

Abstract
Reactive oxygen species (ROS) are signaling factors involved in many intracellular transduction pathways. In the nervous system, ROS are thought to modulate various mechanisms of synaptic plasticity. One important source of ROS production in the brain is the NADPH oxidase complex. Stimulation of NMDA receptors activates NADPH oxidase, which provides selective oxidative responses accompanying the induction of synaptic changes. The activity of NADPH oxidase is known to be crucial for the induction of LTP in the hippocampus. However, the involvement of this complex in cortical synaptic plasticity is still unclear. Here we provide evidence that genetic ablation of NOX2 (the prototypical member of NADPH oxidase family of proteins) suppresses LTP and LTD in the primary visual cortex of the mouse. We also found that the involvement of NOX2 on LTP is partially age-dependent, as the activity of this complex is not critical for mechanisms of synaptic potentiation occurring in immature animals. Furthermore, we show that inhibition of NOX2 reduces the NMDA receptor function, suggesting a possible mechanism that could be the basis of the effects on synaptic plasticity.
Keywords: LTD, LTP, NOX2, reactive oxygen species, synaptic plasticity, visual cortex
Introduction
Reactive oxygen species (ROS) are chemically reactive molecules that modulate signal transduction pathways involved in synaptic plasticity (Klann, 1998; Klann and Thiels, 1999; Knapp and Klann, 2002; Kishida et al., 2005, 2006; Kishida and Klann, 2007). During brain function, one of the main sources of ROS is the NADPH oxidase complex (Infanger et al., 2006; Bedard and Krause, 2007). The prototypical member of this family of proteins, NOX2-based oxidase, consists of six subunits, four cytosolic (p67phox, p47phox, p40phox, and Rac), and the membrane-spanning heterodimer (gp91phox and p22phox) (Sumimoto et al., 2004). The gp91phox is the catalytic subunit that transfers electrons from NADPH to molecular oxygen. The gp91phox and p47phox proteins are present in several areas of the brain, including the cerebral cortex (Serrano et al., 2003).
Production of ROS via the NADPH oxidase system is rapid and specific to particular signaling events (Quinn and Gauss, 2004). Specifically, NADPH oxidase is known to be activated by NMDA receptor (NMDAR) stimulation (Kishida et al., 2005; Brennan et al., 2009). This makes this complex particularly suited to provide high amounts of ROS during the induction of synaptic plasticity. Mutant mice lacking NADPH oxidase subunits show impaired LTP in the hippocampus (Kishida et al., 2006). Application of pharmacological inhibitors of the NADPH oxidase complex also impairs hippocampal LTP (Kishida et al., 2006). However, the role of the NADPH oxidase complex in synaptic modifications of cortical circuitry has not been investigated.
The cerebral cortex is an important site of memory storage and information processing. Long-term memories are thought to be formed within cortical circuitry by activity-dependent changes of synaptic connections (Bear, 1996; Fedelman, 2009).
In the present study, we aimed to elucidate the involvement of the NOX2 isoform of NADPH oxidase in two forms of synaptic plasticity (LTP and LTD) in the primary visual cortex (V1). V1 is recognized as a suited model to illustrate how activity-dependent synaptic modifications contribute to the development of cortical functions (Kirkwood and Bear, 1995; Feldman, 2009). We focused on the layer 4 to layers 2/3 connection, which is thought to be the initial site for receptive field plasticity (Trachtenberg et al., 2000). We hypothesized that ROS production through NOX2 is fundamental for synaptic modifications. To test our hypothesis, we used cortical tissue preparations obtained from knock-out mice for the catalytic subunit of NOX2 (gp91phox−/−). We first assessed whether NOX2 is an important source of ROS production in cortical neurons. Then, we evaluated the effects of genetic ablation of NOX2 on LTP and LTD.
Our results demonstrated that loss of a functional NOX2 oxidase complex impairs LTP and LTD in V1, and these effects are partially dependent on age. Our data are the first to show a role of NOX2 oxidase in cortical plasticity and could provide an important advance in understanding the molecular mechanisms underlying the modifications of synaptic connections in the cerebral cortex.
Materials and Methods
Animals.
Male gp91phox−/− mice (The Jackson Laboratory), along with male wild-type mice (C57BL/6), were used throughout this study. The animals had free access to food and water and were maintained on a 12:12 h light–dark cycle. All animals used in our experiments were 20–24 postnatal days, except for the age-dependent study, in which P9–P12 and P61–P65 animals were also used. All procedures were approved by the Institutional Animal Care Committee of the Institute of Biomedical Sciences, University of São Paulo (CEUA ICB/USP 090, fls. 1°).
Preparation and maintenance of brain slices.
Animals were deeply anesthetized by inhalation of isoflurane (AErrane; Baxter Pharmaceuticals) and then decapitated. The brain was quickly removed and submerged in cooled (0°C) oxygenated (5% CO2–95% O2) dissection buffer (in mm) as follows: 206 sucrose, 25 NaHCO3, 2.5 KCl, 10 MgSO4, 1.25 NaH2PO4, 0.5 CaCl2, and 11 d-glucose. Brains were blocked and vibratome sectioned. Coronal slices (∼300 μm thick) were cut and rapidly transferred to a holding chamber with ACSF (in mm) as follows: 125 NaCl, 25 NaHCO3, 3 KCl, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, and 25 d-glucose) and kept oxygenated at room temperature (20°C–25°C) for at least 1 h before proceeding with dihydroethidium (DHE) imaging or electrophysiological recording.
Pharmacology.
In some DHE imaging experiments, NMDA (100 μm) or NMDA plus the nonspecific NOX2 inhibitor, apocynin (100 μm, Sigma) were used. In some electrophysiological experiments, the NMDA receptor blocker MK-801 (50 μm, Tocris Bioscience), or apocynin (100 μm, Sigma) or hydrogen peroxide (H2O2; 100 μm) were used. In experiments aimed to isolate NMDA receptor responses, we used a magnesium free ACSF containing the AMPA receptor blocker DNQX (50 μm, Tocris Bioscience) and the GABA receptor blocker picrotoxin (50 μm, Tocris Bioscience).
We prepared NMDA stock solutions in distilled water. For MK-801, DNQX, picrotoxin, and apocynin, stock solutions were prepared in DMSO (0.1%). All drugs were diluted in ACSF to their final concentration just before use. In some experiments, apocynin or H2O2 was applied after a stable baseline was achieved. In these cases, the system of tubes providing ACSF to the recording chamber was connected to a 10 ml container of ACSF in a recirculating mode. Drugs were applied by adding the stock solution to the 10 ml container providing ACSF to the recording chamber. The final bath concentration was calculated on the basis of the total volume of the recording chamber, the tubes, and the small container of ACSF.
ROS detection using DHE.
The oxidation of DHE to their fluorescent products was used to estimate intracellular ROS levels. Visual cortical slices were incubated free-floating for 5 min with a solution containing the following: sodium phosphate buffer (PB; 0.2 m), DHE (5 μm), and diethylene triamine pentaacetic acid (DTPA; 100 μm). The brain slices were then washed once with PB and mounted on gelatinized slides. Subsequently, the slices were immediately photographed with a epifluorescence microscope (rhodamine filter) using 480 nm excitation and 586 nm emission. The DHE staining was evaluated in layer 2/3 of the primary visual cortex by measuring the integrated fluorescence area. In experiments aimed to compare ROS production of gp91phox−/− slices with slices obtained from wild-type animals, the fluorescence intensity was normalized with respect to the mean value observed in the wild-type.
In additional experiments, NMDA or NMDA plus apocynin, a nonspecific NOX2 inhibitor, were used. In these experiments, the slices were kept at 30°C for 5 min in oxygenated ACSF (5% CO2–95% O2) containing NMDA or NMDA plus apocynin. Subsequently, the slices were incubated free-floating with the DHE solution for 5 min, mounted, and immediately photographed. The fluorescence intensity was normalized with respect to the controls treated with vehicles.
Electrophysiological recordings.
Visual cortical slices were placed in a submersion-type recording chamber on a modified microscope stage with constant perfusion of oxygenated ACSF (5% CO2–95% O2). Whole-cell recordings were made from layer 2/3 of primary visual cortex (V1). Layer 2/3 was identified by differential interference contrast microscopy: layer 1 was typically thin and aneuronal, whereas layer 4 showed a dense packing of cell bodies. Layers 2/3 can be recognized as located between layers 1 and 4. All recordings were obtained from regular spiking pyramidal-shaped cells. Recording pipettes were fabricated from borosilicate glass (Garner Glass) with input resistances of ∼4–6 mΩ and were filled with intracellular solution (in mm) as follows: 135 K-gluconate, 7 NaCl, 10 HEPES, 2 Na2ATP, 0.3 Na3GTP, 2 MgCl2, and 0.1%–0.5% biocytin, pH of 7.3 obtained with KOH and osmolality of 290 mOsm. All experiments were performed using a visualized slice setup under a differential interference contrast-equipped Nikon Eclipse E600FN microscope. Recordings were made by using a Multiclamp 700B amplifier and pClamp software (Molecular Devices). Only recordings from cells with membrane potentials <−65 mV, access resistance <20 mΩ, and input resistance >100 mΩ and <1000 mΩ were included in our data. Cells were discarded if any of these values changed >15% during the experiment. We injected depolarizing currents to identify regular, tonic, or bursting spike patterns. Postsynaptic responses were recorded in current-clamp mode.
Electrical stimulation.
Synaptic responses were evoked by electrically stimulating inputs to neurons of layer 2/3 (0.1 Hz; 0.2 ms). Stimulation was delivered through a concentric bipolar electrode (125 μm diameter; FHC) placed in the middle of the cortical thickness of layer 4. We only included in our data responses we determined to be monosynaptic using the following criteria: latencies <3 ms, latency jitter <1 ms, and no failures for stimulation levels above threshold. The intensity of stimulation was gradually increased from a subthreshold level by steps of 5 μA until a response was evoked. The intensity was then adjusted to evoke 2–5 mV responses. We used stimulation intensities between 5 and 50 μA.
To induce synaptic plasticity, the recording mode was switched from current clamp to voltage clamp. The induction consisted of a pairing protocol of 200 epochs (1 Hz) during which the voltage membrane was kept at 0 mV for the induction of LTP and −40 mV for the induction of LTD. Changes in synaptic strength were quantified as changes in the initial amplitude of the postsynaptic potential normalized by the mean baseline response obtained during the first 10 min of stable recordings. In all graphs showing the LTP or LTD time course, the EPSP amplitude was normalized with respect to the averaged EPSP value of the last 5 min of baseline. The LTP and LTD magnitude was analyzed by calculating the average EPSP value of the last 5 min (35–40 min) of the postsynaptic period. This value was normalized, and the baseline was subtracted to obtain the relative increase or decrease of EPSP.
In some experiments, we aimed to determinate the relationship between stimulation intensity and the evoked NMDAR-mediated synaptic response. In these experiments, the intensity stimulation values were normalized with respect to the “minimal stimulation” intensity, which is defined as follows: the stimulation level was increased from subthreshold in 5 μA steps until a reliable response was triggered; intensity was then further increased by 5 μA from this point, and this was the minimum level of intensity stimulation.
Statistical analysis.
When we analyzed results obtained from DHE imaging, we compared the fluorescence intensity between the different experimental groups by means of a t test and of the one-way ANOVA with Newman–Keuls multiple-comparison post hoc test. In electrophysiological experiments, LTP and LTD were assessed through a paired t test. We paired the mean values of EPSPs calculated averaging the last 5 min of the baseline with the mean values calculated averaging the last 5 min (35–40 min) of the postinduction period. Differences between experimental groups were analyzed using an unpaired t test. The intensity/response curves of NMDAR responses were compared by means of a two-way ANOVA. The effect of apocynin on NMDAR responses was assessed through a paired t test. We first averaged the EPSP values of the baseline. This averaged value was then compared with the mean values recorded during a 5 min period, which occurred 5 min after the application of the drug.
Results
DHE imaging of NOX2-mediated ROS production
We first investigated the involvement of NOX2 in the ROS production of layers 2/3 neurons of V1. ROS levels were quantified by measuring the intracellular accumulation of oxidized DHE. Visual cortical slices obtained from P21-P24 animals were treated with DHE. This agent is oxidized by ROS to form fluorescent ethidium, which is trapped inside the cells by DNA binding (Bindokas et al., 1996; Zhao et al., 2003). We first evaluated whether gp91phox−/− animals have indeed lower levels of ROS accumulation. We found that gp91phox−/− mice showed reduced levels of ROS compared with their wild-type littermates (Fig. 1A,B; wild-type: 13 slices, 4 animals; gp91phox−/−: 10 slices, 4 animals; p < 0.01 on a t test). Subsequently, we investigated whether NMDAR activation causes an increase in ROS production through NOX2. For this purpose, we used an NMDAR agonist (NMDA; 50 μm) and a nonspecific NOX2 inhibitor apocynin (100 μm). Its mechanism of action involves blocking the translocation of the cytosolic subunit p47phox to the plasma membrane, thereby inhibiting the NOX2 complex activation. In wild-type animals, slices treated with NMDA showed higher levels of DHE fluorescence compared with controls (Fig. 1C,D; control: 7 slices, 4 animals; NMDA: 7 slices, 4 animals; p < 0.05 on a one-way ANOVA and Newman–Keuls multiple-comparison test). Slices treated with apocynin and NMDA were not statistically different from controls (Fig. 1C,D; control: 7 slices, 4 animals; NMDA and apocynin: 7 slices, 4 animals; p > 0.1 on a one-way ANOVA and Newman–Keuls multiple-comparison test). Slices obtained from gp91phox−/− animals were also treated with NMDA and compared with their controls (Fig. 1E,F). In this case, no statistical difference was found (Fig. 1E,F; control: 7 slices, 4 animals; NMDA: 7 slices, 4 animals; p > 0.1 on a t test). These findings suggest that NMDAR activation results in ROS production through NOX2 in the visual cortex. This is consistent with previous studies that were focused on hippocampal neurons (Gunasekar et al., 1995, Bindokas et al., 1996, Kishida et al., 2005; Brennan et al., 2009).
Figure 1.

Pharmacological and genetic suppression of NOX2 results in lower levels of ROS production. A, Representative DHE digital images (grayscale-inverted colors) showing neuronal labeling in layer 2/3 of visual cortical slices obtained from wild-type (i) and gp91phox−/− experimental groups (ii). B, The graph represents fluorescence intensity evaluated by DHE staining in wild-type slices and gp91phox−/− animals. Slices derived from wild-type animals showed higher fluorescence intensity (wild-type: 13 slices, 4 animals; gp91phox−/−: 10 slices, 4 animals; p < 0.01 on a t test). Plotted data are average ± SEM. C, Representative DHE digital images (grayscale-inverted colors) showing neuronal labeling in slices from wild-type animals: treated with vehicle (i), treated with NMDA (ii), and treated with NMDA and apocynin. D, Results of NMDA application in slices obtained from wild-type animals. The graph shows the average values of DHE fluorescence in controls, in slices treated with NMDA, and in slices treated with NMDA and apocynin. Slices treated with NMDA showed higher fluorescence intensity than controls (NMDA: 7 slices, 4 animals; control: 7 slices, 4 animals; p < 0.05 on a one-way ANOVA with Newman–Keuls multiple-comparison test), whereas slices treated with NMDA and apocynin were not statistically different from controls (NMDA + apocynin: 7 slices, 4 animals; p > 0.1 on a one-way ANOVA with Newman–Keuls multiple-comparison test). Plotted data are average ± SEM. E, Examples of DHE fluorescence (grayscale-inverted colors) showing neuronal labeling in slices from gp91phox−/− animals: treated with vehicle (i), treated with NMDA (ii). F, Results of NMDA application in slices derived from gp91phox−/− animals. The graph shows the average values of DHE fluorescence in controls and in slices treated with NMDA. Slices treated with NMDA were not statistically different from controls (NMDA: 9 slices, 4 animals; control: 9 slices, 4 animals; p > 0.1 on a t test). Plotted data are average ± SEM.
Electrophysiological experiments
We performed whole-cell recording experiments obtained from 238 neurons in layer 2/3 of V1. Cells had resting membrane potentials of 70.02 ± 0.25 mV, input resistances of 308.87 ± 9.15 mΩ, and access resistance of 16.34 ± 0.13 mΩ. Current injections were applied and all cells showed a regular spiking pattern.
Synaptic plasticity and NMDA receptor activation
We investigated whether LTP and LTD in layers 2/3 of V1 depend on the activation of NMDAR. For this purpose, a set of experiments was performed in slices derived from P21–P24 animals and treated with the NMDAR blocker MK-801 (50 μm). Slices treated with vehicle were used as controls. LTP could be induced by pairing presynaptic stimulation with 0 mV postsynaptic voltage in control slices (Fig. 2A; 9 slices, 8 animals; p < 0.01 on a paired t test). The same protocol had no effect in the presence of MK-801 (Fig. 2A; 8 slices, 7 animals; p > 0.1 on a paired t test). Pairing presynaptic stimulation with −40 mV postsynaptic voltage induced LTD in controls (Fig. 2B; 9 slices, 7 animals; p < 0.001 on a paired t test) and in slices treated with MK-801 (Fig. 2B; 8 slices, 7 animals; p < 0.05 on a paired t test). However, in the presence of MK-801, the LTD was significantly reduced, compared with controls (p < 0.05 on a t test). These results confirmed that NMDAR is involved in the induction of LTP and LTD in layers 2/3 of V1.
Figure 2.
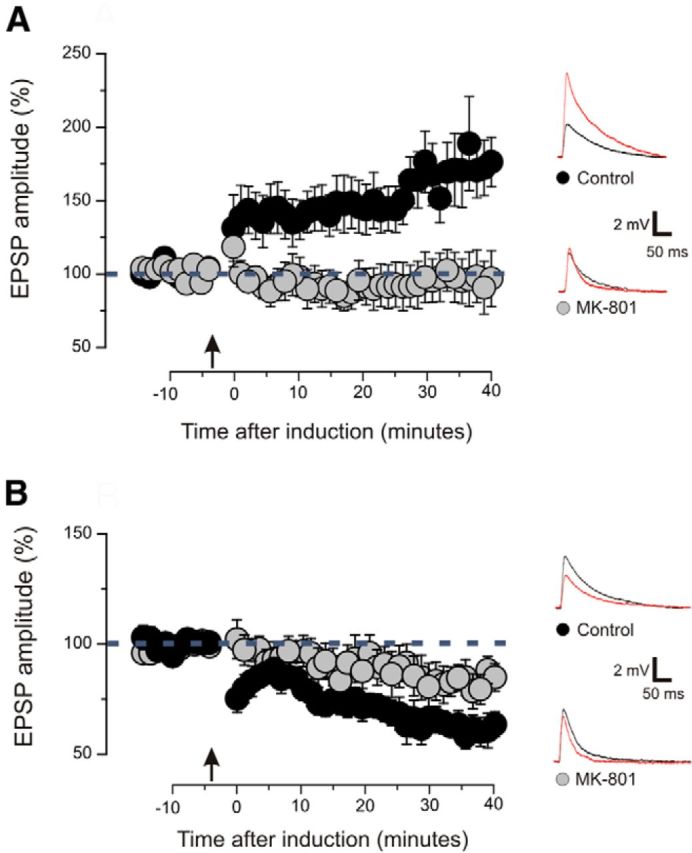
LTP and LTD depend on the activation of NMDA receptors. A, Magnitude of EPSPs before and after the induction of LTP. Arrow indicates the time of induction. Slices treated with the NMDA receptor blocker MK-801 are compared with control slices treated with vehicle. In control slices, a significant LTP was induced (9 slices; 8 animals; p < 0.01 on a paired t test). In slices treated with MK-801, no significant change of postsynaptic response was found (8 slices; 7 animals; p > 0.1 on a paired t test). Examples of EPSP traces showed the amplitude before (black line) and after (red line) the induction. Plotted data are average ± SEM. B, Magnitude of EPSPs before and after the induction of LTD. Arrow indicates the time of induction. Slices treated with the NMDA receptor blocker MK-801 are compared with control slices treated with vehicle. A significant LTD was induced in control slices (9 slices; 7 animals; p < 0.001 on a paired t test) and in slices treated with MK-801 (8 slices; 7 animals; p < 0.05 on a paired t test). The LTD was significantly reduced in slices treated with MK-801 (p < 0.05 on t test). Examples of EPSP traces showed the amplitude before (black line) and after (redline) the induction. Plotted data are average ± SEM.
Genetic suppression of NADPH oxidase and synaptic plasticity
Our data revealed that ROS production via NOX2 is stimulated by NMDAR activity in visual cortical circuitry. It was also shown that synaptic plasticity depends on the activation of NMDAR. We then hypothesized that ROS production via NOX2 is important for cellular mechanisms that are required for NMDAR-dependent synaptic plasticity.
We tested this hypothesis by investigating LTP and LTD in wild-type and gp91phox−/− mice. LTP could be induced in wild-type animals (Fig. 3A; 9 slices, 8 animals; p < 0.05 on a paired t test) but was blocked in gp91phox−/− (Fig. 3A; 9 slices, 7 animals; p > 0.1 on a paired t test). Similarly, LTD was successfully induced in wild-type mice (Fig. 3B; 8 slices, 7 animals; p < 0.0001 on a paired t test) but not in gp91phox−/− (Fig. 3B; 10 slices, 7 animals; p > 0.1 on a paired t test). These findings indicate that NOX2 is required for LTD and LTP in layers 2/3 of the visual cortex.
Figure 3.
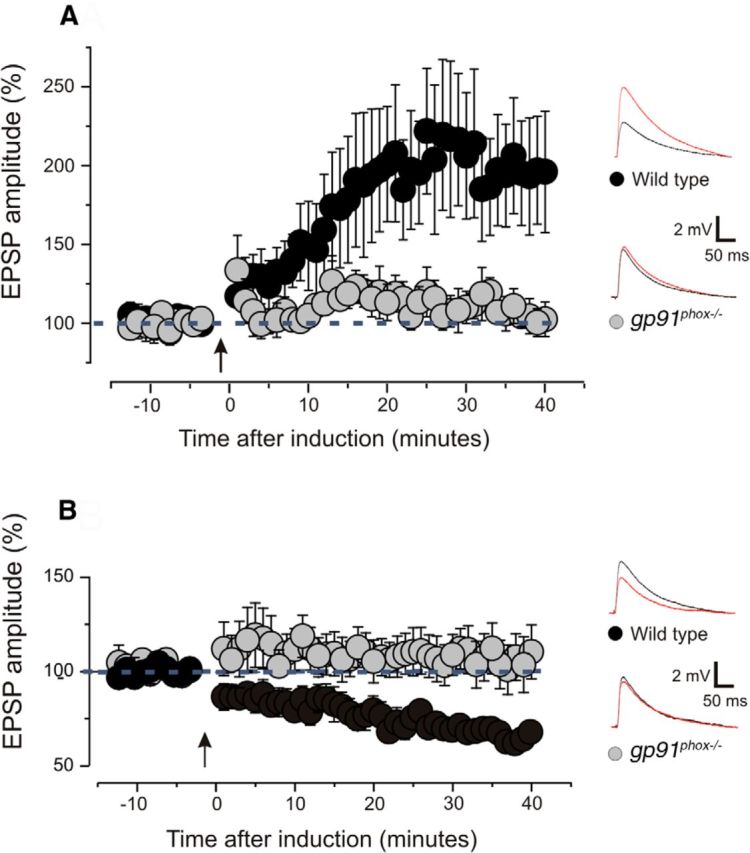
LTP and LTD are impaired in gp91phox−/− mice. A, Magnitude of EPSPs before and after the induction of LTP. Arrow indicates the time of induction. Slices obtained from gp91phox−/− animals are compared with slices obtained from wild-type mice. A significant LTP was induced in wild-type slices (9 slices; 8 animals; p < 0.05 on a paired t test). Slices derived from gp91phox−/− animals showed no LTP (9 slices; 7 animals; p > 0.1 on a paired t test). Examples of EPSP traces showed the amplitude before (black line) and after (red line) the induction. Plotted data are average ± SEM. B, Magnitude of EPSPs before and after the induction of LTD. Arrow indicates the time of induction. Slices obtained from gp91phox−/− animals are compared with slices derived from controls. LTD was successfully induced in wild-type slices (8 slices; 7 animals; p < 0.0001 on a paired t test). LTD could not be induced in slices derived from gp91phox−/− animals (10 slices; 7 animals; p > 0.1 on a paired t test). Examples of EPSP traces showed the amplitude before (black line) and after (red line) the induction. Plotted data are average ± SEM.
Pharmacological inhibition of NADPH and synaptic plasticity
Ours results obtained from gp91phox−/− animals showed that genetic suppression of NOX2 prevents NMDAR-dependent synaptic plasticity. NMDAR activation causes a rapid increase of ROS production via NOX2 (Fig. 1C,D). This rapid increase might be required during the induction of LTP/LTD for triggering intracellular pathways involved in synaptic changes. However, gp91phox−/− animals could have impaired synaptic plasticity because of lower levels of ROS accumulation (Fig. 1A,B). It remains unclear whether plasticity requires a strong rapid increase of ROS during the induction or rather requires a long lasting accumulation of ROS. To address this issue, we evaluated the effects of pharmacological inhibition of NOX2 on LTP and LTD in P21–P24 animals.
We inhibited the activity of NOX2 by treating visual cortical slices with apocynin (100 μm). Bath application of apocynin had no effect on the amplitude of postsynaptic response (Fig. 4A; 9 slices, 6 animals; p > 0.1 on a paired t test). However, the induction of LTP was prevented in slices treated with apocynin (Fig. 4B; 10 slices, 8 animals; p > 0.1 on a paired t test) but not in slices treated with vehicle (Fig. 4B; 9 slices, 8 animals; p < 0.05 on paired t test). Interestingly, LTD could be successfully induced in the control (Fig. 4C; 9 slices, 7 animals; p < 0.001 on a paired t test) and in the presence of apocynin (Fig. 4C; 8 slices, 7 animals; p < 0.001 on a paired t test). The magnitude of LTD was not statistically different compared with slices treated with vehicle (p > 0.1 on a paired t test). These results indicate that pharmacological inhibition of NOX2 specifically blocks LTP in the visual cortex, leaving intact basal synaptic transmission and LTD.
Figure 4.
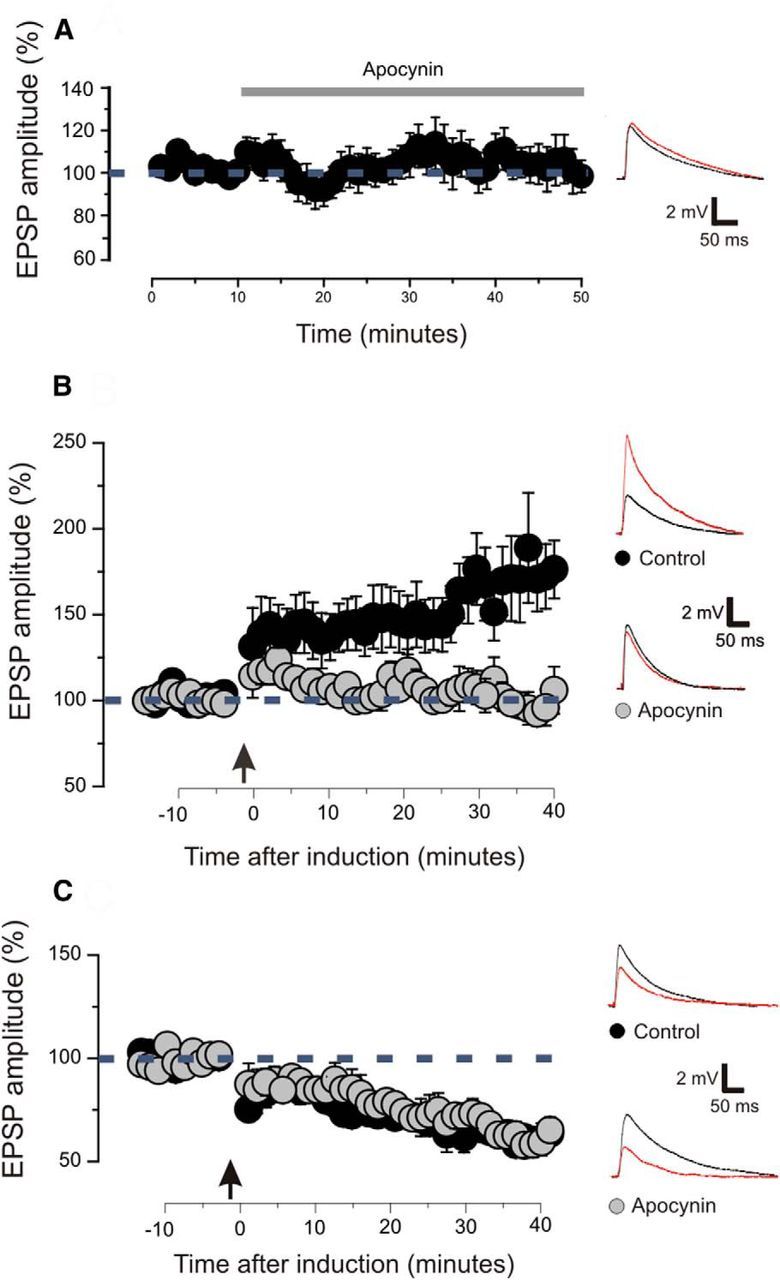
LTP but not LTD is impaired by pharmacological inhibition of NOX2. A, Magnitude of EPSPs before and after the application of apocynin. Arrow indicates the time of induction. Examples of EPSP traces showed the amplitude before (black line) and after (red line) the drug application. Apocynin had no effect on the EPSPs (9 slices; 6 animals; p > 0.1 on a paired t test). Plotted data are average ± SEM. B, Magnitude of EPSPs before and after the induction of LTP. Arrow indicates the time of induction. Slices treated with apocynin are compared with control slices treated with vehicles. LTP was successfully induced in control slices (9 slices; 8 animals; p < 0.01 on a paired t test). Slices treated with apocynin showed no LTP (10 slices; 8 animals; p > 0.1 on a paired t test). Examples of EPSP traces showing the amplitude before (black line) and after (red line) the drug application. Plotted data are average ± SEM. C, Magnitude of excitatory postsynaptic responses before and after the induction of LTD. Arrow indicates the time of induction. Slices treated with apocynin are compared with control slices treated with vehicles. A significant LTD was obtained in control slices (9 slices; 7 animals; p < 0.001 on a paired t test) and in slices treated with apocynin (8 slices; 7 animals; p < 0.001 on a paired t test), and these values were not statistically different (p > 0.1 on a t test). Examples of EPSP traces showed the amplitude before (black line) and after (red line) the induction. Plotted data are average ± SEM.
NMDA receptors mediated postsynaptic potentials and NOX2
Our results showed that the LTP is critically dependent on the activity of NOX2 in the visual cortex. The activation of NOX2 by means of NMDAR may be involved in various intracellular pathways important for synaptic plasticity. Alternatively, ROS production through NOX2 might improve the functionality of NMDAR and therefore facilitate the induction of LTP. To test this hypothesis, we studied the effect of genetic and pharmacological suppression of Nox2 on postsynaptic potentials mediated by NMDAR.
EPSPs evoked by NMDAR activation (NMDAR-EPSPs) were isolated by using a magnesium-free solution, an AMPA receptors blocker (DNQX, 50 μm) and a GABA receptors blocker (picrotoxin, 50 μm). Under these conditions, we observed postsynaptic responses (Fig. 5Ai) that were abolished in the presence of the NMDAR blocker MK-801 (50 μm; Fig. 5Aii).
Figure 5.
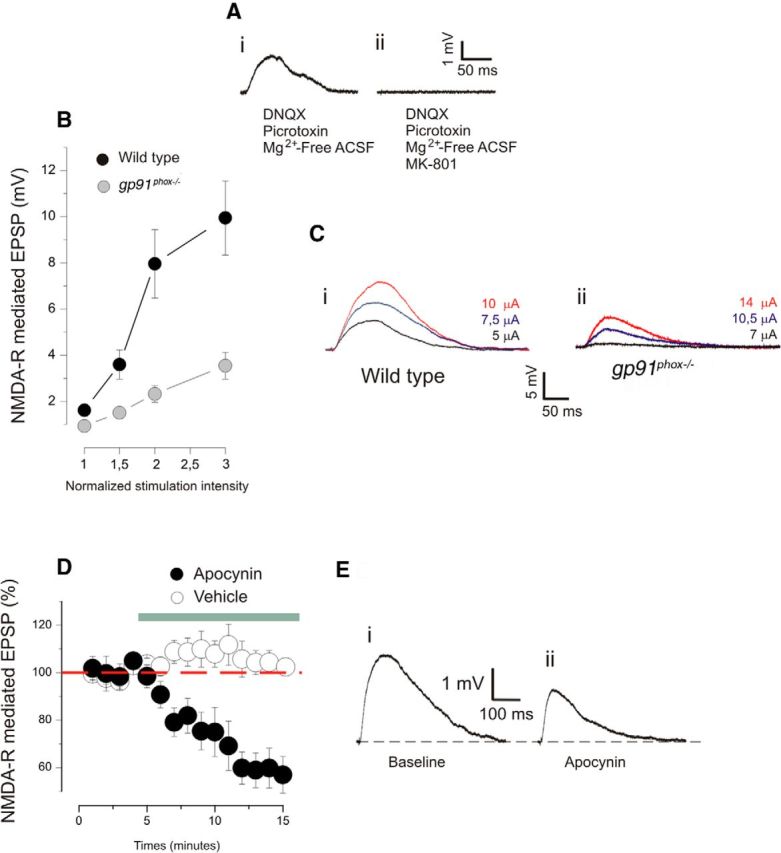
NMDA receptor-mediated postsynaptic responses are reduced by inhibition of NOX2. A, Examples of an NMDAR-EPSP trace before (i) and after (ii) the application of the NMDAR blocker MK-801. B, Magnitude of NMDAR-EPSPs recorded from slices obtained from wild-type and gp91phox−/− animals. The stimulation intensities are normalized with respect to the intensity value evocating the minimal response. Plotted data are average ± SEM. C, Examples of an NMDAR-EPSP recorded in slices derived from wild-type (i) and gp91phox−/− (ii) mice. In gp91phox−/− animals; the amplitude of the NMDAR-EPSPs is significantly reduced (wild-type: 16 slices, 6 animals; gp91phox−/−: 13 cells, 5 animals; p < 0.0001 on a two-way ANOVA), and this effect was more pronounced with higher intensities of stimulation (p < 0.001 on a two-way ANOVA). D, Magnitude of NMDAR-EPSPs before and after the application of apocynin or vehicle in wild-type animals. Plotted data are average ± SEM. E, Examples of NMDAR-EPSPs traces showing the amplitude before (i) and after (ii) the application of apocynin. With the presence of apocynin, the NMDAR-EPSPs are significantly reduced (7 slices, 4 animals; p < 0.01 on a paired t test), whereas the application of vehicle produced no significant change (7 slices, 4 animals; p > 0.1 on a paired t test).
We first tested whether slices obtained from gp91phox−/− animals have reduced NMDAR responses. For this purpose, we recorded intensity-response curves comparing gp91phox−/− and wild-type mice (P21–P24). In gp91phox−/− animals, the amplitude of the NMDAR-EPSPs was significantly lower (Fig. 5B,C; wild-type: 16 slices, 6 animals; gp91phox−/−: 13 cells, 5 animals; p < 0.0001 on a two-way ANOVA), and this effect was more pronounced when the intensity of stimulation was increased (Fig. 5B,C; p < 0.001 on a two-way ANOVA).
Subsequently, we used a group of wild-type animals to investigate the effect of apocynin on the NMDAR-EPSPs. After observing a stable baseline for 5 min, the apocynin was applied in the bath for a 10 min period. The administration of the drug caused a significant reduction of the NMDAR-EPSPs (Fig. 5D,E; 7 slices, 4 animals; p < 0.01 on a paired t test). On the other hand, no significant change was observed in slices treated with vehicle only (Fig. 5D; 7 slices, 4 animals; p > 0.1 on a paired t test). These results demonstrated that Nox2-derived ROS facilitates the activity of NMDAR. These observations, together with the results obtained from DHE experiments, suggest that Nox2 and NMDAR may interact in a process of mutual reinforcement.
Recovery of Nox2-dependent LTD through hydrogen peroxide application
Our findings showed that both LTP and LTD are absent in gp91phox−/− animals, whereas only LTP can be successfully blocked by apocynin. These findings suggest that LTP requires a strong activation of Nox2 during the induction, whereas LTD needs a basal and moderate quantity of ROS accumulation. To test this hypothesis, we tested whether the administration of exogenous ROS in the bath would allow the recovery of LTD in slices obtained from gp91phox−/− mice.
Nox2 produces superoxide, which is instantly converted mainly into hydrogen peroxide (H2O2). Thus, we used a group of gp91phox−/− animals to perform a set of experiments in which H2O2 was added into the bath solution 5 min before the induction of LTD. With the presence of H2O2, the pairing protocol caused a significant reduction of the postsynaptic response (Fig. 6A; 8 slices, 5 animals; p < 0.01 on a paired t test), and this value was statistically different compared with that obtained from untreated slices (p < 0.05 on a t test). Figure 6B summarizes these findings showing the recovery of LTD obtained through H2O2 application. These results pointed out that superoxide production via NOX2 is likely to generate the basal levels of ROS, which are required for the induction of LTD.
Figure 6.
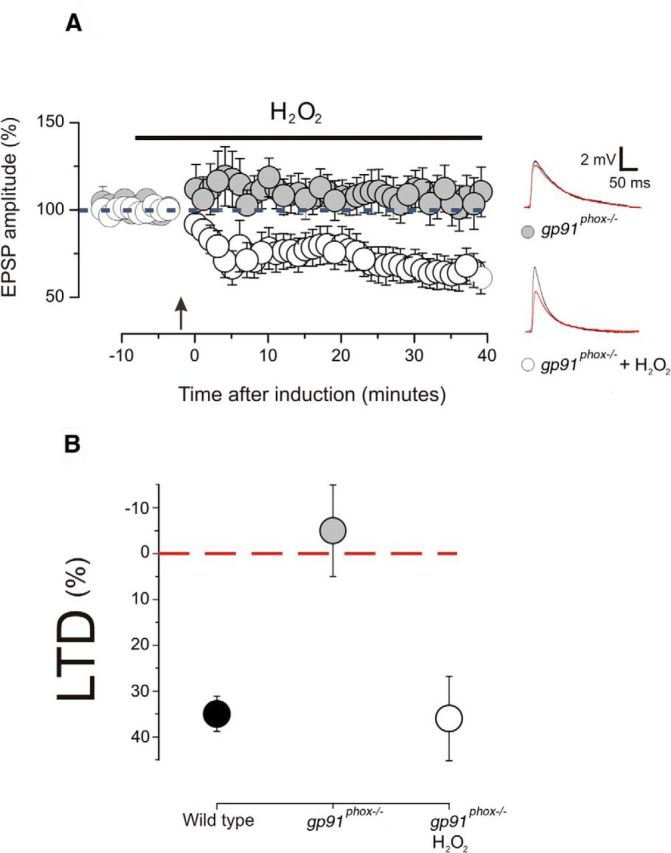
Bath application of H2O2 recovers the LTD in gp91phox−/− animals. A, Magnitude of EPSPs before and after the induction of LTD in gp91phox−/− animals with and without H2O2. LTD is absent in gp91phox−/− animals (10 slices; 7 animals; p > 0.1 on a paired t test), but it was successfully induced in the presence of H2O2 (8 slices, 5 animals; p < 0.01 on a paired t test). Examples of EPSP traces showing the amplitude before (black line) and after (red line) the induction. Plotted data are average ± SEM. B, The graph summarizes the effects of genetic suppression of NOX2 on LTD during and the recovery obtained through H2O2 application. The LTD values derived from wild-type and gp91phox−/− slices are not statistically different (p > 0.1 on a one-way ANOVA with Newman–Keuls multiple-comparison test), but they are different from the values obtained from gp91phox−/− slices treated with H2O2 (p < 0.01 on a one-way ANOVA with Newman–Keuls multiple-comparison test for both comparison). Plotted data are average ± SEM.
NOX2-dependent synaptic plasticity and postnatal age
In the cerebral cortex, ROS production decreases throughout postnatal development and seems to be involved in the maturation of cortical neurons (Tsatmali et al., 2006). ROS potentiate the neuronal capability to develop Ca2+ signaling, and this effect becomes markedly reduced in adult cortical neurons (Yermolaieva et al., 2000). Based on these observations, we evaluated the possibility that ROS production acts on cortical plasticity following a developmental course. To investigate this possible role of NOX2, we tested LTP and LTD at two age periods (P9–P12 and P60–P65) comparing gp91phox−/− mice with their wild-type littermates.
We first used the paired conditioning protocols to assess changes in synaptic plasticity occurring at 9–12 postnatal days. In both wild-type and gp91phox−/− mice, the pairing protocol induced a consistent LTP (Fig. 7A,E; wild-type: 13 slices, 7 animals; gp91phox−/−: 10 slices, 8 animals; p < 0.01 on paired t test in both cases). However, the magnitude of potentiation was not significantly different in the two groups (p > 0.1 on a paired t test). LTD induction caused a significant reduction of the EPSPs in wild-type mice (Fig. 7B,F; 13 slices, 9 animals; p < 0.001 on a paired t test), but not in the gp91phox−/− (Fig. 7B,F; 15 slices, 11 animals; Fig. 6B,F; p > 0.1 on a paired t test).
Figure 7.

LTP and LTD are impaired in mutant mice gp91phox−/− throughout postnatal cortical development. A, Magnitude of postsynaptic responses before and after the induction of LTP in young animals (P9–P12). Arrow indicates the time of induction. Slices obtained from gp91phox−/− animals are compared with slices obtained from wild-type. A significant LTP was obtained in slices obtained from wild-type animals (13 slices; 7 animals; p < 0.001 on a paired t test) and in slices derived from gp91phox−/− animals (10 slices; 8 animals; p < 0.001 on a paired t test), and these values are not statistically different (p > 0.1 on a t test). Examples of EPSP traces showed the amplitude before (black line) and after (red line) the induction. Plotted data are average ± SEM. B, Magnitude of EPSPs before and after the induction of LTD in young animals (P9–P12). Arrow indicates the time of induction. Slices obtained from gp91phox−/− animals are compared with slices obtained from controls. LTD was successfully induced in wild-type slices (13 slices; 9 animals; p < 0.001 on a paired t test). LTD could not be induced in slices derived from gp91phox−/− animals (15 slices; 11 animals; p > 0.1 on a paired t test). Examples of EPSP traces showed the amplitude before (black line) and after (red line) the induction. Plotted data are average ± SEM. C, Magnitude of excitatory postsynaptic responses before and after the induction of LTP in adult animals (P61–P65). Arrow indicates the time of induction. Slices obtained from gp91phox−/− animals are compared with slices obtained from wild-type. In wild-type slices, a significant LTP was observed (15 slices; 14 animals; p < 0.01 on a paired t test). LTP could not be induced in slices derived from gp91phox−/− animals (9 slices; 8 animals; p > 0.1 on a paired t test). Examples of EPSP traces showed the amplitude before (black) and after (red line) the induction. D, Magnitude of excitatory postsynaptic responses before and after the induction of LTD in adult animals (P61–P65). Arrow indicates the time of induction. Slices obtained from gp91phox−/− animals are compared with slices obtained from wild-type. In wild-type slices, a consistent LTD was reported (10 slices; 9 animals; p < 0.001 on a paired t test). Slices derived from gp91phox−/− animals showed no LTD (9 slices; 8 animals; p > 0.1 on a paired t test). Examples of EPSP traces showed the amplitude before (black line) and after (red line) the induction. Plotted data are average ± SEM. E, Summary of the effects of genetic suppression of NOX2 on LTP during postnatal cortical development. Slices obtained from gp91phox−/− animals are compared with slices obtained from wild-type at different ages. In both groups, the LTP decreases with age (p < 0.01 for the age comparison of a two-way ANOVA for both groups). LTP values of wild-type and gp91phox−/− are statistically different (p < 0.01 for the genotype comparison of a two-way ANOVA). Plotted data are average ± SEM. F, The graph summarizes the effects of genetic suppression of NOX2 on LTD during postnatal cortical development. Slices obtained from gp91phox−/− animals are compared with slices obtained from wild-type at different ages. The LTD does not depend on age in both groups (p > 0.1 for the age comparison of a two-way ANOVA). LTD values of wild-type and gp91phox−/− are statistically different (p < 0.001 for the genotype comparison on a two-way ANOVA). Plotted data are average ± SEM.
We next evaluated the expression of LTP and LTD in adult animals. LTP was consistently produced in slices derived from wild-type animals (Fig. 7C,E; 15 slices, 14 animals; p < 0.05 on a paired t test). The LTP magnitude (∼30%) was significantly reduced compared with P9-P12 and P21-P24 animals (Fig. 6E; p < 0.05 on a t test for both comparisons). This suggests that LTP in layer 2/3 follows a developmental decline. In gp91phox−/− mice, paired conditioning caused no synaptic potentiation (Fig. 7C; 9 slices, 8 animals; p > 0.1 on a paired t test). This resembles the effects previously observed at early ages. When LTD was tested, wild-type animals showed a significant decrease in the EPSP amplitude (Fig. 7D; 10 slices, 9 animals; p < 0.001 on a paired t test). On the other hand, no LTD was found in slices from gp91phox−/− animals (Fig. 7D; 9 slices, 8 animals; p > 0.1 on a paired t test). Figure 7E, F summarizes the involvement of NOX2 in LTP and LTD in the three age groups observed. Together, these results confirmed that genetic ablation of NOX2 impairs LTD throughout the period of cortical development. On the other hand, gp91phox−/− mice showed prevented LTP by P21–P24, suggesting the permanence of a developmental decline of synaptic potentiation.
Discussion
The present study demonstrated that ROS production via NOX2 is fundamental for LTP and LTD in the primary visual cortex (V1). We first observed that layer 2/3 neurons of gp91phox−/− animals have lower production of ROS and that stimulation of NMDAR results in the activation of the NOX2 complex. We next found that NOX2 activity is critical for LTP and LTD, suggesting two possible mechanisms. First, we showed that NOX2 facilitates the activation of NMADR, and this process is likely to be fundamental for LTP. Second, we showed that NOX2 could induce ROS production levels that are necessary for the induction of LTD. Finally, we found that the genetic ablation of NOX2 anticipates and increases the developmental decline of LTP.
The activation of NADPH oxidase in response to NMDA receptor activation and calcium influx has been previously documented in hippocampal neurons and cultured cells (Gunasekar et al., 1995; Reynolds and Hastings, 1995; Bindokas et al., 1996; Kishida et al., 2005; Brennan et al., 2009). We demonstrated that similar mechanisms are also active in cortical neurons.
Given that ROS production through NOX2 is critically enhanced by NMDA application, we aimed to confirm that LTP and LTD in the visual cortex are NMDAR-dependent. LTP induction was abolished when the NMDA receptor activity was blocked by MK-801, and this is consistent with previous reports (Fathollahi and Salami, 2001; Yoshimura et al., 2003; Marchena et al., 2008; Kuo and Dringenberg, 2012). LTD was also reduced in slices treated with MK-801. Interestingly, the blockade of NMDA receptor failed to prevent LTD completely. This suggests that part of the expression of LTD derives from mechanisms that do not depend on the activation of NMDA receptors. Our data suggest that the NMDA receptor is critical for the induction of LTP, although it is not essential for inducing LTD in the visual cortex. These findings are in agreement with a previous study showing that NMDAR antagonists do not block completely the electrically induced LTD (Marchena et al., 2008).
Our results showed that genetic ablation of the gp91phox subunit of NOX2 impairs the expression of LTP and LTD in the visual cortex. This is consistent with previous works, which investigated the role played by ROS production in synaptic plasticity in the hippocampus (Klann, 1998; Klann et al., 1998; Thiels et al., 2000; Kishida et al., 2006). The results presented in this study are the first to directly demonstrate that the activity of this complex is necessary for cortical plasticity as well. Furthermore, our work is the first to report that ROS production is also required for LTD. If joined with previous studies, our findings suggest that NOX2 plays important functions in neuronal intracellular transductions leading to synaptic changes. These functions might be general and might be present in several parts of the nervous system.
Interestingly, we observed that pharmacological inhibition of NOX2 blocked LTP but left LTD unaffected. Thus, we found a discrepancy between data obtained through pharmacological inhibition and data obtained through genetic suppression. This discrepancy could result from the existence of two distinct processes affecting synaptic plasticity: a tonic and a phasic production of ROS. Data obtained from DHE experiments showed that gp91phox−/− animals have reduced basal levels of ROS (Fig. 1; tonic effect) and decreased response to NMDA receptor stimulation (Fig. 2; phasic effect). Both LTP and LTD are probably affected by long periods of NOX2 inactivity, which results in low levels of ROS accumulation (tonic effect). On the other hand, only LTP seems to require the rapid increase of ROS caused by NMDA receptor activation during the paired conditioning. This would be the phasic effect, which could be blocked by pharmacological inhibition. This interpretation is consistent with our data showing that: (1) NMDAR activation is essential to induce LTP, and (2) LTD could be rescued when H2O2 is added in the bath and basal levels of ROS are restored. Another alternative explanation could be the occurrence of a partial inhibition of NOX2 by apocynin. This incomplete inhibition could be insufficient to counteract specific mechanisms inducing LTD.
In addition to demonstrating that the activity of NOX2 is critical to induce LTP and LTD in cortical structures, our study provides interesting findings about a probable mechanism involved. Indeed, we showed that NMDAR postsynaptic responses are reduced when NOX2 is inhibited. This observation pointed out that NOX2-mediated ROS production improves the efficacy of NMDA receptors. Together, our data suggest that the interaction between NMDA receptor and NOX2 plays an important role in activating the intracellular pathways resulting in synaptic changes.
We finally investigated the possibility that the involvement of NOX2 in plasticity might follow an age-dependent course. In wild-type animals, we could induce LTD at all ages, and this is in agreement with previous reports (Yoshimura et al., 2003; Jiang et al., 2007). Neurons from young and adult wild-type mice were capable to undergo LTP, but the magnitude of the potentiation was notably reduced at P60–P65. Two previous works already investigated the age dependence of LTP in layer 2/3 in the visual cortex (Yoshimura et al., 2003; Jiang et al., 2007). However, there is a discrepancy resulting from these studies. Jiang et al. (2007) found similar effects of LTP in young and adult mice, whereas Yoshimura et al. (2003) showed no synaptic potentiation after P60. Our results are partially in agreement with both studies and introduce the possibility of a clearer view about this issue.
The experiments performed with gp91phox−/− animals revealed that genetic suppression of NOX2 impairs LTD at all ages. This suggests that the activity of Nox2 is necessary to ensure LTD mechanisms throughout the entire postnatal development. Interestingly, gp91phox−/− animals showed consistent LTP at P9–P12, whereas no potentiation could be induced by P20. Thus, suppression of NOX2 increases and anticipates the overall decline of LTP. This suggests that Nox2 could be important to maintain high the possibility of synaptic potentiation in the immature cortical circuitry.
High levels of ROS are present in brain slices of early postnatal cortex and begin to decrease by P20 (Tsatmali et al., 2005, 2006). ROS production has been proposed to be involved in neuronal developmental plasticity (Mattson, 1996; Coyoy et al., 2013). In young cortical brain, paired application of depolarization and oxidation induces long-lasting facilitation of the Ca2+ signaling (Yermolaieva et al., 2000). This facilitation requires ROS production and markedly decreases in adult cortical neurons. Consistently, our results pointed out that inhibition of Nox2 completely abolished the LTP by P21–P24. NOX2 could be a key factor that facilitates synaptic mechanisms of LTP at early stages of cortical development by potentiating the Ca2+ signaling.
In conclusion, our results demonstrated that ROS production through NOX2 is essential for LTP and LTD in the visual cortex. Our data are the first to show a role of NOX2 in plastic changes occurring in the cerebral cortex. We also provided a probable mechanism involved. NOX2 is likely to promote synaptic plasticity by improving the activity of NMDAR. Furthermore, we introduce the possibility of a differential role of NOX2 on LTP and LTD. The induction of LTP seems to require an acute burst of NOX2-mediated ROS production. On the other hand, the activation of intracellular pathways leading to LTD could also be achieved through a long-term accumulation of ROS. The evidence found in the present study could be an important contribution to the study of the physiological processes involved in the modification of synaptic connections in cortical circuitry.
Footnotes
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo Grants 2011/23874-0 and 2012/07522-0. We thank Adilson da Silva Alves for technical support.
The authors declare no competing financial interests.
References
- Bear MF. A synaptic basis for memory storage in the cerebral cortex. Proc Natl Acad Sci U S A. 1996;93:13453–13459. doi: 10.1073/pnas.93.24.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Jordán J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan M, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyoy A, Olguín-Albuerne M, Martínez-Briseño P, Morán J. Role of reactive oxygen species and NADPH-oxidase in the development of rat cerebellum. Neurochem Int. 2013;62:998–1011. doi: 10.1016/j.neuint.2013.03.009. [DOI] [PubMed] [Google Scholar]
- de Marchena J, Roberts AC, Middlebrooks PG, Valakh V, Yashiro K, Wilfley LR, Philpot BD. NMDA receptor antagonists reveal age-dependent differences in the properties of visual cortical plasticity. J Neurophysiol. 2008;100:1936–1948. doi: 10.1152/jn.90290.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fathollahi Y, Salami M. The role of N-methyl-d-aspartate receptors in synaptic plasticity of rat visual cortex in vitro: effect of sensory experience. Neurosci Lett. 2001;306:149–152. doi: 10.1016/S0304-3940(01)01894-8. [DOI] [PubMed] [Google Scholar]
- Feldman DE. Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci. 2009;32:33–55. doi: 10.1146/annurev.neuro.051508.135516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekar PG, Kanthasamy AG, Borowitz JL, Isom GE. NMDA receptor activation produces concurrent generation of nitric oxide and reactive oxygen species: implication for cell death. J Neurochem. 1995;65:2016–2021. doi: 10.1046/j.1471-4159.1995.65052016.x. [DOI] [PubMed] [Google Scholar]
- Infanger DW, Sharma RV, Davisson RL. NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal. 2006;8:1583–1596. doi: 10.1089/ars.2006.8.1583. [DOI] [PubMed] [Google Scholar]
- Jiang B, Treviño M, Kirkwood A. Sequential development of long-term potentiation and depression in different layers of the mousevisual cortex. J Neurosci. 2007;27:9648–9652. doi: 10.1523/JNEUROSCI.2655-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Bear MF. Elementary forms of synaptic plasticity in the visual cortex. Biol Res. 1995;28:73–80. [PubMed] [Google Scholar]
- Kishida KT, Klann E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid Redox Signal. 2007;9:233–244. doi: 10.1089/ars.2007.9.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida KT, Pao M, Holland SM, Klann E. NADPH oxidase is required for NMDA receptor-dependent activation of ERK in hippocampal area CA1. J Neurochem. 2005;94:299–306. doi: 10.1111/j.1471-4159.2005.03189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida KT, Hoeffer CA, Hu D, Pao M, Holland SM, Klann E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol Cell Biol. 2006;26:5908–5920. doi: 10.1128/MCB.00269-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann E. Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. J Neurophysiol. 1998;80:452–457. doi: 10.1152/jn.1998.80.1.452. [DOI] [PubMed] [Google Scholar]
- Klann E, Thiels E. Modulation of protein kinases and protein phosphatases by reactive oxygen species: implications for hippocampal synaptic plasticity. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:359–376. doi: 10.1016/S0278-5846(99)00002-0. [DOI] [PubMed] [Google Scholar]
- Klann E, Roberson ED, Knapp LT, Sweatt JD. A role for superoxide in protein kinase C activation and induction of long-term potentiation. J Biol Chem. 1998;273:4516–4522. doi: 10.1074/jbc.273.8.4516. [DOI] [PubMed] [Google Scholar]
- Knapp LT, Klann E. Role of reactive oxygen species in hippocampal long-term potentiation: contributory or inhibitory? J Neurosci Res. 2002;70:1–7. doi: 10.1002/jnr.10371. [DOI] [PubMed] [Google Scholar]
- Kuo MC, Dringenberg HC. Comparison of long-term potentiation (LTP) in the medial (monocular) and lateral (binocular) rat primary visual cortex. Brain Res. 2012;1488:51–59. doi: 10.1016/j.brainres.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Calcium and free radicals: mediators of neurotrophic factor and excitatory transmitter-regulated developmental plasticity and cell death. Perspect Dev Neurobiol. 1996;3:79–91. [PubMed] [Google Scholar]
- Quinn MT, Gauss KA. Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. J Leukoc Biol. 2004;76:760–781. doi: 10.1189/jlb.0404216. [DOI] [PubMed] [Google Scholar]
- Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano F, Kolluri NS, Wientjes FB, Card JP, Klann E. NADPH oxidase immunoreactivity in the mouse brain. Brain Res. 2003;988:193–198. doi: 10.1016/S0006-8993(03)03364-X. [DOI] [PubMed] [Google Scholar]
- Sumimoto H, Ueno N, Yamasaki T, Taura M, Takeya R. Molecular mechanism underlying activation of superoxide-producing NADPH oxidases: roles for their regulatory proteins. Jpn J Infect Dis. 2004;57:S24–S25. [PubMed] [Google Scholar]
- Thiels E, Urban NN, Gonzalez-Burgos GR, Kanterewicz BI, Barrionuevo G, Chu CT, Oury TD, Klann E. Impairment of long-term potentiation and associative memory in mice that overexpress extracellular superoxide dismutase. J Neurosci. 2000;20:7631–7639. doi: 10.1523/JNEUROSCI.20-20-07631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg JT, Trepel C, Stryker MP. Rapid extragranular plasticity in the absence of thalamocortical plasticity in the developing primary visual cortex. Science. 2000;287:2029–2032. doi: 10.1126/science.287.5460.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsatmali M, Walcott EC, Crossin KL. Newborn neurons acquire high levels of reactive oxygen species and increased mitochondrial proteins upon differentiation from progenitors. Brain Res. 2005;1040:137–150. doi: 10.1016/j.brainres.2005.01.087. [DOI] [PubMed] [Google Scholar]
- Tsatmali M, Walcott EC, Makarenkova H, Crossin KL. Reactive oxygen species modulate the differentiation of neurons in clonal cortical cultures. Mol Cell Neurosci. 2006;33:345–357. doi: 10.1016/j.mcn.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yermolaieva O, Brot N, Weissbach H, Heinemann SH, Hoshi T. Reactive oxygen species and nitric oxide mediate plasticity of neuronal calcium signaling. Proc Natl Acad Sci U S A. 2000;97:448–453. doi: 10.1073/pnas.97.1.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura Y, Ohmura T, Komatsu Y. Two forms of synaptic plasticity with distinct dependence on age, experience, and NMDA receptor subtype in rat visual cortex. J Neurosci. 2003;23:6557–6566. doi: 10.1523/JNEUROSCI.23-16-06557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Kalivendi S, Zhang H, Joseph J, Nithipatikom K, Vásquez-Vivar J, Kalyanaraman B. Superoxide reacts with hydroethidine, but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radic Biol Med. 2003;34:1359–1368. doi: 10.1016/S0891-5849(03)00142-4. [DOI] [PubMed] [Google Scholar]