

Abstract
DNA replication machinery is responsible for accurate and efficient duplication of the chromosome. Since inhibition of DNA replication can lead to replication fork stalling, resulting in DNA damage and apoptotic death, inhibitors of DNA replication are commonly used in cancer chemotherapy. Ribonucleotide reductase (RNR) is the rate-limiting enzyme in the biosynthesis of deoxyribonucleoside triphosphates (dNTPs) that are essential for DNA replication and DNA damage repair. Gemcitabine, a nucleotide analog that inhibits RNR, has been used to treat various cancers. However, patients often develop resistance to this drug during treatment. Thus, new drugs that inhibit RNR are needed to be developed. In this study, we identified a synthetic analog of resveratrol (3,5,4’-trihydroxy-trans-stilbene), termed DHS (trans-4,4’-dihydroxystilbene), that acts as a potent inhibitor of DNA replication. Molecular docking analysis identified the RRM2 (ribonucleotide reductase regulatory subunit M2) of RNR as a direct target of DHS. At the molecular level, DHS induced cyclin F-mediated down-regulation of RRM2 by the proteasome. Thus, treatment of cells with DHS reduced RNR activity and consequently decreased synthesis of dNTPs with concomitant inhibition of DNA replication, arrest of cells at S-phase, DNA damage, and finally apoptosis. In mouse models of tumor xenografts, DHS was efficacious against pancreatic, ovarian, and colorectal cancer cells. Moreover, DHS overcame both gemcitabine resistance in pancreatic cancer and cisplatin resistance in ovarian cancer. Thus, DHS is a novel anti-cancer agent that targets RRM2 with therapeutic potential either alone or in combination with other agents to arrest cancer development.
Keywords: DHS, DNA replication, pancreatic cancer, RRM2, gemcitabine resistance
Introduction
DNA replication machinery is responsible for accurate and efficient duplication of the chromosome. Inhibition of DNA replication can cause replication fork stalling, resulting in DNA damage and apoptotic death. Therefore, inhibitors of DNA replication proteins have been commonly used in anti-cancer for decades [1, 2]. Indeed, a broad spectrum of DNA-damaging agents, including alkylating agents, platinum drugs, topoisomerase inhibitors (irinotecan and etoposide), gemcitabine, and 5-fluorouracil have been used in the treatment of cancers [3, 4]. Additionally, various chemicals that target DNA damage response, such as Chk1 inhibitors (UCN-01, LY2606368, and AZD7762), and ATR inhibitor (AZD6738), and PARP1 inhibitor (Olaparib) either have been or are being actively developed as anti-cancer agents in monotherapy and in combinatorial therapies [4].
Ribonucleotide reductase (RNR) catalyzes the rate-limiting step in the biosynthesis of dNTPs, which are essential for DNA replication and DNA damage repair [5]. RNR consists of two RRM1 subunits and two RRM2 subunits [6]. The relevance of RRM2 to cancer is underscored by the fact that RRM2 is overexpressed in gastric, ovarian, bladder and colorectal cancers [7–11], and that RRM2 overexpression contributes to gemcitabine resistance in human oral [12] and pancreatic [13] cancer cells. Furthermore, overexpression of RRM2 in breast cancer cells causes resistance to tamoxifen [14]. Therefore, inhibitors of RRM2, such as hydroxyurea (HU), 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) and antisense GTI-2040, are currently used to treat chronic myelogenous leukemia and head and neck cancer [15, 16]. Targeting RRM2 is also a strategy for overcoming gemcitabine resistance [17]. For example, COH29 (a RRM2 inhibitor currently in a phase I clinical trial) inhibits the growth of cancer cells that are resistant to HU and gemcitabine [18, 19]. Another RRM2 inhibitor, GW8510, has been reported to suppress colorectal cancer growth [20]. Together, these studies demonstrate that RRM2 is an important target for the development of novel anticancer drugs [5]. RRM2B is one small subunit of RRM2, which is induced by p53 and required for DNA repair and mtDNA synthesis [21, 22].
Resveratrol (3,5,4’-trihydroxy-trans-stilbene) is a natural stilbene produced by plants in response to stressful insults, such as fungal infection and UV irradiation [23]. It has been shown that resveratrol acts as an antioxidant and inhibits a number of factors, including low density lipoproteins (LDL) [24], insulin-like growth factor-1 (IGF-1), IGF-binding protein-3 (IGFBP-3) [25], Sirt3 [26], mTOR complex 1 (mTORC1) [27], CYP1A1, CYP1A2, and CYP1B1 [28, 29]. Resveratrol is also reported to reduce the activity of ribonucleotide reductase [30]. Since the first demonstration of resveratrol’s anti-cancer activity by Jang et al [31], hundreds of studies have confirmed the anti-cancer activity of resveratrol in cells derived from a wide variety of cancers, including colon cancer, liver cancer, neuroendocrine tumor, multiple myeloma, and prostate cancer [32]. Resveratrol was shown to have synergistic effects with cisplatin or gemcitabine on several cancers, including pancreatic cancer cells [33], non-small cell lung cancer cell [34, 35], and ovarian cancer cells [36]. In addition, synthetic resveratrol analogues have been developed with more efficacious anti-cancer activity. One such analog, DMU-212 (3,4,5,4’-tetramethoxystilbene), has been demonstrated to be effective against colon and ovarian cancer cells [37–39]. Another analog, DHS (4,4’-trans-dihydroxystilbene), is more effective than resveratrol at inhibiting the growth of human promyelocytic leukemia cells, and the transformation of murine fibroblasts, MCF7 human breast cancer cells, human neuroblastoma cells, and the metastasis of human lung cancer cells [40–45]. However, the mechanism by which DHS inhibits cell proliferation and induces apoptosis remains unknown.
In this study, we compared the anti-cancer activity of DHS with that of resveratrol and several synthetic resveratrol analogs against a panel of human cancer cell lines. The results revealed DHS as the most potent growth inhibitory member of this group against a broad spectrum of cancer cells. At the molecular level, DHS binds directly to RRM2 in cells, resulting in cyclin F-mediated degradation of RRM2 by the proteasome. This degradation reduced cellular RNR activity, resulting in decreased synthesis of dNTPs, inhibition of DNA replication, DNA damage, S-phase arrest, and finally apoptosis. In addition, DHS inhibited cancer cell proliferation synergistically with gemcitabine and cisplatin, and DHS was effective against gemcitabine-resistant pancreatic cancer cells and cisplatin-resistant ovarian cancer cells, both in vitro and in vivo. Thus, DHS is a promising anti-cancer agent that inhibits DNA replication by targeting RRM2 directly.
Results
Resveratrol and its analogs inhibit cell proliferation
Resveratrol inhibits cell proliferation in a variety of cell types, but the IC50 values were relatively high. For example, the IC50 values for resveratrol on HCT116 coloncarcinoma, U2OS osteosarcoma and MDA-231adenocarcinoma cell lines were approximately 24.47 ± 3.32 μM, 30.21 ± 4.22 μM, and 42.41 ± 4.90 μM, respectively (Fig. 1b). To identify more potent compounds that inhibit cell proliferation, we synthesized five analogs of resveratrol based on the structure similarity (Fig. 1a). Among these analogs, DHS was consistently and much more inhibitory on cancer cell proliferation (2.30 ± 0.78 μM, 1.23 ± 0.73 μM, 5.32 ± 3.38 μM, respectively) than resveratrol (Fig. 1b and Supplementary Table 1). Interestingly, DHS was also able to inhibit proliferation of non-cancer cells, including hTert-RPE1, hTert-BEAS-2B, and hTert-HCA2 with IC50 values at 19.61 ± 2.23 μM, 10.49 ± 1.23 μM, and 7.53 ± 1.63 μM, respectively.
Fig. 1.
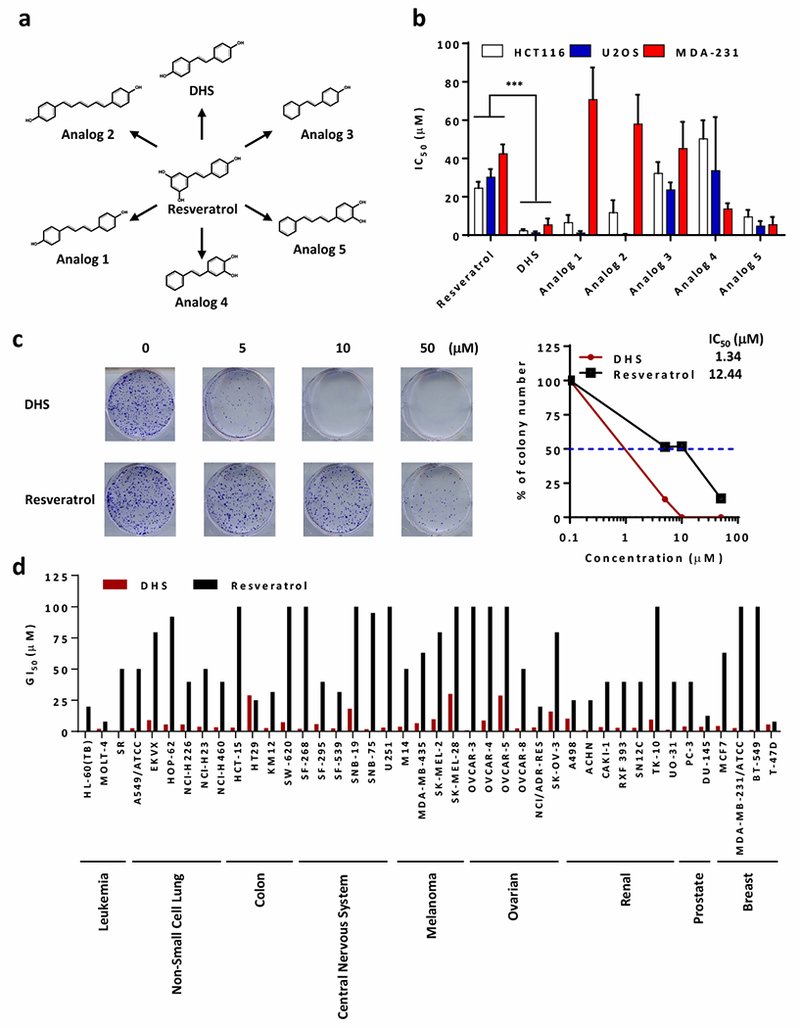
Identification of resveratrol and DHS suppressing cancer cells growth. a Chemical structures of resveratrol, and its analogs including DHS as well as Analog1 to 5. b The IC50 (μM) values of indicated compounds against HCT116, U2OS, and MDA-231 cells. c The colony formation of HCT116 cells treated with DHS or resveratrol as indicated concentration for 14 days. d The GI50 (concentration of 50% growth inhibition, μM) values of DHS and resveratrol against various cancer cell lines from results of NCI-60 Human Tumor Cell Lines Screen (dtp.cancer.gov).
Clonogenic assays on HCT116 cells revealed that DHS was approximately 10 times more effective than resveratrol (Fig. 1c). Although the analogs 1-5 also had inhibitory effect on cell proliferation, their IC50 values vary greatly according to cell types (Fig. 1b). Therefore, inhibition of cell proliferation by DHS became the primary focus of this study.
The inhibitory effect of DHS on the proliferation of 42 cell lines derived from 9 cancer types obtained from the NCI Development Therapeutics Program was markedly 10 to 25-time more effective than resveratrol (Fig. 1d). These results revealed DHS as a potent and general inhibitor of cell proliferation.
DHS inhibits DNA synthesis and blocks cell cycle at S-phase
The effect of DHS on progression of HCT116 cells through the cell cycle were striking. DHS at 10 μM induced a robust arrest of cells in S-phase, whereas resveratrol at the same concentration had no effect (Fig. 2a). These results suggested that DHS inhibited DNA replication. To test this hypothesis, DNA in HCT116 cells was labeled in vivo with BrdU in the presence or absence of DHS. Fluorescence activated cell sorting (FACS) was used to quantify the extent of BrdU incorporation into DNA. These results revealed that a 20-minute treatment with DHS reduced DNA synthesis in a dose-dependent fashion in proportion to the fraction of cells arrested at S-phase (Fig. 2b). Thus, DHS inhibited cell proliferation by inhibiting DNA synthesis at S-phase, thereby arresting cell proliferation.
Fig. 2.
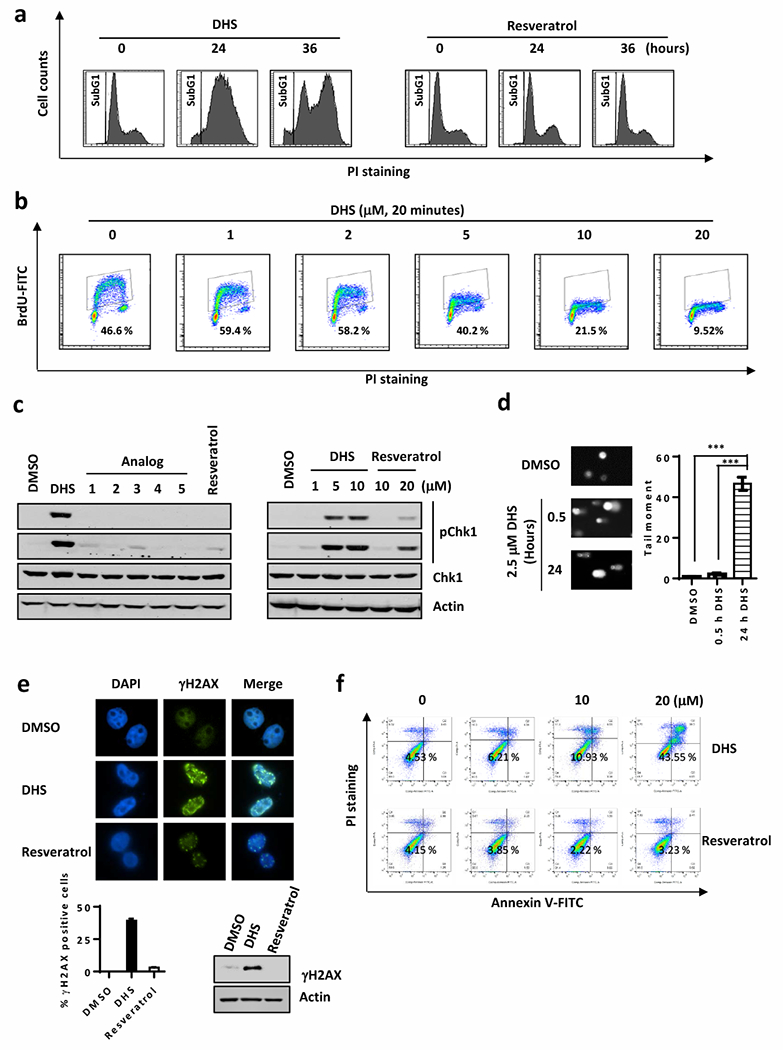
DHS induces DNA damage and inhibits cell cycle. a Cell-cycle arrested by treatments of DHS or resveratrol at 10 μM for 24 or 36 hours. b FACS to detect BrdU incorporation in DHS-treated U2OS cells. c Cells were harvested after treated with DHS, resveratrol, and analog1 to 5 for 20 minutes for Western blotting for indicated antibodies. d Modified comet assay with alkaline single cell electrophoresis to detect double strand break induced in DHS treated cells. Right panel is the quantification of cells with tail moment. e Immunostaining to examine γH2AX in DHS-treated U2OS cells. Lower panels are the quantification of the immunostaining results and Western blotting for γH2AX. f Annexin V staining for flow cytometry to analyze populations of apoptotic cells in HCT116 cells treated with DHS or resveratrol.
DHS induces DNA damage and apoptosis
To determine whether or not DHS also induces DNA damage, we examined the expression of pChk1 (Ser345) by Western blotting. Treatment of HCT116 cells with 5 to 10μM DHS for as brief as 20 minutes resulted in a robust accumulation of pChk1 (Ser345), whereas the same concentrations of resveratrol and resveratrol analogs 1 through 5 (Fig. 1a) had no such effect (Fig. 2c). In addition, the modified comet assay with alkaline single cell electrophoresis revealed that DHS induced a significant number of double-strand DNA breaks (Fig. 2d), which was confirmed by the presence of high levels of γH2AX in the nuclei of HCT116 cells that had been treated with 10 μM DHS for 12 hours (Fig. 2e).
DNA damage in cells if unrepaired generally causes apoptosis. DHS apparently induced apoptosis, as evidenced from our quantitative FACS analysis of DHS-treated cells stained for Annexin V (Fig. 2f). In contrast, treatment of HCT116 cells with resveratrol at the same concentrations, had negligible effects on apoptosis (Fig. 2f). Taking together, these findings demonstrated that DHS induced inhibition of DNA synthesis, which resulted in the blocking of DNA replication, S-phase arrest, DNA damage, and finally apoptosis.
DHS inhibits dNTP synthesis by downregulating RRM2
Since DHS suppresses DNA replication, treatment of cells with DHS might inhibit synthesis of the deoxyribonucleoside triphosphates (dNTPs) needed for DNA replication and DNA damage repair. Therefore, the effect of DHS on the levels of dNTPs was quantified in HCT116 cells. The results revealed that DHS induced a dose-dependent decrease in the dATP level in the cells (Fig. 3a) that was fully evident within two hours (Fig. 3b). Moreover, the levels of dTTP were as sensitive to DHS as were the levels of dATP (Fig. 3b). In contrast, dCTP level was less reduced but the reduction was nevertheless still significant and the reduction in the level of dGTP was only marginal. We further found that DHS (10 μM) significantly suppressed the levels of dATP while resveratrol did not affect the level of dATP at the same dose (10 μM, Fig. 3c). These results suggested that RNR, the enzyme that catalyzes the rate-limiting step in the synthesis of dNTPs, was the target through which DHS inhibited DNA synthesis. RNR is composed of two RRM1 and two RRM2 subunits [6], and DHS did, in fact, reduce the levels of both subunits (Fig. 3d), suggesting that DHS decreased RNR activity, which resulted in decreased synthesis of dNTPs.
Fig. 3.
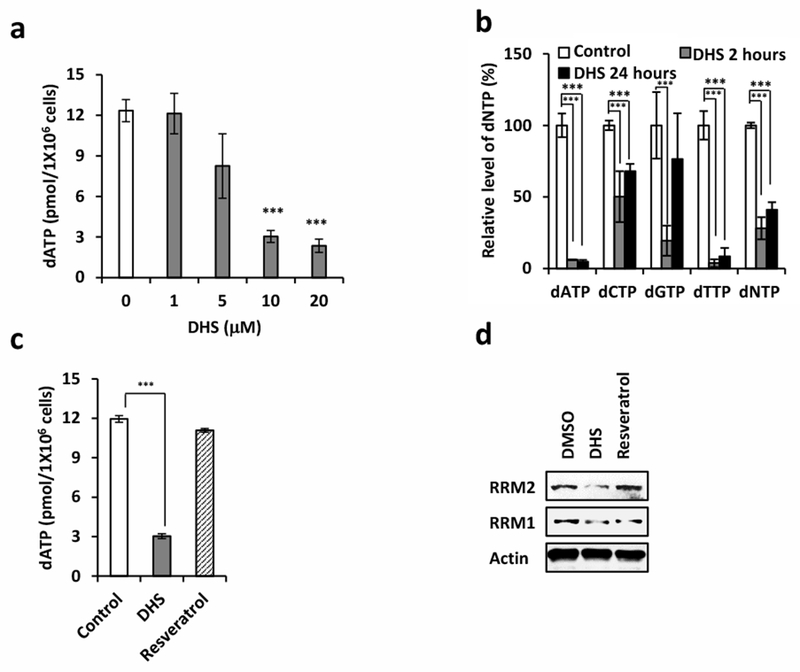
DHS attenuates dNTP synthesis by downregulating RRM2. a Analysis of dATP production in DHS-treated cells. b dNTP pool assay to examine the synthesis of dATP, dCTP, dGTP, and dTTP in DHS-treated cells for 2 or 24 hours. c Analysis of dATP production in DHS or resveratrol-treated cells. d Cells were harvested after treated with DHS or resveratrol at 10 μM for 20 minutes for Western blotting for indicated proteins.
DHS binds to RRM2 directly
Gemcitabine is known to bind RRM1 [46, 47] and HU to RRM2 [48]. To determine whether DHS physically binds to RRM1 or RRM2 or both, cells were subjected to a thermal shift assay [49]. The thermal shift assay compares the thermal stabilization of a purified protein in ligand-free and ligand-bound states, because ligand-free proteins are more readily denatured than ligand-bound proteins at high temperatures. Thus, proteins tightly bound to the ligand of interest are recovered in the soluble fraction after cell lysis and can be detected by quantitative Western blotting [49].
The results from this experiment confirmed that treatment of HCT116 cells with gemcitabine increased the thermal stability of RRM1, and that treatment with HU caused increased thermal stability of RRM2 (Fig. 4a). In contrast, DHS treatment had no effect on the thermal stability of RRM1 and PCNA protein, a loading control in which DNA bound PCNA retained thermal stability (Fig. 4a). However, DHS treatment increased the thermal stability of RRM2 when cells were pre-treated with the proteasome inhibitor MG132 for one hour (Fig. 4a). It appeared that the effect of DHS on RRM2 stability was dose-dependent (Fig. 4b). These results strongly suggested a specific physical interaction between DHS and RRM2.
Fig. 4.
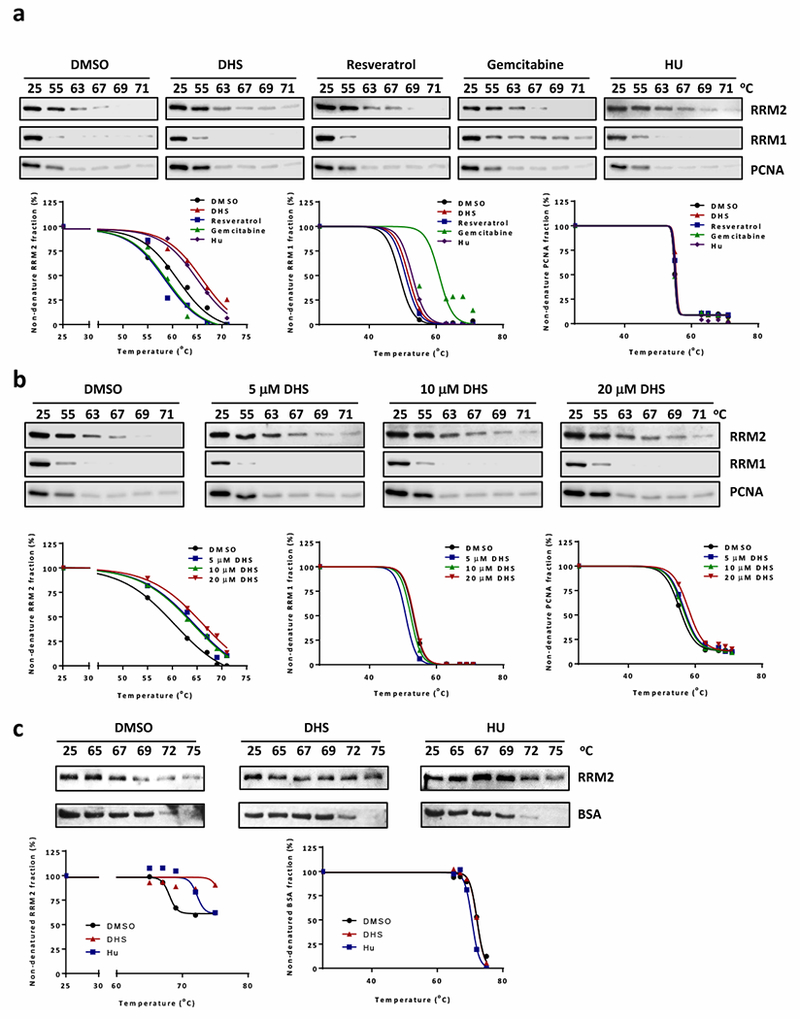
DHS interacts with RRM2. a Cellular thermal shift assay to examine interactions of compounds (10 μM DHS, 10 μM resveratrol, 0.5 μM gemcitabine, or 500 μM HU) with RRM1 and RRM2. Cells were treated with 10 μM MG132 for 1 hour followed by 4-hour incubation with compounds before performing thermal shit assay. Low panel is the charts of percentages of non-denatured protein fraction. b Cellular thermal shift assay to analyze the interaction of DHS (5, 10, or 20 μM) with RRM2. Low panel is the charts of percentages of non-denatured protein fraction. c In vitro thermal shift assay to analyze DHS (10 μM) and RRM2 interaction in vitro. DHS or HU (500 μM) and 400 ng purified RRM2 were incubated for 4 hours. The aliquots were further incubated at 65, 67, 69, 72, or 75 °C for 4 minutes. Low panel is the charts of percentages of non-denatured protein fraction.
The above findings were also validated by applying the thermal shift assay to purified recombinant human RRM2 protein. Purified RRM2 was incubated either with 10 μM DHS or with 500 μM HU for 4 hours before thermal shift assays were performed. Bovine serum albumin (BSA) was used as a loading control. The results demonstrated that both HU and DHS stabilized RRM2 protein, whereas neither HU nor DHS stabilized BSA (Fig. 4c).
A putative binding site in RRM2 protein for DHS was identified through simulation of drug-protein interaction using the virtual docking analysis technique (www.dockingserver.com) and the RRM2 protein structure in the PDB database (2UW2) [50]. Simulation analysis suggested that DHS binding to RRM2 involved residues Val146, Ser150, Gln151, Thr156, Arg159, Cys160, and Ile166 and with an estimated binding free energy of −2.91 kcal/mol (Fig. 5a and Supplementary Fig. 1). This region possesses a ferritin-like superfamily structure and contains 20 amino acid residues from Glu147 to Ile166 (Fig. 5b) that are highly conserved in the human, mouse, rat, chicken, dog, monkey, and fly (Fig. 5c). Therefore, a Flag-tagged mutant human RRM2 gene was constructed in which the codons for the Glu147-Ile166 sequence were deleted. We ectopically expressed these genes in cells and assessed how DHS affected their expression levels by Western blotting and cellular thermal shift assays. DHS treatment reduced the steady-state level of full-length Flag-RRM2 protein but not that of Flag-ΔRRM2 mutant protein (Fig. 5d). Similarly, DHS treatment increased the thermal stability of full-length Flag-RRM2 but not that of the Flag-ΔRRM2 protein (Fig. 5e). To further confirm the interaction sites, we mutated 4 amino acid residues (Val146, Ser150, Gln151, and Ile166) to alanine (donated as Flag-4A-RRM2). DHS treatment did not enhance the thermal stability of the Flag-4A-RRM2 protein (Fig. 5e), indicating that Val146, Ser150, Gln151, and Ile166 residues are critical for DHS to interact with RRM2.
Fig. 5.
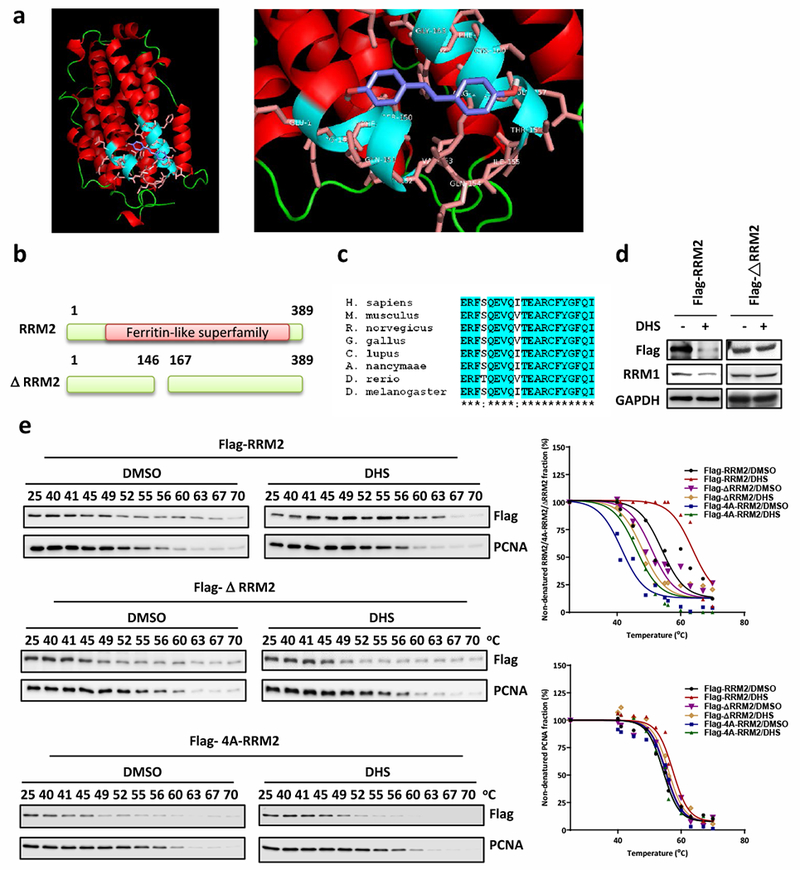
DHS interacts with RRM2 at the ferritin-like superfamily domain. a Computational docking of interaction of DHS and RRM2. b The scheme of RRM2 protein domains. The ΔRRM2 contains a deletion from Val146 to Ala167. c The conserve alignment of RRM2 proteins from various species. d Cells were harvested after transfected Flga-RRM2 or Flag-ΔRRM2 plasmid DNA then treated with DHS for Western blotting for Flag-tag proteins and RRM1. e Cellular thermal shift assay to analyze DHS (10 μM) binding with Flag-RRM2, Flag-ΔRRM2, or Flag4A-RRM2. Right panel is the charts of percentages of non-denatured protein fraction.
DHS induces RRM2 protein degradation via cyclin F-mediated proteasome degradation pathway
To investigate whether the proteasome was involved in DHS-induced down-regulation of RRM2, cells were treated with DHS either in the absence or presence of MG132. The results showed that MG132 prevented DHS from down-regulating both RRM2 and RRM1 (Fig. 6a), indicating DHS-induced degradation of RRM2 by the proteasome. Co-downregulation of RRM1 by DHS suggested that the stability of RRM1 depends on RRM2. To test this hypothesis, cells were transfected with siRNA against either RRM1 or RRM2. The results confirmed that RRM2 knockdown resulted in down-regulation of RRM1 as well, and that RRM1 knockdown also decreased the level of RRM2 at protein level but at not mRNA level (Fig. 6b; Supplementary Fig. 2a). Therefore, the stabilities of RRM2 and RRM1 were interdependent.
Fig. 6.
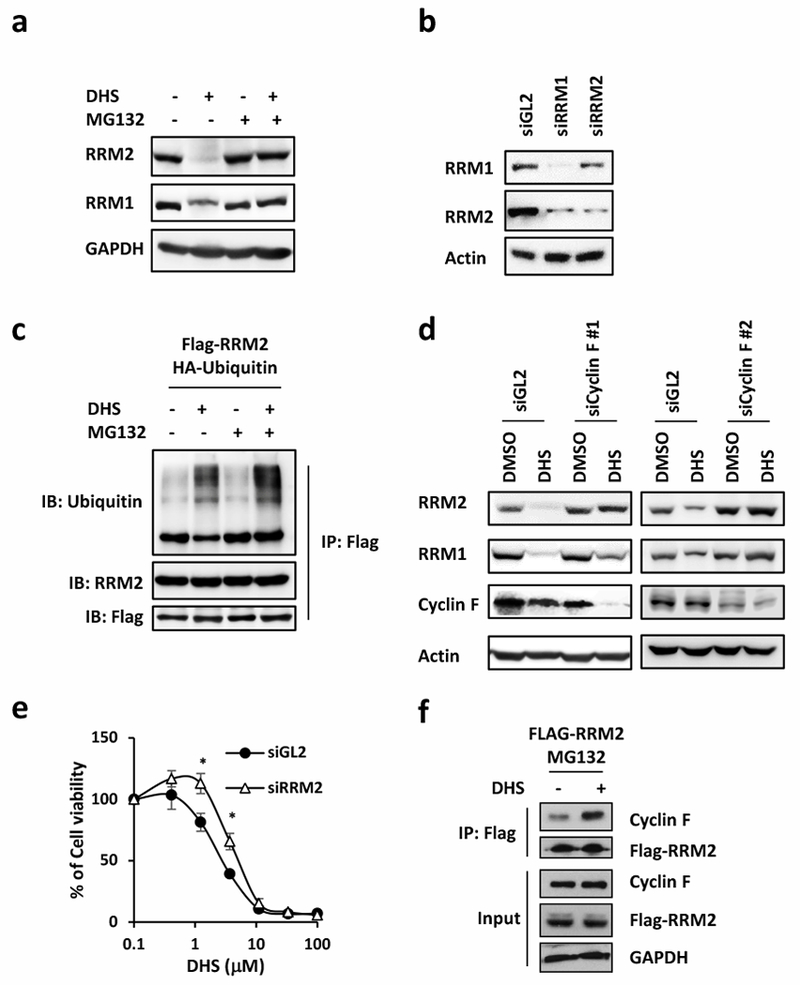
DHS promotes RRM2 degradation through proteasome degradation pathway. a Cells treated with 10 μM MG132 for 1 hour followed by 48-hour incubation with 10 μM DHS for Western blotting for RRM1 and RRM2. b Western blotting to detect RRM1 and RRM2 protein in RRM1 or RRM2 depleted cells. siGL2 is the control. c Cells were transfected with Flag-RRM2 and HA-Ubiquitin. After treated with 10 μM MG132 for 1 hour followed by 48-hour incubation with 10 μM DHS, proteins were immunoprecipitated with Flag beads for Western blotting for ubiquitin, RRM2, and Flag. d Western blotting to detect RRM1 and RRM2 protein amount in cyclin F knockdown cells. e Cell viability of DHS in RRM2-depleted cells. f Proteins were immunoprecipitated with Flag beads from Flag-RRM2 overexpressed cells treated with DHS for Western blotting for RRM1 and Flag.
The ubiquitination status of RRM2 was evaluated in Flag-RRM2 expressing cells. Ubiquitinated FLAG-tagged RRM2 was not detected in the absence of either DHS or MG132, but in the presence of DHS alone, there was significant amount of ubiquitinated FLAG-RRM2 with concomitant reduction in RRM2 (Fig. 6c). Moreover, the amount of ubiquitinated FLAG-RRM2 was higher in the presence of both DHS and MG132 without a reduction in the amount of RRM2 (Fig. 6c), indicating that DHS induced ubiquitination of RRM2 and degradation of RRM2 by the proteasome.
Previous studies have reported that RRM2 was ubiquitinated by Cyclin F, a substrate recognition component of the ubiquitin ligase CRL1 [51], suggesting that cyclin-F might be required for DHS-induced proteasome-mediated degradation of RRM2. In fact, the extent of DHS-induced RRM2 down-regulation was greatly reduced by siRNA induced knockdown of cyclin F (Fig. 6d). Moreover, the viability of RRM2-depleted cells in the presence of DHS increased (Fig. 6e), suggesting that RRM2 is one of the major targets of DHS. Consistently, DHS treatment increased the interaction between cyclin F and RRM2 (Fig. 6f). Taken together, the above results indicated that DHS-induced down-regulation of RRM2 protein required cyclin F-mediated ubiquitination of RRM2 followed by proteasome-dependent degradation of the ubiquitinated RRM2.
Resistance to gemcitabine and HU is overcome by DHS-induced suppression of RRM2
To explore the anti-cancer potential of DHS, the anti-cancer activity of DHS alone and in combination with other anti-cancer drugs was investigated in drug-resistant cancer cells. For example, gemcitabine and HU resistance has been shown to occur during chemotherapy of human oral cancers [12]. The KB-Gem is derived from the human KB oral cancer cell line as a gemcitabine resistant sub-line and the KB-HU as a HU resistant sub-line. Strikingly, both drug resistant cell lines selectively overexpress RRM2 (Fig. 7a). Since we found both KB-Gem and KB-HU overexpressed RRM2 (Fig. 7a), we hypothesized that DHS would overcome gemcitabine and HU resistance by targeting RRM2. To test this hypothesis, we measured the synergy of combined DHS with gemcitabine or HU in resistant cells. The combination of DHS with gemcitabine exhibited high Loewe synergy scores [52] at most of combined dosages, indicating a good synergistic effect (Fig. 7b). Consequently, we examined whether RRM2 protein levels were affected by the combinational treatments in KB-HU and KB-Gem cells by Western blotting. Strikingly, RRM2 was decreased in cells treated with DHS (10 μM), although gemcitabine treatments enhanced RRM2 levels (Fig. 7c). To test whether or not DHS and HU bind to RRM2 at the same site, we conducted cellular thermal shift assay with DHS and/or HU using KB-HU cells. We found that single treatments of DHS or HU stabilized RRM2 protein, while DHS plus HU further enhanced the RRM2 thermal stability (Fig. 7d), suggesting that HU and RRM2 most likely bind to RRM2 at different docking sites.
Fig. 7.
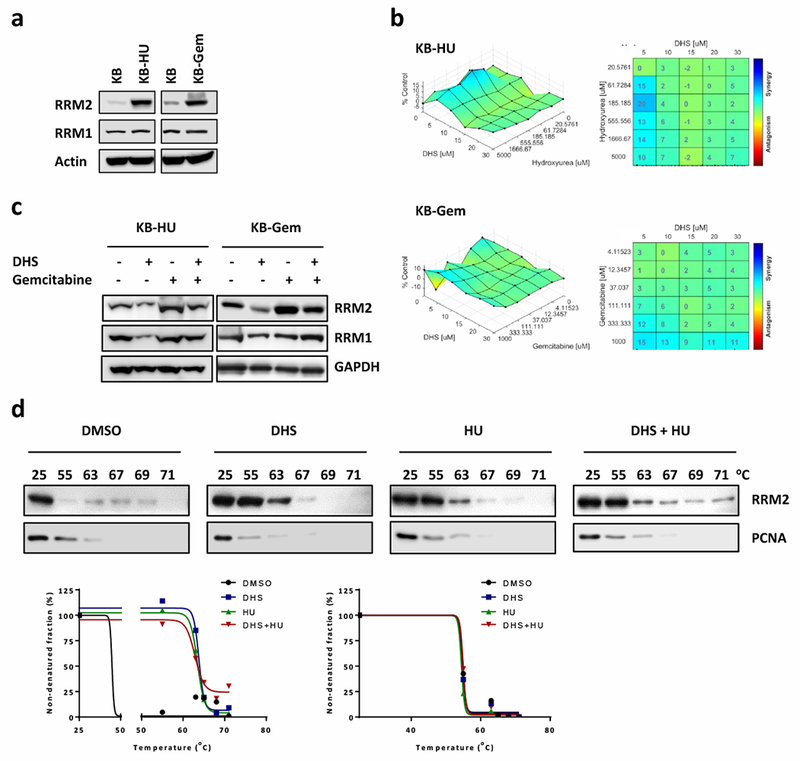
DHS overcomes gemcitabine and HU resistance by downregulating RRM2. a Western blotting to detect RRM1 and RRM2 protein amount in KB, KB-HU, and KB-Gem cells. b Synergy assay of DHS combined with HU against KB-HU (upper panel) and DHS combined with gemcitabine and KB-Gem cells (lower panel). c KB-HU and KB-Gem cells treated with DHS and/or gemcitabine were harvested for Western blotting for RRM1 and RRM2. d Cellular thermal shift assay to examine the interactions of compounds (10 μM DHS, and/or 500 μM HU) with RRM2. Cells treated with 10 μM MG132 for 1 hour and then incubated with indicated compounds for 4-hour before performing thermal shit assay. Low panel is the charts of percentages of non-denatured protein fraction.
Resistance to gemcitabine in pancreatic cancer is overcome by DHS
RRM2 also is overexpressed in gemcitabine resistant pancreatic cancer [13], and survival rates of patients with high RRM2-expressing cancers compared with patients with low RRM2-expressing cancers revealed that patients with high RRM2-expressing cancers had poor survival rates (Fig. 8a). We used two pairs of gemcitabine-resistance pancreatic cancer cells to demonstrate their RRM2 levels. PK-9 and RPK-9 have been report previously [53]. PANC-1-R was generated by long-term gemcitabine treatment from PANC-1 in cell culture and resistant to gemcitabine (Supplementary Fig. 2b). As with human oral cancers, the RRM2 expression levels in PK-9 and PANC-1 cells from pancreatic cancers were substantially lower than RRM2 levels in their gemcitabine-resistant pancreatic cancer counterparts, RPK-9 and PANC-1-R, respectively (Fig. 8b). In contrast, RRM1 levels were upregulated in RPK-9 cells, but not in PANC-1-R cells (Fig. 8b). Significantly, combination of gemcitabine and DHS could overcome gemcitabine resistance by showing high synergy scores in both gemcitabine resistant pancreatic cancer cells (Fig. 8c). In RPK-9 cells, DHS down-regulated RRM2, whereas gemcitabine alone up-regulated RRM2, but together DHS prevented gemcitabine from up-regulating RRM2 (Fig. 8d). Furthermore, combined DHS with gemcitabine enhanced apoptotic cell population in RPK-9 cells (Fig. 8e).
Fig. 8.
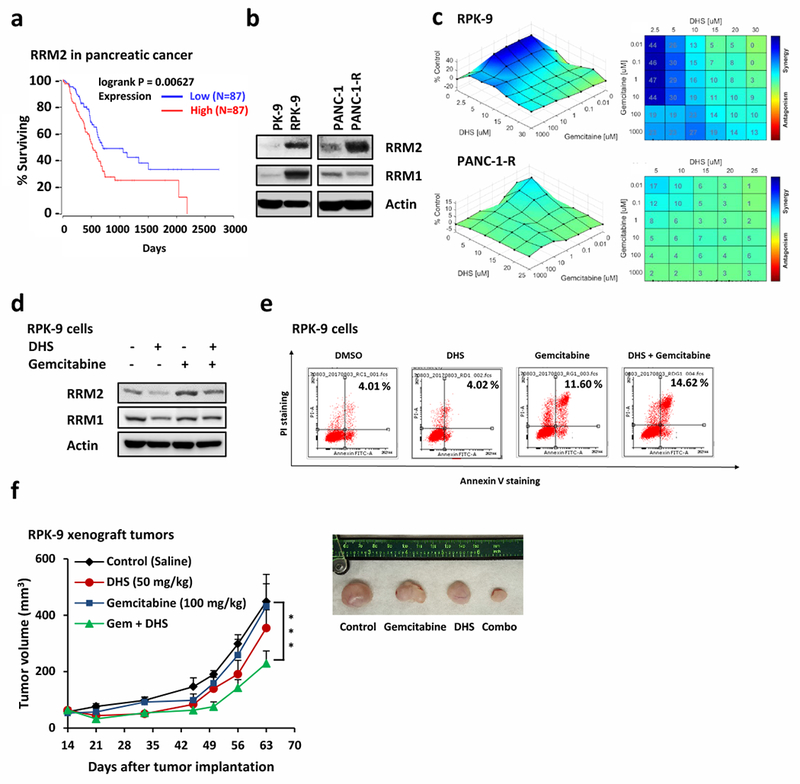
DHS overcomes gemcitabine resistance in pancreatic cancer by targeting RRM2. a Kaplan-Meier curves of overall survival rate based on clinical and molecular data for pancreatic cancer patients. The patients were stratified by the RRM2 expression levels in their tumors. Medium survival, Log-rank (Mantel-Cox) p values and hazard ratios (HR); 95 % confidence interval in parentheses) are shown. Patient information was included in Supplemental Table 2. b Western blotting to detect RRM1 and RRM2 in gemcitabine resistant lines and their parental lines (PK-9/RPK-9 and PANC-1/PANC-1-R). c Synergy assay of combined DHS with gemcitabine against RPK-9 cells (upper panel) and PANC-1-R (lower panel). d RPK-9 cells treated with DHS and/or gemcitabine were harvested for Western blotting for RRM1 and RRM2. e RPK-9 cells treated as indicated were harvested for FACS analysis to examine the apoptosis. f Growth curves of RPK-9 xenograft tumors treated with vehicle, DHS (50 mg/kg IP on days 14 to 28 and 35 to 49), gemcitabine (100 mg/kg IP on days 14, 21, 35 and 49), or DHS plus gemcitabine. Data are represented as means ± SEM, n = 6 mice/group. ***, p < 0.001 by two-way ANOVA. The lower panel is the photograph of the representative tumors from mice on day 63. Ruler scale is in cm.
To determine whether or not DHS and DHS plus gemcitabine had similar effects on pancreatic cancer in vivo, RPK-9 xenograft tumor-bearing mice were treated with vehicle, DHS (50 mg/kg), gemcitabine (100 mg/kg), or DHS plus gemcitabine for 14 consecutive days. The results showed that RPK-9 tumors were not sensitive to either gemcitabine or DHS alone. However, the combination of DHS with gemcitabine significantly inhibited the growth of RPK-9 tumors (Fig. 8f). The body weights of these animals remained constant throughout these treatments, suggesting the absence of collateral toxicity (Supplementary Fig. 3a).
Resistance to cisplatin in ovarian cancer could be overcome by DHS
Cisplatin-resistant human IGROV1 CR ovarian cancer xenografts also were sensitive to DHS, and these tumors were even more sensitive to the combinatorial treatment with DHS plus cisplatin (Supplementary Fig. 3b), suggesting a synergistic effect between DHS and cisplatin. Similar results were obtained with human HCT116 colorectal xenografts. Growth of HCT116 tumors was inhibited either by cisplatin alone or by DHS treatment alone (Supplementary Fig. 3c), but the combination of DHS plus cisplatin was even more effective at inhibiting tumor growth (Supplementary Fig. 3c). Neither the monotherapies nor the combinatorial therapy produced any significant toxicity, as judged by the absence of body weight loss during the treatments (Supplementary Fig. 3c), and by the absence of gross pathological lesions in the liver, heart, lungs, spleen or kidneys (Supplementary Fig. 3d). These results further demonstrated the anti-cancer potential of DHS, especially when applied in combinatorial anti-cancer therapies with other drugs.
Discussion
In this study, we have obtained data indicating that the resveratrol analog, DHS, is a novel inhibitor of RNR via its ability to interact with RRM2. This is an important finding because RNR is composed of two RRM1 and two RRM2 subunits, catalyzes the rate limiting step of the synthesis of dNTPs, which are essential for DNA replication and DNA damage repair [54]. In proliferating cells, RNR activity is regulated in a cell-cycle-dependent fashion to ensure sufficient synthesis of deoxyribonucleotides for DNA replication [55]. Cells maintain a constant level of RRM1 throughout the cell cycle and only up-regulates the level of RRM2 in the S-phase to ensure sufficient RNR activity [55]. Therefore, RNR activity is regulated through the regulation of the expression of RRM2. Based on the data we have presented, we propose that DHS binds to RRM2 and causes ubiquitination of RRM2 by cyclin F and then degradation of the ubiquitinated RRM2 by the proteasome, thereby reducing the cellular RNR activity and impairing the synthesis of deoxynucleotides, which in turn causes inhibition of DNA replication, S-phase arrest and apoptosis of cells.
In comparison to resveratrol and resveratrol analogs 1-5, DHS is unique in that it is a potent inhibitor of the proliferation of a wide spectrum of cancer cell lines (Fig. 1d). The findings of this study attribute the ability of DHS to inhibit cell proliferation to its ability to target and induce down-regulation of RRM2. Our docking simulation analysis identified a binding motif for DHS in RRM2 (Fig. 5a and Supplementary Fig. 1). This motif contains amino acids 146-167, which is critical for DHS binding to RRM2 because deletion of this sequence results in a mutant RRM2 that no longer interacts with DHS (Fig. 5d and e). Interestingly, this amino acid sequence is highly conserved in the RRM2 of different species (Fig. 5c), suggesting functional importance of this sequence. Moreover, this sequence contains a conserved serine, except that it is threonine in zebrafish, and it is a putative phosphorylation site by DNAPK or ATM (http://www.cbs.dtu.dk/services/NetPhos/). Therefore, it raises a possibility that phosphorylation and dephosphorylation of this serine (or threonine) may differentially regulate the stability of RRM2 and hence the activity of RNR.
Over-expression of RRM2 in various cancer types, such as gastric cancer, ovarian cancer, bladder cancer, and colorectal cancers, has been documented [7–11]. Therefore, RRM2 appears to be a target for the development of novel anti-cancer drugs [5]. The anti-cancer drug gemcitabine (2’,2’-difluoro-2’-deoxycytidine; dFdC) is a deoxycytidine analog and is metabolized by cells into the triphosphate form of gemcitabine, dFdCTP (2’,2’-difluoro-2’-deoxycytidine triphosphate), which competes with normal dCTP to be incorporated into DNA during DNA replication [56]. The presence of dFdC in DNA causes DNA replication inhibition, DNA damage and apoptosis [57]. Metabolism of dFdC to dFdCTP depends on dCK (deoxycytidine kinase). Thus deficiency of dCK results in gemcitabine resistance [56]. However, another common mechanism for gemcitabine resistance is RRM2 overexpression [12, 58, 59]. Although RRM2 overexpression in cells does not alter the steady state level of dNTPs, having high RRM2 levels are important for cells under the conditions that limit dNTP synthesis, such as RNR inhibition by HU [60]. It is possible that RRM2 overexpression in gemcitabine-resistant cancer cells is a compensatory mechanism to increase the amount of dCTP to competitively inhibit the incorporation of dFdCTP into DNA, notwithstanding the fact that the expanded dCTP pool causes downregulation of the activity of dCK via a negative-feedback pathway [56]. Given that our results indicate that DHS inhibits growth of cancer cells regardless whether they are sensitive or resistant to gemcitabine, that DHS inhibits RNR-dependent synthesis of dNTPs, that sensitizes gemcitabine-resistant cancer cells to gemcitabine in vitro and in vivo, and that DHS and gemcitabine exhibit synergistic anti-cancer effect in vivo, it is reasonable to suggest that DHS-mediated down-regulation of RRM2 causes reduced dCTP synthesis (Fig. 3b), thereby increasing the amount of dFdC to be incorporated into DNA to cause DNA damage.
Overexpression of RRM2 causes resistance to gemcitabine and other drugs. For example, it has been reported that overexpression of RRM2 in breast cancer cell causes resistance to tamoxifen [61]. This is because overexpression of RRM2 increases NFκB activity and the activities of numerous anti-apoptosis factors. Therefore, the clinical benefit of targeted inhibition of RRM2 by novel agents is beyond sensitization of cancer cells to gemcitabine. Thus, it is also important to recognize the anti-cancer potential of DHS in another way – that is, the ability of DHS to synergize with cisplatin to treat cisplatin resistant and sensitive cancers in vivo. The ability of DHS to synergize with cisplatin to inhibit the growth of cancer can be explained in the following way. By causing RRM2 down-regulation, DHS reduces RNR-dependent synthesis of dNTPs in cancer cells, and consequently it reduces the cells’ ability to repair cisplatin-induced DNA damage, because DNA damage requires deoxyribonucleotides. The implication here is that DHS is most likely to synergize with additional DNA-damaging anti-cancer drugs. Given that it has been reported that DHS exhibits better pharmacokinetic profile than resveratrol [62], we believe it is warranted that the anti-cancer activity of DHS, either in monotherapy or combinatorial therapy, be further investigated.
Materials and Methods
Chemicals and reagents
Resveratrol (3,5,4’-trihydroxy-trans-stilbene), DHS (trans-4,4’-dihydroxystilbene), analog 1 (4,4’-((1E,3E)-buta-1,3-diene-1,4-diyl)diphenol), analog 2 (4,4’-((1E,3E,5E)-hexa-1,3,5-triene-1,6-dyl)diphenol), analog 3 ((E)-4-styrylphenol), analog 4((E)-4-styrylbenzene-1,2-diol), and analog 5 (4-((1E,3E)-4-phenylbuta-1,3-dien-1-yl)benzene-1,2-diol) (Fig. 1a) were synthesized and their structures and purity (>98%) were confirmed by HPLC, MS and 1H NMR spectroscopy, as previously described [63, 64]. Other chemicals and reagents were purchased by Sigma-Aldrich except for where is labeled.
Cell culture
HCT116, U2OS, PANC-1, MDA-231 cells were purchased from the American Type Culture Collection. Cells were cultured in the DMEM medium containing 10% fetal bovine serum, 100 Units/ml penicillin, 100Units/mL streptomycin, 29.2mg/mL L-glutamine at 37°C in a humidified atmosphere with 5% CO2.
PK-9 and its gemcitabine resistant line, RPK-9, were kind gifts from Dr. Yuriko Saiki [53]. KB cells were kind gift from Dr. Yun Yen. IGROV cells were kind of gift from Dr. Wei Zheng. The gemcitabine-resistant PANC-1-R cell line was derived from PANC-1 cell line by continuously treating cultured PANC-1 cells with gemcitabine (from 0.01, 0.03, to 0.1μM for at least two weeks). PK-9, RPK-9, KB, KB-Gem, and KB-Hu were cultured in the RPMI medium. PANC-1, PANC-1-R, and IGROV1 CR [65] cells were cultured with DMEM medium. hTert-RPE1, hTert-BEAS-2B, and hTert-HCA2 cells were kindly provided by Dr. Edward Seto (The George Washington University). These cells were cultured in DMEM medium.
Antibody, plasmid DNA, deletion mutant, protein, and siRNA
Details were provided in Appendix S1 (Supplementary methods).
In Vitro cytotoxicity assay and qPCR
Assays were performed as previously described [65]. Details were provided in Appendix S1 (Supplementary methods).
Western blotting and flow cytometry
Assays were performed as previously described [66].
Modified comet assay
HCT116 cells were incubated with DHS for either 0.5 or 24 hours. Afterward, the cells were irradiated with X-rays at the dose of 20 Gy before being harvested for alkaline single cell electrophoresis as previously described [66].
dNTP assay
dNTP assays were done as previously described [67, 68]. DHS or resveratrol was added at the dose of 10 μM for treatment.
Cellular thermal shift assay and in vitro thermal shift assay
Cellular thermal shift assay was as previously described [49]. Briefly, cells were pretreated with MG132 (10 μM, 1 hour) then incubated with DHS, HU, gemcitabine, or DMSO for 4 hours. After washing with ice-cold PBS (suppled with Protease Inhibitor Cocktail, Roche), cells were aliquoted into PCR tubes (100 μL each) and then incubated at different temperatures (from 25 to 71°C) for 4 minutes. After the cells were frozen and thawed twice using liquid nitrogen, proteins were isolated from the cells after centrifugation and incubated at 70°C for 10 minutes for analysis by Western blotting. In vitro thermal shift assays were performed by mixing purified recombinant human RRM2 protein (400 ng) with either DHS (10 μM) or HU (500 μM) in binding buffer [50 mM Tris (pH 8.0), 150 mM NaCl, 10 mM MgCl2, 0.5 mM DTT, 30 μg/ml bovine serum albumin (BSA), and Protease Inhibitor Cocktail, Roche] for 4 hours at 25°C and then incubated at 25, 65, 67, 69, 72, or 71°C for 4 minutes. Proteins were isolated by freezing and thawing twice in liquid nitrogen and then incubated at 70°C for 10 minutes prior to analysis by Western blotting.
In silico docking
Virtual protein docking was performed online at www.dockingserver.com [69]. RRM2 protein structure was obtained from the PDB database (2UW2) [50]. Detailed information was provided in Appendix S1 (Supplementary methods).
Co-immunoprecipitation, immunostaining, and BrdU labeling
Assays were performed as previously described [70].
Xenografts in nude mice
The guidelines of the GWU Animal Research Facility and the Institutional Animal Care and Use Committee were followed for all animal experiments in this study. Female athymic nude mice at 6-weeks of age were obtained from the Jackson Laboratory (ME, US). Tumors were induced by inoculating RPK-9 (5×106 cells), HCT116 (3×106 cells), or IGROV1 CR (5×106 cells) suspended in 100 μl of phosphate buffered saline (PBS, pH 7.4) subcutaneously into the hind limb of mice (two tumors per mouse). Treatment of animals began at about 10 to 14 days after cell inoculation. The compound to be tested was dissolved in DMSO/Tween 80/saline (10:10:80; v/v/v) and introduced by intraperitoneal (IP) injection. DHS (50 mg/kg) was injected on 14 consecutive days. Cisplatin (8 mg/kg) was injected twice each week. Gemcitabine (100 mg/kg) was injected three times each week. In combinatorial treatments, DHS was injected 15 minutes before injection of gemcitabine or cisplatin. To monitor tumor formation, the longest and shortest diameters of the tumors were measured using calipers. Tumor volume (mm3) was calculated according to the following formula: tumor volume = (length × width2)/2. The whole-body weight of each animal was measured to assess the systemic toxicity of the treatments. When the experiment was terminated, hearts, livers, spleens, lung, and kidneys of the sacrificed animals were collected, and tissue slices were stained with hematoxylin and eosin (H&E).
Supplementary Material
Acknowledgements
This work was partially supported by funding from the National Institutes of Health (CA177898 and CA184717 to W. Z.). W. Zhu was supported by a Research Scholar Grant, RSG-13-214-01-DMC from the American Cancer Society.
Footnotes
Conflict of Interest
Authors declare no conflict of interest.
References
- 1.O’Connor MJ. Targeting the DNA Damage Response in Cancer. Mol Cell. 2015; 60: 547–560. [DOI] [PubMed] [Google Scholar]
- 2.Puigvert JC, Sanjiv K, Helleday T. Targeting DNA repair, DNA metabolism and replication stress as anti-cancer strategies. FEBS J 2016; 283: 232–245. [DOI] [PubMed] [Google Scholar]
- 3.Kitao H, Iimori M, Kataoka Y, Wakasa T, Tokunaga E, Saeki H et al. DNA replication stress and cancer chemotherapy. Cancer Sci 2018; 109: 264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang J, Dai Q, Park D, Deng X. Targeting DNA Replication Stress for Cancer Therapy. Genes (Basel). 2016; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aye Y, Li M, Long MJ, Weiss RS. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene. 2015; 34: 2011–2021. [DOI] [PubMed] [Google Scholar]
- 6.Kohnken R, Kodigepalli KM, Wu L. Regulation of deoxynucleotide metabolism in cancer: novel mechanisms and therapeutic implications. Mol Cancer. 2015; 14: 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morikawa T, Maeda D, Kume H, Homma Y, Fukayama M. Ribonucleotide reductase M2 subunit is a novel diagnostic marker and a potential therapeutic target in bladder cancer. Histopathology. 2010; 57: 885–892. [DOI] [PubMed] [Google Scholar]
- 8.Wang LM, Lu FF, Zhang SY, Yao RY, Xing XM, Wei ZM. Overexpression of catalytic subunit M2 in patients with ovarian cancer. Chin Med J (Engl) 2012; 125: 2151–2156. [PubMed] [Google Scholar]
- 9.Morikawa T, Hino R, Uozaki H, Maeda D, Ushiku T, Shinozaki A et al. Expression of ribonucleotide reductase M2 subunit in gastric cancer and effects of RRM2 inhibition in vitro. Hum Pathol 2010; 41: 1742–1748. [DOI] [PubMed] [Google Scholar]
- 10.Lu AG, Feng H, Wang PX, Han DP, Chen XH, Zheng MH. Emerging roles of the ribonucleotide reductase M2 in colorectal cancer and ultraviolet-induced DNA damage repair. World J Gastroenterol 2012; 18: 4704–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu X, Zhang H, Lai L, Wang X, Loera S, Xue L et al. Ribonucleotide reductase small subunit M2 serves as a prognostic biomarker and predicts poor survival of colorectal cancers. Clin Sci (Lond) 2013; 124: 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goan YG, Zhou B, Hu E, Mi S, Yen Y. Overexpression of ribonucleotide reductase as a mechanism of resistance to 2,2-difluorodeoxycytidine in the human KB cancer cell line. Cancer Res 1999; 59: 4204–4207. [PubMed] [Google Scholar]
- 13.Nakano Y, Tanno S, Koizumi K, Nishikawa T, Nakamura K, Minoguchi M et al. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer. 2007; 96: 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah KN, Mehta KR, Peterson D, Evangelista M, Livesey JC, Faridi JS. AKT-induced tamoxifen resistance is overturned by RRM2 inhibition. Mol Cancer Res 2014; 12: 394–407. [DOI] [PubMed] [Google Scholar]
- 15.Hehlmann R Current CML therapy: progress and dilemma. Leukemia. 2003; 17: 1010–1012. [DOI] [PubMed] [Google Scholar]
- 16.Shewach DS, Lawrence TS. Antimetabolite radiosensitizers. J Clin Oncol 2007; 25: 4043–4050. [DOI] [PubMed] [Google Scholar]
- 17.Minami K, Shinsato Y, Yamamoto M, Takahashi H, Zhang S, Nishizawa Y et al. Ribonucleotide reductase is an effective target to overcome gemcitabine resistance in gemcitabine-resistant pancreatic cancer cells with dual resistant factors. J Pharmacol Sci 2015; 127: 319–325. [DOI] [PubMed] [Google Scholar]
- 18.Zhou B, Su L, Hu S, Hu W, Yip ML, Wu J et al. A small-molecule blocking ribonucleotide reductase holoenzyme formation inhibits cancer cell growth and overcomes drug resistance. Cancer Res 2013; 73: 6484–6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen MC, Zhou B, Zhang K, Yuan YC, Un F, Hu S et al. The Novel Ribonucleotide Reductase Inhibitor COH29 Inhibits DNA Repair In Vitro. Mol Pharmacol 2015; 87: 996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsieh YY, Chou CJ, Lo HL, Yang PM. Repositioning of a cyclin-dependent kinase inhibitor GW8510 as a ribonucleotide reductase M2 inhibitor to treat human colorectal cancer. Cell Death Discov 2016; 2: 16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K et al. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000; 404: 42–49. [DOI] [PubMed] [Google Scholar]
- 22.Yamaguchi T, Matsuda K, Sagiya Y, Iwadate M, Fujino MA, Nakamura Y et al. p53R2-dependent pathway for DNA synthesis in a p53-regulated cell cycle checkpoint. Cancer Res 2001; 61: 8256–8262. [PubMed] [Google Scholar]
- 23.Hosseini A, Ghorbani A. Cancer therapy with phytochemicals: evidence from clinical studies. Avicenna J Phytomed 2015; 5: 84–97. [PMC free article] [PubMed] [Google Scholar]
- 24.Fabbrocini G, Staibano S, De Rosa G, Battimiello V, Fardella N, Ilardi G et al. Resveratrol-containing gel for the treatment of acne vulgaris: a single-blind, vehicle-controlled, pilot study. Am J Clin Dermatol 2011; 12: 133–141. [DOI] [PubMed] [Google Scholar]
- 25.Brown VA, Patel KR, Viskaduraki M, Crowell JA, Perloff M, Booth TD et al. Repeat dose study of the cancer chemopreventive agent resveratrol in healthy volunteers: safety, pharmacokinetics, and effect on the insulin-like growth factor axis. Cancer Res 2010; 70: 9003–9011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gertz M, Nguyen GT, Fischer F, Suenkel B, Schlicker C, Franzel B et al. A molecular mechanism for direct sirtuin activation by resveratrol. PLoS One. 2012; 7: e49761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li G, Rivas P, Bedolla R, Thapa D, Reddick RL, Ghosh R et al. Dietary resveratrol prevents development of high-grade prostatic intraepithelial neoplastic lesions: involvement of SIRT1/S6K axis. Cancer Prev Res (Phila) 2013; 6: 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen ZH, Hurh YJ, Na HK, Kim JH, Chun YJ, Kim DH et al. Resveratrol inhibits TCDD-induced expression of CYP1A1 and CYP1B1 and catechol estrogen-mediated oxidative DNA damage in cultured human mammary epithelial cells. Carcinogenesis. 2004; 25: 2005–2013. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Wang Q, Wu DC, Wang XW, Sun Y, Chen XY et al. Differential regulation of CYP1A1 and CYP1B1 expression in resveratrol-treated human medulloblastoma cells. Neurosci Lett 2004; 363: 257–261. [DOI] [PubMed] [Google Scholar]
- 30.Fontecave M, Lepoivre M, Elleingand E, Gerez C, Guittet O. Resveratrol, a remarkable inhibitor of ribonucleotide reductase. FEBS Lett 1998; 421: 277–279. [DOI] [PubMed] [Google Scholar]
- 31.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997; 275: 218–220. [DOI] [PubMed] [Google Scholar]
- 32.Varoni EM, Lo Faro AF, Sharifi-Rad J, Iriti M. Anticancer Molecular Mechanisms of Resveratrol. Front Nutr 2016; 3: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harikumar KB, Kunnumakkara AB, Sethi G, Diagaradjane P, Anand P, Pandey MK et al. Resveratrol, a multitargeted agent, can enhance antitumor activity of gemcitabine in vitro and in orthotopic mouse model of human pancreatic cancer. International Journal of Cancer. 2010; 127: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma L, Li W, Wang R, Nan Y, Wang Q, Liu W et al. Resveratrol enhanced anticancer effects of cisplatin on non-small cell lung cancer cell lines by inducing mitochondrial dysfunction and cell apoptosis. Int J Oncol 2015; 47: 1460–1468. [DOI] [PubMed] [Google Scholar]
- 35.Hu S, Li X, Xu R, Ye L, Kong H, Zeng X et al. The synergistic effect of resveratrol in combination with cisplatin on apoptosis via modulating autophagy in A549 cells. Acta Biochim Biophys Sin (Shanghai). 2016; 48: 528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nessa MU, Beale P, Chan C, Yu JQ, Huq F. Combinations of resveratrol, cisplatin and oxaliplatin applied to human ovarian cancer cells. Anticancer Res 2012; 32: 53–59. [PubMed] [Google Scholar]
- 37.Sale S, Tunstall RG, Ruparelia KC, Potter GA, Steward WP, Gescher AJ. Comparison of the effects of the chemopreventive agent resveratrol and its synthetic analog trans 3,4,5,4’-tetramethoxystilbene (DMU-212) on adenoma development in the Apc(Min+) mouse and cyclooxygenase-2 in human-derived colon cancer cells. Int J Cancer. 2005; 115: 194–201. [DOI] [PubMed] [Google Scholar]
- 38.Piotrowska H, Myszkowski K, Ziolkowska A, Kulcenty K, Wierzchowski M, Kaczmarek M et al. Resveratrol analogue 3,4,4’,5-tetramethoxystilbene inhibits growth, arrests cell cycle and induces apoptosis in ovarian SKOV-3 and A-2780 cancer cells. Toxicol Appl Pharmacol 2012; 263: 53–60. [DOI] [PubMed] [Google Scholar]
- 39.Piotrowska H, Myszkowski K, Amarowicz R, Murias M, Kulcenty K, Wierzchowski M et al. Different susceptibility of colon cancer DLD-1 and LOVO cell lines to apoptosis induced by DMU-212, a synthetic resveratrol analogue. Toxicol In Vitro. 2013; 27: 2127–2134. [DOI] [PubMed] [Google Scholar]
- 40.Fan GJ, Liu XD, Qian YP, Shang YJ, Li XZ, Dai F et al. 4,4’-Dihydroxy-trans-stilbene, a resveratrol analogue, exhibited enhanced antioxidant activity and cytotoxicity. Bioorg Med Chem 2009; 17: 2360–2365. [DOI] [PubMed] [Google Scholar]
- 41.Maccario C, Savio M, Ferraro D, Bianchi L, Pizzala R, Pretali L et al. The resveratrol analog 4,4’-dihydroxy-trans-stilbene suppresses transformation in normal mouse fibroblasts and inhibits proliferation and invasion of human breast cancer cells. Carcinogenesis. 2012; 33: 2172–2180. [DOI] [PubMed] [Google Scholar]
- 42.Balan KV, Wang Y, Chen SW, Chen JC, Zheng LF, Yang L et al. Proteasome-independent down-regulation of estrogen receptor-alpha (ERalpha) in breast cancer cells treated with 4,4’-dihydroxy-trans-stilbene. Biochem Pharmacol 2006; 72: 573–581. [DOI] [PubMed] [Google Scholar]
- 43.Kimura Y, Sumiyoshi M, Baba K. Antitumor activities of synthetic and natural stilbenes through antiangiogenic action. Cancer Sci 2008; 99: 2083–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saha B, Patro BS, Koli M, Pai G, Ray J, Bandyopadhyay SK et al. trans-4,4’-Dihydroxystilbene (DHS) inhibits human neuroblastoma tumor growth and induces mitochondrial and lysosomal damages in neuroblastoma cell lines. Oncotarget. 2017; 8: 73905–73924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Savio M, Ferraro D, Maccario C, Vaccarone R, Jensen LD, Corana F et al. Resveratrol analogue 4,4’-dihydroxy-trans-stilbene potently inhibits cancer invasion and metastasis. Sci Rep 2016; 6: 19973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Plunkett W, Huang P, Gandhi V. Preclinical characteristics of gemcitabine. Anticancer Drugs. 1995; 6 Suppl 6: 7–13. [DOI] [PubMed] [Google Scholar]
- 47.Chen Z, Zhou J, Zhang Y, Bepler G. Modulation of the ribonucleotide reductase M1-gemcitabine interaction in vivo by N-ethylmaleimide. Biochem Biophys Res Commun 2011; 413: 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thelander M, Graslund A, Thelander L. Subunit M2 of mammalian ribonucleotide reductase. Characterization of a homogeneous protein isolated from M2-overproducing mouse cells. J Biol Chem 1985; 260: 2737–2741. [PubMed] [Google Scholar]
- 49.Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundback T, Nordlund P et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 2014; 9: 2100–2122. [DOI] [PubMed] [Google Scholar]
- 50.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H et al. The Protein Data Bank. Nucleic Acids Res 2000; 28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.D’Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y et al. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell. 2012; 149: 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loewe S The problem of synergism and antagonism of combined drugs. Arzneimittelforschung. 1953; 3: 285–290. [PubMed] [Google Scholar]
- 53.Saiki Y, Yoshino Y, Fujimura H, Manabe T, Kudo Y, Shimada M et al. DCK is frequently inactivated in acquired gemcitabine-resistant human cancer cells. Biochem Biophys Res Commun 2012; 421: 98–104. [DOI] [PubMed] [Google Scholar]
- 54.Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem 2006; 75: 681–706. [DOI] [PubMed] [Google Scholar]
- 55.Chabes A, Thelander L. Controlled protein degradation regulates ribonucleotide reductase activity in proliferating mammalian cells during the normal cell cycle and in response to DNA damage and replication blocks. J Biol Chem 2000; 275: 17747–17753. [DOI] [PubMed] [Google Scholar]
- 56.Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol 1995; 22: 3–10. [PubMed] [Google Scholar]
- 57.Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol 2006; 17 Suppl 5: v7–12. [DOI] [PubMed] [Google Scholar]
- 58.Jung CP, Motwani MV, Schwartz GK. Flavopiridol increases sensitization to gemcitabine in human gastrointestinal cancer cell lines and correlates with down-regulation of ribonucleotide reductase M2 subunit. Clin Cancer Res 2001; 7: 2527–2536. [PubMed] [Google Scholar]
- 59.Scharadin TM, Zhang H, Zimmermann M, Wang S, Malfatti MA, Cimino GD et al. Diagnostic Microdosing Approach to Study Gemcitabine Resistance. Chem Res Toxicol 2016; 29: 1843–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lopez-Contreras AJ, Specks J, Barlow JH, Ambrogio C, Desler C, Vikingsson S et al. Increased Rrm2 gene dosage reduces fragile site breakage and prolongs survival of ATR mutant mice. Genes Dev 2015; 29: 690–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shah KN, Wilson EA, Malla R, Elford HL, Faridi JS. Targeting Ribonucleotide Reductase M2 and NF-kappaB Activation with Didox to Circumvent Tamoxifen Resistance in Breast Cancer. Mol Cancer Ther 2015; 14: 2411–2421. [DOI] [PubMed] [Google Scholar]
- 62.Chen W, Yeo SC, Elhennawy MG, Xiang X, Lin HS. Determination of naturally occurring resveratrol analog trans-4,4’-dihydroxystilbene in rat plasma by liquid chromatography-tandem mass spectrometry: application to a pharmacokinetic study. Anal Bioanal Chem 2015; 407: 5793–5801. [DOI] [PubMed] [Google Scholar]
- 63.Fang JG, Lu M, Chen ZH, Zhu HH, Li Y, Yang L et al. Antioxidant effects of resveratrol and its analogues against the free-radical-induced peroxidation of linoleic acid in micelles. Chemistry. 2002; 8: 4191–4198. [DOI] [PubMed] [Google Scholar]
- 64.Cai YJ, Wei QY, Fang JG, Yang L, Liu ZL, Wyche JH et al. The 3,4-dihydroxyl groups are important for trans-resveratrol analogs to exhibit enhanced antioxidant and apoptotic activities. Anticancer Res 2004; 24: 999–1002. [PubMed] [Google Scholar]
- 65.Zhou W, Sun W, Yung MMH, Dai S, Cai Y, Chen CW et al. Autocrine activation of JAK2 by IL-11 promotes platinum drug resistance. Oncogene. 2018; 37: 3981–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen CW, Wu MH, Chen YF, Yen TY, Lin YW, Chao SH et al. A Potent Derivative of Indolizino[6,7-b]Indole for Treatment of Human Non-Small Cell Lung Cancer Cells. Neoplasia. 2016; 18: 199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sherman PA, Fyfe JA. Enzymatic assay for deoxyribonucleoside triphosphates using synthetic oligonucleotides as template primers. Anal Biochem 1989; 180: 222–226. [DOI] [PubMed] [Google Scholar]
- 68.Zhou BS, Ker R, Ho R, Yu J, Zhao YR, Shih J et al. Determination of deoxyribonucleoside triphosphate pool sizes in ribonucleotide reductase cDNA transfected human KB cells. Biochem Pharmacol 1998; 55: 1657–1665. [DOI] [PubMed] [Google Scholar]
- 69.Bikadi Z, Hazai E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J Cheminform 2009; 1: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu W, Ukomadu C, Jha S, Senga T, Dhar SK, Wohlschlegel JA et al. Mcm10 and And-1/CTF4 recruit DNA polymerase alpha to chromatin for initiation of DNA replication. Genes Dev 2007; 21: 2288–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.