

Abstract
The protein NOS1AP/CAPON mediates signaling from a protein complex of NMDA receptor, PSD95 and nNOS. The only stroke trial for neuroprotectants that showed benefit to patients targeted this ternary complex. NOS1AP/nNOS interaction regulates small GTPases, iron transport, p38MAPK-linked excitotoxicity, and anxiety. Moreover, the nos1ap gene is linked to disorders from schizophrenia, post-traumatic stress disorder, and autism to cardiovascular disorders and breast cancer. Understanding protein interactions required for NOS1AP function, therefore, has broad implications for numerous diseases. Here we show that the interaction of NOS1AP with nNOS differs radically from the classical PDZ docking assumed to be responsible. The NOS1AP PDZ motif does not bind nNOS as measured by multiple methods. In contrast, full-length NOS1AP forms an unusually stable interaction with nNOS. We mapped the discrepancy between full-length and C-terminal PDZ motif to a novel internal region we call the ExF motif. The C-terminal PDZ motif, although neither sufficient nor necessary for binding, nevertheless promotes the stability of the complex. It therefore potentially affects signal transduction and suggests that functional interaction of nNOS with NOS1AP might be targetable at two distinct sites. We demonstrate that excitotoxic pathways can be regulated, in cortical neuron and organotypic hippocampal slice cultures from rat, either by the previously described PDZ ligand TAT-GESV or by the ExF motif-bearing region of NOS1AP, even when lacking the critical PDZ residues as long as the ExF motif is intact and not mutated. This previously unrecognized heterodivalent interaction of nNOS with NOS1AP may therefore provide distinct opportunities for pharmacological intervention in NOS1AP-dependent signaling and excitotoxicity.
Keywords: excitotoxic lesion, NMDA receptor, nNOS/NOS1, NOS1AP/CAPON, protein interaction, target identification
Introduction
NOS1AP (also known as CAPON) recently emerged as a potential player in conditions from cerebral ischemia, anxiety disorders, and schizophrenia to diabetes and sudden cardiac death. Mechanistic studies suggest that interaction of NOS1AP with nNOS contributes to NOS1AP-mediated excitotoxicity (Li et al., 2013; Lai et al., 2014), cardiac action potential duration (Chang et al., 2008), elaboration of neuronal processes (Carrel et al., 2009), and possibly schizophrenia (Xu et al., 2005).
PDZ interactions are well understood; a peptide ligand of 4 or 5 amino acids with specific conserved C-terminal residues docks in the binding pocket of a ∼90 amino acid domain (Doyle et al., 1996; Harris and Lim, 2001). The mechanism of interaction of NOS1AP with nNOS, assumed to be a canonical PDZ interaction, has indeed consistently evaded elucidation. NOS1AP is the most prominent nNOS-PDZ ligand in the literature but terminates in the Class II “IAV” motif (ψXψ-COOH) (Vaccaro and Dente, 2002). In contrast, peptide and two-hybrid screens revealed that nNOS-PDZ binds a distinct “Class III” ligand motif ending G(D/E)xV (Schepens et al., 1997; Stricker et al., 1997). A large-scale systematic analysis revealed only three ligand peptides for nNOS-PDZ (Stiffler et al., 2007). These were not similar to NOS1AP and had affinities weaker (15–78 μm) than other ligand-PDZ pairs studied.
Here we report that the C-terminal peptide from NOS1AP exhibits virtually no affinity for nNOS, and confirm this by multiple methods. In contrast, full-length and large C-terminal fragments of NOS1AP interact extremely tightly in conventional pull-down assay. This was dependent on the PDZ ligand motif as deleting the three C-terminal residues eliminated stable binding. Surprisingly, the PDZ ligand motif was entirely dispensable for binding in interaction assays without washing steps and for interaction between nNOS and NOS1AP fragments in intact cells. Our data suggest a heterodivalent interaction between NOS1AP and the PDZ domain. This involves the following: (1) a previously unidentified site, which may be the primary interaction, as it is sufficient alone; and (2) the C-terminal PDZ ligand motif, which is not sufficient alone but stabilizes binding (∼4-fold difference in koff). An alanine scan of an evolutionary conserved internal motif identified NOS1AP residues Glu429 and Phe431 as critical for this second binding site.
This heterodivalent interaction potentially provides two distinct opportunities for development of drugs to inhibit interaction, whereas targeting both sites might increase selectivity and reduce off-target effects. As nNOS-NOS1AP interaction contributes to p38 activation and excitotoxic lesions (Li et al., 2013), we investigated whether each site alone provides opportunities for intervention. Cell-permeable peptides (1) based on NOS1AP containing both sites, (2) possessing the second or “ExF motif” interaction site but lacking the PDZ motif, or (3) the PDZ ligand we previously developed (TAT-GESV) each inhibited NMDA-evoked p38MAPK activation. In organotypic hippocampal slices, these conferred neuroprotection, which in the case of the internal motif peptides depended on residues 429/431. In summary, we showed that NOS1AP interacts with nNOS via two distinct sites, which cooperate to generate both affinity and stability. This suggests that the heterodivalent form of interaction provides two alternate avenues for interference with signaling processes and tissue lesions in disease models.
Materials and Methods
Peptides
Short peptides were obtained from GENECUST or GenicBio as follows: “EIAV,” NH2-DSLDDEIAV-COOH; “TAT-GESV,” NH2-GRKKRRQRRR-YAGQWGESV-COOH; and “TAT,” NH2-GRKKRRQRRR-COOH.
Longer peptides were expressed as GST fusions from pGEX6P vectors, affinity purified on immobilized GSH, and released from the GST by on-column cleavage with 3C human rhinovirus protease (plasmid generously provided by Professor John Heath, School of Biosciences, University of Birmingham, Birmingham, United Kingdom). These included NOS1AP(400–506), NOS1AP(400–503), NOS1AP(432–506), and NOS1AP(432–503). TAT-fused NOS1AP peptides used were expressed with an additional His-tag from pET28a.
Antibodies
Rabbit polyclonal antibodies against CAPON/NOS1AP(R-300), nNOS (Z-RNN3), and phospho-p38 (“T183/Y185”) were from Santa Cruz Biotechnology, Invitrogen, and Cell Signaling Technology, respectively. Rabbit monoclonal against p38 isoforms (clone D13E1) was from Cell Signaling Technology, and mouse monoclonal against GST (clone B10) was from Santa Cruz Biotechnology. Secondary reagents were from Santa Cruz Biotechnology, Cell Signaling Technology, and Millipore Biotech.
Cloning, plasmids, and plasmid preparation
Constructs encoding pEGFP and pGEX-human nNOS constructs, human NOS1AP fused to luciferase, and unfused luciferase vector were described previously (Li et al., 2013). The construct encoding GST-fused nNOS 1–155 for mammalian expression, pKEBG-nNOS 1–155, was constructed by replacing the CMV promoter and GFP coding sequence of pEGFP-nNOS 1–155 with the EF1 promoter and GST coding sequence from pEBG. NOS1AP fragments incorporating amino acids 1–503, 382–506, 400–506, 432–506, 400–503, and 432–503 were generated by PCR-based methods from full-length human NOS1AP plasmid previously described (Li et al., 2013). Alanine scan point mutants were obtained by insertional overlapping PCR in coding sequences for regions 400–503 and 400–506. Fragments were transferred to corresponding vectors to generate the Luciferase, monomeric Cherry, and GST (pGEX6P) fusion plasmids for assays as indicated. Constructs encoding PSD95-PDZ2 and PDZ3 fused to luciferase were generated by transferring the PDZ2 and PDZ3 sequences to luciferase vectors from corresponding pGEX vectors previously described (Cao et al., 2005). His tagged nNOS 1–108 and 1–133 were described previously (Tochio et al., 1999, 2000). TAT fusions were generated from the pET28a vector into which TAT sequence had been inserted followed by the wild-type or mutant NOS1AP encoding sequence fragments as indicated.
Recombinant protein preparation
GST-tagged PSD95-PDZ3, Tiam1-PDZ, SCRIB-PDZ4, nNOS(1–155)wt (“ePDZ”), and Y77H/D78E and free GST were prepared from BL21(DE3) Escherichia coli strain as described previously (Courtney and Coffey, 1999). Native and denatured His/TAT-tagged proteins were prepared similarly by immobilized metal-affinity chromatography following protocols provided by Steven Dowdy (Becker-Hapak et al., 2001). Purity and size were checked by SDS-PAGE gels by Coomassie staining.
Cell culture, transfection, NMDA stimulation, cell lysis, and phospho-p38 detection
Cortical neuron cultures were prepared from P0 rats of either sex (mixed) as described previously (Semenova et al., 2007). All isolation of cells and tissues from animals was performed in accordance with corresponding local, national, and European Union regulations. The cells were cultured in NeurobasalA/B27 medium (Invitrogen) and stimulated with NMDA after exposure to TAT peptides (denatured to enhance cellular uptake and in-cell renaturation, as recommended by Becker-Hapak et al., 2001) as indicated, to evaluate inhibition of excitotoxic signaling. Excitotoxic stimulation of cortical neurons (8 DIV) was performed as described previously (Li et al., 2013). Briefly, neuroprotectant NeurobasalA/B27 culture medium was replaced with MEM supplemented with glutamine, penicillin/streptomycin, ± peptides 1 h before NMDA as indicated. Activation of p38MAPK was determined by immunoblotting of cleared neuron lysates in Laemmli buffer after 10 min of NMDA treatment.
The 293T and COS7 cell lines were cultured as previously described (Li et al., 2013). COS7 cells were transfected with lipofectamine2000 (Invitrogen) according to the manufacturer's protocol, and 293T cells were transfected with the CaCl2 phosphate method. Transfected cells were rinsed twice in PBS, lysed by scraping into low salt buffer (LSB) supplemented with protease inhibitors and detergent 0.5% Igepal as described previously (Li et al., 2013). Lysates were cleared by centrifugation at 20,000 × g, 4°C for 10 min.
Organotypic hippocampal slice cultures
Organotypic hippocampal slice cultures were prepared according to a previously described procedure with modifications (Kasri et al., 2008). In brief, 7-day-old Wistar rats of either sex (mixed) were decapitated; brains were collected in dissection medium (low Na+ ACSF containing 1 mm CaCl2, 5 mm MgCl2, 4 mm KCl, 26 mm NaHCO3, 8% sucrose, 0.5% phenol red, oxygenated with 95% O2/5% CO2 gas mixture in a beaker chilled on salted ice), and hippocampi were dissected and transversely sliced into 400-μm-thick slices on a McIlwain tissue chopper (Mickle Laboratory). The slices were transferred to culture medium. Only slices with an intact structure of CA and DG regions were selected. Four slices were placed onto each 30 mm Millicell cell culture insert (0.4 μm, Millipore), which were placed in 6-well tissue culture plates with 750 μl of filter-sterilized long-term slice culture medium (MEM-Hanks medium, 1 mm l-glutamine, 1 mm CaCl2, 2 mm MgSO4, 1 mg/L insulin, 1 mm NaHCO3, 20% heat inactivated horse serum, 0.5 mm l-ascorbate, 30 mm HEPES) under the insert. The slices were placed in a humidified chamber with 5% CO2 at 35°C, and medium was exchanged every second day.
Protein interaction assays
Pull-down assays: conventional, no wash, and filter-plate assays
Conventional pull-down assay.
These were performed as previously described by incubating immobilized recombinant GST fusion proteins (2 μg/assay) as indicated with cleared lysates of 293T cells expressing soluble protein as shown in the figures (pLuc-C1 vector expressing only firefly luciferase used as a negative control). To assist with quantification, the expressed soluble proteins were expressed as fusions with firefly luciferase as described previously (Li et al., 2013). Beads were rotated for 1 h at 4°C to allow binding, and beads were washed three times and resuspended in CCLR (Promega) before quantification using luciferase assay reagent. Total “input” amount used in each case was normalized (7000 units per assay of 250 μl, using a Turner TD20/20 10 s measurement). The final amount in the bead pellet was also measured, and pull-down data were expressed as percentage of total.
No-wash assays using purified recombinant proteins.
To identify interacting protein that is lost during the washing steps in a conventional pull-down assay, and avoid possible contributions of other proteins in cell lysates, some experiments were performed with purified recombinant GST-nNOS immobilized on glutathione beads and purified NOS1AP fragments cleaved off from the GST moiety in an on-bead cleavage protocol using GST-rhinovirus 3C protease. Purity was validated by observing a single band on a Coomassie-stained gel. We used residues 1–155 of nNOS as this encompasses the extended PDZ domain, including the flanking β-finger motif, and a recombinant of this length was more readily produced than shorter fragments.
Filter-plate assay.
Alanine scan pulldown: To each well of a 96 well U-bottom plate, 2 μl beads bound to 1 μg GST or GST-fused nNOS protein as indicated were dispensed. Cell lysates containing equal amounts of luciferase fusion proteins were added to each well to a final volume of 100 μl per well. Beads were incubated 60 min at 4°C with gentle shaking and transferred to a multiscreen filter plate (Millipore), cell lysate removed by applying vacuum 30 s. Once pressure had normalized, beads were resuspended in lysis reagent. The 96 channel pipetting and vacuum control were performed with a Biomek NXP96 (Beckman-Coulter). Luciferase assay reagent was added and signal measured using a plate reader (Polarstar, BMG). TAT peptide affinity assay: 5 μl beads bound to 1 μg GST-fused nNOS protein (or GST for background measurement) were rotated with concentrations of native recombinant His-TAT-NOS1AP peptides as shown for 1 h at 4°C and transferred to multiscreen filter plates (Millipore). Unbound peptides were removed by centrifuging at 1000 × g for 5 min. Beads were resuspended in LSB and protein eluted with 4× Laemmli buffer, boiling at 95°C for 10 min. The affinity of each peptide was determined using fluorescent immunoblotting, with DyLight 680-conjugated anti-rabbit secondary antibody (Cell Signaling Technology), quantified with the Odyssey infrared imaging system (LI-COR) and ImageJ as follows. Bound peptide, together with the background binding of each TAT peptide to GST beads, was quantified in each blot using a set of standard concentrations of the same TAT fragment on the same blot. Background binding, barely detected, except at the highest fragment concentration, was subtracted in each case. Fitting was performed for each replicate with Excel using the quadratic solution formula to account for bound ligand, fb = B/L0 = {+(Kd + P0 + L0) − √[(Kd + P0 + L0) ^ 2 − 4L0P0]}/2L0. The affinities of His-TAT-NOS1AP[400–503] Ala-mutant peptides were so weak that their Bmax could not be directly obtained and the Bmax of His-TAT-NOS1AP[400–503] wild-type peptide was assumed to be applicable also to the mutants to estimate their Kd values. Even if this assumption is not correct, it is clear from the results (see Fig. 9C) that the mutants bind extremely weakly.
Figure 9.
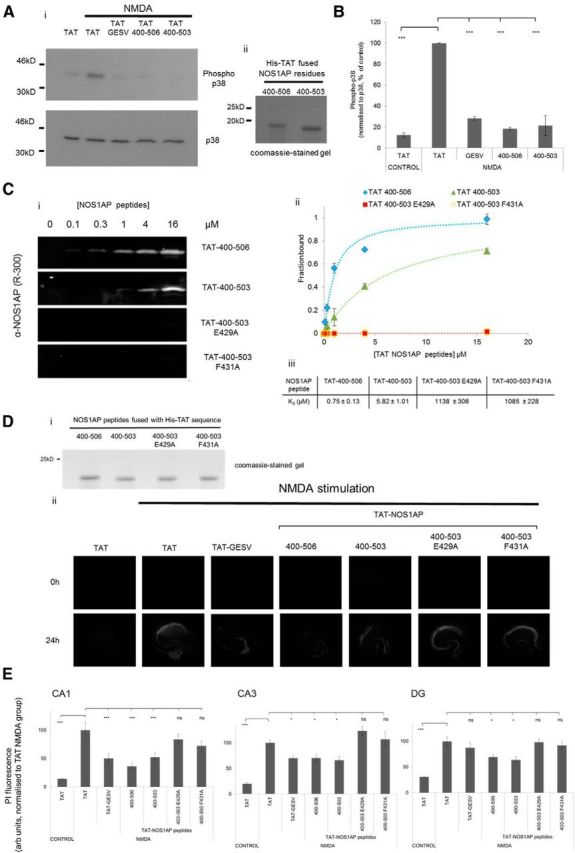
The ExF motif region can be used to reduce excitotoxic lesions in multiple hippocampal regions. Ai, Cortical neuron cultures were incubated with cell-permeant TAT peptides or TAT alone (1 μm) as shown and treated with NMDA (50 μm, 10 min). Activation of p38 was detected by phospho-p38 Western blotting (top), and pan-p38 antibody as loading control (bottom). Aii, The TAT-NOS1AP 400–506 and 400–503, expressed as His-tag fusions purified by immobilized metal-affinity chromatography, were evaluated for purity by Coomassie staining; shorter peptides were synthesized and quality controlled commercially. B, Quantified data show inhibition of NMDA-evoked p38 activation by TAT-GESV as previously reported as well as by NOS1AP 400–506, which contains both ExF motif and PDZ ligand motif; and 400–503, which contains ExF motif but lacks the PDZ ligand motif (n = 3, one-way ANOVA followed by Newman-Keuls Multiple Comparison Post Hoc Test). Ci, Representative fluorescent immunoblot images showing binding of His-TAT-NOS1AP to GST-nNOS[1–155], incubated at increasing concentrations of peptide as indicated. Bound peptide was detected by NOS1AP primary antibody and an Odyssey infrared imaging system. Cii, Binding curves from replicates of experiments shown in Ci, with each data point background-corrected and normalized to internal standards on each gel for each peptide. Data are mean ± SEM; n = 3. Curves were fitted to data as described in Materials and Methods. Ciii, Affinity constants were obtained by fitting each replicate for each peptide. Data are mean ± SEM Kd values (in μm). A value for Bmax could not be reliably obtained for the His-TAT-NOS1AP[400–503] mutants due to weak binding, and the Bmax from His-TAT-NOS1AP[400–503] wild-type was applied in these cases (see Materials and Methods). D, Organotypic hippocampal slice cultures were incubated with TAT peptides as indicated, at 1 μm; Coomassie gel shown in Di. Dii, At 24 h after 50 μm NMDA exposure for 30 min, lesions were visualized by incubation with the live cell-impermeant DNA stain propidium iodide. E, Lesions, determined by propidium iodide staining (see Materials and Methods), were significant in CA1, CA3, and DG. They were reduced by TAT-GESV (CA1 and CA3 only) as well as TAT fusions of NOS1AP peptides, including ExF motif with (400–506) or without (400–503) the PDZ ligand motif (all regions). However, mutation of either E429 or F431 in the ExF motif renders the reduction in lesion to a minimal nonsignificant level in all regions (n = 4–8, one-way ANOVA and Newman-Keuls Multiple Comparison Post Hoc Test). *p < 0.05. ***p < 0.001. ns, Not significant.
Bead-immobilized protein perfusion and semiquantitative kinetic analysis
GST-tagged nNOS 1–155 was prepared as described by affinity chromatography on glutathione agarose beads. A perfusion chamber was developed using a Parafilm spacer (nominal thickness 0.127 mm) into which a rectangular trough was cut, between a glass slide substrate and 25-mm-diameter glass coverslip upper window. A 1 ml pipette tip, beveled at the open end, was attached with Parafilm as the in-port of the perfusion chamber. The chamber was heated to allow the Parafilm to melt and seal. The in-port was further sealed with nail varnish and allowed to dry. Beads were applied from the out-port, driven in by surface tension, and trapped in the chamber. Buffer was allowed to flow-through the in-port past the beads by gravity flow at 0.3 ml/min and effluent leaving the out port removed by open aspiration. NOS1AP 400–503 and 400–506 were affinity-purified as GST-tagged fusions, cleaved on beads with GST-tagged 3C-HRV protease, and labeled with FITC (Sigma) according to standard protocols (Invitrogen). Proteins labeled at low stoichiometry (∼0.1–0.2 dye molecules per protein, by OD494/280) were purified from FITC label and desalted by spin column (Bio-Rad Biospin P6), normalized for label by addition of unlabeled protein, and diluted in buffer to normalize protein levels. Labeled protein (50 μl at 10 μm) was introduced into a sample loop in the gravity flow path and allowed to flow over the beads followed by buffer without protein at a continuous rate (0.3 ml/min). Beads were imaged using a fluorescent microscope (IX70 with 10×N.A.0.3 objective, XLED1 460 nm excitation, 510 nm LP emission filter) with a Hamamatsu Flash4 CMOS camera (500 ms exposure, 4 × 4 binning) using HCimage camera software. Data from bead ROIs were corrected using neighboring background ROIs, subtracting contribution from buffer in the bead ROIs (beads are approximately spherical, whereas ROIs represent cylindrical volumes) and a term representing system dark value and a minor exponential background drift (in the form A0[1-e−kt] where k = 0.0013 s−1 in all cases). Residual bead fluorescence (see Fig. 5A, top) remains but appears relatively constant and therefore will not contribute to decay curves. Decay curves were fitted to the Schuck and Minton two-compartment model for dissociation under flow conditions (Schuck and Minton, 1996; de Mol and Fischer, 2008). This model includes terms to correct for rebinding of released protein. We assumed that the fluorescein dye does not bleach during the imaging time, but this does not affect the observation that 400–506 remains on beads considerably longer and a difference in off-rate is the most likely explanation.
Figure 5.

Visualization of NOS1AP 400–503/506 binding and Kd estimates by fluorescent methods. A, Beads bound to GST-nNOS1–155 as used in previous figures were trapped in a flow chamber, an aliquot of fluoresceinated NOS1AP applied as indicated (middle panel), followed by perfusion of beads with buffer (bottom). Fluorescent images using a fluorescein filter set are shown. B, The background region-corrected intensity of fluorescence from the bead, indicating the level of interacting protein, is shown as pixel counts against time. The perfusion of protein (50 μl at t = 0, flow rate 5 μl/s) is indicated by the dark bar and arrow above the graphs. The loss of binding of 400–503 is clearly faster than loss of binding of 400–506. Average of 5 experiments/curve are shown. Ci, Cii, Replicate curves of five experiments as in A, B are shown. Each curve is an average of ∼3 beads per experiment. Ciii, Civ, The curves from the left panels were fitted (after t = 120 s, to allow for complete washout of unbound protein), to the two-compartment model for dissociation under flow conditions (Schuck and Minton, 1996; de Mol and Fischer, 2008). In the examples shown for 400–503 (top) and 400–506 (bottom), the raw data (blue solid line), the fitted curve (orange dotted line), and the residual between the two (green solid line) are plotted. Background fluorescence from the beads before addition of protein is relatively stable after a brief transience and therefore should not affect the fit to the decay curves starting at 120 s. Cv, Based on curves obtained (A–Cii) fitted to the model (Ciii, Civ), values for koff, kon, and Kd, obtained for binding of nNOS to both NOS1AP 400–503 and 400–506, are shown. D, Fluorescence polarization was used to determine solution binding of fluoresceinated NOS1AP 400–506 to nNOS (1–155), giving small polarization changes as expected for such a large ligand. The data (n = 3, mean ± SEM) were fitted to a curve as described in Materials and Methods, giving a Kd of 0.051 μm.
Solution methods
Protein peptide ligand interaction assay by fluorescence polarization.
Fluorescence polarization assays were performed using a PerkinElmer Life Sciences LS-55 fluorimeter equipped with an automated polarizer (see Fig. 1) or a BMG Polarstar OPTIMA reader (see Fig. 5) set at 25°C in all cases. Synthetic peptide corresponding to the C terminus of NOS1AP (residues 400–506) or nonamer peptides “-EIAV” was labeled with fluorescein-5-isothiocyanate (Molecular Probes) at the N terminus and purified by passing the reaction mixture through a Superdex200 or Superdex Peptide column (GE Healthcare) according to size. In the case of -EIAV (see Fig. 2), fluorescence titration was performed by adding increasing amounts of His-Tagged nNOS core PDZ domain (1–108) to a constant amount of the fluoresceinated peptide (∼1 μm) in 50 mm Tris buffer, pH 7.4, containing 100 mm NaCl, 1 mm EDTA, and 1 mm dithiothreitol. The dissociation constant (Kd) was obtained by fitting the titration curves with the classical one-site binding model ΔmP = mPmax * [nNOS]/(Kd + [nNOS]) using the software GraphPad Prism (GraphPad). Experiments were performed in triplicate. In the case of NOS1AP[400–506] peptide (used at 50 nm) nNOS (1–155) cleaved from a GST fusion was used for titration to be consistent with the bead-based assays in preceding figures. The same buffer as above was used. Fitting was performed with Excel using the quadratic solution formula to account for bound ligand, fb = B/L0 = {+(Kd + P0 + L0) − √[(Kd + P0 + L0) ^ 2 − 4L0P0]}/2L0. ΔmP is calculated from mP − mP0, where mP is the measured polarization value of each titration point and mP0 is the polarization value of the free peptide.
Figure 1.

NOS1AP C terminus does not bind nNOS, but the full-length protein binds strongly. A, Projection of MelR 9-mer peptide docking in the ligand-binding pocket of nNOS-PDZ, a canonical PDZ interaction (1B8Q.pdb NMR structure 1, Tochio et al., 1999; projected with Chimaera, Pettersen et al., 2004). B, The fluoresceinated C-terminal PDZ motif of NOS1AP/Capon (DSLDDEIAV) binds with nNOS (His-tagged residues 1–108) in fluorescence polarization assay at an affinity, determined by curve fitting, of ∼600 μm. This method provides ∼17 μm affinity for Cx43 9mer ligand binding ZO1-PDZ2, for example (Chen et al., 2008). C, D, GST-fused domains of different PDZ classes (shown in Coomassie gel) were used for pull-down assay (2 μg immobilized PDZ/assay, pull-down expressed as percentage of input as described in Materials and Methods), revealing efficient recovery of NOS1AP by nNOS-extended PDZ. This requires the C-terminal Class II-type PDZ ligand motif “IAV” because an equal amount of NOS1AP-ΔIAV, in which the motif is deleted, is not pulled down. The motif does not mediate binding to the Class II PDZ domain, whereas the Class I PDZ domain like Scrib-PDZ4 does not bind detectably. The Class II PDZ domain does bind to NOS1AP, but this binding persists even in the absence of the Class II motif. ***p < 0.001 (Newman-Keuls Multiple Comparison Post Hoc Test after ANOVA). ns, Not significant. E, The efficiency of the well-characterized interaction between nNOS β-finger (distinct from but adjacent to the PDZ binding pocket) and PSD95-PDZ2 is shown for comparison.
Figure 2.

The PDZ ligand-binding pocket of nNOS and the 3 C-terminal PDZ motif residues (IAV) are necessary for NOS1AP-nNOS interaction, but the PDZ motif is not sufficient. A, GST-nNOS residues 1–155 were bead-immobilized and exhibit efficient pull-down of full-length NOS1AP, but not the NOS1AP PDZ motif (9 C-terminal amino acids ending EIAV). The previously described ligand-binding pocket double-mutant Y77H/D78E (see text) exhibits reduced binding to full-length NOS1AP. GST and empty vectors were used as negative controls. Recombinant proteins used are shown in the Coomassie gel under the histogram. ***p < 0.001 (Newman-Keuls Multiple Comparison Post Hoc Test after two-way ANOVA). ns, Not significant. B, The 293T cells were cotransfected with constructs for full-length NOS1AP (luciferase fused for quantification) and nNOS (EGFP-fused to facilitate affinity purification). Cell lysates were incubated with GFP-binding protein beads to immobilize the nNOS, and pulled down using a conventional protocol (see Materials and Methods). The coexpressed NOS1AP was efficiently copurified while controls produced negligible signal. ***p < 0.001. C, COS7 cells were transfected with plasmid encoding Cherry-fused NOS1AP and untargeted EGFP expression vector, and imaged by laser scanning confocal microscopy in xy (bottom panel, merged images) and xz planes (top, image channels and merged channels as shown), taken at the thickest part of the cell to facilitate visualization of membrane localization. The white line on the xy image indicates the location of the xz plane scanned (125 μm width × 25 μm depth). NOS1AP localizes to the edges of the cell, presumably near or at the plasma membrane. D, COS7 cells were transfected with full-length fusion constructs encoding Cherry-NOS1AP and EGFP-nNOS as indicated, and imaged by laser scanning confocal microscopy in xy (bottom panels, merged images) and xz scans (top, image channels and merged channels as shown) as in C. Alone, nNOS resides predominantly in the cytoplasm and is excluded from the nucleus and vesicular structures (left panels). However, when coexpressed with NOS1AP, nNOS translocates to NOS1AP (right panels) at membrane proximal regions, and the negative contrast with the nucleus and vesicular structures is lost. This suggests that the expressed proteins interact to a substantial extent in intact cells.
Isothermal titration calorimetry (ITC).
ITC measurements were performed on a VP-ITC calorimeter (Microcal) at 25°C. All protein and peptide samples were in 20 mm, pH 7.0 Tris-HCl buffer. The titration processes were performed by injecting 10 μl aliquots of the nNOS extended PDZ (1–133) fragments into untagged NOS1AP 9-mer peptide -EIAV at time intervals of 3 min to ensure that the titration peak returned to the baseline. The titration data were analyzed using the program Origin 7.0 from Microcal and fitted using the one-site binding model.
Coprecipitation methods
Coimmunoprecipitation of luciferase fusion proteins coexpressed with GFP fusion proteins was performed as previously described (Li et al., 2013). Precleared HEK-293T lysates cotransfected with pEGFP-nNOS full-length and pLuc-NOS1AP full-length (pEGFP-C1 and pLuc-C1 as controls) in LSB/protease inhibitors and 0.5% Igepal detergent were coimmunoprecipitated by rotation 1 h at 4°C with 5 μl of beads coupled to a single-chain anti-GFP camelid antibody-based protein (“GFP-Trap,” Chromotek). Beads were washed 3 times in LSB/protease inhibitors and 0.5% Igepal detergent, drained beads resuspended in cell culture lysis reagent (Promega), and luciferase in washed bead and input samples were measured and precipitated activity was normalized to initial levels for each sample.
Coprecipitation of luciferase-fused NOS1AP mutants coexpressed with GST-fused nNOS[1–155] was performed similarly, except that glutathione Sepharose 4B (GE Healthcare) beads were used to precipitate the nNOS. Efficiencies of precipitation were measured by immunoblotting with anti-GST (scbt clone B14) and coprecipitation by luciferase assay as above.
Imaging and segmentation of COS7 cells for protein interaction assay
Transfected COS7 cultures were incubated with Hoechst (1 μg/ml, 15 min in 37°C before imaging) to facilitate algorithm-based image segmentation. The cultures were imaged (20× NA 0.45 long-working distance objective, laser-based focus, 4 × 4 fields captured per well) with filter sets to obtain Hoechst, YFPVenus and Cherry images. Data were analyzed with the aid of AttoVision1.7 software (BD Biosciences) using automated background correction (constant value) of each channel followed by segmentation as follows:
Hoechst images were used to seed polygons (preprocessing parameters: median 5 × 5, rolling ball 25 × 25, dilate 3 × 3, automated thresholding level 1, size range 250–3000) eroded by 6 pixels to define nuclear regions of interest without potential contamination from cytoplasmic signals. The uneroded polygons were expanded using corresponding Cherry images (preprocessing: median 5 × 5, automated thresholding level 2) to define cytoplasmic regions. Dilation was limited to 10 pixels to ensure the cytoplasmic regions belonged to the corresponding nuclei and not neighboring cells, and erosion 1 pixel to increase discrimination from corresponding nuclear ROIs. Untransfected cells (intensity of cytoplasmic region in Cherry channel < threshold value of 100) were excluded from analysis. The translocation index was defined for each cell as the ratio of YFP channel average pixel intensity in cytoplasmic regions to the corresponding nuclear regions of the same cell. For each well, an average of the ratio of all transfected cells (mean 40 ± 4 per well) was obtained, and the data were graphed as mean ± SEM of well averages (n = 3 wells).
Protein structure visualization and prediction and evolutionary conservation analysis
Protein structure data presentations were rendered with UCSF-Chimera and POVray (Pettersen et al., 2004). Evolutionary conservation analysis was performed using the Consurf server (Ashkenazy et al., 2010). Secondary structure prediction tools used were JPRED (Cole et al., 2008) and PSIPRED (Buchan et al., 2013).
Excitotoxic stimulation and imaging of slice cultures for tissue lesion assay
Following the protocol of Bunk et al. (2010), at 8–10 d in vitro, cultured slices were selected by propidium iodide staining (2 μg/ml for 15 min in conditioned medium) to exclude damaged cultures. Slices were rinsed in experimental buffer containing 120 mm NaCl, 3.5 mm KCl, 0.4 mm KH2PO4, 5 mm NaHCO3, 20 mm HEPES, and 1.2 mm Na2SO4 supplemented with 15 mm glucose and 1.2 mm CaCl2, and exposed to 50 μm NMDA for 30 min at 35°C in the same medium. Control slices were treated with experimental buffer alone. After treatment, the slices were washed in the same buffer (without NMDA) and conditioned medium was added. Slices were analyzed for propidium iodide staining after 24 h. Images were acquired with a BD pathway 855 imager (2.5× objective, 555 nm/28 nm excitation, 595 nm LP dichroic, 645 nm/75 nm emission).
Quantification of cell death in slice culture images
This was performed as described previously (Bunk et al., 2010; D'Orsi et al., 2012). Briefly, fluorescence of propidium iodide staining in CA1, CA3, and DG subfields (three 0.027 mm2 areas per subfield measured and averaged) of the slices (4–8/condition) were quantified from images taken 24 h after addition of NMDA using ImageJ software. For each image, background fluorescence was calculated similarly from image regions from the filter support neighboring the slice. This signal was subtracted from each subfield value. One-way ANOVA was performed followed by Newman-Keuls Multiple Comparison Post Hoc Test. p < 0.05 was considered significant.
Confocal imaging of COS7 cells for protein localization
COS7 cells were imaged through a 40× NA 1.3 objective (Nikon) on a Diaphot configured with a Biorad Radiance 2100 laser confocal scanner. GFP fusions were imaged with the 488 nm argon laser line for excitation, a 515/30 emission filter, and a multialkali cathode photomultiplier. Cherry fusions were imaged with the 543 nm HeNe laser line for excitation, a 600LP emission filter, and a gallium-arsenide phosphide photomultiplier. Laser intensities and gain settings were adjusted to avoid saturation of the detectors (typically 1%–10% for the 488 nm line and 100% for the HeNe line). xy images of dimension 324 × 256 pixels were obtained with 0.38 μm interval. xz images on the line showed in the figures were obtained with 0.5 μm interval over a z range of 25 μm. Green and red images were merged with ImageJ; and when both red and green proteins were present, the image contrasts were linearly adjusted so that both red and green (yellow when overlapping) were clearly visible in the merged images.
Statistical analysis
Data were analyzed by one- or two-way ANOVA followed by Newman-Keuls Multiple Comparison Post Hoc Test.
Results
The isolated PDZ motif of NOS1AP does not interact with nNOS but contributes to binding in a pull-down assay
The binding of NOS1AP to nNOS has been suggested as potential target for development of therapeutic agents in some of the most common causes of death and disability (Li et al., 2013; Weber et al., 2014; Zhu et al., 2014). NMR structure analysis showed that nNOS-PDZ forms a canonical PDZ interaction with MelR peptide (Tochio et al., 1999) (Fig. 1A), and NOS1AP was also thought to interact with nNOS in this way (Jaffrey et al., 1998; Tochio et al., 1999). Canonical PDZ interactions involve the docking of a short motif, typically 4 or 5 C-terminal amino acids, into the binding pocket of a PDZ domain (Doyle et al., 1996; Harris and Lim, 2001). Surprisingly, however, we found the affinity of the nNOS-PDZ domain for the C-terminal peptide from NOS1AP was exceedingly low (“-EIAV,” Kd > 600 μm, Fig. 1B). An independent method, ITC, was applied to this peptide, giving a similar estimate of Kd (data not shown). Notably, this level of affinity is consistent with a recent report that found it necessary to use 50–500 nmol of a 12 amino acid C-terminal NOS1AP peptide by intrahippocampal injection to disrupt nNOS-NOS1AP interactions (Zhu et al., 2014). This approximates to 2–20 mm peptide, as mouse hippocampus has a total volume ∼25 μl (Persson et al., 2014).
Such a low affinity is not considered significant binding. The NOS1AP C terminus conforms to a Class II motif, and yet nNOS is a Class III PDZ domain (Vaccaro and Dente, 2002). We asked whether the PDZ class mismatch was responsible for the weak interaction observed. NOS1AP-nNOS interaction has been reported in pull-down assays (Jaffrey et al., 1998). Therefore, we used a panel of recombinant immobilized PDZ domains of different classes for pull-down assays (Fig. 1C). Full-length luciferase-tagged NOS1AP was recovered from cell lysates by the extended nNOS-PDZ domain immobilized on beads (Fig. 1D). Recovery was extremely efficient, 10-fold better than obtained with the well-established PDZ domain interaction between PSD95-PDZ2 and nNOS β-finger (Fig. 1E) (Hillier et al., 1999; Christopherson et al., 1999; Tochio et al., 2000). Pull-down of NOS1AP with nNOS is dependent on the last three amino acids of NOS1AP, Ile-Ala-Val, as a construct lacking this motif (“ΔIAV”) was not recovered in the pull-down (Fig. 1D). This is a characteristic property of PDZ interactions. We find that this Class II PDZ motif (ψXψ type) (Vaccaro and Dente, 2002) did not mediate binding of NOS1AP to the Class II PDZ domain tested (from Tiam1), whereas the Class I PDZ domain (from PSD95) did not bind detectably under any conditions tested. Scrib-PDZ4 is of unknown specificity (Zhang et al., 2006) but was included as it is reported to interact with NOS1AP PTB domain (Richier et al., 2010). However, we do not observe an interaction in this assay.
Thus full-length NOS1AP forms a stable complex with the extended PDZ domain in nNOS in a manner dependent on the 3 C-terminal residues (Fig. 1D). However, the interaction was not detectable by conventional solution interaction assays using the PDZ domain and the 9 residue C-terminal peptide (Fig. 1B). We next investigated this discrepancy with series of pull-down assays using NOS1AP fragments.
Mutation of key residues in the nNOS-PDZ pocket reduces pull-down of NOS1AP
Mutating critical residues in the PDZ ligand-binding pocket to corresponding residues in a Class I PDZ domain (PSD95-PDZ3) strongly reduced binding to the nNOS extended PDZ domain (1–155) but did not eliminate it (WT vs Y77H/D78E; Fig. 2A). This is a subtle mutation, not expected to eliminate PDZ function but merely change its selectivity (Stricker et al., 1997; Christopherson et al., 1999). The results suggest that NOS1AP does interact with the PDZ domain ligand-binding pocket, as expected for a canonical PDZ interaction (Fig. 1A). Contradicting the assumption of a canonical interaction, however, we found the C-terminal motif of NOS1AP (“-EIAV”) showed no binding whether wild-type or Class I-like mutant domain is used (Fig. 2A). This result is consistent with lack of binding of this sequence as an isolated peptide in fluorescence polarization or ITC (Fig. 1B; and data not shown). This suggests the binding is indeed not a canonical PDZ interaction after all.
We have found that nNOS and NOS1AP can form a very stable complex compared with related interactions, such as that between nNOS and PSD95PDZ2, in cell-free assays using exogenous immobilized nNOS fragments (Fig. 1D,E). To consider intact full-length proteins, we coexpressed constructs for full-length nNOS and NOS1AP in 293T cells. Immunoprecipitation of nNOS from lysates coprecipitated ∼10% of total NOS1AP expressed, suggesting that these proteins also form stable complexes in intact cells (Fig. 2B). xz scans of COS7 cells expressing full-length NOS1AP alone (Fig. 2C, red) showed that it localizes to the plasma membrane, whereas full-length nNOS alone does not (Fig. 2D, left, green). Coexpression of both (Fig. 2D, right, lower cell) showed that the presence of NOS1AP relocalizes nNOS to the plasma membrane. This suggests that these proteins interact quantitatively in intact cells.
NOS1AP residues between 400 and 431 are required for interaction with nNOS
Full-length NOS1AP exhibits substantial binding to nNOS, but the C-terminal PDZ ligand motif alone does not bind (Figs. 1B, 2A). We performed progressively larger N-terminal deletions to map the binding requirements. Stable interaction was retained until NOS1AP residues 400–431 were removed, which resulted in precipitous loss of signal (Fig. 3A). The requirement for both region 400–431 and C-terminal motif (amino acids 504–506) for stable binding in pull-down (Figs. 1D, 3A) suggests one of three scenarios: (1) region 400–431 contributes to binding nNOS indirectly via undefined components in lysates of cells expressing NOS1AP fragments (Fig. 3Bi); (2) 400–431 is required for correct folding and presentation of the PDZ ligand motif 504–506 (Fig. 3Bii), like the salt bridge between residues Asp62 and Gln121 in nNOS is required for a correctly presented β-finger (Tochio et al., 2000; Karlsson et al., 2012); or (3) 400–431 contributes a second, previously unidentified, contact with the nNOS-PDZ domain that is critical for interaction (Fig. 3Biii).
Figure 3.

Binding of NOS1AP to nNOS beads requires residues 400–431 but not residues preceding 400. A, N-terminal deletion mutants of NOS1AP as shown were used as luciferase fusions in a pull-down assay as in Figures 1 and 2 with 2 μg immobilized GST-fused nNOS 1–155 or immobilized GST alone as negative control. NOS1AP pull-down is maintained at a similar level to measurements with full length (Figs. 1, 2) even when residues 1–399 are deleted, but deleting residues 400–431 almost eliminates pull-down (n = 3; one-way ANOVA, Newman-Keuls Multiple Comparison Post Hoc Test). ***p < 0.001. ns, Not significant. B, Three models are proposed to explain the role of NOS1AP residues 400–431 in the nNOS-NOS1AP interaction: (1) Model 1 proposes that an additional protein present in the cell lysate used as a source of NOS1AP in the pull-down experiments serves as a required interaction platform, with both nNOS and NOS1AP residues 400–431 docking with the platform such that both the PDZ motif between nNOS and NOS1AP as well as the platform interaction are required for stable interaction and thus efficient pull-down; (2) Model 2 proposes that residues 400–431 are required for the NOS1AP C-terminal PDZ ligand motif to adopt the appropriate conformation to dock effectively in the nNOS ligand-binding pocket; and (3) Model 3 proposes that nNOS possesses two distinct interaction sites; one is the ligand-binding pocket that forms a conventional PDZ interaction that is by itself extremely weak (affinity ∼1 mm as in Fig. 1) but is complemented by a distinct interaction involving NOS1AP residues 400–431 such that both together are required to form a stable interaction and efficient pull-down.
nNOS-NOS1AP interaction occurs independent of other cell components
To address the first scenario (i.e., that undefined components from 293T lysates used in pull-downs mediate the interaction), we generated recombinant immobilized GST-nNOS[1–155] and NOS1AP[400–506] purified to homogeneity (by Coomassie, Fig. 4A) after proteolytically removing the GST tag. NOS1AP[400–506] formed stable interactions with immobilized nNOS[1–155] and was efficiently retained on washed beads (Fig. 4Bi), whereas NOS1AP[400–503] was almost completely lost even though 20-fold more nNOS was used compared with Figures 1, 2, and 3. This indicates that NOS1AP[400–506] exhibits stable binding to nNOS[1–155] on beads in the absence of other detectable proteins, dependent on residues 504–506.
Figure 4.

NOS1AP residues 400–431 are necessary for interaction, whereas PDZ motif residues 504–506 are not required for interaction but stabilize the complex. A, Recombinant GST-nNOS and tag-free NOS1AP 400–506, 400–503, 432–506, and 432–503 purified to near homogeneity, are shown by Coomassie staining. B, GST-nNOS was immobilized on beads in a conventional pull-down with tag-free NOS1AP. Input samples, unbound supernatant, and washed beads were probed for NOS1AP by Western blotting with a polyclonal antibody R300 raised against the C-terminal region of NOS1AP. Using 40 μg immobilized nNOS/sample, recovery of NOS1AP 400–506 was quantitative (Bi), but barely detectable with NOS1AP 400–503 (Bii). However, 400–503 was greatly sequestered from supernatant when exposed to nNOS beads (middle lanes weaker than inputs). Equal amounts of GST control beads (Biii, Biv) did not sequester protein from supernatants (signals in middle lanes similar to inputs). Representative blots are shown (n = 3). C, Modification of the pull-down assay to separate flow-through and drained beads is shown schematically. Unbound “supernatant” is separated from beads by spinning through a spin-filter, providing flow-through samples and drained beads, which can be resuspended and run directly (“drained beads”) or washed in a conventional manner. D, Results of the modified binding assay as in C are shown. NOS1AP 400–506 is completely depleted from flow-through, and washing beads does not appreciably remove bound protein. In contrast, 400–503 is detected in drained beads but almost completely lost from washed beads. NOS1AP proteins lacking residues 400–432 are not retained on drained beads to any detectable extent. Representative blots are shown (n = 3).
Residues 504–506 are dispensable for interaction but required for stable retention on nNOS beads
NOS1AP was greatly depleted from the supernatant even in the case of NOS1AP[400–503] (Fig. 4Bii), suggesting that binding indeed occurred but was lost during washing steps. Conventional pull-down assays require both affinity and slow off-rates so that captured protein is not lost during washes. Off-rates for signaling molecule interactions vary widely. Thus complexes of Ras with Nore1 have ∼100-fold longer lifetimes than Ras-Raf complexes (Avruch et al., 2009), whereas moderate-affinity interactions may be barely detected if they are very transient. We modified the procedure to remove supernatant from beads by rapid centrifugation through a filter without washing steps (Fig. 4C). This revealed that even [400–503] exhibited substantial binding, which is lost in subsequent washing steps (Fig. 4Dii), as we suspected. In contrast, peptides lacking residues 400–431 were not retained even in drained unwashed beads (Fig. 4Diii–Div).
These results with purified proteins indicate the following: (1) nNOS[1–155] can bind NOS1AP[400–506] directly, in the absence of cellular proteins (Fig. 4Bi); and (2) PDZ motif residues 504–506 are not obligatory for binding but are required for a stable interaction (Fig. 4Di,Dii). This suggests the canonical PDZ interaction (C-terminal residues docking in the PDZ domain pocket) (Doyle et al., 1996) is dispensable for nNOS:NOS1AP protein complex to form. However, it does provide a secondary contribution to the interaction, enhancing lifetime of the complex. Short-lived complexes are revealed as such upon washing. Consistent with this, all pull-down experiments using washing steps show no binding when the PDZ motif, residues 504–506, is absent. The data do not support a role for NOS1AP residues 400–431 in merely maintaining conformation of the C-terminal PDZ motif 504–506 for PDZ binding (Fig. 3Bii) as the motif can be removed without eliminating binding, unlike residues 400–431 (Fig. 4D). Instead, a previously uncharacterized nNOS interaction site overlapping with NOS1AP region 400–431 appears critical for interaction affinity. The PDZ motif merely increases stability of the nNOS:NOS1AP complex. In conclusion, maximal stable binding of NOS1AP with the nNOS-PDZ domain depends on a heterodivalent interaction.
To directly visualize in real time whether release of NOS1AP from nNOS depends on the PDZ motif as inferred above, we established a system in which nNOS protein beads used in Figures 123-4 are trapped in a perfusion chamber. We applied FITC-labeled NOS1AP fragments and monitored loss of fluorescence under buffer flow (Fig. 5A; see Materials and Methods). The curves obtained clearly show that deletion of the PDZ motif substantially reduces the apparent rate of loss of label from nNOS beads (Fig. 5B). We applied the Schuck and Minton two-compartment model for dissociation under flow conditions (which takes account of rebinding) (Schuck and Minton, 1996; de Mol and Fischer, 2008), which fitted well to the decay phases of the data (Fig. 5Ci–Civ). Both fitted curves have on-rates ∼5 μm−1 s−1, consistent with a previously described PDZ motif interaction (Chi et al., 2009). However, the fitted off-rates are extremely low. NOS1AP[400–506] shows an ∼4-fold lower off-rate and therefore a corresponding higher apparent affinity (Fig. 5Cv). As solid-phase assays with GST fusions can overestimate affinities, we measured affinity by fluorescence polarization after proteolytically removing GST tags. This indicated Kd ∼50 nm (Fig. 5D), lower affinity than in the perfused bead assay, but still >104 fold higher than the affinity of the NOS1AP 9 residue C-terminal peptide (Fig. 1B).
Residues 504–506 are dispensable for interaction of NOS1AP with nNOS in intact cells
Pull-down assays typically include washing steps to minimize background binding. This experimental manipulation has no direct counterpart in living cells; nevertheless, lifetimes of protein complexes may greatly impact signaling. If NOS1AP[400–503] binds and dissociates rapidly, leading to loss of binding under washing conditions, then in intact cells we should expect an interaction with nNOS without a strong dependency on the PDZ motif, even though this would be counterintuitive based on current literature. Thus, we predict that NOS1AP[400–503] interacts with nNOS in intact cells. We expressed in COS7 cells Cherry-fused NOS1AP[400–506], which was retained in the cytoplasm (Fig. 6Ai,Bi), or EGFP-fused nNOS[1–155], which diffused to both cytoplasmic and nuclear compartments (Fig. 6Av,Bii) like unfused EGFP and Cherry moieties alone (Fig. 6Aii,Aiv). The presence of NOS1AP[400–506] resulted in relocalization of coexpressed nNOS to the cytoplasm (Fig. 6Aviii,Biii), whereas [432–506] or [432–503], which showed no interaction in cell-free assays (Figs. 3A, 4D), failed to relocalize nNOS (Fig. 6Axiv,Biv,Cv). Notably, NOS1AP[400–503] also relocalized nNOS to the cytoplasm (Fig. 6Axi), although the interaction it forms in a purified system is detected only when the complexes are not washed (Fig. 4). This suggests that the NOS1AP-nNOS interaction in intact cells does not require the PDZ motif even though the lifetime of the complex may be affected by this motif (Figs. 4, 5).
Figure 6.
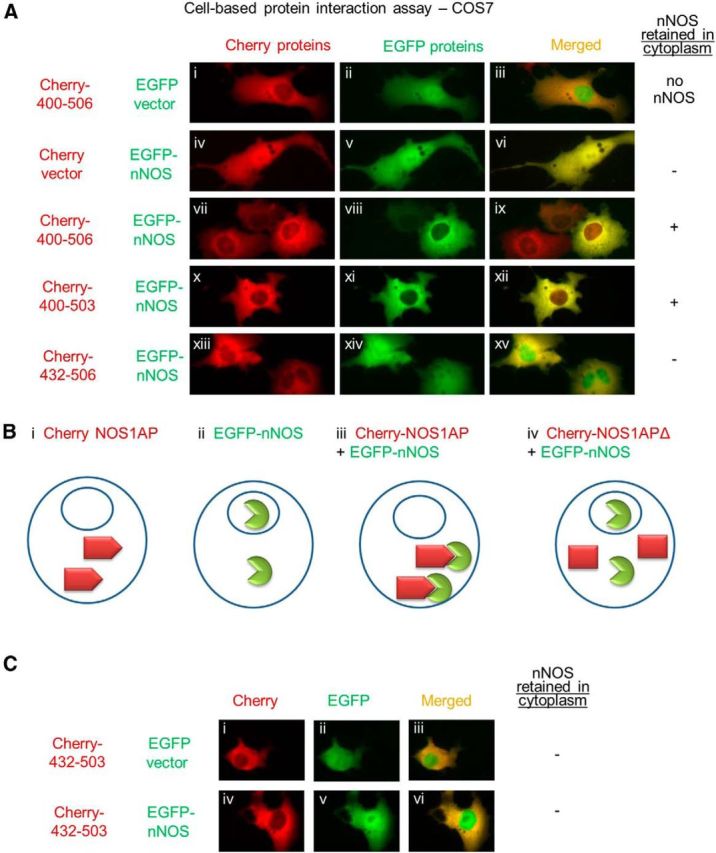
NOS1AP translocates nNOS in intact cells in a manner dependent on residues 400–431 but not the PDZ motif 504–506. A, COS7 cells were transfected with Cherry and EGFP vectors or plasmids encoding Cherry fusions with NOS1AP sequences (400–506, 400–503, 432–506) and EGFP fusions with nNOS 1–155 as indicated. B, Schematic model depicting the behavior of proteins used. Bi, All C-terminal fragments of NOS1AP used, in fusion with Cherry, reside predominantly outside the nucleus. Bii, EGFP-nNOS 1–155 enters both cytoplasmic and nuclear compartments. Biii, When both are expressed, NOS1AP localization is unchanged, but nNOS 1–155 is largely excluded from the nuclei, suggesting the formation of nNOS-NOS1AP complexes that, like NOS1AP, do not freely enter the nucleus. Biv, Deleting regions of NOS1AP that disrupt nNOS interaction is predicted to allow nNOS to enter the nucleus. Thus, deleting NOS1AP residues 400–431 eliminates the effect of NOS1AP on nNOS relocalization, whereas NOS1AP lacking the PDZ motif, residues 504–506, retains nNOS relocalization. C, Experiments were performed as in A using NOS1AP residues 432–503, lacking both PDZ motif 504–506 and the 400–432 region that appears to be required for interaction with nNOS, fused to Cherry fluorescent protein and EGFP vector (Ci–Ciii) or EGFP-nNOS1–155 (Civ–Cvi). Cherry-NOS1AP 432–503 resided predominantly in cytoplasm (Ci, Civ), but EGFP-nNOS was present in both nuclear and cytoplasmic compartments (Cv, Cvi).
Residues E429/F431 are required for PDZ motif independent NOS1AP interaction with nNOS in intact cells
The results above suggest that a region within NOS1AP 400–431 is required to form a second contact with nNOS (Fig. 6Axii,Biii). Evolutionary conservation analysis of NOS1AP[400–506] reveals three conservation hotspots: the C-terminal PDZ ligand motif, a central region, and residues ∼420–430 (Fig. 7A), the latter of which secondary structure prediction algorithms suggests may form a β-strand (data not shown). To determine the contributions of residues 421–431 to NOS1AP/nNOS interaction, we applied an alanine scan approach in which consecutive residues were replaced by alanine (Fig. 7B). A pull-down using this panel of NOS1AP[400–506] mutants revealed that E429 and F431 were most critical for interaction (Fig. 7C). We used the same alanine scan panel with the in-cell interaction assay of Figure 6, but with NOS1AP[400–503] because deleting the PDZ ligand motif removes any ambiguity about its involvement and ensures maximal dependence of interaction on the region 400–431 (Fig. 3B). This revealed NOS1AP-induced translocation of nNOS was most compromised by mutation of residue E429 or F431 (Fig. 7D) or double mutation of K427/L428. This finding was further confirmed by performing an independent coprecipitation experiment of NOS1AP alanine mutants with coexpressed nNOS[1–155] (Fig. 7E). It is notable that amino acids K427, L428, E429, and F431 are particularly highly conserved residues throughout evolution (Fig. 8). Thus, whether the PDZ motif (residues 504–506) is present or not, mutating residues E429 or F431 reduces the interaction with nNOS, indicating a requirement for these side chains independent of the PDZ motif. We refer to this second interaction site as the ExF motif.
Figure 7.
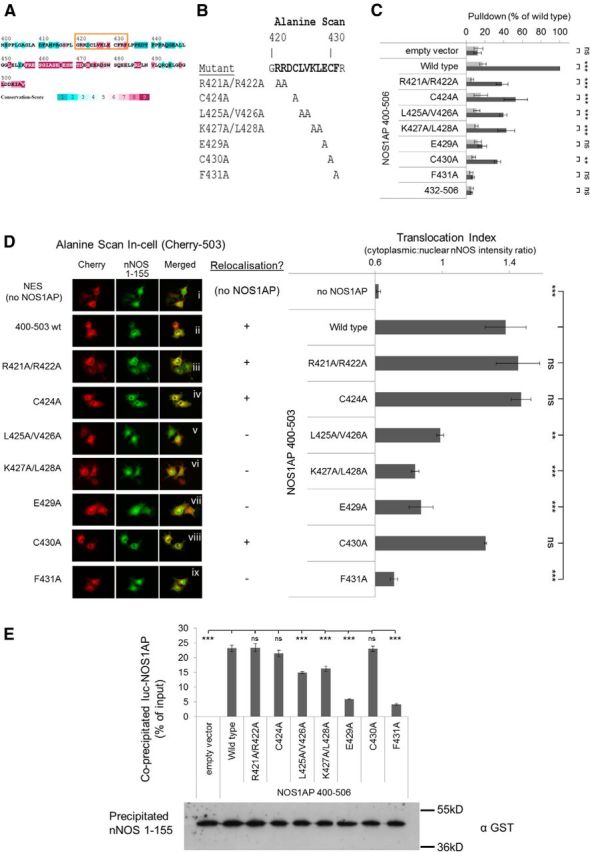
Alanine scans reveal a critical ExF motif for interaction of NOS1AP with nNOS in pull-down and cell-based protein translocation assays. A, Evolutionary conservation analysis (using ConSurf, Ashkenazy et al., 2010) of NOS1AP region 400–506 shows hotspots of evolutionary conservation at the PDZ ligand motif (503–506), a central zone, and a region between 420 and 432. The latter shows as a predicted β sheet using several secondary structure prediction tools (JPRED3, PSIPRED). B, Alignment of conserved region 420–432 showing the panel of alanine mutants generated in NOS1AP constructs. C, The alanine scan panel of NOS1AP shown in B was used in a modified pull-down using a filter plate and aspiration manifold to facilitate the handling of larger numbers of samples. NOS1AP 400–506 was defined as 100% binding; and as expected, the negative control NOS1AP 432–506 failed to interact. This analysis revealed the clearest loss of binding in NOS1AP 400–506 mutants E429A and F431A (n = 4, two-way ANOVA followed by Newman-Keuls Multiple Comparison Post Hoc Test). D, The COS7 cell-based assay shown in Figure 6 was used with the alanine mutants shown in B, C, and nuclear translocation indices (ratio of pixel intensities from segmented cytoplasmic and nuclear compartments defined as described in Materials and Methods) obtained from transfected cells in the images. Once again, the clearest loss of translocating effect was observed with NOS1AP 400–503 mutants E429A and F431A (n = 4, one-way ANOVA followed by Newman-Keuls Multiple Comparison Post Hoc Test). E, The alanine mutant panel (in luciferase-fused NOS1AP[400–506]) was coexpressed with GST-fused nNOS[1–155]. Cell lysates were incubated with glutathione beads to immobilize nNOS, and pulled down using a conventional protocol. Coprecipitated NOS1AP was measured by luciferase assay of washed beads and normalized to initial levels in each sample. Equal precipitation of nNOS was confirmed by immunoblotting. This revealed loss of binding across the alanine mutant panel as in D (n = 3, one-way ANOVA followed by Newman-Keuls Multiple Comparison Post Hoc Test). **p < 0.01. ***p < 0.001. ns, Not significant.
Figure 8.

Evolutionary conservation of ExF motif among NOS1AP homologs possessing C-terminal ψxψ* motif. NOS1AP homologs containing the C-terminal PDZ motif were identified by blast search, and evolutionary diverse examples are included with mammalian sequences for alignment. This revealed identity (*) was retained in only 4 residues surrounding the ExF motif, including E429 (human numbering), although F431 only exhibited a conservative substitution to Y in birds and some reptiles. We found no NOS1AP homologs possessing only the PDZ motif, without ExF motif.
TAT peptides fused to PDZ ligand motif or ExF motif containing region each inhibit excitotoxic activation of p38MAPK
Our data from cell-free and heterologous systems suggest nNOS:NOS1AP interaction requires a novel interface between nNOS and a cluster of residues around and including NOS1AP αα429/431, together with a PDZ ligand that stabilizes the interaction but alone has virtually no affinity. Nevertheless, loss of the PDZ motif does destabilize the interaction; and, as previously reported (Li et al., 2013), competition at this site alone with a synthetic ligand (TAT-GESV) is sufficient to inhibit NMDA-induced recruitment of NOS1AP to nNOS and downstream signaling in cultured cortical neurons. This suggests that, in intact cells, a productive signaling interaction, in the latter case sufficient to evoke excitotoxic pathways, requires the stability of the heterodivalent interaction conferred by the PDZ ligand motif. We therefore predicted that competition at the ExF motif would also inhibit signaling, and simultaneous competition at both sites might be at least as neuroprotective as either alone. We stimulated cortical neurons with NMDA after exposure to recombinant cell-permeable TAT peptides comprising NOS1AP residues 400–503, which lacks the PDZ ligand motif but includes the ExF containing region, or residues 400–506, which contains both PDZ ligand motif and the ExF motif. Both recombinant peptides (with PDZ motif present or deleted) were able to inhibit NMDA-evoked activation of p38 at least as much as the PDZ ligand TAT-GESV, which was used as a positive control for inhibition (Li et al., 2013) (Fig. 9A,B).
Residues E429/F431 are critical for affinity of TAT-NOS1AP[400–503] to the N terminus of nNOS
To explore whether the ExF motif might be a basis for neuroprotection, we first determined the affinities of the TAT-fused fragments of NOS1AP for their target in the N terminus of nNOS. Increasing concentrations of recombinant TAT peptides were incubated with immobilized GST-nNOS[1–155] on beads, revealing saturable binding (Fig. 9Ci–Cii) with Kd values of <1 μm for His-TAT-NOS1AP[400–506] and ∼5 × lower affinity for the NOS1AP[400–503] form lacking the PDZ ligand motif (Fig. 9Ciii). This difference is consistent with that determined with fragments lacking His/TAT using the bead binding assay (Fig. 5), although the actual affinities are somewhat lower. In contrast, engineering inactivating mutations in the ExF site of TAT-NOS1AP[400–503], at either E429 or F431, respectively (Fig. 7), almost abolished binding (Fig. 9C). This TAT-NOS1AP[400–503] was effective at 1 μm on neuronal p38MAPK activation (Fig. 9A,B) but has ∼5 μm affinity for nNOS[1–155] (Fig. 9C). This apparent discrepancy may merely represent the known accumulation of TAT peptide in cells (Wadia and Dowdy, 2005) and/or the increased activity after in-cell refolding (Becker-Hapak et al., 2001).
Neuroprotection in organotypic slice cultures can be achieved with TAT-fused peptides lacking the PDZ ligand if the ExF motif is intact
Next, we addressed the impact of the TAT peptides on the negative consequences of NMDA receptors in a more physiological setting than dissociated culture as used in Figure 9A, B. We evaluated neuroprotection from NMDA-induced lesions in organotypic hippocampal slice cultures, which possess an intact network of neurons retaining some characteristic in vivo architecture of cell arrangement and morphology and has been useful for studying excitotoxicity (D'Orsi et al., 2012). A substantial lesion, visualized by propidium iodide, was detected 24 h after NMDA treatment (50 μm, 30 min) in CA1, CA3, and DG (Fig. 9D). Pretreatment with PDZ motif ligand TAT-GESV demonstrated significant reductions in CA1/CA3 staining but not in DG. Peptide TAT-NOS1AP[400–503], lacking the PDZ ligand motif, significantly reduced staining not only in CA1/CA3 but also in DG (Fig. 9D,E). TAT-NOS1AP[400–506], possessing intact ExF and PDZ ligand motifs, showed the greatest CA1 protection, as well as reducing staining of CA3/DG (Figs. 9D,E). As controls to evaluate whether the ExF motif is truly relevant to the protection in multiple regions evoked by the peptide lacking the PDZ ligand motif, we compared the effects of the two NOS1AP[400–503] peptides bearing mutations at either E429 or F431 that ablate binding of the TAT peptides to nNOS (Fig. 9C). Consistent with our proposal that two sites are required for excitotoxic lesions, the ExF mutant peptides did not significantly reduce NMDA-induced propidium iodide of any slice region evaluated.
Discussion
We have shown here that NOS1AP interacts with the N-terminal region of nNOS via a novel internal “ExF” motif, in contrast to the classical PDZ docking assumed to be responsible. The affinity of the NOS1AP C-terminal motif for the PDZ binding pocket is nearly 1 mm, which is not considered a significant interaction. However, in the presence of an internal region containing the “ExF” motif, the C-terminal PDZ ligand motif increases the lifetime of the nNOS-NOS1AP complex, which appears to be a critical parameter to initiate signaling.
Interaction between NOS1AP and nNOS is increasingly cited as a potential drug target in a growing number of diseases, including cardiovascular and psychiatric conditions, such as schizophrenia and anxiety disorders (Weber et al., 2014; Zhu et al., 2014). It is a downstream component of the excitotoxic NR-PSD95-nNOS pathway (Cao et al., 2005; Li et al., 2013). This pathway is the target of the only neuroprotectant that has been successful in a clinical trial for stroke (NA-1) (Hill et al., 2012). It has furthermore been implicated in models of neuropathic pain and depression (Florio et al., 2009; LeBlanc et al., 2010; Doucet et al., 2013) and neurodegenerative diseases from Alzheimer's and Huntington's to Parkinson's disease (Ittner et al., 2010; Fan et al., 2012; Hu et al., 2014), as has p38MAPK activation (LeBlanc et al., 2010; Fan et al., 2012; Xu et al., 2014; Zhou et al., 2014). Most known actions of NOS1AP, from activation of DexRas, iron transport, and p38MAPK downstream of NMDA receptor activation (Fang et al., 2000; Cheah et al., 2006; Li et al., 2013), to regulation of cardiac action potential (Chang et al., 2008; Kapoor et al., 2014) and neurite outgrowth (Carrel et al., 2009) are thought to depend on interaction with nNOS (for review, see Courtney et al., 2014). It is therefore essential to understand the mechanisms of interaction between nNOS and NOS1AP.
NMR structural analysis of the MelR ligand docked in the nNOS-PDZ domain clearly shows a canonical PDZ interaction with three C-terminal ligand residues apposed to residues lining the PDZ pocket formed by the βB sheet and αB helix (Tochio et al., 1999, shown here in Fig. 1A; Harris and Lim, 2001). NOS1AP was also thought to bind nNOS via a canonical PDZ interaction (Christopherson et al., 1999; Tochio et al., 1999). However, three different binding methods clearly show the C-terminal PDZ motif of NOS1AP, which does not even conform to the nNOS class of PDZ motifs, has little or no affinity for nNOS (Figs. 1B, 2; and ITC data not shown). However, full-length NOS1AP protein quantitatively relocalizes nNOS in intact cells. Furthermore, NOS1AP binds even low levels of immobilized nNOS, yielding high recoveries compared with other PDZ interactions, after repeated washing. Paradoxically, this recovery is highly dependent on the last three C-terminal residues of NOS1AP (Fig. 1). Residues upstream of the three C-terminal ones often contribute to PDZ interactions, typically due to additional interaction energies conferred by residues 4–7 from the C terminus docking into a β2-β3 loop (Birrane et al., 2003; Thomas et al., 2008; Lee and Zheng, 2010; Luck et al., 2012). However, even the 9 residue peptide from the C terminus of NOS1AP completely fails to show affinity for nNOS. In contrast, we detect interaction even when deleting the last three residues. This is quite contrary to expectation for a PDZ interaction.
We addressed this paradox by deletion mapping, revealing a critical requirement for a binding region of NOS1AP far upstream of the C terminus. This does not merely stabilize the PDZ motif nor act via a third binding partner but is alone sufficient for interaction with the N-terminal region of nNOS in the complete absence of the PDZ motif. The PDZ motif requirement in conventional pull-down assays is explained by the secondary contribution of the PDZ motif to stability of the complex by reducing off-rate. Nevertheless, in intact cells, this stability is not required to visualize the functional consequences of interaction, specifically the relocalization of nNOS N terminus by NOS1AP fragments bearing the internal binding region. Alanine scanning an evolutionary conserved stretch of NOS1AP within the internal binding region revealed Glu-429 and Phe-431 as critical for interaction with the N terminus of nNOS. All NOS1AP homologs we could find containing the C-terminal PDZ ligand motif also possessed an ExF/Y motif (Fig. 8), suggesting a functional relationship between these two motifs. Furthermore, predicted mammalian NOS1AP variants, such as XP_007974555, possess an alternate C terminus starting immediately after amino acid 419, further suggesting that residues starting close to 420 and ending at 506 might cooperate to confer a discrete function.
The results suggests that the primary interaction between NOS1AP and nNOS is not the PDZ motif interaction but a separate interaction involving sequences around and including residues 429/431 (“ExF motif”) sufficient to mediate binding, whereas the PDZ motif increases the interaction lifetime. Therefore, the effects of deleting C-terminal residues from NOS1AP (e.g., Carrel et al., 2009) may not merely implicate nNOS but the requirement for a long-lived interaction, a significant factor in signal propagation. We showed that a ligand for the nNOS-PDZ ligand-binding pocket inhibits coimmunoprecipitable nNOS:NOS1AP induced by excitotoxic stimulation and that, in this case, the competition at the binding pocket is sufficient to inhibit downstream signaling (p38MAPK activation and excitotoxic death) (Li et al., 2013).
Thus, PDZ ligand-binding pocket interaction is not necessary for visually detectable recruitment of nNOS N terminus in living cells (Fig. 7), but competition at the PDZ interaction interface is effective at inhibiting neuronal kinase pathways (Li et al., 2013). This apparent discrepancy most likely depends on the time required for activation of the kinase pathway, which takes some minutes to reach a peak (Cao et al., 2005). Multivalent binding, a more general form of the heterodivalent binding described here, is common in tyrosine kinase signaling and is thought to result in highly specific yet dynamic binding (de Mol and Fischer et al., 2008). The same might therefore be predicted for nNOS-NOS1AP interaction and, consistent with this, the interaction is highly inducible (Li et al., 2013). This kinetic property influences and may facilitate opportunities for development of inhibitory strategies.
These results suggest that the PDZ-binding motif is not the only nNOS interaction available for targeting to achieve disruption of excitotoxic or other nNOS/NOS1AP-dependent signaling events, which may contribute to a variety of disorders for which there are insufficient treatments available (Zhou and Sheng, 2013; Weber et al., 2014; Zhu et al., 2014). The ExF motif interaction mapped here may present additional opportunities for pharmacological manipulation in some or all of these conditions. Here we find evidence that fragments of NOS1AP containing the ExF motif but lacking the PDZ ligand motif can indeed be used as pharmacological agents inhibiting excitotoxic lesions in hippocampal tissue, suggesting the ExF interaction should be considered as a novel drug target.
Recruitment of NOS1AP to nNOS is inducible (Li et al., 2013), and the ability to disrupt signaling by competition at either by the PDZ ligand motif or the ExF-containing region alone (Fig. 8) indicates that both interaction sites are required for signal transduction in this case. However, among NMDA signaling-related conditions for which new interventions should be developed, there may be cases where simultaneous competition at both sites (and perhaps even more sites) is necessary to prevent downstream signaling. Furthermore, specific examples exist where combinatorial inhibition at two sites on a target protein provides substantial increase in inhibitor potency (Petros et al., 2010). This possibility has also been proposed for proteins containing multiple PDZ domains (Klosi et al., 2007). An increase in the number of potential sites for therapeutic intervention can only help the development of future therapeutic approaches. One may speculate that a multitarget approach may even provide the opportunity to build in redundancy into an inhibitory strategy to increase the chances of improved outcomes.
Footnotes
This work was supported by the University of Eastern Finland, the Academy of Finland (Grants 127717, 134893, and 135073), the European Union-FP7, the Doctoral Programme in Molecular Medicine, the Finnish Cultural Foundation North Savo regional fund, Turku Centre for Biotechnology, and the Magnus Ehrnooths Foundation. The Biocentre Finland imaging facility MUIC at the University of Eastern Finland was used for this work. We thank John Heath (School of Biosciences, University of Birmingham, Birmingham, United Kingdom) for plasmid pGEX-HRV 3C protease; Steven Dowdy for providing a protocol for preparation of TAT fusion proteins from bacteria; Aila Seppänen and Veera Pevgonen for technical assistance; and Katryna Cisek for critical reading of the manuscript.
The authors declare no competing financial interests.
References
- Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–W533. doi: 10.1093/nar/gkq399. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avruch J, Xavier R, Bardeesy N, Zhang XF, Praskova M, Zhou D, Xia F. Rassf family of tumor suppressor polypeptides. J Biol Chem. 2009;284:11001–11005. doi: 10.1074/jbc.R800073200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker-Hapak M, McAllister SS, Dowdy SF. TAT-mediated protein transduction into mammalian cells. Methods. 2001;24:247–256. doi: 10.1006/meth.2001.1186. [DOI] [PubMed] [Google Scholar]
- Birrane G, Chung J, Ladias JA. Novel mode of ligand recognition by the Erbin PDZ domain. J Biol Chem. 2003;278:1399–1402. doi: 10.1074/jbc.C200571200. [DOI] [PubMed] [Google Scholar]
- Buchan DW, Minneci F, Nugent TC, Bryson K, Jones DT. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013;41:W349–W357. doi: 10.1093/nar/gkt381. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunk EC, König HG, Bonner HP, Kirby BP, Prehn JH. NMDA-induced injury of mouse organotypic hippocampal slice cultures triggers delayed neuroblast proliferation in the dentate gyrus: an in vitro model for the study of neural precursor cell proliferation. Brain Res. 2010;1359:22–32. doi: 10.1016/j.brainres.2010.05.024. [DOI] [PubMed] [Google Scholar]
- Cao J, Viholainen JI, Dart C, Warwick HK, Leyland ML, Courtney MJ. The PSD95-nNOS interface: a target for inhibition of excitotoxic p38 stress-activated protein kinase activation and cell death. J Cell Biol. 2005;168:117–126. doi: 10.1083/jcb.200407024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrel D, Du Y, Komlos D, Hadzimichalis NM, Kwon M, Wang B, Brzustowicz LM, Firestein BL. NOS1AP regulates dendrite patterning of hippocampal neurons through a carboxypeptidase E-mediated pathway. J Neurosci. 2009;29:8248–8258. doi: 10.1523/JNEUROSCI.5287-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KC, Barth AS, Sasano T, Kizana E, Kashiwakura Y, Zhang Y, Foster DB, Marbán E. CAPON modulates cardiac repolarization via neuronal nitric oxide synthase signaling in the heart. Proc Natl Acad Sci U S A. 2008;105:4477–4482. doi: 10.1073/pnas.0709118105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah JH, Kim SF, Hester LD, Clancy KW, Patterson SE, 3rd, Papadopoulos V, Snyder SH. NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron. 2006;51:431–440. doi: 10.1016/j.neuron.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Pan L, Wei Z, Zhao Y, Zhang M. Domain-swapped dimerization of ZO-1 PDZ2 generates specific and regulatory connexin43-binding sites. EMBO J. 2008;27:2113–2123. doi: 10.1038/emboj.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi CN, Bach A, Engström A, Wang H, Strømgaard K, Gianni S, Jemth P. A sequential binding mechanism in a PDZ domain. Biochemistry. 2009;48:7089–7097. doi: 10.1021/bi900559k. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Hillier BJ, Lim WA, Bredt DS. PSD-95 assembles a ternary complex with the N-methyl-d-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J Biol Chem. 1999;274:27467–27473. doi: 10.1074/jbc.274.39.27467. [DOI] [PubMed] [Google Scholar]
- Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney MJ, Coffey ET. The mechanism of Ara-C-induced apoptosis of differentiating cerebellar granule neurons. Eur J Neurosci. 1999;11:1073–1084. doi: 10.1046/j.1460-9568.1999.00520.x. [DOI] [PubMed] [Google Scholar]
- Courtney MJ, Li LL, Lai YY. Mechanisms of NOS1AP action on NMDA receptor-nNOS signaling. Front Cell Neurosci. 2014;8:252. doi: 10.3389/fncel.2014.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mol NJ, Fischer MJ. Kinetic and thermodynamic analysis of ligand–receptor interactions: SPR applications in drug development. In: Schasfoort RBM, Tudos AJ, editors. Handbook of surface plasmon resonance. Cambridge: RSC; 2008. pp. 123–172. [Google Scholar]
- D'Orsi B, Bonner H, Tuffy LP, Düssmann H, Woods I, Courtney MJ, Ward MW, Prehn JH. Calpains are downstream effectors of bax-dependent excitotoxic apoptosis. J Neurosci. 2012;32:1847–1858. doi: 10.1523/JNEUROSCI.2345-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet MV, Levine H, Dev KK, Harkin A. Small-molecule inhibitors at the PSD-95/nNOS interface have antidepressant-like properties in mice. Neuropsychopharmacology. 2013;38:1575–1584. doi: 10.1038/npp.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Lee A, Lewis J, Kim E, Sheng M, MacKinnon R. Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell. 1996;85:1067–1076. doi: 10.1016/S0092-8674(00)81307-0. [DOI] [PubMed] [Google Scholar]
- Fan J, Gladding CM, Wang L, Zhang LY, Kaufman AM, Milnerwood AJ, Raymond LA. P38 MAPK is involved in enhanced NMDA receptor-dependent excitotoxicity in YAC transgenic mouse model of Huntington disease. Neurobiol Dis. 2012;45:999–1009. doi: 10.1016/j.nbd.2011.12.019. [DOI] [PubMed] [Google Scholar]
- Fang M, Jaffrey SR, Sawa A, Ye K, Luo X, Snyder SH. Dexras1: a G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron. 2000;28:183–193. doi: 10.1016/S0896-6273(00)00095-7. [DOI] [PubMed] [Google Scholar]
- Florio SK, Loh C, Huang SM, Iwamaye AE, Kitto KF, Fowler KW, Treiberg JA, Hayflick JS, Walker JM, Fairbanks CA, Lai Y. Disruption of nNOS-PSD95 protein-protein interaction inhibits acute thermal hyperalgesia and chronic mechanical allodynia in rodents. Br J Pharmacol. 2009;158:494–506. doi: 10.1111/j.1476-5381.2009.00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris BZ, Lim WA. Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci. 2001;114:3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- Hill MD, Martin RH, Mikulis D, Wong JH, Silver FL, Terbrugge KG, Milot G, Clark WM, Macdonald RL, Kelly ME, Boulton M, Fleetwood I, McDougall C, Gunnarsson T, Chow M, Lum C, Dodd R, Poublanc J, Krings T, Demchuk AM, Goyal M, Anderson R, Bishop J, Garman D, Tymianski M, ENACT trial investigators Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012;11:942–950. doi: 10.1016/S1474-4422(12)70225-9. [DOI] [PubMed] [Google Scholar]
- Hillier BJ, Christopherson KS, Prehoda KE, Bredt DS, Lim WA. Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science. 1999;284:812–815. doi: 10.1126/science.284.5415.812. [DOI] [PubMed] [Google Scholar]
- Hu W, Guan LS, Dang XB, Ren PY, Zhang YL. Small-molecule inhibitors at the PSD-95/nNOS interface attenuate MPP(+)-induced neuronal injury through Sirt3 mediated inhibition of mitochondrial dysfunction. Neurochem Int. 2014;79:57–64. doi: 10.1016/j.neuint.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Snowman AM, Eliasson MJ, Cohen NA, Snyder SH. CAPON: a protein associated with neuronal nitric oxide synthase that regulates its interactions with PSD95. Neuron. 1998;20:115–124. doi: 10.1016/S0896-6273(00)80439-0. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Sekar RB, Hansen NF, Fox-Talbot K, Morley M, Pihur V, Chatterjee S, Brandimarto J, Moravec CS, Pulit SL, Pfeufer A, Mullikin J, Ross M, Green ED, Bentley D, Newton-Cheh C, Boerwinkle E, Tomaselli GF, Cappola TP, Arking DE, et al. An enhancer polymorphism at the cardiomyocyte intercalated disc protein NOS1AP locus is a major regulator of the QT interval. Am J Hum Genet. 2014;94:854–869. doi: 10.1016/j.ajhg.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson OA, Chi CN, Engström A, Jemth P. The transition state of coupled folding and binding for a flexible β-finger. J Mol Biol. 2012;417:253–261. doi: 10.1016/j.jmb.2012.01.042. [DOI] [PubMed] [Google Scholar]
- Kasri NN, Govek EE, Van Aelst L. Characterization of oligophrenin-1, a RhoGAP lost in patients affected with mental retardation: lentiviral injection in organotypic brain slice cultures. Methods Enzymol. 2008;439:255–266. doi: 10.1016/S0076-6879(07)00419-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klosi E, Saro D, Spaller MR. Bivalent peptides as PDZ domain ligands. Bioorg Med Chem Lett. 2007;17:6147–6150. doi: 10.1016/j.bmcl.2007.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157–188. doi: 10.1016/j.pneurobio.2013.11.006. [DOI] [PubMed] [Google Scholar]
- LeBlanc BW, Iwata M, Mallon AP, Rupasinghe CN, Goebel DJ, Marshall J, Spaller MR, Saab CY. A cyclic peptide targeted against PSD-95 blocks central sensitization and attenuates thermal hyperalgesia. Neuroscience. 2010;167:490–500. doi: 10.1016/j.neuroscience.2010.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Zheng JJ. PDZ domains and their binding partners: structure, specificity, and modification. Cell Commun Signal. 2010;8:8. doi: 10.1186/1478-811X-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LL, Ginet V, Liu X, Vergun O, Tuittila M, Mathieu M, Bonny C, Puyal J, Truttmann AC, Courtney MJ. The nNOS-p38MAPK pathway is mediated by NOS1AP during neuronal death. J Neurosci. 2013;33:8185–8201. doi: 10.1523/JNEUROSCI.4578-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck K, Charbonnier S, Travé G. The emerging contribution of sequence context to the specificity of protein interactions mediated by PDZ domains. FEBS Lett. 2012;586:2648–2661. doi: 10.1016/j.febslet.2012.03.056. [DOI] [PubMed] [Google Scholar]
- Persson A, Sim SC, Virding S, Onishchenko N, Schulte G, Ingelman-Sundberg M. Decreased hippocampal volume and increased anxiety in a transgenic mouse model expressing the human CYP2C19 gene. Mol Psychiatry. 2014;19:733–741. doi: 10.1038/mp.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petros AM, Huth JR, Oost T, Park CM, Ding H, Wang X, Zhang H, Nimmer P, Mendoza R, Sun C, Mack J, Walter K, Dorwin S, Gramling E, Ladror U, Rosenberg SH, Elmore SW, Fesik SW, Hajduk PJ. Discovery of a potent and selective Bcl-2 inhibitor using SAR by NMR. Bioorg Med Chem Lett. 2010;20:6587–6591. doi: 10.1016/j.bmcl.2010.09.033. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF chimera: a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Richier L, Williton K, Clattenburg L, Colwill K, O'Brien M, Tsang C, Kolar A, Zinck N, Metalnikov P, Trimble WS, Krueger SR, Pawson T, Fawcett JP. NOS1AP associates with Scribble and regulates dendritic spine development. J Neurosci. 2010;30:4796–4805. doi: 10.1523/JNEUROSCI.3726-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepens J, Cuppen E, Wieringa B, Hendriks W. The neuronal nitric oxide synthase PDZ motif binds to -G(D, E)XV* carboxyterminal sequences. FEBS Lett. 1997;409:53–56. doi: 10.1016/S0014-5793(97)00481-X. [DOI] [PubMed] [Google Scholar]
- Schuck P, Minton AP. Analysis of mass transport-limited binding kinetics in evanescent wave biosensors. Anal Biochem. 1996;240:262–272. doi: 10.1006/abio.1996.0356. [DOI] [PubMed] [Google Scholar]
- Semenova MM, Mäki-Hokkonen AM, Cao J, Komarovski V, Forsberg KM, Koistinaho M, Coffey ET, Courtney MJ. Rho mediates calcium-dependent activation of p38alpha and subsequent excitotoxic cell death. Nat Neurosci. 2007;10:436–443. doi: 10.1038/nn1869. [DOI] [PubMed] [Google Scholar]
- Stiffler MA, Chen JR, Grantcharova VP, Lei Y, Fuchs D, Allen JE, Zaslavskaia LA, MacBeath G. PDZ domain binding selectivity is optimized across the mouse proteome. Science. 2007;317:364–369. doi: 10.1126/science.1144592. [Erratum (2007) 318:393] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker NL, Christopherson KS, Yi BA, Schatz PJ, Raab RW, Dawes G, Bassett DE, Jr, Bredt DS, Li M. PDZ domain of neuronal nitric oxide synthase recognizes novel C-terminal peptide sequences. Nat Biotechnol. 1997;15:336–342. doi: 10.1038/nbt0497-336. [DOI] [PubMed] [Google Scholar]
- Thomas M, Dasgupta J, Zhang Y, Chen X, Banks L. Analysis of specificity determinants in the interactions of different HPV E6 proteins with their PDZ domain-containing substrates. Virology. 2008;376:371–378. doi: 10.1016/j.virol.2008.03.021. [DOI] [PubMed] [Google Scholar]
- Tochio H, Zhang Q, Mandal P, Li M, Zhang M. Solution structure of the extended neuronal nitric oxide synthase PDZ domain complexed with an associated peptide. Nat Struct Biol. 1999;6:417–421. doi: 10.1038/8216. [DOI] [PubMed] [Google Scholar]
- Tochio H, Mok YK, Zhang Q, Kan HM, Bredt DS, Zhang M. Formation of nNOS/PSD-95 PDZ dimer requires a preformed beta-finger structure from the nNOS PDZ domain. J Mol Biol. 2000;303:359–370. doi: 10.1006/jmbi.2000.4148. [DOI] [PubMed] [Google Scholar]
- Vaccaro P, Dente L. PDZ domains: troubles in classification. FEBS Lett. 2002;512:345–349. doi: 10.1016/S0014-5793(02)02220-2. [DOI] [PubMed] [Google Scholar]
- Wadia JS, Dowdy SF. Transmembrane delivery of protein and peptide drugs by TAT-mediated transduction in the treatment of cancer. Adv Drug Deliv Rev. 2005;57:579–596. doi: 10.1016/j.addr.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Weber H, Klamer D, Freudenberg F, Kittel-Schneider S, Rivero O, Scholz CJ, Volkert J, Kopf J, Heupel J, Herterich S, Adolfsson R, Alttoa A, Post A, Grußendorf H, Kramer A, Gessner A, Schmidt B, Hempel S, Jacob CP, Sanjuán J, et al. The genetic contribution of the NO system at the glutamatergic post-synapse to schizophrenia: further evidence and meta-analysis. Eur Neuropsychopharmacol. 2014;24:65–85. doi: 10.1016/j.euroneuro.2013.09.005. [DOI] [PubMed] [Google Scholar]
- Xu B, Wratten N, Charych EI, Buyske S, Firestein BL, Brzustowicz LM. Increased expression in dorsolateral prefrontal cortex of CAPON in schizophrenia and bipolar disorder. PLoS Med. 2005;2:e263. doi: 10.1371/journal.pmed.0020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Cao DH, Wu GM, Hou XY. Involvement of P38MAPK activation by NMDA receptors and non-NMDA receptors in amyloid-β peptide-induced neuronal loss in rat hippocampal CA1 and CA3 subfields. Neurosci Res. 2014;85:51–57. doi: 10.1016/j.neures.2014.05.011. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yeh S, Appleton BA, Held HA, Kausalya PJ, Phua DC, Wong WL, Lasky LA, Wiesmann C, Hunziker W, Sidhu SS. Convergent and divergent ligand specificity among PDZ domains of the LAP and zonula occludens (ZO) families. J Biol Chem. 2006;281:22299–22311. doi: 10.1074/jbc.M602902200. [DOI] [PubMed] [Google Scholar]
- Zhou F, Xu Y, Hou XY. MLK3-MKK3/6-P38MAPK cascades following N-methyl-d-aspartate receptor activation contributes to amyloid-β peptide-induced apoptosis in SH-SY5Y cells. J Neurosci Res. 2014;92:808–817. doi: 10.1002/jnr.23354. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Sheng M. NMDA receptors in nervous system diseases. Neuropharmacology. 2013;74:69–75. doi: 10.1016/j.neuropharm.2013.03.030. [DOI] [PubMed] [Google Scholar]
- Zhu LJ, Li TY, Luo CX, Jiang N, Chang L, Lin YH, Zhou HH, Chen C, Zhang Y, Lu W, Gao LY, Ma Y, Zhou QG, Hu Q, Hu XL, Zhang J, Wu HY, Zhu DY. CAPON-nNOS coupling can serve as a target for developing new anxiolytics. Nat Med. 2014;20:1050–1054. doi: 10.1038/nm.3644. [DOI] [PubMed] [Google Scholar]