocation, and the acute-phase response. J Virol. 2008;82:7100–7110. doi: 10.1128/JVI.00403-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coupé B, Ishii Y, Dietrich MO, Komatsu M, Horvath TL, Bouret SG. Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab. 2012;15:247–255. doi: 10.1016/j.cmet.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Croci C, Brändstatter JH, Enz R. ZIP3, a new splice variant of the PKC-zeta-interacting protein family, binds to GABAC receptors, PKC-zeta, and Kv beta 2. J Biol Chem. 2003;278:6128–6135. doi: 10.1074/jbc.M205162200. [DOI] [PubMed] [Google Scholar]
- Durán A, Serrano M, Leitges M, Flores JM, Picard S, Brown JP, Moscat J, Diaz-Meco MT. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev Cell. 2004;6:303–309. doi: 10.1016/S1534-5807(03)00403-9. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Bjørbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998;395:535–547. doi: 10.1002/(SICI)1096-9861(19980615)395:4<535::AID-CNE9>3.3.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. 1999;22:221–232. doi: 10.1016/S0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Fader CM, Sánchez D, Furlan M, Colombo MI. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic. 2008;9:230–250. doi: 10.1111/j.1600-0854.2007.00677.x. [DOI] [PubMed] [Google Scholar]
- Friedman JM. The function of leptin in nutrition, weight, and physiology. Nutr Rev. 2002;60:S1–14. doi: 10.1301/002966402320634878. discussion S68–84, 85–17. [DOI] [PubMed] [Google Scholar]
- Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, Fu XY. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci U S A. 2004;101:4661–4666. doi: 10.1073/pnas.0303992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geetha T, Wooten MW. Association of the atypical protein kinase C-interacting protein p62/ZIP with nerve growth factor receptor TrkA regulates receptor trafficking and Erk5 signaling. J Biol Chem. 2003;278:4730–4739. doi: 10.1074/jbc.M208468200. [DOI] [PubMed] [Google Scholar]
- Gutierrez MG, Munafó DB, Berón W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117:2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Sasaki M, Miyahara T, Hashimoto C, Matsuo S, Yoshii M, Ozawa K. Endoplasmic reticulum stress induces leptin resistance. Mol Pharmacol. 2008;74:1610–1619. doi: 10.1124/mol.108.050070. [DOI] [PubMed] [Google Scholar]
- Howe CL. Modeling the signaling endosome hypothesis: why a drive to the nucleus is better than a (random) walk. Theor Biol Med Model. 2005;2:43. doi: 10.1186/1742-4682-2-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T, Yanagawa T, Kawane T, Yuki K, Seita J, Yoshida H, Bannai S. Murine peritoneal macrophages induce a novel 60-kDa protein with structural similarity to a tyrosine kinase p56lck-associated protein in response to oxidative stress. Biochem Biophys Res Commun. 1996;226:456–460. doi: 10.1006/bbrc.1996.1377. [DOI] [PubMed] [Google Scholar]
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- Jiang J, Parameshwaran K, Seibenhener ML, Kang MG, Suppiramaniam V, Huganir RL, Diaz-Meco MT, Wooten MW. AMPA receptor trafficking and synaptic plasticity require SQSTM1/p62. Hippocampus. 2009;19:392–406. doi: 10.1002/hipo.20528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung I, Strominger JL, Shin J. Molecular cloning of a phosphotyrosine-independent ligand of the p56lck SH2 domain. Proc Natl Acad Sci U S A. 1996;93:5991–5995. doi: 10.1073/pnas.93.12.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Arias E, Kwon H, Lopez NM, Athonvarangkul D, Sahu S, Schwartz GJ, Pessin JE, Singh R. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep. 2012;13:258–265. doi: 10.1038/embor.2011.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Bao YC, Nomura Y, Moon Y, Tonozuka Y, Minoshima Y, Hatori T, Tsuchiya A, Kiyono M, Nosaka T, Nakajima H, Williams DA, Kitamura T. Rac1 and a GTPase-activating protein, MgcRacGAP, are required for nuclear translocation of STAT transcription factors. J Cell Biol. 2006;175:937–946. doi: 10.1083/jcb.200604073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Bao YC, Minoshima Y, Nomura Y, Hatori T, Hori T, Fukagawa T, Fukada T, Takahashi N, Nosaka T, Inoue M, Sato T, Kukimoto-Niino M, Shirouzu M, Yokoyama S, Kitamura T. A Rac GTPase-activating protein, MgcRacGAP, is a nuclear localizing signal-containing nuclear chaperone in the activation of STAT transcription factors. Mol Cell Biol. 2009;29:1796–1813. doi: 10.1128/MCB.01423-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermorgant S, Parker PJ. Receptor trafficking controls weak signal delivery: a strategy used by c-Met for STAT3 nuclear accumulation. J Cell Biol. 2008;182:855–863. doi: 10.1083/jcb.200806076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim OJ, Ariano MA, Namkung Y, Marinec P, Kim E, Han J, Sibley DR. D2 dopamine receptor expression and trafficking is regulated through direct interactions with ZIP. J Neurochem. 2008;106:83–95. doi: 10.1111/j.1471-4159.2008.05348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Komatsu M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 2002;70:1582–1588. doi: 10.1086/340731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, McBride KM, Reich NC. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc Natl Acad Sci U S A. 2005;102:8150–8155. doi: 10.1073/pnas.0501643102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q, Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J Biol Chem. 2011;286:32324–32332. doi: 10.1074/jbc.M111.254417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT, Wooten MW. Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci. 2007;32:95–100. doi: 10.1016/j.tibs.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Munafo DB, Colombo MI. Induction of autophagy causes dramatic changes in the subcellular distribution of GFP-Rab24. Traffic. 2002;3:472–482. doi: 10.1034/j.1600-0854.2002.30704.x. [DOI] [PubMed] [Google Scholar]
- Münzberg H, Flier JS, Bjørbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- Myers MG, Cowley MA, Münzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol. 2008;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- Nakaso K, Kitayama M, Ishii T, Bannai S, Yanagawa T, Kimura K, Nakashima K, Ohama E, Yamada K. Effects of kainate-mediated excitotoxicity on the expression of rat counterparts of A170 and MSP23 stress proteins in the brain. Brain Res Mol Brain Res. 1999;69:155–163. doi: 10.1016/S0169-328X(99)00093-5. [DOI] [PubMed] [Google Scholar]
- Nakaso K, Kitayama M, Fukuda H, Kimura K, Yanagawa T, Ishii T, Nakashima K, Yamada K. Oxidative stress-related proteins A170 and heme oxygenase-1 are differently induced in the rat cerebellum under kainate-mediated excitotoxicity. Neurosci Lett. 2000;282:57–60. doi: 10.1016/S0304-3940(00)00836-3. [DOI] [PubMed] [Google Scholar]
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG, Jr, Ozcan U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9:35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic; 2005. [DOI] [PubMed] [Google Scholar]
- Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- Puls A, Schmidt S, Grawe F, Stabel S. Interaction of protein kinase C zeta with ZIP, a novel protein kinase C-binding protein. Proc Natl Acad Sci U S A. 1997;94:6191–6196. doi: 10.1073/pnas.94.12.6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan W, Kim HK, Moon EY, Kim SS, Choi CS, Komatsu M, Jeong YT, Lee MK, Kim KW, Kim MS, Lee MS. Role of hypothalamic pro-opiomelanocortin neuron autophagy in the control of appetite and leptin response. Endocrinology. 2012;153:1817–1826. doi: 10.1210/en.2011-1882. [DOI] [PubMed] [Google Scholar]
- Ramesh Babu J, Lamar Seibenhener M, Peng J, Strom AL, Kemppainen R, Cox N, Zhu H, Wooten MC, Diaz-Meco MT, Moscat J, Wooten MW. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem. 2008;106:107–120. doi: 10.1111/j.1471-4159.2008.05340.x. [DOI] [PubMed] [Google Scholar]
- Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006;6:602–612. doi: 10.1038/nri1885. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Durán A, Selloum M, Champy MF, Diez-Guerra FJ, Flores JM, Serrano M, Auwerx J, Diaz-Meco MT, Moscat J. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006;3:211–222. doi: 10.1016/j.cmet.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Sahu A. Minireview: a hypothalamic role in energy balance with special emphasis on leptin. Endocrinology. 2004;145:2613–2620. doi: 10.1210/en.2004-0032. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- Sehgal PB. Paradigm shifts in the cell biology of STAT signaling. Semin Cell Dev Biol. 2008;19:329–340. doi: 10.1016/j.semcdb.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M, Patel K, Mukhopadhyay S, Xu F, Guo G, Sehgal PB. Membrane-associated STAT3 and PY-STAT3 in the cytoplasm. J Biol Chem. 2006;281:7302–7308. doi: 10.1074/jbc.M508527200. [DOI] [PubMed] [Google Scholar]
- Shin J. P62 and the sequestosome, a novel mechanism for protein metabolism. Arch Pharm Res. 1998;21:629–633. doi: 10.1007/BF02976748. [DOI] [PubMed] [Google Scholar]
- Sugimoto R, Warabi E, Katayanagi S, Sakai S, Uwayama J, Yanagawa T, Watanabe A, Harada H, Kitamura K, Noguchi N, Yoshida H, Siow RC, Mann GE, Ishii T. Enhanced neointimal hyperplasia and carotid artery remodelling in sequestosome 1 deficient mice. J Cell Mol Med. 2010;14:1546–1554. doi: 10.1111/j.1582-4934.2009.00914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale EJ, Azizi F, Artalejo CR. Rab2 utilizes glyceraldehyde-3-phosphate dehydrogenase and protein kinase Cι to associate with microtubules and to recruit dynein. J Biol Chem. 2009;284:5876–5884. doi: 10.1074/jbc.M807756200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uotani S, Bjørbaek C, Tornøe J, Flier JS. Functional properties of leptin receptor isoforms: internalization and degradation of leptin and ligand-induced receptor downregulation. Diabetes. 1999;48:279–286. doi: 10.2337/diabetes.48.2.279. [DOI] [PubMed] [Google Scholar]
- Ushijima R, Sakaguchi N, Kano A, Maruyama A, Miyamoto Y, Sekimoto T, Yoneda Y, Ogino K, Tachibana T. Extracellular signal-dependent nuclear import of STAT3 is mediated by various importin alphas. Biochem Biophys Res Commun. 2005;330:880–886. doi: 10.1016/j.bbrc.2005.03.063. [DOI] [PubMed] [Google Scholar]
- Vadlamudi RK, Joung I, Strominger JL, Shin J. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem. 1996;271:20235–20237. doi: 10.1074/jbc.271.34.20235. [DOI] [PubMed] [Google Scholar]
- Vaisse C, Halaas JL, Horvath CM, Darnell JE, Jr, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet. 1996;14:95–97. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- Valassi E, Scacchi M, Cavagnini F. Neuroendocrine control of food intake. Nutr Metab Cardiovasc Dis. 2008;18:158–168. doi: 10.1016/j.numecd.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]