

Abstract
The antimicrobial agent triclosan (TCS) is used in products such as toothpaste and surgicalsoaps and is readily absorbed into oral mucosa and human skin. These and many other tissues contain mast cells, which are involved in numerous physiologies and diseases. Mast cells release chemical mediators through a process termed degranulation, which is inhibited by TCS. Investigation into the underlying mechanisms led to the finding that TCS is a mitochondrial uncoupler at non-cytotoxic, low-micromolar doses in several cell types and live zebrafish. Our aim was to determine the mechanisms underlying TCS disruption of mitochondrial function and of mast cell signaling. We combined super-resolution (fluorescence photoactivation localization) microscopy and multiple fluorescence-based assays to detail triclosan’s effects in living mast cells, fibroblasts, and primary human keratinocytes. TCS disrupts mitochondrial nanostructure, causing mitochondria to undergo fission and to form a toroidal, “donut” shape. TCS increases reactive oxygen species production, decreases mitochondrial membrane potential, and disrupts ER and mitochondrial Ca2+ levels, processes that cause mitochondrial fission. TCS is 60x more potent than the banned uncoupler 2,4-dinitrophenol. TCS inhibits mast cell degranulation by decreasing mitochondrial membrane potential, disrupting microtubule polymerization, and inhibiting mitochondrial translocation, which reduces Ca2+ influx into the cell. Our findings provide mechanisms for both triclosan’s inhibition of mast cell signaling and its universal disruption of mitochondria. These mechanisms provide partial explanations for triclosan’s adverse effects on human reproduction, immunology, and development. This study is the first to utilize super-resolution microscopy in the field of toxicology.
Keywords: triclosan, mast cell, mitochondria, super-resolution microscopy, RBL-2H3, NIH-3T3, keratinocyte, Ca2+, fluorescence photoactivation localization microscopy (FPALM), reactive oxygen species, membrane potential, degranulation, microtubules
Graphical Abstract
Introduction
Triclosan (TCS) is a broad spectrum antimicrobial (Lyman and Furia 1968) recently banned from some soap products following the FDA’s 2016 risk assessment (Kux 2016). However, TCS remains in many high-production consumer products (e.g., toothpaste, hand sanitizer) and in hospital soaps (Cooney 2010).
TCS is readily absorbed through the skin (Moss et al. 2000), oral mucosa (Gilbert et al. 1987; Lin 2000), and gastrointestinaltract (Sandborgh-Englund et al. 2006). Consumer products contain high concentrations of TCS, up to ~10 mM, and 3–25% of applied TCS is rapidly absorbed into the oral mucosa or skin (Gilbert 1987; Lin 2000; Moss et al. 2000), where it remains, largely in the parent form, for at least 24 hours (Manevski et al. 2015; Moss et al. 2000). Approximately 0.4 to 64 nmol of TCS per milligram of human skin tissue protein is found in human skin following one hour exposure to a typical consumer product (Manevski et al. 2015; Moss et al. 2000; Weatherly and Gosse 2017). In the current study, exposure of cells to micromolar TCS levels results in approximately 0.8–2.8 nmol TCS/mg cell protein (Weatherly and Gosse 2017), on the low end of levels in exposed human tissue and similar to the 2.5 nmol TCS/mg protein dosing which caused mitochondrial toxicity in isolated rat mitochondria (Newton et al. 2005). In addition to high TCS levels within human tissues, high TCS concentrations have been found in human body fluids: 1 μM in blood following ingestion of ~1 tablespoon of TCS mouthwash (Sandborgh-Englund et al. 2006) and up to 13.1 μM in urine (Calafat et al. 2008). Thus, in this manuscript, we utilize TCS doses equivalent to levels found in human tissues directly exposed to TCS-containing consumer products (Weatherly and Gosse 2017).
While TCS is used in hospitals and in consumer products due to its positive clinical effects as an antibacterial (Daoud et al. 2014) and anti-gingival (Rover and Leu-Wai-See 2014) agent and also potentially could be used as a topical treatment for atopic dermatitis (Tan et al. 2010), recent epidemiological reports have linked TCS to a variety of human health problems. TCS exhibits detrimental effects on human thyroid function and reproduction (Koeppe et al. 2013; Velez et al. 2015; Wang and Tian 2015; Wang et al. 2015). TCS is also associated with increased occurrence of allergies and asthma in humans (Spanier et al. 2014). In boys, prenatal TCS exposure is associated with decreased head circumference (Philippat et al. 2014). However, the molecular mechanisms underlying these human effects are not yet known.
Resulting from efforts to determine the molecular effects of TCS on mammalian cells, our lab previously discovered both that TCS inhibits the functions of mast cells (Palmer et al. 2012; Weatherly et al. 2013), which are ubiquitous immune cells, and that TCS is a mitochondrial toxicant (Shim et al. 2016; Weatherly et al. 2016). However, the underlying mechanisms and the ultrastructural changes underlying these effects remain unknown, and this current study aimed to investigate these processes.
Recently, we and others (Ajao et al. 2015; Raftery et al. 2017) have reported that, at low micromolar doses, TCS is a proton ionophore mitochondrial uncoupler (which inhibits ATP production and increases oxygen consumption rate) in multiple types of living cells (Weatherly et al. 2016), including primary human keratinocytes (a major cell type in the outer layer of skin), and in zebrafish (Shim et al. 2016). The doses that cause this mitochondrial toxicity are non-cytotoxic (Palmer et al. 2012; Weatherly et al. 2016) and also non-lethal to zebrafish embryos (Shim et al. 2016). TCS possesses an ionizable proton as does other small-molecule mitochondrial uncouplers such as carbonyl cyanide 3-chlorophenylhydrazone (CCCP), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), and 2,4-dinitrophenol (DNP).
Along with inhibition of ATP production and enhancement of respiration, mitochondrial uncouplers also can affect mitochondrial (Rottenberg and Wu 1998) and plasma (Mohr and Fewtrell 1987b) membrane potential, Ca2+ levels (Mohr and Fewtrell 1987b), mitochondrial translocation (Theoharides et al. 2013), and reactive oxygen species (ROS) production (Okuda et al. 1992; Przygodzki et al. 2005). Donut-shaped or toroid mitochondria are observed in multiple cell types after exposure to CCCP and FCCP (Ding et al. 2012; Giedt et al. 2012; Liu and Hajnoczky 2011). Uncouplers also can disrupt the mitochondrial fusion/fission balance, leading to fragmentation (Giedt et al. 2012; Griparic et al. 2007). However, TCS effects on mitochondrial ultrastructure, translocation, and Ca2+ dynamics are not yet known.
Another cell type in which we documented TCS-induced mitochondrial uncoupling is mast cells (Weatherly et al. 2016): immune effector cells involved in neurological, immunological, and cancer-related functions (Metcalfe et al. 1997; Silver and Curley 2013). We have previously shown that TCS inhibits a variety of functions of mast cells, both in rat (Palmer et al. 2012; Weatherly et al. 2013) and human models (Weatherly et al. 2016). Mast cells are found in nearly all human tissues, including the oral mucosa and skin (Walsh et al. 1995; Walsh 2003), making them prime targets for TCS exposure. TCS inhibits multiple mast cell functions, including degranulation (Palmer et al. 2012; Weatherly et al. 2016), which is the release of chemical mediators (e.g., histamine, serotonin, β-hexosaminidase) from the cell. Degranulation is initiated when antigen (Ag) binds to and crosslinks IgE-bound FcεRI receptors, leading to phosphorylation of kinases including Lyn and PLCγ (Kinet 1999). Inositol 1,4,5-triphosphate (IP3) is generated by PLCγ and binds to its receptor on the endoplasmic reticulum (ER) membrane, instigating a flood of Ca2+ out of the ER (Berridge 1993). Depletion of ER Ca2+ stores causes the ER Ca2+ sensor STIM-1 to bind to the Orai1 subunit of the Ca2+ release-activated Ca2+ (CRAC) channel in the plasma membrane (Vig et al. 2006), resulting in an influx of Ca2+ across the plasma membrane (Hogan et al. 2010) (store-operated calcium entry), SOCE (Putney 1986). Influx of Ca2+ across the plasma membrane permits reuptake of Ca2+ into the ER through sarco/endoplasmic Ca2+-ATPase (SERCA) pumps (Ma and Beaven 2011). In mast cells, mitochondria support degranulation by acting as Ca2+ buffers, taking up Ca2+ from both the ER and the cytosol (Furuno et al. 2015; Takekawa et al. 2012). Cytosolic Ca2+, along with ROS production (Swindle et al. 2004), activates protein kinase C (PKC), a key event leading to degranulation (Ozawa et al. 1993). Granules are transported to the plasma membrane via microtubules (Guo et al. 1998), for degranulation (Smith et al. 2003). Mitochondria also rely on microtubules for transport (Iqbal and Hood 2014), and degranulation requires translocation of mitochondria from around the nucleus to exocytotic sites on the plasma membrane (Zhang et al. 2011). Together, all of these processes lead to degranulation. However, TCS effects on ER/mitochondrial/cytosolic Ca2+ levels, mitochondrial translocation, ROS, and microtubules are not yet known, and the mechanism(s) underlying TCS inhibition of degranulation are not yet known.
Numerous critical biological processes and structures occur at lengths that conventional microscopy techniques cannot resolve. In conventional fluorescence microscopy, large numbers of fluorescent molecules are visible at once, and diffraction blurs molecules closer than 200–250 nm apart, obscuring fine details. Fluorescence photoactivation localization microscopy (FPALM) is a super-resolution microscopy technique that circumvents the diffraction limit, allowing for ~10X improved spatial res olution (Hess et al. 2006). FPALM uses photoactivatable fluorescent probes, which are initially non-fluorescent (inactive). A low-intensity activation laser converts a small subset of inactive fluorophores into active ones, which are then imaged, localized to precisely determine their positions, and then photobleached, turning them off permanently. The remaining inactive fluorophores undergo the process of activation, imaging, localization, and photobleaching. This process is repeated until enough molecules have been localized to reveal a super-resolved image of the sample.
In the first usage of super-resolution microscopy in the field of toxicology, we have utilized FPALM’s 10X higher resolution to demonstrate that TCS disrupts mitochondrial nanostructure in multiple cell types including mast cells and primary human keratinocytes. We also show that TCS disrupts multiple other cellular functions including ROS production, Ca2+ mobilization, membrane potential, mitochondrial translocation, and microtubule formation. Together, these results illustrate a mechanism by which triclosan inhibits mast cell degranulation and causes universal dysfunction of mitochondria.
Methods
Chemicals and reagents
TCS (99%; Sigma-Aldrich) and CCCP (VWR) were dissolved into aqueous buffers to deliver concentrations (5–20 μM TCS) previously proven to be mitotoxic and inhibitory of mast cells, while not cytotoxic, in Weatherly et al. 2013 and 2016 and in Palmer et al. 2012. DNP (Sigma-Aldrich) was freshly prepared daily, in cell culture water with 3.7 g/L sodium bicarbonate buffer (VWR), pH 7.4, followed by filtration (0.22 μm). DNP concentration was determined via UV-Vis spectrophotometry (extinction coefficient 12,100 M/L/cm at 400 nm (Peralta et al. 2010)). The components of Tyrodes buffer (which contains glucose) are those of Weatherly et al., 2013.
Cell culture
Primary human keratinocytes (LifeLine Cell Technology), NIH-3T3 mouse fibroblasts, and RBL-2H3 (“RBL”) mast cells were cultured as in Weatherly et al., 2016. Phenol red-free media (Gibco) contains no glutamine, phenol red, HEPES, or sodium pyruvate; fetal bovine serum, gentamicin, and L-glutamine were added, and complete media was filtered (0.22 μm).
Our previous experiments showing that TCS is a mitochondrial uncoupler (Weatherly et al. 2016) were performed in the absence of glucose. Glucose was omitted from those experiments because, in the presence of glucose, cells (both immortalized (Crabtree 1928; Ibsen 1961) and primary (Wang et al. 1976)) rely on glycolysis to generate ATP and, therefore, bypass mitochondrial oxidative phosphorylation (the “Crabtree effect”). Thus, in order to probe effects of mitochondrial toxicants on oxidative phosphorylation (Marroquin et al. 2007), cells must be forced to undergo oxidative phosphorylation; this effect is accomplished experimentally by omitting glucose, which is replaced by glutamine plus galactose (“galactose media”). Under these conditions, virtually all cellular ATP is generated by oxidative phosphorylation fueled by catabolism of glutamine (an appropriate fuel source because it is abundant in plasma (Van Slyke et al. 1943)). The galactose (which is catabolized much more slowly than glutamine (Lowry and Passonneau 1969)) is included in the media in order to supply the pentose phosphate pathway, to support biosynthesis and detoxification (Reitzer et al. 1979).
Preparation of Dendra2-TOM20 construct to image the mitochondrial outer membrane
The Dendra2-TOM20 (translocase of the outer membrane) construct was used to image the outer mitochondrial membrane. Into a p-Dendra2-N plasmid (Clontech), a TOM20 localizer was inserted into the multiple cloning site upstream from the Dendra2 open reading frame to create a TOM20-Dendra2-N plasmid. A forward primer containing a new HindIII site and a reverse primer containing a new Age1 site were developed and used to clone the TOM20 localizer, via PCR, from 15 day old C57BL/6 mouse hind limb DNA. The PCR product underwent a double-digest with HindIII and Age1, followed by ligation of the purified PCR product into the Dendra2 plasmid and by sequencing for confirmation.
Fluorescence photoactivation localization microscopy (FPALM) imaging and processing
NIH-3T3 cells were transfected with the Dendra2-TOM20 construct via Lipofectamine 3000 (ThermoFisher Scientific) as per manufacturer’s instructions, and RBLs and keratinocytes were transfected via Amaxa Nucleofection. The next day, cells were exposed to TCS in galactose media-bovine serum albumin (BSA) or Tyrodes-BSA (BT; recipe in Hutchinson et al. 2011).
Imaging began immediately after exposure and continued for up to one hour. Imaging was performed on an Olympus IX71 microscope, with a 561 nm laser (Sapphire, Coherent) used to activate and excite Dendra2 fluorescence. For details, see Supplement. Data from the EMCCD were analyzed using custom-built MATLAB (MathWorks Inc.) scripts for single color localization (Hess et al. 2006; Smith et al. 2010).
FPALM mitochondria analysis
To quantify the presence or absence of mitochondrial donuts, localized images were analyzed using a second custom-built MATLAB script. For detail, see Supplement.
Image analysis was also performed using NIH Fiji ImageJ software mitochondria calculator plug-in (Dagda et al. 2009). The images were extracted to binary (mitochondria-specific fluorescence was shown as black pixels), thresholded, and smoothed to resolve individual mitochondria. ImageJ’s “analyze particles” option was used and uniformly thresholded for pixel size to decrease background. Measures of mitochondrial fission (mitochondrial major axis, perimeter, and elongation) were 1.) graphed over 1 hr with 20 minute binning (Fig. 3A–I) and 2.) cells exposed between 30 and 60 minutes were combined with normalization to control (Fig. 3J).
Fig. 3.

TCS effects on mitochondrial perimeter, major axis, and elongation measurements in primary human keratinocytes (A-C, J), RBL cells (D-F, J), and NIH-3T3 cells (G-I, J). Responses were measured in RBL cells exposed to ± 15μM TCS in galactose media-BSA, keratinocytes exposed to ± 10 μM TCS in galactose media-BSA, and NIH-3T3 cells exposed to ± 10 μM TCS in glucose Tyrodes -BSA buffer. In (A-I), data were binned in 20 minute intervals from images which had been collected at exposure times from 0–20 mins, 21–40 mins, and 41–60 mins--plotted as three data points per treatment. In (J), data from cells imaged between 30 and 60 mins after TCS exposure were combined for each cell type. Each cell type within each parameter was first normalized to the average of the corresponding control of that same cell type. These normalized values were then combined into a single control for each parameter measured. Values presented are means ± SEM; each binned interval includes data from 4 to 15 (A-I) or 10 to 22 (J) cells combined from at least 2 independent days of imaging. In (A-I), statistically significant results, as compared to a slope of zero, are represented by ** p < 0.01, as determined by linear regression. In (J), statistically significant results, as compared to control, are represented by ** p < 0.01 and *** p < 1.1, as determined by one-way ANOVA followed by Tukey’s post-hoc test.
Reactive Oxygen Species Detection Assay
Reactive oxygen species (ROS) levels were assayed via instructions of the OxiSelect Intracellular ROS Assay Kit (Cell Biolabs, Inc.). Briefly, 50,000 cells were plated per well of a black, clear bottom 96-well plate in phenol red-free media or keratinocyte media and incubated overnight at 37°C/5% CO2. RBL cells were exposed to 0.1 μg/mL monoclonal anti-DNP mouse immunoglobulin E (IgE; Sigma-Aldrich) in media for 1 hr at 37°C/5% CO2. Cells received 100 μL of either ROS dye (50 μM final concentration) or methanol vehicle (0.25% final concentration) in BT and were incubated for 30 mins at 37°C/5% CO2. Following dye incubation, cells were washed with BT, then exposed to ± TCS or CCCP and ± DNP-BSA antigen (Ag) in BT. Using a microplate reader (Synergy 2, Biotek), fluorescence (485 ± 10 nm excitation, 528 ± 10 nm emission) was monitored immediately after exposure, at 37°C. With keratinocytes, the same assay was conducted, except without IgE and Ag exposure. Data were analyzed first by averaging the replicates for each chemical concentration, and then the baseline value for 0 μM treatment without Ag was subtracted from all data points. Next, these values were normalized to the last time point for 0 μM TCS without Ag. The final values were plotted as a time series. Area under the curve (AUC) for each individual experiment was determined with Graphpad Prism, averaged, and normalized to 0 μM TCS (no Ag).
Mitochondrial Ca2+ assay
In order to measure mitochondrial Ca2+ levels, RBL cells were transfected with pCMV CEPIA2mt (a gift from Masamitsu Iino; Addgene plasmid #58218) (Suzuki et al. 2014) via electroporation using Amaxa cell line Nucleofector kit T. After transfection, 100,000 cells/well were plated in phenolred-free media in 8-well plates (ibidi USA inc.) and incubated at 37°C/5% CO2 overnight. The following day, cells were sensitized with 0.1 μg/mL IgE for 1 hr in phenolred-free media (100 μL/well). Next, cells were washed with BT and imaged in 200 μL BT for baseline, followed by addition of 200 μL of 2X Ag ± 2X TCS in BT. Images were taken with a confocal microscope, at 37°C via ibidi heating system(Olympus FV-1000, with an Olympus IX-81 inverted microscope, an oil-immersion 100X objective [NA 1.4], and a 30 milliwatt multi-argon laser [488 nm excitation, emission 505–605 nm bandpass filter]). In ImageJ, background was subtracted before F/F0 normalization of fluorescence values: baseline readings were normalized to the first baseline reading (time point = −6), and Ag ± TCS values were normalized to the first image immediately following Ag ± TCS addition (time point = 0). Values were plotted as percent increase over baseline. Number of oscillations was determined by the following guidelines: an oscillation must have at least a 15% increase on both sides of the low point, and the low and high point must be separated by at least 4 data points (2 mins).
Endoplasmic Reticulum Ca2+ assay
To measure ER Ca2+ levels, RBL cells were transfected with ER-GCaMP6–210 (Juan-Sanz et al. 2017), via electroporation using Amaxa cell line Nucleofector kit T. In phenol red-free media, 100,000 cells/well were plated into a black, clear bottom 96-well plate (Greiner Bio-One) after transfection and incubated at 37°C/5% CO2 overnight. The following day, cells were sensitized with 0.1 μg/mL IgE for 1 hr in phenol red-free media. Next, cells were washed with BT, and exposed to TCS ± Ag in Ca2+-free BT. Fluorescence was monitored with 485 ± 10 nm excitation, 528 ± 10 nm emission. Background was subtracted out by averaging all values from the non-transfected cells and using that average for the y-axis baseline value.
Mitochondrial Membrane Potential Assay
Mitochondrial membrane potential (MMP) was assayed with MitoTracker Red FM (ThermoFisher Scientific). In galactose media, 30,000 RBL cells/well were plated in a black, clear bottom 96-well plate and incubated 1 hr at 37°C/5% CO2. MitoTracker Red FM dye was diluted to 8 nM (0.16% DMSO vehicle) or 1.25 nM (0.02% DMSO) in both galactose media-BSA and a 30 μM TCS galactose media-BSA solution; these two solutions were used to prepare TCS dilutions. CCCP solutions were also made in MitoTracker dye-containing galactose media-BSA. Cells were exposed to TCS or CCCP with 8 nM dye for 1 hr at 37°C. In the microplate reader, fluorescence was monitored at 530 ± 25 nm and 645 ± 15 nm emission. For further analysis details, see Supplement.
ATP production and cytotoxicity assays
ATP production was measured in BT as described in Weatherly et al. 2016. For ATP experiments with DNP treatment, DNP was prepared in galactose media-BSA, and then cells were exposed to DNP for 1 hr at 37°C. DNP interfered with fluorescence of the cytotoxicity assay reagent provided by the Toxglo manufacturer, so the trypan blue assay was used to assess DNP cytotoxicity.
Cytosolic Ca2+ assay
To measure cytosolic Ca2+ levels in mast cells, RBL cells were transfected with pGP-CMV-GCaMP6f (a gift from Douglas Kim; Addgene #40755 (Chen et al. 2013)) via electroporation using Amaxa cell line Nucleofector kit T (Cohen et al. 2015). In phenol red-free media, 100,000 cells/well were plated in a black, clear bottom 96-well plate after transfection and incubated at 37°C/5% CO2 overnight. The following day, cells were sensitized with 0.1 μg/mL IgE for 1 hr in phenolred-free media. Next, cells were washed and exposed to TCS ± Ag in BT. Fluorescence was monitored with 485 ± 10 nm excitation, 528 ± 10 nm emission. A lactate dehydrogenase assay for cytotoxicity was conducted immediately after the 1 hr read, following the protocolin (Hutchinson et al. 2011). To determine AUC, the y-axis baseline was set to the average of the no-Ag wells in order to measure fluorescence due to Ag and TCS exposure only.
Confocal imaging of mitochondrial translocation
RBL cells (52,500/well) were plated in phenolred-free media in μ-Slide 8-well glass bottom plates (ibidi USA inc.) overnight at 37°C/5% CO2. The following day, cells were exposed to 0.1 μg/mL IgE in galactose media for 1 hr. Cells were washed, then incubated with 8nM MitoTracker Red FM and 100 nM Hoechst 33342. Next, cells were washed, then exposed to galactose media-BSA ± Ag and ±10 μM TCS. After exposure, an aliquot of supernatant was taken to assay for degranulation as described in Weatherly et al., 2013. Next, cells were washed, then exposed to galactose media-BSA or BT for imaging. After imaging, another aliquot of supernatant was taken to assay for β-hexosaminidase release as a marker for cell death.
Mitochondrial translocation was imaged on the confocal microscope described in the “Mitochondrial Ca2+ assay” sub-section, with the following changes: Hoechst fluorescence (excitation 405 nm, emission 430–470 nm) and MitoTracker Red fluorescence (excitation 543 nm, emission 560–660 nm) were measured. Image acquisition speed was 10 μs/pixel. Cells were analyzed using a Fiji ImageJ software mitochondria plug-in (Dagda et al. 2009), to determine total fluorescence of Mitotracker Red around the nucleus. This number was divided by total Mitotracker Red FM fluorescence within that entire cell, subtracted from 1, and multiplied by 100%, to calculate the percentage of mitochondria translocated away from nucleus.
Confocal imaging of microtubules
RBL mast cells were transfected via Amaxa Nucleofection (as described in “FPALM imaging and processing”) with pIRESneo-EGFP-a lpha Tubulin, a gift from Patricia Wadsworth (Addgene plasmid # 12298) (Rusan et al. 2001). To ibidi μ-Slide 8 well glass bottom plates were added 100,000 transfected RBL cells/well in phenol red-free media at 37°C/5% CO2. The following day, cells were exposed to 0.1 μg/mL IgE in phenol red-free media for 1 hr. Cells were washed, then exposed to ± Ag and ± 10 μM TCS in BT for 15 minutes at 37°C/5% CO2. Images were taken via Olympus FV-1000 laser scanning confocal, using the same settings as in “Mitochondrial Ca2+ assay.”
Tubulin Polymerization Assay
In vitro tubulin polymerization assays were performed according to the manufacturer’s instructions (Cytoskeleton, Inc).
Trypan Blue Exclusion Cytotoxicity Assay
Trypan blue assays were conducted as in Hutchinson et al. 2011. Cells were plated (2–3 million/well) in a 6-well plate and incubated for 1 hr at 37°C, 5% CO2. Next, 1 mL of each 2X DNP solution (in galactose media-BSA) was added to the 1 mL galactose media in each well and exposed for 1 h at 37°C, 5% CO2.
Statistical analyses
All analyses were done in Graphpad Prism. In experiments where triplicates were used (as in data from figures 4, 7, 8, and 12) data were scanned using a Q-test with a 95% confidence interval and, if present, outliers were excluded, leaving duplicates that were averaged. In case of samples with duplicates (as in data from figures 6, 9, and 11 d), raw data were always within 5% of each other. The biological replicates from at least three different experiments were averaged and used to determine the SEM across the three experiments. Significance levels for most experiments were assessed via one-way ANOVA with Tukey’s post-hoc test. Significance for toroid mitochondrial morphology was determined via a two sample t-test. Mitochondrial fission time series analysis was conducted via linear regression, and significance was determined compared to a slope of zero. Significance of mitochondrial Ca2+ “highest percentage increase” and “number of oscillations” was assessed via unpaired one-tailed t-test.
Fig. 4.

Intracellular reactive oxygen species production. ROS response was measured in RBL cells in a plate reader during stimulation with 0.01 μg/mL Ag with 0, 5, or 20 μM TCS (A) or 0, 1, or 4 μM CCCP (C) and in keratinocytes with 0, 5, or 20 μM TCS exposure only (B, D), for 1 hr in Tyrodes-BSA. For (D), mean fluorescence intensity was determined by subtracting out the appropriate controls and normalizing to 0 μM TCS. The area under the curve was obtained from raw fluorescence values corresponding to ROS levels measured over a 1 hr period via Graphpad Prism software. AUC was plotted as a bar graph, normalized to control (no Ag, no TCS). Values presented are means ± SEM of at least three independent experiments; three replicates per treatment per experiment. Statistically significant results of ± Ag within a given toxicant dose are represented by * p < 0.05, ***p < 0.001 (A, C); of Ag + no CCCP vs. Ag + CCCP by ## p < 0.01, ### p < 0.001 (C); and of no-Ag control vs. no-Ag + TCS by †† p < 0.01, ††† p < 0.001 (A, B, D), as determined by one-way ANOVA followed by Tukey’s post-hoc test.
Fig. 7.

Mitochondrial membrane potential of RBL cells. MMP response was measured in RBL cells after exposure to 0, 5, or 10 μM TCS or ± 1 μM CCCP for 1 hr in galactose media-BSA. Mean fluorescence intensity was determined by subtracting out appropriate controls and normalizing to 0 μM Control. Values presented are means ± SEM of at least four independent experiments; three to four replicates per experiment. Statistically significant results, as compared to the appropriate control (0 μM TCS or 0 μM CCCP + DMSO vehicle), are represented by * p < 0.05, ** p < 0.01, as determined by one-way ANOVA followed by Tukey’s post-hoc test.
Fig. 8.

ATP, degranulation, and cytotoxicity responses in RBL cells after TCS exposure in Tyrodes-BSA buffer. RBL cell responses were measured after 60 min total exposure to 0.0004 μg/mL Ag ± TCS in Tyrodes -BSA. TCS concentrations ranged between 0 and 20 μM. The x-axis is a three-segment line to clarify data interpretation. Percentage of untreated control (y-axis) was determined by dividing the fluorescence or luminescence values of the sample by the average of the untreated control (ATP and cytotoxicity). These values were then normalized to control (0 μM TCS + Ag). Data presented are means ± SEM of at least three independent experiments; three replicates per experiment. The degranulation data is reproduced, with permission, from (Weatherly et al. 2013). Statistically significant results, as compared to control, are represented by * p < 0.05; *** p < 0.001, as determined by one-way ANOVA followed by Tukey’s post-hoc test.
Fig. 12.

Cytotoxicity and ATP levels were measured in RBL cells after 60 min of DNP exposure in galactose media-BSA. DNP concentrations ranged from 0 to millimolar levels as indicated on the x-axis. The x-axis is a two-segment line to clarify data interpretation. Percentage of untreated control (y -axis) was determined by dividing the sample’s signal by the average signal from the untreated control. These values were then normalized to control (0 μM DNP). Data presented are means ± SEM of at least five independent experiments; three replicates per experiment. Statistically significant results, as compared to control, are represented by ** p < 0.01 and ** * p < 0.001, determined by one-way ANOVA followed by Tukey’s post-hoc test.
Fig. 6.

TCS effects on ER Ca2+ levels in Ag-stimulated RBL mast cells in plate reader. RBL cells were transfected with the ER-GCaMP6–210 construct. Cells were exposed to ± 0.005 μg/mL Ag and ± 10 μM TCS in Ca2+-free BT. Fluorescence was recorded for 1 hr immediately following Ag/TCS exposure: representative time-series (A). Average background fluorescence (from untransfected cells) was subtracted from each time point, and values presented are means ± SD of one representative experiment with three replicates per treatment (A). Area under this fluorescence curve (AUC) was determined using the raw fluorescence of the time-series graph, processed via background subtraction and normalization to 0 μM TCS/0 Ag (B). Values presented are means ± SEM of 3 independent experiments; two to three replicates for each experiment (B). Statistically significant results, as compared to 0 μM TCS + Ag, are represented by * p < 0.05, and, as compared to control (no stimulation), are represented by ### p < 0.001, determined by one-way ANOVA followed by Tukey’s post-hoc test.
Fig. 9.

TCS effects on cytosolic Ca2+ levels in Ag-stimulated RBL mast cells. RBL cells were transfected with the GCaMP6 construct to allow measurement of cytosolic Ca2+, using a plate reader. Cells were exposed to ± 0.005 μg/mL Ag and 0, 10, or 20 μM TCS in glucose Tyrodes-BSA buffer for 1 hr. (A) Representative graphs of GCaMP6 fluorescence, which was recorded for 1 hr immediately following Ag/TCS exposure. Average background fluorescence (from untransfected cells) was subtracted from each time point (A). Area under this fluorescence curve (AUC) was determined using the raw fluorescence of the time-series graph, normalized to 0 μM TCS (B). Values presented in (A) are means ± SD of one experiment; three replicates per treatment. Values presented in (B) are means ± SEM of at least 3 independent experiments; two to three replicates for each experiment. Statistically significant results, as compared to control, are represented by *** p < 0.001, determined by one-way ANOVA followed by Tukey’s post-hoc test.
Fig. 11.

TCS effects on tubulin dynamics. Live cell confocal fluorescence microscopy of α-tubulin-EGFP transfected RBL cells (A-C). RBL cells were exposed to 0.005 μg/mL Ag (B and C) and 10 μM TCS (C) for 15 min with glucose Tyrodes-BSA, then imaged. One representative image from three independent days of imaging is shown. Scale bar, 10 μm. Time-series graph of an in vitro tubulin polymerization assay, ± 8.3 μM TCS or paclitaxel (D). Mean fluorescence intensity was determined by subtracting out the appropriate controls and normalizing to 0 μM TCS. Values presented are means ± SEM of three independent experiments with two replicates per treatment per experiment. RFU values on the y-axis correspond to fluorescence values obtained from plate reader upon tubulin polymerization.
Results
Triclosan alters mitochondrial morphology in multiple cell types
FPALM nanoscopy (Hess et al. 2006), was utilized to provide precise images of mitochondrial morphology. A construct linking the photoactivatable fluorescent protein Dendra 2 to the outer mitochondrial membrane protein TOM20 was used. TCS, dissolved in galactose media, induces donut morphology in NIH-3T3 cells (Fig. 1). Analysis reveals a significant increase in the fraction of donut-shaped mitochondria over time with TCS exposure, compared to control (Fig. 1C).
Fig. 1.

TCS exposure induces toroid morphology of NIH-3T3 mitochondria. Live cell super resolution FPALM microscopy of Dendra2-TOM20 labeled mitochondrial outer membrane. NIH-3T3 cells were exposed to 10 μM TCS in galactose media-BSA for 1 hr. Shown are representative images (Control [A] and TCS [B]) taken from three independent days of imaging. Scale bars, 1μm. Analysis shows over time, 10 μM TCS induces a toroid morphology in NIH-3T3 cell mitochondria compared to control (C). On the y-axis, ξ represents the fraction of regions that possessed a radial distribution function peak between 200 and 500 nm. Cells were binned in 21 minute intervals where exposure from 0–21 mins, 22–43 mins, and 44–60 mins were combined and plotted, as three data points per treatment. Values presented are means ± SEM of three independent experiments, which yielded a total of 9–13 cells tested per condition per time point. Two sample t-tests were performed, comparing control cells imaged within a specific time range to TCS-treated cells imaged within the same specific time range, for each of the three binned time points. Statistically significant results are represented by * p < 0.05 and *** < 0.001.
These TCS/galactose experiments were repeated with RBL mast cells and primary human keratinocytes (Fig. 2C–F). While donut morphology is not widely seen, TCS causes fragmentation of mitochondria in these cell types (Fig. 2C–F). Mitochondrial perimeter, major axis, and elongation were quantified. Mitochondrial elongation is the inverse of circularity; a value of “1” corresponds to a round mitochondrion whereas a higher number corresponds to a more tubular structure (Dagda et al. 2009). TCS decreases all three of these parameters over time in RBLs and keratinocytes, compared to no changes in control, indicating mitochondrial fission (Fig. 3A–F). Data from cells exposed to TCS for 30 to 60 mins were combined, showing significant decreases in all mitochondrial shape parameters compared to control (Fig. 3J).
Fig. 2.

Super resolution FPALM microscopy of Dendra2-TOM20 labeled mitochondrial outer membrane in NIH-3T3 cells in glucose Tyrodes-BSA (A-B), RBL cells in galactose media-BSA (C-D), and primary human keratinocytes in galactose media-BSA (E-F). Live cell imaging was conducted the day following transfection of cells with the construct Dendra2-TOM20. Cells were exposed to ± 10 μM TCS (NIH-3T3 cells and keratinocytes) or ± 15 μM TCS (RBL cells). Shown are representative images of single cells taken from at least two independent days of imaging: control (A, C, E) and 10 μM (B, F) or 15 μM (D) TCS. Scale bars, 1μm.
In order to determine whether a concomitant decrease in ATP is necessary to induce changes in mitochondrial morphology, these experiments were repeated with NIH-3T3 cells exposed to TCS in BT buffer containing glucose, which allows glycolysis to produce normal levels of ATP regardless of mitochondrial health (Weatherly et al. 2016). In the presence of glucose, TCS does not induce donut morphology in NIH-3T3 cell mitochondria (Fig. 2A–B), as it does in the presence of galactose (Fig. 1). However, in glucose-treated NIH-3T3 cells, TCS-exposed mitochondria appear shortened and rounded compared to control (Fig. 2A–B), with decreases in mitochondrial perimeter, major axis, and elongation (Fig. 3G–J). This result, that TCS induces mitochondrial fragmentation even in the presence of glucose, indicates that inhibition of ATP production is not the cause of triclosan’s induction of mitochondrial fragmentation.
We utilized clonogenic assays (Komissarova et al. 2005) under the experimental conditions used to generate the data in Figures 1–3. TCS causes neither apoptosis nor necrosis in NIH-3T3 cells or keratinocytes (Fig. S1) or RBL cells (Weatherly et al. 2016) even while it induces mitochondrial morphology deformations in these cells.
Triclosan increases reactive oxygen species production in multiple cell types
An increase in intracellular ROS is associated with mitochondrial fragmentation (Deheshi et al. 2015; Fan et al. 2010), leading us to hypothesize that TCS could induce an increase in ROS, resulting in mitochondrial fragmentation. Using RBL mast cells, we examined effects of TCS (for 1 hr) on ROS production both with and without Ag stimulation. Ag increases ROS production in RBL cells (Fig. 4A, C; significance indicated by *, Fig. S2A). TCS also increases ROS production in RBLs, in the absence of Ag stimulation (Fig. 4A, Fig. S2A), and in primary human keratinocytes (Fig. 4B and D). The fluorescence traces (Fig. 4D) from which summary AUC bar graphs (Fig. 4B) were calculated are shown for keratinocytes. TCS appears to mildly increase ROS production of Ag-stimulated cells, but this effect is not statistically significant (Fig. 4A, Fig. S2A).
Using RBL mast cells and the same conditions as in the TCS experiments, we also examined effects of the canonical uncoupler CCCP on ROS production (Fig. 4C, Fig. S2B). Interestingly, CCCP produces the opposite effect on ROS production, compared to TCS: CCCP does not affect ROS production of unstimulated mast cells, but CCCP dramatically reduces intracellular ROS of Ag-stimulated RBLs (Fig. 4C; significance indicated by #).
We confirmed that this increase in ROS production is caused neither by TCS interference with the fluorescent probe (Fig. S3A) nor by cytotoxicity in either RBL cells (Fig. S2C) or keratinocytes (Fig. S2D). We also confirmed that the ROS probe fluorescence is not photobleached during the measurements (Fig. S3B).
Triclosan affects mitochondrial Ca2+ in Ag-stimulated RBL-2H3 mast cells
Uncouplers affect mitochondrial Ca2+ levels (Rao et al. 2015; Samanta et al. 2014; Suzuki et al. 2014). We measured fluorescence of the construct CEPIA2mt (Suzuki et al. 2014) in RBL mast cells, with Ag stimulation, using confocal microscopy. The baseline fluorescence reading of the unstimulated cells fluctuated ~10–15% but remained essentially flat (Fig. 5A). After Ag stimulation, fluorescence readings increase 40% above baseline immediately after Ag addition, with the first peak at 2 min after Ag. Ag stimulation also causes mitochondrial Ca2+ levels to undergo slow oscillations, with about 8 mins between peaks of each oscillation (Fig. 5A, Ag). Upon Ag + 10 μM TCS exposure, initial peak fluorescence readings increase to 65%. Interestingly, beyond that first peak, the presence of TCS abolishes the oscillations that are seen with Ag alone (Fig. 5A, Ag + TCS).
Fig. 5.

TCS effects on mitochondrial Ca2+ levels in Ag-stimulated RBL mast cells. RBL cells were transfected with the CEPIA2mt construct to allow visualization of mitochondrial Ca2+ in individual cells, using confocal microscopy. Cells were exposed to ± 0.005 μg/mL Ag and ± 10 μM TCS in glucose Tyrodes -BSA buffer. Mitochondrial Ca2+ was analyzed as percentage increase in fluorescence after F/F0 normalization (A). Baseline readings were taken for 6 minutes followed by simultaneous addition of Ag ± 10 μM TCS (at time point 0). Percent increase from lowest to highest fluorescence data point following Ag ±TCS addition was calculated for each individual cell, data were averaged, and significance of the TCS effect was determined via one-tailed t-test (B, black squares). Number of oscillations within a 15 minute period after Ag addition was determined as stated in the Methods section and analyzed via one-tailed t-test (B, blue triangles). Values presented are means ± SEM of three independent days of imaging, with 14–22 cells analyzed per treatment. Statistically significant results compared to control are represented by ** p<0.01, ***p<0.001.
Next, the CEPIA2mt fluorescence traces for individual cells (rather than the averaged data shown in Fig. 5A) were analyzed. The highest percentage increase in mitochondrial Ca2+ level (“highest peak”) of each individual cell trace was determined. TCS increases this “highest peak” value, compared to Ag alone (Fig. 5B). The highest percentage increase graph (Fig. 5B) has higher values compared to the peaks shown in the combined time-series graph due to variability in the timing of the peaks from cell to cell, as expected from previous mast cell studies (Millard et al. 1989). Also, TCS decreases the number of oscillations compared to Ag alone (Fig. 5B). Thus, TCS disrupts mitochondrial Ca2+ buffering capacity.
Triclosan alters ER Ca2+ levels in Ag-stimulated RBL-2H3 mast cells
To determine whether TCS affects ER Ca2+, we transfected the fluorescent construct ER-GCaMP6–210 (Juan-Sanz et al. 2017) into RBL mast cells. In the absence of extracellular Ca2+, Ag stimulation alone causes an immediate and robust decrease in ER Ca2+ (as shown in the representative fluorescence trace in Fig. 6A), as expected (Berridge 1993). The decrease reaches the lowest point at ~10 mins after Ag addition, followed by a slow, gradual increase in fluorescence, which indicates re-filling of ER stores. TCS causes a more rapid and sustained decrease in ER Ca2+ level compared to Ag alone (Fig. 6A). Triclosan’s enhanced depletion of ER Ca2+ continues throughout the 1 hr measurement period: TCS suppresses the gradualincrease in ER-GCaMP6 fluorescence during the 10–60 min time points following Ag addition (Fig. 6A). AUC analysis quantifies this decrease in ER Ca2+ with Ag stimulation compared to no-Ag control and the further decrease in ER Ca2+ with TCS and Ag (Fig. 6B). Thus, TCS enhances Ag-stimulated depletion of ER Ca2+ stores.
To measure ER pH levels, we transfected the fluorescent construct ER-pHluorin into RBL mast cells (de Juan-Sanz et al. 2017). Neither Ag nor Ag + TCS affected ER-pHlourin fluorescence (data not shown), in stark contrast to the large effects caused by Ag and TCS on ER-GCaMP6–210 fluorescence (Fig. 6A, B). Thus, neither Ag nor Ag+TCS exposure changes the pH of the ER, compared to control, supporting the conclusion that triclosan’s effects on ER-GCaMP6–210 fluorescence are caused by changes in ER Ca2+ levels and not by a corruption of the fluorescent probe due to a change in ER pH.
Triclosan decreases mitochondrial membrane potential
To examine whether TCS affects MMP, we used MitoTracker Red FM dye, which accumulates in the mitochondria in an MMP-dependent fashion. A significant decrease in MMP is seen after a 1 hr exposure to 10 μM TCS (Fig. 7). CCCP (1 μM for 1 hr), a positive control for MMP inhibition (Rottenberg and Wu 1998) also decreases MMP (Fig. 7). Both 1 μM CCCP and 10 μM TCS decrease MMP by ~20%. This result is similar to our previous findings with ATP production and oxygen consumption rate, showing that the canonical mitochondrial poison CCCP is ~10X more potent than TCS as a mitochondrial uncoupler (Weatherly et al. 2016). Careful controls were conducted (see Methods) to overcome triclosan’s and CCCP’s interference with MitoTracker Red fluorescence (similar to CCCP’s known interference with a plasma membrane potential probe (Mohr and Fewtrell 1987b)).
Triclosan modestly decreases ATP production in Ag-stimulated RBL-2H3 mast cells in the presence of glucose
In light of the discovery that TCS is a mitochondrial uncoupler in multiple cell types (Weatherly et al. 2016), including in RBL mast cells, the question arises of whether triclosan’s inhibition of degranulation is solely caused by its mitochondrial inhibition. In order to determine what percentage of triclosan’s inhibition of degranulation observed in glucose-containing BT (Weatherly et al. 2013) is due to ATP inhibition, we performed the ATP production experiment under the same conditions as in a degranulation assay using BT buffer (with glucose). Under these conditions, TCS (20 μM) causes an ~8% decrease in ATP, compared to an ~80% decrease in degranulation. Thus, only ~10% of the inhibition of degranulation is due to a decrease in ATP when glucose is present in BT buffer (Fig. 8).
Triclosan inhibits the cytosolic Ca2+ response to Ag stimulation in RBL-2H3 mast cells
Cytosolic Ca2+ mobilization is inhibited by other known mitochondrial uncouplers (Mohr and Fewtrell 1987b). We transfected the fluorescent construct GCaMP6 into RBL mast cells in order to measure cytosolic Ca2+ levels. We confirmed successful transfection (Fig. S4A), and we confirmed that TCS does not interfere with GCaMP fluorescence (Fig. S4B). GCaMP6 fluorescence was measured in the plate reader immediately after Ag stimulation ± TCS and graphed (Fig. 9A), from which AUC was calculated. Antigen stimulation increased GCaMP fluorescence in RBL cells by a factor similar to that found by other researchers (Furuno et al. 2015). Both 10 and 20 μM TCS decrease the Ca2+ response compared to control (Fig. 9B). TCS at 10 μM decreases AUC to 0.6 ± 0.1 (SEM) of the 0 μM control level, and 20 μM TCS decreases AUC to 0.51 ± 0.04 (SEM). There is a rapid initial rise in fluorescence immediately after Ag stimulation (Fig. 9A, 0 μM TCS), due to release from ER stores (compare first 10 min of “0 μM TCS + Ag” data from each of Figures 6A and 9A) (Holowka et al. 2012; Mohr and Fewtrell 1987a), followed by a decrease and plateau phase (Fig. 9A, 0 μM TCS) due to influx of Ca2+ across the plasma membrane (Mohr and Fewtrell 1987a). Triclosan greatly reduces this plateau (Fig. 9A, +TCS). Interestingly, the initial rise in Ca2+ is only slightly inhibited by TCS (Fig. 9A, +TCS). This observation aligns both with our finding that TCS does not inhibit release of Ca2+ from the ER (Fig. 6) and with published data showing that CCCP exhibits similar effects on cytosolic Ca2+ (Mohr and Fewtrell 1987b). GCaMP transfection, TCS treatment, and/or Ag treatment cause no cytotoxicity (Fig. S5A, B), confirming that the decrease in GCaMP fluorescence in Fig. 9 is not due to cell death. These data indicate that TCS inhibits the influx of Ca2+ across the plasma membrane.
Triclosan inhibits mitochondrial translocation in Ag-stimulated RBL-2H3 mast cells
We used MitoTracker Red to label mitochondria and Hoechst dye to label nuclei of RBL mast cells in galactose media-BSA (Fig. 10). In resting RBL mast cells, the majority of mitochondria are located around the nuclei (Fig. 10Ai, Bi). In contrast, Ag stimulation causes mitochondria to translocate away from the nucleus (Fig. 10Aii, Bii). Addition of 10 μM TCS (with Ag stimulation) inhibits the translocation of the mitochondria, leaving the majority of the mitochondria localized around the nucleus (Fig. 10Aiii). Analysis shows that ~ 17% of the mitochondria are located away from the nucleus in control cells, compared to ~50% of the mitochondria translocated away from the nucleus in Ag-stimulated cells after 30 min Ag exposure (Fig. 10Aiv). TCS causes the percentage of mitochondria that translocate in Ag-stimulated cells to drop back to control levels, ~23% translocation (Fig. 10Aiv).
Fig. 10.

TCS effects on mitochondrial translocation. Representative images of mitochondrial translocation in RBL mast cells with ± Ag and ± 10 μM TCS (A) and ± Ag and ± 2.4 μM CCCP (B) exposure in galactose media-BSA (images are pseudo-colored green for Mitotracker dye-labeled mitochondria and purple for Hoechst-stained nuclei). Cells were exposed to media only (Ai, Bi), media with 0.0004 μg/mL Ag (Aii, Bii), or media with Ag and 10 μM TCS or 2.4 μM CCCP (Aiii, Biii) for 30 minutes at 37°C/5% CO2. Scale bar, 10 μm. Percentage of translocated mitochondria in control, Ag-stimulated, and Ag-stimulated + 10 μM TCS or 2.4 μM CCCP (Aiv, Biv) samples was quantified. Values presented (Aiv, Biv) are means ± SEM of three independent days of imaging. Figure Aiv combines data from control cells (total n=98), Ag-stimulated cells (n=73), and Ag-stimulated cells + TCS (n=81). Figure Biv combines data from control cells (n=84), Ag-stimulated cells (n=84), and Ag-stimulated cells + CCCP (n=90). Statistically significant results compared to Ag are represented by ***p < 0.001, statistically significant results compared to control are represented by ### p < 0.001, as determined by one-way ANOVA followed by Tukey’s post-hoc test.
With glucose present, control and Ag-stimulated translocation is ~10% higher than in galactose experiments (Fig. S6 vs. Fig. 10Aiv). When glucose is present, TCS decreases Ag-stimulated mitochondrial translocation; however, this decrease is not as pronounced as in galactose media (Fig. S6 vs. Fig. 10Aiv). Mitochondrial translocation requires ATP, so a more pronounced decrease in translocation in conditions (+galactose, no glucose) that decrease ATP is expected. Thus, TCS inhibits mitochondrial translocation, whether or not glucose is present. These data indicate that a large decrease in ATP is not required for a significant decrease in mitochondrial translocation. Mitochondrial translocation is not required for de novo cytokine release (Zhang et al. 2012); indeed, TCS does not inhibit cytokine release (Fig. S7) even though it inhibits both mitochondrial translocation (Fig. 10) and degranulation.
We confirmed that robust degranulation responses occurred and were inhibited by TCS under the experimental conditions used in the mitochondrial translocation assays (Fig. S8A). Also, triclosan’s inhibition of mitochondrial translocation is not due to cell death (Fig. S8B).
To compare the TCS effects to those of a canonical mitochondrial uncoupler, translocation experiments were performed with 2.4 μM CCCP (a dose equally effective at inhibiting ATP as 10 μM TCS (Weatherly et al. 2016)). CCCP inhibits Ag-stimulated mitochondrial translocation, but to a lesser extent than TCS (Fig. 10, compare Aiv to Biv).
Triclosan disrupts tubulin polymerization in Ag-stimulated RBL-2H3 mast cells
We hypothesized that TCS disrupts microtubule polymerization, thereby inhibiting degranulation and mitochondrial translocation. We utilized confocal microscopy and α-tubulin-EGFP to image microtubule formation in transfected RBL mast cells. In control resting cells, α-tubulin-EGFP is mainly located near the plasma membrane and in the cytoplasm near the nucleus in a microtubule organization center (Joshi 1994) (Fig. 11A). Ag stimulation leads to drastic microtubule filamentation (Fig. 11B) in all 30 cells imaged (1 representative cell in Fig. 11B). TCS causes a drastic inhibition of microtubule formation in Ag-stimulated cells (Fig. 11C).
An in vitro (no cell) assay was conducted to determine whether TCS directly inhibits tubulin polymerization. No significant difference in tubulin polymerization is seen between control and 8.3 μM TCS (Fig. 11D); similar results were obtained with 1, 5, and 12 μM TCS (data not shown). A paclitaxel (inhibitor of microtubule disassembly) positive control (Fig. 11D) was included for comparison. These in vitro data show that TCS does not inhibit microtubule polymerization by directly binding to tubulin.
Triclosan is a more potent inhibitor of ATP production compared to the banned uncoupler 2,4-dinitrophenol
Assays of DNP effects on ATP production in RBL mast cells were performed, using the same experimental conditions used for TCS assays (glucose-free, galactose-containing media) (Weatherly et al. 2016). DNP disrupts ATP production in RBL cells with an EC50 value of 389–677 μM (95% CI) (Fig. 12). These concentrations are not cytotoxic (Fig. 12). Under these same conditions, we previous ly showed that TCS disrupts ATP production with an EC50 value of 7.5–9.7 μM (95% CI) (Weatherly et al. 2016). Thus, in RBL cells, TCS is ~60X more toxic to mitochondria than DNP. Analogous experiments were also conducted in primary human keratinocytes, leading to the conclusion that TCS is ~33X more mitotoxic than DNP in these cells (DNP EC50 109–128 μM [95% CI]; TCS EC50 3.0–4.1 μM [95% CI]).
Discussion
Triclosan is a proton ionophore mitochondrial uncoupler in numerous mammalian cell types (from different species and tissue types), including primary human skin cells, and in living zebrafish (Ajao et al. 2015; Shim et al. 2016; Weatherly et al. 2016), a well-accepted model for human toxicology (Planchart et al. 2016). By dissipating electrochemical gradients (Mohr and Fewtrell 1987b), proton ionophores interfere with both mitochondrial ATP production and a wide range of other cellular functions. In this study, we employed super-resolution microscopy among other techniques to detail cellular effects of TCS, at concentrations relevant to human exposures to consumer products, on mitochondria and on cellular signal transduction.
Using live-cell super-resolution microscopy for the first time in the field of toxicology, we show that micromolar doses of TCS dramatically and rapidly disrupt mitochondrial ultrastructure. TCS induces toroidal morphology and increases mitochondrial fission. Our results with TCS are supported by the fact that other known mitochondrial uncouplers also induce “donut” formation, by initiating mitochondrial swelling, detachment from microtubules, and auto-fusion (Liu and Hajnoczky 2011). FCCP and CCCP also increase fission (Giedt et al. 2012; Griparic et al. 2007). Although triclosan’s effects on mitochondrial morphology appear similar to changes caused during apoptosis (Sun et al. 2007), TCS does not cause apoptosis or necrosis under our experimental conditions (see Supplement), and other uncouplers (FCCP and CCCP), likewise, do not cause apoptosis under conditions that cause fission/donuts (Griparic et al. 2007; Liu and Hajnoczky 2011). In fact, Liu and Hajnoczky also showed that mitochondria recover quickly after removal of FCCP (donut formation reversed), ind icating that FCCP does not instigate an irreversible pathway leading to apoptosis under conditions in which it does distort mitochondrial shape (Liu and Hajnoczky 2011).
Mitochondrial shape is crucial for proper mitochondrial functioning, which directly impacts many diseases (Youle and van der Bliek 2012). For example, increases in occurrence of mitochondrial donuts in monkey presynaptic boutons are associated with cognitive decline (Hara et al. 2014). TCS has been detected in brain tissue of exposed fish (Escarrone et al. 2016) at 0.49 nmol TCS/mg protein, which lies within the 0.4–64 nmol TCS/mg protein levels found in human tissue following consumer product exposure (Weatherly and Gosse 2017). Certain genes linked to Parkinson’s disease also increase the occurrence of mitochondrial donuts (Bhandari et al. 2014; Cui et al. 2010). In skeletal muscle, mitochondrial fission is associated with insulin resistance (Jheng et al. 2012). Mitochondrial fusion is essentialfor embryonic development (Chen et al. 2003), and a recent study found increased TCS levels in blood from mothers of infants with birth defects, compared to blood from mothers of healthy babies (Wei et al. 2017). Together, these findings suggest that triclosan’s deformation of mitochondrial morphology could be a mechanism underlying disease.
In mast cells and keratinocytes, TCS increases ROS production, as found by other researchers investigating other species (Binelli et al. 2009; Riva et al. 2012; Tamura et al. 2012; Yueh et al. 2014). Because TCS increases ROS production in the presence of glucose, which ablates triclosan’s inhibition of ATP production (in unstimulated cells) (Weatherly et al. 2016), triclosan’s ROS production is not directly caused by mitochondrial uncoupling. A recent human epidemiology study shows a correlation between increased TCS in urine and an increase in a marker for oxidative stress (Lv et al. 2016). Triclosan’s ability to increase ROS levels stands in contrast to other uncouplers, such as CCCP, which actually decrease ROS under some conditions (Przygodzki et al. 2005).
ROS production is required for mast cell degranulation (Swindle et al. 2004). Because TCS does not dampen mast cell ROS, manipulation of ROS is not the mechanism underlying triclosan’s inhibition of degranulation (Weatherly et al. 2013). It should be noted that Ag stimulation causes a very strong stimulation of ROS levels, so an additional increase caused by TCS may be occurring, though difficult to measure. In fact, data in Fig. S2A do suggest a modest increase in Ag-stimulated ROS due to TCS.
We show, for the first time, that Ag stimulation induces slow oscillations in mast cell mitochondrial Ca2+ levels, as revealed by individual-cell analysis. TCS alters mast cell mitochondrial Ca2+ in an unexpected manner: TCS enhances the initial Ag-stimulated rise in mitochondrial Ca2+ but then abolishes subsequent oscillations. Thus, TCS may inhibit cytosolic Ca2+ levels and, hence, degranulation, by causing mitochondria to immediately sequester Ca2+ released by the ER following Ag stimulation. In contrast, FCCP and other uncouplers reduce mitochondrial Ca2+ (Rao et al. 2015; Samanta et al. 2014; Suzuki et al. 2014). This contrast between triclosan’s and other uncouplers’ effects on mitochondrial Ca2+ may be due to the fact that, unlike other uncouplers, TCS increases ROS, which is linked to increased mitochondrial Ca2+ levels (Jornot et al. 1999; Peng and Jou 2010). ROS increases mitochondrial Ca2+ levels by disrupting the Na+/Ca2+ ion exchanger, which is responsible for Ca2+ efflux from mitochondria (Jornot et al. 1999) (Palty et al. 2010). TCS-stimulated ROS may inhibit this exchanger and cause the initial enhancement of mitochondrial Ca2+. The TCS-induced large initial mitochondrial Ca2+ peak may subsequently trigger the opening of the mitochondrial permeability transition pore (mPTP), leading to efflux of mitochondrial Ca2+ (Crompton 1999). The opening of the mPTP would also explain the lack of oscillations with TCS exposure. FCCP causes mPTP opening (Liu and Hajnoczky 2011). This FCCP-induced mPTP opening also may be a mechanism behind the mitochondrial donut formation (Liu and Hajnoczky 2011).
Mitochondria frequently are localized in close proximity to the ER (Mazel et al. 2009) in order to form mitochondrial/ER junctions (Pacher et al. 2000) that allow for Ca2+ transfer between mitochondria and the ER (Ma and Beaven 2011; Mazel et al. 2009). Recently, a role for the VAP family of ER membrane proteins in the initial rise in mitochondrial Ca2+ and for subsequent SOCE due to Ag stimulation was discovered (Holowka et al. 2016). Ag stimulation rapidly (within 2–4 min) lowers mast cell ER Ca2+ levels, as expected (Berridge 1993). TCS further drives down the Ag-stimulated decrease in ER Ca2+, just as it enhances the Ag-stimulated increase in mitochondrial Ca2+, indicating that TCS could be enhancing the Ca2+ exchange from the ER to the mitochondria. A possible explanation for this phenomenon is that TCS affects the ER’s IP3 receptor, leading to enchanced ER Ca2+ efflux. TCS increases production of ROS, which stimulates the IP3 receptor (Suzuki and Ford 1992), enhancing Ca2+ release from the ER. Another possible mechanism behind triclosan’s enhanced dampening of ER Ca2+ is that TCS-induced ROS production may inhibit the ER SERCA pump (Feissner et al. 2009), thereby inhibiting Ca2+ re-entry into the ER.
Triclosan’s inhibition of MMP and enhancement of mitochondrial Ca2+, together, illustrate a mechanism for TCS-induced mitochondrial fission. Lowered MMP leads to mitochondrial fragmentation (Giedt et al. 2012; Guillery et al. 2008). Elevation of mitochondrial Ca2+ induces mitochondrial fission in prostate cancer cells (Kaddour-Djebbar et al. 2010) and in human lung cells (Ahmad et al. 2013). Increases in cytosolic ROS correspond to mitochondrial fragmentation (Deheshi et al. 2015; Fan et al. 2010) and may also partially explain the TCS-induced mitochondrial fission.
In the presence of glucose, 20 μM TCS inhibits degranulation by 82% while inhibiting ATP by only 8%, indicating that mitochondrial uncoupling is not the only mechanism causing triclosan’s inhibition of degranulation. Thus, TCS must target other components within the degranulation pathway. During mast cell degranulation, depletion of ER Ca2+ stores leads to CRAC channel activation and SOCE (Hogan et al. 2010). CRAC channel activation triggers mitochondrial translocation to the plasma membrane along microtubules (Quintana et al. 2006; Zhang et al. 2011). This mitochondrial translocation then, in turn, keeps the CRAC channels active, continuing the Ca2+ influx (Quintana et al. 2006). Our data indicate that TCS dampens Ag-stimulated degranulation, even in the presence of glucose, by decreasing cytosolic Ca2+ levels, via disruption of SOCE across the plasma membrane. Triclosan’s disruption of SOCE may be caused by its inhibition of mitochondrial translocation to the plasma membrane, through a decrease in microtubule polymerization. While TCS strongly inhibits microtubule polymerization in living cells, TCS does not directly interfere with tubulin polymerization in vitro. Inhibition of microtubule polymerization also impedes antigen-stimulated granule movement to the plasma membrane, thereby dampening degranulation (Smith et al. 2003). Because it is a proton ionophore, triclosan-induced disruption of the plasma membrane potential (PMP) is another potential contributor to the decrease in Ca2+ influx (Mohr and Fewtrell 1987b); however, we were unable to test PMP because TCS interferes with the fluorescence of plasma membrane potential probes. Triclosan’s disruption of microtubules is similar to FCCP’s microtubule disruption, caused by FCCP’s inhibition of MMP (Maro et al. 1982); TCS could be acting in a similar manner. Any cell type that depends on calcium signaling, mitochondrial function, and cytoskeleton function (e.g., T cells, muscle cells) are subject to TCS effects (Feske 2007; Hill-Eubanks et al. 2011).
It is interesting to note that, under identical experimental conditions in RBL mast cells, TCS is a 60-fold more potent uncoupler, compared to the potent weight loss drug and uncoupler DNP (Tainter et al. 1934). Due to numerous adverse health effects, pharmacological usage of DNP was banned in the U.S. in 1938 (Harris and Corcoran 1995).
Several recent studies have found that TCS causes negative effects on human reproduction. One found decreased fecundity with increased TCS urine concentration (Velez et al. 2015). Other studies linked increasing TCS concentration in urine to miscarriage (Wang and Tian 2015; Wang et al. 2015). TCS reduces pregnancy weight gain and increases miscarriage in rats (Feng et al. 2016). Proper mitochondrial function is essentialin reproduction (Ramalho-Santos et al. 2009), so triclosan’s mitochondrial toxicity could partly explain these adverse reproduction effects. Human developmental defects associated with TCS exposure (Philippat et al. 2014; Wei et al. 2017) could be caused by triclosan’s inhibition of mitochondrial fusion, which is essentialfor embryonic development (Chen et al. 2003; Chen and Chan 2010). Furthermore, a human study found an inverse relationship between urinary TCS concentration and body mass index (Li et al. 2015), which could be explained by triclosan’s mitochondrial uncoupling. TCS-induced mitochondrial defects may also explain reduced energy and movement in exposed freshwater mussels (Goodchild et al. 2016). Triclosan’s mitochondrial toxicity may underlie triclosan’s adverse effects on reproduction; additional animal or human studies are required to prove a connection between triclosan’s mitochondrial and human health effects.
In conclusion, using super-resolution microscopy, we have shown that TCS, a proton ionophore mitochondrial uncoupler (Weatherly et al. 2016), disrupts mitochondrial morphology in a rat mast cell line, a mouse fibroblast line, and primary human keratinocytes. We provide biochemical mechanisms underlying these effects, which occur at doses relevant to human exposure via consumer products. Also, TCS inhibits degranulation and other mast cell functions (Weatherly et al. 2013; Weatherly et al. 2016), and we have determined the mechanisms underlying this inhibition. This disruption of mitochondrial morphology is related to TCS-induced decrease in MMP and disruption of mitochondrial Ca2+ (Fig. 13). TCS-induced increases in ROS may disrupt the mitochondrial Na+/Ca2+ ion exchanger, leading to increased mitochondrial Ca2+ levels, which, in turn, trigger the opening of the mPTP, leading to mitochondrial donut formation. Increased mitochondrial Ca2+ is also caused by enhanced ER Ca2+ depletion, which is caused by TCS. Mitochondrial uncoupling/dysfunction caused by TCS explains a portion but not most of triclosan’s inhibition of mast cell degranulation. We additionally show that TCS inhibits mast cell degranulation via inhibition of Ag-stimulated influx of Ca2+ into the cytosol, enhancement of mitochondrial Ca2+ sequestration, and interference with mitochondrial Ca2+ oscillations (and possibly due to triclosan’s interference with PMP). TCS also inhibits mast cell degranulation by decreasing MMP, which leads to disruption of microtubule polymerization, which causes inhibition of mitochondrial translocation, which ablates sustained activation of the CRAC channels (Fig. 13, adapted in part from Holowka et al. 2016). In summary, we show that TCS is a mitotoxicant that is much more potent than the banned drug DNP and also is a robust modulator of signal transduction in mammalian cells.
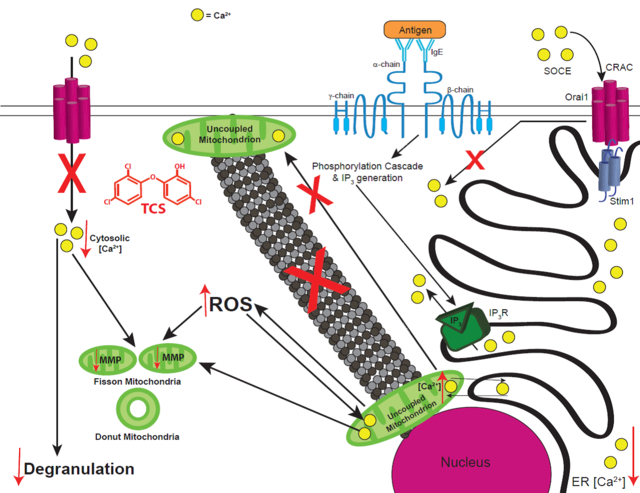
Fig. 13.

Schematic of TCS effects on mitochondria and on Ag-stimulated mast cell signaling. Crosslinking of IgE-bound FcεRI receptors by multivalent Ag stimulates a phosphorylation cascade that leads to IP3 generation. IP3 then binds to its receptor on the ER membrane, causing efflux of Ca2+ from the ER, a process which is enhanced by TCS. While, in healthy cells, depletion of Ca2+ stores from the ER causes Stim1-Orai1 interaction and the activation of SOCE through plasma membrane CRAC channels, TCS instead leads to enhanced uptake of Ca2+ into mitochondria and inhibition of SOCE, leading to decreased cytosolic Ca2+ levels. Also, TCS inhibits microtubule polymerization and, thus, decreases mitochondrial translocation from the perinuclear region to the plasma membrane, further inhibiting SOCE. All these effects lead to inhibition of degranulation due to TCS exposure. TCS also decreases mitochondrial membrane potential (MMP), increases intracellular ROS, and modulates mitochondrial Ca2+ levels, all of which lead to fragmented mitochondria morphology. Red arrows indicate increases or decreases in various parameters, due to TCS exposure, and “X” marks indicate processes inhibited by TCS.
Supplementary Material
Highlights.
Antimicrobial triclosan harms mast cells, fibroblasts, primary human keratinocytes
Super-resolution microscopy reveals mitochondrial nanostructure deformation
Triclosan causes mitochondrial fission via ROS, membrane potential, Ca2+ changes
Triclosan inhibits mast cells via microtubule, mitochondrial, and Ca2+ changes
Acknowledgements
We thank Drs. David Holowka and Barbara Baird for the RBL-2H3 cells and very helpful scientific discussions; Molly Caron, Logan Gerchman, Nicholas Doucette, and Talya Briana for lab assistance; Dr. Carol Kim for use of the Amaxa Nucleofector equipment; Dr. Robert Wheeler and Allison Scherer for use of and help with the ibidi heating system; and Tyler McGathey for confocal training.
Funding information: This research was supported by the National Institute of Environmental Health Sciences of the National Institutes of Health under the Award Number R15ES24593; by the 2015–2016 Radke Research Fellowship (University of Maine), and by an Institutional Developmental Award (IDeA) from the National institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103423. L.M.W. was supported in part by the Chase Distinguished Research Assistantship (University of Maine) and by the Michael J. Eckardt Dissertation Fellowship (University of Maine).
Disclaimer: This research was supported by the National Institute of Environmental Health Sciences of the National Institute of Health under the Award Number R15ES24593. The content is solely the responsibility o f the authors and does not necessarily represent the official views of the National Institute of Health.
Glossary of abbreviations
- Ag
antigen
- ATP
adenosine triphosphate
- AUC
areaunder the curve
- BSA
bovineserum albumin
- BT
Tyrodes-bovine serum albumin
- CCCP
carbonyl cyanide 3-chlorophenylhydrazone
- CRAC
Ca2+ release-activated Ca2+
- DNP
2,4-dinitrophenol
- ER
endoplasmic reticulum
- FCCP
carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
- FPALM
fluorescence photoactivation localization microscopy
- IgE
immunoglobulin E
- IP3
inositol 1,4,5-triphosphate
- MMP
mitochondrial membrane potential
- Mptp
mitochondrial permeability transition pore
- PKC
protein kinase C
- PMP
plasmamembrane potential
- RBL
rat basophilic leukemia cells, clone 2H3
- ROS
reactive oxygen species
- SERCA
sarco/endoplasmic Ca2+-ATPase
- SOCE
store-operated calcium entry
- TCS
triclosan
- TOM
translocase of the outer membrane
- SEM
standard error of the mean
Footnotes
The authors have no financial interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad T, Aggarwal K, Pattnaik B, Mukherjee S, Sethi T, Tiwari BK, et al. 2013. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis 4:e461, 10.1038/cddis.2012.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajao C, Andersson MA, Teplova VV, Nagy S, Gahmberg CG, Andersson LC, et al. 2015. Mitochondrial toxicity of triclosan on mammalian cells. Toxicology Reports 2:624–637, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. 1993. Inositol trisphosphate and calcium signalling. Nature 361:315–325, [DOI] [PubMed] [Google Scholar]
- Bhandari P, Song M, Chen Y, Burelle Y, Dorn GW 2nd. 2014. Mitochondrial contagion induced by parkin deficiency in drosophila hearts and its containment by suppressing mitofusin. Circ Res 114:257–265, 10.1161/circresaha.114.302734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binelli A, Cogni D, Parolini M, Riva C, Provini A. 2009. In vivo experiments for the evaluation of genotoxic and cytotoxic effects of triclosan in zebra mussel hemocytes. Aquat Toxicol 91:238–244, 10.1016/j.aquatox.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Calafat AM, Ye X, Wong LY, Reidy JA, Needham LL. 2008. Urinary concentrations of triclosan in the u.S. Population: 2003–2004. Environ Health Perspect 116:303–307, 10.1289/ehp.10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. 2003. Mitofusins mfn1 and mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160:189–200, 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC. 2010. Physiological functions of mitochondrial fusion. Ann N Y Acad Sci 1201:21–25, 10.1111/j.1749-6632.2010.05615.x. [DOI] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, et al. 2013. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499:295–300, 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen R, Holowka DA, Baird BA. 2015. Real-time imaging of ca(2+) mobilization and degranulation in mast cells. Methods Mol Biol 1220:347–363, 10.1007/978-1-4939-1568-2_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney CM. 2010. Triclosan comes under scrutiny. Environ Health Perspect 118:A242, 10.1289/ehp.118-a242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree HG. 1928. The carbohydrate metabolism of certain pathological overgrowths. Biochem J 22:1289–1298, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M 1999. The mitochondrial permeability transition pore and its role in cell death. Biochem J 341 ( Pt 2):233–249, [PMC free article] [PubMed] [Google Scholar]
- Cui M, Tang X, Christian WV, Yoon Y, Tieu K. 2010. Perturbations in mitochondrial dynamics induced by human mutant pink1 can be rescued by the mitochondrial division inhibitor mdivi-1. J Biol Chem 285:11740–11752, 10.1074/jbc.M109.066662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT. 2009. Loss of pink1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 284:13843–13855, 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoud FC, Edmiston CE Jr., Leaper D. 2014. Meta-analysis of prevention of surgical site infections following incision closure with triclosan-coated sutures: Robustness to new evidence. Surg Infect (Larchmt) 15:165–181, 10.1089/sur.2013.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Juan-Sanz J, Holt GT, Schreiter ER, de Juan F, Kim DS, Ryan TA. 2017. Axonal endoplasmic reticulum ca2+ content controls release probability in cns nerve terminals. Neuron 93:867–881.e 866, 10.1016/j.neuron.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deheshi S, Dabiri B, Fan S, Tsang M, Rintoul GL. 2015. Changes in mitochondrial morphology induced by calcium or rotenone in primary astrocytes occur predominantly through ros-mediated remodeling. J Neurochem 133:684–699, 10.1111/jnc.13090. [DOI] [PubMed] [Google Scholar]
- Ding WX, Li M, Biazik JM, Morgan DG, Guo F, Ni HM, et al. 2012. Electron microscopic analysis of a spherical mitochondrial structure. J Biol Chem 287:42373–42378, 10.1074/jbc.M112.413674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escarrone AL, Caldas SS, Primel EG, Martins SE, Nery LE. 2016. Uptake, tissue distribution and depuration of triclosan in the guppy poecilia vivipara acclimated to freshwater. Sci Total Environ 560–561:218–224, 10.1016/j.scitotenv.2016.04.039. [DOI] [PubMed] [Google Scholar]
- Fan X, Hussien R, Brooks GA. 2010. H2o2-induced mitochondrial fragmentation in c2c12 myocytes. Free Radic Biol Med 49:1646–1654, 10.1016/j.freeradbiomed.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feissner RF, Skalska J, Gaum WE, Sheu SS. 2009. Crosstalk signaling between mitochondrial ca2+ and ros. Front Biosci (Landmark Ed) 14:1197–1218, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Zhang P, Zhang Z, Shi J, Jiao Z, Shao B. 2016. Endocrine disrupting effects of triclosan on the place nta in pregnant rats. PLoS One 11:e0154758, 10.1371/journal.pone.0154758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S 2007. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol 7:690–702, 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]
- Furuno T, Shinkai N, Inoh Y, Nakanishi M. 2015. Impaired express ion of the mitochondrial calcium uniporter suppresses mast cell degranulation. Mol Cell Biochem 410:215–221, 10.1007/s11010-015-2554-4. [DOI] [PubMed] [Google Scholar]
- Giedt RJ, Pfeiffer DR, Matzavinos A, Kao CY, Alevriadou BR. 2012. Mitochondrial dynamics and motility inside living vascular endothelial cells: Role of bioenergetics. Ann Biomed Eng 40:1903–1916, 10.1007/s10439-012-0568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert RJ. 1987. The oral clearance of zinc and triclosan after delivery from a dentifrice. J Pharm Pharmacol 39:480–483, [DOI] [PubMed] [Google Scholar]
- Gilbert RJ, Fraser SB, van der Ouderaa FJ. 1987. Oral disposition of triclosan (2,4,4’-trichlo ro-2’-hydro xydiphenyl ether) delivered from a dentifrice. Caries Res 21:29–36, [DOI] [PubMed] [Google Scholar]
- Goodchild CG, Frederich M, Zeeman SI. 2016. Is altered behavior linked to cellular energy regulation in a freshwater mussel (elliptio complanata) exposed to triclosan? Comp Biochem Physiol C Toxicol Pharmacol 179:150–157, 10.1016/j.cbpc.2015.10.008. [DOI] [PubMed] [Google Scholar]
- Griparic L, Kanazawa T, van der Bliek AM. 2007. Regulation of the mitochondrial dynamin -like protein opa1 by proteolytic cleavage. J Cell Biol 178:757–764, 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillery O, Malka F, Frachon P, Milea D, Rojo M, Lombes A. 2008. Modulation of mitochondrial morphology by bioenergetics defects in primary human fibroblasts. Neuromuscul Disord 18:319–330, 10.1016/j.nmd.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Guo Z, Turner C, Castle D. 1998. Relocation of the t-snare snap-23 from lamellipodia-like cell surface projections regulates compound exocytosis in mast cells. Cell 94:537–548, [DOI] [PubMed] [Google Scholar]
- Hara Y, Yuk F, Puri R, Janssen WG, Rapp PR, Morrison JH. 2014. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proc Natl Acad Sci U S A 111:486–491, 10.1073/pnas.1311310110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MO, Corcoran JJ. 1995. Toxicological profile for dinitriphenols. 205–93-0606.Agency for Toxic Substances and Disease Registry. [Google Scholar]
- Hess ST, Girirajan TP, Mason MD. 2006. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J 91:4258–4272, 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill-Eubanks DC, Werner ME, Heppner TJ, Nelson MT. 2011. Calcium signaling in smooth muscle. Cold Spring Harb Perspect Biol 3:a004549, 10.1101/cshperspect.a004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Lewis RS, Rao A. 2010. Molecular basis of calcium s ignaling in lymphocytes: Stim and orai. Annu Rev Immunol 28:491–533, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowka D, Calloway N, Cohen R, Gadi D, Lee J, Smith NL, et al. 2012. Roles for ca(2+) mobilization and its regulation in mast cell functions. Front Immunol 3:104, 10.3389/fimmu.2012.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowka D, Wilkes M, Stefan C, Baird B. 2016. Roles for ca2+ mobilization and its regulation in mast cell functions: Recent progress. Biochem Soc Trans 44:505–509, 10.1042/bst20150273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson LM, Trinh BM, Palmer RK, Preziosi CA, Pelletier JH, Nelson HM, et al. 2011. Inorganic arsenite inhibits ige receptor-mediated degranulation of mast cells. J Appl Toxicol 31:231–241, 10.1002/jat.1585. [DOI] [PubMed] [Google Scholar]
- Ibsen KH. 1961. The crabtree effect: A review. Cancer Res 21:829–841, [PubMed] [Google Scholar]
- Iqbal S, Hood DA. 2014. Cytoskeletal regulation of mitochondrial movements in myoblasts. Cytoskeleton (Hoboken) 71:564–572, 10.1002/cm.21188. [DOI] [PubMed] [Google Scholar]
- Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, et al. 2012. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol 32:309–319, 10.1128/mcb.05603-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jornot L, Maechler P, Wollheim CB, Junod AF. 1999. Reactive oxygen metabolites increase mitochondrial calcium in endothelial cells: Implication of the ca2+/na+ exchanger. J Cell Sci 112 ( Pt 7):1013–1022, [DOI] [PubMed] [Google Scholar]
- Joshi HC. 1994. Microtubule organizing centers and gamma-tubulin. Curr Opin Cell Biol 6:54–62, [DOI] [PubMed] [Google Scholar]
- Juan-Sanz Jd, Holt GT, Schreiter ER, Juan Fd, Kim DS, Ryan TA. 2017. Axonaol endoplasmic reticulum ca2+ content controls release probability in cns nerve terminals. Neuron 93:867–881, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaddour-Djebbar I, Choudhary V, Brooks C, Ghazaly T, Lakshmikanthan V, Dong Z, et al. 2010. Specific mitochondrial calcium overload induces mitochondrial fission in prostate cancer cells. Int J Oncol 36:1437–1444, [DOI] [PubMed] [Google Scholar]
- Kinet JP. 1999. The high-affinity ige receptor (fc epsilon ri): From physiology to pathology. Annu Rev Immunol 17:931–972, [DOI] [PubMed] [Google Scholar]
- Koeppe ES, Ferguson KK, Colacino JA, Meeker JD. 2013. Relationship between urinary triclosan and paraben concentrations and serum thyroid measures in nhanes 2007–2008. Sci Total Environ 445–446:299–305, 10.1016/j.scitotenv.2012.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komissarova EV, Saha SK, Rossman TG. 2005. Dead or dying: The importance of time in cytotoxicity assays using arsenite as an example. Toxicol Appl Pharmacol 202:99–107, 10.1016/j.taap.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Kux L 2016. Federal register vol. 81 no. 126 Available: https://www.gpo.gov/fdsys/pkg/FR-2016-06-30/pdf/2016-15410.pdf. [Google Scholar]
- Li S, Zhao J, Wang G, Zhu Y, Rabito F, Krousel-Wood M, et al. 2015. Urinary triclosan concentrations are inversely associated with body mass index and waist circumference in the us general population: Experience in nhanes 2003–2010. Int J Hyg Environ Health 218:401–406, 10.1016/j.ijheh.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YJ. 2000. Buccal absorption of triclosan following topical mouthrinse application. Am J Dent 13:215–217, [PubMed] [Google Scholar]
- Liu X, Hajnoczky G. 2011. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell Death Differ 18:1561–1572, 10.1038/cdd.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Passonneau JV. 1969. Phosphoglucomutase kinetics with the phosphates of fructose, glucose, mannose, ribose, and galactose. J Biol Chem 244:910–916, [PubMed] [Google Scholar]
- Lv Y, Rui C, Dai Y, Pang Q, Li Y, Fan R, et al. 2016. Exposure of children to bpa through dust and the association of urinary bpa and triclosan with oxidative stress in guangzhou, china. Environ Sci Process Impacts 18:1492–1499, 10.1039/c6em00472e. [DOI] [PubMed] [Google Scholar]
- Lyman FL, Furia TE. 1968. Toxicology of 2,4,4’-trichloro-2’-hydro xyphenyl ether. IMS Ind Med Surg 37:546, [PubMed] [Google Scholar]
- Ma HT, Beaven MA. 2011. Regulators of ca(2+) signaling in mast cells: Potential targets for treatment of mast cell-related diseases? Adv Exp Med Biol 716:62–90, 10.1007/978-1-4419-9533-9_5. [DOI] [PubMed] [Google Scholar]
- Manevski N, Swart P, Balavenkatraman KK, Bertschi B, Camenisch G, Kretz O, et al. 2015. Phase ii metabolism in human skin: Skin explants show full coverage for glucuronidation, sulfation, n -acetylation, catechol methylation, and glutathione conjugation. Drug Metab Dispos 43:126–139, 10.1124/dmd.114.060350. [DOI] [PubMed] [Google Scholar]
- Maro B, Marty MC, Bornens M. 1982. In vivo and in vitro effects of the mitochondrial uncoupler fccp on microtubules. EMBO J 1:1347–1352, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. 2007. Circumventing the crabtree effect: Replacing media glucose with galactose increases susceptibility of hepg2 cells to mitochondrial toxicants. Toxicol Sci 97:539–547, 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- Mazel T, Raymond R, Raymond-Stintz M, Jett S, Wilson BS. 2009. Stochastic modeling of calcium in 3d geometry. Biophys J 96:1691–1706, 10.1016/j.bpj.2008.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe DD, Baram D, Mekori YA. 1997. Mast cells. Physiol Rev 77:1033–1079, [DOI] [PubMed] [Google Scholar]
- Millard PJ, Ryan TA, Webb WW, Fewtrell C. 1989. Immunoglobulin e receptor cross -linking induces oscillations in intracellular free ionized calcium in individual tumor mast cells. J Biol Chem 264:19730–19739, [PubMed] [Google Scholar]
- Mohr FC, Fewtrell C. 1987a. Depolarization of rat basophilic leukemia cells inhibits calcium uptake and exocytosis. J Cell Biol 104:783–792, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr FC, Fewtrell C. 1987b. The relative contributions of extracellular and intracellular calcium to secretion from tumor mast cells. Multiple effects of the proton ionophore carbonyl cyanide m-chlorophenylhydrazone. J Biol Chem 262:10638–10643, [PubMed] [Google Scholar]
- Moss T, Howes D, Williams FM. 2000. Percutaneous penetration and dermal metabolism of triclosan (2,4, 4’-trichloro-2’-hydroxydiphenyl ether). Food Chem Toxicol 38:361–370, [DOI] [PubMed] [Google Scholar]
- Newton AP, Cadena SM, Rocha ME, Carnieri EG, Martinelli de Oliveira MB. 2005. Effect of triclosan (trn) on energy-linked functions of rat liver mitochondria. Toxicol Lett 160:49–59, 10.1016/j.toxlet.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Okuda M, Lee HC, Kumar C, Chance B. 1992. Comparison of the effect of a mitochondrial uncoupler, 2,4-dinitrophenol and adrenaline on oxygen radical production in the isolated perfused rat liver. Acta Physiol Scand 145:159–168, 10.1111/j.1748-1716.1992.tb09351.x. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Yamada K, Kazanietz MG, Blumberg PM, Beaven MA. 1993. Different isozymes of protein kinase c mediate feedback inhibition of phospholipase c and stimulatory signals for exocytosis in rat rbl-2h3 cells. J Biol Chem 268:2280–2283, [PubMed] [Google Scholar]
- Pacher P, Csordas P, Schneider T, Hajnoczky G. 2000. Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J Physiol 529 Pt 3:553–564, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RK, Hutchinson LM, Burpee BT, Tupper EJ, Pelletier JH, Kormendy Z, et al. 2012. Antibacterial agent triclosan suppresses rbl-2h3 mast cell function. Toxicol Appl Pharmacol 258:99–108, [DOI] [PubMed] [Google Scholar]
- Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, et al. 2010. Nclx is an essential component of mitochondrial na+/ca2+ exchange. Proc Natl Acad Sci U S A 107:436–441, 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng TI, Jou MJ. 2010. Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci 1201:183–188, 10.1111/j.1749-6632.2010.05634.x. [DOI] [PubMed] [Google Scholar]
- Peralta RA, Bortoluzzi AJ, de Souza B, Jovito R, Xavier FR, Couto RA, et al. 2010. Electronic structure and spectro-structuralcorrelations of fe(iii)zn(ii) biomimetics for purple acid phosphatases: Relevance to DNA cleavage and cytotoxic activity. Inorg Chem 49:11421–11438, 10.1021/ic101433t. [DOI] [PubMed] [Google Scholar]
- Philippat C, Botton J, Calafat AM, Ye X, Charles MA, Slama R. 2014. Prenatal exposure to phenols and growth in boys. Epidemiology 25:625–635, 10.1097/ede.0000000000000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planchart A, Mattingly CJ, Allen D, Ceger P, Casey W, Hinton D, et al. 2016. Advancing toxicology research using in vivo high throughput toxicology with small fish models. Altex 33:435–452, 10.14573/altex.1601281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przygodzki T, Sokal A, Bryszewska M. 2005. Calcium ionophore a23187 action on cardiac myocytes is accompanied by enhanced production of reactive oxygen species. Biochim Biophys Acta 1740:481–488, 10.1016/j.bbadis.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Putney JW Jr. 1986. A model for receptor-regulated calcium entry. Cell Calcium 7:1–12, [DOI] [PubMed] [Google Scholar]
- Quintana A, Schwarz EC, Schwindling C, Lipp P, Kaestner L, Hoth M. 2006. Sustained activity of calcium release-activated calcium channels requires translocation of mitochondria to the plasma membrane. J Biol Chem 281:40302–40309, 10.1074/jbc.M607896200. [DOI] [PubMed] [Google Scholar]
- Raftery TD, Jayasundara N, Di Giulio RT. 2017. A bioenergetics assay for studying the effects of environmental stressors on mitochondrial function in vivo in zebrafish larvae. Comp Biochem Physiol C Toxicol Pharmacol 192:23–32, 10.1016/j.cbpc.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalho-Santos J, Varum S, Amaral S, Mota PC, Sousa AP, Amaral A. 2009. Mitochondrial functionality in reproduction: From gonads and gametes to embryos and embryonic stem cells. Hum Reprod Update 15:553–572, 10.1093/humupd/dmp016. [DOI] [PubMed] [Google Scholar]
- Rao W, Zhang L, Peng C, Hui H, Wang K, Su N, et al. 2015. Downregulation of stim2 improves neuronal survival after traumatic brain injury by alleviating calcium overload and mitochondrial dysfunction. Biochim Biophys Acta 1852:2402–2413, 10.1016/j.bbadis.2015.08.014. [DOI] [PubMed] [Google Scholar]
- Reitzer LJ, Wice BM, Kennell D. 1979. Evidence that glutamine, not sugar, is the major energy source for cu ltured hela cells. J Biol Chem 254:2669–2676, [PubMed] [Google Scholar]
- Riva C, Cristoni S, Binelli A. 2012. Effects of triclosan in the freshwater mussel dreissena polymorpha: A proteomic investigation. Aquat Toxicol 118–119:62–71, 10.1016/j.aquatox.2012.03.013. [DOI] [PubMed] [Google Scholar]
- Rottenberg H, Wu S. 1998. Quantitative assay by flow cytometry of the mitochondrial membrane potential in intact cells. Biochim Biophys Acta 1404:393–404, [DOI] [PubMed] [Google Scholar]
- Rover JA, Leu-Wai-See P. 2014. Role of colgate total toothpaste in helping control plaque and gingivitis. Am J Den t 27:167–170, [PubMed] [Google Scholar]
- Rusan NM, Fagerstrom CJ, Yvon AM, Wadsworth P. 2001. Cell cycle-dependent changes in microtubule dynamics in living cells expressing green fluorescent protein-alpha tubulin. Mol Biol Cell 12:971–980, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta K, Douglas S, Parekh AB. 2014. Mitochondrial calcium uniporter mcu supports cytoplasmic ca2+ oscillations, store-operated ca2+ entry and ca2+-dependent gene expression in response to receptor stimulation. PLoS One 9:e101188, 10.1371/journal.pone.0101188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandborgh-Englund G, Adolfsson-Erici M, Odham G, Ekstrand J. 2006. Pharmacokinetics of triclosan following oral ingestion in humans. J Toxicol Environ Health A 69:1861–1873, [DOI] [PubMed] [Google Scholar]
- Shim J, Weatherly LM, Luc RH, Dorman MT, Neilson A, Ng R, et al. 2016. Triclosan is a mitochondrial uncoupler in live zebrafish. J Appl Toxicol 36:1662–1667, 10.1002/jat.3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver R, Curley JP. 2013. Mast cells on the mind: New insights and opportunities. Trends Neurosci 36:513–521, [DOI] [PubMed] [Google Scholar]
- Smith AJ, Pfeiffer JR, Zhang J, Martinez AM, Griffiths GM, Wilson BS. 2003. Microtubule-dependent transport of secretory vesicles in rbl-2h3 cells. Traffic 4:302–312, [DOI] [PubMed] [Google Scholar]
- Smith CS, Joseph N, Rieger B, Lidke KA. 2010. Fast, single-molecule localization that achieves theoretically minimum uncertainty. Nat Methods 7:373–375, 10.1038/nmeth.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanier AJ, Fausnight T, Camacho TF, Braun JM. 2014. The associations of triclosan and paraben exposure with allergen sensitization and wheeze in children. Allergy Asthma Proc 35:475–481, 10.2500/aap.2014.35.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MG, Williams J, Munoz-Pinedo C, Perkins GA, Brown JM, Ellisman MH, et al. 2007. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat Cell Biol 9:1057–1065, 10.1038/ncb1630. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Kanemaru K, Ishii K, Ohkura M, Okubo Y, Iino M. 2014. Imaging intraorganellar ca2+ at subcellular resolution using cepia. Nat Commun 5:4153, 10.1038/ncomms5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki YJ, Ford GD. 1992. Superoxide stimulates ip3-induced ca2+ release from vascular smooth muscle sarcoplasmic reticulum. Am J Physiol 262:H114–116, [DOI] [PubMed] [Google Scholar]
- Swindle EJ, Metcalfe DD, Coleman JW. 2004. Rodent and human mast cells produce functionally significant intracellular reactive oxygen species but not nitric oxide. J Biol Chem 279:48751–48759, [DOI] [PubMed] [Google Scholar]
- Tainter ML, Cutting WC, Stockton AB. 1934. Use of dinitrophenol in nutritional disorders : A critical survey of clinical results. Am J Public Health Nations Health 24:1045–1053, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekawa M, Furuno T, Hirashima N, Nakanishi M. 2012. Mitochondria take up ca2+ in two steps dependently on store-operated ca2+ entry in mast cells. Biol Pharm Bull 35:1354–1360, [DOI] [PubMed] [Google Scholar]
- Tamura I, Kanbara Y, Saito M, Horimoto K, Satoh M, Yamamoto H, et al. 2012. Triclosan, an antibacterial agent, increases intracellular zn(2+) concentration in rat thymocytes: Its relation to oxidative stress. Chemosphere 86:70–75, 10.1016/j.chemosphere.2011.09.009. [DOI] [PubMed] [Google Scholar]
- Tan WP, Suresh S, Tey HL, Chiam LY, Goon AT. 2010. A randomized double-blind controlled trial to compare a triclosan-containing emollient with vehicle for the treatment of atopic dermatitis. Clin Exp Dermatol 35:e109–112, [DOI] [PubMed] [Google Scholar]
- Theoharides TC, Asadi S, Panagiotidou S, Weng Z. 2013. The “missing link” in autoimmunity and autism: Extracellular mitochondrial components secreted from activated live mast cells. Autoimmun Rev 12:1136–1142, 10.1016/j.autrev.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Van Slyke DD, Phillips RA, Hamilton PB, Archibald RM, Futcher PH, Hiller A. 1943. Glutamine as source material of urinary ammonia. J Biol Chem 150:481–482, [Google Scholar]
- Velez MP, Arbuckle TE, Fraser WD. 2015. Female exposure to phenols and phthalates and time to pregnancy: The maternal-infant research on environmental chemicals (mirec) study. Fertil Steril 103:1011–1020.e1012, 10.1016/j.fertnstert.2015.01.005. [DOI] [PubMed] [Google Scholar]
- Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, et al. 2006. Cracm1 is a plasma membrane protein essentialfor store-operated ca2+ entry. Science 312:1220–1223, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh LJ, Davis MF, Xu LJ, Savage NW. 1995. Relationship between mast cell degranulation and inflammation in the oral cavity. J Oral Pathol Med 24:266–272, [DOI] [PubMed] [Google Scholar]
- Walsh LJ. 2003. Mast cells and oral inflammation. Crit Rev Oral Biol Med 14:188–198, [DOI] [PubMed] [Google Scholar]
- Wang CF, Tian Y. 2015. Reproductive endocrine-disrupting effects of triclosan: Population exposure, present evidence and potential mechanisms. Environ Pollut 206:195–201, 10.1016/j.envpol.2015.07.001. [DOI] [PubMed] [Google Scholar]
- Wang T, Marquardt C, Foker J. 1976. Aerobic glycolysis during lymphocyte proliferation. Nature 261:702–705, [DOI] [PubMed] [Google Scholar]
- Wang X, Chen X, Feng X, Chang F, Chen M, Xia Y, et al. 2015. Triclosan causes spontaneous abortion accompanied by decline of estrogen sulfotransferase activity in humans and mice. Sci Rep 5:18252, 10.1038/srep18252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherly LM, Kennedy RH, Shim J, Gosse JA. 2013. A microplate assay to assess chemical effects on rbl-2h3 mast cell degranulation: Effects of triclosan without use of an organic solvent. J Vis Exp:e50671, 10.3791/50671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherly LM, Shim J, Hashmi HN, Kennedy RH, Hess ST, Gosse JA. 2016. Antimicrobial agent triclosan is a proton ionophore uncoupler of mitochondria in living rat and human mast cells and in primary human keratinocytes. J Appl Toxicol 36:777–789, 10.1002/jat.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherly LM, Gosse JA. 2017. Triclosan exposure, transformation, and human health effects. J Toxicol Environ Health B Crit Rev 20:447–469, 10.1080/10937404.2017.1399306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Qiao P, Shi Y, Ruan Y, Yin J, Wu Q, et al. 2017. Triclosan/triclocarban levels in maternal and umbilical blood samples and their association with fetal malformation. Clin Chim Acta 466:133–137, 10.1016/j.cca.2016.12.024. [DOI] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. 2012. Mitochondrial fission, fusion, and stress. Science 337:1062–1065, 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yueh MF, Taniguchi K, Chen S, Evans RM, Hammock BD, Karin M, et al. 2014. The commonly used antimicrobial additive triclosan is a liver tumor promoter. Proc Natl Acad Sci U S A 111:17200–17205, 10.1073/pnas.1419119111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Alysandratos KD, Angelidou A, Asadi S, Sismanopoulos N, Delivanis DA, et al. 2011. Human mast cell degranulation and preformed tnf secretion require mitochondrial translocation to exocytosis sites: Relevance to atopic dermatitis. J Allergy Clin Immunol 127:1522–1531.e 1528, 10.1016/j.jaci.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Weng Z, Sismanopoulos N, Asadi S, Therianou A, Alysandratos KD, et al. 2012. Mitochondria distinguish granule-stored from de novo synthesized tumor necrosis factor secretion in human mast cells. Int Arch Allergy Immunol 159:23–32, 10.1159/000335178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.