

SUMMARY
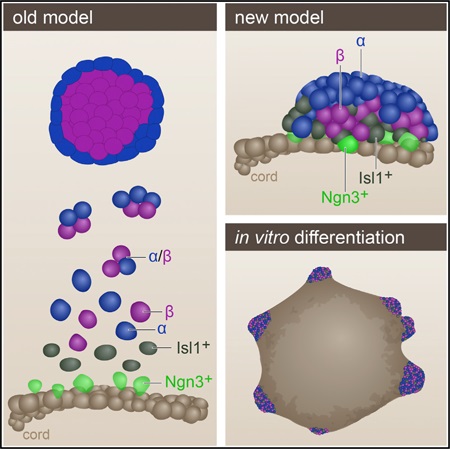
The pancreatic islets of Langerhans regulate glucose homeostasis. The loss of insulin-producing β cells within islets results in diabetes, and islet transplantation from cadaveric donors can cure the disease. In vitro production of whole islets, not just β cells, will benefit from a better understanding of endocrine differentiation and islet morphogenesis. We used single-cell mRNA sequencing to obtain a detailed description of pancreatic islet development. Contrary to the prevailing dogma, we find islet morphology and endocrine differentiation to be directly related. As endocrine progenitors differentiate, they migrate in cohesion and form bud-like islet precursors, or “peninsulas” (literally “almost islands”). α cells, the first to develop, constitute the peninsular outer layer, and β cells form later, beneath them. This spatiotemporal collinearity leads to the typical core-mantle architecture of the mature, spherical islet. Finally, we induce peninsula-like structures in differentiating human embryonic stem cells, laying the ground for the generation of entire islets in vitro.
In Brief
Single-cell spatiotemporal studies of the developing pancreas reveal a tight link between morphology and endocrine cell differentiation, with α and β cells forming layered peninsular structures.
Graphical abstract
INTRODUCTION
Type 1 diabetes (T1D) is caused by the destruction of insulin-producing β cells. These cells reside within the pancreatic islets of Langerhans, which also contain glucagon-generating α cells and small numbers of δ, ε, and PP cells that produce the hormones somatostatin, ghrelin, and pancreatic polypeptide, respectively. Islet transplantation from cadavers has proved highly effective in alleviating T1D (Shapiro et al., 2000), but donor scarcity makes in vitro-generated islets an attractive substitute. Recent success with in vitro generation of insulin-producing β cells and a few other endocrine cell types from human pluripotent cells lends hope to establishing such an alternative source (Pagliuca et al., 2014; Rezania et al., 2011, 2014; Russ et al., 2015). However, increasing evidence indicates that proper glucose regulation requires coordination between the various islet cell types, and the islet’s internal arrangement supports that requirement (Johnston et al., 2016; van der Meulen et al., 2015). It may therefore be advantageous to produce whole islets in vitro rather than differentiating cells into a specific cell type. However, this requires recapitulating tissue morphogenesis in addition to cell differentiation.
In mice, endocrine progenitors appear around E12.5, and their production rate peaks around E15.5, concurrent with an overall expansion of the primordial pancreas termed “the secondary transition” (Pan and Wright, 2011; Zhou et al., 2007). The endocrine progenitors emerge from a network of epithelial tubules, or “cords,” found in the core of the developing pancreas (Bankaitis et al., 2015; Zhou et al., 2007). New progenitors form in a continuous flux and differentiate toward their specific fate in an asynchronous manner (Johansson et al., 2007; Miyatsuka et al., 2009). As cells differentiate, they are thought to undergo epithelial-to-mesenchymal transition (EMT), which renders them motile and allows them to migrate away from the cords, dispersing into the surrounding mesenchyme. Once they have acquired their particular fate and activated their specific hormones, the cells are believed to aggregate into small clusters that later constitute the complete islet (Gouzi et al., 2011; Larsen and Grapin-Botton, 2017; Pan and Wright, 2011; Rukstalis and Habener, 2007; Villasenor et al., 2012).
This dispersal-aggregation model describes how islets form and assemble away from the pancreatic ducts, which are likewise descendants of the epithelial cords. However, the model fails to explain how the differentiated endocrine cells find one another and assemble in the surrounding mesenchyme. Equally important, it does not explain how the islet acquires its final unique architecture. Mature islets in mice are built as an internal core of β cells surrounded by a mantle of α cells. In humans, this core-mantle architecture is maintained in small islets, whereas larger islets are built as composites of small core-mantle components (Bonner-Weir et al., 2015).
With the ultimate goal of reconstructing islet formation in vitro, we set out to examine the changes in gene expression that accompany in vivo islet formation. Because of the asynchronous nature of endocrine differentiation, at any single time point the developing pancreas contains a mixture of cells at varying stages of commitment. Thus, sampling bulks of cells at consecutive time points has a limited ability to tell early gene expression from late.
Therefore, rather than sampling bulks of cells, we sequenced mRNA from single cells as they progress from endocrine progenitors to fully differentiated, hormone-expressing cells. Using computational tools, we constructed a trajectory that, like a map, describes the sequential transcriptional changes that occur as endocrine progenitors form the islet. Contrary to previous models, this map shows that endocrine differentiation and islet morphogenesis are closely linked. By combining the transcriptional map with visual analysis of the developing pancreas, we present a new explanation for the process that shapes the islets of Langerhans.
RESULTS
Mapping Early Pancreatic Islet Formation Using Single-Cell RNA Sequencing
Endocrine differentiation from epithelial cords is first marked by expression of the bHLH transcription factor Neurogenin3 (Ngn3) (Gu et al., 2002). To isolate differentiating islet precursors for single-cell mRNA sequencing (scRNA-seq), we used Ngn3-eGFP reporter mice in which the Ngn3 coding region was replaced with eGFP (Ngn3-eGFP+/−) (Lee et al., 2002). Homozygous mice lack islets and die close to birth, but heterozygous mice are viable and form a normal endocrine system. Ngn3 expression is transient and has a short half-life, so eGFP protein persists in the cells well after endogenous Ngn3 is no longer detected (Miyatsuka et al., 2009). Thus, eGFP+ cells include not only the early Ngn3+ endocrine progenitors but also older Ngn3− cells that have recently expressed the gene (Figure 1A).
Figure 1. Mapping Pancreatic Islet Formation Using Single-Cell RNA Sequencing.
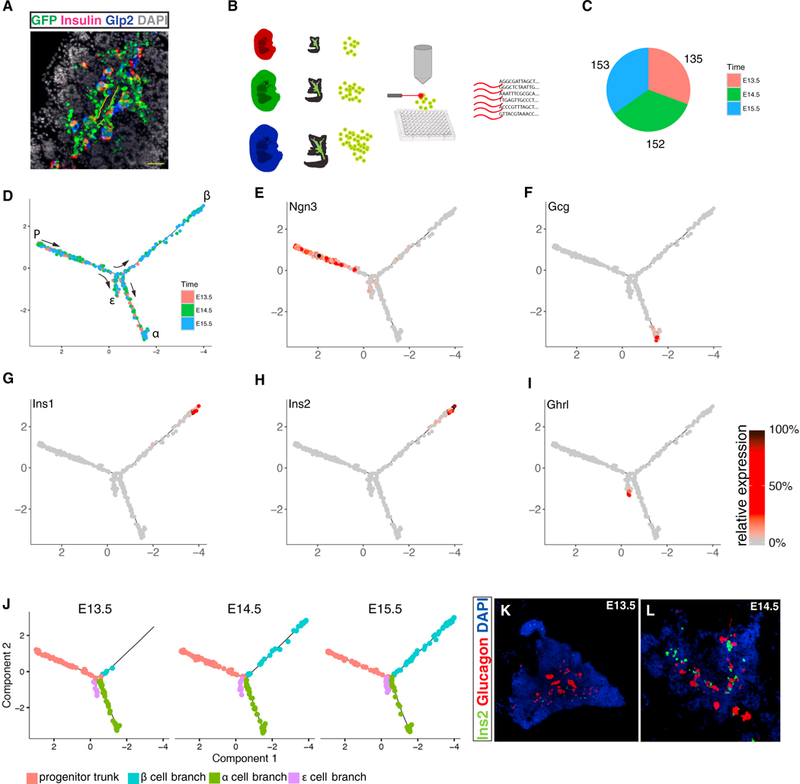
(A) Immunostaining of a cross section through E15.5 pancreas from a Ngn3-eGFP+/− embryo. Notice that eGFP first appears where Ngn3+ cells emerge, at the periphery of the epithelial cords (outlined with yellow dashed line), and persists long enough to be co-expressed with Insulin or Glucagon (detected by an antibody to its processing byproduct, Glp2).
(B) Experimental outline: a pancreas from a single Ngn3-eGFP+/− mouse embryo was removed on E13.5 (red), E14.5 (green), and E15.5 (blue) days post-coitum. Single eGFP+ cells were sorted into 96-well plates, and single-cell cDNA libraries were generated.
(C) Altogether, 440 single cells were suitable for further analysis. Colors represent the developmental day of cell collection.
(D) Monocle arranged the sequenced cells into a tree-shaped branched trajectory. Colors represent the developmental day of cell collection.
(E–I) Each cell is colored according to its relative expression level of the indicated gene: (E) Ngn3, (F) Gcg, (G) Ins1, (H) Ins2, and (I) Ghrl.
(J) Skeletons of the developmental tree dressed only with the cells collected on a given day. Notice the absence of insulin-expressing cells at E13.5.
(K and L) In situ hybridization of sections through embryonic pancreata at E13.5 (K) and E14.5 (L) shows that Ins+ cells start becoming abundant at E14.5. Tiling of multiple high-resolution images.
Pancreata were extracted from heterozygous Ngn3-eGFP+/− embryos on E13.5, E14.5, and E15.5 post-coitum. Following dissociation, single Ngn3-eGFP+/− cells were fluorescence-activated cell sorted (FACS) into 96-well plates and sequenced (Figure 1B). After applying various filtering steps (see STAR Methods), a total of 440 cells were retained for subsequent analysis (Figure 1C).
It was previously shown that single-cell trajectory analysis can accurately recover gene expression kinetics of differentiating cells even when they are highly asynchronous (Trapnell et al., 2014). Accordingly, we used Monocle 2 (Qiu et al., 2017a, 2017b) to draw a developmental map of the islet on the basis of the scRNA-seq data (Figure 1D). Briefly, each cell’s multi-dimensional representation is projected into a two-dimensional (2D) plane so that the distance between cells correlates with their similarity in gene expression. Next, in an unbiased fashion, Monocle finds a tree that fits within the data such that each cell is placed close to some segment of the tree. Tracking the cells along the tree from “root” to “leaves” provides a stepwise description of the minute consecutive transcriptional changes a cell undergoes as it differentiates.
Monocle arranged the cells in a trajectory that contains a trunk and three branches (Figure 1D). Ngn3, which marks early progenitors, was highly expressed exclusively in what was defined as the trunk (Figure 1E). In the three branches, cells at the distant tips expressed a distinct hormone: Glucagon (Figure 1F), Insulin (Figures 1G and 1H), or Ghrelin (Figure 1l). We therefore interpret the trajectory as a developmental map that describes how the Ngn3+ progenitors in the trunk proceed along one of three optional paths toward α, β, or ε cell fates (Figure 1D). The area of the tree where the branches emerge from the trunk represents the developmental window of lineage specification. PP+ and Sst+ cells, which are relatively rare in adult islets (Baron et al., 2016), were not detected in sufficient numbers to confidently generate additional distinct branches.
α Cells Appear Earlier than β Cells
Cells collected at all 3 days are found in both the α and ε branches, whereas the β cell branch is occupied only from day 14.5 onward (Figure 1J). Accordingly, RNA fluorescent in situ hybridization detects large numbers of insulin+ cells on day 14.5 but not on E13.5 (Figures 1K and 1L). Thus, the branched trajectory agrees with previous reports showing that α cells form earlier than β cells (Johansson et al., 2007) and sets a time frame of ~24 h for this interval.
Topic Modeling Reveals Temporal Gene Expression Programs
The trajectory arranges the cells according to their relative position along a presumed developmental path, regardless of the age of the embryo from which they were collected (Figure 1D). This establishes a virtual timescale, or “pseudotime.” Monocle assigns pseudotime values to each cell, from 0 for the first cell at the base of the trunk to 10 for the cell farthest away (Figure 2A). Around pseudotime 5, the trajectory bifurcates, and cells progress concurrently along one of three independent pseudotime axes. The transcriptional expression of a gene can now be plotted as a function of pseudotime, either across all cells (Figure 2B) or specifically along each of the three developmental branches (Figure 2C). In actual time, we estimate the entire process described by the map to last no more than 24 hr, in agreement with previous reports (Figure S1) (Bankaitis et al., 2015).
Figure 2. Topic Modeling Reveals Pseudotime-Dependent Gene Expression Programs.
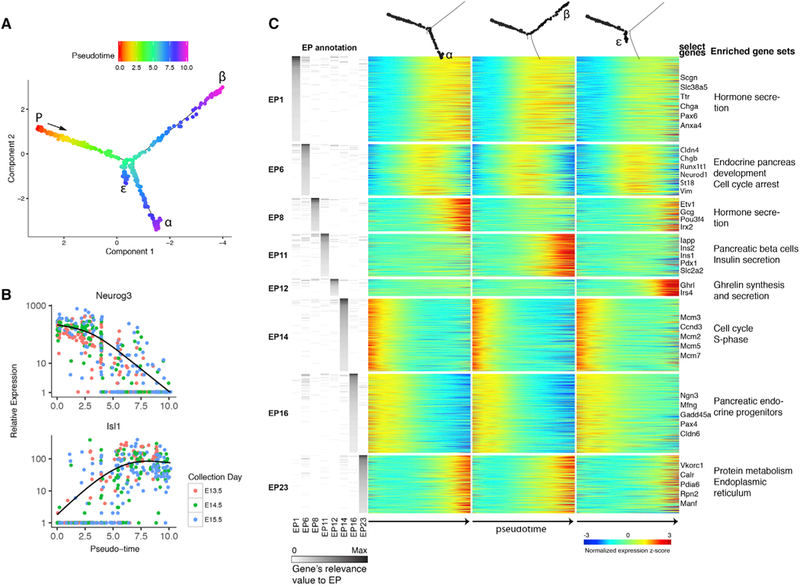
(A) Pseudotemporal annotation of the pancreatic endocrine developmental tree. “P” (progenitor): earliest cell In pseudotime. Notice bifurcation around pseudotime = 5.
(B) Expression level of Ngn3 (top) and Isl1 (bottom) in each cell is plotted against the cell’s position in pseudotime. Cells are colored by the actual time of collection. Negative binomial regression curves illustrate the gene’s expression trend over pseudotime.
(C) Topic modeling analysis identifies eight pseudotime-dependent gene expression programs (EPs). A multi-way heatmap shows pseudotemporal gene expression changes separately for each of the three branches (columns). Rows represent genes with non-zero relevance values for either of the eight pseudotemporal EPs. Left: genes’ relevance values to EPs. Right: representative genes and select enriched gene sets. See also Figures S1 and S2.
To characterize gene expression changes that occur during endocrine differentiation, we combined the pseudotime framework generated by Monocle with Topic Modeling (TM), a probabilistic, unsupervised learning algorithm (Blei, 2012; Gerber et al., 2007; Teh et al., 2006). In TM, genes that are frequently expressed together in a cell are grouped into “expression programs” (EPs). For each EP, every gene receives a “relevance value” that describes how well it represents the entire group. The relevance value increases in correlation with the gene’s tendency to appear with other members of the EP in the same cells. Conversely, if a gene is frequently expressed together with genes from other EPs, its relevance value for the first EP will decrease. Cells, on the other hand, receive “usage values” according to their tendency to use genes from a given EP. This way, rather than limiting each gene or cell to a single cluster, TM allows a gene that appears in different contexts to take part in defining several EPs. Accordingly, each cell may use several EPs.
TM recognized 22 EPs used by groups of more than 50 cells (Figure S1D; Table S1). Of these, 8 were used by cells in a pseudotime-dependent manner (Figures S1E and S2A). These “pseudotemporal EPs” represent groups of genes that are coordinately regulated as a cell differentiates (Figure 2C).
A Switch between Gene EPs Marks the Onset of Cell Specification and Cellular Translocation away from Epithelial Cords
Five of the pseudotemporal EPs (1, 6, 14, 16, and 23) include genes with a similar expression pattern along the three branches (Figures 2C and S2A). EP #14 genes were usually detected in the very early cells and downregulated thereafter. Gene set enrichment analysis (GSEA) showed that this EP contains many cell cycle-related genes, including multiple members of the pre-S-phase Mcm family (Figures 2C and S2A; Table S2). Conversely, genes in EP #23 are activated at the highest levels only in the very late pseudotime cells and are enriched for factors related with the endoplasmic reticulum (Figures 2C and S2A; Table S2). Together, these two EPs demarcate a developmental map of endocrine differentiation from its onset, soon after the exit from the last division of the parent cell (Miyatsuka et al., 2011), through the formation of the secretory mechanisms in the differentiated hormonal cells.
Combined, EPs #1, #6, and #16 are used by all cells analyzed (Figure 3A). EP#16 is primarily used in early cells (Figure 3A) and is enriched for genes related with the endocrine progenitor state, including Ngn3 and Pax4 (Figures 3B, S2A, and S2B). Conversely, EP #1 is used by all cells in late pseudotime (Figures 3A, S2A, and S2B) and is defined by pan-endocrine genes relevant to hormone secretion, including Scgn, Isl1, Chga, and Pax6 (Figures 3B and S2A; Table S2). EP #6 shows an intermediate temporal pattern and peaks in cells that are positioned in pseudotime close to the bifurcation point (Figure 3A). The gene with the highest relevance value for this EP is the transitionally expressed Cldn4, but most top genes that define EP #6 do not show an expression pattern unique to the intermediate stage (Figures 3B, S2A, and S2B). Rather, most of the genes in EP #6 are either early genes that are still not completely downregulated or late genes in their initial stages of activation. This way, some of the genes that define EP #6 appear also in the early EP #16 (e.g., Pax4, Stmn1) or the late EP #1 (e.g., Chga, Pax6).
Figure 3. Switch from Endocrine-Progenitor to Pan-endocrine EPs.
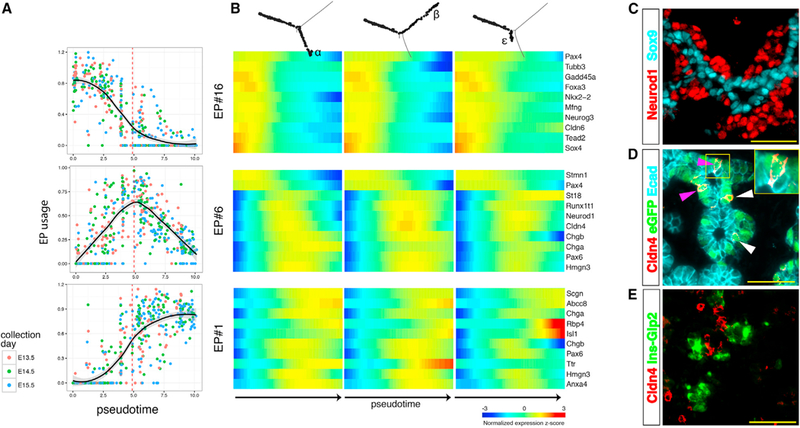
(A) Each cell’s usage values for the major pseudotemporal EPs is plotted against the cell’s position in pseudotime. A vertical dashed line marks bifurcation pseudotime.
(B) A heatmap describes the expression pattern of representative genes from EPs #16, #6, and #1.
(C–E) Immunofluorescent staining of E15.5 pancreas.
(C) Neurod1+ cells are outside of the Sox9+ epithelial cords.
(D) Cldn4 is expressed in GFP+ cells on the rim of the cord (white arrowheads) and in GFP+ cells embedded in the epithelial cord (purple arrowhead; see inset), as indicated by proximity to the lumen.
(E) Cldn4 is not expressed in cells that express either insulin or glucagon. Scale bars, 50 μm. See also Figures S1, S2, and S3.
The paucity of exclusively intermediate genes indicates that endocrine differentiation involves an abrupt transcriptional switch from a progenitor EP to a pan-endocrine one. In pseudotime it corresponds to the bifurcation point in the Monocle tree that marks the beginning of cell specification (Figures 2A and 3A). Altogether, this means that progenitor cells have a uniform expression signature regardless of their prospective fate, and cell heterogeneity first appears together with the switch to the pan-endocrine state.
Last, cells expressing Ngn3 are found in the epithelial cords (Figure S3A). However, by the end of the differentiation process, the cells occupy a region outside of the cords (Figures 3C and S3B). We identified Cldn4 expression in cells still embedded in the epithelial cords and in cells expressing GFP on the outer rim of the cord (Figures 3D and S3C). However, Cldn4 is never detected together with either Ins or Gcg (Figures 3E, S3D, and S3E). Thus, the transitional state marked by Cldn4 expression delimits the physical translocation of the differentiating cells, from within the ductal system toward the surrounding mesenchyme.
Gradual Accumulation of Cell-Type-Specific Gene EPs
Each of the remaining three pseudotemporal EPs (#8, #11, and #12) is used by cells positioned along a specific branch (Figure 4A). EP#11 is used by cells that follow the path toward β cells (Figures 4A and S2A), and it includes Ins1, Ins2, lapp, and Pdx1 (Figures 2C and S2C). Cells that use EP #8 are positioned along the α cell path and express Gcg, Gast, Etv1, and Pou3f4 (Figures 2C and S2C). EP #12 is enriched in genes that mark ε cells, including Ghrl and Mboat4 (Figures 2C and S2C; Tables S1 and S2), and it is used by cells along the equivalent branch (Figures 4A and S2A). Additionally, we identified genes previously unknown to be related with specific cell types. These include Arhgap36 and the long non-coding RNA GM27033, which are enriched in the β cell path; the α cell-specific genes Pou6f2 and Nxph1; and Irs4, which is ε cell specific (Figure S2C).
Figure 4. Analysis of Branch-Specific EPs Reveals Sequential Gene Activation during Endocrine Cell Specification.
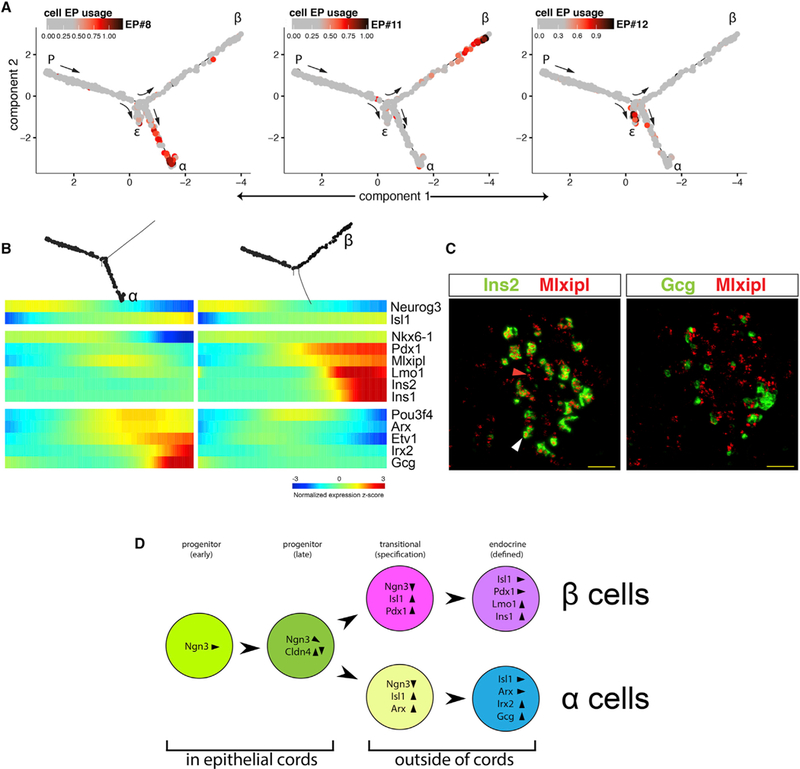
(A) Each cell in the developmental trajectory is colored according to the EP usage value for a given cell-type-specific EP.
(B) Pseudotemporal heatmaps of select α cell (left) and β cell (right) branch-specific genes. Notice the additive activation of factors with time.
(C) In situ hybridization of E15.5 embryonic pancreas. Mlxipl transcripts are detected in Ins2+ β cells (white arrowhead) and in cells that are still Ins2− (red arrowhead). Mlxipl is not expressed in Gcg+ cells. Scale bar, 50 μm.
(D) Stepwise endocrine differentiation. Early progenitors express high levels of Ngn3. While still embedded in the epithelial cords, Ngn3 expression declines, and Cldn4 is expressed transiently. As Ngn3 is downregulated, the pan-endocrine marker Isl1 is upregulated. Concurrently, lineage-specific patterns emerge, the cells leave the epithelial cord, and Cldn4 is downregulated. In the α cell lineage Arx is upregulated, whereas in β cells Pdx1 expression is dramatically increased. Finally, the cell-specific hormones appear together with additional lineage-specific transcription factors. See also Figure S2C.
The pseudotemporal behavior of the identified genes revealed a lapse between the first appearance of cell-type-specific genes and the expression of hormones (Figure 4B). In the β cell path, both Pdx1 and Mlxipl expression levels rise prior to those of Lmo1 and Insulin (Figures 4B and 4C). In α cells, Pou3f4, Etv1, and Arx are upregulated first, while Irx2 appears later, together with high Gcg expression (Figure 4B). In both α and β lineages, but not in ε cells, Ripply3 marks the final, hormone+ phase (Figure S2C). Thus, unlike the abrupt transition to the endocrine state, activation of the cell-type-specific gene EP is gradual.
In all, the pseudotemporal analysis provides a detailed profile of the changes in gene expression undertaken by differentiating endocrine cells. Ngn3 expression levels are high in the early progenitors and decline as specification commences and the cells enter a transitional phase. As cells acquire their new fate, they turn on pan-endocrine markers such as Chga and Isl1. Simultaneously, they upregulate early lineage-specific genes followed by activation of lineage-specific hormones (Figure 4D).
Differentiating Endocrine Cells Migrate in a Coordinated Fashion and Form a Peninsula
Once fully differentiated, hormone+ cells are thought to continue migrating as individual mesenchymal cells and aggregate into islets. To analyze this migration in depth, we set out to determine the pseudotemporal window for the expression of the canonical EMT marker vimentin (Vim) (Nieto et al., 2016; Thiery et al., 2009). Unexpectedly, Vim mRNA expression pattern was found to be similar to that of Cldn4, the marker of the pre-hormonal intermediate cells leaving the cords (Figures 5A and 3D). Vim expression peaks during the transitional phase and is lost right before hormones are turned on. Accordingly, Vim protein shows partial overlap with the EP #6-related intermediate to late marker Syp (Figure S4B), but both Vim mRNA and protein are absent from cells expressing either insulin or glucagon (Figures 5B, 5C, and S4A). The absence of Vim from hormone+ cells suggests that by the time the cells are fully differentiated, they are no longer mesenchymal and are thus unable to migrate freely and aggregate into islets. If so, this is not consistent with the notion that islet formation is mediated through EMT and the aggregation of fully differentiated hormone+ cells.
Figure 5. Islets Form as Peninsulas, Not through EMT.
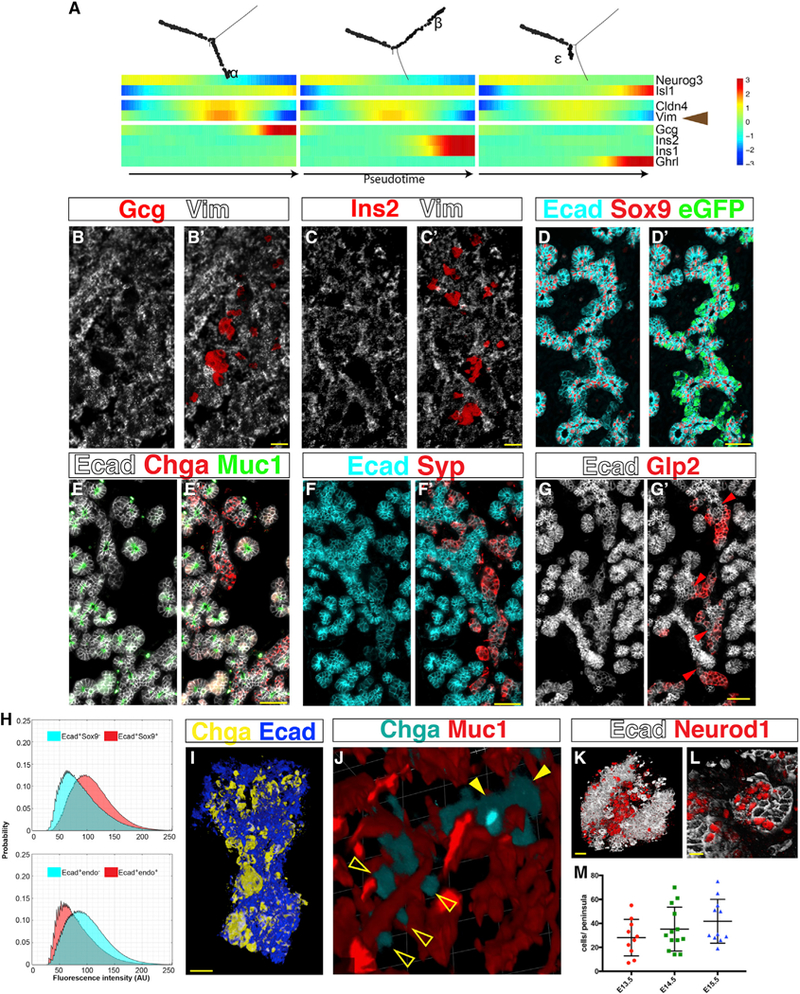
(A–G) Islet formation does not involve EMT.
(A) A multi-way heatmap shows that Vim mRNA transcripts disappear right before hormones are expressed.
(B and C) In situ hybridization of an E14.5 embryonic pancreas shows that Vim transcripts are absent from Gcg+ (B) and Ins2+ (C) cells. Scale bars, 25 μm.
(D–G) Immunofluorescent staining shows that differentiating endocrine precursors maintain Ecad expression and remain cohesive.
(D) E14.5 pancreas. Ecad is high in Sox9+ epithelial cords and low in Sox9−eGFP+ regions. Notice no spatial gaps between the two populations.
(E) E15.5 pancreas. Ecad is high in the epithelial cords, identified by Muc1 on their apical surface (the inner lining of the tubular structure), and low in the Chga+ endocrine domain.
(F) E16.5 pancreas. Notice low Ecad expression in the Syp+ endocrine domain and continuity between Ecadhigh and Ecadlow regions.
(G) E14.5 pancreas. Gcg+ (Glp2+) cells express Ecad and are connected to the epithelial cords via an Isthmus (red arrowheads), marked by continued expression of Ecad. Scale bars, 50 μm.
(H) Average intensity distributions of Ecad immunofluorescent labeling. Top: Ecad pixel intensity in the Sox9+ cords is higher than in the Sox9− regions (summary of images taken at E13.5, E14.5 and E15.5. p = 0.014, n = 3). Bottom: Ecad pixel intensity in areas that express an endocrine marker (Chga, Syp, or Glp2) is lower compared with non-endocrine areas (p = 0.022, n = 3).
(I–M) Spatial analysis of endocrine bud (“peninsula”) organization; see also accompanying 3D panoramas in Video S1.
(I) Three-dimensional image of whole-mount immunostaining of E15.5 pancreas followed by tissue clearing. Chga+ peninsulas bud from the epithelial cord. Scale bar, 30 μm.
(J) Three-dimensional rendering of whole-mount stained E16.5 pancreas shows the distribution of Chga+ peninsulas along the Muc1+ interior lining of the epithelial cords. Empty arrowheads, individual peninsulas. Filled arrowheads, aggregates of peninsulas.
(K and L) Three-dimensional visualization of cleared embryonic pancreata shows clusters of Neurod1+Ecadlow endocrine cells forming peninsulas that bud out of the Ecadhigh epithelial cords.
(K) E13.5 embryo. Scale bar, 30 μm.
(L) E14.5 embryo. Scale bar, 20 μm.
(M) Peninsula size was estimated by the number of Neurod1+ cells identified within clusters in whole-mount stained and cleared embryonic pancreata. N ≥ 10. See also Figures S4, S5, and S6 and Video S1.
In contrast to Vim, E-cadherin (Ecad) marks the epithelial state and is downregulated during EMT (Nieto et al., 2016; Thiery et al., 2009). As expected, high levels of Ecad are detected on the membranes of the epithelial cords (Figures 5D, 5H, and S5A). Furthermore, in agreement with previous reports (Gouzi et al., 2011), we found that Ecad is not completely lost during endocrine differentiation. Rather, islet precursors continue presenting low levels of Ecad on their membranes throughout differentiation, and regions that express advanced endocrine markers are connected to the epithelial cords through an Ecad+ isthmus (Figures 5D–5H and S5). Finally, transcripts of other EMT-related genes (Zeb1, Ncad, αSMA, and Slug) did not correlate with Vim’s pseudotemporal pattern, and their protein was detected mostly outside of Ngn3-GFP+ cells (Figure S4C).
Altogether, these data show that EMT is not a part of the islet-forming process, and the persistence of Ecad expression means that neither dispersal nor aggregation of individual cells in the surrounding mesenchyme occurs during islet formation.
In the absence of dispersal and aggregation, we hypothesized that each islet forms as differentiating cells leave the epithelial cords while maintaining cell-to-cell contacts, thus creating a cohesive bud-like structure. To test this hypothesis, we visualized embryonic pancreata in three dimensions using whole-mount immunostaining and tissue clearing. Indeed, we noticed that emerging endocrine precursors remain cohesive and form Ecad+ buds (Figure 5I). These structures are distributed non-uniformly along the cords throughout the pancreas, mainly at its core (Figure S6A). In some areas, adjacent buds create a continuous sheath wrapping around the forming ducts (Figure 5J), and occasionally endocrine buds originating from two separate cords merge at the tip (Figure S6B; Video S1). The size of Neurod1+ clusters shows that buds grow as differentiation proceeds: E13.5 buds are made of fewer than 30 cells on average, whereas an individual cluster at E15.5 may contain more than 70 cells (Figures 5K–5M and S6C–S6E). We suggest naming these transitional structures “endocrine peninsulas” (from Latin, “almost island”).
Spatiotemporal Collinearity in the Forming Peninsula Shapes the Internal Architecture of the Islet
The realization that islets start as cohesive peninsulas necessitates reevaluation of the hypothesis that islet architecture is established through aggregation of individually migrating cells. Combining our recent findings with previous reports by others, we created an inclusive model to describe islet development (Figure 6A). The basic premises of the model are as follows: (1) Islets form in a budding process. Cells that recently activated Ngn3 leave the epithelial cord and remain attached to one another. (2) The forming bud, or peninsula, remains attached to the cord as it develops. (3) Differentiating endocrine precursors do not divide, and therefore peninsular expansion relies on the recruitment of new cells from the epithelial cord. (4) In each peninsula, α cells appear before β cells. (5) Different endocrine cell types have different adhesion properties (Esni et al., 1999; Rouiller et al., 1991). (6) Before birth and in the first days afterward, the rate at which new Ngn3+ progenitors appear wanes (Solar et al., 2009), while immature differentiated cells proliferate (Georgia and Bhushan, 2004).
Figure 6. Spatiotemporal Collinearity during Peninsula Growth.
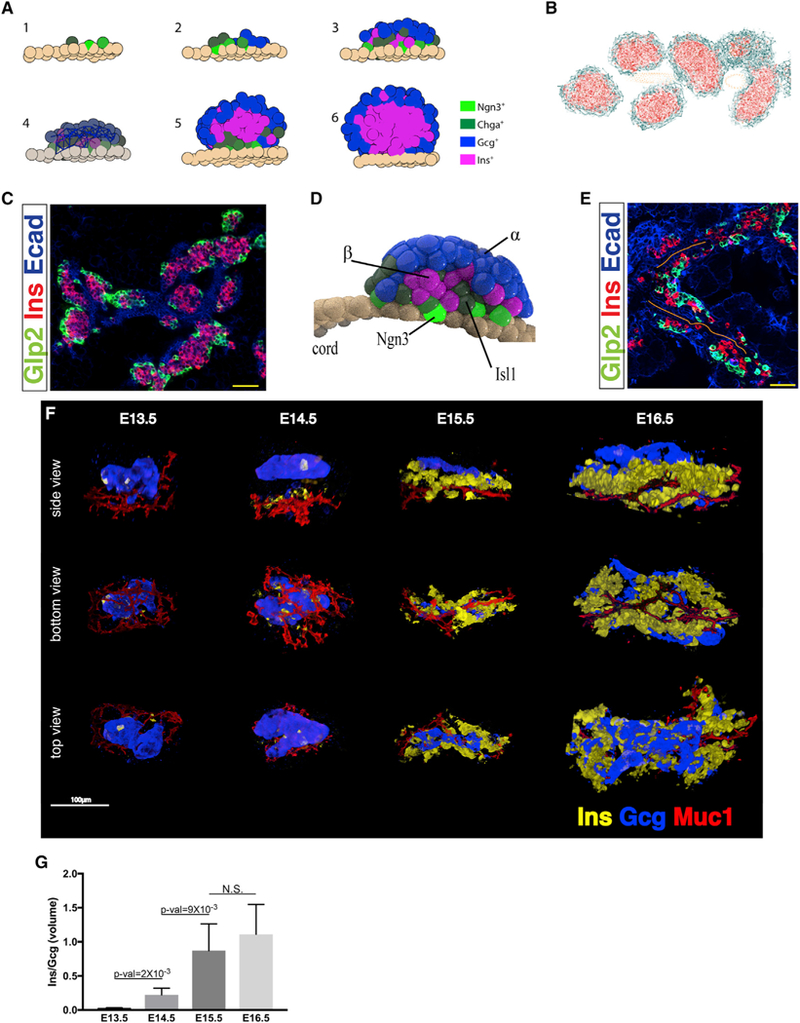
(A) Comprehensive model for islet development. (1)Ngn3+ cells emerge from dividing cells in the epithelial cord and remain attached to its outer surface. (2) Newly formed endocrine precursors push the older cells outward, expanding the peninsula. (3) α cells appear first at the tip of the peninsula. β cells form later and create a layer between the α cells and the cord. (4) Differential adhesion properties maintain the initial cell-type segregation throughout islet development. (5) Close to birth, the production rate of new Ngn3+ precursors wanes. (6) Around birth cell division is resumed in differentiated endocrine cells, and the islets acquire a spherical shape.
(B) Simulation of the peninsular model “grows” 3D islets in silico. A virtual cross section at the end of the simulation shows islets (red, β cells; cyan, α cells) closely aggregated around the cords (beige). Lines mark pairs of connected cells. See also Video S2.
(C) Immunofluorescence staining of a cross section through a postnatal pancreas (P0) shows aggregates of islets appearing as pearls on a necklace. Scale bar, 50 μm.
(D) The model-based simulation predicts that peninsulas will acquire a flattened-cap shape, with β cells sandwiched between α cells and the epithelial cord.
(E) Immunofluorescent staining of a thick (100 μm) section through an E16.5 pancreas shows stratification of the forming peninsula. α cells appear at the outer edge, whereas β cells are closer to the epithelial cords. Orange lines mark the center of the tubular epithelial cords. Scale bar, 50 μm.
(F) Whole-mount staining of embryonic pancreata was followed by tissue clearing and multi-photon microscopy imaging. Side, bottom, and top views of a peninsula at days 13.5–16.5. Muc1 marks the inner lining of the epithelial cord. The cell bodies of the cord are therefore missing. Notice β cells sandwiched between the cords and α cells. See also 3D panoramas in Videos S3, S4, S5, and S6.
(G) Volumetric analysis of 3D imaged peninsulas shows a daily increase in the volume of β cells relative to α cells. Student’s t test, n ≥ 3. See also Figure S6 and Videos S2, S3, S4, S5, and S6.
The limited movement of adhesive cells means that in the growing peninsula, older cells are pushed outward by emerging progenitors (Figure 6A3). Thus, the spatial position of a cell is expected to reflect its temporal state, and the first cells to emerge from the cord (α cells) will constitute the outer layer of the peninsula. Differential cell adhesion properties are assumed to maintain this initial segregation throughout development.
To test this model, we simulated the dynamics of peninsula development in time and space using three-dimensional (3D) animation software (Video S2; STAR Methods). On the basis of the model’s assumptions, the simulation was able to create whole islets in silico, starting with dividing cells in the epithelial cords and ending with spherical islets with the typical coremantle architecture (Figure 6B). Because the model claims that islets form in vicinity to the epithelial cords, the simulation gave rise to multiple islets arranged densely as “pearls on a necklace,” in accordance with islet arrangement around birth (Miller et al., 2009) (Figures 6B and 6C).
A major prediction of the peninsular model, as presented by the simulation, is that nascent peninsulas form a bi-layered structure, with α cells lying on top of β cells and facing away from the epithelial cords (Figure 6D). This arrangement can be observed in histological sections of embryonic pancreata (Figure 6E), but their detection depends on the angle of sectioning. To properly test peninsular architecture, we analyzed the 3D structure of embryonic pancreata using whole-mount immunostaining followed by tissue clearing. In agreement with the early appearance of α cells, E13.5 peninsulas are composed almost entirely of α cells, and β cells appear sporadically (Figures 6F, 6G, S6F, and S6G; Video S3). By the next day, the number of β cells in peninsulas increases significantly, but they still constitute less than 25% of peninsular volume (Figures 6F, 6G, S6F, and S6G; Video S4). In the following 2 days there is a further increase in β cell volume, which in some of the analyzed peninsulas exceeded that of α cells (Figures 6F, 6G, S6F, S6G; Videos S5 and S6).
In agreement with the model, peninsulas appear cap-shaped and not spherical like mature islets (Figure 6F; Videos S3, S4, S5, and S6). Furthermore, as predicted, nascent peninsulas have a bi-layered structure, which appears first with the detection of β cells at day 14 and becomes more prominent at day 15, when β cell mass increases. Cell populations are segregated, with β cells sandwiched between the epithelial cords on one side and α cells on the exterior of the peninsula (Figure 6F).
Altogether, this model ties together the order of cell fate determination and islet architecture and accurately predicts spatiotemporal collinearity during islet formation: the earlier a cell differentiates, the more peripheral it will be.
Generation of hESC-Derived Human Endocrine Peninsulas In Vitro
The peninsular organization of islet formation in vivo raises the question of whether a similar structure can be generated in vitro from pluripotent cells. We used a 3D protocol for the differentiation of human embryonic stem cells (hESCs) into β-like cells (Pagliuca et al., 2014) and focused on the stage when endocrine cells first appear in the spheroid clusters (stage 4). Activation of the TGF-β pathway was previously reported to induce endocrine bud formation from epithelial rudiments of rat embryonic pancreata. These rudiments were embedded in collagen, and the buds were assumed to result from re-aggregation of migrating cells (Miralles et al., 1998). However, addition of the TGF-β family member ActivinA to hESC-derived clusters differentiating in suspension results in the shedding of cells from the spheroids (Figure 7A). We speculated that these are indeed differentiating endocrine cells, which, once removed from the bulk of the cluster, do not re-aggregate. Considering its effect on cell adhesion, we added the Rho kinase inhibitor (ROCKi) Y-27632 (Riento and Ridley, 2003) to the ActivinA treated clusters. This prevented the shedding of cells and promoted the appearance of bud-like protrusions on the otherwise smooth surface of the clusters (Figure 7A; Video S7). These buds express the pan-endocrine marker CHGA (Figure 7B) and contain cells expressing the hormones SST, GCG, and INS (Figures 7C and 7D). Many of the hormone+ cells are polyhormonal, which indicates they are not fully differentiated. In the absence of ActivinA, ROCK inhibition did not induce extensive bud formation (Figure 7A). Altogether, this shows that hESCs can be induced to recapitulate formation of endocrine peninsulas in vitro and that this process requires maintenance of cell adhesion rather than re-aggregation of dissociated cells.
Figure 7. Induction of Human Peninsula-like Structures In Vitro by Directed Differentiation of Human Embryonic Stem Cells.
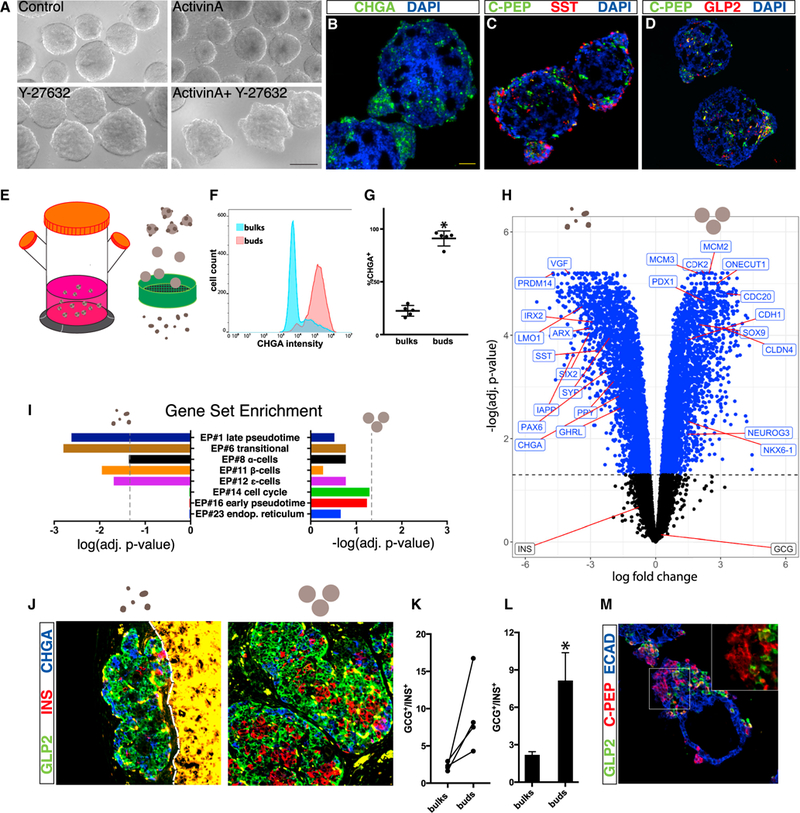
(A) HUES8 cells differentiated to pancreatic progenitors form smooth spheroids at stage 4, when the first endocrine cells appear. ActivinA alone induces cell shedding. ActivinA together with Y-27632 induces buds. Scale bar, 200 μm.
(B–D) Immunofluorescence staining of cross-sections through clusters derived from the directed differentiation of hESCs into β-like cells (SC-β): (B) CHGA,
(C) C-PEP (byproduct of insulin processing) and SST, and (D) C-PEP and GLP2.
(E) SC-β clusters at early stage 5 were mechanically agitated to break the buds off. Buds and bulks were size separated using sieves.
(F and G) Flow cytometry analysis on bulks and buds immediately after mechanical separation.
(F) Representative distribution of CHGA intensity in cells dissociated from either bulks or buds.
(G) Relative abundance of CHGA+ cells in bulks and buds. P = 4.97e−5, n = 5, Student’s t test.
(H) RNA-seq comparison of isolated buds and bulks (n = 3 and n = 5, respectively, from five separate differentiation batches). Significantly differentially expressed genes are colored in blue (adjusted p < 0.05). Genes enriched in buds are to the left, genes enriched in bulks are to the right.
(I) Gene set enrichment analysis on the differentially expressed genes was performed using gene sets defined based on the top 100 genes of each EP. Significance of enrichment in buds and bulks is illustrated to the left and right, respectively. Dashed lines, adjusted p = 0.05. False discovery rate (FDR) < 10%.
(J–L) Continued differentiation of isolated cluster compartments after transplantation to the kidney capsule of immunodeficient mice.
(J) Immunofluorescent staining of a section through a kidney transplanted with bulks (right) or buds (left) grown in culture priorto transplantation. A dashed line marks the boundary between the kidney and the transplant (kidney appears yellow because of autofluorescence).
(K and L) Quantification of the ratios between GCG+ α-like cells and INS+ β-like cells in grafts.
(K) Buds and bulks from the same in vitro batch are matched.
(L) Summary of transplantations (geometric mean ± SE). p = 0.028, n = 4, Student’s t test on the log of GCG/INS ratios.
(M) Immunofluorescent staining of a cross-section through a bulk grown in Matrigel. Notice a bud containing multiple monohormonal INS+ (C-PEP+) cells. Inset: INS+ cells are arranged densely, as in normal islets. See also Figure S6 and Video S7.
Isolation and Characterization of In Vitro-Generated Human Endocrine Peninsulas
At the next stage in the differentiation protocol (stage 5), we separated the buds (20–100 μm diameter) from the bulk of the spheroids (>200 μm diameter) by mechanical agitation followed by size separation (Figures 7E and S6H). Flow cytometry analysis revealed that on average, CHGA is expressed in ~25% of the cells in the bulks and in more than 90% of the cells in the buds (Figures 7F and 7G). Similarly, the buds contained higher proportions of cells expressing INS and GCG (Figure S6I). RNA sequencing (RNA-seq) shows that the buds express higher levels of general endocrine markers (PAX6, SYP) and of lineage-specific transcripts (LMO1, ARX). Conversely, the bulks of the clusters express elevated levels of genes associated with pancreatic progenitors in the epithelial cords, including SOX9 and ONECUT1, alongside cell cycle-related genes such as CDK2 and MCM3 (Figure 7H).
Gene set enrichment analysis (GSEA) shows that the human buds are significantly enriched for transcripts that have high relevance values for EPs #1, #6, #8, #11, and #12, which characterize the intermediate and late steps of endocrine differentiation in mouse (Figure 7I). Conversely, the bulk clusters showed higher enrichment (albeit with marginal p values) for genes that are relevant to EPs #14 and #16, representing cell cycle genes and early endocrine genes, respectively (Figure 7I). This shows that the buds represent a more advanced endocrine state, while the bulks include both proliferating progenitors and early endocrine precursors. Indeed, CHGA, which marks the advanced endocrine state, is enriched in the buds, whereas NEUROG3, which marks emerging endocrine precursors, is enriched in the bulks (Figure 7H).
To test the full potential of the two compartments, we transplanted either bulks or buds under the kidney capsule of SCID beige mice and extracted the grafts after 6–17 weeks. In both cases, development in the mouse environment resulted in the formation of monohormonal endocrine cells (Figures 7J and S6J). However, when comparing the ratios between β-like cells and α-like cells in the grafts, we noticed that the proportion of β-like cells generated by the bulks was more than 3-fold higher, on average, than that generated by transplanted buds (Figures 7K and 7L). This shows that similar to normal embryonic development, later forming endocrine cells in vitro have a higher tendency to differentiate into β-like cells.
The larger proportion of INS+ β-like cells observed after transplantation of the CHGA-poor bulks corroborates the finding from the transcriptional analysis, which indicated that bulks contain cord-like progenitor cells capable of giving rise to new endocrine cells. Indeed, when we embedded the spheroid bulks in Matrigel for the remainder of the protocol, new buds appeared. Histological examination revealed these buds contain both INS+ and GCG+ monohormonal cells (Figure 7M). Altogether, this shows that as in mouse development in vivo, hESCs can be induced in vitro to generate hormone+ cells within a peninsula-like structure.
DISCUSSION
More than half a century ago, Hard (1944) showed that forming islets are connected to the epithelial tubules from which they derive. However, with the advent of more sophisticated molecular analysis tools, the notion that islets bud from the epithelium as a unit was rejected in favor of a model whereby EMT and subsequent islet cell aggregation is the mechanism of islet formation. Our analysis supports the hypothesis offered by Hard (1944). Through scRNA-seq we construct a detailed map of endocrine differentiation and find that islet formation does not involve migration of multiple individual cells. Rather, differentiating endocrine precursors maintain cell contacts throughout their development, and the islet forms as a growing “peninsula.” Early differentiating α cells appear at the tip of the peninsula, and the later differentiating β cells form in their rear. This generates spatiotemporal collinearity during differentiation and lays the ground for final islet architecture: the cells at the tip of the peninsula end up on the periphery of islets, whereas β cells, which appear at the base of the peninsula, form the islet’s core. It is noted that the human islets are built as a composite of small core-mantle components, rather than the single core and mantle observed in each mouse islet (Bonner-Weir et al., 2015). It seems plausible that the human structure forms when several small peninsulas coalesce.
Several reasons led to the acceptance of the EMT-aggregation model. Foremost was the identification of individual cells expressing either Ins or Gcg in histological cross-sections of embryonic pancreata, suggesting a mesenchymal state. However, staining with the epithelial marker Ecad shows that all endocrine precursors remain attached to one another throughout differentiation (Figure 5). Thus, cells that apparently seem to be “floating” in the mesenchyme are in fact part of a cohesive structure and do not move freely. Our 3D visualization of cleared pancreata reveals the complex structures that form during islet development, which are difficult to detect by conventional 2D histology. As for the mesenchymal state itself, we do observe expression of the mesenchymal marker Vim during endocrine differentiation, but it is absent from cells that express either of the hormones, indicating that by that stage, they are no longer mesenchymal. Furthermore, Vim expression correlates with that of the tight-junction protein Cldn4, which has been shown to attenuate EMT (Lin et al., 2013). Thus, the expression of Vim does not indicate full EMT. Rather, we believe it to have a role in the release of the differentiating cells from the epithelial cord into the peninsula.
Another support for the EMT-aggregation model is the oligoclonal nature of islets (Swenson et al., 2009), which suits the notion of multiple cells coming together from distant places. However, our transcriptional analysis confirms, as found by others (Miyatsuka et al., 2009), that endocrine precursors do not divide. Expansion of the growing peninsula is therefore based on the recruitment of new precursors from the dividing cells of the epithelial cord. Thus, islet oligoclonality merely reflects that of the adjacent cord. Because the aggregation model also assumes that individual cells aggregate in vicinity to the cords that produced them, it too assumes cord oligoclonality.
Last, the fulcrum of the aggregation model is the ability of dissociated islets to re-aggregate in vitro and re-acquire architecture similar to that of the original islet (Esni et al., 1999; Rouiller et al., 1991). However, the ability of cells to re-aggregate in vitro does not show that their embryonic development involves a similar process. In fact, there are clear differences between the two processes. For example, enzymatic digestion of Ecad prevents re-aggregation of dissociated cells in vitro (Rouiller et al., 1991), but knockout of the gene does not affect islet morphogenesis in vivo (Wakae-Takada et al., 2013).
To test our model, we simulated peninsular development using 3D animation tools. The nature of the simulation allowed us to adjust parameters such as proliferation probability, adhesion intensity, and time in each transitional state. For example, we noticed that the size of the α cell mantle surrounding the islet could be determined by modulating an α cell’s chances to divide according to the identity of its neighbors (In Silico Morphogenetic Modeling in Data S1). In agreement with this, a recent report showed that deletion of roundabout receptors in β cells of mature islets disrupted the core-mantle architecture and caused an increase in α cell proportion (Adams et al., 2018). This suggests that α cell proliferation is regulated by neighboring β cells.
Since the derivation of hESCs, efforts have been made to differentiate them into fully functional β cells. Using in vitro-generated β cells as an unlimited source for transplantation to treat T1D is considered feasible because of the nature of T1D, in which only β cells are lost. Because β cells were thought to form independently of islet morphogenesis, it was further assumed that recapitulating their specification alone would be sufficient to create them in vitro. Our findings connect islet shape acquisition with endocrine specification and point to the potential importance of recapitulating morphogenesis for the proper formation of β cells and islets in vitro. Indeed, our ability to induce endocrine buds in 3D spheres of hESC-derived pancreatic progenitors shows that the in vitro clusters resemble organoids in their ability to coordinate differentiation and morphogenesis.
Altogether, this work provides a new framework for understanding how islets develop and offers novel ways to harness this understanding in the search for clinical alternatives to treat diabetes.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Doug Melton (dmelton@harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals and animal handling
For the generation of Ngn3-eGFP+/− embryos, Neurog3tm1(EGFP)Khk mice (Lee et al., 2002) were crossed with C57BL/6 mice (Jackson laboratory, Bar Harbor, ME). A heterozygous colony of Neurog3tm1(EGFP)Khk mice was maintained by either inbreeding or crossing with C57BL/6 mice. Noon of the day when vaginal plugs first appeared was determined as 0.5 days post coitum (dpc). Pregnant females were euthanized through CO2 inhalation. For transplantation experiments, we used immunodeficient SCID-Beige mice aged 8–10 weeks (Charles River).
Animal studies were performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All of the animals were handled according to approved institutional animal care and use committee (IACUC) protocol number [16–05-269]. The protocol was approved by the Committee on the Use of Animals in Research and Teaching of Harvard University Faculty of Arts & Sciences (HU/FAS). The HU/FAS animal care and use program is AAALAC International accredited, has a PHS Assurance (A3593–01) on file with NIH’s Office of Laboratory Animal Welfare, and is registered with the USDA (14-R-0128).
HESC culture and differentiation
HUES8 human embryonic stem cells were differentiated into β-like cells in suspension as described previously (Pagliuca et al., 2014), with slight modifications. Briefly, 150e6 cells were suspended in 300ml of supplemented mTeSR1 and grown in spinning flasks (70rpm, 37°C, 5%CO2) for 48 hours prior to the beginning of a stepwise differentiation protocol:
Stage 1 – 24 hours in S1 medium supplemented with ActivinA (100ng/ml) and CHIR99021 (14μg/ml), followed by 48 hours without CHIR99021.
Stage 2 – 72 hours in S2 medium supplemented with KGF (50ng/ml).
Stage 3 – 48 hours in S3 medium supplemented with KGF (50ng/ml), LDN193189 (200nM), Sant1 (0.25μM), retinoic acid (2μM), PDBU (500nM) and Y-27632 (10μM).
Stage 4 – 5 days in S3 medium supplemented with KGF (50ng/ml), Sant1 (0.25μM) and retinoic acid (0.1 μM). Bud induction was achieved by adding Y-27632 (10μM) and ActivinA (5ng/ml).
Stage 5 – 7 days in BE5 medium supplemented with Beta Cellulin (20ng/ml), XXI (1μM), Alk5iII (10 μM) and T3 (1μM). Sant1 (0.25μM) was added in the first three days, and retinoic acid was added at 0.1 μM in the first three days, then at 0.025μM.
Stage 6 – CMRLS medium supplemented with Alk5iII (10μM) and T3 (1μM).
Factors and media are as described in (Pagliuca et al., 2014).
METHOD DETAILS
Single cell isolation
Embryos were removed and the pancreas extracted from each using fine tweezers. To avoid noise from inter-embryonic asynchrony, all the cells sequenced at each time point were derived from a single embryo. Ngn3-eGFP+/− pancreata were visually identified using fluorescent microscopy. Each pancreas was placed in an Eppendorf tube containing PBS, and then spun down at 1100 RPM for 2 minutes. PBS was aspirated, and the pancreas was dissociated by 5min incubation in TrypLE Express (GIBCO) at 37°C, followed by mechanical agitation through pipetting. TrypLE activity was terminated by washing with PBS+1% FCS. Shortly prior to cell sorting, Propidium Iodide (BD Biosciences, Billerica, MA) was added to eliminate dead cells. Cells were sorted using MoFlo FACS sorter into 96 well plates containing 5μl of RLT lysis buffer (QIAGEN, Hilden, Ge) with 0.55mM 2-Mercaptoethanol (GIBCO).
Single cell library sequencing
Whole transcriptome amplification of the mRNA was done using the Smart-seq protocol (Ramsköld et al., 2012). Briefly, Agencourt RNAClean beads were used to isolate the mRNA, followed by reverse transcription reaction using SMARTScribe (Clontech) enzyme. cDNA was isolated using Agencourt AMPure XP beads, followed by PCR amplification using the Advantage2 kit (Clontech). After another wash with the AMPure beads, DNA quality and concentration were measured on randomly selected wells using High Sensitivity DNA Chips for Bioanalyzer (Agilent technologies). This information was used to estimate DNA concentrations in the other wells, and to eliminate empty wells. Cells were labeled and sequencing libraries were prepared using Nextera XT DNA library preparation kit (Illumina), after wells were diluted in TE according to their estimated concentration. After all wells from a given plate were pooled and washed using AMPure XP beads, samples were sequenced using Rapid flowcell on an Illumina HiSeq2500 sequencer.
2D tissue analysis
Isolated pancreata were fixed overnight in 4% paraformaldehyde, washed in PBS, embedded in paraffin and sectioned on a microtome. Paraffin was removed using Histo-Clear (National Diagnostics) and serial washes in declining ethanol concentrations. After antigen retrieval, samples were blocked in PBS with 5% donkey serum and 0.1% Triton x-100 for 1 hour at room temperature. Primary antibody was diluted in the blocking solution and incubated at room temperature for 1hr, followed by washing. Secondary antibody was incubated for 1 hour at room temperature, washed, and DAPI staining was added.
In situ hybridization was performed using ViewRNA ISH tissue assay kit, according to the manufacturer’s protocol. Probes used: Ins2-VB6-13064; Gcg-VB1-11255; Vim-VB1-14320; Mlxipl-VB6-20923.
3D tissue imaging
Wholemount immunostaining presented in Figure 5J and in Figure S6A was performed after tissue clearing using the Clarity protocol (Cronan et al., 2015). The cleared pancreas was then blocked in PBS with 5% donkey serum, 5% BSA and 0.3% Triton x-100. Primary antibodies were incubated at 4°C for at least 24 hours, and similar incubation time was used with the secondary antibody. Samples were imaged using either Lightsheet or confocal microscopy. For all other figures and videos, pancreata were incubated in primary antibodies for 3 days, then with secondary antibodies for 3 more days, and only then put through the Clarity protocol with the following adjustments: hydrogel solution contained 4% PFA, 3% acrylamide, 0.05% bis-acrylamide and 0.25% VA-044. Following 3 days of incubation in the hydrogel solution at 4°C, tissue was placed in 37°C for 3 hours to allow gel consolidation. Then, samples were washed for 3 days in 8% SDS solution at 37°C, followed by 1–3 days in PBS-triton (0.3%). After an additional wash in PBS, tissues were incubated overnight in RIMS solution (20mM phosphate buffer with Histodenz) at 4°C, and imaged the following day using multiphoton microscopy (LSM 880 NLO multi-photon microscope, Zeiss).
Image processing
Multiphoton images were deconvoluted using Zen2 (Zeiss, Germany), and processed and analyzed using IMARIS (Bitplane, UK). The images produced in each fluorescent channel were shifted manually relative to each other to achieve maximal overlap between the channels. To produce the movies, 3D images were first prepared by applying filtering masks to remove background noise. Next, segments of the 3D reconstructed image were removed in silico to expose individual peninsulas.
In vitro peninsula isolation
Differentiating clusters were collected at the third day of stage 5, and buds were broken off by vigorous pipetting. Size fractionation was performed using a series of pluriSelect cell strainers. After separation, cluster bodies (“bulks”) and buds were further differentiated according to the remainder of the protocol. Clusters between 200μm and 300μm in diameter were put back in the spinner flask, and buds with diameters between 20μm and 100μm were placed in low adhesion 6-well plates on a rocker.
Bulk RNA sequencing
mRNA was extracted from the isolated buds and bulks using Direct-zol RNA mini kit (Thermo Fisher Scientific). Polyadenylated mRNAs were selected from 500ng total RNA samples using oligo-dT-conjugated magnetic beads on an Apollo324 automated workstation (PrepX PolyA mRNA isolation kit, Takara Bio USA). Entire poly-adenylated RNA samples were immediately converted into stranded Illumina sequencing libraries using 200bp fragmentation and sequential adaptor addition on an Apollo324 automated workstation following manufacturer’s specifications (PrepX RNA-seq for Illumina Library kit, Takara Bio USA). Libraries were enriched and indexed using 14 cycles of amplification (LongAmpTaq 2x MasterMix, New England BioLabs Inc.) with PCR primers which included a 6bp index sequence to allow for multiplexing (custom oligo order from Integrated DNA Technologies). Excess PCR reagents were removed using magnetic bead-based cleanup on an Apollo324 automated workstation (PCR Clean DX beads, Aline Biosciences). Resulting libraries were assessed using a 2200 TapeStation (Agilent Technologies) and quantified by qPCR (Kapa Biosystems). Libraries were pooled and sequenced on an Illumina NextSeq 500 mid output flow cell using single end, 75bp reads.
Animal transplantation
Isolated buds and bulks were placed in Costar ultra-low attachment 6-well plates (Corning, ME, USA) with stage 5 differentiation medium. The plate was placed on a rocker to ensure constant agitation. The separated components of the clusters were grown according to the remaining protocol until transplantation, either at stages 5 or 6. The size-separated components were transplanted under the kidney capsule of immunodeficient SCID-Beige mice. 6–17 weeks later the mice were euthanized and the kidneys extracted for immunohistological analysis.
Data Analysis
Read alignment and processing
Reads were aligned to the mouse reference genome build mm10 with TopHat (v2.0.13), which was provided with gene annotations from GENCODE (vM2). Gene expression profiles for each cell were computed using Cufflinks (2.2.0) as previously described in Trapnell et al., 2014. Per-cell expression profiles were normalized prior to downstream analyses such as trajectory reconstruction, Topic Modeling, and differential expression using a prototype of the Census algorithm (Qiu et al., 2017b) included in Monocle 2.
Cell filtering and trajectory reconstruction
The sequencing data was further analyzed using Monocle (Qiu et al., 2017a, 2017b; Trapnell et al., 2014). Further information about Monocle can be found in Monocle’s manual and vignette on Bioconductor. For this analysis, we used an early version of Monocle2. The following strategy was used to filter away putative low-quality cells such as cell fragments or multiplets: After applying Census, we visualized the cells’ distribution along the range of total mRNA per cell. We first did this for all cells and removed the outliers based on visual appearance of the histogram. We then repeated this procedure separately for each sample. This approach automatically accounted for all cells lying outside of 2 standard deviations of the mean of each sample’s log-transformed total mRNA count per cell. As a final step, we removed cells expressing more than one of the hormones insulin, glucagon and ghrelin, using an arbitrary mRNA count threshold of 30.
The developmental trajectory was reconstructed using a semi-supervised approach. After defining a CellTypeHierarchy with Neurog3 high, Gcg high, Ins½ high, and Ghrl high cells, the top 15 markers of each cell type were used as ordering genes. DDRTree was used as the dimensionality reduction method, and the trajectory was plotted in 2D using the first 2 of 4 dimensions. Pseudotime was defined by setting the branch with Neurog3 high cells as the root. The 3-way heatmaps and expression plots were generated using modified Monocle functions after manually subsetting the CellDataSet into 3 distinct CellDataSets, each containing the progenitor cells and one of the three differentiated branches.
Inference of EPs via Topic Modeling
TM is a probabilistic unsupervised learning algorithm which is designed to discover latent topics present in a set of text documents, based solely on the frequency at which each word appears in each document (Blei, 2012). For text data, a topic is defined as a set of (a priori unspecified) related words which tend to probabilistically co-occur. For example, statistical co-occurrence of words such as “banks,” “market,” and “GDP” within the same document provides strong evidence that atopic such as finance is being discussed. Once the topics have been inferred, a document can be more interpretably represented in terms of the relative presence of each topic rather than a massive collection of word frequencies. In our analysis, we employ the topic model and inference algorithm used in the GeneProgram software tool (Gerber et al., 2007), which employs a Hierarchical Dirichlet Process (HDP) prior. This enables automatic inference of the number of latent topics (along with the topics themselves) from the data (Teh et al., 2006).
TM is adapted to scRNA-seq data by treating each cell as a document and each gene asa word. Within the cell, the unit amount of mRNA from the gene is therefore the genomic equivalent of a word’s frequency within a document. In this context, we refer to a latent topic as an EP, which represents a set of genes that behave coordinately in particular cells. Unlike traditional clustering techniques, our TM analysis allows many EPs to be active within a given cell, and a single gene may also be used by different EPs (one gene can regulate multiple different biological processes depending on the context in which it is expressed). For each EP, a gene receives a relevance value (quantifying how active the gene is within the EP) and a cell is assigned a usage value (quantifying how active the EP is within the cell). After inferring the EPs underlying our scRNA-seq measurements via Markov-Chain Monte Carlo (Gerber et al., 2007), we can interpretably represent and classify each cell in terms of its relative usage of different EPs, rather than the raw expression values of many thousand genes. Similarly, each EP is itself represented as a collection of genes along with their “relevance” in defining the EP. The biological significance of an EP can thus be elucidated by investigating which genes are the most actively used (Gerber et al., 2007).
The TM software used to infer the EPs in our analysis is available at http://groups.csail.mit.edu/cgs/geneprogram.html. TM analysis was performed using all cells that passed quality control, including those removed by the last filter (removal of polyhormonal cells). TM was run with the following settings: 4 levels of discretization for the expression values, 100 posterior samples (taken every 1,000 steps of the MCMC-chain with an initial burn-in of 30,000 steps), minimum of 2 genes per EP with a minimum usage value of 0.1, with the default prior HDP concentration and minimum-similarity/merging parameters used in (Gerber et al., 2007). See (Gerber et al., 2007) for detailed descriptions of these parameters.
Defining pseudotemporal EPs and GSEA
After importing the Topic Modeling data, i.e., the genes’ relevance and the cells’ usage values for each EP, into R, we eliminated EPs used by less than 50 cells in order to maintain biological relevance. Using Monocle-based tools, we generated a CellDataSet where we used EPs and EP values instead of genes and their expression. This allowed us to plot each EP’s behavior by pseudotime, akin to pseudotemporal gene expression plots. We used LOESS regression to visualize the trend separately for each branch and defined “pseudotemporal EPs” by setting an arbitrary threshold difference of 0.5 between the minimal and maximal cellular usage values of the smoothed LOESS curves across all 3 branches. Heatmaps were annotated with each gene’s relevance value for the 8 pseudotemporal EPs. Each gene was assigned to the EP for which it has the highest relevance value.
Gene set enrichment analysis (GSEA) was performed as previously described using the piano package (Väremo et al., 2013) and the MSigDB v6.0 (Subramanian et al., 2005). Mouse gene short names were capitalized to be compatible with the human annotations of the MSigDB gene lists. All gene sets with an adjusted p value of < 0.01 of the distinct-directional / upregulated class were defined as significantly enriched.
Bulk RNA-seq analysis
For RNA-seq alignment, adaptor sequences were removed from the paired-end raw sequence fastq files with TrimGalore (http://www.bioinformatics.babraham.ac.uk/projects/trim galore/) and then aligned to the transcript sequences of the human genome. Reference transcript sequences for alignment with RSEM (Li and Dewey, 2011) were built from the GRCh38 assembly, human GeneBank Assembly ID GCA 000001405_ 27, and the corresponding gtf annotation file. Genomic sequences and annotations were downloaded from the Ensembl database (Aken et al., 2017). The STAR software (Dobin et al., 2013) was set as the alternative aligner for alignment with the RSEM software. The gene expression estimates in the form of expected counts were used in the subsequent differential expression analysis.
For differential expression analysis, the table of expected counts was read into R as a list data structure using the R package edgeR (Robinson et al., 2010) from Bioconductor (Gentleman et al., 2004). Expected counts were converted to cpm and only those genes with more than one count per sample and more than 6 counts over all samples retained. The final step before statistical analysis was to normalize the cpm data, which was done by scaling according to the method of (Robinson and Oshlack, 2010). Differentially expressed genes were determined with the linear modeling pipeline as implemented in the Bioconductor package limma (Ritchie et al., 2015). This required that each gene was multiplied by a weight term estimated from the mean-variance relation as performed with the voom function (Law et al., 2014).
Gene set enrichment analysis was performed with the piano package (Väremo et al., 2013). Gene sets were curated based on the mouse-derived Topic Modeling analysis. Of the 2180 genes that have a value > 0 for at least one of the pseudotime-dependent EPs, 1724 appeared in the analyzed hESC data. From these, the top 100 genes from each EP were used to determine a gene set.
In silico morphogenetic modeling
The simulation was created using Houdini FX (www.sidefx.com), a tool commonly used in the visual-effects (VFX) industry for its flexibility and strong simulation capabilities. The basis of the simulation is that of a particle system. Cells are represented as points in space, each containing attributes that describe the state of that particular cell at a given time point. These attributes are constantly updated in a stepwise manner. This setup allowed the use of Houdini’s grain solver-a position based dynamics system. Even though the grain solver was created to simulate effects such as sand, it possesses several built-in features that make it particularly attractive as a platform upon which to build the cell simulation: stability, native clumping and explicit constraints. The full details on the simulation process is provided in Data S1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Ngn3 cell counting
Dissociated Ngn3-GFP+/− individual pancreata were analyzed with flow cytometry using BD LSR-II. The entire sample was run to evaluate the total number of GFP+ cells in each pancreas.
Immunofluorescent labeling quantification
Quantitative analysis of Ecad protein intensity was calculated based on immunofluorescent staining, using MATLAB image analysis toolbox. An Ecad+ pixel mask was determined using several rounds of squaring and local histogram equalization followed by Otsu’s thresholding. Similarly, a logical mask for endocrine+ regions was created using Otsu’s thresholding. Combining both masks identified regions that either express or do not express one of the additional markers tested, and the intensity of Ecad in each pixel was calculated into histograms. The plots in Figure S5 represent the calculated distribution plot for each image, and in cases where several fields were analyzed from a single sample, the average distribution plot is presented. Notice that the actual panels present only a portion of the entire field used for histogram calculation, for image resolution purposes. To generate the average endocrine distribution plot presented in Figure 5H, the average distribution plots of Chga (average of days 13.5–15.5), Syp (average of days 13.5–16.5), and Glp2 (average of day 15.5) were combined. The average of the daily median expression level in Ecad+ and Ecad- regions was calculated for each endocrine marker separately, and Student’s t test was performed on these values.
To quantify transplanted bulks/buds composition, sections at least 7 slices apart were analyzed to avoid counting the same cell twice. From each kidney, INS+ and GLP2+ cells were manually counted in at least 10 sections, and more than 1,400 positive cells were counted in each kidney analyzed.
DATA AND SOFTWARE AVAILABILITY
Single-cell RNA-sequencing data was deposited to the NCBI Gene Expression Omnibus (GEO) under accession number GEO: GSE121416.
Bulk RNA-sequencing data (buds versus bulks of spheroids) was deposited to the NCBI Gene Expression Omnibus (GEO) under accession number GEO: GSE121679.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti C-peptide | DSHB | Cat#GN-ID4 |
| Rabbit anti Chga | Abcam | Cat#Ab15160 |
| Rabbit anti Cldn4 | Lifespan BioSciences | Cat#LS-B8382 |
| Mouse anti E-cadherin | BD Transduction Laboratories | Cat#610181 |
| Chicken anti GFP | Aves Labs inc. | Cat#GFP-1020 |
| Goat anti glp2 | Santa Cruz Biotechnology | Cat#Sc-7781 |
| Guinea pig anti Insulin | DAKO | Cat#A0564 |
| Hamster anti Muc1 | NeoMarkers | Cat#HM-1630-P1 |
| Rabbit anti Ncad | Invitrogen | Cat#MA5–16324 |
| Goat anti Neurod1 | R&D systems | Cat#AF2746 |
| Rabbit anti Neurogenin3 | EMD Millipore | Cat#Ab10535 |
| Mouse anti Nkx6.1 | DSHB | Cat#F55A12 |
| Rabbit anti proglucagon | Cell Signaling Technology | Cat#D16G10 |
| Rabbit anti Slug | Cell Signaling Technology | Cat#95855 |
| Rabbit anti Sox9 | Abcam | Cat#Ab-5535 |
| Goat anti somatostatin | Santa Cruz Biotechnology | Cat#Sc-7819 |
| Rabbit anti Synaptophysin | ProteinTech Group | Cat#17785–1-AP |
| Chicken anti Vimentin | Encor Biotechnologies | Cat#CPCA-Vim |
| Mouse anti Zeb1 | eBioscience | Cat#14–9741-82 |
| Rabbit anti αSMA | Abcam | Cat#Ab5694 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ActivinA | R&D Systems | Cat#338-AC |
| Rock Inhibitor Y-27632 | DNSK | Cat#DNSK-Kl15–02 |
| Critical Commercial Assays | ||
| ViewRNA ISH Tissue Assay Kit (2-plex) | ThermoFisher | Cat#QVT0012 |
| SMARTScribe Reverse Transcriptase | Clontech | Cat#639537 |
| Advantage2 Polymerase mix | Clontech | Cat#639202 |
| Agencourt Rnaclean | ThermoFisher | Cat#NC0068576 |
| Agencourt AMPure | Beckman Coulter | Cat#A63881 |
| Direct-zol | ThermoFisher | Cat#50444622 |
| Deposited Data | ||
| Raw and analyzed single cell sequencing data | This paper | GEO: GSE121416 |
| Raw and analyzed bulk sequencing data | This paper | GEO: GSE121679 |
| Experimental Models: Cell Lines | ||
| HUES8 hESCS | HSCI | hES Cell line: HUES-8 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Neurog3tm1(EGFP)Khk | Raised in house | RRID:MMRRC_000344-UCD |
| Mouse: CB17.Cg-PrkdcscidLystbg-J/Crl | Charles River | RRID:IMSR_CRL:250 |
| Software and Algorithms | ||
| Houdini | Side Effects Software | www.sidefx.com |
| Topic modeling | This paper | http://groups.csail.mit.edu/cgs/geneprogram.html |
| Monocle | Trapnell lab | http://cole-trapnell-lab.github.io/monocle-release/ |
Highlights.
Mapping changes in gene expression during pancreatic islet formation using scRNA-seq
Islets form as budding “peninsulas,” not through aggregation of dispersed cells
Islet architecture reflects the temporal order of α and β cell appearance
Peninsula-like buds generated in vitro from differentiating pluripotent cells
ACKNOWLEDGMENTS
N.S. is partly supported by the Gruss-Lipper Charitable Foundation. R.C. is supported by an Early Postdoctoral Mobility Fellowship from the Swiss National Science Foundation. J.M. received funding from the NIH (T32HG004947). C.T. is supported by the NIH (grants DP2HD088158 and U54 DK107979), the W. M. Keck Foundation, the Dale F. Frey Award for Break-through Scientists, and an Alfred P. Sloan Foundation Research Fellowship. D.M. is an Investigator of the Howard Hughes Medical Institute. This work was supported by grants from the Harvard Stem Cell Institute, Helmsley Charitable Trust, the JDRF, and the JPB Foundation. This research was performed using resources and/or funding provided by the NIDDK-supported Human Islet Research Network (HIRN; RRID:SCR_014393, UC4 DK104165–04, and UC4 DK104159–03). We thank Prof. Aviv Regev from the Broad Institute and Dr. Alex Shalek, currently at MIT, for help with technical aspects of scRNA-seq; Mr. Juerg Straubhaar from Harvard for his help with data analysis; Dr. Aharon Helman from Harvard for critically reading the manuscript; Dr. Doug Richardson from the Harvard Center for Biological Imaging (CBI) and Mr. Sven Terclavers from Zeiss for assistance with imaging; Mss. Jennifer Hyoje-Ryu Kenty and Deanne Watson for mouse transplantations; and Mss. Cathy MacGillvary and Diane Faria for histological preparations.
D.M. is a founder and Scientific Advisory Board (SAB) member for Semma Therapeutics.
Footnotes
DECLARATION OF INTERESTS
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures, two tables, seven videos and one data file and can be found with this article online at https://doi.org/10.1016/j.cell.2018.12.003.
REFERENCES
- Adams MT, Gilbert JM, Hinojosa Paiz J, Bowman FM, and Blum B (2018). Endocrine cell type sorting and mature architecture in the islets of Langerhans require expression of Roundabout receptors in β cells. Sci. Rep. 8, 10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, Billis K, Carvalho-Silva D, Cummins C, Clapham P, et al. (2017). Ensembl 2017. Nucleic Acids Res. 45 (D1), D635–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankaitis ED, Bechard ME, and Wright CV (2015). Feedback control of growth, differentiation, and morphogenesis of pancreatic endocrine progenitors in an epithelial plexus niche. Genes Dev. 29, 2203–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, Ryu JH, Wagner BK, Shen-Orr SS, Klein AM, et al. (2016). A single-cell transcriptomic map of the human and mouse pancreas reveals inter-and intra-cell population structure. Cell Syst. 3, 346–360.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blei DM (2012). Probabilistic topic models. Commun. ACM 55, 77–84. [Google Scholar]
- Bonner-Weir S, Sullivan BA, and Weir GC (2015). Human islet morphology revisited: human and rodent islets are not so different after all. J. Histochem. Cytochem. 63, 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronan MR, Rosenberg AF, Oehlers SH, Saelens JW, Sisk DM, Jurcic Smith KL, Lee S, and Tobin DM (2015). CLARITY and PACT-based imaging of adult zebrafish and mouse for whole-animal analysis of infections. Dis. Model. Mech. 8, 1643–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esni F, Täljedal IB, Perl AK, Cremer H, Christofori G, and Semb H (1999). Neural cell adhesion molecule (N-CAM) is required for cell type segregation and normal ultrastructure in pancreatic islets. J. Cell Biol. 144, 325–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgia S, and Bhushan A (2004). Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J. Clin. Invest. 114, 963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber GK, Dowell RD, Jaakkola TS, and Gifford DK (2007). Automated discovery of functional generality of human gene expression programs. PLoS Comput. Biol. 3, e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouzi M, Kim YH, Katsumoto K, Johansson K, and Grapin-Botton A (2011). Neurogenin3 initiates stepwise delamination of differentiating endocrine cells during pancreas development. Dev. Dyn. 240, 589–604. [DOI] [PubMed] [Google Scholar]
- Gu G, Dubauskaite J, and Melton DA (2002). Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 129, 2447–2457. [DOI] [PubMed] [Google Scholar]
- Hard WL (1944). The origin and differentiation of the alpha and beta cells in the pancreatic islets of the rat. Am. J. Anat. 75, 369–403. [PubMed] [Google Scholar]
- Johansson KA, Dursun U, Jordan N, Gu G, Beermann F, Gradwohl G, and Grapin-Botton A (2007). Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev. Cell 12, 457–465. [DOI] [PubMed] [Google Scholar]
- Johnston NR, Mitchell RK, Haythorne E, Pessoa MP, Semplici F, Ferrer J, Piemonti L, Marchetti P, Bugliani M, Bosco D, et al. (2016). Beta cell hubs dictate pancreatic islet responses to glucose. Cell Metab. 24, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen HL, and Grapin-Botton A (2017). The molecular and morphogenetic basis of pancreas organogenesis. Semin. Cell Dev. Biol. 66, 51–68. [DOI] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Perreault N, Brestelli JE, and Kaestner KH (2002). Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes Dev. 16, 1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Shang X, Manorek G, and Howell SB (2013). Regulation of the epithelial-mesenchymal transition by claudin-3 and claudin-4. PLoS ONE 8, e67496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller K, Kim A, Kilimnik G, Jo J, Moka U, Periwal V, and Hara M (2009). Islet formation during the neonatal development in mice. PLoS ONE 4, e7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles F, Battelino T, Czernichow P, and Scharfmann R (1998). TGF-beta plays a key role in morphogenesis of the pancreatic islets of Langerhans by controlling the activity of the matrix metalloproteinase MMP-2. J. Cell Biol. 143, 827–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatsuka T, Li Z, and German MS (2009). Chronology of islet differentiation revealed by temporal cell labeling. Diabetes 58, 1863–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatsuka T, Kosaka Y, Kim H, and German MS (2011). Neurogenin3 inhibits proliferation in endocrine progenitors by inducing Cdkn1a. Proc. Natl. Acad. Sci. USA 108, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA, and Thiery JP (2016). Emt: 2016. Cell 166, 21–45. [DOI] [PubMed] [Google Scholar]
- Pagliuca FW, Millman JR, Gürtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, and Melton DA (2014). Generation of functional human pancreatic β cells in vitro. Cell 159, 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan FC, and Wright C (2011). Pancreas organogenesis: from bud to plexus to gland. Dev. Dyn. 240, 530–565. [DOI] [PubMed] [Google Scholar]
- Qiu X, Hill A, Packer J, Lin D, Ma YA, and Trapnell C (2017a). Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 14, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu XMQ, Tang Y, Wang L, Chawla R, Pliner H, and Trapnell C (2017b). Reversed graph embedding resolves complex single-cell developmental trajectories. Nat. Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsköld D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, Daniels GA, Khrebtukova I, Loring JF, Laurent LC, et al. (2012). Full-length mRNA-seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 30, 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezania A, Riedel MJ, Wideman RD, Karanu F, Ao Z, Warnock GL, and Kieffer TJ (2011). Production of functional glucagon-secreting α-cells from human embryonic stem cells. Diabetes 60, 239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, et al. (2014). Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 32, 1121–1133. [DOI] [PubMed] [Google Scholar]
- Riento K, and Ridley AJ (2003). Rocks: multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 4, 446–456. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bio-conductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouiller DG, Cirulli V, and Halban PA (1991). Uvomorulin mediates calcium-dependent aggregation of islet cells, whereas calcium-independent cell adhesion molecules distinguish between islet cell types. Dev. Biol. 148, 233–242. [DOI] [PubMed] [Google Scholar]
- Rukstalis JM, and Habener JF (2007). Snail2, a mediator of epithelial-mesenchymal transitions, expressed in progenitor cells of the developing endocrine pancreas. Gene Expr. Patterns 7, 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ HA, Parent AV, Ringler JJ, Hennings TG, Nair GG, Shveygert M, Guo T, Puri S, Haataja L, Cirulli V, et al. (2015). Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBOJ. 34, 1759–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro AM, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL, Kneteman NM, and Rajotte RV (2000). Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 343, 230–238. [DOI] [PubMed] [Google Scholar]
- Solar M, Cardalda C, Houbracken I, Martín M, Maestro MA, De Medts N, Xu X, Grau V, Heimberg H, Bouwens L, and Ferrer J (2009). Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryo-genesis but not after birth. Dev. Cell 17, 849–860. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson ES, Xanthopoulos J, Nottoli T, McGrath J, Theise ND, and Krause DS (2009). Chimeric mice reveal clonal development of pancreatic acini, but not islets. Biochem. Biophys. Res. Commun. 379, 526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh YW, Jordan MI, Beal MJ, and Blei DM (2006). Hierarchical Dirichlet processes. J. Am. Stat. Assoc. 101, 1566–1581. [Google Scholar]
- Thiery JP, Acloque H, Huang RY, and Nieto MA (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen T, Donaldson CJ, Cáceres E, Hunter AE, Cowing-Zitron C, Pound LD, Adams MW, Zembrzycki A, Grove KL, and Huising MO (2015). Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat. Med. 21, 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väremo L, Nielsen J, and Nookaew I (2013). Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 41, 4378–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villasenor A, Marty-Santos L, Dravis C, Fletcher P, Henkemeyer M, and Cleaver O (2012). EphB3 marks delaminating endocrine progenitor cells in the developing pancreas. Dev. Dyn. 241, 1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakae-Takada N, Xuan S, Watanabe K, Meda P, and Leibel RL (2013). Molecular basis for the regulation of islet beta cell mass in mice: the role of E-cadherin. Diabetologia 56, 856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Law AC, Rajagopal J, Anderson WJ, Gray PA, and Melton DA (2007). A multipotent progenitor domain guides pancreatic organogenesis. Dev. Cell 13, 103–114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-sequencing data was deposited to the NCBI Gene Expression Omnibus (GEO) under accession number GEO: GSE121416.
Bulk RNA-sequencing data (buds versus bulks of spheroids) was deposited to the NCBI Gene Expression Omnibus (GEO) under accession number GEO: GSE121679.