

Abstract
Nipah virus (NiV) is an emerging zoonotic pathogen, reported for the recent severe outbreaks of encephalitis and respiratory illness in humans and animals, respectively. Many antiviral drugs have been discovered to inhibit this pathogen, but none of them were that much efficient. To overcome the complications associated with this severe pathogenic virus, we have designed a multi-epitope subunit vaccine using computational immunology strategies. Identification of structural and nonstructural proteins of Nipah virus assisted in the vaccine designing. The selected proteins are known to be involved in the survival of the virus. The antigenic binders (B-cell, HTL, and CTL) from the selected proteins were prognosticated. These antigenic binders will be able to generate the humoral as well as cell-mediated immunity. All the epitopes were united with the help of suitable linkers and with an adjuvant at the N-terminal of the vaccine, for the enhancement of immunogenicity. The physiological characterization, along with antigenicity and allergenicity of the designed vaccine candidates, was estimated. The 3D structure prediction and its validation were performed. The validated vaccine model was then docked and simulated with the TLR-3 receptor to check the stability of the docked complex. This next-generation approach will provide a new vision for the development of a high immunogenic vaccine against the NiV.
Introduction
Nipah virus is the recently emerging zoonotic pathogen, belongs to the Paramyxoviridae family has created the foremost threat among public and animal health.1 This zoonotic pathogen causes severe encephalitis in humans and respiratory problems in animals, mainly pig.2 The fruit bat (genus-Pteropus) is responsible for spreading this infection by secreting the body fluid-like saliva or urine on the fruit (raw date plum), which in turn, consumed by animals and humans. According to the WHO report, the mortality rate is higher in the Asian population as compared to other people (http://www.who.int/news-room/fact-sheets/detail/nipah-virus).3 As per the National Centre for Disease Control report, the first significant outbreak of the virus was seen in Malaysia and Singapore between the year 1998 and 1999 with 276 cases (https://ncdc.gov.in/showfile.php?lid=229).4 Further, the next outbreak was observed in 2001 in Bangladesh and West Bengal (Siliguri). Recently in 2018, Nipah virus outbreak was seen in Kerala, an Indian state (mainly Kozhikode and Malappuram districts) in which 17 people got clutched into the hand of this severe infection and hence lost their life (https://www.who.int/csr/don/07-august-2018-nipah-virus-india/en/).5 It was identified that disease caused in Malaysia and Bangladesh was due to two different Nipah virus strains, which are termed as NiV M and NiV B, respectively. Further, investigations showed that the NiV B strain is the most pathogenic as this strain is responsible for more recent outbreaks.6 The clinical signs and symptoms associated with this viral infection include fever, headache trailed by encephalitis, nausea, giddiness, muscular pain, and respiratory illness.7 Subsequently, if the symptoms of the disease last for a long time, it may lead to coma within 48 h.8 So far, no effective vaccine or drug treatment is existing for the cure of this disease in humans; the last option is the supportive care and avoids close contact to infected ones.7 Many researchers are conscientious to find an effective treatment for this severe zoonotic infection. An open-label clinical trial was conducted with ribavirin, an antiviral drug, which showed a reduction in the mortality rate, but the drug was not effective in the prevention of the disease.9 Due to the unavailability of treatment, the human and animal population is affecting severely. There is an indispensable need to develop an effective regimen against infectious diseases, which are the severe cause of fatality and mortality worldwide. The postgenomic era supports the progression of computational based vaccine deigning approaches to fight against the infectious diseases. The aim of the rational vaccine approach is to identify the antigenic epitopes, which have the potential to generate the robust immune responses. Recently, some studies reported by Pandey et al. in which they have shown the construction of a multi-epitope subunit vaccine against the vector-borne diseases like malaria and visceral leishmaniasis by exploring the salivary protein of the vectors (sandfly or mosquito).10−12 However, a subunit vaccine against NiV was reported utilizing immunoinformatic approaches where they have utilized three NiV proteins for vaccine designing.13 Further, the same approach was applied by Mohammed et al. (2019) in which they have used the NiV G protein.14 Both studies have exploited one or three proteins for NiV subunit vaccine designing, but this study included more number of proteins, and the epitopes were also vary from each other. Therefore, we can say that the designed vaccine will be more immunogenic and efficacious against the NiV infection.
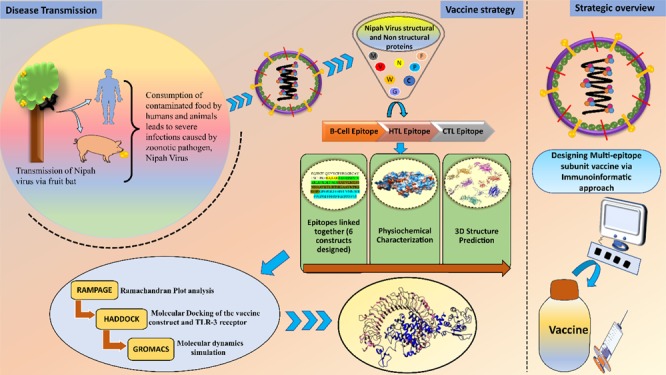
The multi-epitope subunit vaccine consists of highly antigenic epitopes (B-cell, HTL, and CTL epitopes), and these MHC-restricted binders were identified by multiple clones of T-cell receptors, which in turn induce the potent humoral and cell-mediated immune responses.15 The multi-epitope vaccine is a mixture of antigenic epitopes derived from the proteins, which are highly pathogenic and aid pathogen to cause disease. For a more potent immune response, this vaccine was adjuvanted with the suitable adjuvant at the N-terminal. Immunogenic adjuvant leads to more antibody production and helps in providing the long-established protection. In comparison to the traditional vaccine, the multi-epitope vaccines will be highly immunogenic with less or no adverse events. The immunoinformatic approach makes the path more comfortable and helps in the screening of antigenic epitopes as well as the vaccine construct analyzed by various parameters, supports the best candidate for the treatment of the disease (Figure 1).
Figure 1.

Schematic representation of Nipah virus transmission and strategies applied for the subunit vaccine designing against Nipah virus.
Here, in this study, we exploited the viral proteins, which consist of antigenic determinants and would be able to elicit the immune response in the host body against the infection. P gene of Nipah virus encodes for four proteins, which include phosphoprotein P (UniProt ID: H6V860) as well as three accessory proteins, namely, protein W (UniProt ID: H6V848), nonstructural protein V (UniProt ID: H6V847), and protein C (UniProt ID: Q4VCP8).16 At the time of transient expression, these proteins show the IFN-antagonist activity. Due to which, they are found to be the most important virulence factors and suitable for vaccine designing. Among them, the protein W acts as the inhibitor of the TLR-3 pathway, whereas proteins V and C function on the IFN signaling by interacting with STAT1 and STAT2.17 Nucleocapsid protein N (UniProt ID: H6V857) is responsible for the encapsidation of the virus genome and hence protects nuclease activity. Further, the matrix protein, namely, protein M (UniProt ID: Q4VCP7), is accountable for the structural integrity of the virion. Fusion glycoprotein F0 (UniProt ID: D2DEC0) has shown a promising role in the fusion of the viral membrane to the host cell membrane results in the delivery of all the contents into the host cytoplasm. Glycoprotein G (UniProt ID: Q4VCP5) modulates the exo-alpha-sialidase activity and contributes to the attachment of the virus to the host cell surface receptors18 (Figure 2). To devise a potential and antigenic vaccine candidate, we have designed a multi-epitope subunit vaccine by using immunoinformatic approaches and also proved its potency to bind with the TLR-3 receptor by molecular docking.
Figure 2.

Representation of selected Nipah virus antigenic proteins and their functional aspects.
Results and Discussion
Retrieval of Structural and Nonstructural Nipah Virus Protein Sequences for Vaccine Designing
The reported and experimentally validated 8 pathogenic proteins of Nipah virus were selected. The sequences of proteins [matrix protein (M), nucleoprotein (N), fusion glycoprotein F0 (F), and glycoprotein (G) and nonstructural proteins named phosphoprotein (P), nonstructural protein (V), protein (W), and Protein (C)] were retrieved from the UniProt database in a FASTA format.
Homology Assessment of Selected NiV Proteins
The similarity between the host and pathogen epitopes can lead to autoimmune diseases (chances of cross reactivity19) and molecular mimicry.20 In this study, a protein BLAST analysis was done by using the selected NiV protein, and their homology was checked against the human proteins. It was found that the chosen proteins have shown less than 40% homology with the human proteins and hence can be proved as excellent vaccine candidates for vaccine construction.
Continuous B-Cell Epitopes Mapping
B-cell epitopes are responsible for the generation of humoral immune response by producing class-switched antibodies against the pathogen. The ABCpred server was used for the B-cell epitope prediction, which envisages the antigenic epitopes based on the artificial neural network with a precision rate of 65.99%. The epitope selection was based on the highest score secured by the epitopes with a default threshold of 0.51. So, from 8 proteins, the single most top scorer epitope got selected (overall 8 B-cell epitopes). These epitopes were further utilized to produce multi-epitope vaccine candidates (Table S1).
Helper T-Lymphocyte Epitopes Mapping
Helper T-cells are the essential cell of the immune system and are responsible for adaptive immunity. The IDEB server was used for the prediction of MHC II antigenic binders. The HTL epitopes were sorted based on the lowest percentile rank and IC50 value. The epitopes obtained with the lowest percentile rank has an excellent binding affinity; therefore, for each selected protein, the score with the lowest percentile rank was selected. The next parameter includes the sorting of peptides based on the IC50 value: the peptides <50 nM considered to have the highest binding affinity, <500 nM intermediate affinity, and <5000 nM have the lowest affinity for the T-cell receptor (Table S2).
HTL Epitopes Selection Based on HLA-Restricted Alleles
The human leucocyte antigen (HLA) ligand-binding step is usually considered to be extra specific other than succeeding levels of the antigen processing pathways and thus crucial for vaccine designing. After literature survey and WHO report, it was found that the areas that are massively affected by NiV infection, for instance, Australia, Africa, Bangladesh, Cambodia, China, India, Indonesia, Madagascar, Malaysia, Papua New Guinea, Thailand, and so on, by considering this data, we have selected the alleles as per the distribution of the disease. All the alleles have different binding preferences; therefore, the allelic selection was made with the help of the Allelefrequencies.net database. Here are the selected alleles: “HLA-DRB1*07:01, HLA-DRB5*01:01, HLA-DRB1*13:02, and HLA-DRB1*15:01”, which found to be circulated over these countries and cover the specific population (Table S2).
Cytotoxic T-Lymphocyte Epitopes Mapping
CTL epitopes prediction is the pivotal step in vaccine designing as they are necessary for the clearance of intracellular pathogens by arbitrating cell-mediated immunity. Here, the potential CTL epitope for 8-sorted protein sequences was predicted as per 3 supertypes, namely, A2, A3, and B7 with a threshold score of 0.75. These selected supertypes provide maximum population coverage of 88.3% of the entire world population.21 Further, the antigenic binders were selected based on the highest obtained score. This way for each input protein, three epitopes were selected belongs to 3 supertypes, and overall, 24 CTL epitopes were sorted for the construction of the subunit vaccine (Table S3).
Devising Potential Multi-Epitope Vaccine Candidate
Next, the immunogenic epitopes were united with the help of linkers B-cell (KK linker), HTL (AAY linker), and CTL (GPGPG linker) along with adjuvant (β-defensin- TLR-3 agonist). For the formulation of a subunit vaccine, adjuvants are the significant aspects, which improve the immunogenicity of vaccine by enhancing the immunogenic responses analogous to the natural immune response. The designed constructs were composed of 8 B-cell epitopes, 8 HTL epitopes, and 24 CTL epitopes. Total 6 constructs were developed with different combinations, by keeping adjuvant at the N-terminal of the sequence (Scheme S1).
Physiochemical Characterization, Antigenicity, and Allergenicity Prediction of Designed Vaccine
Investigation of physiochemical properties of the 6-designed vaccine candidates was predicted with the help of the ExPASy ProtParam server (Table S4). The estimated values have shown that all the developed vaccine candidates were stable and met entirely the standards, which are necessary for the formulation of the vaccine. Second, the obtained score from the VaxiJen server v2.0 was higher than 0.5 for each vaccine candidate, which signifies that the designed candidates could be a probable antigen and may have the potential to provoke the immune response inside the host body. Allergenicity prediction was achieved with the help of the AllerTOP server, which predicted that the nature of the vaccine protein was nonallergenic (not able to cause any side effects) for all 6 vaccine candidates. Hence, after predicting the overall properties of the vaccine candidates, it was concluded that all designed vaccines constructs were stable, antigenic, and nonallergenic.
Vaccine Protein Tertiary Structure Prediction and Refinement
The folding and unfolding patterns of the tertiary structure were obtained with the help of the RaptorX server. All the designed 6 vaccine candidates were subjected to tertiary structure prediction. RaptorX predicted the tertiary structure in the PDB file along with various parameters (Figure 3A).
Figure 3.

(A) (a)–(f) Tertiary structures of different subunit vaccine candidate obtained from template-based homology modelling. (B) Representation and comparative analysis of the structural difference between the refined and nonrefined tertiary structures of designed vaccine candidates.
Among predicted 6 structures, the best vaccine candidate was chosen based on the P value. As per the selection norms, smaller P value (i.e., values less than 10–3) indicates a good model standard. The predicted P value for the best model (model-1, presented as Figure 3A(a)) was 1.48 × 10–3, which was more modest in comparison to other predicted models. The obtained uGDT (unnormalized global distance test) and GDT score of the selected model were 148 and 25, respectively. The uGDT score >50 exemplifies the good model quality. The 3D structure obtained from RaptorX was then subjected for the refinement to the ModRefiner server. That server shaped the predicted tertiary structure to a refined tertiary structure. Further, the refined and nonrefined models (selected model) were superimposed on each other to identify the differences between them (Figure 3B).
Avouchment of Predicted 3D Model of the Vaccine Candidate
The validation of the refined model was achieved with the help of the Ramachandran plot assessment servers: RAMPAGE and PROCHECK. After the comparative analysis, it was found that the RAMPAGE server peculiarly predicted 92.7% residues in the energetically favored region, whereas 2.4% residues in the disallowed area and 4.9% in the allowed part. PROCHECK data showed that 86.9% of residues lies in the favored area, while 10.8% residues and 1.8% were found in the additional and generously allowed area, respectively, followed by 0.6% in the disallowed area. This data represents that the structure obtained after refinement has high resolution and structural quality with minimum steric atomic clashes between the residues (Figure 4).
Figure 4.

(A, B) Ramachandran plot assessment by two different softwares: (A) RAMPAGE and (B) PROCHECK servers for the validation of the designed tertiary structure of selected subunit vaccine candidate.
Tertiary Structure Fixation of Immune Cell Receptor and Immunogenic Ligand
The tertiary structure of the target immune cell receptor, that is, Toll-Like Receptor 3 (TLR-3), was derived from the Protein Data bank (PDB ID: 2a0Z). Both the receptor (TLR-3) and ligand (vaccine candidate) were subjected to the PDB Hydro server for structure fixation and solvation removal. This server provides the more refined structures of receptor and ligand, which were fixed and have no solvent molecules. Further, the model was selected for docking and dynamics simulation.
Molecular Docking of Antigenic Vaccine Candidate with TLR-3 Receptor
Protein–Protein docking was performed between the receptor (TLR-3) and ligand (vaccine candidate) with the help of the HADDOCK server. This server needs ambiguous interaction restraints (AIRs), which denotes the active and passive residue numbers. Active residues are involved in the interaction between the molecules and accessible solvent, while passive residues are surface neighbor active residues, which are solvent accessible. Here, TLR-3 (PDB ID: 2A0Z) was conjugated with the vaccine candidate to make the stable protein complex. The server predicted 7 clusters and 42 structures. The result was presented with their respective HADDOCK scores, cluster size, van der Waals energy, electrostatic energy, desolvation energy, restraints violation energy, buried surface area, and Z score. Among them, cluster 1 showed the lowest Z score value, that is, −1.5, HADDOCK scores −11.2, cluster size 13, van der Waals energy −81.5, electrostatic energy −515.2, desolvation energy −18.4, restraints violation energy 1917.8, and buried surface area 3617.9. For more precise selection, the finest structure of cluster 1 was chosen for molecular dynamics simulation (Figures 5 and 6).
Figure 5.
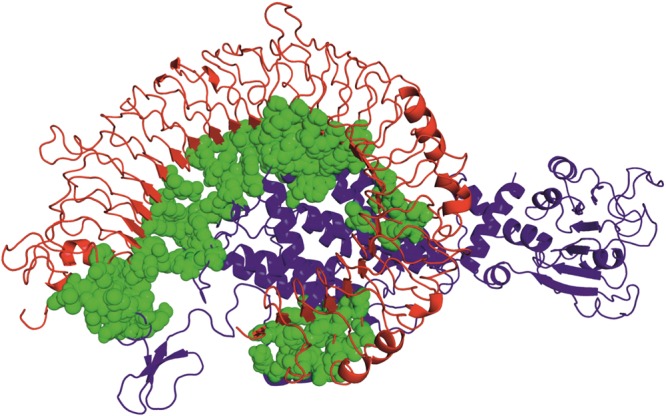
Molecular docking of the TLR-3 receptor and vaccine candidate (ligand) via HADDOCK server. The domains of the receptor have shown in blue color, whereas red color represents the vaccine candidate, and green color represents the interaction between receptor and vaccine candidate.
Figure 6.

(a)–(f) Graphical representation of docking parameters of molecular-docked 7 clusters. (a) Ambiguous interaction restraints (AIRs) against ligand RMSD. (b) Electrostatic energy of docked molecule against interface-RMSD. (c) van der Waals energy against interface RMSD. (d) Haddock score against interface RMSD. (e) Haddock score against ligand RMSD. (f) Haddock score against a fraction of frequent contacts.
Molecular Dynamics Simulation of the Docked Complex
For the evaluation of stability and physical interactions between the atoms and molecules, the molecular dynamics simulation was performed. Temperature and pressure were equilibrated by applying NVT and NPT ensemble with the period of 100 ps. The estimated potential energy for stabilizing the system was −2 × 106 kJ mol–1. The stability of the final vaccine candidate was appraised by using the GROMOS 96a force field. The RMSD plot obtained for the backbone of the docked complex represents an initial deviation at 0.5 nm. Gradually, it increases with time until 6 ns, afterward it becomes stable until 10 ns with an RMSD value of 0.9 nm. The RMSF plot showed that the highest fluctuation among the residues ranges from residue numbers 400–600 and becomes steady after 600 residues with an average RMSF value of ∼0.7 nm. The RMSD and RMSF levels of the complex suggested that the docked complex was pliable and stable (Figure 7).
Figure 7.

(a)–(d)Molecular dynamics simulation. The plots representing the interaction and stability of the atoms and molecules between receptor (TLR-3) and ligand (vaccine candidate); plot (a) root mean square deviation (RMSD) of the receptor–ligand complex shows infinitesimal deviation and becomes stable at the period of 6 ns, which concludes the steady nature of the complex. (b) Root mean square fluctuation plot (RMSF) representing the fluctuation of side chain residues. (c)–(d) Pressure and temperature plots concerning time.
Discussion
Nipah virus is the recently emerging deadliest zoonotic pathogen, which is severely affecting the worldwide population. Its pathogenesis ranges from acute infection to chronic encephalitis. The Southeast Asia region is severely affected by the NiV infection with an estimated fatality rate of 40–77% (2001–2018 WHO report: https://www.who.int/news-room/fact-sheets/detail/nipah-virus).3 The health of the world population is the main apprehension as the disease can cause high virulence in humans.22 Until now, no approved medication is available for the treatment of the disease.
As discussed in the Introduction part, ribavirin, an antiviral drug, was introduced during the first significant outbreak, and there was a reduction in the mortality rate, but the drug was not effective in complete prevention of the disease.23 Further, the same drug was given in combination with chloroquine, and it was found that the drug was unable to prevent death in the hamster model infected by Nipah and Hendra viruses.24 In 2012, a subunit vaccine was reported by Bossaert Katharine N. to protect the African green monkeys from Nipah virus infection.25 But later in 2015, the detailed analysis was performed on African green monkey against NiV infection by Johnston et al., and they recognize 100% chances of relapse encephalitis in the animals.26 However, during the recent outbreaks, the previously developed drug favipiravir was used as a regimen for human use.27 The ability of the drug to inhibit the full range of RNA viruses makes it remarkable, but certain complications have been observed, such as contraction in the lungs.
Apart from this, many vectored vaccines, for instance, the vesicular stomatitis virus vaccine recombinant28 and live attenuated,29 recombinant measles virus vaccine expressing the NiV G protein,30 and VLP-based vaccines have shown good immune response in animal models: African green monkeys and hamsters.31 The bivalent rabies virus vaccine is in pipeline against the NiV and rabies virus, as it has shown good immune response in animal model.32 The described vaccines were mainly tested on animals and not licensed for the NiV treatment in humans. All reported vaccines primarily composed of single viral protein, and it is a well-known phenomenon that, RNA viruses have a high mutation rate, to prompt alteration of viral genome and proteome. Therefore, constant antigenic changes contribute to the increased pathogenicity or virulence and early ineffectiveness of vaccine, which may be a drawback for a single viral protein vaccine.33 However, on the other hand, the multisubunit vaccine acquires the most antigenic part of the viral proteins to become more potent, and it can fight against RNA virus evolution efficiently. Epitopes identified from the viral proteins will activate the APCs and hence induces the costimulatory molecules to produce a robust immune response. As per the literature survey, TLR-3 was involved in TRIF/TRAM-mediated Th1 signaling activation, which in turn provide defense against the viral diseases. This approach can be validated by the use of a TLR-3 agonist as an adjuvant, which helps the host to fight against the intracellular microbes.34
In 2005, it was reported that the NiV protein W stimulates the TLR-3 receptor, which inhibits IFN beta promotor activation.35 Further, in 2010, it was shown that during Nipah virus infection in the endothelial cell majority of the NiV protein W, localize in cytoplasm and generate an IFN beta response, whereas in M17 neuronal cell (infected with Nipah virus) NiV protein W, tend to localize in the nucleus and blocks TLR-3 signaling.36 In both conditions, TLR-3 expression increases and acts as a significant contributing factor in NiV infection. The selected Nipah virus proteins used for this study have already been reported previously and experimentally validated.37 In this study, we have applied the next-generation vaccine designing approach to design a multi-epitope-based subunit vaccine, which can be potent enough to generate the immunological responses against the pathogen.38 The selected viral proteins have shown no similarity with the human proteins and hence prove to be the potential vaccine candidates. When the antigenic part from B-cell, HTL, and CTL of different proteins joined with the adjuvant, they displayed high antigenicity. The physiochemical parameter data suggested that the designed vaccine was stable with half of >30 h in mammalian reticulocytes. The Log P value of the construct was positive, indicating the hydrophobic nature and hence suggested that a liposomal delivery system will the right way for vaccine delivery. The predicted tertiary structure with the minimum P value indicates the high model quality. From the ramachandran plot analysis, higher amount of amino acids was found to be in the favored region, which was considerable and defined the protein stability. The docked complex of the vaccine construct and TLR-3 receptor gives a lower score, confer the highest binding energy. The molecular dymanics simulation calculated the fluctuation and deviation of amino acids in the docked complex, and with this data, it can be concluded that the complex was flexible and stable. All the findings from the imunoinformatic approach suggested that the intended vaccine candidate may further undergo in vitro and in vivo experimental analyses for the development of a potential vaccine against Nipah virus infection.
Methodology
Selection of Nipah Virus Structural and Nonstructural Proteins To Design Multi-Epitope Subunit Vaccine
The Nipah virus structural and nonstructural proteins were identified with the help of a literature survey. The sequences of the 8 experimentally validated pathogenic viral proteins (UniProt Proteome ID: UP000103103)39 named as the matrix protein (M), nucleoprotein (N), fusion glycoprotein F0 (F), and glycoprotein (G) as well as nonstructural proteins called phosphoprotein (P), nonstructural protein (V), protein (W), and protein (C) were retrieved from the UniProt database in the FASTA format. Apart from this protein above sequences, β-defensin, a 45 amino acid long chain retrieved from NCBI (National Centre for Biotechnology Information:https://www.ncbi.nlm.nih.gov/) had been used as an adjuvant, which was imperative to generate the efficient or potential immunogenic responses.40
Homology Assessment of Nipah Virus Proteins
Protein blast of each selected protein was done to find out the homology of the chosen protein sequence with the human proteome by using NCBI Protein–Protein Blast (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome). Homology of viral protein with human proteome will interfere with the immunogenic response of the designed vaccine. Therefore, there should be no or least homology (<40%) with the human proteome.20
Continuous/Linear B-Cell Epitopes Mapping
In this study, B-cell epitopes for the selected antigenic protein sequences were predicted with the help of the publicly available ABCpred server (http://crdd.osdd.net/raghava/abcpred/). This server utilizes artificial and recurrent neural network based on machine learning techniques for the prediction of B-cell epitopes with fixed length patterns. The obtained output was verified by 5-fold cross-corroboration and provided an overall or maximum accuracy of 65.93%.41,42
Helper T-Lymphocyte (Th-Cells) Epitopes Mapping
Prediction of immunogenic HTL epitopes was achieved with the help of the easily accessible online IEDB server (Immune Epitope Database and Analysis Resources: http://www.iedb.org/). With the assistance of this server, experimentally validated epitopes (from different species) involved in infection or immune related diseases can be investigated.43 Currently, the updated single-data repository encompasses around 260,000 epitopes, which help in the designing of vaccines. Consequently, the prediction of HTL epitopes was accomplished by utilizing consensus ANN and SMM methods. The sorting of antigenic epitopes with the highest binding affinity was done based on their lowest percentile rank and IC50 value.
Identification of HLA-Restricted CTL Epitopes
After the prediction of HTL epitopes, the sorting depends on the specific criteria as discussed above, and allele selection was one of them. For the assortment of allele diversity and geographical distribution, The Allele Frequency Net Database (http://www.allelefrequencies.net/default.asp) was used. This freely available database is a complete suite for the deposition of allelic frequency information from diverse polymorphic regions in the human genome. The Asia-Pacific region is profoundly affected by this severe infection (https://www.cdc.gov/vhf/nipah/outbreaks/distribution-map.html).44
Cytotoxic T-Lymphocyte Epitopes Mapping
The online and freely available server NetCTL 2.0 (http://www.cbs.dtu.dk/services/NetCTL/) was used to predict the antigenic CTL epitopes for the selected Nipah virus proteins. The current version of server provides an integrative output by combining proteasomal cleavage (C-terminal), TAP transport efficiency, and MHC class I affinity, which restricted to 12 MHC I supertypes. This server exploits the artificial neural network and hence offers the most antigenic epitopes with their respective scores.45
Devising Multi-Epitope Vaccine by Combining Immune Cell Epitopes
For devising the multi-epitope subunit vaccine, the antigenic epitopes of B-cell, HTL, and CTL were fused with the assistance of linkers. The N-terminal of the designed vaccine sequence was adjuvanted the TLR-3 agonist, β-defensin. Defensins possess the antimicrobial as well as antiviral activity and have the ability to recruit the antigen-presenting immune cells having MHC-I and MHC-II. Hence, it can be concluded that β-defensin can induce potential immunogenic responses similar to natural immune responses.46 EAAAK,47 the helix forming linker, was added, which was used to link the adjuvant and first epitope of the sequence. Linkers are requisite for the enhanced expression, stability, and folding of the protein, and they do it so by parting the functional domains.48 For each immune cell, different linkers were used such as to link B-cell epitopes together, the KK49 linker was used; for linking HTL epitopes together, the GPGPG linker50 was added; and concurrently, for connecting CTL epitopes, the AAY linker was appended.51
Determination of Antigenicity and Allergenicity of the Designed Vaccine Candidate
The antigenicity prediction was achieved with the help of VaxiJen server v2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html), for the robust validation of antigenicity. The difference between antigens and nonantigens was done by utilizing five training sets of antigens with a prediction accuracy of 70–89%.52 Allergenicity of all the six combinations was checked by the AllerTOPv.2.0 server; this was performed to analyze that the designed vaccine candidate will not cause any allergy upon administration. This server predicts the allergenicity with 86.7% sensitivity, 90.7% specificity, and 88.7% accuracy (AllerTOP v.2: a server for in silico prediction of allergens).
Physicochemical Characterizations of the Designed Vaccine Candidate
The physicochemical characterization of the developed vaccine was achieved through the help of the freely available ExPASy (Expert Protein Analysis System) ProtParam server (https://web.expasy.org/protparam/). This server envisages the properties of the vaccine candidate, which includes the amino acid composition, molecular weight, extension coefficient, theoretical isoelectric point, instability index, the estimated half-life of the candidate, as well as aliphatic index and the GRAVY (grand average of hydropathicity) score.53 The property prediction was performed for all designed vaccine candidates.
Tertiary Structure Prediction of the Designed Vaccine along with the Structure Refinement and Avouchment
The structure prediction was done with the help of the readily accessible RaptorX server (http://raptorx.uchicago.edu/) server. This server predicted the model quality by calculating the solvent accessibility, P value, uGDT (GDT), disordered regions, and pocket multiplicity.54 After obtaining the modeled structure with the aid of RaptorX, the structure was subjected to refinement, and this process was achieved with the help of the ModRefiner (https://zhanglab.ccmb.med.umich.edu/ModRefiner/) online server. This server produces an initial model by predicting backbone topology, a hydrogen bond, and a side chain position closer to the initial structure and hence generated the refined model, which is not restrained by the input model.55 Despite that, validation of the predicted refined structure was done with the help of two different servers: RAMPAGE (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php) and PROCHECK (https://www.ebi.ac.uk/thornton-srv/software/PROCHECK/). These servers predicted the overall geometry of the residues by examining the stereochemical quality of protein. Hence, it provides the distributive data of amino acids in favored, allowed, and sterically disallowed regions (exception glycine and proline residue), which confirm the stability of the modeled structure.56
Fixation of Refined Tertiary Structures of Vaccine Candidate and Immune Cell TLR-3 Receptor
Before proceeding to dock, fixation of PDB structure was done to remove the solvent molecule and fix the missing atoms. The PDB structure of the receptor TLR-3 was obtained from NCBI (PDB ID: 2A0Z). This PDB structure, along with the vaccine candidates, was fixed by a server PDB Hydro (http://lorentz.immstr.pasteur.fr/pdb_hydro.php). This server generates the rotamer of the lowest van der Waals energy of given the PDB file and also identifies missing side chains. It works on an algorithm, which fits alanine in the place of the lost side chain to get an ideal confirmation of peptide bonds.57
Assessment of Molecular Interaction of Immunogenic Vaccine Candidate with Immune Cell Receptor TLR-3 by Performing Molecular Docking
Molecular docking between the TLR-3 (receptor) and vaccine (ligand) was presented with the help of online accessible HADDOCK: High Ambiguity Driven protein–protein DOCKing server (https://milou.science.uu.nl/services/HADDOCK2.2/haddockserver-easy.html). This online server combines the information from biochemical bioinformatics or biophysical method to improve scoring and sampling. This server produces a docking score along with other parameters like van der Waals energy, Z score, etc. The Z score denotes the standard deviations of a given cluster concerning the mean of all the groups generated. The best-docked multi-epitope is one with the minimum Z score. HADDOCK also produces several plots for better understanding of the docking result. These plots display the comparison of the best-docked structure with all generated structures concerning docking score, RMSD, etc.58
Molecular Dynamics Simulation of Vaccine-Immuned Cell Receptor-Docked Complex
The molecular dynamics simulation analyzed the nonbonded interactions between the protein molecules with the help of the Groningen Machine for Chemical Simulations (GROMACS) molecular dynamic package.59 Here, the molecular dynamics simulation was performed for the docked complex of the vaccine candidate and TLR-3 receptor, obtained from the HADDOCK server. The atomistic GROMOS 96a force field was employed for the simulation process.60 The system was well equilibrated, and charge neutralization was done using suitable ions. The system temperature and pressure were equilibrated at 300 k and 1 bar for an equilibration period of 100 ns, respectively. Minimization of energy was done using the steepest descent algorithm. Last, MD simulation was set for 10 ns, and respective RMSD (backbone residues) and RMSF (fluctuation of side chain residues) were determined to achieve a stable and flexible vaccine complex.60−62
Acknowledgments
R.O. is thankful to the Central University of Rajasthan for providing research fellowship. R.K.P. is thankful to DST India for providing INSPIRE fellowship. V.K.P. and D.P. are thankful to the Central University of Rajasthan for providing the computational facility.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b00944.
Designed vaccine sequences; predicted B-cell epitopes for the selected Nipah virus proteins with their respective peptide sequences and scores; predicted HTL epitopes for the selected Nipah virus proteins with their respective peptide sequences, percentile rank, IC50 value, allele, and geographical distribution; predicted CTL epitopes by considering 3 supertypes along with their respective scores; and physiochemical properties along with Antigenicity of designed vaccine constructs (PDF)
Author Contributions
The protocol was designed by R.O. and V.K.P. Methodology was performed by R.O., A.P., R.K.P., and V.K.P. The manuscript was written by R.O., D.P., and V.K.P.
The authors declare no competing financial interest.
Notes
The data generated during the study has been added to the supporting information file.
Supplementary Material
References
- Sawatsky B.; Grolla A.; Kuzenko N.; Weingartl H.; Czub M. Inhibition of henipavirus infection by Nipah virus attachment glycoprotein occurs without cell-surface downregulation of ephrin-B2 or ephrin-B3. J. Gen. Virol. 2007, 88, 582–91. 10.1099/vir.0.82427-0. [DOI] [PubMed] [Google Scholar]
- Luby S. P.; Gurley E. S.; Hossain M. J. Transmission of human infection with Nipah Virus. Clin. Infect. Dis. 2009, 49, 1743–1748. 10.1086/647951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nipah Virus (http://www.who.int/news-room/fact-sheets/detail/nipah-virus).
- NCDC . Nipah Virus Infection (https://ncdc.gov.in/showfile.php?lid=229).
- WHO . Nipah virus – India ( https://www.who.int/csr/don/07-august-2018-nipah-virus-india/en/).
- DeBuysscher B. L.; de Wit E.; Munster V. J.; Scott D.; Feldmann H.; Prescott J. Comparison of the pathogenicity of Nipah virus isolates from Bangladesh and Malaysia in the Syrian hamster. PLoS Neglected Trop. Dis. 2013, 7, e2024 10.1371/journal.pntd.0002024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly S.; Sunita V. K.; Pagrut N.; Faran N. K. Nipah virus: An update on prevention and control strategies with special reference to the latest outbreak in India. Int. J. Vet. Sci. Anim. Husb. 2018, 20. [Google Scholar]
- Geisbert T. W.; Mire C. E.; Geisbert J. B.; Chan Y.-P.; Agans K. N.; Feldmann F.; Fenton K. A.; Zhu Z.; Dimitrov D. S.; Scott D. P.; Bossart K. N.; Feldmann H.; Broder C. C. Therapeutic Treatment of Nipah Virus Infection in Nonhuman Primates with a Neutralizing Human Monoclonal Antibody. Sci. Transl. Med. 2014, 6, 242ra82. 10.1126/scitranslmed.3008929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broder C. C.; Xu K.; Nikolov D. B.; Zhu Z.; Dimitrov D. S.; Middleton D.; Pallister J.; Geisbert T. W.; Bossart K. N.; Wang L.-F. A treatment for and vaccine against the deadly Hendra and Nipah viruses. Antiviral Res. 2013, 100, 8–13. 10.1016/j.antiviral.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey R. K.; Prajapati V. K. Exploring sand fly salivary proteins to design multi-epitope subunit vaccine to fight against visceral leishmaniasis. J. Cell. Biochem. 2019, 120, 1141–1155. 10.1002/jcb.26719. [DOI] [PubMed] [Google Scholar]
- Pandey R. K.; Bhatt T. K.; Prajapati V. K. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci. Rep. 2018, 8, 1125. 10.1038/s41598-018-19456-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatoon N.; Pandey R. K.; Ojha R.; Aathmanathan V. S.; Krishnan M.; Prajapati V. K. Exploratory algorithm to devise multi-epitope subunit vaccine by investigating Leishmania donovanimembrane proteins. J. Biomol. Struct. Dyn. 2019, 2381–2393. 10.1080/07391102.2018.1484815. [DOI] [PubMed] [Google Scholar]
- Ravichandran L.; Venkatesan A.; Febin Prabhu Dass J. Epitope-based immunoinformatics approach on RNA-dependent RNA polymerase (RdRp) protein complex of Nipah virus (NiV). J. Cell. Biochem. 2019, 120, 7082–7095. 10.1002/jcb.27979. [DOI] [PubMed] [Google Scholar]
- Mohammed A. A.; Shantier S. W.; Mustafa M. I.; Osman H. K.; Elmansi H. E.; Osman I.-A. A.; Mohammed R. A.; Abdelrhman F. A.; Elnnewery M. E.; Yousif E. M.; Mustafa M. M.; Elfadol N. M.; Abdalla A. I.; Mahmoud E.; Eltay A. A.; Ahmed y. A.; Hassan M. A. Epitope - based peptide vaccine against glycoprotein G of Nipah henipavirus using immunoinformatics approaches. bioRxiv 2019, 678664. 10.1101/678664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shey R. A.; Ghogomu S. M.; Esoh K. K.; Nebangwa N. D.; Shintouo C. M.; Nongley N. F.; Asa B. F.; Ngale F. N.; Vanhamme L.; Souopgui J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci. Rep. 2019, 9, 4409. 10.1038/s41598-019-40833-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua K. B.; Bellini W. J.; Rota P. A.; Harcourt B. H.; Tamin A.; Lam S. K.; Ksiazek T. G.; Rollin P. E.; Zaki S. R.; Shieh W.-J.; Goldsmith C. S.; Gubler D. J.; Roehrig J. T.; Eaton B.; Gould A. R.; Olson J.; Field H.; Daniels P.; Ling A. E.; Peters C. J.; Anderson L. J.; Mahy B. W. J. Nipah virus: a recently emergent deadly paramyxovirus. Science 2000, 288, 1432–1435. 10.1126/science.288.5470.1432. [DOI] [PubMed] [Google Scholar]
- Yoneda M.; Guillaume V.; Sato H.; Fujita K.; Georges-Courbot M.-C.; Ikeda F.; Omi M.; Muto-Terao Y.; Wild T. F.; Kai C. The nonstructural proteins of Nipah virus play a key role in pathogenicity in experimentally infected animals. PLoS One 2010, 5, e12709 10.1371/journal.pone.0012709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan Y. P.; Chua K. B.; Koh C. L.; Lim M. E.; Lam S. K. Complete nucleotide sequences of Nipah virus isolates from Malaysia. J. Gen. Virol. 2001, 82, 2151–2155. 10.1099/0022-1317-82-9-2151. [DOI] [PubMed] [Google Scholar]
- Kanduc D. Peptide cross-reactivity: the original sin of vaccines. Front. Biosci. 2012, 4, 1393–1401. 10.2741/S341. [DOI] [PubMed] [Google Scholar]
- Rojas M.; Restrepo-Jiménez P.; Monsalve D. M.; Pacheco Y.; Acosta-Ampudia Y.; Ramírez-Santana C.; Leung P. S. C.; Ansari A. A.; Gershwin M. E.; Anaya J. M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123. 10.1016/j.jaut.2018.10.012. [DOI] [PubMed] [Google Scholar]
- Sette A.; Sidney J. Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics 1999, 50, 201–12. 10.1007/s002510050594. [DOI] [PubMed] [Google Scholar]
- Siddique A. B.; Fardows J.; Farhana N.; Mazumder M. Nipah Virus: A Public Health Concern. J. Enam Med. Coll. 2016, 6, 101–105. 10.3329/jemc.v6i2.27766. [DOI] [Google Scholar]
- Chong H. T.; Kamarulzaman A.; Tan C. T.; Goh K. J.; Thayaparan T.; Kunjapan S. R.; Chew N. K.; Chua K. B.; Lam S. K. Treatment of acute Nipah encephalitis with ribavirin. Ann. Neurol. 2001, 49, 810–813. 10.1002/ana.1062. [DOI] [PubMed] [Google Scholar]
- Freiberg A. N.; Worthy M. N.; Lee B.; Holbrook M. R. Combined chloroquine and ribavirin treatment does not prevent death in a hamster model of Nipah and Hendra virus infection. J. Gen. Virol. 2010, 91, 765–772. 10.1099/vir.0.017269-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossart K. N.; Rockx B.; Feldmann F.; Brining D.; Scott D.; LaCasse R.; Geisbert J. B.; Feng Y.-R.; Chan Y.-P.; Hickey A. C.; Broder C. C.; Feldmann H.; Geisbert T. W. A Hendra virus G glycoprotein subunit vaccine protects African green monkeys from Nipah virus challenge. Sci. Transl. Med. 2012, 4, 146ra107. 10.1126/scitranslmed.3004241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston S. C.; Briese T.; Bell T. M.; Pratt W. D.; Shamblin J. D.; Esham H. L.; Donnelly G. C.; Johnson J. C.; Hensley L. E.; Lipkin W. I.; Honko A. N. Detailed analysis of the African green monkey model of Nipah virus disease. PLoS One 2015, 10, e0117817. 10.1371/journal.pone.0117817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawes B. E.; Kalveram B.; Ikegami T.; Juelich T.; Smith J. K.; Zhang L.; Park A.; Lee B.; Komeno T.; Furuta Y.; Freiberg A. N. Favipiravir (T-705) protects against Nipah virus infection in the hamster model. Sci. Rep. 2018, 8, 7604. 10.1038/s41598-018-25780-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mire C. E.; Versteeg K. M.; Cross R. W.; Agans K. N.; Fenton K. A.; Whitt M. A.; Geisbert T. W. Single injection recombinant vesicular stomatitis virus vaccines protect ferrets against lethal Nipah virus disease. Virol. J. 2013, 10, 353. 10.1186/1743-422X-10-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott J.; DeBuysscher B. L.; Feldmann F.; Gardner D. J.; Haddock E.; Martellaro C.; Scott D.; Feldmann H. Single-dose live-attenuated vesicular stomatitis virus-based vaccine protects African green monkeys from Nipah virus disease. Vaccine 2015, 33, 2823–9. 10.1016/j.vaccine.2015.03.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda M.; Georges-Courbot M. C.; Ikeda F.; Ishii M.; Nagata N.; Jacquot F.; Raoul H.; Sato H.; Kai C. Recombinant measles virus vaccine expressing the Nipah virus glycoprotein protects against lethal Nipah virus challenge. PLoS One 2013, 8, e58414 10.1371/journal.pone.0058414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walpita P.; Cong Y.; Jahrling P. B.; Rojas O.; Postnikova E.; Yu S.; Johns L.; Holbrook M. R. A VLP-based vaccine provides complete protection against Nipah virus challenge following multiple-dose or single-dose vaccination schedules in a hamster model. npj Vaccines 2017, 2, 21–21. 10.1038/s41541-017-0023-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshwara R.; Shiels T.; Postnikova E.; Kurup D.; Wirblich C.; Johnson R. F.; Schnell M. J. Rabies-based vaccine induces potent immune responses against Nipah virus. npj Vaccines 2019, 4, 15. 10.1038/s41541-019-0109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R.; Domingo-Calap P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. 10.1007/s00018-016-2299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duthie M. S.; Windish H. P.; Fox C. B.; Reed S. G. Use of defined TLR ligands as adjuvants within human vaccines. Immunol. Rev. 2011, 239, 178–196. 10.1111/j.1600-065X.2010.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M. L.; Cardenas W. B.; Zamarin D.; Palese P.; Basler C. F. Nuclear localization of the Nipah virus W protein allows for inhibition of both virus- and toll-like receptor 3-triggered signaling pathways. J. Virol. 2005, 79, 6078–88. 10.1128/JVI.79.10.6078-6088.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo M. K.; Miller D.; Aljofan M.; Mungall B. A.; Rollin P. E.; Bellini W. J.; Rota P. A. Characterization of the antiviral and inflammatory responses against Nipah virus in endothelial cells and neurons. Virology 2010, 404, 78–88. 10.1016/j.virol.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Harcourt B. H.; Tamin A.; Ksiazek T. G.; Rollin P. E.; Anderson L. J.; Bellini W. J.; Rota P. A. Molecular characterization of Nipah virus, a newly emergent paramyxovirus. Virology 2000, 271, 334–349. 10.1006/viro.2000.0340. [DOI] [PubMed] [Google Scholar]
- Kumar Pandey R.; Ojha R.; Mishra A.; Kumar Prajapati V. Designing B-and T-cell multi-epitope based subunit vaccine using immunoinformatics approach to control Zika virus infection. J. Cell. Biochem. 2018, 119, 7631–7642. 10.1002/jcb.27110. [DOI] [PubMed] [Google Scholar]
- Lo M. K.; Lowe L.; Hummel K. B.; Sazzad H. M. S.; Gurley E. S.; Hossain M. J.; Luby S. P.; Miller D. M.; Comer J. A.; Rollin P. E.; Bellini W. J.; Rota P. A. Characterization of Nipah virus from outbreaks in Bangladesh, 2008-2010. Emerg. Infect. Dis. 2012, 18, 248–255. 10.3201/eid1802.111492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan T.; Mitra D.; Rao D. N. Nasal delivery of PLG microparticle encapsulated defensin peptides adjuvanted gp41 antigen confers strong and long-lasting immunoprotective response against HIV-1. Immunol. Res. 2014, 58, 139–53. 10.1007/s12026-013-8428-5. [DOI] [PubMed] [Google Scholar]
- Saha S.; Raghava G. P. S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 2006, 65, 40–48. 10.1002/prot.21078. [DOI] [PubMed] [Google Scholar]
- Pandey R. K.; Ojha R.; Aathmanathan V. S.; Krishnan M.; Prajapati V. K. Immunoinformatics approaches to design a novel multi-epitope subunit vaccine against HIV infection. Vaccine 2018, 36, 2262–2272. 10.1016/j.vaccine.2018.03.042. [DOI] [PubMed] [Google Scholar]
- Fleri W.; Paul S.; Dhanda S. K.; Mahajan S.; Xu X.; Peters B.; Sette A. The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front. Immunol. 2017, 8, 278. 10.3389/fimmu.2017.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breed A. C.; Meers J.; Sendow I.; Bossart K. N.; Barr J. A.; Smith I.; Wacharapluesadee S.; Wang L.; Field H. E. The distribution of henipaviruses in Southeast Asia and Australasia: is Wallace’s line a barrier to Nipah virus?. PLoS One 2013, 8, e61316 10.1371/journal.pone.0061316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M. V.; Lundegaard C.; Lamberth K.; Buus S.; Lund O.; Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinf. 2007, 8, 424. 10.1186/1471-2105-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani K.; Murphy W. J.; Chertov O.; Salcedo R.; Koh C. Y.; Utsunomiya I.; Funakoshi S.; Asai O.; Herrmann S. H.; Wang J. M.; Kwak L. W.; Oppenheim J. J. Defensins act as potent adjuvants that promote cellular and humoral immune responses in mice to a lymphoma idiotype and carrier antigens. Int. Immunol. 2000, 12, 691–700. 10.1093/intimm/12.5.691. [DOI] [PubMed] [Google Scholar]
- Wei H.; Lenz S. D.; Thompson D. H.; Pogranichniy R. M. DNA-vaccine platform development against H1N1 subtype of swine influenza A viruses. Viral Immunol. 2012, 25, 297–305. 10.1089/vim.2011.0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamriz S.; Ofoghi H.; Moazami N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput. Biol. Med. 2016, 76, 24–9. 10.1016/j.compbiomed.2016.06.015. [DOI] [PubMed] [Google Scholar]
- Rana A.; Akhter Y. A multi-subunit based, thermodynamically stable model vaccine using combined immunoinformatics and protein structure based approach. Immunobiology 2016, 221, 544–557. 10.1016/j.imbio.2015.12.004. [DOI] [PubMed] [Google Scholar]
- Saadi M.; Karkhah A.; Nouri H. R. Development of a multi-epitope peptide vaccine inducing robust T cell responses against brucellosis using immunoinformatics based approaches. Infect., Genet. Evol. 2017, 51, 227–234. 10.1016/j.meegid.2017.04.009. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Sun W.; Guo J.; Zhao G.; Sun S.; Yu H.; Guo Y.; Li J.; Jin X.; Du L.; Jiang S.; Kou Z.; Zhou Y. In silico design of a DNA-based HIV-1 multi-epitope vaccine for Chinese populations. Hum. Vaccines Immunother. 2015, 11, 795–805. 10.1080/21645515.2015.1012017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova I. A.; Flower D. R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinf. 2007, 8, 4. 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E.; Gattiker A.; Hoogland C.; Ivanyi I.; Appel R. D.; Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–8. 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J.; Xu J. RaptorX: exploiting structure information for protein alignment by statistical inference. Proteins 2011, 79, 161–171. 10.1002/prot.23175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D.; Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–34. 10.1016/j.bpj.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell S. C.; Davis I. W.; Arendall W. B. III; de Bakker P. I.; Word J. M.; Prisant M. G.; Richardson J. S.; Richardson D. C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins: Struct., Funct., Bioinf. 2003, 50, 437–450. 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Azuara C.; Lindahl E.; Koehl P.; Orland H.; Delarue M. PDB_Hydro: incorporating dipolar solvents with variable density in the Poisson-Boltzmann treatment of macromolecule electrostatics. Nucleic Acids Res. 2006, 34, W38–W42. 10.1093/nar/gkl072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert G. C. P.; Rodrigues J. P. G. L. M.; Trellet M.; Schmitz C.; Kastritis P. L.; Karaca E.; Melquiond A. S. J.; van Dijk M.; de Vries S. J.; Bonvin A. M. J. J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- Lindahl E.; Hess B.; Van Der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. Mol. Model. Ann. 2001, 7, 306–317. 10.1007/s008940100045. [DOI] [Google Scholar]
- Shukla R.; Shukla H.; Sonkar A.; Pandey T.; Tripathi T. Structure-based screening and molecular dynamics simulations offer novel natural compounds as potential inhibitors of Mycobacterium tuberculosis isocitrate lyase. J. Biomol. Struct. Dyn. 2018, 36, 2045–2057. 10.1080/07391102.2017.1341337. [DOI] [PubMed] [Google Scholar]
- Ojha R.; Khatoon N.; Prajapati V. K. Conglomeration of novel Culex quinquefasciatus salivary proteins to contrive multi-epitope subunit vaccine against infections caused by blood imbibing transmitter. Int. J. Biol. Macromol. 2018, 118, 834–843. 10.1016/j.ijbiomac.2018.06.112. [DOI] [PubMed] [Google Scholar]
- Shukla R.; Shukla H.; Kalita P.; Sonkar A.; Pandey T.; Singh D. B.; Kumar A.; Tripathi T. Identification of potential inhibitors of Fasciola gigantica thioredoxin1: computational screening, molecular dynamics simulation, and binding free energy studies. J. Biomol. Struct. Dyn. 2018, 36, 2147–2162. 10.1080/07391102.2017.1344141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.