

Abstract
The growth of axons is an intricately regulated process involving intracellular signaling cascades and gene transcription. We had previously shown that the stimulus-dependent transcription factor, serum response factor (SRF), plays a critical role in regulating axon growth in the mammalian brain. However, the molecular mechanisms underlying SRF-dependent axon growth remains unknown. Here we report that SRF is phosphorylated and activated by GSK-3 to promote axon outgrowth in mouse hippocampal neurons. GSK-3 binds to and directly phosphorylates SRF on a highly conserved serine residue. This serine phosphorylation is necessary for SRF activity and for its interaction with MKL-family cofactors, MKL1 and MKL2, but not with TCF-family cofactor, ELK-1. Axonal growth deficits caused by GSK-3 inhibition could be rescued by expression of a constitutively active SRF. The SRF target gene and actin-binding protein, vinculin, is sufficient to overcome the axonal growth deficits of SRF-deficient and GSK-3-inhibited neurons. Furthermore, short hairpin RNA-mediated knockdown of vinculin also attenuated axonal growth. Thus, our findings reveal a novel phosphorylation and activation of SRF by GSK-3 that is critical for SRF-dependent axon growth in mammalian central neurons.
Keywords: axon growth, filopodia, GSK-3, neurite outgrowth, serum response factor
Introduction
In developing CNS, neurite initiation and outgrowth are essential to ensure proper development of axons and to establish functional neuronal connections (Zhou and Snider, 2006; Polleux and Snider, 2010). SRF is a MADS-domain-containing transcription factor that is critical for expression of several stimulus-activated and cytoskeletal genes, including actin, vinculin, c-fos, and egr-1 (Treisman, 1987; Miano, 2003; Ramanan et al., 2005). Recent studies, including our own, using conditional knock-out mice have shown that SRF plays a critical cell-autonomous role in axonal growth both in vitro and in vivo (Knöll et al., 2006; Lu and Ramanan, 2011). Despite these findings, the post-translational mechanisms by which SRF activity is regulated in neurons as well as the downstream target genes that are critical for mediating axon growth remain poorly understood.
Among the intracellular signaling mechanisms, GSK-3 signaling has gained particular prominence in the regulation of both neuronal polarity and axon outgrowth. GSK-3 is a serine/threonine kinase first characterized for its role in glycogen metabolism (Cohen and Frame, 2001; Doble and Woodgett, 2003). Recent studies, using both pharmacological and genetic approaches to block GSK-3 activity, have shown that GSK-3 is necessary for axon growth (Kim et al., 2006; Garrido et al., 2007; Alabed et al., 2010; Hur et al., 2011). Complete inactivation of GSK-3 kinases resulted in strong inhibition of axon growth, which may be the result of dampened microtubule dynamics (Kim et al., 2006; Alabed et al., 2010). A widely studied mechanism by which GSK-3 regulates axon growth is by regulating microtubule reorganization in the growth cone (Zhou et al., 2004; Yoshimura et al., 2005; Kim et al., 2006). Despite these advances, whether GSK-3 activates transcriptional mechanisms critical for axon outgrowth remains known. It is possible that axonal growth deficits caused by inhibition of GSK-3 activity could result, at least in part, from inhibition of transcriptional programs critical for axon growth.
In this study, we identify SRF as a novel transcriptional target of GSK-3 and show that phosphorylation of SRF by GSK-3 is necessary for SRF transcriptional activity. Specifically, this phosphorylation is required for SRF interaction with the myocardin family cofactors, MKL1 and MKL2, but not with the TCF-family cofactor, ELK1. In addition, blockade of GSK-3 activity attenuates SRF target gene expression. Furthermore, SRF is sufficient to promote axonal growth in the absence of GSK-3 signaling. We also found that SRF is essential for filopodia formation, a necessary first step for Stage 1 axon growth. Finally, we demonstrate that vinculin (VCL), an actin-binding protein and previously identified SRF target gene, can promote axonal growth in SRF-deficient and GSK-3 inhibited neurons. Together, our findings suggest that SRF is likely a critical transcription factor for GSK-3-mediated axonal growth.
Materials and Methods
Animals.
The generation of Srf-f/f mice was previously described (Ramanan et al., 2005). These mice were bred to Nestin-Cre transgenic mice (The Jackson Laboratory). Srf-NestinCKO mutant mice were obtained from crosses between Srf-f/f and Srf-f/+;NestinCre double heterozygotes. Mice of either sex were used for preparing neuronal cultures. All experiments were approved by Animals Studies Committee, Division of Comparative Medicine, Washington University School of Medicine.
Immunohistochemistry.
Immunohistochemistry was performed as previously described (Ramanan et al., 2005; Lu and Ramanan, 2011). Briefly, P0.5 animals were fixed by transcardial perfusion. The brains were cryopreserved in 30% sucrose, frozen, and stored at −80°C until use. For staining, 12 to 16 μm cryosections were made and incubated in blocking/permeabilization solution containing 3% goat serum + 0.3% Triton-X in PBS. The antibodies used are listed below (see Immunocytochemistry).
Neuronal culture, transfection, and pharmacology.
Hippocampus and neocortex were dissected from postnatal 0.5 d (P0.5) mice, digested with papain for 30 min at 30°C, and dissociated with micropipette tips in culturing medium (Neurobasal-A medium supplemented with 0.5 mm l-glutamine and B27 supplements, Invitrogen). Neurons were then plated on coverglasses (Glaswarenfabrik Karl Hecht) coated with poly-d-lysine or poly-d-lysine and laminin at a density of ∼125 neurons/mm2. Neuronal cultures were incubated at 37°C with 5% CO2. After neurons attached to poly-d-lysine coverglass (∼15 min after plating), the medium was gently exchanged to new neuronal culturing medium supplemented with 33 U/ml penicillin/streptomycin. After 4 d in culture, neurons were fixed with 4% PFA/4% sucrose in PBS for 20 min. Neurons were transfected using Amaxa Nucleofection (Lonza) according to the manufacturer's instructions. A total of 3–5 μg of DNA was used for each transfection. The inhibitors, 6-bromoindirubin-3′-acetoxime (500 nm, Calbiochem) or lithium chloride (2 mm, Sigma-Aldrich) were directly added to neuronal culture medium ∼15 min after plating at the initial medium exchange.
Phalloidin staining.
Neuronal cultures were rinsed in PBS and fixed in 1–2% paraformaldehyde in PBS for 15 min at room temperature; 0.1 m glycine was added to quench excess aldehyde for 5 min. The cultures were then permeabilized with 0.1% Triton X-100 in PBS for 5 min and incubated in 1:125 dilution of rhodamine-phalloidin (Cytoskeleton) for 15 min. The cultures were washed with PBS for 5 min, thrice, and mounted on slides.
Immunocytochemistry and quantification of axon growth.
Neurons were fixed at different time points. Fixed neurons on coverglass were blocked and permeabilized with blocking solution (10% goat serum and 0.1% Triton-X in PBS) for 30 min with gentle rocking, incubated with primary antibody diluted in blocking solution for 45 min with gentle rocking, washed with PBS for 2 min thrice, incubated with secondary antibody diluted in blocking solution for 25 min with gentle rocking, and then washed with PBS before mounting with vector shield mounting medium supplemented with DAPI (Vector Laboratories). The following primary antibodies were used: anti-SRF (1:2000, Santa Cruz Biotechnology), anti-MAP2 (1:1000, Sigma), anti-β-III tubulin (Tuj1, 1:1000, Sigma), anti-Tau (1:1000, Millipore), anti-pan sodium channel, clone K58/35 (1:500, Sigma), anti-HA (1:1000, Sigma), and anti-Myc (1:100, Developmental Hybridoma). The secondary antibodies were AlexaFluor-488 and -594–conjugated anti-mouse or anti-rabbit at 1:1000 dilution (Invitrogen). Only live neurons, based on the characteristic uncondensed DAPI staining, were chosen for analysis in a double-blinded manner. Neurite growth was analyzed by measuring the length of the longest neurite of Tuj1-positive neurons. ImageJ software and NeuronJ plug-in were used to measure neurite length, growth cone and cell soma areas, etc.
Immunoprecipitation.
P0.5 mouse brains were dissected and homogenized in ice-cold protein extraction buffer: 20 mm HEPES, 125 mm NaCl, 5 mm MgCl2, 0.2 mm EDTA, pH 8, 1 mm EGTA, 1 mm NaF, 5 mm DTT, glycerol, NP40, protease inhibitors (Roche), and phosphatase inhibitors (Roche). The homogenate was centrifuged at 12,000 × g at 4°C for 10 min, precleared with beads, and supernatant was quantitated with Bradford reagent (Fermentas). A total of 10 μl of protein-A magnetic beads was washed with 0.1% BSA in PBS twice and 3 times with PBS. The antibody of choice (1–3 μg) was conjugated to the beads by incubating with the beads on a rotary shaker for 1–3 h. The beads were then washed 3 times with PBS; 3 mg of total protein was mixed with the antibody-conjugated beads and incubated overnight at 4°C on the rotary shaker. The following day, the magnetic beads were washed with protein extraction buffer 3 times. The beads were boiled with Laemmli sample buffer before separation by SDS-PAGE electrophoresis. In addition to epitope antibodies listed above, GSK-3α/β antibody (Santa Cruz Biotechnology), GSK-3β, and GSK-3β pSer9 antibody (Cell Signaling Technology) were also used.
Site-directed mutagenesis.
Site-specific mutants were generated using QuikChange Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions. Briefly, complementary primers of ∼40 bp were designed with the mutation site in the middle of the sequence and 25 ng of template DNA was used along with 25 pmol each of the mutant primers. A 2 min extension for every 1 kb of template was used. PCR for a total of 18 cycles was set as follows: 95°C for 3 min; 68°C for (X-kb × 2 min); 18 cycles; 72°C for 10 min; end at 4°C. The reaction mixture was digested with DpnI to remove template DNA and ligation set with T4 DNA ligase for 1 h at room temperature. A small aliquot of the reaction was transformed into DH10B, and transformants were selected on appropriate antibiotic plates. The success of the site-directed mutation was confirmed by DNA sequencing of both strands of at least 2 independent clones.
Bacterial expression of proteins.
SRF and SRF-S224A were cloned into pGEX-4T-1 (GST tag vector) (BD Biosciences). BL21(DE3) strain of Escherichia coli was transformed with the target constructs. Transformants were grown in LB medium until OD600 was ∼0.6 and induced with 1 mm IPTG at 16°C overnight. Bacteria were harvested the following day, and GST-tagged target protein was purified using TALON affinity cobalt column (Clontech) as per the manufacturer's instructions. Purified proteins were quantitated, snap-frozen in liquid nitrogen, and stored as single-use aliquots at −80°C.
In vitro kinase assay.
The different mutants of GSK-3β were expressed and immunopurified from HEK293T cells as previously described (Cho and Johnson, 2003). Briefly, HA-GSK-3β-CA and HA-GSK-3β-R96A constructs were transfected into HEK293T cells by Amaxa Nucleofection (Lonza). Immunoprecipitation assays were performed using anti-HA antibody as described above with the following modification. In the final step, beads were washed 3 times with 2× kinase buffer (40 mm Tris, pH 7.4, 20 mm MgCl2, and 2.5 mm CaCl2) and resuspended in the 2× kinase buffer supplemented with 20 μm ATP. The beads were incubated with the same volume of bacterially purified GST-SRF, GST-SRFS224A, or GST alone (1 μg of protein used for SRF and S224A proteins and 0.373 μg of GST) along with 5 μCi of [γ-32P]ATP. The samples were incubated at 37°C for 10 min, and the reaction was stopped by the addition of Laemmli buffer. The samples were then separated by SDS-PAGE gel, and the radioactive signal was analyzed using a Phosphoimager.
In vivo kinase assay.
HEK293T cells were cotransfected with HA-tagged SRF or HA-SRFS224A along with Myc-GSK-3β-WT, Myc-GSK-3β-KD, or Myc-GSK-3β-CA. After 48 h, total lysate was prepared; SRF was immunoprecipitated using anti-HA antibody and run on a 10% SDS-PAGE gel. The Western blot was probed using a phospho-serine antibody, anti-MPM2 (Invitrogen). The blot was then striped and probed for total SRF.
Luciferase reporter gene assays.
Luciferase assays were performed in HEK293T. After transfection, cells were grown in serum-starved condition for 36 h; the medium was changed to that supplemented with 10% FBS. Luciferase assay was performed using Dual Luciferase Assay kit (Promega) 6 h after serum induction. Transfection efficiency was normalized using a second reporter gene, Renilla luciferase, and compared with unstimulated controls.
Statistical analyses.
Statistical differences were determined with one-way ANOVA followed by Tukey's post hoc test or with two-tailed Student's t test using GraphPad Prism software. Error bars in figures are SEM. “n” within the bars denotes the number of cells counted for analysis.
Microarray analysis.
Total RNA was isolated from the forebrain of P0.5 Srf-f/f and Srf-NestinCKO mice (n = 2) using PureLink RNA mini kit (Ambion). A total of 5 μg of total RNA was submitted to the Multiplexed Gene Analysis Core Facility, Washington University School of Medicine (St. Louis, Missouri) for quality control, labeling, hybridization, scanning, and software services. Affymetrix Mouse Gene 1.0 ST was used for the microarray, and data mining was performed using Partek Genomics Suite software. Principal component analysis was performed to assess the quality of the data. Genes that showed a fold change of at least 1.5 and a present call in all 2 chips were considered. In addition, a two-tailed t test with p < 0.05 was applied. From the list of downregulated genes, only those that were either part of, or predicted to interact with, the cytoskeleton were used for further analysis. Figure 7 shows genes that were downregulated in Srf-NestinCKO mutant brains compared with control. After validation using qRT-PCR, these genes were then screened for their ability to promote axon growth in SRF-deficient hippocampal neurons.
Figure 7.

MAP1B expression is attenuated in SRF-deficient neurons. A, Lack of expression of MAP1B in SRF-deficient neurons. SemiqRT-PCR of total RNA isolated from brains of Srf-NestinCKO mice and Srf-f/f control littermates (n = 3). RPS29 expression served as the control. B, MAP1B expression partially promotes axon growth in SRF-null neurons. Hippocampal neurons from neonatal Srf-NestinCKO mice were transfected with either control myc vector or myc-tagged MAP1B along with farnesylated GFP (F-GFP) and grown for 4 d on poly-d-lysine and laminin. MAP1B was able to partially increase axonal length in SRF-deficient cells. However, MAP1B was not efficient in rescuing the enlarged growth cone (arrows) and abnormal cell soma (arrowheads) morphologies observed in SRF-deficient neurons. Expression of MAP1B did not increase axonal growth in WT neurons. Scale bar, 50 mm. C, Quantitation of axon length and growth cone area from B. ***p < 0.001 (one-way ANOVA and Tukey post-test analysis). n.s., Not significant.
Image acquisition.
Fluorescent and bright-field images were acquired using Nikon 80i and Nikon 90i microscopes (Nikon Instruments), Roper H2 cooled CCD camera (Photometrics), and Optigrid Structured Illumination Confocal with a Prior (Rockland) motorized translation stage. Pseudo-colored images were acquired in MetaMorph (Molecular Devices), filtered, and levels were modified in ImageJ (National Institutes of Health, Bethesda, Maryland) and Photoshop (Adobe) for clarity.
Results
GSK-3β phosphorylates SRF on a highly conserved serine reside
SRF has been shown to regulate axonal growth in cultured hippocampal and DRG neurons (Knöll et al., 2006; Wickramasinghe et al., 2008). Our recent findings using conditional SRF mutant mice revealed that SRF is cell-autonomously required for growth of several major axonal tracts, including corticostriatal and entorhinal–hippocampal axonal projections in vivo (Lu and Ramanan, 2011). To understand the molecular mechanisms by which SRF regulates axonal growth, we first sought to determine how SRF activity is regulated in neurons. Transcription factors, in general, are regulated by at least two mechanisms: post-translational modifications and association with specific cofactors. SRF has been shown to be phosphorylated upon synthesis, but the regulation of its activity in neurons by phosphorylation has not been reported (Misra et al., 1991; Rivera et al., 1993). We took an in silico approach to identify phosphorylation sites on SRF using Scansite (Obenauer et al., 2003). Among several residues identified as potential recognition motifs of different kinases, serine 224 (ser228 in human SRF) was predicted to be the single target site for GSK-3β. This serine residue lies within previously described GSK-3 consensus sequence, S/T-X-X-X-S/T, in which the first Ser or Thr is the phosphorylated residue (Doble and Woodgett, 2003) (Fig. 1A). Ser224 is highly conserved across species, and structural analyses suggested that this residue should be distant from target DNA binding and accessible for modification (Fig. 1A,B) (Pellegrini et al., 1995). Interestingly, GSK-3-inhibited wild-type and SRF-deficient neurons exhibit similar morphologies, including highly attenuated axon growth, suggesting a possible functional link between SRF and GSK-3 (Goold et al., 1999; Owen and Gordon-Weeks, 2003; Kim et al., 2006; Stern et al., 2009; Hur et al., 2011).
Figure 1.
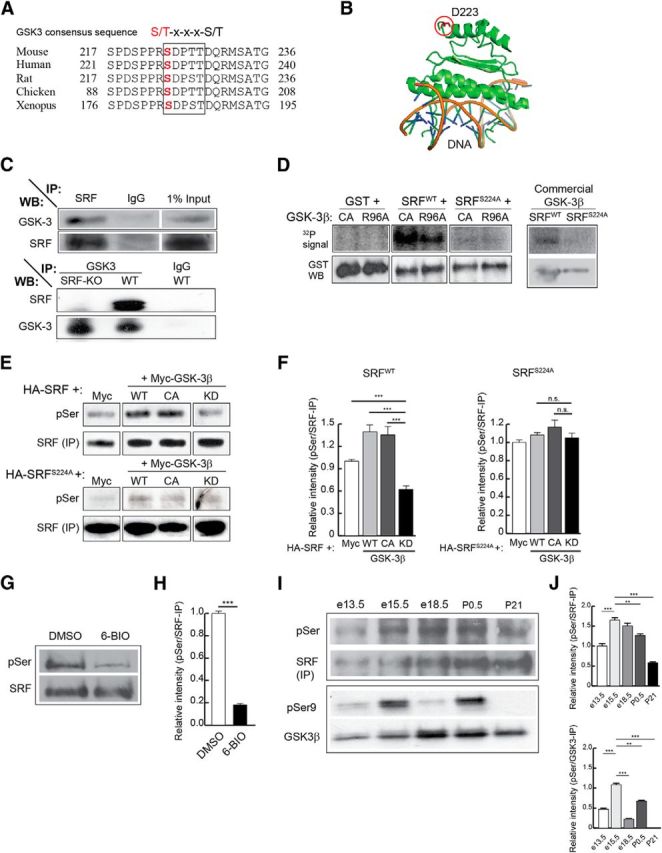
SRF interacts with and is phosphorylated by GSK-3 on ser224. A, Protein alignment shows that ser224 in mouse SRF (ser228 in human) is highly conserved among different species. The boxed region represents the suggested GSK-3 consensus sequence, S/T-x-x-x-S/T. B, Analysis of the solved x-ray crystal structure of the N-terminus 1–223 amino acids of human SRF bound to DNA shows that the aspartate residue at 223 is far away from the DNA binding region of SRF. Therefore, the target serine 228 (human), located five amino acids further c terminus, is unlikely to interfere with DNA binding. C, SRF and GSK-3 interact in the brain. SRF was immunoprecipitated from whole-brain lysate of P0.5 wild-type mice and immunoblotted for GSK-3 and SRF (top). GSK-3α and GSK-3β were efficiently pulled down only with the SRF antibody and not with anti-IgG control antibody. Likewise, GSK-3 was able to pull down SRF only from wild-type but not from Srf-NestinCKO brain lysate (bottom). Pull down using IgG alone served as control. D, SRF is efficiently phosphorylated by GSK-3β in vitro. Kinase assays ware performed using bacterially generated GST, GST-SRF, or GST-SRFS224A along with either GSK-3β-CA, GSK-3β-R96A mutant kinases or commercial recombinant wild-type GSK-3β in the presence of [γ-32P]ATP. GST-SRF alone was phosphorylated by GSK-3β-CA, GSK-3β-R96A, and wild-type GSK-3β. In contrast, SRFS224A mutant and GST alone showed no signal suggesting that there are no other GSK-3β sites on SRF. E, SRF is also phosphorylated by GSK-3β in vivo. HEK293T cells were transfected with HA-tagged SRFWT or HA-SRFS224A along with empty Myc vector or with each of the different Myc-tagged GSK-3β kinases. SRF was pulled down with anti-HA antibody and immunoblotted using anti-phosphoserine (anti-MPM2) and anti-SRF antibodies. The level of phosphorylation of HA-SRFWT increased in the presence of active GSK-3β kinases (WT and CA), whereas phosphorylation is attenuated in the presence of GSK-3β-KD mutant kinase. SRFS224A mutant did not exhibit any changes in phosphorylation levels. F, Densitometry analysis of results from E shows changes in phosphorylation levels relative to that for SRFWT or SRFS224A protein levels. Error bars indicate SEM. ***p < 0.001 (one-way ANOVA, Tukey post-test analysis). n.s., Not significant. G, SRF is phosphorylated by GSK-3 in neurons. Hippocampal neurons from neonatal pups were grown for 3 d in the presence of either DMSO (vehicle) or 500 nm 6-BIO to inhibit GSK-3. Endogenous SRF was immunoprecipitated with anti-SRF antibody and immunoblotted with anti-phosphoserine antibody. The level of serine phosphorylation decreased in the presence of 6-BIO compared with DMSO-treated control. H, Densitometric analysis of phospho-SRF to total SRF levels from G shows significant decrease in SRF serine phosphorylation in the presence of 6-BIO relative to DMSO control. Error bars indicate SEM. I, Changes in SRF and GSK-3 phosphorylation during brain development. SRF was immunoprecipitated from whole-brain lysates of wild-type embryos and pups at the indicated stages and immunoblotted with pSer antibody. GSK-3β activity was measured by immunoblotting for GSK-3β-Ser9 phosphorylation. SRF expression increased during development, whereas the level of pSer phosphorylation peaked ∼e15.5-e18.5. GSK-3β-Ser9 phosphorylation decreased between e15.5 and e18.5 and postnatally, suggesting GSK-3β activation during this time period. J, Densitometric analysis of pSer levels and GSK-3β-pSer9 from I shows increase in pSer phosphorylation of SRF and GSK-3β ∼e15.5 to e18.5. Error bars indicate SEM. ***p < 0.001 (one-way ANOVA, Tukey post-test analysis). **p < 0.01 (one-way ANOVA, Tukey post-test analysis).
Based on this prediction, we first examined whether SRF and GSK-3 physically interact in the brain. Using pull-down assays from wild-type brain lysates, we found that SRF and GSK-3 were able to coimmunoprecipitate with each other (Fig. 1C). This was further confirmed using Srf-NestinCKO and control brain lysates, in which GSK-3 coimmunoprecipitated with SRF from control lysate alone but not from Srf-NestinCKO lysate (Fig. 1C). Together, these experiments suggest that GSK-3 either directly interacts with SRF or associates with the SRF transcriptional complex.
To address the functional relevance of the predicted phosphorylation and the coimmunoprecipitation results, we tested whether GSK-3β directly phosphorylates SRF on ser224 residue using in vitro kinase assay. Bacterially expressed and purified GST, GST-SRFWT, or GST-SRFS224A was transiently incubated with GSK-3β-CA (constitutively active) or GSK-3β-R96A (active on unprimed substrates alone) or commercial GSK-3β kinase in the presence of radioisotopic [γ-32P]ATP (Cho and Johnson, 2003). SRFS224A was generated by mutating serine 224 to alanine by site-directed mutagenesis. We found that SRFWT was efficiently phosphorylated by all three GSK-3 kinases (Fig. 1D). In contrast, GST alone and SRFS224A mutant exhibited no phosphorylation signal in the presence of these GSK-3 kinases, suggesting that the phosphorylation occurs specifically on ser224 (Fig. 1D). GSK-3 has been shown to phosphorylate two classes of substrates, primed and unprimed, depending on the requirement of a priming phosphorylation on a discrete residue c-terminal to the target site (Frame et al., 2001; Doble and Woodgett, 2003). The unprimed status of a substrate can be determined using the GSK-3β-R96A mutant, which can bind to and phosphorylate only unprimed substrates (Frame et al., 2001; Kim et al., 2006; Twomey and McCarthy, 2006). The phosphorylation of bacterially generated SRFWT, which should lack any priming phosphorylation, by GSK-3β-R96A suggests that SRF is an unprimed substrate for GSK-3β.
We next asked whether GSK-3 can also phosphorylate SRF in vivo. This was studied using in vivo phosphorylation assays, in which HEK293T cells were cotransfected with HA-SRFWT or HA-SRFS224A along with GSK-3β-WT, GSK-3β-CA, or GSK-3β-KD. GSK-3β-KD is a catalytically inactive mutant that functions as a dominant-negative kinase and inhibits both GSK-3α and GSK-3β (He et al., 1995; Kim et al., 2006; Kang et al., 2008). The HA-SRFs were immunoprecipitated with anti-HA antibody and phosphorylation was studied using a phospho-serine (pSer)-specific antibody, anti-MPM2. We observed that SRFWT expressed alone exhibited a basal level of serine phosphorylation and this is likely the result of phosphorylation on ser224 by endogenous GSK-3 or on serine residues by other kinases (Fig. 1E) (Misra et al., 1991; Rivera et al., 1993). Expression of GSK-3β-CA or GSK-3β-WT increased pSer levels, whereas GSK-3β-KD expression attenuated pSer signal (Fig. 1E,F). Interestingly, the pSer signal on SRFWT in the presence of GSK-3β-KD expression was lower than that seen when SRFWT alone was expressed, confirming the hypothesis that endogenous GSK-3 contributes to basal serine phosphorylation on SRF (Fig. 1F, left; compare SRFWT + Myc vs SRFWT + GSK-3β-KD). In contrast, SRFS224A point mutant exhibited neither any increase in phosphorylation in the presence of GSK-3β-CA or GSK-3β-WT nor any attenuation of pSer phosphorylation when coexpressed with GSK-3β-KD (Fig. 1E,F).
Given that SRF is efficiently phosphorylated by GSK-3 both in vitro and in 293T cells, we asked whether similar GSK-3-mediated phosphorylation of SRF also occurs in neurons. We inhibited GSK-3 kinases using the specific inhibitor, 6-bromoindirubin-3′-acetoxamine (6-BIO) (500 nm) and then probed for SRF phosphorylation with anti-pSer antibody. 6-BIO is a cell-permeable small-molecule inhibitor that is highly selective for GSK-3 but not for other kinases, including cyclin-dependent kinases (Meijer et al., 2003; Kim et al., 2006). We found that SRF was robustly phosphorylated in cultured hippocampal neurons, and this phosphorylation was significantly attenuated in the presence of 6-BIO (Fig. 1G,H). Together, these experiments demonstrate that SRF is a novel target of GSK-3 and is phosphorylated in an unprimed manner on ser224 both in vitro and in cells.
Because GSK-3 phosphorylates SRF, we therefore examined whether SRF phosphorylation is regulated in the brain during development. SRF was immunoprecipitated from whole-brain lysates at E13.5, E15.5, E18.5, P1, and P21, and status of serine phosphorylation was probed using anti-pSer antibody (Fig. 1I). SRF expression increased during development and remained higher postnatally (Fig. 1I). In contrast, SRF phosphorylation levels increased and peaked between E15.5 and E18.5. We then assessed for GSK-3 activation by probing for GSK-3β-Ser9 phosphorylation, a measure of its inactivation (Cohen and Frame, 2001; Doble and Woodgett, 2003). We observed that GSK-3β-Ser9 phosphorylation decreased between E15.5 and E18.5, suggesting that GSK-3β activity increases during this time, similar to that observed for SRF-Ser224 phosphorylation (Fig. 1I,J). This increase in SRF phosphorylation appears to coincide with active axonal growth that occurs after neurogenesis and is likely mediated by GSK-3.
Serine 224 phosphorylation by GSK-3 is critical for SRF function
To assess whether ser224 phosphorylation is important for SRF function, we asked whether SRFS224A mutant can rescue axonal growth deficits of Srf-NestinCKO neurons. Reintroduction of SRFWT into Srf-deficient neurons rescued axonal growth. In contrast, SRFS224A mutant was unable to rescue axonal growth in Srf-NestinCKO neurons (Fig. 2A,B). Interestingly, SRFS224A acted as a dominant-negative mutant when expressed in wild-type neurons and attenuated axonal growth (Fig. 2C,D). We compared this inhibitory effect of SRFS224A mutant on axonal growth with two other dominant-negative SRF mutants, SRF-I and SRF-En. SRF-I contains the N terminus homodimerization and DNA binding domains alone but lacks the C terminus transactivation domain. SRF-I has been previously shown to be very efficient in blocking activation of SRF-promoters in cultured cells and is thought to function by either saturating SRF binding sites on promoters or by sequestering endogenous SRF by forming nonactive heterodimers (Kemp and Metcalfe, 2000). SRF-En is a chimeric protein generated by fusing SRF-I with the repressor domain (amino acids 2–298) of the engrailed gene from Drosophila (Jaynes and O'Farrell, 1991). Engrailed is a homeodomain protein and is a potent repressor of transcription. This repressor domain, when fused to other transcription factors, has been shown to function as dominant-negative regulators of transcription (Badiani et al., 1994; Vickers and Sharrocks, 2002; Vickers et al., 2004). Expression of SRF-I and SRF-En in wild-type neurons also resulted in attenuated axonal growth similar to that of SRFS224A mutant (Fig. 2C,D).
Figure 2.

Serine 224 is required for SRF function. A, SRFS224A mutant cannot rescue axonal growth deficits of SRF-deficient neurons. P0.5 hippocampal neurons from Srf-NestinCKO mice were transfected with IRES-EGFP empty vector, HA-SRFWT, or HA-SRFS224A. Cells were fixed and immunostained for GFP or HA (green) to visualize transfected cells and anti-Tuj1 (red) at 4 DIV. Axonal growth deficits exhibited by SRF-deficient neurons were rescued by SRFWT alone and not by empty vector or SRFS224A (arrows). Arrowhead indicates an untransfected cell. Scale bar, 50 μm. B, Quantitation of axon length from A. n = 4 mice; “n” in the bars indicates the number of cells measured for statistics. Error bars indicate SEM. ***p < 0.001 (one-way ANOVA, Tukey post-test analysis). n.s., Not significant. C, SRFS224A functions as a dominant mutant when expressed in wild-type hippocampal neurons. Cells were transfected with HA-SRFWT or HA-SRFS224A and grown for 4 DIV. Cells were fixed and immunostained with anti-HA (green) and anti-Tau (red) antibodies. HA-SRFS224A expression attenuated axonal growth (arrows). Scale bar, 50 μm. D, Data quantified from C. Error bars indicate SEM. ***p < 0.001 (one-way ANOVA, Tukey post-test analysis). E, Inhibiting GSK-3 activity or mutating serine 224 to alanine abolishes SRF transcriptional activity. HEK293T cells were transfected with 5×-SRE luciferase and Renilla luciferase (for transfection efficiency) and treated with either 6-BIO or cotransfected with control vector, SRFWT, SRF-S224A, or SRF-VP16. 6-BIO treatment blocked serum-induced SRF-transcriptional activity. Whereas vector and SRFWT transfected cells showed normal activation of an SRE reporter, the SRFS224A mutant was severely compromised in activating transcription. Expression of constitutively active SRF-VP16 served as the control. Error bars indicate mean ± SEM.
We next determined the effect of GSK-3 inhibition and SRF-S228A mutation on SRF-dependent transcription. For this, HEK293T cells were transfected with SRE-luciferase reporter constructs and treated with either 6-BIO to inhibit GSK-3 activity or cotransfected with SRFWT, SRFS224A, SRF-VP16, SRF-I, and SRF-En (Fig. 2E). SRF-VP16 is a constitutively active chimeric protein, which is generated by fusing 260 amino acids of SRF N terminus with the strong transactivation domain of VP16 protein from herpes simplex virus. SRF-VP16 transcriptional activity has not been shown to depend on cofactor requirement or post-translational modifications. Expression of SRF-VP16 results in increased reporter activation, even in the absence of any inducing stimulus (Fig. 2E). Inhibition of GSK-3 activity blocked serum-induced SRF-dependent transcription (Fig. 2E). Unlike SRFWT but similar to SRF-I and SRF-En, SRFS224A mutant was also incapable of activating SRF-dependent transcription (Fig. 2E). Together, these experiments suggest that GSK-3 activity and SRF-Ser224 phosphorylation is critical for SRF function in neurons.
Phosphorylation of SRF by GSK-3 is necessary for MKL cofactor binding
Given that Ser224 phosphorylation by GSK-3 is necessary for SRF function, a critical question is how Ser224 phosphorylation contributes to SRF-dependent transcription. One of the widely described regulatory mechanisms for SRF-dependent transcription is its association with specific cofactors (Sharrocks, 2001; Cen et al., 2004). We therefore asked whether SRFS224A mutant can interact with TCF-family member ELK1 and myocardin-family members MKL1 and MKL2 using coimmunoprecipitation assays. HEK293T cells were cotransfected with HA-tagged SRFWT or SRFS224A along with Flag-MKL1 or Myc-MKL2. SRF was immunoprecipitated with anti-HA antibody and immunoblotted for Flag or Myc epitopes. S224A mutation specifically attenuated SRF binding to MKL1 and MKL2 (Fig. 3A,B). To probe ELK1 interaction, the ability of SRFWT and SRFS224A mutant to immunoprecipitate endogenous activated phospho-ELK1 (pELK1) was analyzed. In contrast to MKL1 and MKL2, the interaction of SRFS224A mutant with pELK1 was unaffected (Fig. 3C,D), suggesting that phosphorylation of ser224 by GSK-3 is necessary for the recruitment of MKLs, but not TCF-family cofactors, onto SRF.
Figure 3.
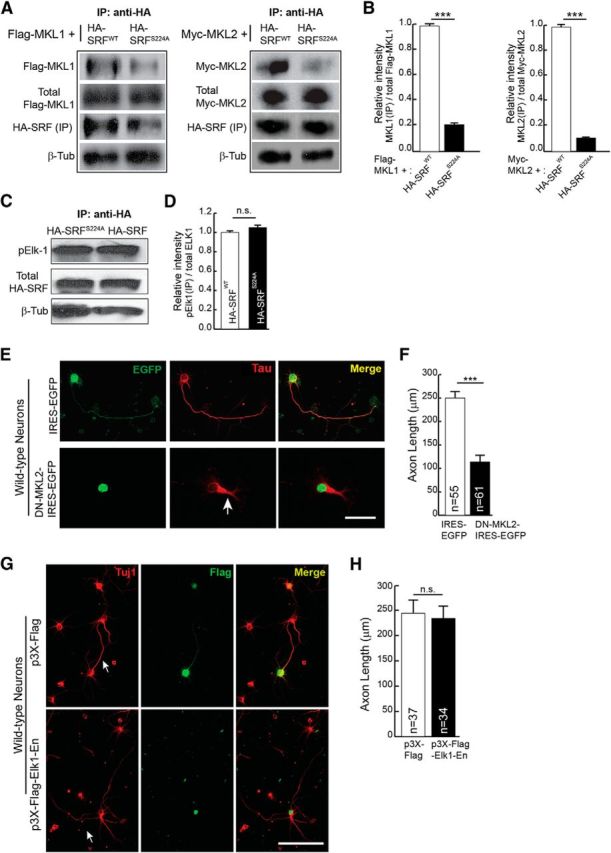
Serine 224 phosphorylation is required for SRF interaction with MKL-family cofactors and not with TCF-family cofactor ELK1. A, HEK293T cells were cotransfected with HA-SRFWT or HA-SRFS224A along with Flag-MKL1 or Myc-MKL2. Immunoprecipitation with anti-HA antibody pulled down significantly less MKL1 (left) and MKL2 (right) from HA-SRF224A-transfected cells. The expression levels of Flag-MKL1, Myc-MKL2, and HA-SRF were the same in both groups of transfected cells. β-Tubulin levels in equal amounts of lysates used for IP served as loading control. B, Densitometric analysis of immunoblot in A. Error bars indicate SEM. C, Serine 224 to alanine mutation does not affect SRF interaction with the TCF-family member ELK1. HEK293T cells were transfected with HA-SRFWT or HA-SRFS224A. Immunoprecipitation with anti-HA antibody and immunoblotting for endogenous activated ELK1 using anti-phosphoELK1 (pELK1) antibody showed that a similar amount of pELK1 was immunoprecipitated with HA-SRFWT and HA-SRFS224A. Levels of SRF in the immunoprecipitates were similar and β-tubulin levels in equal amounts of lysates used for IP served as loading control. D, Densitometric analysis of immunoblot in C. Error bars indicate SEM. n.s., Not significant. E, MKL1 and MKL2 are required for axon growth. Neonatal wild-type hippocampal neurons were transfected with IRES-EGFP vector or DN-MKL2-IRES-EGFP. Expression of DN-MKL2 blocked axonal growth (arrow) as visualized with anti-Tau (red) and anti-GFP (green). Scale bar, 50 μm. F, Quantitation of D. Error bars indicate SEM; n = 3 mice. ***p < 0.001 (two-tailed t test analysis). G, Inhibition of TCF-family cofactors does not affect axonal growth. Hippocampal neurons from wild-type neonatal pups were transfected with dominant-negative Elk1-En (bottom) or empty vector (top). Cells were stained for Tuj1 (red) and Flag epitope (green) at 4 DIV. Expression of Elk1-En, which inhibits all three members of the TCF family, does not affect axonal growth. Scale bar, 50 μm. H, Quantitation of axon length in G. Error bars indicate SEM; n = 3 mice. p = 0.8087 (two-tailed t test analysis).
Previous studies have shown that the MKL-family cofactors are required for axonal growth (Knöll et al., 2006; Wickramasinghe et al., 2008; Mokalled et al., 2010; O'Sullivan et al., 2010). Consistent with these studies, we found that expression of a dominant-negative MKL2 (DN-MKL2), which inhibits the activities of both MKL1 and MKL2, can block axonal growth in hippocampal neurons (Selvaraj and Prywes, 2003) (Fig. 3E,F). Thus far, the requirement for TCF-family cofactors in regulating axon growth has not been studied. Elk1-null mice are viable and do not exhibit any structural abnormalities in the brain, and this is attributed to compensation by family members, Sap1 and Sap2 (Cesari et al., 2004). To address the requirement of TCF-family cofactors for axon growth, we expressed ELK1-Engrailed (ELK1-En), a chimeric protein generated by fusing ELK1 coding region with the strong transcriptional repressor domain of the Drosophila gene, Engrailed. ELK1-En has been shown to efficiently inhibit the activities of all three members of the TCF family (Vickers and Sharrocks, 2002; Vickers et al., 2004). Expression of ELK1-En in hippocampal neurons did not affect axon outgrowth (Fig. 3G,H). Together, these data suggest that phosphorylation of ser224 by GSK-3 and subsequent binding of MKLs and not TCF-family members contribute to SRF-dependent axon growth.
SRF can rescue axon outgrowth deficits caused by GSK-3 inhibition
Based on the above findings, we hypothesized that SRF likely functions downstream of GSK-3 signaling and that activation of SRF-dependent gene expression should be able to promote axonal growth when GSK-3 kinases are inhibited. To test this, SRF-dependent gene expression was activated using constitutively active SRF-VP16, whereas GSK-3 kinases were inhibited by LiCl (2 mm) or 6-BIO (500 nm) (Meijer et al., 2003; Kim et al., 2006). 6-BIO at 300 nm concentrations was found to be efficient in blocking GSK-3 activity and axon outgrowth in DRG neurons (Kim et al., 2006; Alabed et al., 2010). However, we found that 500 nm of 6-BIO was more efficient in blocking axonal growth in hippocampal neurons without affecting neuronal survival (data not shown). Consistent with earlier reports, culturing hippocampal neurons in the presence of LiCl or 6-BIO severely attenuated axonal growth (Kim et al., 2006; Alabed et al., 2010) (Fig. 4A,B; and data not shown). SRF-VP16 expression in neurons grown in the presence of 6-BIO or LiCl was able to significantly promote axon growth compared with IRES-EGFP control vector (Fig. 4A,B; and data not shown). Expression of SRF-VP16 alone in neurons did not produce any additional growth-promoting effects under normal conditions (Fig. 4D). We next asked whether this rescue specifically requires SRF or whether other transcription factors can promote axonal growth under conditions of GSK-3 inhibition. Previous studies using knock-out mice have shown that CREB is required for axonal growth in peripheral neurons (Lonze et al., 2002; Gao et al., 2004). Expression of constitutively active CREB (VP16-CREB) was unable to rescue the growth deficits resulting from GSK-3 inhibition, indicating that CREB-dependent gene expression might be insufficient to rescue axonal growth when GSK-3 is inhibited (Fig. 4A,B). In addition to pharmacological inhibition, we also expressed GSK-3β-KD, which acts as a dominant-negative mutant and has been shown to attenuate axon growth in cultured hippocampal neurons (Fig. 4C) (Kim et al., 2006). Similar to that seen for 6-BIO-mediated inhibition, expression of GSK-3β-KD in hippocampal attenuated axonal growth, and this was rescued by SRF-VP16 expression (Fig. 4C,D). Together, these findings suggest that SRF functions downstream of GSK-3 and that activation of SRF-dependent expression can promote axon growth when GSK-3 activity is inhibited.
Figure 4.

SRF can rescue axonal growth deficits caused by GSK-3 inhibition. A, P0.5 hippocampal neurons from wild-type mice were transfected with either empty IRES-EGFP vector or constitutively active SRF (SRF-VP16-IRES-EGFP) or constitutively active CREB (VP16-CREB) and grown in the presence of the GSK-3-specific inhibitor 6-BIO (500 nm) or DMSO (vehicle). Cells were fixed and immunostained using anti-Tau (red) and GFP or VP16 (green) at 4 DIV. GSK-3 inhibition by 6-BIO results in attenuation of axonal growth (top, arrowhead), and this was rescued by expression of SRF-VP16 (middle). Expression of a constitutively active CREB (VP16-CREB) was unable to promote axonal growth under similar growth conditions (bottom). Arrows indicate dead cells/debris. Scale bar, 50 μm. B, Quantitation of A. Error bars indicate SEM. **p < 0.01 (two-tailed t test analysis). ***p < 0.001 (two-tailed t test analysis). n.s., Not significant. C, Hippocampal neurons were cotransfected with GSK-3β constructs and SRF-VP16 as indicated above and grown for 4 d. Expression of GSK-3β-KD attenuated axon growth (middle). This attenuation was rescued by SRF-VP16 expression (bottom). Expression of GSK-3β-WT alone had no influence on axon outgrowth. Arrows indicate dead cells/debris. Scale bar, 50 μm. D, Axon length quantified from C. Error bars indicate SEM. ***p < 0.0001 (one-way ANOVA, Tukey post-test analysis).
SRF is required during Stage 1 neurite growth and axon extension
Given the importance of SRF for axon growth in vitro and in vivo, it is critical to know when SRF-dependent transcription is required during axon development. In cultures, hippocampal and neocortical neurons follow a stereotypic pattern of growth (Craig and Banker, 1994; de Lima et al., 1997). When dissociated and cultured, neurons form extensive filopodial and lamellipodial protrusions at the periphery (Stage 1) followed by extension of several neurites (Stage 2). Subsequently one of these neurites elongates rapidly in an asymmetric manner to become the axon (Stage 3) followed by dendritic specialization and growth (Stage 4) (Craig and Banker, 1994; de Lima et al., 1997). Hippocampal neurons cultured from neonatal Srf-NestinCKO mice and control littermates were fixed and stained for Tuj1 and MAP2 at the indicated time points and axonal length (Tuj1+/MAP2− process) was determined at each time point (Fig. 5A). We found that neurons cultured from both control and knock-out mice extended a single Tuj1+/MAP2− process, which alone was positive for the axonal marker, Tau (data not shown). In contrast, neurons from Srf-NestinCKO mice displayed no or very few processes, which exhibited almost no growth in the first 2 d (Fig. 5A,B). Subsequently, the average length of the axon in SRF-deficient neurons increased slightly but still was significantly lower than that of control neurons (Fig. 5A,B). At 5 d, Srf-NestinCKO neurons exhibited significantly attenuated axon growth along with an enlarged growth cone compared with Srf-f/f neurons (Fig. 5A,B).
Figure 5.

SRF-deficient neurons are deficient in Stage 1 neurite growth and exhibit attenuated axonal growth in vitro. A, Hippocampal neurons from Srf-NestinCKO mice exhibit delayed and attenuated axonal growth as visualized by immunostaining with Tuj1 (red) and MAP2 (green). SRF-deficient neurons produced fewer neurites at 12 h (arrows, left second row panels) and, at 5 d, had shorter axons and enlarged growth cone (right bottom, arrow). Control neurons extended longer axon (right middle, arrow) and multiple dendrites. Arrowheads indicate dead cells. Scale bar, 50 μm. B, Axonal growth in A was studied by measuring the length of the Tuj1+/MAP2− process at the indicated time points. Error bars indicate SEM. C, Srf-NestinCKO neurons are deficient in filopodia formation. Rhodamine-conjugated phalloidin reveals several filopodia (12 h, arrows) and distinct neurites (48 h) in control neurons. In contrast, mutant neurons had fewer or no filopodia (arrow) and an enlarged cell body. Scale bar, 12.5 μm. D, Histograms of the total filopodia counted from C. p < 0.0001 (two-tailed t test analysis). E, Immunostaining with axon-specific PanNaV (green) and Tuj1 (red) antibodies shows normal polarization of Srf-NestinCKO neurons as observed for Srf-f/f neurons (n = 32). Scale bar, 25 μm.
Analysis of axonal growth strongly suggested that the earliest deficits exhibited by Srf-NestinCKO neurons are in Stage 1 growth. During Stage 1, cell symmetry is breached by filopodial protrusions from cell soma, which is a necessary first step in the generation of neurites (Craig and Banker, 1994; Dent et al., 2007). To study Stage 1 growth in detail, hippocampal neurons from Srf-NestinCKO and control littermates were fixed and stained with phalloidin to visualize F-actin-rich filopodial protrusions at 12, 24, and 48 h after plating. We found that SRF-deficient neurons had very few or no filopodia at 12 and 24 h compared with numerous filopodial protrusions exhibited by control neurons (Fig. 5C,D; and data not shown). At 48 h, control neurons had extended neurites with many filopodia at the tips. In contrast, the SRF-deficient neurons exhibited few or no distinguishable neurites or filopodia (Fig. 5A–D). SRF-deficient neurons also exhibited an enlarged growth cone as previously reported (Knöll et al., 2006) and an enlarged cell soma. It has been reported that SRF-deficient neurons lack filopodia in growth cones of extending axons (Stern et al., 2009). Because filopodia formation is a critical step in neuronal growth, it is reasonable to speculate that the severe Stage 1 growth deficits observed in Srf-NestinCKO neurons are likely the result of inability of mutant neurons to form or stabilize filopodia. Together, these findings demonstrate that SRF-dependent transcription plays a critical role in filopodia formation during early stages of axon growth.
Although SRF-deficient neurons exhibited highly attenuated axonal growth, these neurons extended a single Tau-positive axon indicating that cell polarity is not affected. To further confirm this, we immunostained hippocampal neurons from Srf-NestinCKO mice and control littermates with a pan sodium channel antibody (Pan-NaV) directed against the α subU (intracellular III–IV loop) of all vertebrate channels. This Pan-NaV antibody labels only the proximal part of the axon (Garrido et al., 2003; Kress et al., 2010). We found that SRF-deficient neurons extended a single Pan-NaV-positive neurite suggesting that neuronal polarity is not impaired in the absence of SRF (Fig. 5E).
Actin-binding protein, vinculin, can rescue growth defects of SRF-deficient neurons
We next sought to identify the downstream target genes of SRF that are important for axon growth. We performed a microarray analysis to identify genes differentially expressed in the forebrain of neonatal Srf-NestinCKO mutant mice and control littermates (Fig. 6A). Because SRF-deficient neurons exhibited deficits in filopodia formation, we reasoned that proteins that bind actin and stabilize the actin cytoskeleton might be good candidates to rescue axon growth. We therefore considered only those downregulated genes that are either part of, or interact with, the cytoskeleton and validated their expression using qRT-PCR (Fig. 6B). We next asked which of these genes could rescue axonal growth in SRF-deficient neurons. Expression of cortexin, nexillin, and Rusc1 did not promote axonal growth in Srf-NestinCKO neurons (data not shown). However, expression of VCL and microtubule-associated protein 1B (MAP1B) was able to promote axon outgrowth in SRF-deficient neurons to varying extents. VCL is a previously identified SRF target gene, and earlier studies using chromatin immunoprecipitation and gene expression assays have shown that Vcl is a direct target of SRF and MKL1 (Moiseyeva et al., 1993; Schratt et al., 2002; Miralles et al., 2003; Cooper et al., 2007). VCL is an actin-binding protein and a component of cell–matrix junctions. In cultured CNS neurons, VCL is localized in cell soma, at the ends of neurites and along with focal adhesion kinase (Stevens et al., 1996; Contestabile et al., 2003). Studies from cultured cells have shown that VCL functions as a bridge between actin cytoskeleton and extracellular matrix (Ziegler et al., 2006; Humphries et al., 2007). Despite these findings, the functions of VCL in the nervous system remain poorly understood because of early embryonic lethality of Vcl-null mice due to cardiac and neural tube defects (Xu et al., 1998). MAP1B is a member of a large class of microtubule-associated proteins that function in stabilizing the microtubule (Dehmelt and Halpain, 2004). It is important for axon growth, and its function is regulated by phosphorylation by GSK-3β (Dehmelt and Halpain, 2004; Gonzalez-Billault et al., 2004).
Figure 6.
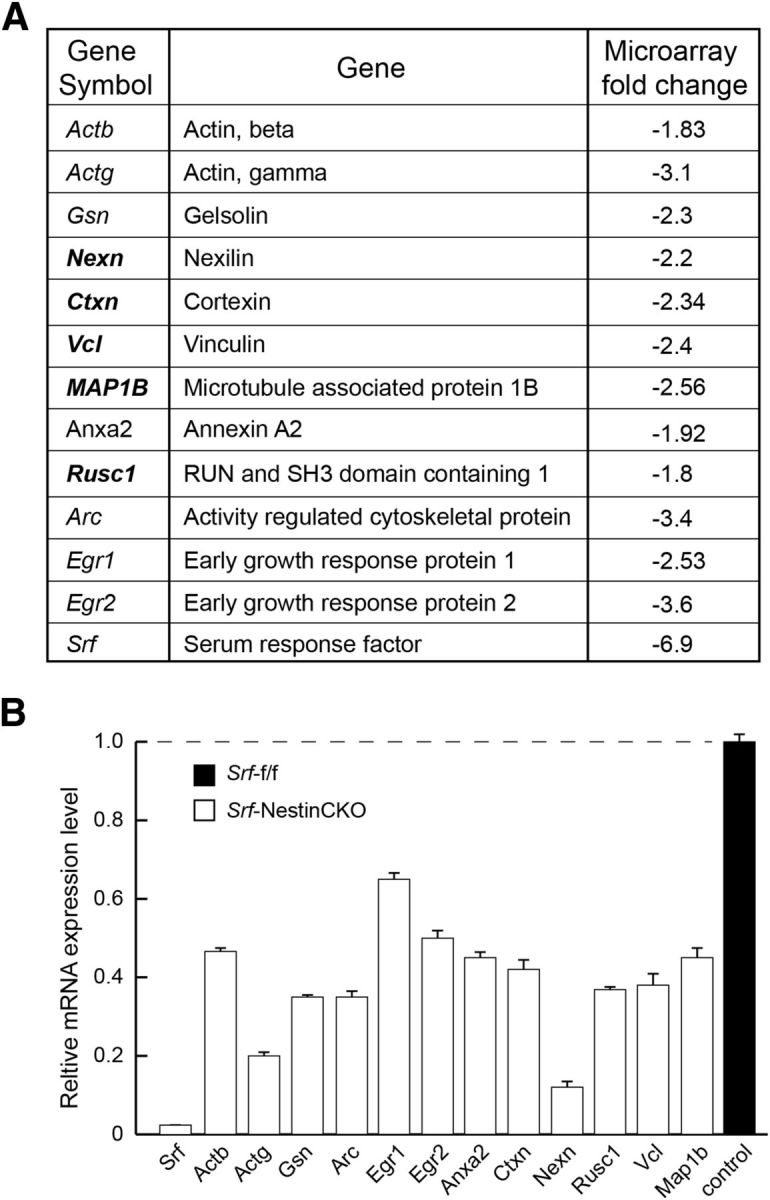
Putative SRF target genes identified from forebrain. A, Table represents Srf target genes identified from forebrain of neonatal Srf-f/f and Srf-NestinCKO mice. The genes in bold were further analyzed for their ability to rescue axonal growth deficits of Srf-NestinCKO mutant neurons. B, mRNA expression of Srf target genes in A. Relative mRNA expression levels were assessed by real-time qRT-PCR. The expression level in control sample was set to 1 (n = 3 experiments).
Expression of both Vcl and Map1b was severely attenuated in the brains of Srf-NestinCKO mice (Figs. 6, 7A, and 8A). We found that expression of Map1b partially rescued axonal growth but failed to rescue the enlarged growth cone and abnormal cell soma phenotypes observed in SRF-deficient neurons when cells were grown on poly-d-lysine and laminin (Fig. 7B,C). In contrast, Vcl expression was able to rescue axonal growth as well as enlarged growth cone and cell soma phenotypes exhibited by Srf-NestinCKO neurons when neurons were grown on poly-d-lysine and laminin (Fig. 8B,C). Because VCL is a critical player in coupling integrin signaling to the dynamic actin cytoskeleton, we tested the ability of VCL to rescue axonal growth deficits of SRF-deficient neurons grown in the absence of laminin. We found that Vcl expression only partially rescued axonal length of SRF-deficient neurons grown on poly-d-lysine alone in the absence of laminin (Fig. 8D,E). This suggested that efficient rescue of growth deficits exhibited by SRF-deficient neurons by VCL likely involves laminin and integrin signaling. Expression of VCL in wild-type neurons did not have any additional growth-promoting effects (Fig. 8C,E). Together, these data suggest that both VCL and MAP1B are capable of rescuing axon growth in SRF-deficient neurons, but VCL is more efficient and VCL alone can rescue the enlarged growth cone and cell soma morphologies.
Figure 8.
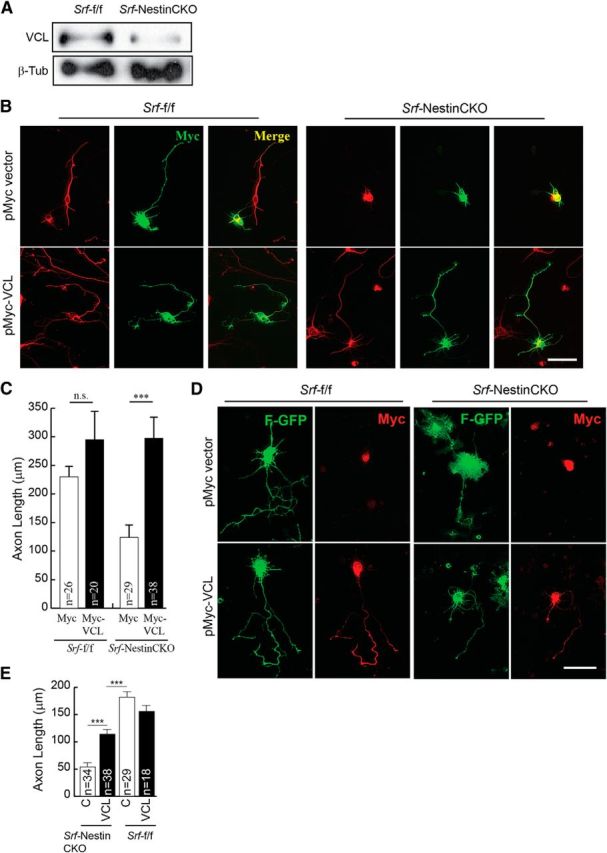
Vinculin is sufficient to promote axonal growth in SRF-deficient neurons. A, Western blot of total brain lysate showing reduced Vcl expression in Srf-NestinCKO mice compared with control littermates. B, P0.5 hippocampal neurons from Srf-NestinCKO and Srf-f/f mice were cotransfected with empty vector (pMyc) or pMyc-VCL and cultured for 4 DIV on poly-d-lysine and laminin. Cells were fixed and immunostained for Tuj1 (red) and Myc (green). Expression of Vcl but not empty vector was able to promote axon growth. Scale bar, 25 μm. C, Quantitation of B. Error bars indicate SEM. ***p < 0.001 (one-way ANOVA, Tukey post-test analysis). n.s., Not significant. D, Vinculin only partially rescues axonal growth in SRF-deficient neurons in the absence of laminin. P0.5 hippocampal neurons from Srf-NestinCKO and Srf-f/f mice were cotransfected with empty vector (pMyc) or pMyc-VCL along with farnesylated-GFP (F-GFP) and cultured for 4 DIV on poly-d-lysine alone without laminin. Cells were fixed and immunostained for GFP (green) to visualize neuronal morphology and Myc (red) for transfected cells. Expression of VCL, but not empty vector, could partially rescue axonal growth in the absence of laminin, suggesting a role for laminin–integrin signaling downstream of VCL in promoting axon growth. Scale bar, 25 μm. E, Quantitation of axon length in D. ***p < 0.001 (one-way ANOVA, Tukey post-test analysis).
Phosphorylation of SRF by GSK-3 promotes SRF-MKL1 binding, and SRF and MKL1 regulate Vcl expression (Schratt et al., 2002; Miralles et al., 2003). Based on these observations, we asked whether VCL can also promote axon growth when GSK-3 is inhibited. We first assessed the status of Vcl expression under conditions of GSK-3 inhibition and subsequent rescue by SRF-VP16 (Fig. 9A). Hippocampal neurons treated with 6-BIO exhibited significant reduction in Vcl mRNA expression as determined by qRT-PCR, and expression of SRF-VP16 in the presence of 6-BIO resulted in increased Vcl mRNA (Fig. 9A). We next expressed VCL in wild-type hippocampal neurons grown in the presence of 6-BIO and found that VCL expression significantly increased axon length (Fig. 9B,C). Last, we asked whether VCL is necessary for axon growth. Using shRNA specific for mouse Vcl, we found that Vcl knockdown resulted in attenuated axonal growth similar to that observed in SRF-deficient neurons (Fig. 9D,E; and data not shown). Coexpression of a chicken homolog of Vcl, which is functionally similar to mouse Vcl, rescued axonal growth (Fig. 9D,E) (Grashoff et al., 2010). The chicken Vcl is resistant to mouse shVcl, suggesting that axonal growth deficits caused by shVcl are not the result of off-target effects. Together, these observations demonstrate that SRF promotes axon outgrowth downstream of GSK-3 partly through regulation of MAP1B and VCL expression.
Figure 9.
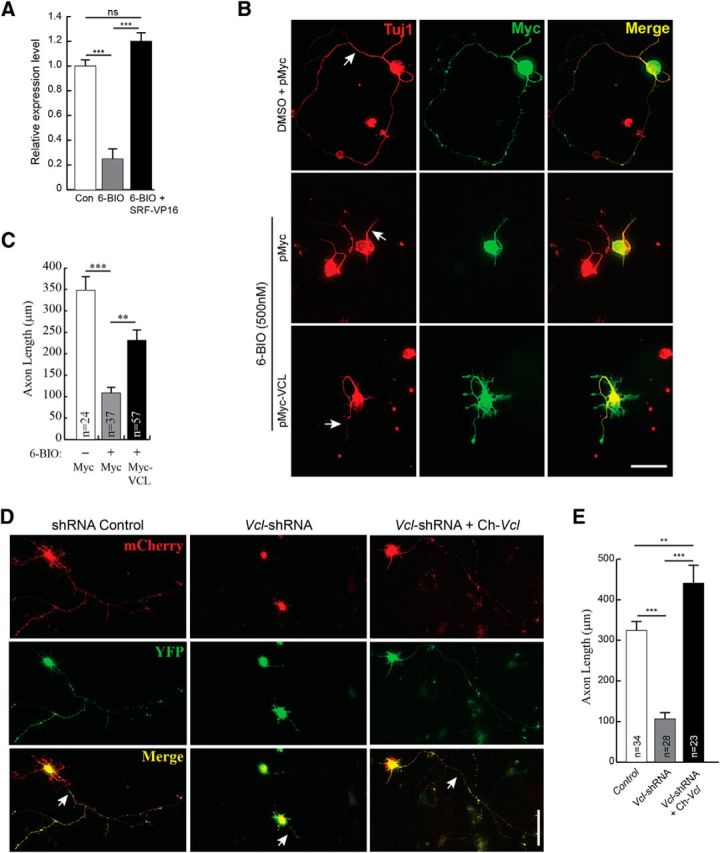
Vinculin can partially rescue axonal growth in the absence of GSK-3 signaling. A, Vcl mRNA expression is attenuated in neurons when GSK-3 is inhibited. Vcl mRNA expression was assessed by real-time qRT-PCR from total RNA isolated from hippocampal neurons grown in the presence of DMSO (con), 500 nm 6-BIO or 6-BIO+SRF-VP16 at 3 DIV. Vcl mRNA levels decreased in the presence 6-BIO and expression of SRF-VP16 rescued Vcl levels. ***p < 0.001 (one-way ANOVA, Tukey post-test analysis). n.s., Not significant. B, VCL expression was also able to promote axonal growth when GSK-3 is blocked by 6-BIO. Wild-type hippocampal neurons were transfected with either empty Myc vector or Myc-VCL and cultured in the presence of DMSO (vehicle) or 500 nm 6-BIO for 4 DIV on poly-d-lysine and laminin. Cells were fixed and immunostained for Myc (green) and Tuj1 (red). VCL expression alone promoted axonal growth (arrow). Scale bar, 25 μm. C, Quantitation of B. Error bars indicate SEM. **p = 0.005 (6-BIO+Myc vs 6-BIO+Myc-VCL) (two-tailed t test analysis). ***p = 0.0002 (6-BIO+Myc vs 6-BIO+Myc-VCL) (two-tailed t test analysis). D, shRNA knockdown of VCL expression in hippocampal neurons attenuates axonal growth. Wild-type hippocampal neurons were transfected with shRNA targeting mouse Vcl (Vcl-shRNA-CMV-mCherry) or a control shRNA (shRNA-CMV-mCherry) along with YFP and grown for 4 d. Cells were fixed and stained for mCherry encoded from within the shRNA vector and YFP. For rescue experiment, neurons were transfected with Vcl-shRNA along with the chicken homolog, venus-chicken Vcl (Ch-Vcl). Ch-Vcl is resistant to mouse Vcl-shRNA, and its expression rescued axonal growth (arrow). Scale bar, 25 μm. E, Quantitation of D. Error bars indicate SEM. **p < 0.01 (one-way ANOVA, Tukey post-test analysis). ***p < 0.001 (one-way ANOVA, Tukey post-test analysis).
Discussion
In this study, we report a novel phosphorylation of SRF, mediated by GSK-3, that is required for SRF transcriptional activity and for SRF-dependent axon growth. SRF is an unprimed transcriptional substrate of GSK-3, and GSK-3 inhibition results in reduced SRF phosphorylation and SRF activity, which attenuates axon growth. Phosphorylation of SRF on serine 224 is necessary for SRF to interact with its cofactors MKL1 and MKL2. Moreover, SRF is sufficient to promote axonal growth in the absence of GSK-3 signaling in cultured hippocampal neurons. Furthermore, SRF regulates axonal growth through regulation of VCL and MAP1B expression. Finally, knockdown of VCL attenuates axonal growth, and VCL can promote axonal growth in the absence of SRF and GSK-3 signaling. Together, our findings suggest that SRF promotes axon growth by likely activating gene expression downstream of GSK-3 signaling in hippocampal neurons.
Given the importance of SRF for neural development, activity-dependent gene expression, and synaptic plasticity, surprisingly little is known about the post-translational modifications that activate SRF downstream of diverse cellular signals. Previous studies have shown that SRF is phosphorylated on several serine and threonine residues, but the importance of this phosphorylation for SRF transcriptional activity remains unclear (Misra et al., 1991; Rivera et al., 1993; Iyer et al., 2006). In this study, we have identified a novel phosphorylation mechanism by which SRF is activated in neurons and how this contributes to SRF-dependent axon growth (Fig. 10). SRF is directly phosphorylated by GSK-3 on ser224 that lies outside the MADS-domain required for DNA binding and SRF homodimerization (Pellegrini et al., 1995). Furthermore, global inhibition of GSK-3 activity in cultured neurons attenuated SRF phosphorylation and SRF transcriptional activity (Figs. 1 and 9). However, currently it remains unknown whether the decrease in SRF phosphorylation is the result of GSK-3β inhibition in the nucleus or axons and whether nucleus-specific GSK-3 inhibition alone can block axon growth. A recent study revealed a novel mechanism by which cytoplasmic SRF promoted axon regeneration after peripheral nerve injury in a transcription-independent manner through interactions with cofilin (Stern et al., 2013). It is possible that pSer224-SRF might have a similar cytoplasmic role during axon growth. However, this is unlikely because the SRFS224A point mutant acted in a dominant-negative manner and attenuated both transcription and axon growth. Future experiments will address the effect of nucleus- and axon-specific inhibition of GSK-3 on SRF phosphorylation and axon growth.
Figure 10.

Summary model. Model of GSK-3-SRF signaling pathway in regulation of axon growth. Phosphorylation by GSK-3 on serine 224 results in the activation of SRF and subsequent recruitment of MKL-family cofactors. The SRF-MKL transcriptional complex activates genes, such as VCL and MAP1B. MAP1B regulates microtubule dynamics, whereas VCL tethers F-actin to focal adhesion junctions to promote neurite growth downstream of the GSK-3-SRF pathway. Absence of SRF or GSK-3 activity results in inhibition or attenuation of neurite growth.
Previous studies have shown that GSK-3β can shuttle between cytoplasm and nucleus and that GSK-3β is present in the nucleus where it interacts with transcription factors (Bijur and Jope, 2003; Caspi et al., 2008; Bechard and Dalton, 2009). In the context of GSK-3/SRF interaction, it remains unclear whether SRF is constitutively phosphorylated by GSK-3 in the nucleus or whether their interaction is triggered by growth-promoting signals and/or actin-cytoskeletal changes. Future studies will address whether extrinsic factors, such as neurotrophins and actin cytoskeletal changes, result in SRF ser224 phosphorylation. These studies will provide additional insights into how extracellular signals are conveyed to the nucleus for efficient axonal growth.
During development, we observed that ser224 phosphorylation on SRF increased between E15.5 and E18.5 and decreased subsequently. In contrast, at E15.5, we found relatively higher GSK-3β-Ser9 phosphorylation, a measure of its inactivation. Thus, it seems to appear that SRF-Ser224 phosphorylation does not closely correlate temporally with GSK-3β activity in the brain. A parsimonious explanation for this is that we measured total GSK-3β activity in whole-brain lysates, and this might not accurately reflect the activity of nuclear GSK-3β. However, a previous study had shown that GSK-3β activity increases during brain development when assessed for GSK-3β targets, such as c-Myc and β-catenin (Kim et al., 2009), and these findings appear to be consistent with our observations for SRF ser224 phosphorylation.
The ser224 phosphorylation is specifically required for SRF to form a functional transcriptional complex with the cofactors MKL1 and MKL2 and not with ELK1. Among these two groups of cofactors, the MKLs alone have been shown to regulate axonal and dendritic growth in hippocampal, neocortical, and embryonic DRG neurons (Shiota et al., 2006; Wickramasinghe et al., 2008; Mokalled et al., 2010; O'Sullivan et al., 2010). The role for TCF-family members in neuronal development remains unclear. We found that, unlike dominant-negative MKL2, expression of a dominant-negative ELK1-En, which inhibits all TCF-family members, in cultured hippocampal neurons did not affect axon growth (Fig. 3G,H). Therefore, it appears that TCF-target genes are unlikely to regulate axon growth in hippocampal neurons. Based on these observations, we speculate that SRFS224A acts as a dominant-negative mutant by forming heterodimers with endogenous wild-type SRF, thereby preventing the formation of a functional SRF-MKL complex. Currently, it is unclear whether the ser224 phosphorylation is also required for other functions of SRF, including activity-dependent immediate early gene expression and synaptic plasticity (Ramanan et al., 2005).
Previous studies have shown that pharmacological inhibition of the MEK pathway or expression of dominant-negative ELK1-En did not affect Vcl expression (Gineitis and Treisman, 2001; Vickers et al., 2004). Instead, expression of DN-MKL1, which blocks both MKL1 and MKL2 or inhibition of actin polymerization by latrunculin-B blocks Vcl expression (Gineitis and Treisman, 2001; Cen et al., 2003). Together, these findings imply that Vcl expression is independent of Elk1/TCF-family members and ERK/MEK pathway activation. Consistent with these findings, we found that ELK1-En expression did not inhibit axon outgrowth, whereas DN-MKL2 expression profoundly inhibits growth likely by inhibiting genes, including Vcl.
Interestingly, hippocampal neurons lacking SRF or inhibited for GSK-3 activities exhibit similar growth phenotypes in culture. Both these groups of neurons exhibit a delay in Stage 1 growth observed in the first 12–24 h (Fig. 5; and data not shown). However, SRF-deficient neurons were able to extend a single but significantly shorter axon at 5 DIV. In contrast, consistent with previous studies, complete inhibition of GSK-3 exerts a more profound effect and neurons fail to extend an axon (Kim et al., 2006; Garrido et al., 2007).
Our work identifies VCL as a critical mediator of axon growth downstream of GSK-3/SRF signaling. VCL is a major component of cell-matrix adhesions and acts as a critical link between focal adhesions and the actin cytoskeleton through direct interaction with several binding partners, including talin, paxillin, and actin (Ziegler et al., 2006; Humphries et al., 2007). Previous findings have shown that β-actin is a transcriptional target of SRF, suggesting that neurite outgrowth deficits exhibited by SRF-deficient neurons are likely the result of reduced actin levels (Ramanan et al., 2005; Knöll et al., 2006). However, expression of full-length actin or various mutant forms of actin did not rescue growth deficits in SRF-deficient hippocampal neurons (Knöll et al., 2006; Stern et al., 2009). Our findings suggest that it is likely that the axonal defects observed in SRF-deficient neurons are not the result of reduced actin expression, but rather the absence of proteins, such as VCL, which are required for mechanical coupling of the actin cytoskeleton to focal adhesion sites. Therefore, restoring VCL expression in SRF-deficient neurons may suffice to assemble the actin cytoskeleton and provide the necessary driving force to promote axonal growth.
We observed that, whereas VCL expression could rescue axonal growth in SRF-deficient neurons, VCL ability to rescue axonal growth in GSK-3-inhibited neurons was only partial, suggesting the involvement of other GSK-3 targets and/or pathways. It has been shown that GSK-3 regulates axon growth by regulating the activity of several microtubule-associated proteins, including MAP1B, CRMPs, and APC (Zhou et al., 2004; Goold and Gordon-Weeks, 2005; Kim et al., 2006; Alabed et al., 2010). MAP1B is activated by GSK-3 phosphorylation and contributes to microtubule dynamics (Goold and Gordon-Weeks, 2005). Therefore, in the absence of these critical GSK-3 targets, VCL could promote axon growth in GSK-3 inhibited neurons only to a limited extent.
GSK-3 inhibitory drugs are commonly used to treat neuropsychiatric and neurological disorders, including bipolar disorder, Parkinson's disease, and Alzheimer's disease (Bhat et al., 2004; Cohen and Goedert, 2004). SRF has been shown to play a critical role in immediate early gene expression, synaptic plasticity, and learning and memory, whereas SRF cofactors MAL/MKLs are important for cerebellar long-term depression (Ramanan et al., 2005; Etkin et al., 2006; Smith-Hicks et al., 2010). It is likely that targeting GSK-3 in the treatment of the above disorders might also affect SRF-dependent neuronal development and plasticity. Future studies will probe the effect of anti-GSK-3 therapeutics on SRF transcriptional activity and its critical functions in the nervous system.
Footnotes
This work was supported by Andrew B. and Virginia C. Craig Faculty Fellowship (N.R.) and the Whitehall Foundation (N.R.). We thank members of the N.R. laboratory for comments on the manuscript; X. He and H. Piwnica-Worms for GSK-3β constructs; D. Ginty for VP16-CREB and ELK1-En; R. Prywes for Flag-MKL1; N. Cowan for mouse MAP1B full-length cDNA clone; S. Mennerick for Pan-NaV antibody; and E. Anthony and S. Harmon for help with the in vitro kinase assay.
The authors declare no competing financial interests.
References
- Alabed YZ, Pool M, Ong Tone S, Sutherland C, Fournier AE. GSK-3 beta regulates myelin-dependent axon outgrowth inhibition through CRMP4. J Neurosci. 2010;30:5635–5643. doi: 10.1523/JNEUROSCI.6154-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badiani P, Corbella P, Kioussis D, Marvel J, Weston K. Dominant interfering alleles define a role for c-Myb in T-cell development. Genes Dev. 1994;8:770–782. doi: 10.1101/gad.8.7.770. [DOI] [PubMed] [Google Scholar]
- Bechard M, Dalton S. Subcellular localization of glycogen synthase kinase 3beta controls embryonic stem cell self-renewal. Mol Cell Biol. 2009;29:2092–2104. doi: 10.1128/MCB.01405-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Budd Haeberlein SL, Avila J. Glycogen synthase kinase 3: a drug target for CNS therapies. J Neurochem. 2004;89:1313–1317. doi: 10.1111/j.1471-4159.2004.02422.x. [DOI] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Glycogen synthase kinase-3 beta is highly activated in nuclei and mitochondria. Neuroreport. 2003;14:2415–2419. doi: 10.1097/00001756-200312190-00025. [DOI] [PubMed] [Google Scholar]
- Caspi M, Zilberberg A, Eldar-Finkelman H, Rosin-Arbesfeld R. Nuclear GSK-3beta inhibits the canonical Wnt signalling pathway in a beta-catenin phosphorylation-independent manner. Oncogene. 2008;27:3546–3555. doi: 10.1038/sj.onc.1211026. [DOI] [PubMed] [Google Scholar]
- Cen B, Selvaraj A, Burgess RC, Hitzler JK, Ma Z, Morris SW, Prywes R. Megakaryoblastic leukemia 1, a potent transcriptional coactivator for serum response factor (SRF), is required for serum induction of SRF target genes. Mol Cell Biol. 2003;23:6597–6608. doi: 10.1128/MCB.23.18.6597-6608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen B, Selvaraj A, Prywes R. Myocardin/MKL family of SRF coactivators: key regulators of immediate early and muscle specific gene expression. J Cell Biochem. 2004;93:74–82. doi: 10.1002/jcb.20199. [DOI] [PubMed] [Google Scholar]
- Cesari F, Brecht S, Vintersten K, Vuong LG, Hofmann M, Klingel K, Schnorr JJ, Arsenian S, Schild H, Herdegen T, Wiebel FF, Nordheim A. Mice deficient for the ets transcription factor elk-1 show normal immune responses and mildly impaired neuronal gene activation. Mol Cell Biol. 2004;24:294–305. doi: 10.1128/MCB.24.1.294-305.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Johnson GV. Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites: differential impact on microtubule binding. J Biol Chem. 2003;278:187–193. doi: 10.1074/jbc.M206236200. [DOI] [PubMed] [Google Scholar]
- Cohen P, Frame S. The renaissance of GSK-3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- Cohen P, Goedert M. GSK-3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- Contestabile A, Bonanomi D, Burgaya F, Girault JA, Valtorta F. Localization of focal adhesion kinase isoforms in cells of the central nervous system. Int J Dev Neurosci. 2003;21:83–93. doi: 10.1016/S0736-5748(02)00126-0. [DOI] [PubMed] [Google Scholar]
- Cooper SJ, Trinklein ND, Nguyen L, Myers RM. Serum response factor binding sites differ in three human cell types. Genome Res. 2007;17:136–144. doi: 10.1101/gr.5875007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AM, Banker G. Neuronal polarity. Annu Rev Neurosci. 1994;17:267–310. doi: 10.1146/annurev.ne.17.030194.001411. [DOI] [PubMed] [Google Scholar]
- Dehmelt L, Halpain S. Actin and microtubules in neurite initiation: are MAPs the missing link? J Neurobiol. 2004;58:18–33. doi: 10.1002/neu.10284. [DOI] [PubMed] [Google Scholar]
- de Lima AD, Merten MD, Voigt T. Neuritic differentiation and synaptogenesis in serum-free neuronal cultures of the rat cerebral cortex. J Comp Neurol. 1997;382:230–246. doi: 10.1002/(SICI)1096-9861(19970602)382:2<230::AID-CNE7>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Dent EW, Kwiatkowski AV, Mebane LM, Philippar U, Barzik M, Rubinson DA, Gupton S, Van Veen JE, Furman C, Zhang J, Alberts AS, Mori S, Gertler FB. Filopodia are required for cortical neurite initiation. Nat Cell Biol. 2007;9:1347–1359. doi: 10.1038/ncb1654. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkin A, Alarcón JM, Weisberg SP, Touzani K, Huang YY, Nordheim A, Kandel ER. A role in learning for SRF: deletion in the adult forebrain disrupts LTD and the formation of an immediate memory of a novel context. Neuron. 2006;50:127–143. doi: 10.1016/j.neuron.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK-3 and its inactivation by phosphorylation. Mol Cell. 2001;7:1321–1327. doi: 10.1016/S1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- Gao Y, Deng K, Hou J, Bryson JB, Barco A, Nikulina E, Spencer T, Mellado W, Kandel ER, Filbin MT. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron. 2004;44:609–621. doi: 10.1016/j.neuron.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Garrido JJ, Giraud P, Carlier E, Fernandes F, Moussif A, Fache MP, Debanne D, Dargent B. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science. 2003;300:2091–2094. doi: 10.1126/science.1085167. [DOI] [PubMed] [Google Scholar]
- Garrido JJ, Simón D, Varea O, Wandosell F. GSK-3 alpha and GSK-3 beta are necessary for axon formation. FEBS Lett. 2007;581:1579–1586. doi: 10.1016/j.febslet.2007.03.018. [DOI] [PubMed] [Google Scholar]
- Gineitis D, Treisman R. Differential usage of signal transduction pathways defines two types of serum response factor target gene. J Biol Chem. 2001;276:24531–24539. doi: 10.1074/jbc.M102678200. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Billault C, Jimenez-Mateos EM, Caceres A, Diaz-Nido J, Wandosell F, Avila J. Microtubule-associated protein 1B function during normal development, regeneration, and pathological conditions in the nervous system. J Neurobiol. 2004;58:48–59. doi: 10.1002/neu.10283. [DOI] [PubMed] [Google Scholar]
- Goold RG, Gordon-Weeks PR. The MAP kinase pathway is upstream of the activation of GSK-3beta that enables it to phosphorylate MAP1B and contributes to the stimulation of axon growth. Mol Cell Neurosci. 2005;28:524–534. doi: 10.1016/j.mcn.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Goold RG, Owen R, Gordon-Weeks PR. Glycogen synthase kinase 3beta phosphorylation of microtubule-associated protein 1B regulates the stability of microtubules in growth cones. J Cell Sci. 1999;112:3373–3384. doi: 10.1242/jcs.112.19.3373. [DOI] [PubMed] [Google Scholar]
- Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, Yang MT, McLean MA, Sligar SG, Chen CS, Ha T, Schwartz MA. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 2010;466:263–266. doi: 10.1038/nature09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Saint-Jeannet JP, Woodgett JR, Varmus HE, Dawid IB. Glycogen synthase kinase-3 and dorsoventral patterning in Xenopus embryos. Nature. 1995;374:617–622. doi: 10.1038/374617a0. [DOI] [PubMed] [Google Scholar]
- Humphries JD, Wang P, Streuli C, Geiger B, Humphries MJ, Ballestrem C. Vinculin controls focal adhesion formation by direct interactions with talin and actin. J Cell Biol. 2007;179:1043–1057. doi: 10.1083/jcb.200703036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur EM, Saijilafu, Lee BD, Kim SJ, Xu WL, Zhou FQ. GSK-3 controls axon growth via CLASP-mediated regulation of growth cone microtubules. Genes Dev. 2011;25:1968–1981. doi: 10.1101/gad.17015911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer D, Chang D, Marx J, Wei L, Olson EN, Parmacek MS, Balasubramanyam A, Schwartz RJ. Serum response factor MADS box serine-162 phosphorylation switches proliferation and myogenic gene programs. Proc Natl Acad Sci U S A. 2006;103:4516–4521. doi: 10.1073/pnas.0505338103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaynes JB, O'Farrell PH. Active repression of transcription by the engrailed homeodomain protein. EMBO J. 1991;10:1427–1433. doi: 10.1002/j.1460-2075.1991.tb07663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung MC, Piwnica-Worms H. GSK-3 beta targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3 beta inactivation correlates with Cdc25A overproduction in human cancers. Cancer Cell. 2008;13:36–47. doi: 10.1016/j.ccr.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp PR, Metcalfe JC. Four isoforms of serum response factor that increase or inhibit smooth-muscle-specific promoter activity. Biochem J. 2000;345:445–451. doi: 10.1042/0264-6021:3450445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WY, Zhou FQ, Zhou J, Yokota Y, Wang YM, Yoshimura T, Kaibuchi K, Woodgett JR, Anton ES, Snider WD. Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron. 2006;52:981–996. doi: 10.1016/j.neuron.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WY, Wang X, Wu Y, Doble BW, Patel S, Woodgett JR, Snider WD. GSK-3 is a master regulator of neural progenitor homeostasis. Nat Neurosci. 2009;12:1390–1397. doi: 10.1038/nn.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöll B, Kretz O, Fiedler C, Alberti S, Schütz G, Frotscher M, Nordheim A. Serum response factor controls neuronal circuit assembly in the hippocampus. Nat Neurosci. 2006;9:195–204. doi: 10.1038/nn1627. [DOI] [PubMed] [Google Scholar]
- Kress GJ, Dowling MJ, Eisenman LN, Mennerick S. Axonal sodium channel distribution shapes the depolarized action potential threshold of dentate granule neurons. Hippocampus. 2010;20:558–571. doi: 10.1002/hipo.20667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Riccio A, Cohen S, Ginty DD. Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron. 2002;34:371–385. doi: 10.1016/S0896-6273(02)00686-4. [DOI] [PubMed] [Google Scholar]
- Lu PP, Ramanan N. Serum response factor is required for cortical axon growth but is dispensable for neurogenesis and neocortical lamination. J Neurosci. 2011;31:16651–16664. doi: 10.1523/JNEUROSCI.3015-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CA, Brivanlou A, Dajani R, Crovace C, Tarricone C, Musacchio A, Roe SM, Pearl L, Greengard P. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Miano JM. Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol. 2003;35:577–593. doi: 10.1016/S0022-2828(03)00110-X. [DOI] [PubMed] [Google Scholar]
- Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–342. doi: 10.1016/S0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- Misra RP, Rivera VM, Wang JM, Fan PD, Greenberg ME. The serum response factor is extensively modified by phosphorylation following its synthesis in serum-stimulated fibroblasts. Mol Cell Biol. 1991;11:4545–4554. doi: 10.1128/MCB.11.9.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseyeva EP, Weller PA, Zhidkova NI, Corben EB, Patel B, Jasinska I, Koteliansky VE, Critchley DR. Organization of the human gene encoding the cytoskeletal protein vinculin and the sequence of the vinculin promoter. J Biol Chem. 1993;268:4318–4325. [PubMed] [Google Scholar]
- Mokalled MH, Johnson A, Kim Y, Oh J, Olson EN. Myocardin-related transcription factors regulate the Cdk5/Pctaire1 kinase cascade to control neurite outgrowth, neuronal migration and brain development. Development. 2010;137:2365–2374. doi: 10.1242/dev.047605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan NC, Pickering M, Di Giacomo D, Loscher JS, Murphy KJ. Mkl transcription cofactors regulate structural plasticity in hippocampal neurons. Cereb Cortex. 2010;20:1915–1925. doi: 10.1093/cercor/bhp262. [DOI] [PubMed] [Google Scholar]
- Owen R, Gordon-Weeks PR. Inhibition of glycogen synthase kinase 3beta in sensory neurons in culture alters filopodia dynamics and microtubule distribution in growth cones. Mol Cell Neurosci. 2003;23:626–637. doi: 10.1016/S1044-7431(03)00095-2. [DOI] [PubMed] [Google Scholar]
- Pellegrini L, Tan S, Richmond TJ. Structure of serum response factor core bound to DNA. Nature. 1995;376:490–498. doi: 10.1038/376490a0. [DOI] [PubMed] [Google Scholar]
- Polleux F, Snider W. Initiating and growing an axon. Cold Spring Harb Perspect Biol. 2010;2:a001925. doi: 10.1101/cshperspect.a001925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan N, Shen Y, Sarsfield S, Lemberger T, Schütz G, Linden DJ, Ginty DD. SRF mediates activity-induced gene expression and synaptic plasticity but not neuronal viability. Nat Neurosci. 2005;8:759–767. doi: 10.1038/nn1462. [DOI] [PubMed] [Google Scholar]
- Rivera VM, Miranti CK, Misra RP, Ginty DD, Chen RH, Blenis J, Greenberg ME. A growth factor-induced kinase phosphorylates the serum response factor at a site that regulates its DNA-binding activity. Mol Cell Biol. 1993;13:6260–6273. doi: 10.1128/MCB.13.10.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt G, Philippar U, Berger J, Schwarz H, Heidenreich O, Nordheim A. Serum response factor is crucial for actin cytoskeletal organization and focal adhesion assembly in embryonic stem cells. J Cell Biol. 2002;156:737–750. doi: 10.1083/jcb.200106008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj A, Prywes R. Megakaryoblastic leukemia-1/2, a transcriptional co-activator of serum response factor, is required for skeletal myogenic differentiation. J Biol Chem. 2003;278:41977–41987. doi: 10.1074/jbc.M305679200. [DOI] [PubMed] [Google Scholar]
- Sharrocks AD. The ETS-domain transcription factor family. Nat Rev Mol Cell Biol. 2001;2:827–837. doi: 10.1038/35099076. [DOI] [PubMed] [Google Scholar]
- Shiota J, Ishikawa M, Sakagami H, Tsuda M, Baraban JM, Tabuchi A. Developmental expression of the SRF co-activator MAL in brain: role in regulating dendritic morphology. J Neurochem. 2006;98:1778–1788. doi: 10.1111/j.1471-4159.2006.03992.x. [DOI] [PubMed] [Google Scholar]
- Smith-Hicks C, Xiao B, Deng R, Ji Y, Zhao X, Shepherd JD, Posern G, Kuhl D, Huganir RL, Ginty DD, Worley PF, Linden DJ. SRF binding to SRE 6.9 in the Arc promoter is essential for LTD in cultured Purkinje cells. Nat Neurosci. 2010;13:1082–1089. doi: 10.1038/nn.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern S, Debre E, Stritt C, Berger J, Posern G, Knöll B. A nuclear actin function regulates neuronal motility by serum response factor-dependent gene transcription. J Neurosci. 2009;29:4512–4518. doi: 10.1523/JNEUROSCI.0333-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern S, Haverkamp S, Sinske D, Tedeschi A, Naumann U, Di Giovanni S, Kochanek S, Nordheim A, Knöll B. The transcription factor serum response factor stimulates axon regeneration through cytoplasmic localization and cofilin interaction. J Neurosci. 2013;33:18836–18848. doi: 10.1523/JNEUROSCI.3029-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens GR, Zhang C, Berg MM, Lambert MP, Barber K, Cantallops I, Routtenberg A, Klein WL. CNS neuronal focal adhesion kinase forms clusters that co-localize with vinculin. J Neurosci Res. 1996;46:445–455. doi: 10.1002/(SICI)1097-4547(19961115)46:4<445::AID-JNR6>3.3.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Treisman R. Identification and purification of a polypeptide that binds to the c-fos serum response element. EMBO J. 1987;6:2711–2717. doi: 10.1002/j.1460-2075.1987.tb02564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey C, McCarthy JV. Presenilin-1 is an unprimed glycogen synthase kinase-3beta substrate. FEBS Lett. 2006;580:4015–4020. doi: 10.1016/j.febslet.2006.06.035. [DOI] [PubMed] [Google Scholar]
- Vickers ER, Sharrocks AD. The use of inducible engrailed fusion proteins to study the cellular functions of eukaryotic transcription factors. Methods. 2002;26:270–280. doi: 10.1016/S1046-2023(02)00031-2. [DOI] [PubMed] [Google Scholar]
- Vickers ER, Kasza A, Kurnaz IA, Seifert A, Zeef LA, O'Donnell A, Hayes A, Sharrocks AD. Ternary complex factor-serum response factor complex-regulated gene activity is required for cellular proliferation and inhibition of apoptotic cell death. Mol Cell Biol. 2004;24:10340–10351. doi: 10.1128/MCB.24.23.10340-10351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe SR, Alvania RS, Ramanan N, Wood JN, Mandai K, Ginty DD. Serum response factor mediates NGF-dependent target innervation by embryonic DRG sensory neurons. Neuron. 2008;58:532–545. doi: 10.1016/j.neuron.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Baribault H, Adamson ED. Vinculin knockout results in heart and brain defects during embryonic development. Development. 1998;125:327–337. doi: 10.1242/dev.125.2.327. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Snider WD. Intracellular control of developmental and regenerative axon growth. Philos Trans R Soc Lond B Biol Sci. 2006;361:1575–1592. doi: 10.1098/rstb.2006.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron. 2004;42:897–912. doi: 10.1016/j.neuron.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Ziegler WH, Liddington RC, Critchley DR. The structure and regulation of vinculin. Trends Cell Biol. 2006;16:453–460. doi: 10.1016/j.tcb.2006.07.004. [DOI] [PubMed] [Google Scholar]