

Abstract
Formation of proper neuromuscular connections is a process coordinated by both motoneuron-intrinsic and target-dependent programs. Under these programs, motoneurons innervate target muscles, escape programmed cell death during fetal development, and form neuromuscular junctions (NMJ). Although a number of studies have revealed molecules involved in axon guidance to target muscles and NMJ formation, little is known about the molecular mechanisms linking intramuscular innervation and target-derived trophic factor-dependent prevention of motoneuron apoptosis. Here we studied the physiological function of CLAC-P/collagen XXV, a transmembrane-type collagen originally identified as a component of senile plaque amyloid of Alzheimer's disease brains, by means of generating Col25a1-deficient (KO) mice. Col25a1 KO mice died immediately after birth of respiratory failure. In Col25a1 KO mice, motor axons projected properly toward the target muscles but failed to elongate and branch within the muscle, followed by degeneration of axons. Failure of muscular innervation in Col25a1 KO mice led to excessive apoptosis during development, resulting in almost complete and exclusive loss of spinal motoneurons and immaturity in skeletal muscle development. Bax deletion in Col25a1 KO mice rescued motoneurons from apoptosis, although motor axons remained halted around the muscle entry site. Furthermore, these motoneurons were positive for phosphorylated c-Jun, an indicator of insufficient supply of target-derived survival signals. Together, these observations indicate that CLAC-P/collagen XXV is a novel essential factor that regulates the initial phase of intramuscular motor innervation, which is required for subsequent target-dependent motoneuron survival and NMJ formation during development.
Introduction
Collagens comprise a large family of proteins involved in a variety of functions ranging from the formation of fibrillar networks of the extracellular matrix to cell adhesion. Recently, it has been shown that some collagens play important roles in the nervous system (e.g., axon outgrowth, targeting, and synapse formation) (Fox, 2008).
We previously identified CLAC (collagenous Alzheimer amyloid plaque component) as a component of senile plaques, the latter being a hallmark pathology of Alzheimer's disease (AD) brains (Hashimoto et al., 2002; Kowa et al., 2004). CLAC corresponds to an ectodomain fragment of the transmembrane precursor, CLAC-P, which is cleaved by furin convertase. CLAC-P consists of three collagenous triple-helical domains flanked by four short noncollagenous domains and therefore has been classified as type XXV collagen (COL25A1) as well. CLAC-P/collagen XXV mRNA is strongly expressed in the adult CNS both in human and mouse, where the expression is exclusively observed in neurons. CLAC binds to amyloid-β fibrils, the primary constituent of senile plaques, through the positively charged amino acid cluster in the first collagenous domain, indicating that CLAC may regulate amyloid formation during the development and progression of AD (Osada et al., 2005). In contrast to the pathological role in AD, the physiological function of CLAC-P/collagen XXV remained unknown.
CLAC-P/collagen XXV is a member of the collagen subfamily termed membrane-associated collagens with interrupted triple helices (MACIT), together with type XIII, XVII, and XXIII collagens (Pihlajaniemi and Rehn, 1995). Emerging evidence suggests that MACIT collagens are involved in cell adhesion. The most well-studied member, collagen XVII/BP180, is a structural component of the hemidesmosome, which anchors epidermal keratinocytes to the basement membranes (Powell et al., 2005). Among the three MACIT collagens, collagen XIII shares the highest homology to CLAC-P/collagen XXV. Collagen XIII is broadly expressed in various tissues, predominantly in mesenchymal cells (Heikkinen et al., 2012). Collagen XIII is suggested to be localized to cell–cell and cell–matrix interaction sites as a component of adherens junctions and focal adhesions, respectively (Hägg et al., 2001). Knock-out mouse study revealed that collagen XIII is a regulator for the neuromuscular junction (NMJ) maturation, implicating a function in muscle–nerve connectivity (Latvanlehto et al., 2010). We therefore speculated that CLAC-P/collagen XXV might also exert a role either in developing and adult motoneurons.
To study the in vivo function of CLAC-P/collagen XXV in the nervous system, we developed CLAC-P/collagen XXV-deficient mice. Col25a1 KO mice died immediately after birth due to respiratory failure. During the development of neuromuscular system in Col25a1 KO mice, motor axon bundles properly reached the target skeletal muscle but failed to elongate and branch within the muscle, followed by degeneration and retraction of axons. Defects in the intramuscular innervation were followed by excessive apoptosis during the period of naturally occurring motoneuron death, resulting in almost complete loss of spinal motoneurons in Col25a1 KO mice. Our findings suggest that CLAC-P/collagen XXV plays an essential role in the initial phase of intramuscular motor innervation, which is required for the subsequent target-dependent motoneuron survival and NMJ formation during development.
Materials and Methods
Generation of Col25a1-deficient mice.
A Col25a1 gene targeting vector was constructed from a 12.1 kb NcoI-XhoI fragment of mouse genomic DNA containing the first and second exons of Col25a1. The short arm, a 1 kb genomic fragment containing exon 1 and intron 1, was amplified by PCR. NotI and BamHI sites were introduced at 5′- and 3′-ends of the short arm, respectively. The short arm fragment, an oligonucleotide linker containing BamHI-SalI-SacII-BamHI sites, and a 7.5 kb BamHI-XhoI fragment (long arm) were ligated into pBluescript. A PGK-βgeo (Neo-LacZ) cassette was inserted in a reverse orientation with SalI-XhoI compatible ends and SacII. The diphtheria toxin A (DT-A) fragment cDNA was inserted into NotI site for negative selection. Targeted ES clones were identified by Southern blot analysis using 357 bp fragment ∼3 kb upstream of exon 1 as a probe, and were injected into C57BL/6 blastocysts to obtain chimeric mice. Col25a1+/geo mice were crossed with actin-flippase transgenic mice (ACT-FLPe; The Jackson Laboratory) to obtain Col25a1+/− mice. Col25a1+/− mice were backcrossed using SNP marker-assisted speed congenic breeding strategy to obtain mice on a pure C57BL/6 background. The two Col25a1 KO lines exhibited the same phenotypic abnormalities. Hb9-GFP transgenic mice and Bax KO mice were obtained from The Jackson Laboratory. All experimental procedures were conducted according to the institutional guidelines for animal experiments and safety of the University of Tokyo. Pups and embryos of either sex were used. The genotypes of Col25a1 mutant mice were determined by PCR with specific oligonucleotide primers, 5′-TTTACTTCCAATGGAGAGGC-3′ and 5′-ATATTTCCATATGGTGACG-3′, which amplify a 1965 bp fragment in wild-type allele and a 500 bp fragment in targeted allele, respectively.
RT-PCR analyses of mouse tissues and C2C12 cells.
For RT-PCR, total RNA was extracted from the embryonic mouse brains by ISOGEN (Nippon Gene) and was converted to cDNA using ReverTra Ace (TOYOBO). For quantitative RT-PCR analysis in mouse tissues, total RNA was extracted and reverse transcribed from the muscles, spinal cords, and brains of E12.5 and 10-week-old mice. Whole limbs were used to analyze the expression in the muscle at E12.5. C2C12 myoblasts (ATCC) or C2C12 myotubes after a 5 d induction with 2% horse serum were cultured and extracted RNA was reverse transcribed. Quantitative real-time PCR was performed using Thunderbird SYBR qPCR mix (TOYOBO) and analyzed by LightCycler480 (Roche). Expression values were normalized to GAPDH mRNA. The primer sequences used for PCR were as follows: 5′-GAACAAGGTGATCAGGGACC-3′ and 5′-CCTTGGTCACCCTTAATCAG-3′ for CLAC-P/collagen XXV, and 5′-AACGACCCCTTCATTGAC-3′ and 5′-GAAGACACCAGTAGACTCCAC-3′ for GAPDH.
Histological analysis and immunohistochemistry.
For preparation of paraffin sections, organs were fixed in 4% PFA for 16 h before embedding in paraffin. Sections were cut at 5 μm thickness and stained with hematoxylin and eosin (H&E) or by immunohistochemistry. For immunohistochemistry, sections were incubated with rabbit anti-GFP (1:2000, Invitrogen) in PBS containing 10% calf serum (Invitrogen) for 16 h. Biotinylated goat anti-rabbit antibody (1:200; Vector Laboratories) was used as a secondary antibody, followed by a standard avidin-biotin-DAB visualization procedure (Vector Laboratories) and hematoxylin counterstaining.
For preparation of cryosections, organs were fixed in 4% PFA for 6 h and then cryoprotected in 30% sucrose in PBS at 4°C. Fixed organs were embedded in OCT compound (Sakura Finetek), and 16-μm-thick sections were cut on a cryostat. Immunohistochemistry was performed as follows: cryosections were permeabilized in MeOH for 20 min at −20°C (except for immunostaining for choline acetyltransferase), and blocked in PBS containing 10% calf serum. Sections were immunostained with mouse anti-Islet1/2 (39.4D5, 1:100, DSHB), anti-neurofilament (2H3, 1:2000, DSHB), goat anti-choline acetyltransferase (ChAT) (1:200, Millipore), rabbit anti-GFP, rabbit anti-TrkA, anti-TrkB, anti-TrkC (1:1000, Abcam), or rabbit anti-phospho-c-Jun (1:100, Cell Signaling Technology) antibodies, followed by labeling by AlexaFluor dye-conjugated secondary antibodies (1:1000; Invitrogen) and DAPI.
Quantification of motoneurons.
The number of motoneurons was quantified as previously described with minor changes (Uetani et al., 2006). Quantification of motoneurons in Hb9-GFP transgenic mice was conducted on 5-μm-thick serial paraffin sections of upper cervical (C3–C5) and brachial (C6–T1) spinal cords from E11.5 and E13.5 embryos. The relative level of the transverse sections within the cervical cord was determined by the morphology of the surrounding tissues. The number of GFP-positive motoneurons was counted in every 10th section in a total of 5 cross sections from each animal, and the mean value of these counts was statistically analyzed by Student's t test. The numbers of motoneurons immunostained for ChAT at E18.5, Islet-1/2 at E14.5, and p-c-Jun at E14.5 were quantified on 16-μm-thick serial cryostat sections from embryos. The number of immunopositive motoneurons was counted in every fourth section in a total of 5 cross sections from each animal and analyzed with one-way ANOVA followed by Tukey-Kramer post hoc analysis.
In situ hybridization.
Digoxigenin-labeled RNA probes for in situ hybridization were created using MegaScript T7/T3 kits (Ambion) and DIG RNA Labeling Mix (Roche). Each synthesized probe corresponded to the sense and antisense nucleotide sequence of murine CLAC-P/collagen XXV (nucleotides −265 to 294); 6-μm-thick paraffin sections were treated with 100 μg/ml proteinase K in PBS for 6 min at 37°C and then washed for 5 min by PBS. The sections were acetylated with freshly prepared 0.25% acetic anhydride in 100 mm triethanolamine, pH 8.0, for 20 min, then washed in 4 × saline-sodium citrate (1 × SSC; 150 mm NaCl, 15 mm sodium citrate, pH 7.0). The sections were prehybridized by hybridization buffer (4 × SSC, 40% deionized formamide, 10% dextran sulfate, 1 × Denhardt's solution, 10 mm DTT, 1 mg/ml yeast tRNA) for 1 h at 50°C. For hybridization, the slides were probed with hybridization buffer containing 2 ng/ml DIG-labeled RNA probe for 16 h at 50°C in humidified chamber. After washing with 4 × SSC at 50°C, 2 × SSC containing 50% formamide for 1 h at 50°C, and twice with 0.1 × SSC for 1 h at 50°C, the sections were placed in blocking solution containing 100 mm maleic acid, pH 7.5, 150 mm NaCl and 1% blocking reagent (Roche). Each section was incubated with an anti-DIG antibody conjugated with alkaline phosphatase (1:10,000, Roche) for 2 h at room temperature. The sections were washed with TBS containing 0.1% Tween 20, and then with detection buffer (100 mm Tris-HCl and 100 mm NaCl, pH 9.5), and visualized by incubating in a nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolyl-phosphate substrate solution overnight at 4°C.
TUNEL staining.
TUNEL assay was performed using in situ cell death detection Kit (Roche) according to the manufacturer's instructions. TUNEL-positive cells per each ventral spinal cord were counted in every 10th section in E13.5 upper cervical (C3–C5) and brachial (C6–T1) spinal cords, and the mean value of those counts was used as an individual data point for statistical analysis by Student's t test.
Whole-mount immunohistochemistry.
Diaphragms were fixed in 4% PFA in 100 mm phosphate buffer, pH 7.4, for 5 min at 4°C, rinsed with PBS, and permeabilized with 1% Triton X-100 for 2 h. The diaphragms were incubated with anti-neurofilament or anti-GFP antibodies in PBS containing 10% calf serum for 2 d at 20°C. After washing three times for 1 h each with PBS, the diaphragms were incubated with anti-rabbit/mouse IgG conjugated with AlexaFluor-488 (1:1000; Invitrogen) and α-bungarotoxin (αBTX) conjugated with AlexaFluor-594 (1:1000; Invitrogen) in the diluent for 16–24 h. The analysis of forelimbs was performed after the tissues were finally made semitransparent by immersion in glycerol.
Spinal cord cell culture.
Spinal cord cells were isolated as described previously (Nishimune et al., 2004) with some modifications, without further purification of motor neurons. Spinal cords were dissected from E11.5 embryos and chopped into pieces and incubated in Hank's Balanced Salt Solution supplemented with 0.02% Trypsin for 8 min at 37°C. The spinal cords were gently triturated in 0.4% BSA and 0.1 mg/ml DNase in Neurobasal Medium. The cell suspension of the spinal cord was centrifuged on 4% BSA cushion for 5 min at 480 × g. The cells were resuspended and plated on poly-d,l-ornithine/laminin precoated multiwall plates in the culture media (Neurobasal Medium supplemented with 2% B27 Supplement, 2% horse serum, 0.5 mm l-glutamine, 21 μm β-mercaptoethanol, 50 U/ml penicillin, and 50 mg/ml streptomycin). Cultures were maintained in a humidified atmosphere of 5% CO2 and 95% air at 37°C
Trophic factor-dependent survival of motoneuron was assessed as described previously (Aebischer et al., 2011). Briefly, cells were plated at a density of 2.0 × 103 cells/cm2 in culture media supplemented with neurotrophic factors (+NTF) or without (−NTF) 1.0 ng/ml BDNF, 10 ng/ml CNTF, and 0.1 ng/ml GDNF (R&D Systems). The number of Hb9-GFP-positive cells was counted under a fluorescent microscope. The number of motoneurons 5 h after plating was defined as that of 100% survival. Analysis was performed with one-way ANOVA followed by Tukey's post hoc analysis.
Immunocytochemistry.
For immunocytochemistry, spinal cord cells were plated at a density of 4.0 × 104 cells/cm2 in culture media supplemented with NTF. At 3 d in vitro, cells were cultured in nonsupplemented Neurobasal Medium for 2.5 h, followed by treatment with 5.0 ng/ml BDNF, 50 ng/ml CNTF, and 0.5 ng/ml GDNF (+NTF) or PBS (−NTF) for 10 min before fixation. Cultures were fixed with 4% PFA for 20 min. The cells were permeabilized with 0.2% Triton X-100 for 15 min, blocked with 3% BSA for 30 min, and incubated with the primary antibodies (mouse anti-GFP, 1:1000, Roche, or rabbit anti-phospho-ERK1/2, 1:200, Cell Signaling Technology) for 3 h at room temperature or overnight at 4°C. The cells were incubated with Alexa-conjugated secondary antibody for 1 h at room temperature and mounted in PermaFluor (Thermo).
Results
Targeted deletion of CLAC-P/collagen XXV results in early neonatal death due to respiratory failure
To determine the physiological function of CLAC-P/collagen XXV, we generated CLAC-P/collagen XXV knock-out (Col25a1 KO) mice by deleting exon 2 of the murine Col25a1 gene, which contains the furin cleavage site (Fig. 1A). Targeted ES clones were identified by Southern blot analysis, and Col25a1+/− mice were obtained (Fig. 1B). Pups were genotyped using PCR with gene-specific primers (Fig. 1C). Heterozygous Col25a1+/− mice were viable and fertile with neither developmental nor gross physical abnormalities compared with their wild-type littermates. Col25a1+/− mice were interbred to generate mice homozygous for the Col25a1 gene disruption. Resulting homozygous Col25a1 KO mice died shortly after birth displaying cyanosis and abnormal body curvature (Fig. 1D). Caesarean sections at embryonic day 18.5 (E18.5) revealed that approximately one-fourth of embryos were homozygous mutants (28.2%; n = 110), indicating that the Col25a1 KO were viable by birth. RT-PCR analysis of E18.5 brains indicated that levels of CLAC-P/collagen XXV mRNA were decreased in the heterozygous embryo and diminished in Col25a1 KO as expected (Fig. 1E). Histological examination revealed that the lung alveoli of newborn Col25a1 KO mice remained collapsed (Fig. 1F). We therefore conclude that neonatal lethality of Col25a1 KO mice was attributable to respiratory failure due to incapability of ventilation after birth.
Figure 1.

Targeted disruption of the Col25a1 gene resulted in early neonatal death due to respiratory failure. A, Strategy for targeted disruption of the Col25a1 locus. 5′-UTR (open boxes) and coding regions (filled boxes) are represented. B, H, X, N, and Sc denote restriction sites of BamHI, HindIII, XhoI, NotI, and SacII, respectively. The primers for genotyping PCR are indicated by the arrows. The probe used for Southern blotting is indicated in the diagram. B, Southern blot analysis of targeted ES cell clones. Genomic DNA from parental embryonic stem cells (WT) and homologous recombinant clones (mt) were digested with SacII for the hybridization probe. A 15.3 kb fragment is detected only in the mt alleles. C, PCR genotyping of wild-type (+/+), heterozygous (+/−), and Col25a1 KO mice. D, Col25a1 KO mice exhibited cyanosis and died immediately after birth. E, RT-PCR analysis of CLAC-P/collagen XXV mRNA using total RNAs from E18.5 brains. GAPDH served as a control. F, H&E-stained lung sections from newborn pups at P0. G, H&E-stained cross-sections of the diaphragm from Col25a1 KO and littermate wild-type mice at P0. Arrowheads indicate centrally located nuclei. Representative images from multiple animals are shown. H, H&E-stained transverse sections of the E18.5 spinal cord (top) and an enlarged view of the anterior horn (bottom) at the brachial level. Representative images from multiple animals are shown. I, Motoneurons immunostained by an anti-ChAT antibody in the brachial spinal cord at E18.5. Nuclei were stained with DAPI (blue). J, Quantification of ChAT-positive motoneurons (MNs) in the brachial spinal cord of wild-type, heterozygous, and Col25a1 KO mice. Data are mean ± SEM. ***p < 0.001. n = 3.
Immature skeletal muscle development and loss of spinal motoneurons in Col25a1 KO mice at birth
To examine the effects of the mutation in the Col25a1 gene on development of various tissue structures, histological examination was performed. Compared with wild-type and Col25a1+/− littermates, the thickness of the skeletal muscles of Col25a1 KO mice was generally thinner, and the muscle fibers were smaller in diameter, with their nuclei centrally localized along the fiber, indicating that the skeletal muscle remained immature with their development halted at an early stage of myotube formation (Fig. 1G).
Because respiratory failure at birth is often associated with defects in development of the neuromuscular system, we then explored the possibility that development of motoneurons was disrupted by loss of CLAC-P/collagen XXV function. Whereas motoneurons in the spinal cord were readily identified by their large cell bodies and nucleoli in wild-type embryos at E18.5, only a few morphologically abnormal motoneurons of smaller size were found in Col25a1 KO mice (Fig. 1H). Immunostaining for a motoneuron marker ChAT revealed that motoneurons almost totally disappeared by E18.5 (Fig. 1I,J). In contrast, Col25a1+/− mice showed no decrease in the number of motoneuron. The gross anatomy of the other part of the CNS as well as of other organs appeared normal in Col25a1 KO and Col25a1+/−. We obtained and examined two Col25a1 KO lines, which exhibited the same phenotypic abnormalities.
CLAC-P/collagen XXV is expressed both in developing spinal motoneurons and myotubes
It is important to determine which cell types could be the origin of CLAC-P/collagen XXV required for the proper neuromuscular development. We therefore analyzed the pattern of Col25a1 gene expression by in situ hybridization. CLAC-P/collagen XXV mRNA was expressed in neurons in the spinal cord, including motoneurons and in DRG neurons at E11.5 (Fig. 2A). We also observed strong signals in developing skeletal muscles (Fig. 2B) and relatively weak signals in some other organs, including heart, lung and epidermis (data not shown). As we reported previously (Hashimoto et al., 2002), CLAC-P/collagen XXV mRNA was not detected in adult skeletal muscles (Fig. 2B). Quantitative RT-PCR analyses revealed almost no expression of CLAC-P/collagen XXV mRNA in 10-week-old adult skeletal muscles, whereas relatively high-level expression was detected in the embryonic limbs (Fig. 2C). In contrast, expression of CLAC-P/collagen XXV mRNA in the nervous tissues was constantly detected both in embryos and adults. The presence of CLAC-P/collagen XXV mRNA in developing muscles prompted us to examine whether the expression is regulated during the process of myoblast maturation. To this aim, we extracted RNA fractions from a mouse muscle cell line, C2C12, and measured mRNA levels by quantitative RT-PCR (Fig. 2D). In undifferentiated C2C12 myoblasts, the expression of CLAC-P/collagen XXV mRNA was extremely low. Interestingly, after inducing myoblast fusion into myotubes, the level of CLAC-P/collagen XXV expression was significantly upregulated by >40 times compared with that in myoblasts. These results suggest that CLAC-P/collagen XXV derived either from motoneurons or differentiating muscle tissues could regulate the neuromuscular development in mice. Immunocytochemistry of cultured neurons or muscular cells, immunohistochemistry or immunoblot analyses of embryonic or adult nervous or muscular tissues, did not clearly detect the CLAC-P/collagen XXV polypeptides, presumably because of low level of endogenous protein expression.
Figure 2.

CLAC-P/collagen XXV mRNA is expressed in both developing spinal motoneurons and myotubes. A, In situ hybridization analysis was performed to detect expression of CLAC-P/collagen XXV mRNA in the spinal cord and DRG of wild-type embryo at E11.5. Motoneurons in the ventral horn (MN) and DRG neurons are encircled by dashed lines. B, In situ hybridization analysis in the developing (E11.5, top) and adult (bottom) skeletal muscles. C, Expression of CLAC-P/collagen XXV mRNA in muscles, spinal cord, and brain was analyzed by quantitative RT-PCR at E12.5 and in 10-week-old mice. GAPDH mRNA was used as a reference. Data are mean ± SEM. ***p < 0.001. **p < 0.01. n = 8 (E12.5) and n = 4 (adult). D, Expression of CLAC-P/collagen XXV mRNA in C2C12 myoblasts (MB) and C2C12 myotubes (MT) was measured by quantitative RT-PCR, with GAPDH mRNA as a reference. Results are presented as fold change over MB. Data are mean ± SEM. *p < 0.05. n = 4.
Progressive loss of postmitotic motoneurons by apoptosis in Col25a1 KO mice
We then asked whether loss of motoneurons in CLAC-P/collagen XXV-deficient mice resulted from differentiation failure at an earlier stage or due to progressive loss of postmitotic motoneurons. By crossing with Hb9-GFP transgenic mice, which express GFP protein specifically in postmitotic motoneurons (Wichterle et al., 2002), we examined the ventral pool of motoneurons in the spinal cord from E11.5 through E14.5. At E11.5 when motor axons exit the spinal cord and at E12.5 when motor axons project to target muscles, the number as well as the morphology of GFP-positive motoneurons at the brachial (lower cervical) level of Col25a1 KO mice were comparable with those of wild-type mice (Fig. 3A,B). However, the number of motoneurons both in the medial and lateral motor columns of the brachial spinal cord was reduced in Col25a1 KO mice at E13.5 (Fig. 3A,C), and only few motoneurons remained at E14.5. Consistent with this observation, morphometric analysis of the upper cervical spinal cord revealed that the number of motoneurons did not differ between wild-type and Col25a1 KO mice at E11.5 but decreased to ∼35% of that in wild-type mice in Col25a1 KO at E13.5 (Fig. 3B,C). This reduction was observed throughout different levels of the spinal cord, including thoracic and lumbosacral cords (Fig. 3D,E). Considering that most of the ChAT-positive motoneurons were lost at birth (Fig. 1I,J), these results suggest that Col25a1-deficient motoneurons differentiated normally and progressively disappeared thereafter.
Figure 3.

Motoneurons in Col25a1 KO mice underwent massive apoptosis during development. A, Anti-GFP immunostaining of the brachial spinal cords of Hb9-GFP;Col25a1+/+ (top) and Hb9-GFP;Col25a1 KO (bottom) embryos from E11.5 to E14.5. B, C, Quantitative analyses of GFP-positive motoneurons in the upper cervical (C3–C5) and brachial (C6–T1) spinal cord at E11.5 (B) and E13.5 (C). Data are mean ± SEM. ***p < 0.001. **p < 0.05. n = 3–5. D, Immunoperoxidase-labeled GFP-positive motoneurons (arrowheads) in the ventral horn and sympathetic preganglionic neurons (arrows) in the lateral horn of the thoracic spinal cord at E14.5. Representative images in multiple animals are shown. E, Immunoperoxidase-labeled GFP-positive motoneurons (arrowheads) in the ventral horn of the lumbosacral spinal cord at E14.5. Representative images in multiple animals are shown. F, TUNEL staining of the brachial spinal cord of Col25a1+/+ and Col25a1 KO embryos at E13.5. Arrowheads indicate TUNEL-positive apoptotic nuclei in the ventral horn. Nuclei were stained with DAPI (blue). G, Quantification of TUNEL-positive motoneurons in the brachial spinal cord at E13.5. Data are mean ± SEM. ***p < 0.001. n = 10. H, Overview (top) and high-magnification views (bottom) of H&E-stained sections of the DRG at E18.5. Representative images in multiple animals are shown. I, Immunostaining of DRG neurons with anti-TrkA, TrkB, and TrkC antibodies at E13.5. Nuclei were stained with DAPI (blue).
It is known that ∼50% of postmitotic motoneurons undergo apoptotic programmed cell death (PCD) starting at E12.5 in mice (Oppenheim, 1991). To determine whether the loss of motoneurons in Col25a1 KO embryos was due to apoptosis, we performed TUNEL staining. At E13.5, Col25a1 KO mice exhibited larger numbers of apoptotic neurons in the spinal cord compared with control littermates (Fig. 3F). Quantitative analysis demonstrated that the number of TUNEL-positive motoneurons in Col25a1 KO mice was significantly increased both in the upper cervical and brachial levels of the spinal cord (Fig. 3G), supporting the notion that the genetic deletion of CLAC-P/collagen XXV promotes massive apoptosis during the wave of PCD. In contrast to the motoneuron loss, cholinergic preganglionic column neurons that innervate the sympathetic ganglia were completely conserved in Col25a1 KO mice (Fig. 3D, arrows). In addition, all three major subpopulations of sensory neurons in the DRG: TrkA-positive nociceptive, TrkB-positive mechanoreceptive, and TrkC-positive proprioceptive neurons were normally preserved in Col25a1 KO mice (Fig. 3H,I), indicating that loss of Col25a1 causes a motoneuron specific defect during development.
Aberrant intramuscular innervation of the motor axon bundles in Col25a1 KO mice
Accumulating evidence suggests that survival of motoneurons during the embryonic period of PCD is predominantly dependent on the accessibility to target-derived trophic support, which is achieved by successful innervation of motor axons (Banks and Noakes, 2002). We therefore examined the motor nerve innervation of the diaphragm at various developmental stages by whole-mount immunostaining. As early as at E12.5, axon bundles of the phrenic nerve reached the muscle tissues of diaphragm: upon reaching the diaphragm, axon bundles split into three main branches. Although axon bundles directly contacted the diaphragm at E12.5 in Col25a1 KO mice, branches did not emanate from the main nerve trunk (Fig. 4A). As development proceeded, the phrenic nerve of control embryos ramified highly and ran toward the center of the myotubes, forming secondary and tertiary finer branches. In contrast, in the Col25a1 KO mice, the tip of the nerve bundle remained near the entry site displaying signs of withdrawal as represented by the formation of retraction bulb-like structures (Fig. 4A,B). It has been known that synaptic AChRs cluster in the central region of developing muscle fibers independently of motor innervation. At E12.0, both control and Col25a1 KO mice showed the prepatterned AChR clusters on the myotubes (Fig. 4C), suggesting that an intrinsic capacity to initiate postsynaptic differentiations was not affected in the absence of CLAC-P/collagen XXV. At E13.5, however, AChR clusters became significantly immature in Col25a1 KO diaphragm compared with those in control mice (Fig. 4D), as has been observed in Hb9 KO mice, in which motor axons fail to form (Lin et al., 2001). To ask whether defects in intramuscular innervation are a general consequence of CLAC-P/collagen XXV deficiency, we examined the motor nerves projecting to dorsal muscles of the forelimb. Motor axon guidance into the limb bud along the routes of major nerve trunks took place accurately in Col25a1 KO at E11.5 (Fig. 4E). However, terminal arborization was severely disturbed in the mutants starting at E12.5. At E13.5, main nerve trunk became thinner, presumably reflecting axonal degeneration. We also observed similar defects in branching in intercostal motor nerves (data not shown). Motoneurons projecting to the diaphragm, intercostal muscles, and limb muscles form different columns in the spinal cord, indicating that CLAC-P/collagen XXV may be required in neuromuscular development in general. Together, in Col25a1 KO mice, motor axon bundles successfully reach the target muscles but fail to form intramuscular branches. Shortly after, axons start to degenerate and retract concomitantly with the massive motoneuron apoptosis observed in the spinal cord, which in turn might delay maturation of muscles.
Figure 4.

Failure of intramuscular innervation by motor axons in Col25a1 KO mice. A, Whole-mount immunohistochemistry of the diaphragm at E12.5 and the hemidiaphragm at E13.5. Motor axons were labeled with anti-GFP. Arrowheads indicate the muscle entry sites of the motor axons. B, Immunostaining of the motor axons in the diaphragm of E13.5 Col25a1 KO with an anti-neurofilament (NF) antibody. Arrows indicate retraction bulb-like structures. C, Intramuscular innervation and AChR cluster formation in E12.5 diaphragms were evaluated by double staining with anti-NF (green) and αBTX (red). Arrowheads show prepatterned AChR clusters. D, Postsynaptic maturation of the diaphragm was assessed at E13.5 by labeling with αBTX. E, Whole-mount immunohistochemistry of the dorsal forelimb from E11.5 to E13.5. GFP-positive motor axons and neurofilament (NF) were visualized in green and red, respectively. Areas bounded by the dashed lines show intramuscular arborization. Arrowheads indicate degenerating axon bundles in Col25a1 KO mice.
Intramuscular innervation failure is a cause of excessive motoneuron apoptosis in Col25a1 KO mice
In Col25a1 KO mice, massive apoptosis and aberrant intramuscular innervation of motoneurons occurred simultaneously around E13.5. To study the causal relationship between these events, we blocked PCD in Col25a1 KO mice by deleting the proapoptotic gene, Bax (Knudson et al., 1995). It has been shown that Bax deletion dramatically decreased the naturally occurring cell death of postmitotic motoneurons (Deckwerth et al., 1996; White et al., 1998). Indeed, the number of Islet-1/2-positive motoneurons in Bax KO;Col25a1+/− mice was approximately twice that of control mice (Fig. 5A,B). We confirmed that motoneuron death of Bax KO;Col25a1 KO was totally rescued to the levels of Bax KO;Col25a1+/−.
Figure 5.
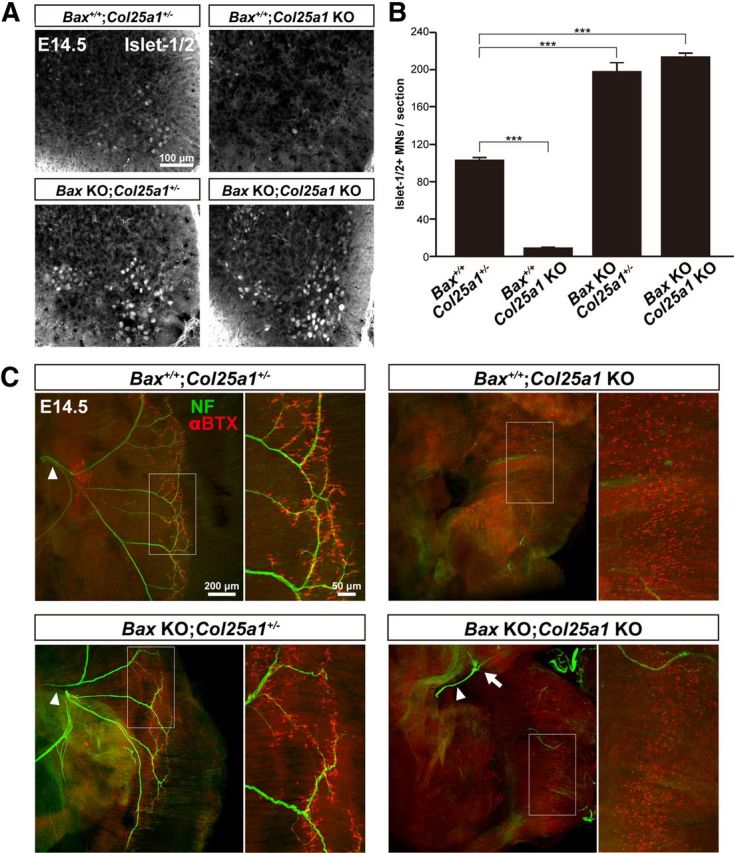
Preventing apoptosis by knock-out of Bax gene failed to rescue the innervation of diaphragm by motor axons in Col25a1 KO mice. A, Immunostaining of Islet-1/2-positive motoneurons in the brachial spinal cord at E14.5. B, Quantification of Islet-1/2-positive motoneurons indicated in A. Data are mean ± SEM. ***p < 0.001. n = 3. C, Intramuscular innervation and neuromuscular junction formation in E14.5 diaphragms were evaluated by double staining with anti-NF (green) and αBTX (red). Right, Enlarged views of the motor endplate. Arrowheads indicate the sites of motor axon entry. Representative images in multiple animals are shown.
To assess the intramuscular innervation of the rescued motoneurons, we stained the phrenic nerve with antineurofilament antibody and postsynaptic AChR clusters with α-bungarotoxin (Fig. 5C). By E14.5, motoneurons reached the end-plate band area and formed NMJ as observed in control Bax+/+;Col25a1+/− mice. The diameter of phrenic nerves of Bax KO;Col25a1+/− mice was slightly larger compared with that of wild-type mice, presumably because of the increased numbers of motoneurons, but the innervation pattern of the diaphragm was displayed similarly to the control mice. In contrast, motor axons of the Col25a1 KO mice were retracted from the diaphragm already at E14.5, with scattered AChR clusters throughout the myotubes. Although Bax deficiency spared projection of motoneurons to the diaphragm at E14.5 in Bax KO;Col25a1 KO mice, these axons failed to innervate the muscle fibers further and remained halted close at the entry site to the diaphragm (Fig. 5C, arrow). These observations suggest that the massive apoptosis of motoneurons observed in CLAC-P/collagen XXV-deficient spinal cord is a consequence, but not the cause, of the failure of intramuscular innervation.
Inadequate target-derived trophic support impairs survival of developing motoneurons in Col25a1 KO mice
A previous report indicates that an insufficient supply of target-derived survival factors triggers motoneuron apoptosis during PCD period (E13-E18) via activation of c-Jun (Sun et al., 2005). These survival factors termed neurotrophic factors include BDNF, NT-3, NT-4, GDNF, and CNTF. To ask whether the excessive apoptosis of motoneurons in Col25a1 KO mice was dependent on c-Jun activation, we stained tissue sections from the spinal cord of E14.5 embryos with an antibody against phosphorylated c-Jun (p-c-Jun) (Fig. 6A). The number of p-c-Jun-positive neurons was significantly increased in Bax KO;Col25a1+/− compared with that of control Col25a1+/− mice (Fig. 6B). The number of p-c-Jun-positive neurons was further increased in Bax KO;Col25a1 KO mice compared with that in Bax KO, suggesting that excessive apoptosis of motoneurons was executed via c-Jun phosphorylation in Col25a1 KO.
Figure 6.

Accelerated programmed cell death of motoneurons in Col25a1 KO mice despite preservation of response to trophic support in vitro. A, Immunostaining of p-c-Jun-positive motoneurons in the brachial spinal cord of apoptosis-rescued (Bax KO) and control mice at E14.5. B, Quantification of p-c-Jun-positive motoneurons indicated in A. Data are mean ± SEM. ***p < 0.001. n = 3. C, Survival rate of primary cultured motoneurons derived from Col25a1+/− (n = 7) and Col25a1 KO (n = 4) mice on an Hb9-GFP background at 3 d in vitro, treated with (+) or without (−) NTFs (mixture of BDNF, CNTF, GDNF). Data are mean ± SEM. ***p < 0.001. *p < 0.05. n.s., Not significant. D, Primary cultured motoneurons derived from Hb9-GFP;Col25a1+/+ or Hb9-GFP;Col25a1 KO mice, treated with (+) or without (−) NTF, immunostained by anti-phosphorylated ERK1/2 (red) and anti-GFP (green).
One possible explanation for these observations included an inability of CLAC-P/collagen XXV-deficient motoneurons to receive trophic factors. We therefore examined the trophic factor-dependent survival and activation of the signaling pathway using primary cultures of motoneuron dissociated from E11.5 CLAC-P/collagen XXV deficient spinal cord. Survival of Col25a1 KO motoneurons was significantly enhanced in the presence of the neurotrophic factors (BDNF, CNTF, and GDNF) after 3 d in culture, and the extent of the increment in survival rate was comparable with that in motoneurons derived from control littermates (Fig. 6C). Furthermore, application of neurotrophic factors elicited activation of ERK signaling as indicated by a clear enhancement of phosphorylated ERK1/2 immunostaining at similar levels in Col25a1 KO and control motoneurons (Fig. 6D). These results indicate that, despite the preservation of ability to respond to trophic support-dependent anti-apoptotic signal, Col25a1 KO motoneurons failed to receive the survival signal in vivo.
Discussion
In this study, we demonstrated that motoneurons fail to innervate the target muscles, and subsequently undergo excessive apoptosis, during the period of PCD in the absence of CLAC-P/collagen XXV. We postulate that, among a series of axon guidance events, CLAC-P/collagen XXV regulates extension and/or arborization of the motor axon bundles after entering its target muscles.
The striking phenotype of Col25a1 KO mice was the almost complete loss of motoneurons throughout different levels of the spinal cord, which has been found very rarely in other genetically modified mouse models. Based on the results using Col25a1 KO mice crossed with Bax KO mice that showed significant increase in phosphorylation of c-Jun in rescued motoneurons, we concluded that the lack of target-derived trophic support resulted in massive apoptosis of motoneurons in Col25a1 KO mice (Fig. 6A,B).
Survival of motoneurons during development has been shown to depend on the target-derived signals induced by several factors (e.g., BDNF, NT-3, NT-4/5, GNDF, and CNTF) (Sendtner et al., 2000). None of the mice lacking any single survival factors or its receptors showed complete death of motoneurons (Snider, 1994). In contrast, a near-complete absence of motoneuron has been observed in mice lacking the two key myogenic regulatory transcription factors, MyoD and Myf-5, in which mature skeletal muscle is completely absent (Kablar and Rudnicki, 1999). This model recapitulates the early limb-bud ablation experiment in chick embryos, where a lack of target-derived survival factors triggers excessive apoptosis of limb-innervating motoneurons (Hamburger, 1934; Oppenheim et al., 1978). These indicate that the survival signals are required to work in a coordinated manner. Possible mechanisms that may underlie motoneuron loss in Col25a1 KO mice would be as follows: (1) defect in trophic factor receipt in Col25a1 KO motoneurons, (2) abnormal secretion/expression of neurotrophic factors due to immature development of muscles, (3) lack of trophic factor-like effect of CLAC-P/collagen XXV, and (4) incomplete motor axon extension into the muscles. Similar degrees of increase in survival rate and activation in downstream signaling in response to neurotrophic factors between control and Col25a1 KO primary motoneurons excluded the first possibility (Fig. 6C,D). We observed no significant decrease in the number of TrkC-positive proprioceptive sensory neurons in Col25a1 KO mice (Fig. 3I), whereas proprioceptive neurons have been shown to be affected in the absence of muscle-derived neurotrophic support (Oakley et al., 1997), ruling out the second hypothesis. The third possibility that CLAC-P/collagen XXV acts on motoneurons as a trophic factor was excluded because recombinant CLAC-P/collagen XXV proteins did not promote survival of motoneurons in vitro (data not shown). Together with the observation that inhibition of apoptosis in Col25a1 KO mice failed to rescue the severe stall of phrenic nerve bundles (Fig. 5C), we speculate that premature death of motoneurons induced by lack of antiapoptotic signals in Col25a1 KO mice results from incomplete motor axon extension within the muscle, rather than global abnormalities in trophic factor-dependent antiapoptotic signaling per se.
Similar innervation defects as in Col25a1 KO mice have been reported in mice lacking damage-induced neuronal endopeptidase (DINE) (Nagata et al., 2010). DINE-deficient mice exhibit a significant decrease in nerve terminal arborization, which seems less severe compared with that in Col25a1 KO mice; the phrenic nerve trunk of DINE-deficient mice is capable of growing to some extent and forming NMJs at the regions proximal to the nerve entry site in the muscles, without branching. This may enable motoneurons to receive sufficient trophic support, preventing from a significant decrease in motoneurons in DINE-deficient mice. Striking differences in the motoneuron survival between DINE- and Col25a1 KO mice indicate that there is a threshold level of the concentration of trophic factors for the survival of developing motoneurons that might be determined by the spatially graded distribution of trophic factors. Together, it is likely that CLAC-P/collagen XXV is required at the very early phase of intramuscular axonal extension.
CLAC-P/collagen XXV mRNA is widely expressed in various types of neurons, including motoneurons, as well as in skeletal muscles from early embryonic stages (Fig. 2B). The phenotype observed in Col25a1 KO mice in the present study was particularly prominent in motoneurons and skeletal muscles. Furthermore, in Col25a1 KO mice, axon guidance from the spinal cord to the target muscles takes place correctly (data not shown), and the innervation is disturbed only after the axon bundles penetrate inside the muscle tissues. These observations suggest that CLAC-P/collagen XXV may be specifically involved in the communication between the motor nerve and muscles in early development. It is not clear at this stage what cell types contribute to the function of CLAC-P/collagen XXV to support the motor innervation. Expression of the CLAC-P/collagen XXV mRNA both in the embryonic and adult CNSs may suggest a role in postmitotic neurons. In contrast, its expression in the skeletal muscle was specifically detected in early developmental stages as well as in differentiating cultured myotubes (Fig. 2C,D), corresponding to the time window of the intramuscular innervation. In addition, no decrease in sensory neurons was observed in Col25a1 KO mice despite the expression of CLAC-P/collagen XXV in DRG in wild-type embryos (Fig. 2A). These observations prompt us to speculate that CLAC-P/collagen XXV expressed on the muscle cell surface, or its ectodomain released from muscles, contributes to the interaction with motor axons, resulting in axonal extension and branching. In Col25a1 KO mice, myotube formation and accumulation of prepatterned aneural AChR microclusters occurred normally at the stage when motor axon entry took place (Fig. 4C). Because progressive delay in postsynaptic maturation became obvious after E13.5 (Fig. 4D), which is similar to the phenotype observed in mice lacking motor innervation, the delay might be a consequence of defects in innervation (Lin et al., 2001).
As MACIT family collagens are suggested to play roles at cell–cell or cell–matrix adhesion sites (Franzke et al., 2003), the identification of interacting molecules of CLAC-P/collagen XXV is crucial. Collagen XIII, which belongs to the MACIT family and is homologous to CLAC-P/collagen XXV, has been shown to be involved in the NMJ development (Latvanlehto et al., 2010). Because of the early defects in motor innervation, we were not able to examine the possible involvement of CLAC-P/collagen XXV in NMJ formation in our KO mice. The striking resemblance of the phenotypes of mice lacking the receptor protein tyrosine phosphatases (RPTP) δ and σ (Uetani et al., 2006), exhibiting a dramatic defect in intramuscular branching and total loss of motoneurons, to our Col25a1 KO mice should provide an important key to this question. Type IIa RPTPs, RPTP-δ, RPTP-σ, and LAR are implicated in axonal growth as well as in axonal regeneration after injury both in the PNS and CNS (McLean et al., 2002; Johnson and Van Vactor, 2003; Thompson et al., 2003; Sapieha et al., 2005). Although the molecular mechanisms underlying the phenotypes of RPTP-δ−/−/σ−/− mice remain unclear, it is worth noting that RPTPs interact with heparan sulfate proteoglycans and chondroitin sulfate proteoglycans, which promote and inhibit axon extension, respectively (Aricescu et al., 2002; Shen et al., 2009). Interestingly, it has previously been demonstrated that the ectodomain of CLAC-P/collagen XXV binds to heparin through the collagenous domain highly enriched in positively charged amino acid residues (Osada et al., 2005; Söderberg et al., 2005). It is tempting to speculate that CLAC-P/collagen XXV is involved in muscle–nerve interaction through contact with the extracellular matrix, such as proteoglycans, in a manner cross talking to the mechanism involving RPTPs.
In conclusion, we demonstrated that CLAC-P/collagen XXV is a novel essential factor that regulates the initial phase of intramuscular motor nerve branching and innervation, which is required for the motoneuron survival during development. Our study offers strong evidence for the existence of a previously unknown molecular mechanism that regulates motor nerve-muscle interaction during development. As CLAC-P/collagen is also expressed throughout the CNS and PNS, further study is required to elucidate its roles in differentiated neurons, which will facilitate our understanding of the pathological mechanism of Alzheimer's disease as well.
Footnotes
This work was supported by Core Research for Evolutional Science and Technology of the Japan Science and Technology Agency and the Japan Society for the Promotion of Science (Grant-in-Aid for Young Scientists B 24700373). We thank Drs. J. R. Sanes (Harvard University), J. Kay (Duke University), H. Nishimune (University of Kansas), and Ms. H. Munezane (University of Tokyo) for helpful suggestions.
The authors declare no competing financial interests.
References
- Aebischer J, Sturny R, Andrieu D, Rieusset A, Schaller F, Geib S, Raoul C, Muscatelli F. Necdin protects embryonic motoneurons from programmed cell death. PLoS One. 2011;6:e23764. doi: 10.1371/journal.pone.0023764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aricescu AR, McKinnell IW, Halfter W, Stoker AW. Heparan sulfate proteoglycans are ligands for receptor protein tyrosine phosphatase σ. Mol Cell Biol. 2002;22:1881–1892. doi: 10.1128/MCB.22.6.1881-1892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks GB, Noakes PG. Elucidating the molecular mechanisms that underlie the target control of motoneuron death. Int J Dev Biol. 2002;46:551–558. [PubMed] [Google Scholar]
- Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/S0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Fox MA. Novel roles for collagens in wiring the vertebrate nervous system. Curr Opin Cell Biol. 2008;20:508–513. doi: 10.1016/j.ceb.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Franzke CW, Tasanen K, Schumann H, Bruckner-Tuderman L. Collagenous transmembrane proteins: collagen XVII as a prototype. Matrix Biol. 2003;22:299–309. doi: 10.1016/S0945-053X(03)00051-9. [DOI] [PubMed] [Google Scholar]
- Hägg P, Väisänen T, Tuomisto A, Rehn M, Tu HM, Huhtala P, Eskelinen S, Pihlajaniemi T. Type XIII collagen: a novel cell adhesion component present in a range of cell–matrix adhesions and in the intercalated discs between cardiac muscle cells. Matrix Biol. 2001;19:727–742. doi: 10.1016/S0945-053X(00)00119-0. [DOI] [PubMed] [Google Scholar]
- Hamburger V. The effects of wing bud extirpation on the development of the central nervous system in chick embryos. J Exp Zool. 1934;68:449–494. doi: 10.1002/jez.1400680305. [DOI] [Google Scholar]
- Hashimoto T, Wakabayashi T, Watanabe A, Kowa H, Hosoda R, Nakamura A, Kanazawa I, Arai T, Takio K, Mann DM, Iwatsubo T. CLAC: a novel Alzheimer amyloid plaque component derived from a transmembrane precursor, CLAC-P/collagen type XXV. EMBO J. 2002;21:1524–1534. doi: 10.1093/emboj/21.7.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkinen A, Tu H, Pihlajaniemi T. Collagen XIII: a type II transmembrane protein with relevance to musculoskeletal tissues, microvessels and inflammation. Int J Biochem Cell Biol. 2012;44:714–717. doi: 10.1016/j.biocel.2012.01.024. [DOI] [PubMed] [Google Scholar]
- Johnson KG, Van Vactor D. Receptor protein tyrosine phosphatases in nervous system development. Physiol Rev. 2003;83:1–24. doi: 10.1152/physrev.00016.2002. [DOI] [PubMed] [Google Scholar]
- Kablar B, Rudnicki MA. Development in the absence of skeletal muscle results in the sequential ablation of motor neurons from the spinal cord to the brain. Dev Biol. 1999;208:93–109. doi: 10.1006/dbio.1998.9184. [DOI] [PubMed] [Google Scholar]
- Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- Kowa H, Sakakura T, Matsuura Y, Wakabayashi T, Mann DM, Duff K, Tsuji S, Hashimoto T, Iwatsubo T. Mostly separate distributions of CLAC- versus Aβ40- or thioflavin S-reactivities in senile plaques reveal two distinct subpopulations of β-amyloid deposits. Am J Pathol. 2004;165:273–281. doi: 10.1016/S0002-9440(10)63295-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latvanlehto A, Fox MA, Sormunen R, Tu H, Oikarainen T, Koski A, Naumenko N, Shakirzyanova A, Kallio M, Ilves M, Giniatullin R, Sanes JR, Pihlajaniemi T. Muscle-derived collagen XIII regulates maturation of the skeletal neuromuscular junction. J Neurosci. 2010;30:12230–12241. doi: 10.1523/JNEUROSCI.5518-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410:1057–1064. doi: 10.1038/35074025. [DOI] [PubMed] [Google Scholar]
- McLean J, Batt J, Doering LC, Rotin D, Bain JR. Enhanced rate of nerve regeneration and directional errors after sciatic nerve injury in receptor protein tyrosine phosphatase σ knock-out mice. J Neurosci. 2002;22:5481–5491. doi: 10.1523/JNEUROSCI.22-13-05481.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Kiryu-Seo S, Maeda M, Yoshida K, Morita T, Kiyama H. Damage-induced neuronal endopeptidase is critical for presynaptic formation of neuromuscular junctions. J Neurosci. 2010;30:6954–6962. doi: 10.1523/JNEUROSCI.4521-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimune H, Sanes JR, Carlson SS. A synaptic laminin-calcium channel interaction organizes active zones in motor nerve terminals. Nature. 2004;432:580–587. doi: 10.1038/nature03112. [DOI] [PubMed] [Google Scholar]
- Oakley RA, Lefcort FB, Clary DO, Reichardt LF, Prevette D, Oppenheim RW, Frank E. Neurotrophin-8 promotes the differentiation of muscle spindle afferents in the absence of peripheral targets. J Neurosci. 1997;17:4262–4274. doi: 10.1523/JNEUROSCI.17-11-04262.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Chu-Wang IW, Maderdrut JL. Cell death of motoneurons in the chick embryo spinal cord: III. The differentiation of motoneurons prior to their induced degeneration following limb-bud removal. J Comp Neurol. 1978;177:87–111. doi: 10.1002/cne.901770107. [DOI] [PubMed] [Google Scholar]
- Osada Y, Hashimoto T, Nishimura A, Matsuo Y, Wakabayashi T, Iwatsubo T. CLAC binds to amyloid β peptides through the positively charged amino acid cluster within the collagenous domain 1 and inhibits formation of amyloid fibrils. J Biol Chem. 2005;280:8596–8605. doi: 10.1074/jbc.M413340200. [DOI] [PubMed] [Google Scholar]
- Pihlajaniemi T, Rehn M. Two new collagen subgroups: membrane-associated collagens and types XV and XVIII. Prog Nucleic Acid Res Mol Biol. 1995;50:225–262. doi: 10.1016/S0079-6603(08)60816-8. [DOI] [PubMed] [Google Scholar]
- Powell AM, Sakuma-Oyama Y, Oyama N, Black MM. Collagen XVII/BP180: a collagenous transmembrane protein and component of the dermoepidermal anchoring complex. Clin Exp Dermatol. 2005;30:682–687. doi: 10.1111/j.1365-2230.2005.01937.x. [DOI] [PubMed] [Google Scholar]
- Sapieha PS, Duplan L, Uetani N, Joly S, Tremblay ML, Kennedy TE, Di Polo A. Receptor protein tyrosine phosphatase σ inhibits axon regrowth in the adult injured CNS. Mol Cell Neurosci. 2005;28:625–635. doi: 10.1016/j.mcn.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Pei G, Beck M, Schweizer U, Wiese S. Developmental motoneuron cell death and neurotrophic factors. Cell Tissue Res. 2000;301:71–84. doi: 10.1007/s004410000217. [DOI] [PubMed] [Google Scholar]
- Shen Y, Tenney AP, Busch SA, Horn KP, Cuascut FX, Liu K, He Z, Silver J, Flanagan JG. PTPσ is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science. 2009;326:592–596. doi: 10.1126/science.1178310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Söderberg L, Kakuyama H, Möller A, Ito A, Winblad B, Tjernberg LO, Näslund J. Characterization of the Alzheimer's disease-associated CLAC protein and identification of an amyloid beta-peptide-binding site. J Biol Chem. 2005;280:1007–1015. doi: 10.1074/jbc.M403628200. [DOI] [PubMed] [Google Scholar]
- Sun W, Gould TW, Newbern J, Milligan C, Choi SY, Kim H, Oppenheim RW. Phosphorylation of c-Jun in avian and mammalian motoneurons in vivo during programmed cell death: an early reversible event in the apoptotic cascade. J Neurosci. 2005;25:5595–5603. doi: 10.1523/JNEUROSCI.4970-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson KM, Uetani N, Manitt C, Elchebly M, Tremblay ML, Kennedy TE. Receptor protein tyrosine phosphatase sigma inhibits axonal regeneration and the rate of axon extension. Mol Cell Neurosci. 2003;23:681–692. doi: 10.1016/S1044-7431(03)00120-9. [DOI] [PubMed] [Google Scholar]
- Uetani N, Chagnon MJ, Kennedy TE, Iwakura Y, Tremblay ML. Mammalian motoneuron axon targeting requires receptor protein tyrosine phosphatases σ and δ. J Neurosci. 2006;26:5872–5880. doi: 10.1523/JNEUROSCI.0386-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J Neurosci. 1998;18:1428–1439. doi: 10.1523/JNEUROSCI.18-04-01428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/S0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]