

Abstract
Mitral cells express low-voltage activated Cav3.3 channels on their distal apical tuft dendrites (McKay et al., 2006; Johnston and Delaney, 2010). They also discharge Na+-dependent dendritic action potentials and release glutamate from these dendrites. Around resting membrane potentials, between −65 and −50 mV, Cav3.x channels are a primary determinant of cytoplasmic [Ca2+]. In this study using C57 mice, we present evidence that subthreshold Cav3.x-mediated Ca2+ influx modulates action potential evoked transmitter release and directly drives asynchronous release from distal tuft dendrites. Presynaptic hyperpolarization and selective block of Cav3.x channels with Z941 (Tringham et al., 2012) reduce mitral-to-mitral EPSP amplitude, increase the coefficient of variation of EPSPs, and increase paired-pulse ratios, consistent with a reduced probability of transmitter release. Both hyperpolarization and Cav3.x channel blockade reduce steady-state cytoplasmic [Ca2+] in the tuft dendrite without reducing action potential evoked Ca2+ influx, suggesting that background [Ca2+] modulates evoked release. We demonstrate that Cav3.x-mediated Ca2+ influx from even one mitral cell at membrane potentials between −65 and −50 mV is sufficient to produce feedback inhibition from periglomerular neurons. Deinactivation of Cav3.x channels by hyperpolarization increases T-type Ca2+ influx upon repolarization and increases feedback inhibition to produce subthreshold modulation of the mitral–periglomerular reciprocal circuit.
Keywords: asynchronous release, basal Ca2+ level, dendrodendritic release, olfactory mitral cells, subthreshold potential, T-channel (Cav3)
Introduction
Olfactory afferents synapse in glomeruli in the olfactory bulb. When an odor stimulus is excitatory for a particular mitral cell, in vivo recordings reveal slow membrane potential oscillations (Charpak et al., 2001; Cang and Isaacson, 2003). High-frequency γ oscillations are superimposed on these slow oscillations producing phase-locked action potential (AP) firing. Although it is known that intraglomerular synchronization is dependent on glutamate release from the apical tuft (Nicoll and Jahr, 1982; Aroniadou-Anderjaska et al., 1999; Isaacson, 1999; Friedman and Strowbridge, 2000; Schoppa and Westbrook, 2001; Pimentel and Margrie, 2008), the precise mechanism of glutamate release and whether it is modulated by subthreshold voltages or presynaptic cytoplasmic [Ca2+] are unknown. Odor-evoked subthreshold membrane potential oscillations result in significant dendritic Ca2+ influx in vivo (Charpak et al., 2001). These can be mimicked in vitro to open low-voltage activated (LVA) Cav3.3, T-type channels (Johnston and Delaney, 2010). This manuscript addresses effects of these subthreshold changes in dendritic membrane potential on transmitter release from the apical tuft dendrite.
Neurotransmitter release shows [Ca2+] dependency in two distinct ways. An AP produces a large, brief Ca2+ influx through high-voltage activated (HVA) channels, which produces [Ca2+] of 10s to 100s of μm local to vesicles that directly trigger exocytosis through interaction with low-affinity binding site(s) (Llinás et al., 1972; Schneggenburger and Neher, 2000). AP-evoked release shows a highly nonlinear dependence upon the [Ca2+] at these low-affinity binding sites so an effective way for neurons to modulate release is to alter the magnitude of the HVA Ca2+ influx. At many synapses AP-evoked release is also modulated by persistent elevation of cytoplasmic [Ca2+] in the 0.1–1 μm range (Delaney and Tank, 1994; Regehr et al., 1994; Zucker and Regehr, 2002). Submicromolar accumulations of Ca2+ in the terminal can result as a “residuum” following AP evoked influx (Zucker and Regehr, 2002), from ligand-gated channels (e.g., Kalappa et al., 2011), or as a result of small changes in resting membrane potential that alter voltage-gated calcium channel activity (Awatramani et al., 2005). These latter two processes may be particularly relevant to transmitter-releasing dendrites, which receive an array of synaptic inputs. Submicromolar increments of [Ca2+] increase asynchronous transmitter release from many synapses (Zucker and Regehr, 2002). There is growing evidence for functional physiological effects of asynchronous release at a variety of synapses (Chávez et al., 2006; Best and Regehr, 2009; Jin et al., 2012), including accessory mitral apical tuft (Castro and Urban, 2009).
Using 2-photon Ca2+ imaging and dual whole-cell recordings of synaptically connected olfactory mitral cells, we found that AP-evoked, glutamate release from the distal apical tuft dendrite is reduced by presynaptic hyperpolarization and by block of T-type channels, both of which reduce cytoplasmic [Ca2+]. AP-evoked Ca2+ influx in the tuft was not reduced by preceding hyperpolarizing potentials or by T-channel block, suggesting that release is modulated by changes in basal presynaptic Ca2+, controlled by T-channels. Additionally, we found that subthreshold T-type Ca2+ influx could increase asynchronous release from tuft dendrites by amounts sufficient to activate inhibitory feedback from periglomerular neurons.
Materials and Methods
Ethical approval.
All experiments were reviewed and approved by the University of Victoria Institutional Animal Care and Use Committee in accordance with the Canadian Council for Animal Care and Use guidelines.
Acute brain slice preparation.
Acute horizontal slices (320–350 μm) were prepared from main olfactory bulbs of 65, 12- to 21-d-old C57 mice of either sex. After decapitation under deep anesthesia (isoflurane), olfactory bulbs were placed into ice-cold cutting solution (containing in mm: 207 sucrose, 2.5 KCl, 0.1 CaCl2, 4 MgCl2, 1.25 NaH2PO4, 3 myo-inositol, 10 glucose, 2 Na-ascorbate, 26 NaHCO3) and sliced on a Leica VT1000S vibratome. Slices were transferred to artificial CSF (containing in mm: 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 3 myo-inositol, 10 glucose, 26 NaHCO3, pH = 7.4), incubated for 1 h (36°C-38°C), and then stored at room temperature until use. Solutions were oxygenated with 95% O2 and 5% CO2.
Electrophysiology.
Slices were perfused at 34°C–36°C (1.5–2.5 ml/min) in a JG-23 imaging chamber attached to a PM-1 resistive heating platform (Warner Instruments). Olfactory mitral cells were visualized by an Olympus BX51WI microscope and identified by their shape, size, and location. To confirm the identity of mitral cells, we always included Alexa-594 (20–30 μm) in the pipette. Pipettes were pulled from borosilicate glass with filament (Sutter Instruments). For whole-cell current-clamp recordings, pipettes (4–8 MΩ) were filled with (in mm): 110 KCH3SO3, 8 KCl, 10 glutamate, 10 HEPES, 10 Na2phosphocreatine, 2 MgATP, 0.3 Na2ATP, 0.15 Ca2+-buffer (EGTA or Ca2+ Green-1) and pH 7.2 was set by KOH (∼290 mOsm). For whole-cell voltage-clamp experiments, we loaded pipettes (3–5 MΩ) with the same intracellular solution. In some experiments, we also included low concentration of QX-314 Cl− (0.5 mm) to suppress voltage-gated Na+ channels and to increase the threshold for AP generation while minimizing potential drug effect on T-type Ca2+ channels (Talbot and Sayer, 1996). For whole-cell voltage-clamp recordings from periglomerular cells (PGs), we increased the intrapipette [Cl−] to mimic the physiological Cl− reverse potential (Smith and Jahr, 2002; (ECl− = −52 mV at 36°C). PG cell solution composed of (in mm): 101 KCH3SO3, 19 KCl, 10 GABA, 10 HEPES, 10 Na2phosphocreatine, 2 MgATP, 0.3 Na2ATP, 0.15 EGTA, pH 7.2 with KOH. A 3M KCl agarose bridge reference electrode was used in all experiments. All stated voltages have been corrected for liquid junction potentials (−14 mV). Series resistances were <25 MΩ and compensated by the bridge balance circuitry in current-clamp recordings. In voltage-clamp experiments, series resistance was kept <15 MΩ and compensated >80% with 10 μs lag. Input resistance was continuously monitored, and experiments were ceased if it increased >25%. Cells requiring holding currents >−150 pA at −64 mV were discarded. Data were sampled (20 kHz) and low-pass filtered (3–5 kHz, four-pole Bessel filter) using an EPC10 dual headstage amplifier (HEKA).
For experiments to test the role of presynaptic subthreshold potentials on the modulation of synaptic strength, presynaptic mitral cells were either held at resting (Vrest; − 60 to −65 mV) or hyperpolarized membrane potentials (Vhyper; − 80 to −95 mV). Vhyper was applied 3 s before stimulating APs to allow basal [Ca2+] to settle. To assess changes in the strength of olfactory mitral–mitral connections, we evoked two APs, with a 50 ms interval, with brief somatic current injections (1.8–2.4 nA, 2 ms). Postsynaptic potentials were recorded at Vrest (see Fig. 2A). Sets of 5 AP pairs at Vrest were alternated with sets of 5 at Vhyper while continuously monitoring cell quality. Paired-pulse ratios (PPRs) were calculated dividing the second EPSP by the first. We used 10 s sweep intervals to avoid carryover of short-term synaptic plasticity between tests (see Fig. 2D), and trials were discarded if spontaneous, presynaptic APs preceded the evoked-APs. Individual EPSPs were baseline subtracted and averaged (10 ≤sweeps). This presynaptic and postsynaptic protocol was modified for other experiments (as described). We determined the electrical coupling coefficient (CC) between pairs of mitral cells with a hyperpolarizing current step applied to the presynaptic cell (−300 to −500 pA, 800 ms). To extract spikelets, the traces from the postsynaptic cell were aligned using the peak of the AP measured in the presynaptic cell and averaged. Coefficient of variation (CV) was calculated for each EPSP according to the following equation: CV2 = SD2EPSP − SD2Bg/Mean2Amp, where SDEPSP is the SD of the EPSP amplitude, SDBg is the SD of the background noise, and MeanAmp is the average EPSP amplitude. Data acquisition and analysis were performed with PatchMaster (HEKA) and Igor Pro (WaveMetrics) using the Neuromatic module. To stimulate lateral olfactory tract (LOT), a bipolar electrode was placed on the slice surface ∼100 μm internal to the mitral cell layer, caudal to the test mitral cell. We delivered brief (0.1 ms) isolated current pulses (SIU90, Cygnus Technology). The interstimulus interval between stimuli was ≥10 s.
Figure 2.

Hyperpolarization of the presynaptic olfactory mitral cells decreases the strength of mitral–mitral cell connections. A, Pairs of olfactory mitral cells (M1, presynaptic; M2, postsynaptic) projecting to the same glomerulus were found by electroporation with Alexa-594. Dashed circles in glomerular layer indicate glomeruli and projecting pairs of mitral cells. ML, Mitral cell layer; EPL, external plexiform layer; GL, glomerular layer. See the schematic representation of experimental design on the right. Pairs of somatic APs (20 Hz) were evoked by current injection (1.8–2.4 nA, 2 ms) in M1 either at Vrest (black) or Vhyper (gray). B, Representative experiment shows a pair of APs in M1 delivered either at Vrest (black) or Vhyper (gray) and the average of evoked EPSPs in M2 recorded at Vrest. Colors sign corresponding recordings in M1 and M2. C1, EPSPs were recorded in cohorts of five sweeps by alternating presynaptic membrane potential between Vrest (black) and Vhyper (gray). C2, EPSP amplitudes (left) and PPRs (right) are shown at presynaptic Vrest (rest) and Vhyper (hyper). Black circles connected with thick black lines represent the mean of eight individual experiments. ***p < 0.001. D, Recovery of the mitral–mitral synapse from depression was fitted with a single exponential (inter-stimulus interval [ISI]). E1, The amplitude of EPSP1 (filled triangles) and EPSP2 (open triangles) plotted as a function of coupling coefficient (CC). E2, PPRrest evoked at Vrest plotted as a function of coupling coefficient.
Focal puff application.
For focal application of GABAA receptor blocker (picrotoxin [PTX], 50 μm) or high concentration of KCl (2 s puff of 65 mm KCl, susbstituted for NaCl), a puffer pipette was placed gently over the target glomerulus or the mitral cell layer. Fluorescein (Na+ salt, 300 μm) included in the puffing solution monitored the spread of the drug. Appropriate direction of saline flow and puffing pipette ensured a focal effect (see Fig. 9A,B1, drawings).
Figure 9.

Periglomerular cells are major sources of T-type Ca2+ channel-sensitive asynchronous GABA transmission onto mitral cells. A, Puff application of 50 μm PTX on the apical tuft of a recorded mitral cell (M1) reduced the frequency of IPSCs at −50 mV test potential after a 4 s hyperpolarizing prestep to −100 mV (n = 4). QX-314 (0.5 mm) was included in the pipette. PTX was directed away from the external plexiform layer with a laminar perfusion flow (see arrow on the drawing). Puffer pipette was positioned on the surface of the glomerulus. *p < 0.05. B1, A 2 s puff of 65 mm KCl solution from above the mitral cell layer (30 or 90 μm) depolarizes mitral cells (Rec B1, flow was directed away from external plexiform layer). B2, The high K+ solution puffed onto the mitral cell layer increased the EPSC rate in periglomerular cells (PG, Rec B2) as shown in the example current trace and the mini rate histogram (top and middle). This increase was abolished by 500 μm Ni2+ (bottom; n = 6, 6 trials before and after Ni2+ application in each cell). Again, high K+ solution was directed away from the glomerular layer with a laminar perfusion flow (see arrow on the drawing). TTX (1 μm), MK-801 (20 μm), and bicuculline (20 μm) were present throughout the mini rate measurements. **p < 0.01.
Single-cell electroporation.
To visualize mitral cells projecting to the same glomerulus for paired recording, we electroporated single mitral cells with borosilicate glass electrodes (5–8 MΩ; Harvard Apparatus) filled with Alexa-594 (1–3 mm). We used 1–2 square pulses (10 μA, 10 ms, 0.5 Hz) to label mitral cell apical dendrite in ∼5–10 min. Pulses were generated by a stimulation isolation unit (SIU90, Cygnus Technology) triggered by EPC10 amplifier (HEKA).
Calcium imaging.
Calcium imaging was performed using 810 nm pulsed laser light generated by a Nd:YaG pumped Ti:sapphire laser system (Verdi 10W; MIRA Optima 900F; Coherent Laser) and a custom-made two-photon laser scanning microscope as described previously (Johnston and Delaney, 2010). An x-y galvonometer mirror assembly scanned the beam across the sample through a 40×, 0.8 numerical aperture water-dipping lens (Olympus). A customized Olympus BX51WI microscope was fitted with two H7422P-40 photomultipliers (Hamamatsu) to detect epifluorescence and one H8224–40 photomultiplier below a 0.9 NA oblique illumination condenser to detect transmitted fluorescence. Ca2+ dye epifluorescence and transmitted fluorescence (Ca2+ Green-1, 150 μm) was summed, whereas Ca2+-insensitive epifluorescence (Alexa-594, 30 μm) was simultaneously detected to obtain a green/red (G/R) ratiometric measurement. Fluorescence signals were spectrally separated using appropriate combinations of filters and a dichroic mirror. All recordings were performed > 20 min after breakthrough to allow for dialysis of the extended dendritic compartments. Pixel intensity within a polygonal region of interest was averaged for each frame, and both red and green dye fluorescence was corrected for background fluorescence (adjacent nonfilled tissue). G/R ratio was calculated for each frame to follow changes in Ca2+ level. This method allowed easy discrimination of the changes in Ca2+ transients and basal [Ca2+]. A PCI6111 A/D converter (National Instruments) was used to acquire fluorescence signals (600 kHz to 1.25 MHz sampling rate) and to output scan-wave signals simultaneously with acquisition. Hardware control, image acquisition, and analysis were written in-house using Igor Pro (Wavemetrics). External C++ routines were linked to the Igor interface to accelerate some display and analysis functions. Imaging was performed by scanning small rectangular scan areas that encompassed a length of dendrite and an area of background. Dendrites were aligned along the horizontal axis of the scan using a hardware-based scan rotation device with bidirectional scanning in the horizontal axis to increase scanning rates (Tsai et al., 2002). Frame rates in the range of 40–200 Hz were typically used. Excitation laser light was always kept as low as required to attain a sufficient signal-to-noise ratio and minimal photodamage. To determine back-propagating AP (bAP)-evoked Ca2+ transients and basal [Ca2+], we averaged at least 10 scans. The first Ca2+ transient amplitude (Amp1) was calculated as a difference of the first G/R peak (20 ms around the peak) and the basal G/R (50–100 ms before bAP). The second Ca2+ transient amplitude (Amp2) was a difference of the second G/R peak (20 ms around the peak) and the G/R between the two peaks (10–15 ms before Amp2). Ca2+ imaging data were acquired in cohorts of five scans that alternated data collection between Vrest and Vhyper. Image data were analyzed using the Igor Pro Neuromatic package.
Statistics.
For statistical analyses of significance, paired t test, Pearson correlation calculation, two-way repeated-measures ANOVA, and one-way ordinary ANOVA followed by Bonferroni post hoc test were used always with two-tailed probabilities. Averages are expressed as mean ± SEM. Number of experiments (n) shows the number of individual cells, each from a different slice and animal. Differences were considered significant at p < 0.05, except in the case of Bonferroni post hoc test where the significance levels were corrected by the number of comparisons. Statistics were calculated using GraphPad Prizm 4.
Materials.
AlexaFluor-594 hydrazide and Ca2+ Green-1 hexapotassium salt were purchased from Invitrogen. We obtained AP-5 and NNC 55–0396 dihydrochloride from Tocris Bioscience, isoflurane from Abbott Laboratories, KCl, NaCl, sucrose, CaCl2, MgCl2, NaH2PO4, glucose, and NaHCO3 from Merck, PTX from Abcam, N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium chloride (QX-314, Cl−) from Alomone Labs, and Na-ascorbate from ICN Biochemical. Z941 was a kind gift from Dr. Terrence Snutch (University of British Columbia, Vancouver, British Columbia, Canada). All other chemicals (4-aminopyridine (4-AP), agar, bicuculline, EGTA, potassium methanesulfonate (KCH3SO3), glutamate, HEPES, phosphocreatine, MgATP, Na2ATP, myo-inositol, MK-801, and TTX) were ordered from Sigma.
Results
Strong activation of the olfactory sensory afferent axons produces a long-lasting depolarization (LLD) in mitral cells (Schoppa and Westbrook, 2001). LLD is the result of glutamate released by primary afferents and mitral dendrites within the glomerulus acting through a combination of binding to NMDA-R and activation of LVA T-type channels (Cav3.x) via an mGlu-R dependent pathway (De Saint Jan and Westbrook, 2007; Johnston and Delaney, 2010). In our previous study (Johnston and Delaney, 2010), T-type channel blockade did not attenuate the initial AMPA-dependent, monosynaptic EPSPs from the olfactory afferents to the mitral cells; its effects were specific to the LLD. In the current study, we tested for a further role of T-type channels in controlling olfactory network function by stimulating population postsynaptic potentials with antidromic stimulation of LOT axons (Fig. 1). LOT stimulation fires many mitral and tufted cells (but not olfactory afferents), which propagate APs into their apical and tuft dendrites. This results in a fast-onset EPSP (due to mitral–mitral and tufted-mitral synapses) followed by an LLD. We applied a pan-T-type channel blocker Z941 (10 μm) (Tringham et al., 2012) in the presence of NMDA-R and GABAA-R blocker (AP-5, 50 μm; PTX, 50 μm, respectively; Fig. 1A) and, as expected, found it attenuated the LOT-stimulated LLD, but unexpectedly it also reduced the short latency component of the population PSP (two-way repeated-measures ANOVA, F(1,8) = 7.4, p = 0.0262; EPSP1 control = 3.24 ± 0.71 mV, EPSP1 Z941 = 2.56 ± 0.75 mV, n = 5, p = 0.0093; EPSP2 control = 2.32 ± 0.66 mV, EPSP2 Z941 = 2.23 ± 0.71 mV, n = 5, p = 0.6680) and increased its the paired-pulse ratio (PPR) (PPR control = 0.73 ± 0.16, PPR Z941 = 0.90 ± 0.17, n = 5, p = 0.0071, paired t test; Fig. 1B1,B2). Previous recordings from mitral cells have shown that block of T-channels does not affect the initial EPSPs evoked by olfactory nerve stimulation (De Saint Jan and Westbrook, 2007; Johnston and Delaney, 2010), indicating that postsynaptic T-channels do not boost the initial EPSP. Thus, after observing that Z941 reduced short-latency LOT-evoked synaptic responses in mitral cells, we hypothesized that T-type channels might serve to modulate spike-evoked transmitter release from glomerular tuft dendrites.
Figure 1.

Lateral excitation between mitral cell dendrites in the glomerulus is modulated by T-type Ca2+ channels. A, Antidromic stimulation of the LOT by a bipolar electrode (see schematic drawing on the left) evoked an LLD in olfactory mitral cells. Inhibition of GABAA-R (middle; PTX, 50 μm) and NMDA-R (right; AP-5, 50 μm) revealed a fast-onset EPSP. B1, Z941, a specific T-type channel antagonist (10 μm, gray), reduces the initial EPSP amplitude compared with control (black) elicited by a pair of brief LOT stimulation (20 Hz). Inset, Second EPSPs adjusted according to the baseline. Both traces are averages of multiple trials from a representative experiment. B2, Summary chart shows the Z941 effect on the EPSP amplitudes and the paired-pulse ratio (PPR). Black circles connected by thick black lines represent the mean of five experiments. PTX (50 μm) and AP-5 (50 μm) were present in all experiments. *p < 0.05. **p < 0.01.
Because T-channel activation varies substantially around mitral cell resting membrane potentials, we first examined whether mitral-to-mitral EPSP amplitude was sensitive to presynaptic membrane potential. We obtained dual whole-cell recordings from connected pairs. After electroporation of 20–25 mitral cells per slice, we identified 1–3 pairs that each projected to the same glomerulus and were therefore potentially connected (Fig. 2A) (Schoppa and Westbrook, 2001). Chemically connected pairs were confirmed by observing EPSPs at constant latency of <2.5 ms after a current-evoked AP. Using this dye-filling strategy, we found that 62% (27 of 44) of pairs projecting to the same glomerulus were connected in at least one direction by a chemical synapse, the same probability of connection observed by Pimentel and Margrie (2008). Nineteen of the chemical connections were unidirectional, 6 were bidirectional, and 2 were only tested in one direction. Chemically connected pairs were always electrically coupled, and the coupling coefficient (CC) was always equal between pairs (tested in 25 of the 27 pairs) similar to previous reports (Pimentel and Margrie, 2008). We quantified both the coupling coefficient and the EPSP amplitudes along with the PPRs in 13 of the chemically connected pairs. We plotted each cell's EPSP amplitude versus its coupling coefficient and found that these were not significantly correlated (EPSP1 rest, p = 0.0725; EPSP2 rest, p = 0.0893; Fig. 2E1), whereas the PPRs were clearly uncorrelated (PPR rest, p = 0.9629; Fig. 2E2), indicating that the measured EPSPs were due to lateral chemical transmission (Pimentel and Margrie, 2008) rather than electrical coupling of self-excitation (Schoppa and Westbrook, 2002).
To test whether AP-evoked transmitter release varies with subthreshold potential changes, we recorded evoked EPSPs in the postsynaptic mitral cell (M2) at Vrest (between −60 and −65 mV) while holding the presynaptic neuron (M1) at either Vrest or Vhyper (3 s of hyperpolarization between −80 and −95 mV; Fig. 2A). We evoked APs with 2 ms suprathreshold depolarizing steps from rest or hyperpolarized holding potentials. We collected EPSPs in groups of 5, alternating the presynaptic (M1) membrane potential between Vrest and Vhyper (n = 8; Fig. 2C1) for each group. The 10 s intervals were given between trials to avoid carryover of short-term depression observed at this synapse (Fig. 2D). With this experimental paradigm, we used two measures of presynaptic release probability: the PPR and the coefficient of variation (CV).
Substantial paired-pulse depression and low initial failure rate at Vrest (4.9 ± 3.5%, n = 8) suggest that mitral–mitral connections have a high release probability. APs evoked from a hyperpolarized potential produced smaller initial EPSPs (two-way repeated-measures ANOVA, F(1,14) = 22.67, p = 0.0003; EPSP1 rest = 0.76 ± 0.19 mV, EPSP1 hyper = 0.54 ± 0.15 mV, n = 8, p < 0.0001; EPSP2 rest = 0.33 ± 0.05 mV, EPSP2 hyper = 0.32 ± 0.05 mV, n = 8, p = 0.6624) and resulted in a significant increase in the PPR (PPR rest = 0.50 ± 0.07, PPR hyper = 0.70 ± 0.07, n = 8, p = 0.0009; Table 3; Fig. 2C2). Together, the decrease in EPSP amplitude and increase in PPR suggest a decreased probability of transmitter release with hyperpolarization.
Table 3.
Comparison of changes (%) in EPSP amplitude and PPR caused by hyperpolarization of the presynaptic mitral cells or by block of T-type channels with Z941 (10 μm)
| Control (n = 8) | Hyperpolarized (n = 8) | % change | Pre-Z941 (n = 5) | Z941 (n = 5) | % change | |
|---|---|---|---|---|---|---|
| EPSP1 (mV) | 0.76 ± 0.19 | 0.54 ± 0.15 | −28.9% | 1.28 ± 0.36 | 0.92 ± 0.27 | −28.1 |
| p < 0.0001 | p = 0.0040 | |||||
| EPSP2 (mV) | 0.33 ± 0.05 | 0.32 ± 0.05 | −3.0% | 0.34 ± 0.08 | 0.37 ± 0.07 | 8.8 |
| p = 0.6624 | p = 0.7510 | |||||
| PPR | 0.50 ± 0.07 | 0.70 ± 0.07 | 40.0% | 0.35 ± 0.08 | 0.50 ± 0.09 | 42.9 |
| p = 0.0009 | p = 0.0032 |
Next, we determined the CV of EPSPs with and without presynaptic hyperpolarization. The CV is known to inversely correlate with quantal content and to be independent of postsynaptic sensitivity (Malinow and Tsien, 1990). As expected, a reduction in EPSP1 with hyperpolarization was accompanied by an increase in CV, consistent with a presynaptic mechanism of modulation (CV1 rest = 0.31 ± 0.04, CV1 hyper = 0.44 ± 0.06, n = 8, p = 0.0489, paired t test). Thus, the changes in PPR and CV that accompany AP-evoked release from a hyperpolarized baseline are all consistent with a decline in presynaptic release probability.
We considered two possible sources for the decline in AP-evoked transmitter release resulting from presynaptic hyperpolarization: a reduction in the HVA Ca2+ influx that drives release or a decline in basal [Ca2+] modulating evoked release.
Mitral cells express A-type K+ channels (IA) (Schoppa and Westbrook, 1999; Christie and Westbrook, 2003; Davison et al., 2004; Kollo et al., 2008). A period of hyperpolarization increases IA channel availability by relieving inactivation, potentially narrowing the AP or reducing AP amplitude, which could lead to reduced Ca2+ influx. Indeed, the width at half-amplitude of somatic APs decreased with hyperpolarization (two-way repeated-measures ANOVA, F(1,14) = 59.15, p < 0.0001; AP1 rest, 0.59 ± 0.04 ms; AP1 hyper, 0.46 ± 0.02 ms; n = 8, p = 0.0001; AP2 rest, 0.59 ± 0.04 ms; AP2 hyper, 0.46 ± 0.02 ms; n = 8, p = 0.0001; Fig. 3A).
Figure 3.

Electrically coupled spikelets and bAP-evoked Ca2+ transients in the apical tuft of mitral cells are not decreased with somatic hyperpolarization. A, Somatic APs recorded from the presynaptic mitral cell (M1) narrow at Vhyper (representative average traces on the left; black, Vrest; gray, Vhyper). AP traces are overlaid by the AP generation threshold (dashed line). M2, postsynaptically recorded spikelets were aligned with the peak of the presynaptic AP and overlaid by the spikelet generation threshold (representative average traces on the right; black, Vrest; gray, Vhyper). Arrows indicate AP and spikelet initiation. Summary charts show AP width at half amplitude (bottom left; n = 8) and spikelet amplitude and width at half amplitude (bottom right). Black dots connected with thick black lines represent the means of eight individual experiments. ***p < 0.001. B1, Representative traces of a pair of bAPs (top) and Ca2+ transients (20 Hz, bottom) were measured in the apical tuft at Vrest (black) and Vhyper (gray). Sets of five sweeps at Vrest were alternated with sets of five sweeps at Vhyper. Inset, bAP-evoked Ca2+ transients are adjusted according to the basal [Ca2+]. B2, Summary chart shows Ca2+ transient amplitudes (Amp1, Amp2) at Vrest and Vhyper in the tuft and apical dendrite (Ad). Ca2+ transient amplitudes in the tuft (n = 7) are increased with hyperpolarization, although there is no significant change in the apical dendrite (n = 5). *p < 0.05.
Although IA can shape somatically recorded APs and delay the initiation of an AP to somatically injected current, our measurements do not provide information about the AP at the release sites in the apical tuft. Because the release sites on the apical tufts of mitral cells are inaccessible for patch pipettes, we took advantage of the fact that mitral cells are electrically coupled through gap junctions formed within the glomerulus (Christie et al., 2005). An AP in one mitral cell will evoke a spikelet in an electrically coupled neighbor (Fig. 3A). This spikelet reflects an attenuated version of the AP occurring within the vicinity of the gap junction. We examined these spikelets for changes in shape upon hyperpolarization (Fig. 3A). Although the somatically recorded AP was narrowed (Fig. 3A), the electrically coupled spikelets were unaffected by presynaptic hyperpolarization (amplitude, Vrest, 0.097 ± 0.014 μV; Vhyper, 0.125 ± 0.013 μV; n = 8, p = 0.1106, paired t test; width at half-amplitude, Vrest, 0.22 ± 0.02 ms; Vhyper, 0.20 ± 0.01 ms; n = 8, p = 0.4857, paired t test; Fig. 3A). Although no difference in the spikelets was observed upon hyperpolarization, the strongly attenuated spikelets may be insufficiently sensitive to detect subtle changes in the AP shape. Therefore, to corroborate these findings, we also measured the Ca2+ transients in the tuft and apical dendrite, evoked by somatically generated bAPs from the different membrane potentials. Again, in this set of experiments, the width at half-amplitude of somatic APs was decreased with hyperpolarization (two-way repeated-measures ANOVA, F(1,12) = 44.11, p < 0.0001; AP1, p = 0.0005; AP2, p < 0.0006; Table 1; Fig 3B1,B2), but the bAP-evoked Ca2+ transients in the tuft dendrite increased slightly (two-way repeated-measures ANOVA, F(1,12) = 18.35, p = 0.0011) whereas the transient in the apical dendrite declined (two-way repeated-measures ANOVA, F(1,8) = 4.58, p = 0.0648). These data indicate that the reduction in AP-evoked transmitter release with hyperpolarization (Fig. 2) is not due to changes in the AP waveform within the tuft dendrites (Fig. 3). The data are also consistent with the subcellular distribution of Kv4 channels, which mediate IA. Kv4 channels are present on the cell body and proximal apical dendrite but are absent from the tuft dendrites, as shown with immunohistochemical electron microscopy (Kollo et al., 2008).
Table 1.
Effect of membrane potential on somatic APs and back-propagating AP-evoked Ca2+ transients in the apical tuft and main apical dendrite of mitral cells
| AP width at soma (ms) | Tuft dendrite |
Apical dendrite |
|||
|---|---|---|---|---|---|
| Ca (ΔG/R) n = 7 | p | ΔCa (ΔG/R) n = 5 | p | ||
| AP1 rest | 0.61 ± 0.08 | 0.070 ± 0.015 | 0.0143 | 0.061 ± 0.015 | 0.3133 |
| AP1 hyper | 0.53 ± 0.09 | 0.079 ± 0.016 | 0.058 ± 0.016 | ||
| AP2 rest | 0.61 ± 0.08 | 0.054 ± 0.012 | 0.0077 | 0.055 ± 0.013 | 0.0870 |
| AP2 hyper | 0.53 ± 0.09 | 0.064 ± 0.011 | 0.049 ± 0.013 | ||
As previously reported (Johnston and Delaney, 2010), we found that hyperpolarization significantly lowered cytoplasmic (basal) [Ca2+] in the tuft (Ca2+ rest, G/R = 0.47 ± 0.06, Ca2+ hyper, G/R = 0.40 ± 0.04, n = 7, p = 0.0085, paired t test; Fig. 4A,B). Also consistent with our previous study, hyperpolarization induced a reduction in basal [Ca2+] in the distal main apical dendrite (Ca2+ rest, G/R = 0.55 ± 0.07, Ca2+ hyper, G/R = 0.50 ± 0.07, n = 5, p = 0.0170, paired t test; Fig. 4B). The time course of the change in [Ca2+] in the tuft during hyperpolarization was well fitted with a single exponential function (τdecay = 1.20 ± 0.37 s, n = 5; Fig. 4A).
Figure 4.

T-type Ca2+ channels modulate the basal [Ca2+] at subthreshold potentials in the primary dendrite of mitral cells. A, Mitral cells were loaded with Alexa-594 and Ca2+ Green-1 as shown on the fluorescent images (left). Rectangle represents the scanned area; red polygon represents the region of interest. Basal [Ca2+] was expressed as G/R. A 4-s-long hyperpolarizing step from Vrest to Vhyper caused a reduction in basal [Ca2+] (τdecay = 1.20 s, n = 5). B, A 4-s-long hyperpolarizing step significantly decreased the basal [Ca2+] in the apical dendrite and tuft. Black dots connected with thick black lines indicate the mean of 7 and 5 individual experiments (thin lines) in the apical tuft and apical dendrite (Ad), respectively. *p < 0.05; **p < 0.01. C1, C2, Sinusoidal modulation of membrane potential above and below normal resting potential modulates basal [Ca2+]. Voltage-dependent increases in basal [Ca2+] are attenuated by T-type channel blockers NNC 55–0396 (50 μm) and Z941 (10 μm). Bar charts on the right represent the population effect of Z941 on the amplitudes of subthreshold Ca2+ influx (control, black, n = 4; Z941, gray, n = 4, 5–20 min bath application). ***p < 0.001.
Previously, we investigated the source of subthreshold Ca2+ influx in mitral cells (Johnston and Delaney, 2010). To simulate in vivo odor-evoked oscillations (Charpak et al., 2001), we sinusoidally modulated somatic membrane potential between −70 and −50 mV and detected corresponding [Ca2+] changes. In apical dendrites and tufts, we observed [Ca2+] oscillations in the presence of the NMDA receptor antagonist AP-5. These subthreshold [Ca2+] oscillations were Ni2+-sensitive (500 μm), and we showed that Cav3.3 T-type Ca2+ channels are responsible for them. In the current study, we tested other T-type Ca2+ channel blockers because, in addition to NMDA-Rs (Mayer and Westbrook, 1985), Ni2+ is reported to have effects on R-type Ca2+ channels (Randall and Tsien, 1995; Zamponi et al., 1996), Na+/Ca2+ exchangers (Iwamoto and Shigekawa, 1998), and voltage-gated sodium channels (VGSCs) (Coste et al., 2007).
First, we applied NNC 55–0396, a nonhydrolyzable analog of mibefradil, which is more specific for T-type channels because it lacks the effects of the mibefradil metabolite on HVA Ca2+ channels (Huang et al., 2004); 50 μm NNC 55–0396 applied for >15 min inhibited LVA Ca2+ influx (Fig. 4C1), reduced the amplitude of the somatic APs (Fig. 5A), and reduced bAP-evoked Ca2+ transients (n = 3; data not shown). Thus, although an effective blocker of the LVA Ca2+ influx, it also, over time, adversely affected VGSCs similar to the reported effect of the parent compound mibefradil (Eller et al., 2000; McNulty and Hanck, 2004; McNulty et al., 2006). Although this effect excluded the use of NNC 55–0396 in our later experiments where AP amplitude needed to be unaffected over extended time periods, they confirmed that T-type Ca2+ channels are the primary source of Ca2+ influx at voltages below −50 mV in olfactory mitral cells.
Figure 5.
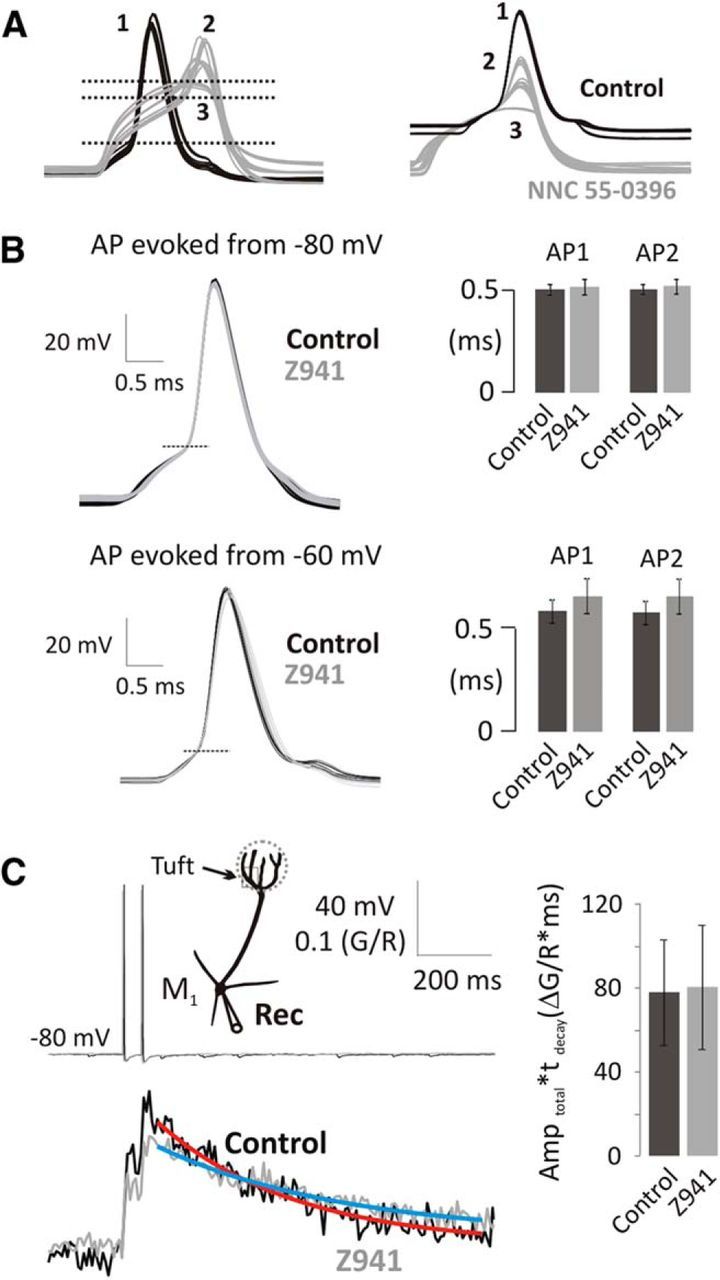
bAP-evoked Ca2+ transients are independent of T-type Ca2+ channels in mitral cells. A, NNC 55–0396 (50 μm) raises threshold (left; horizontal lines; 1, pre-NNC; 2, 9 min NNC; 3, 22 min NNC) and attenuates AP amplitude (right; overlaid at the AP generation threshold). B, Representative overlaid traces of somatic APs (threshold at horizontal line) and summary data indicate that the somatic AP shape is unchanged by Z941. Column charts represent the width at half-the threshold to peak amplitude of somatic APs in control (black) and in the presence of Z941 (10 μm; gray) delivered either at −80 (top; n = 5) or −60 mV (bottom; n = 5). C, bAPs (20 Hz, top) were delivered to mitral cells at Vhyper, and Ca2+ transients were recorded in the apical tuft in control (black) and in the presence of Z941 (gray). See location in the schematic drawing. Fitted single exponential curves indicate τdecay. Integrals of Ca2+ transients were measured as Amptotal × τdecay (n = 5, 5–20 min application).
Next, we tested the specificity of Z941 (10 μm), a novel pan-T-type Ca2+ channel antagonist (Tringham et al., 2012). We confirmed that Z941 reduced subthreshold Ca2+ increases (two-way repeated-measures ANOVA, F(1,6) = 243.2, p < 0.0001; sine1, ΔG/R = 0.190 ± 0.015, sine1 in Z941, ΔG/R = 0.093 ± 0.014, n = 4, p < 0.0001; sine2, ΔG/R = 0.170 ± 0.008, sine2 in Z941, ΔG/R = 0.083 ± 0.013, n = 4, p < 0.0001; Fig. 4C2). However, in contrast to NNC 55–0396, Z941 had no effect on the somatically recorded APs (Fig. 5B); this demonstrates that Z941 does not affect ion channels involved in the AP waveform, such as Nav and Kv channels.
We then used Z941 to determine the contribution of Cav3 channels to AP evoked Ca2+ influx. We held mitral cells continuously at Vhyper, to minimize the Cav3 influx contributing to basal [Ca2+] (Fig. 5C), and delivered a pair of AP before and during application of Z941. Ca2+ influx was tested during the 5–20 min application of Z941. Over this period of time, buffer capacity tends to increase slightly as dye continues to diffuse into distal dendrites (Yasuda et al., 2004). Increased buffer capacity attenuates Ca2+ transient peaks so we estimated the Ca2+ influx using the integral of the Ca2+ transient, a parameter that is less dependent on Ca2+ buffer capacity (Helmchen et al., 1996; Yasuda et al., 2004). We estimated that the total bAP-evoked Ca2+ influx from a pair of APs was not reduced by Z941 (Table 2; pre Z941 Amptotal × τdecay = 76.8 ± 25.1, Z941, Amptotal × τdecay = 79.2 ± 29.4, n = 5, p = 0.6790, paired t test). From these results, we conclude that Cav3.3 T-type voltage-gated Ca2+ channels, present on mitral cells, modulate subthreshold Ca2+ influx but not the bAP-evoked Ca2+ transients in the apical tuft of olfactory mitral cells. This result is consistent with the slow activation kinetics of Cav3.3 channels (τact = 6–10 ms) (Klöckner et al., 1999; Chemin et al., 2002; Hildebrand et al., 2009) compared with the width at half-amplitude of somatic APs at 36°C (∼0.5 ms; Figs. 3 and 5). Furthermore, these data demonstrate that the T-type blocker Z941 selectively blocks LVA Ca2+ entry in mitral cells, leaving both the AP waveform and HVA Ca2+ entry unaffected.
Table 2.
Effect of Z941 on back-propagating AP-evoked Ca2+ transient
| ΔCa AP1 (ΔG/R) | ΔCa AP2 (ΔG/R) | τ decay (ms) | ΔCa total (ΔG/R) | ΔCa total*τ (ΔG/R*ms) | |
|---|---|---|---|---|---|
| Pre-Z941 | 0.124 ± 0.031 | 0.105 ± 0.029 | 319.6 ± 37.0 | 0.218 ± 0.059 | 76.8 ± 25.1 |
| Z941 | 0.093 ± 0.031 | 0.080 ± 0.024 | 395.5 ± 56.5 | 0.178 ± 0.052 | 79.2 ± 29.4 |
Our data suggest that the modulation of release probability, with changes in membrane potential, is likely a result of changes in the subthreshold [Ca2+]. To directly test this, we obtained recordings from pairs of connected mitral cells and applied Z941. We delivered two APs (20 Hz) at Vrest to the presynaptic cell (M1) and recorded EPSPs at Vrest from the postsynaptic cell (M2; Fig. 6A1,A2). EPSPs were collected with cohorts of five sets of stimulation in control and in the presence of Z941 (10 μm; Fig. 6B1). Z941 was applied for >5 min before testing EPSPs. We found a significant decrease in the initial EPSP amplitude (two-way repeated-measures ANOVA, F(1,8) = 6.71, p = 0.0321; control, EPSP1 = 1.28 ± 0.36 mV, Z941, EPSP1 = 0.92 ± 0.27 mV, n = 5, p = 0.0040; control, EPSP2 = 0.34 ± 0.08 mV, Z941, EPSP2 = 0.37 ± 0.07 mV, n = 5, p = 0.7510) along with an increase in PPR with application of Z941 (control, PPR = 0.35 ± 0.08, Z941, PPR = 0.50 ± 0.09, n = 5, p = 0.0032, paired t test). The reduction in EPSP amplitude along with the increase in PPR suggest that block of Cav3 channels decreases the probability of glutamate release from mitral cell apical tufts (Table 3; Fig. 6B2).
Figure 6.

Inhibition of T-type Ca2+ channels decreases the strength of mitral–mitral cell connections. A1, Mitral cell pairs were visualized by Alexa-594. Representative experiment shows an example presynaptic stimulation in M1 at Vrest (20 Hz; top) and the averages of evoked EPSPs in M2 at Vrest in the absence (control, black) and presence of Z941 (10 μm, gray). Inset, Overlaid averages of EPSP2. A2, Individual EPSP1 traces are shown after baseline subtraction. Arrows indicate the peak of average EPSP1 under control conditions and in the presence of Z941. B1, EPSPs were recorded in cohorts of five sweeps in control (black, n = 5) and in the presence of Z941 (gray, n = 5, 5–15 min bath application). Distances between the dots do not represent exact time, only the sequence of recordings. B2, EPSP amplitudes (thin lines, left) and PPRs (thin lines, right) are shown in control and in the presence of Z941. Black dots connected with thick black lines indicate the means of five individual experiments. **p < 0.01.
We observed that the effects of hyperpolarization and Z941 were quantitatively equivalent, consistent with their acting via a similar mechanism (Table 3). Hyperpolarization decreased EPSP1 by 28.9% (n = 8), compared with a decrease of 28.1% (n = 5) with Z941. Hyperpolarization decreased EPSP2 by 3.0% (n = 8), whereas EPSP2 was increased by 8.8% following Z941 application (n = 5). Accordingly, the increase in PPR was also similar, 40.0% for hyperpolarization versus 42.9% for Z941 application. These data suggest that T-type VGCCs, most probably Cav3.3 (Johnston and Delaney, 2010), are able to modulate the strength of mitral–mitral connections as a function of subthreshold membrane potential, most likely acting through the modulation of cytoplasmic [Ca2+].
T-type channels display high levels of steady-state inactivation around resting potentials (Chemin et al., 2002). Hyperpolarization removes this inactivation leading to a transient increase in T-type current upon returning to rest. This manifests as a rebound in [Ca2+] in the apical tuft that may or may not be accompanied by a rebound depolarization or rebound spiking (Johnston and Delaney, 2010). In this study, we confirmed the ubiquitous occurrence of a substantial rebound Ca2+ influx and observed instances of rebound depolarization, which were sometimes sufficient to evoke spiking (Fig. 7A). Rebound Ca2+, and when present, rebound depolarization, were blocked by Z941, consistent with influx through deinactivated T-type channels (Fig. 7B1,B2; number of APs per first second, control, 1.99 ± 0.56, Z941, 0 ± 0, n = 5, p = 0.0244, paired t test; number of low-threshold oscillations, control, 1.42 ± 0.27, Z941, 0.04 ± 0.04, n = 5, p = 0.0059, paired t test). During the course of these experiments, we also detected barrages of membrane potential fluctuations during the rebound depolarization, which were also eliminated by Z941 (Fig. 7, inset). These fluctuations occurred at membrane potentials several mV hyperpolarized to those that activate the TTX-sensitive membrane oscillations described by Desmaisons et al. (1999) and Balu and Strowbridge (2007) and persisted when VGSCs were blocked by internal 0.5 mm QX-314 (Fig. 8). These fluctuations were sensitive to picrotoxin (50 μm, n = 4, p < 0.0001, paired t test; Fig. 8A1, inset), so are likely a result of feedback IPSPs similar to those described by Castro and Urban (2009).
Figure 7.

Rebound depolarizations in mitral cells are dependent on T-type channels. A, Hyperpolarizing prepulse (4 s) evokes a rebound depolarization in mitral cells. APs are truncated. The rebound was dependent on the size of the hyperpolarizing prepulse. B1, Inhibition of the T-type channels eliminates the rebound depolarization as indicated by the representative traces (control, black, n = 5; Z941, 10 μm, gray, n = 5). Inset, Large Ca2+ influx into the apical tuft of a mitral cell (G/R, upper left, black) accompanied by a subthreshold rebound depolarization (lower left, black). Both are sensitive to Z941 (gray). The jitters on the crest of the control voltage trace are inhibited by Z941 while the trains of APs (lower right, gray) and the train-evoked Ca2+ influx into the tuft (upper right, gray) are preserved. B2, Number of APs (first seconds after the hyperpolarizing prestep) and low-threshold-evoked oscillations significantly decreased in the presence of Z941. *p < 0.05. **p < 0.01.
Figure 8.
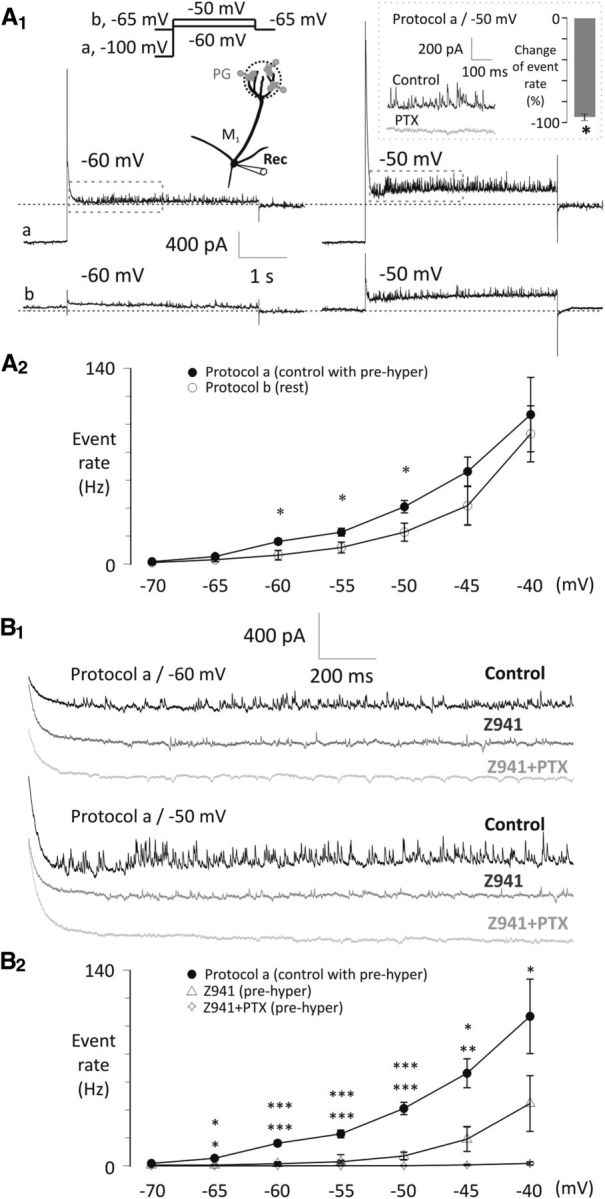
Asynchronous IPSC frequency onto mitral cells is modulated by T-type Ca2+ channels and subthreshold potentials of mitral cells. A1, Example current traces show that voltage steps to −60 or −50 mV after either a 4 s hyperpolarizing prestep to −100 mV (Protocol a/control with prehyper) or keeping the mitral cell at −65 mV (Protocol b/rest, data are not leak subtracted) evoke asynchronous IPSCs onto mitral cells, with a rate dependent on the voltage of the prestep. Segments of control traces in the rectangles are magnified in B1. Inset, Inhibitory effect of bath-applied PTX (50 μm, n = 4) on asynchronous IPSCs at −50 mV test potential after applying Protocol a. *p < 0.05. A2, Summary diagram of the effects of Protocol a (control with prehyper, n = 6) or Protocol b (rest, n = 6) tested with voltage steps between −70 and −40 mV with 5 mV increments. Frequency of the IPSCs was calculated during a 1.5 s interval following the decay of the step-evoked A-current and was modulated by the prestep. *p < 0.05. B1, Example traces of the drug effect on the asynchronous IPSC frequency (control, black; Z941, 10 μm, middle gray; Z941 + PTX, 50 μm; light gray) at −60 and −50 mV test potentials after delivering Protocol a. B2, Summary diagram of the inhibitory drug effects (control, n = 6; Z941, n = 5; Z941 + PTX, n = 4) tested with voltage steps between −70 and −40 mV with 5 mV increments after applying Protocol a. QX-314 (0.5 mm) was included in the pipette throughout. Significance levels indicate comparisons of Protocol a versus Z941 (top row) and Protocol a versus Z941 + PTX (bottom row). *p < 0.05. **p < 0.01. ***p < 0.001.
To quantify the feedback IPSCs evoked by subthreshold depolarizations, we applied small depolarizing voltage-clamp steps from either −100 mV (voltage Protocol a/control with prehyper) or −65 mV (voltage Protocol b/rest; Fig. 8A1,A2). We included QX-314 in the recording pipette at a concentration (0.5 mm) that is low enough to avoid interfering with T-type channels but sufficient to minimize AP generation and block Na+ channel-dependent subthreshold voltage oscillations (Desmaisons et al., 1999). Voltage steps to potentials from −60 to −50 mV evoked higher IPSC frequencies when steady-state inactivation of T-type channels was relieved with voltage Protocol a (Fig. 8A1,A2). This suggests that LVA T-type Ca2+ influx is responsible for driving release of glutamate, which then excites feedback from inhibitory neurons (Fig. 8A1,A2). We confirmed this with Z941, which effectively blocked 80% to 90% of these events for steps up to −50 mV (e.g., step to −50 mV test potential, one-way ANOVA, F(3,17) = 14.6, p < 0.0001; IPSC rate: Protocol a, 41.1 ± 6.44 Hz, n = 6, Protocol b, 22.9 ± 4.42 Hz, n = 6, Z941, 7.0 ± 2.84 Hz, n = 5, Z941 + PTX, 0.15 ± 0.1 Hz, n = 4; Protocol a vs Protocol b, p = 0.0097; Protocol a vs Z941, p < 0.0001; Protocol a vs Z941 + PTX, p < 0.0001; Fig. 8B1,B2). Hyperpolarizing presteps were proportionately less effective at increasing IPSC frequency for depolarizations >−50 mV (Fig. 8A2). Similarly, the relative effectiveness of Z941 to block increased IPSC frequency declined with depolarization beyond −50 mV (Fig. 8B2). This suggests either some saturation of the asynchronous release rate or the recruitment of other HVA Ca2+ channels that is driving release above −50 mV (see also Castro and Urban, 2009; Jerome et al., 2012).
Feedback IPSCs can potentially originate from granule cells that synapse onto secondary dendrites or periglomerular cells that synapse onto the distal tuft. Bath-applied PTX blocks all IPSCs evoked by subthreshold depolarization (Fig. 8A) but does not identify the location of the synaptic input. We focally applied PTX with a laminar perfusion flow directed away from the external plexiform layer through a small-diameter puffer pipette positioned on the surface of the glomerulus. Fluorescent dye in the pipette confirmed the restriction of PTX to the glomerular region. We observed that 76.2 ± 14.0% (n = 4, p = 0.0123, paired t test) of IPSCs in mitral cells elicited by steps from −100 to −50 mV were blocked by application of PTX to the sites of periglomerular synapses (Fig. 9A), indicating that the majority of the feedback IPSCs originate from periglomerular rather than granule cells. Finally, we used localized K+ application (2 s puff of 65 mm KCl, susbsituted for NaCl) to give subthreshold depolarization to groups of mitral cell somata; again, flow was directed away from the external pexiform layer. To isolate fast glutamatergic inputs, 20 μm MK801 and 20 μm bicuculline were included in the bath along with 1 μm TTX to prevent spiking. Under these conditions, a 2 s K+ puff resulted in an average 8.9 ± 1.3 mV depolarization in mitral cells measured directly adjacent to the puffer pipette (n = 5; Fig. 9B1). The amount of depolarization fell off sharply for mitral cells further from the puffer, so it is reasonable to assume that depolarizations of <10 mV were produced in the tuft. We then recorded from periglomerular neurons directly above the population of depolarized mitral cells. The depolarization of mitral cells caused a 19.96 ± 6.6% increase in the rate of EPSCs measured in 6 trials each from 6 cells (rate measured in the 20 s before puff vs 20 s after puff). This increase in EPSC rate was completely blocked by 500 μm Ni+ (n = 6, p = 0.0074, paired t test; Fig. 9B2).
Discussion
Whole-cell recordings and Ca2+ imaging were performed on individual olfactory mitral cells and synaptically connected mitral pairs. We modulated the small but persistent T-type window current (i.e., the overlap of steady-state activation and inactivation curve around resting membrane potential) by changing voltage or antagonizing T-type channels. We demonstrated the selectivity of Z941 for T-type channels in mitral cells (Figs. 4C and 5B,C) and found that a similar decrease in the strength of connection and an increase in PPR between pairs of mitral cells were induced by Z941 and hyperpolarization to −80 mV. Furthermore, we demonstrate that subthreshold depolarizations that activate T-type Ca2+ channels can drive sufficient dendritic glutamate release to evoke feedback IPSCs from periglomerular neurons.
Distal apical mitral dendrites are well known for their ability to initiate and support back-propagation of APs that invade the distal apical tuft. APs evoked from a hyperpolarized baseline result in smaller EPSPs in connected mitral cells. Simultaneous but opposite changes in PPR and CV are consistent with reduced release probability. Imaging reveals that hyperpolarizing current injected into the soma for several seconds slowly reduces basal [Ca2+] in the tuft dendrite. It also deinactivates transient Kv4 channels as revealed by delayed spike initiation and spike narrowing for APs generated by current injected into the soma from hyperpolarized potentials. Transient K+ channels have been shown to play a significant role in controlling AP amplitude and HVA Ca2+ influx into secondary dendrites and as a consequence modulate transmitter release from mitral-to-granule synapses located there (Christie and Westbrook, 2003; Davison et al., 2004). However, our electrically coupled spikelet measurements and tuft Ca2+ imaging suggest that the AP invading the tuft dendrites is unaffected by Kv4 channel activity. With respect to modulation of spike-evoked release at mitral–mitral tuft synapses, our data support a primary role for changes in voltage-dependent basal [Ca2+] rather than modulation of AP-evoked Ca2+ influx by voltage-dependent transient K+ channels. This is consistent with immunoelectron microscopy that indicates that Kv4.2 channels are present in the soma and secondary dendrites but are absent from the distal apical and tuft dendrites (Kollo et al., 2008). Although deinactivation of transient K+ channels by hyperpolarization narrows somatic APs and increases the current needed to initiate bAPs, it does not seem to affect bAP-evoked Ca2+ influx into the tuft. Thus, it does not explain the reduced transmitter release from tuft dendrite that follows a period of hyperpolarization.
On the other hand AP-evoked release was reduced by two manipulations that reduce basal [Ca2+]: hyperpolarization, which closes T-type channels, and specific block of T-type channels with Z941. Thus, as with many other synapses, small changes in [Ca2+] at the presynaptic dendritic release site appear to enhance release evoked by a brief high Ca2+ influx through HVA Ca2+ channels (Delaney and Tank, 1994; Regehr et al., 1994; Zucker and Regehr, 2002).
In most presynaptic terminals, the increased basal [Ca2+] that enhances evoked release is the consequence of residual Ca2+ that remains in the terminal after a preceding period of AP firing. However, at calyx of Held terminals, a small amount of Cav2.x current, on the order of 1–2 pA, alters cytoplasmic [Ca2+] by < 100 nm in a voltage-dependent manner around resting membrane potential to modulate spike-evoked release (Awatramani et al., 2005). Immunohistochemistry reveals a concentration of Cav3.3 reactivity in the apical tuft and distal portions of the apical dendrite with little staining in somata (Johnston and Delaney, 2010). Ca2+ imaging shows a corresponding pattern of [Ca2+] modulation by subthreshold voltage changes: largest in the tuft, diminishing toward the proximal apical dendrite and soma. The predominance of T-type channels to this subthreshold influx is confirmed by its sensitivity to Ni2+, Z941, NNC 55–0396, and the increased influx that follows a period of hyperpolarization characteristic of deinactivation of Cav3.x channels. Thus, we conclude that both cytoplasmic [Ca2+] and AP-evoked release probability vary with subthreshold membrane potential, primarily due to the activity of LVA Ca2+ channels, most likely Cav3.3. Cav3.3 channels are particularly suitable for controlling [Ca2+] around resting membrane potential since the largest window currents are produced by this subtype (Chemin et al., 2002; Perez-Reyes, 2003).
Submicromolar increases in resting [Ca2+] lead to increased frequency of asynchronous release at many synapses (Zucker and Regehr, 2002). Some synapses are designed specifically to communicate with their targets through high rates of asynchronous release, notably vertebrate photoreceptors, bipolar neurons, and auditory/vestibular hair cells, which possess specialized presynaptic ribbon and disc active zones. At many other synapses (e.g., neuromuscular junctions, numerous hippocampal and cortical synapses), small increases in presynaptic [Ca2+] cause an increase in asynchronous release, but there seems to be little postsynaptic electrophysiological consequence of this increased “leak” of quanta because the duration of quantal EPSPs is too short for effective temporal summation unless average release rates are >100 Hz. There is a growing body of evidence directly linking T-type channel mediated Ca2+ influx to increased vesicle fusion (Pan et al., 2001; Carbone et al., 2006; Weiss et al., 2012; Rozanski et al., 2013). In retinal bipolar cells, axonal release driven by subthreshold T-type channel Ca2+ influx has been directly demonstrated, and this release is sufficient to stimulate feedback inhibition from amacrine cells with demonstrable consequences for circuit function (Pan et al., 2001; Singer and Diamond, 2003). We demonstrate a similar process of T-type channel-mediated local feedback inhibition between mitral and periglomerular neurons. The high-input impedance of periglomerular neurons, typically ≥1 GΩ and membrane time constants of several 10s of milliseconds provide the scope for efficient integration of asynchronous EPSPs from mitral cells. Consequently, we can observe a 6- to 7-fold increase in the rate of feedback IPSCs as a mitral cell's membrane potential rises from −65 to −50 mV. The observation that a hyperpolarizing prestep in mitral cells increases the rate of feedback IPSCs evoked at subsequent depolarized potentials indicates that Cav3.x channels are providing the Ca2+ that is driving glutamate release at these potentials. As well, Z941 reduces feedback IPSCs consistent with a role for Cav3.x driving mitral dendrite glutamate release. Feedback IPSC frequency increases significantly with increased steps to −45 and −40 mV, but hyperpolarizing presteps and Z941 are proportionally less effective at these depolarized voltages. This may reflect partial saturation of asynchronous release or recruitment of additional sources of Ca2+ influx, such as L-type channels that are not enhanced by hyperpolarizing presteps. Because immunohistochemistry indicates that T-type channels are present in periglomerular neurons (Johnston and Delaney, 2010), there may be additional effects of this blocker on periglomerular excitability or Ca2+ influx that contributes to reduced GABA release from them in the presence of Z941.
The capacity for subthreshold mitral cell depolarization to activate feedback inhibition has potentially interesting consequences for odor processing. In a traditional circuit model where communication between elements requires spike threshold be attained, inhibitory feedback from periglomerular to mitral cells would not occur in weakly activated glomeruli. Feedforward inhibition (olfactory afferent to periglomerular to mitral) has been proposed to occur in weakly activated glomeruli by virtue of the higher impedance of periglomerular compared with mitral cells resulting in the selective suppression of weakly activated mitral cells (Gire and Schoppa, 2009). T-type channel-mediated, subthreshold release from mitral dendrites expands the scope of periglomerular inhibition of mitral cells to include feedback inhibition, which would further help to stabilize mitral membrane potential in the face of weak, i.e., off target inputs. By working to prevent mitral spiking in weakly activated glomeruli, subthreshold intraglomerular inhibition would also eliminate inappropriate feedforward, lateral inhibition via secondary dendrite activation of granule cells. In conclusion, dendrodendritic release driven by subthreshold Ca2+ fluctuations, combined with high impedance interneurons with long membrane time constants, provides a mechanism for spatial compartmentalization of signaling between the output regions of the mitral cell, namely, the tuft dendrite versus the secondary dendrite and axon.
To limit contribution of HVA Ca channels to basal cytoplasmic [Ca2+], we restricted our evaluation of the effect of presynaptic membrane potential on asynchronous and AP-evoked release to subthreshold potentials in quiescent acute slices. During odor activation, mitral cells undergo oscillations in membrane potential that are often suprathreshold and mitral tuft dendrites are exposed to glutamate levels that activate metabotropic receptors known to enhance T-channel function (Johnston and Delaney, 2010). These conditions would be expected to significantly increase T-channel opening; thus, the contribution of T-channel modulation of evoked and asynchronous release is likely to be greater during odor stimulation than we have demonstrated here.
Footnotes
This work was supported by the Mobility Grant (Marie Curie Action, NKTH-OTKA-EU FP7) of the Hungarian Government (managed by the National Development Agency, financed by the Research and Technology Innovation Fund) to A.F. and the Canadian Institutes for Health Research Operating Grant MOP 86473 to K.R.D. The authors thank Dr. Terence Snutch for his generous donation of compound Z941.
The authors declare no competing financial interests.
References
- Aroniadou-Anderjaska V, Ennis M, Shipley MT. Dendrodendritic excitation in mitral cells of the rat olfactory bulb. J Neurophysiol. 1999;82:489–494. doi: 10.1152/jn.1999.82.1.489. [DOI] [PubMed] [Google Scholar]
- Awatramani GB, Price GD, Trussell LO. Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron. 2005;48:109–121. doi: 10.1016/j.neuron.2005.08.038. [DOI] [PubMed] [Google Scholar]
- Balu R, Strowbridge BW. Opposing inward and outward conductances regulate rebound discharges in olfactory mitral cells. J Neurophysiol. 2007;97:1959–1968. doi: 10.1152/jn.01115.2006. [DOI] [PubMed] [Google Scholar]
- Best AR, Regehr WG. Inhibitory regulation of electrically coupled neurons in the inferior olive is mediated by asynchronous release of GABA. Neuron. 2009;62:555–565. doi: 10.1016/j.neuron.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang J, Isaacson JS. In vivo whole-cell recording of odor-evoked synaptic transmission in the rat olfactory bulb. J Neurosci. 2003;23:4108–4116. doi: 10.1523/JNEUROSCI.23-10-04108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone E, Marcantoni A, Giancippoli A, Guido D, Carabelli V. T-type channels-secretion coupling: evidence for a fast low-threshold exocytosis. Pflügers Arch. 2006;453:373–383. doi: 10.1007/s00424-006-0100-7. [DOI] [PubMed] [Google Scholar]
- Castro JB, Urban NN. Subthreshold glutamate release from mitral cell dendrites. J Neurosci. 2009;29:7023–7030. doi: 10.1523/JNEUROSCI.5606-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpak S, Mertz J, Moreaux L, Beaurepaire E, Delaney K. Odour-evoked calcium signals in dendrites of rat mitral cells in vivo. Proc Natl Acad Sci U S A. 2001;98:1230–1234. doi: 10.1073/pnas.98.3.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez AE, Singer JH, Diamond JS. Fast neurotransmitter release triggered by Ca2+ influx through AMPA-type glutamate receptors. Nature. 2006;443:705–708. doi: 10.1038/nature05123. [DOI] [PubMed] [Google Scholar]
- Chemin J, Monteil A, Perez-Reyes E, Bourinet E, Nargeot J, Lory P. Specific contribution of human T-type calcium channel isotypes (alpha(1G), alpha(1H) and alpha(1I)) to neuronal excitability. J Physiol. 2002;540:3–14. doi: 10.1113/jphysiol.2001.013269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Westbrook GL. Regulation of backpropagating action potentials in mitral cell lateral dendrites by A-type potassium currents. J Neurophysiol. 2003;89:2466–2472. doi: 10.1152/jn.00997.2002. [DOI] [PubMed] [Google Scholar]
- Christie JM, Bark C, Hormuzdi SG, Helbig I, Monyer H, Westbrook GL. Connexin 36 mediates spike synchrony in olfactory bulb glomeruli. Neuron. 2005;46:761–772. doi: 10.1016/j.neuron.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Coste B, Crest M, Delmas P. Pharmacological dissection and distribution of NaN. Nav1.9, T-type Ca2+ currents, and mechanically activated cation currents in different populations of DRG neurons. J Gen Physiol. 2007;129:57–77. doi: 10.1085/jgp.200609665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison IG, Boyd JD, Delaney KR. Dopamine inhibits mitral/tufted to granule cell synapses in the frog olfactory bulb. J Neurosci. 2004;24:8057–8067. doi: 10.1523/JNEUROSCI.2138-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Saint Jan D, Westbrook GL. Disynaptic amplification of metabotropic glutamate receptor 1 responses in the olfactory bulb. J Neurosci. 2007;29:2043–2052. doi: 10.1523/JNEUROSCI.2439-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney KR, Tank DW. A quantitative measure of the dependence of short-term synaptic enhancement on presynaptic residual calcium. J Neurosci. 1994;14:5885–5902. doi: 10.1523/JNEUROSCI.14-10-05885.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmaisons D, Vincent JD, Lledo PM. Control of action potential timing by intrinsic subthreshold oscillations in olfactory neurons. J Neurosci. 1999;19:10727–10737. doi: 10.1523/JNEUROSCI.19-24-10727.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eller P, Berjukov S, Wanner S, Huber I, Hering S, Knaus HG, Toth G, Kimball SD, Striessnig J. High affinity interaction of mibefradil with voltage-gated calcium and sodium channels. Br J Pharmacol. 2000;130:669–677. doi: 10.1038/sj.bjp.0703352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman D, Strowbridge BW. Functional role of NMDA autoreceptors in olfactory mitral cells. J Neurophysiol. 2000;84:39–50. doi: 10.1152/jn.2000.84.1.39. [DOI] [PubMed] [Google Scholar]
- Gire DH, Schoppa NE. Control of on/off glomerular signaling by a local GABAergic microcircuit in the olfactory bulb. J Neurosci. 2009;29:13454–13464. doi: 10.1523/JNEUROSCI.2368-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F, Imoto K, Sakmann B. Ca2+ buffering and action potential-evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys J. 1996;70:1069–1081. doi: 10.1016/S0006-3495(96)79653-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand ME, Isope P, Miyazaki T, Nakaya T, Garcia E, Feltz A, Schneider T, Hescheler J, Kano M, Sakimura K, Watanabe M, Dieudonné S, Snutch TP. Functional coupling between mGluR1 and Cav3.1 T-type calcium channels contributes to parallel fiber-induced fast calcium signaling within Purkinje cell dendritic spine. J Neurosci. 2009;29:9668–9682. doi: 10.1523/JNEUROSCI.0362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Keyser BM, Tagmose TM, Hansen JB, Taylor JT, Zhuang H, Zhang M, Ragsdale DS, Li M. NNC 55–0396 [(1S,2S)-2-(2-N-[3-benzimidazol-2-yl)propyl]-N-methylamino)ethyl)-6-fluoro-1,2,3,4-tetrahydro-1-isopropyl-2-naphtyl cyclopropanecarboxylate dihydrochloride]: a new selective inhibitor of T-type calcium channels. J Pharmacol Exp Ther. 2004;309:193–199. doi: 10.1124/jpet.103.060814. [DOI] [PubMed] [Google Scholar]
- Isaacson JS. Glutamate spillover mediates excitatory transmission in the rat olfactory bulb. Neuron. 1999;23:377–384. doi: 10.1016/S0896-6273(00)80787-4. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Shigekawa M. Differential inhibition of Na+/Ca2+ exchanger isoforms by divalent cation and isothiourea derivative. Am J Physiol. 1998;275:C423–C430. doi: 10.1152/ajpcell.1998.275.2.C423. [DOI] [PubMed] [Google Scholar]
- Jerome D, Hou Q, Yuan Q. Interaction of NMDA receptors and L-type calcium channels during early odor preference learning in rats. Eur J Neurosci. 2012;36:3134–3141. doi: 10.1111/j.1460-9568.2012.08210.x. [DOI] [PubMed] [Google Scholar]
- Jin I, Udo H, Rayman JB, Puthanveettil S, Kandel ER, Hawkins RD. Spontaneous transmitter release recruits postsynaptic mechanisms of long-term and intermediate-term facilitation in Aplysia. Proc Natl Acad Sci U S A. 2012;109:9137–9142. doi: 10.1073/pnas.1206846109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J, Delaney KR. Synaptic activation of T-type Ca2+ channels via mGluR activation in the primary dendrite or mitral cells. J Neurophysiol. 2010;103:2557–2569. doi: 10.1152/jn.00796.2009. [DOI] [PubMed] [Google Scholar]
- Kalappa BI, Feng L, Kem WR, Gusev AG, Uteshev VV. Mechanisms of facilitation of synaptic glutamate release by nicotinic agonists in the nucleus of the solitary tract. Am J Physiol Cell Physiol. 2011;301:C347–C361. doi: 10.1152/ajpcell.00473.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klöckner U, Lee JH, Cribbs LL, Daud A, Hescheler J, Pereverzev A, Perez-Reyes E, Schneider T. Comparison of the Ca2+ currents induced by expression of three cloned alpha1 subunits, alpha1G, alpha 1H and alpha1I, of low-voltage-activated T-type Ca2+ channels. Eur J Neurosci. 1999;11:4171–4178. doi: 10.1046/j.1460-9568.1999.00849.x. [DOI] [PubMed] [Google Scholar]
- Kollo M, Holderith N, Antal M, Nusser Z. Unique clustering of A-type potassium channels on different cell types of the main olfactory bulb. Eur J Neurosci. 2008;27:1686–1699. doi: 10.1111/j.1460-9568.2008.06141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Blinks JR, Nicholson C. Calcium transients in presynaptic terminal of squid giant synapse: detection with aequorin. Science. 1972;176:1127–1129. doi: 10.1126/science.176.4039.1127. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL. The action of N-methyl-D-aspartic acid on mouse spinal neurons in culture. J Physiol. 1985;361:65–90. doi: 10.1113/jphysiol.1985.sp015633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay BE, McRory JE, Molineux ML, Hamid J, Snutch TP, Zamponi GW, Turner RW. Ca(v)3 T-type calcium channel isoforms differentially distribute to somatic and dendritic compartments in rat central neurons. Eur J Neurosci. 2006;24:2581–2594. doi: 10.1111/j.1460-9568.2006.05136.x. [DOI] [PubMed] [Google Scholar]
- McNulty MM, Hanck DA. State-dependent mibefradil block of Na+ channels. Mol Pharmacol. 2004;66:1652–1661. doi: 10.1124/mol.66.6.1652. [DOI] [PubMed] [Google Scholar]
- McNulty MM, Kyle JW, Lipkind GM, Hanck DA. An inner pore residue (Asn406) in the Nav1.5 channel controls slow inactivation and enhances mibefradil block to T-type Ca2+ channel levels. Mol Pharmacol. 2006;70:1514–1523. doi: 10.1124/mol.106.027177. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Jahr CE. Self-excitation of olfactory bulb neurons. Nature. 1982;296:441–444. doi: 10.1038/296441a0. [DOI] [PubMed] [Google Scholar]
- Pan ZH, Hu HJ, Perring P, Andrade R. T-type Ca2+ channels mediate neurotransmitter release in retinal bipolar cells. Neuron. 2001;32:89–98. doi: 10.1016/S0896-6273(01)00454-8. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- Pimentel DO, Margrie TW. Glutamatergic transmission and plasticity between olfactory bulb mitral cells. J Physiol. 2008;586:2107–2119. doi: 10.1113/jphysiol.2007.149575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Delaney KR, Tank DW. The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapses. J Neurosci. 1994;14:523–537. doi: 10.1523/JNEUROSCI.14-02-00523.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozanski GM, Nath AR, Adams ME, Stanley EF. Low voltage-gated calcium channels gate transmitter release at the dorsal root ganglion sandwich synapse. J Physiol. 2013;591:5575–55783. doi: 10.1113/jphysiol.2013.260281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- Schoppa NE, Westbrook GL. Regulation of synaptic timing in the olfactory bulb by an A-type potassium current. Nat Neurosci. 1999;2:1106–1113. doi: 10.1038/16033. [DOI] [PubMed] [Google Scholar]
- Schoppa NE, Westbrook GL. Glomerulus-specific synchronization of mitral cells in the olfactory bulb. Neuron. 2001;31:639–651. doi: 10.1016/S0896-6273(01)00389-0. [DOI] [PubMed] [Google Scholar]
- Schoppa NE, Westbrook GL. AMPA autoreceptors drive correlated spiking in olfactory bulb glomeruli. Nat Neurosci. 2002;5:1194–1202. doi: 10.1038/nn953. [DOI] [PubMed] [Google Scholar]
- Singer JH, Diamond JS. Sustained Ca2+ entry elicits transient postsynaptic currents at a retinal ribbon synapse. J Neurosci. 2003;23:10923–10933. doi: 10.1523/JNEUROSCI.23-34-10923.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TC, Jahr CE. Self-inhibition of olfactory bulb neurons. Nat Neurosci. 2002;5:760–766. doi: 10.1038/nn882. [DOI] [PubMed] [Google Scholar]
- Talbot MJ, Sayer RJ. Intracellular QX-314 inhibits calcium currents in hippocampal CA1 pyramidal neurons. J Neurophysiol. 1996;76:2120–2124. doi: 10.1152/jn.1996.76.3.2120. [DOI] [PubMed] [Google Scholar]
- Tringham E, Powell KL, Cain SM, Kuplast K, Mezeyova J, Weerapura M, Eduljee C, Jiang X, Smith P, Morrison JL, Jones NC, Braine E, Rind G, Fee-Maki M, Parker D, Pajouhesh H, Parmar M, O'Brien TJ, Snutch TP. T-type calcium channel blockers that attenuate thalamic burst firing and suppress absence seizures. Sci Transl Med. 2012;4:121ra19. doi: 10.1126/scitranslmed.3003120. [DOI] [PubMed] [Google Scholar]
- Tsai PS, Nishimura N, Yoder EJ, White A, Dolnick E, Kleinfeld D. Principles, design and construction of a two photon scanning microscope for in vitro and in vivo studies. In: Frostig R, editor. Methods for in vivo optical imaging. Boca Raton, FL: CRC; 2002. pp. 113–171. [Google Scholar]
- Weiss N, Zamponi GW, De Waard M. How do T-type calcium channels control low-threshold exocytosis? Commun Integr Biol. 2012;5:377–380. doi: 10.4161/cib.19997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda R, Nimchinsky EA, Scheuss V, Pologruto TA, Oertner TG, Sabatini BL, Svoboda K. Imaging calcium concentration dynamics in small neuronal compartments. Sci STKE. 2004;2004:pl5. doi: 10.1126/stke.2192004pl5. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Snutch TP. Nickel block of a family of neuronal calcium channels: subtype- and subunit-dependent action at multiple sites. J Membr Biol. 1996;151:77–90. doi: 10.1007/s002329900059. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]