

Abstract
Spinal muscular atrophy (SMA) is a neuromuscular disease characterized by the selective loss of spinal motor neurons due to the depletion of the survival of motor neuron (SMN) protein. No therapy is currently available for SMA, which represents the leading genetic cause of death in childhood. In the present study, we report that insulin-like growth factor-1 receptor (Igf-1r) gene expression is enhanced in the spinal cords of SMA-like mice. The reduction of expression, either at the physiological (through physical exercise) or genetic level, resulted in the following: (1) a significant improvement in lifespan and motor behavior, (2) a significant motor neuron protection, and (3) an increase in SMN expression in spinal cord and skeletal muscles through both transcriptional and posttranscriptional mechanisms. Furthermore, we have found that reducing IGF-1R expression is sufficient to restore intracellular signaling pathway activation profile lying downstream of IGF-1R, resulting in both the powerful activation of the neuroprotective AKT/CREB pathway and the inhibition of the ERK and JAK pathways. Therefore, reducing rather than enhancing the IGF-1 pathway could constitute a useful strategy to limit neurodegeneration in SMA.
SIGNIFICANCE STATEMENT Recent evidence of IGF-1 axis alteration in spinal muscular atrophy (SMA), a very severe neurodegenerative disease affecting specifically the motor neurons, have triggered a renewed interest in insulin-like growth factor-1 (IGF-1) pathway activation as a potential therapeutic approach for motor neuron diseases. The present study challenges this point of view and brings the alternative hypothesis that reducing rather than enhancing the IGF-1 signaling pathway exerts a neuroprotective effect in SMA. Furthermore, the present data substantiate a newly emerging concept that the modulation of IGF-1 receptor expression is a key event selectively determining the activation level of intracellular pathways that lie downstream of the receptor. This aspect should be considered when designing IGF-1-based treatments for neurodegenerative diseases.
Keywords: gene expression, IGF-1R, model mice, signaling pathways, SMN, spinal muscular atrophy
Introduction
Spinal muscular atrophy (SMA) is an autosomal-recessive disease occurring in early childhood for which no specific therapy exists. SMA is characterized by the specific loss of spinal motor neurons, leading to a progressive muscular weakness and subsequent death when vital muscles are affected (Crawford and Pardo, 1996). SMA is caused by mutation of the Survival of Motor Neuron 1 (SMN1) gene (Lefebvre et al., 1995), leading to a defective SMN protein expression. The disease severity is modulated by the expression of SMN2, a SMN1 gene copy. SMN2 produces only a small amount of full-length SMN transcripts due to the alternative splicing of exon 7, leading to a small amount of stable SMN protein (Lorson and Androphy, 2000). Despite two decades of research, the specific sensitivity of motor neurons to defects of the ubiquitous SMN protein that is involved in essential cell processes such as mRNA metabolism (Fischer et al., 1997; Pellizzoni et al., 1998) remains poorly understood. Microarray analyses unexpectedly failed to identify the specific misexpression of some essential genes in motor neurons after SMN depletion (Zhang et al., 2008). This may suggest that the selective degeneration of motor neurons in SMA results from alterations of systemic pathways that would ultimately target motor neurons.
Although multiple neurotrophic factors are implicated in neuron survival, insulin-like growth factor-1 (IGF-1) is a critical factor for the maintenance and the survival of motor neurons (Neff et al., 1993; Ozdinler and Macklis, 2006). Interestingly, several recent investigations pointed toward an IGF-1 signaling pattern-alteration in SMA. First, the levels of circulating IGF-1 and Igf-1 hepatic mRNA expression were shown to display a marked reduction in a severe type of SMA-like model mice (Hua et al., 2011; Murdocca et al., 2012). Second, in skeletal muscles from type 1 SMA patients (Millino et al., 2009) or from SMA mice (Bosch-Marcé et al., 2011), the IGF-1 receptor (IGF-1R) was found to be overexpressed. The level of this overexpression in human biopsies correlated with the severity of the disease (Millino et al., 2009). Interestingly, increasing IGF-1 availability for mouse SMA mouse muscles had no effect on IGF-1R expression (Bosch-Marcé et al., 2011), suggesting that IGF-1R overexpression was constitutive and not the result of a compensatory mechanism for a putative decrease in serum IGF-1 concentration. Unfortunately, data on the IGF-1R expression in SMA mouse CNS are presently lacking. Third, among the three intracellular signaling pathways known to lie downstream of IGF-1R, the ERK1/2, JAK/STAT3, and AKT pathways (D'Mello et al., 1997; Párrizas et al., 1997; Yadav et al., 2005), the ERK cascade was found to be constitutively overactivated in the spinal cord and muscles (Millino et al., 2009; Biondi et al., 2010). In contrast, the AKT pathway was found to be underactivated in SMA mouse spinal cords (Biondi et al., 2010; Kye et al., 2014).
Interestingly, IGF-1R overexpression was reported in the CNS and muscles from patients with amyotrophic lateral sclerosis (ALS) (Wilczak et al., 2003; Lunetta et al., 2012), another fatal motor neuron disease, contrasting with nearly identical serum IGF-1 concentration (Corbo et al., 2010). Moreover, recent data demonstrated that the reduction of IGF-1 signaling through Igf-1r gene partial inactivation (Igf-1r+/−) resulted in an increase in lifespan; a greater resistance to oxidative stress (Holzenberger et al., 2003); and a reduction in behavioral impairment, neuroinflammation, and neuronal and synaptic loss in a mouse model of Alzheimer's disease (Cohen et al., 2009). Therefore, one could ask whether enhancing or depressing IGF-1 related signaling pathway results in a beneficial or detrimental effect on SMA motor neurons. To address this question directly, we generated an SMA mouse model harboring only one functional allele for Igf-1r (Igf-1r+/−) and analyzed the hallmarks of SMA pathogenesis.
Materials and Methods
Mice and treatments.
The knock-out transgenic type 2 SMA-like mice (FVB/NRj-SmnΔ7/Δ7 SMN2) derived from mice obtained from the Institute of Molecular Biology (Hsieh-Li et al., 2000; Academia Sinica). To standardize the type 2 phenotype, male and female mutant mice with a weight ranging from 3 to 4 g at 8 d of age were used for this study, as described previously (Biondi et al., 2008). We generated a new mouse strain by crossing FVB/NRj-Smn+/Δ7 SMN2 mice with Igf1r+/− mice (B6.129-Igf1rtm1.2Mhz/Orl, strain EM00115 from the EMMA mouse repository; (Holzenberger et al., 2003). After the crossing between FVB/NRj-Smn+/Δ7 SMN2 and B6.129-Igf1rtm1.2Mhz/Orl strains, the double mutant carriers were backcrossed for 10 generations to the FVB/NRj strain (Janvier Labs) to purify the genetic background of the mice. Among the different genotypes generated in offspring, the original parental genotypes (SmnΔ7/+ SMN2 Igf-1r+/+; n = 74) mice and (SmnΔ7/+ SMN2 Igf-1r+/−; n = 65) mice were referred to as “control” and “Igf-1r+/− control,” respectively, and the SMA-like mice (SmnΔ7/Δ7 SMN2 Igf-1r+/+; n = 62) and SmnΔ7/Δ7 SMN2 Igf-1r+/− (n = 61) mice were designated as “SMA” and “Igf-1r+/− SMA,” respectively. Male and female were used for this study. Body weight and lifespan recording were performed daily until animal death. The animals were considered to be dead when they were no longer able to stand up after having been placed on their sides for 1 min. The care and treatment of animals followed the national authority (Ministère de l'Enseignement Supérieur et de la Recherche, France) guidelines for the detention, use, and ethical treatment of laboratory animals.
Training protocol.
Exercise was performed in a wheel with a controlled speed as described previously (Grondard et al., 2005). The protocol was started from 8 d of age (postnatal day 8, P8). The mice in the trained group were progressively accustomed to wheel training for 2 d (the first day, 4 periods of 5 min running with a rest period of 30 min; the second day, 2 periods of 10 min running with a rest period of 30 min). Next, mice were individually and continuously trained for 20 min/d. The speed was fixed at 1 m/min for the first 3 d and then increased by successive steps of 2 m/min, finally reaching 5 m/min (maximal speed).
Behavioral evaluation.
The grip strength was performed in the forelimb of Igf-1r+/+ and Igf-1r+/− control mice and Igf-1r+/+ and Igf-1r+/− type 2 SMA-like mice from P8 to death. The time spent holding onto a thin metal rod suspended in midair was calculated. Each mouse was subjected to five successive attempts separated by a 10 min rest period.
The ambulatory behavior was assessed in an open-field test for all groups. The apparatus consisted of a wooden box measuring 28 × 28 × 15 cm. The floor of the arena was divided into 16 7 × 7 cm squares. The mice were tested individually and the open field was washed after each session. Each mouse initially placed in the center of the open field was allowed to move freely for 5 min. The behavioral measures recorded manually during these 5 min were the number of peripheral and central square crossings.
IGF-1 ELISA.
Mice were anesthetized with 1% pentobarbital solution (6 μl/g) diluted in 0.9% saline buffer by intraperitoneal injection. A 400 μl blood sample was taken from the right ventricle with a 1 ml syringe mounted with a 22 gauge needle, both coated with heparin (5000 UI/ml; Pan Pharma Luitré). The blood sample was centrifuged at 1500 × g for 10 min at +4°C. The serum was frozen, stored at −80°C, and used within 1 month. The quantity of IGF-1 was determined on 100 μl of serum in duplicate using the Murine IGF-1 ELISA kit (900-K170; Peprotech) and following the manufacturer's instructions.
In situ hybridization.
To detect Igf-1r mRNAs, antisense oligonucleotide probes were synthesized as described previously (Oshima et al., 2002; Fukaya et al., 2005): forward 5′-CACAGTCATCCGTGGCTGGA-3′ and reverse 5′-CGCTTCCCACACACACTTGG-3′. Under deep pentobarbital (40 mg/kg) anesthesia, spinal cords were obtained from P12 SMA-like mice. Frozen sections (14 μm thickness) were mounted on glass slides coated with 3-aminopropyltriethoxysilane. Each section was fixed immediately after cutting by immersion for 20 min in 2% paraformaldehyde in phosphate buffer (0.1 m, pH 7.4). Sections were then rinsed three times in PBS, dipped in water, and dehydrated with a graded ethanol series (70%, 95%, and 100%). The sections were then air-dried and stored at −80°C until further use. The hybridization mixture contained 50% formamide, 4× standard saline citrate (SSC), 1× Denhardt's solution, 0.25 mg/ml yeast tRNA, 0.25 mg/ml sheared salmon sperm, 0.25 mg/ml polyA, 10% dextran sulfate, 100 mm DTT, and [35S]-dATP-labeled oligonucleotide probes (4.5 × 106 dpm/ml). One hundred microliters of hybridization mixture was placed on each section. The sections were then covered with a Parafilm coverslip and incubated in a humidified chamber at +43°C for 20 h. After hybridization, the sections were washed successively twice in 1× SSC containing 0.01 m DTT and twice in 0.5× SSC containing 0.01 m DTT for 15 min at +55°C and finally in 0.5× SSC containing 0.1 m DTT for 15 min at room temperature. Sections were exposed to Nuclear Track emulsion (NTB-2; Kodak) for 8 weeks.
Histological and immunohistochemical analysis.
For motor neuron studies, anesthetized animals were submitted to an intracardial perfusion of 40 ml of PBS solution at 1.2 ml/min followed by 40 ml of 4% PFA solution diluted in PBS solution at the same flow. Then, spinal cords were dissected, incubated overnight in 4% PFA solution, and washed twice for 2 h with PBS. The lumbar spinal cords (L1 to L5) were cut using a vibratome VT-1000S (Leica) on the whole length of the sample at 50-μm-thick cross sections. One of every five sections was processed for immunohistochemical analysis as described previously (Branchu et al., 2013).
For SMN immunostaining, spinal cords were dissected from anesthetized animals and immediately frozen in cold isopentane solution (−80°C). The lumbar spinal cords (L1 to L5) were cut selectively using a cryostat microtome (CM3050S; Leica) at 14-μm-thick cross sections. One of every 10 sections (40 sections examined) was processed for immunohistochemical analysis as described previously (Grondard et al., 2005).
All types of sections were then incubated for 1 h at room temperature in a blocking solution (7% normal donkey serum with 1% Triton X-100 and 0.5% Tween 20 in TBS). Incubations in corresponding primary antisera, namely polyclonal goat anti-choline acetyltransferase (ChAT; 1:400; Millipore) and polyclonal rabbit anti-SMN clone 502 (1:400; obtained from Suzie Lefebvre, Paris, France) primary antibodies, were then performed for 3 d at +4°C in the blocking solution. Sections were washed in TBS buffer with 0.5% Tween 20 and subsequently incubated with polyclonal Cy3-conjugated donkey anti-goat (1:400; Jackson ImmunoResearch) and/or polyclonal Alexa Fluor 488-conjugated donkey anti-rabbit (1:400; Jackson ImmunoResearch) antibodies for 1 h at room temperature in the blocking solution. The sections were washed in TBS and mounted in Vectashield mounting medium with DAPI (Vector Laboratories). The staining specificity was checked incubating sections in absence of the primary antibody.
For motor end-plate labeling, whole-mount preparations of muscle fibers were stained using Alexa Fluor 568-conjugated α-bungarotoxin (4 mg/ml in PBS with 4% bovine serum albumin), monoclonal rabbit anti-neurofilament light protein (NN18, 1:500; Millipore), and synaptophysin (1:5; Invitrogen Life Technologies). The whole-mount preparations were subsequently incubated with an Alexa Fluor 488 goat anti-rabbit IgG (1:400; Invitrogen) for 1 h at room temperature as described previously (Branchu et al., 2013). The different developmental synapse shapes were determined according to Sanes and Lichtman (1999) and visualized using α-bungarotoxin staining for innervated neuromuscular junctions (NMJs). The acetylcholine receptor (AChR) clusters appearing elliptical and uniform or with some low AChR density but without perforation were defined as “uniform-plaques.” Elliptical AChR clusters displaying at least one perforation but maintaining a peripheral continuity were defined as “perforated-plaques.” Finally, AChR cluster displaying an important number of perforations and a loss of peripheral continuity were defined as “pretzel-like plaques,” the only shape for which we could observe a more precise spatial correspondence between nerve terminals and AChR clusters. The proportions of each type of plaque were determined from a minimum of 100 NMJs per muscle and per mouse and expressed as percentage of total counted NMJs.
For muscle typology, cryostat sections (one transversal medial section per muscle and per animal, which represented ∼4000, 6000, and 12,000 muscle fibers in the soleus, the plantaris, and the tibialis, respectively, for each experimental point) were either stained or processed for immunochemistry. For the staining protocol, muscles sections were dried for 30 min at room temperature and then stained with an H&E classical protocol. For the immunochemistry protocol, muscles sections were fixed with acetone for 20 min at room temperature and washed three times in PBS containing 0.1% Tween 20 (PBST). Slices were then incubated with mouse monoclonal antibodies raised against myosin heavy chains (MyHCs): (1) embryonic (F1.652; University of Iowa, Iowa City, IA), (2) neonatal (N3.36; University of Iowa), (3) slow type I (NCL-MyHCs), (4) all fast type II (NCL-MyHCf; Leica), and (5) intermediate type IIa MyHC isoforms (A4.74) at a dilution of 1:20 for 1 h at room temperature and washed three times in PBST. Sections were then incubated at room temperature with a rabbit anti-mouse Ig conjugated with FITC (1:20; Sigma-Aldrich) for 1 h. After washing with PBS, sections were mounted in Vectashield mounting medium (Vector Laboratories). The percentage of fast IIx/b fibers were determined as the difference between the total number of type II and type IIa fibers.
All counts were performed using ImageJ software version 1.46. Color images were tinted, also using ImageJ, and identical brightness, contrast, and color balance adjustments were applied to all groups.
Microscopy.
All immunofluorescence images were collected with a CMOS camera (ORCA Flash 2.8; Hamamatsu Photonics) mounted on a Zeiss Z1 AxioObserver using ZEN 2012 software with 100× [10× Zeiss Plan NeoFluar numerical aperture (NA) 0.3], 200× (20× Zeiss EC-Plan-Apo NA 0.8), and 630× (63× Zeiss Plan-Apo Oil NA 1.4) magnifications.
Silencing of IGF-1R with siRNA in cell cultures.
MN-1 (Motoneuron-1) cells have been derived from the spinal cords of E18 control mice and immortalized (Salazar-Grueso et al., 1991). These cells only harbor the two alleles of the murine Smn genes. MN-1 cells have been provided by J. Côté (Ottawa University, Canada) and were plated at 5000 cells/cm2 at +37°C in 5% CO2 in a 6 well plate with DMEM (Invitrogen) supplemented with 10% FBS and 1% of penicillin/streptomycin (P/S) growth medium (GM).
Human primary culture of myogenic precursor cells was derived from SMA patient biopsies. Muscle biopsies were obtained from the Bank of Tissues for Research (a partner in the European Union network EuroBioBank) in accordance with European recommendations and French legislation. Satellite cells were isolated from biopsies and grown as described previously (Bigot et al., 2009). The myogenic purity of the population was monitored by immunocytochemistry using desmin as a marker. Myoblast cells were plated at 10,000 cells/cm2 in 6 well plates at +37°C in 5% CO2 in DMEM Glutamax (Invitrogen) supplemented with 20% FBS (Invitrogen), 1% Ultroser G (Pall), and 1% P/S (5000 U; Invitrogen) for 2 d. Then, differentiation was induced with DMEM supplemented with 5% horse serum (Invitrogen) and 1% P/S (5000 U; Invitrogen) differentiation medium (DM) for 10 d.
MN-1 cells at 50% confluence were transfected with Lipofectamine 2000 transfection reagent (Invitrogen) in antibiotic-free GM with either ON-TARGETPlus nontargeting siRNA (D-001810–01; GE Healthcare) as a negative control or ON-TARGETPlus mouse Igf-1r siRNA (L-056843; GE Healthcare) at 50 nm, following the manufacturer's instructions. Human myotubes were transfected with Dharmafect 3 transfection reagent (GE Healthcare) in antibiotic-free DM with either the same nontargeting siRNA or with ON-TARGETPlus human IGF-1R siRNA (L-003012; GE Healthcare) at 50 nm following the manufacturer‘s instructions. Transfection medium was replaced by GM or DM after 16 h. Cells were harvested 56 h later in cold 1× PBS (Invitrogen) and centrifuged at 200 × g for 3 min. Then, supernatants were removed and pellets frozen at −80°C.
Protein and Western blot analysis.
The ventral part of frozen spinal cords was selectively dissected by razor blade at −20°C. A furrow appears in the middle of the ventral part and dorsal root ganglia and can be seen on the dorsal part of the spinal cord. Frozen pellets of MN-1 and human primary culture myotubes were homogenized in 50 μl of ice-cold RIPA buffer [50 mm Tris HCl, pH 8.0, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP40, 5 mm EDTA, pH 8.0, 2 mm PMSF (Sigma-Aldrich), 50 μg/ml leupeptin, 50 μg/ml pepstatin A, and 50 μg/ml aprotinin] for 2 wells of a 6 well plate; ventral lumbar spinal cord samples were homogenized in 100 μl of ice-cold RIPA buffer for 5 mg of tissue. Protein concentration of the clarified homogenates (+4°C, 15 min, 13,500 revolutions/min) was determined on all samples using the Bradford protein assay (Bio-Rad). Then, 10 μg protein samples for SMN analysis and 30 μg samples for other analysis of each homogenate were submitted to 12.5% SDS-PAGE electrophoresis (1.5 m Tris, pH 8.3, 12.5% acrylamide, 0.07% Bis, 0.1% SDS, 0.05% APS, 0.06% TEMED). The separated proteins were transferred on PVDF membranes (Bio-Rad; Towbin et al., 1984). Equal loading of samples was checked by Ponceau dye staining of the transferred gels. Western blot analysis was performed on membranes incubated overnight at +4°C in 4% BSA, 0.1% Tween 20, and TBS, pH 7.4. Each of the following primary antibodies, including monoclonal mouse anti-SMN (1:5000; Santa Cruz Biotechnology), polyclonal rabbit anti-Ser473 phospho-AKT (1:1000; Cell Signaling Technology), polyclonal rabbit anti phospho-ERK1/2 (1:500; Cell Signaling Technology), polyclonal rabbit anti-Ser133 phospho-CREB (1:1000; Millipore), monoclonal mouse anti-Ser183 phospho-Elk-1 (1:1000; Santa Cruz Biotechnology), monoclonal mouse anti-Tyr705 phospho-STAT3 (1:500; Santa Cruz Biotechnology), monoclonal rabbit anti-cleaved caspase 3 (ref.9661, 1:1000; Cell Signaling Technology), and polyclonal rabbit anti-IGF-1Rβ (3027; 1:1000; Cell Signaling Technology), was incubated overnight at +4°C in the above blocking medium. Membranes were rinsed in 0.1% Tween 20 in TBS for 3 × 10 min at room temperature and then incubated in horseradish peroxidase-conjugated goat secondary antibody directed against mouse immunoglobulins (1:5000; Bio-Rad) and directed against rabbit immunoglobulins (1:20,000; Jackson ImmunoResearch) in 0.1% Tween 20 in TBS for 1 h at room temperature. Bound antibody complexes were developed using the ECL system (GE Healthcare) and exposed to hyperfilm ECL-plus x-ray film (GE Healthcare).
In some instances, membranes were stripped after immunoblotting with phospho-AKT, phospho-ERK1/2, phospho-CREB, phospho-Elk-1, and phospho-STAT3 by incubation in stripping buffer (100 mm β-mercaptoethanol, 2% SDS, 62.5 mm Tris-HCl, pH 6.7) for 30 min at +55°C with agitation and membranes were then blocked and reprobed with polyclonal rabbit anti-AKT (1:1000; Cell Signaling Technology), polyclonal rabbit anti-ERK1/2 (1:500; Cell Signaling Technology), polyclonal rabbit anti-CREB (1:1000; Millipore), monoclonal mouse anti-Elk-1 (1:1000; Santa Cruz Biotechnology), polyclonal rabbit anti-STAT3 (1:500; Cell Signaling Technology) and monoclonal mouse anti-GAPDH (1:2000, sc-32233, Santa Cruz Biotechnology). The optical density (OD) of each specific band was quantified with ImageJ, subtracted to the background, and divided by the OD of corresponding GAPDH bands. The values for respective controls were set as 1 and the values for the other groups were normalized by the control and expressed as fold change. The number of animals in each group (n) was obtained by performing independent experiments using different membranes in which each group was compared with the control.
ChIP.
Ventral lumbar spinal cord samples were chopped into small pieces with a scalpel and fixed for 15 min in 1% formaldehyde. Tissues were washed 3 times in cold PBS containing protease inhibitors (2 mm PMSF, 50 μg/ml leupeptin, 50 μg/ml pepstatin A, and 50 μg/ml aprotinin) and collected by centrifugation. Cells were pelleted by centrifugation and resuspended in 300 μl of 85 mm KCl, 0.5% NP-40, 5 mm piperazine-N,N′-bis(2-ethanosulfonic acid; PIPES), pH 8.0, in the presence of protease inhibitors (2 mm PMSF, 50 μg/ml leupeptin, 50 μg/ml pepstatin A, and 50 μg/ml aprotinin). After incubation on ice for 10 min, cells were sonicated 6 times for 30 s using Bioruptor (Diagenode). Lysates were cleared by centrifugation and DNA concentration was determined by nanodrop spectrophotometer. ChIP-Adembeads (Ademtech) were incubated for 15 min at room temperature with blocking buffer on a rotating wheel. Beads were resuspended in 125 μl of ChIP dilution buffer (0.01% SDS, 1% Triton X100, 1.2 mm EDTA, 16.7 mm Tris-HCl, pH 8.1) and, after 1 h of incubation, equal amounts of DNA diluted 10 times in dilution buffer were added. DNA was incubated overnight at +4°C on a rotating wheel with 1 μg of the following antibodies: polyclonal rabbit anti-Ser133 phospho-CREB (Millipore), monoclonal mouse anti-Ser183 phospho-Elk-1 (Santa Cruz Biotechnology), polyclonal rabbit anti-acetyl-histone H3 (Millipore), and polyclonal rabbit-acetyl-histone H4 Lys8 (Millipore). Beads were washed sequentially in 150 mm NaCl TSE (0.1% SDS, 1% Triton X100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, with 500 mm NaCl TSE), buffer A (0.25 m LiCl, 1% NP-40, 1% deoxycholate, 1 mm EDTA, 10 mm Tris-Hcl, pH 8.1), and 2 times with Tris-EDTA, pH 8.1, and then selectively eluted with 200 μl of 1% SDS and 0.1 m NaHCO3. Cross-links were reversed by heating at +65°C for 4 h after adding NaCl at a 200 mm final concentration. After treatment with proteinase K (50 μg/ml) for 1 h at +37°C, DNA was purified using Geneclean Turbo Kit (Q-Biogene; MP Biomedicals). Real-time qRT-PCR analysis of inputs or immunoprecipitated DNAs was performed.
qRT-PCR analysis.
Ventral lumbar spinal cord samples were chopped into small pieces with a scalpel and RNA was extracted using TRIzol (Invitrogen). Each RNA preparation was treated with RQ1 RNase-Free DNase (Promega). One microgram was reverse transcribed with random primers (Promega) and reverse transcriptase M-MLV-RT (Invitrogen).
qRT-PCR was performed with standard protocols using SYBRGreen ROX Mix (ABgene) as a fluorescent detection dye in an ABI Prism 7000 in a final volume of 10 μl, which also contained 300 nm of the following primers (Operon): SMN2 exon 4–exon 5 segment forward: 5′-TGTGTTGTGGTTTACACTGG-3′; SMN2 exon 4–exon 5 segment reverse: 5′-TATTTCCAGGAGACCTGGAG-3′; SMN2 exon 7–exon 8 segment forward: 5′-AAAAAGAAGGAAGGTGCTCAC-3′; SMN2 exon 7–exon 8 segment reverse: 5′-GCCTCACCACCGTGCTGG-3′; Igf-1r forward: 5′-GCATCTGATCATTGCTCTG-3′; Igf-1r reverse: 5′-GCCCAACCTGCTGTTATTTC-3′; 18S forward: 5′-GTAACCCGTTGAACCCCATT-3′; 18S reverse: 5′-CCATCCAATCGGTAGTAGCG-3′; SMN2 promoter site 1 [−2667nu to −2401nu] forward: 5′-GAGAGAGTTCCAGGAGTCAA-3′; SMN2 promoter site 1 [−2667nu to −2401nu] reverse: 5′-GTCTCAAACTCGGTTGCTT-3′; SMN2 promoter site 2 [+160nu to +503nu] forward: 5′-TCGTAGAAAGCGTGAGAAGT-3′; SMN2 promoter site 2 [+160nu to +503nu] reverse: 5′-AAAACGCGGACCACAACTC-3′. The relative amounts of DNA in samples were determined on the basis of the threshold cycle for each PCR product (Ct).
Statistical analysis.
All values are displayed as mean ± SEM within each group (Systat version 8.0; SPSS). A Kolmogorov–Smirnov normal distribution analysis was performed on all data, followed by either a Student's t test for normally distributed data or a nonparametric Kruskal–Wallis test to verify significant differences between groups. Survival analysis was performed using Kaplan–Meier analysis.
Results
The IGF-1R is overexpressed in the spinal cords of SMA-like mice
We first analyzed the distribution of Igf-1r transcripts using in situ hybridization in the spinal cords of SMA-like and control mice at P12 (Fig. 1A, left). We found that Igf-1r mRNAs were significantly overexpressed in the spinal cords of SMA-like mice throughout the cord, including the motor column cells. We next analyzed the Igf-1r expression level by qRT-PCR in the ventral horn of the lumbar spinal cords of SMA-like mice and controls. There was a 2.5-fold increase in Igf-1r expression in SMA spinal cords compared with controls (Fig. 1B). Igf-1r mRNA overexpression in SMA mouse spinal cords was confirmed at the protein level with a 1.6-fold increase (Fig. 1C,D).
Figure 1.

The IGF-1 axis is altered in the spinal cords of severe SMA-like mice. A, In situ hybridization on Igf-1r mRNA for the localization of expression in the lumbar spinal cords (L1–L5) of untrained control and type 2 SMA-like mice (left) compared with trained control and type 2 SMA-like mice (right) at 12 d of age (n = 4). Scale bar, 100 μm. B, Quantification by real-time qRT-PCR of Igf-1r transcripts normalized by 18S transcripts in the ventral lumbar spinal cords of untrained control and type 2 SMA-like mice compared with trained control and type 2 SMA-like mice at 12 d of age (n = 10). C–E, Western blot analysis (C) and quantification of IGF-1Rβ (D) and SMN (E) protein expression in the ventral lumbar spinal cords of untrained control and type 2 SMA-like mice compared with trained control and type 2 SMA-like mice at 12 d of age (n = 4). F, Quantification of circulating IGF-1 concentration by ELISA in the serum of untrained control and type 2 SMA-like mice compared with trained control and type 2 SMA-like mice at 12 d of age (n = 4). G, Schematic representation of the crossings yielding to Igf-1r+/− SMA mice. H, Quantification by real time qRT-PCR of Igf-1r transcripts normalized by 18S transcripts in the ventral lumbar spinal cords of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 10). I, J, Western blot analysis (I) and quantification (J) of IGF-1Rβ protein expression in the ventral lumbar spinal cords of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 5). K, Quantification of circulating IGF-1 concentration by ELISA in the serum of IGF-1R+/+ and IGF-1R+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 4). Data are represented as mean ± SEM and significance is reported versus untrained or Igf-1r+/+ control mice, respectively (*p < 0.05, **p < 0.01, ***p < 0.001).
We then recorded the evolution of IGF-1R expression levels in a physiological neuroprotective paradigm for SMA-like mice based on physical exercise (Grondard et al., 2005). Submitting control and SMA-like mice to a daily running protocol led to a significant reduction in IGF-1R overexpression in the spinal cords of SMA-like mice (Fig. 1A, right, B–D). Because the reduction in IGF-1R expression could be the direct consequence of the exercise-induced enhancement of the ligand expression, we measured the circulating IGF-1 concentrations through ELISA quantifications (Fig. 1F). We found that, in striking contrast to controls, the level of circulating IGF-1 remained significantly lower in SMA compared with control mice, with no detectable change in trained compared with untrained SMA-like mice.
Most interestingly, SMN protein expression levels were inversely proportional to IGF-1R expression levels in the spinal cords of sedentary and trained SMA-like mice (Fig. 1C–E).
Strategy for genetically reducing IGF-1R expression in SMA-like mice
To determine whether the IGF-1R expression level might be involved in SMA pathogenesis, we created a new SMA-like mouse model in which one allele of the Igf-1r gene was inactivated (SmnΔ7/Δ7 SMN2 Igf-1r+/−; Fig. 1G). In this new mouse strain, IGF-1R expression was reduced by 4-fold in the SMA spinal cord, reaching a level comparable to Igf-1r+/− controls, as evidenced by qRT-PCR and Western blot (Fig. 1H–J). We next investigated whether altering IGF-1R expression would change circulating IGF-1 concentrations through ELISA quantifications (Fig. 1K). We found that, in striking contrast to controls, the level of circulating IGF-1 remained significantly lower in SMA regardless of the IGF-1R expression level.
Genetically reducing IGF-1R expression strikingly prolongs survival and improves the motor behavior of SMA mice
The impact of IGF-1R reduction on the life span of SMA mice was then investigated. As expected in this new strain, SMA mice lived no longer than 15 d (Fig. 2A,B), as reported for the original severe SMA-like mouse strains (Hsieh-Li et al., 2000). Reducing IGF-1R expression significantly increased survival of SMA mice (Fig. 2A,B). The mean survival increased from 10.9 ± 2.2 d for SMA mice to 21.5 ± 5.3 d for the Igf-1r+/− SMA, which represented a 2-fold increase in lifespan. Furthermore, the IGF-1R reduction, in striking contrast to controls (data not shown), led to a significant and progressive increase in the body weight of Igf-1r+/− SMA mice until death (Fig. 2C). All these effects were similar regardless of sex.
Figure 2.

Genetic reduction of IGF-1R expression extends survival and motor behavior and promotes neuroprotection in SMA mice. A, Phenotype of Igf-1r+/− control mice compared with SMA mice and Igf-1r+/− SMA mice at 12 d of age. Scale bar, 1 cm. B, Lifespan of Igf-1r+/+ compared with Igf-1r+/− control and SMA mice (n = 40). C, Weight curve of Igf-1r+/+ and Igf-1r+/− control (left) and SMA (right) mice until SMA death (n = 40). D, Grip time of Igf-1r+/+ and Igf-1r+/− control (left) and SMA (right) mice until SMA death (D; n = 20). E, Total number of squares crossings during 5 min in the open-field test for Igf-1r+/+ and Igf-1r+/− control (left) and SMA (right) mice until SMA death (E; n = 20). F–I, Immunodetection of ChAT-positive motor neurons in the lumbar spinal cord (L1–L5) of Igf-1r+/+ (F) and Igf-1r+/− control mice (G) compared with Igf-1r+/+ (H) and Igf-1r+/− SMA mice (I) at 12 d of age (n = 10. Scale bar, 50 μm. J, K, Quantitative analysis of the number (J) and the cell body area (K) of motor neurons per ventral horn in the ventral lumbar spinal cord of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 10). L, M, Western blot analysis (L) and quantification (M) of cleaved caspase-3 protein expression in the ventral lumbar spinal cord of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 4). Dotted lines on Western blot images symbolize some removed interspacing lanes for a side-by-side display of samples from all groups. Data are represented as mean ± SEM and significance is reported versus Igf-1r+/+ control mice (*p < 0.05, **p < 0.01, ***p < 0.001).
We then questioned whether the reduction in IGF-1R expression would improve the motor behavior of the SMA mice. We tested all groups of mice on both the grip strength and the open field tests. We found that, in contrast to SMA mice, which were unable to grip the metal rod efficiently, the grip time of Igf-1r+/− SMA progressively increased up to 17 ± 3 s at P19 and then stabilized until death (Fig. 2D). Again, whereas SMA mice displayed a strongly reduced exploratory activity compared with age-matched controls, the Igf-1r+/− SMA mice displayed a progressive increase in exploratory behavior until P20, decreasing thereafter until death (Fig. 2E).
Reducing IGF-1R expression protects motor neurons in SMA mice
We next compared the number of ChAT-positive motor neurons in the ventral spinal cords of SMA, Igf-1r+/− SMA, control and Igf-1r+/− control mice at P12 (Fig. 2F–J). As observed in the original SMA mouse strain (Hsieh-Li et al., 2000), we found a 34% loss of motor neurons in spinal cords of SMA mice compared with controls (Fig. 2J). In contrast, Igf-1r+/− SMA mice only showed a 10% reduction in the number of motor neurons, suggesting that reducing IGF-1R expression significantly reduced the extent of motor neuron death in SMA mice. Furthermore, the evaluation of motor neuron cell body area provided evidence of a persistent atrophy of the motor neurons in the ventral horn of the spinal cords in SMA mice compared with control mice at P12 (Fig. 2K). In contrast, the extent of atrophy was significantly reduced in Igf-1r+/− SMA mice. No difference could be observed in the number and size of motor neurons in control and Igf-1r+/− control mice at the same age (Fig. 2J,K).
To determine whether the reduction of IGF-1R level interfered with the apoptotic process in the spinal cords of SMA mice, we evaluated the activation of the caspase-3 in the ventral spinal cords of SMA and Igf-1r+/− SMA mice. As shown in Fig. 2, L and M, the IGF-1R reduction reduced the extent of the apoptotic process in SMA mouse spinal cords.
Reducing IGF-1R expression enhances SMN protein expression in the spinal cords of SMA mice and in human myotubes
We next analyzed the impact of IGF-1R reduction on the SMN expression profile in the spinal cords of all four groups at P12. The increase in SMN protein level induced by IGF-1R reduction was clearly evidenced by the Western immunoblot analysis performed at the lumbar spinal cord level (Fig. 3A,B). A 2-fold increase in SMN expression level was observed in Igf-1r+/− SMA mice compared with SMA mice, but it remained 50% lower than controls. Unexpectedly, IGF-1R reduction was unable to enhance SMN expression in controls, as shown in the spinal cords of Igf-1r+/− control mice compared with Igf-1r+/+ control mice, but also in MN1 culture, which arbors only the two alleles of the murine Smn gene and for which Igf-1r depletion by a specific siRNA resulted in a slight decrease in SMN (Fig. 3C–E).
Figure 3.
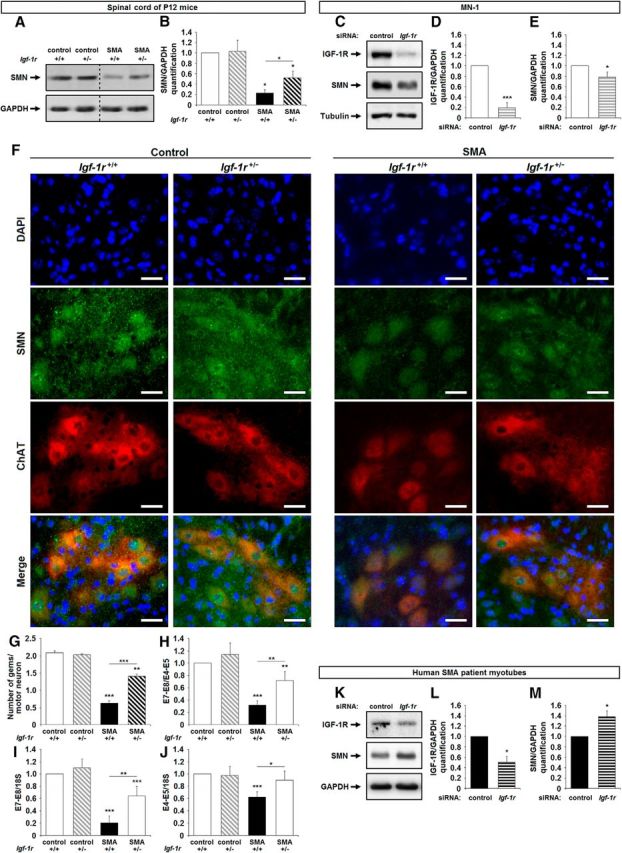
Genetic reduction of IGF-1R expression promotes SMN expression in mouse SMA spinal cords. A, B, Western blot analysis (A) and quantification (B) of SMN protein expression in the ventral lumbar spinal cords of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 4). C–E, Western blot analysis (C) and quantification of IGF-1Rβ (D) and SMN (E) protein expression in MN-1 cell culture transfected with either a nonrelevant siRNA (siRNA Control) or an siRNA against Igf-1r (siRNA Igf-1r; n = 3). F, Immunodetection of SMN protein (SMN, green) in the nucleus (DAPI, blue) of ChAT-positive motor neurons (red) in the lumbar spinal cord (L1–L5) of Igf-1r+/+ and Igf-1r+/− control mice (left) compared with Igf-1r+/+ and Igf-1r+/− SMA mice (right) at 12 d of age (n = 4). Scale bar, 25 μm. G, Number of Gems per nucleus of ChAT-positive motor neurons in the ventral lumbar spinal cord of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (100 motor neurons per mouse; n = 4). H–J, Quantification by real-time 1RT-PCR of the E7–E8 segment containing SMN transcripts normalized either by the E4–E5 segment containing SMN transcripts (H) or by 18S transcripts (I) and of total E4–E5 segment SMN transcripts normalized by 18S transcripts (J) in the ventral lumbar spinal cord of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 10). K–M, Western blot analysis (K) and quantification of IGF-1Rβ (L) and SMN (M) protein expression in primary culture of myotubes from human SMA patient transfected with either a nonrelevant siRNA (siRNA Control) or an siRNA against IGF-1R (siRNA IGF-1R; n = 3). Dotted lines on Western blot images symbolize some removed interspacing lanes for a side-by-side display of samples from all groups. Data are represented as mean ± SEM and significance is reported versus Igf-1r+/+ control mice or siRNA control MN-1 or siRNA control myotube-transfected cells, respectively (*p < 0.05, **p < 0.01, ***p < 0.001).
To investigate whether the IGF-1R reduction-induced increase in SMN expression in SMA spinal cords occurred in motor neurons, we compared the number of Gemini of coiled-bodies (Gems) per nucleus in ChAT-positive neuron within the ventral spinal cords of SMA, Igf-1r+/− SMA, control and Igf-1r+/− control mice at P12 (Fig. 3F,G). Consistent with the values we found in the original SMA mouse strain (Hsieh-Li et al., 2000; Grondard et al., 2005), from 0 to 1 Gems per nucleus (0.6 ± 0.1 on the average) could be counted in SMA, contrasting with up to 2.0 ± 0.1 in control. Therefore, reducing IGF-1R expression increased the number of Gems per nucleus by almost 1, reaching 1.4 ± 0.1 in Igf-1r+/− SMA motor neurons (Fig. 3G).
To determine whether the increased SMN protein level resulted from an increase in SMN transcript steady-state levels and/or from a modulation of the exon-7 inclusion in SMN transcripts, we quantified the fraction of exon-7-containing mRNA inside the population of SMN transcripts using qRT-PCR aimed at amplifying either the exon 4–exon 5 segment (E4–E5) or the E7–E8 segment in the ventral spinal cords of SMA, Igf-1r+/− SMA, controls and Igf-1r+/− control mice at P12 (Fig. 3H–J). We found that the reduction of IGF-1R did increase both the steady-state level of SMN transcripts (Fig. 3J) and also the relative level of exon-7-containing SMN transcripts (Fig. 3H,I).
Finally, in agreement with our findings in mice, the siRNA-induced depletion of IGF-1R transcripts in a myotube culture of paravertebral muscles from type 2 SMA patient resulted in a significant SMN expression increase (Fig. 3K–M).
Genetically reducing IGF-1R expression shifts the equilibrium among ERK, JAK, and AKT signaling pathways in SMA spinal cords
To investigate whether the modulation of IGF-1R expression level affected the activation profile of the intracellular pathways downstream of the receptor, we analyzed the phosphorylation status of ERK, STAT3, and AKT in the spinal cords in all four groups at P12 (Fig. 4A–F). We found that, in the spinal cords of SMA mice, ERK (Fig. 4A,B) and STAT3 (Fig. 4E,F) phosphorylation status were consistently higher and those of AKT (Fig. 4C,D) were consistently lower compared with controls. Reducing IGF-1R expression in the spinal cords of SMA mice resulted in a >2-fold increase in AKT phosphorylation (Fig. 4C,D), a 25% decrease of ERK phosphorylation (Fig. 4A,B), and a 2-fold decrease of STAT3 phosphorylation (Fig. 4E,F). Accordingly, the p-AKT/total AKT ratio increased and the p-ERK/total ERK, pSTAT-3/total STAT3 ratios decreased in the spinal cords of Igf-1r+/− SMA mice, reflecting an increase in the activity level of AKT and conversely a decrease in the activity level of ERK and JAK pathways. Unexpectedly, IGF-1R reduction had no effect on the activation profile of ERK and AKT pathways in control spinal cords, contrasting with the previous data in mouse Igf-1r+/− fibroblasts (Holzenberger et al., 2003).
Figure 4.

Genetic reduction of IGF-1R expression restores intracellular pathway activation profile and promotes phospho-CREB-binding on SMN2 promoter in the spinal cords of SMA mice. A, B, Western blot analysis (A) and quantification (B) of ERK protein phosphorylation in the ventral lumbar spinal cords of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 5). C, D, Western blot analysis (C) and quantification (D) of AKT protein phosphorylation in the ventral lumbar spinal cords of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 5). E, F, Western blot analysis (E) and quantification (F) of STAT3 protein phosphorylation in the ventral lumbar spinal cords of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 5). G, H, Western blot analysis (G) and quantification (H) of Elk-1 protein phosphorylation in the ventral lumbar spinal cords of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 5). I, J, Western blot analysis (I) and quantification (J) of CREB protein phosphorylation in the ventral lumbar spinal cords of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 5). K–N, ChIP analysis of phospho-Elk-1 (K, L) and phospho-CREB (M, N) in the ventral lumbar spinal cords of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age. Real-time qRT-PCR was performed to detect the SMN2 promoter site 1 (K, M) and site 2 (L, N; n = 12). O–R, ChIP analysis of Histone H3 (O, P) and H4 (Q, R) acetylation in the ventral lumbar spinal cords of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age. Real-time qRT-PCR was performed to detect SMN2 promoter site 1 (O, Q) and site 2 (P, R; n = 12). Dotted lines on Western blot images symbolize some removed interspacing lanes for a side-by-side display of samples from all groups. Data are represented as mean ± SEM and significance is reported versus Igf-1r+/+ control mice (*p < 0.05, **p < 0.01, ***p < 0.001).
We next investigated whether this shift from ERK to AKT, induced by IGF-1R reduction, could influence SMN expression. Indeed, phospho-ERK and phospho-AKT activate by phosphorylation the transcription factors Elk-1 and CREB, respectively, resulting in their binding to their specific sites on SMN2 gene promoter and thus altering SMN expression in SMA spinal cords (Branchu et al., 2013). In the SMA background, activated Elk-1 was thought to exert a repressive role on SMN2 gene transcription, contrasting with the activating role of CREB (Branchu et al., 2013). As expected, we found that the activation level of Elk-1 and CREB paralleled those of their respective activating kinases in the spinal cords of all four groups (Fig. 4G–J). Furthermore, the level of Elk-1 binding on the SMN2 promoter, which was originally found to be higher in SMA compared with controls, was significantly lowered by IGF-1R reduction, as evidenced by ChIP analysis (Fig. 4K,L). In contrast, the level of CREB binding on the SMN2 promoter, originally found to be lower in SMA compared with controls, was significantly increased by IGF-1R reduction (Fig. 4M,N). Expectedly, the CREB binding on the SMN2 promoter was correlated with a significant increase in histone H3 and H4 acetylation (Fig. 4O–R), suggesting a chromatin remodeling at the SMN2 promoter level.
Reducing IGF-1R expression improves the NMJ phenotype in SMA mice
Because alteration of the NMJ development is at present considered to be a hallmark of SMA pathogenesis in patients (Kariya et al., 2008) and in mouse models (Biondi et al., 2008; Kariya et al., 2008; Kong et al., 2009), we analyzed shape and surface of the NMJs, as described previously (Sanes and Lichtman, 1999; Biondi et al., 2008). The developmental changes in the synapse shape from an immature, uniform plaque of AChRs (Fig. 5A) to a perforated large plaque of AChRs (Fig. 5B) that eventually adopts a pretzel-like form (Fig. 5C) were determined for innervated NMJs using α-bungarotoxin staining of the two extensor muscles, the slow-twitch soleus (Fig. 5D–G) and fast-twitch plantaris and the flexor fast-twitch tibialis in all four groups of mice at P12. We found that up to 63% of the NMJs were elliptical, made of uniform plaques of AChRs, in the two extensors muscles of SMA mice at P12 (Fig. 5H,J). As expected for a flexor muscle at this age (Biondi et al., 2008), NMJs in the tibialis were more advanced in terms of maturation, with only 34% of immature NMJs in SMA mice (Fig. 5L). In contrast, only 32% and 11% of immature NMJs were found in extensor and flexor muscles of controls, respectively, with a number of pretzel-like form NMJs between 2% and 5% for the extensors and 10% for the flexor, as found previously in the former SMA-like mice (Biondi et al., 2008; Fig. 5H,J,L). IGF-1R reduction improved NMJ morphology in the three muscles in SMA mice, as demonstrated by the increase in NMJ perforation (Fig. 5H,J,L) and enlargement (Fig. 5I,K,M) of the NMJs on the muscle fibers. These effects of IGF-1R reduction on NMJ maturation were only evidenced in SMA mice.
Figure 5.

Genetic reduction of IGF-1R expression accelerates NMJ maturation in SMA mice. A–G, Motor end-plate labeling with α-bungarotoxin (left) and anti-neufilament plus anti-synaptophysin antibodies (right), representing “Uniform-plaque” (A), “Perforated-plaque” (B), and “Pretzel-like plaque” (C), in the soleus of Igf-1r+/+ control mice (D), Igf-1r+/− control mice (E), Igf-1r+/+ SMA mice (F), and Igf-1r+/− SMA mice (G) at 12 d of age. Scale bars, 10 μm for high magnifications, 25 μm for low magnifications, and 5 μm for crops. H, I, Determination of the NMJ maturation (H) and area (I) in the soleus muscle from Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 5). J, K, Determination of the NMJ maturation (J) and area (K) in the plantaris muscle from Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 5). L, M, Determination of the NMJ maturation (L) and area (M) in the tibialis muscle from Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 5). Data are represented as mean ± SEM and significance is reported versus Igf-1r+/+ control mice (*p < 0.05, **p < 0.01, ***p < 0.001).
Reducing IGF-1R expression improves the muscle phenotype in SMA mice
Finally, we examined in all four groups of mice at P12 whether the IGF-1R reduction could alleviate SMA-induced muscular impairments, including muscle aplasia, atrophy, and typology alteration. As evidenced by the phenotypic analysis of H&E-stained soleus, plantaris, and tibialis (Fig. 6A–D), reducing IGF-1R expression in SMA mice was sufficient to counteract muscle aplasia (Fig. 6E) and to reduce muscle atrophy significantly by 50% in soleus and plantaris and by 100% in tibialis (Fig. 6F). We then looked at the muscle maturation rate through the analysis of the different MyHC distribution, including the embryonic; neonatal; and adult I, II IIa, and IIx/IIb isoforms (Fig. 6G–L). In SMA mice, consistent with our previous results in the original SMA mouse strain (Biondi et al., 2008), we found an increase in embryonic (20%, 13%, and 10% for soleus, plantaris, and tibialis, respectively) and type I (5% and 13% for soleus and plantaris, respectively) MyHC-expressing myofibers compared with control mice. These alterations were associated with a decrease in myofibers expressing the neonatal (15%, 20%, and 12% for soleus, plantaris, and tibialis, respectively) and type II (5% and 15% for soleus and plantaris, respectively) isoforms, suggesting, as expected, an overall muscle maturation delay (Fig. 6I,J). Reducing IGF-1R expression in SMA mice resulted in a rebalancing of the percentage of embryonic versus neonatal MyHC-expressing myofibers (Fig. 6K) and of type I versus type II, as well as among the different type II-MyHC-expressing myofibers (Fig. 6L). These data suggested that IGF-1R reduction in SMA muscles is sufficient to override the major characteristic muscular impairments of the disease.
Figure 6.

Genetic reduction of IGF-1R expression limits muscle impairments and accelerates muscle maturation in SMA mice. A–D. H&E staining on the tibialis of Igf-1r+/+ control mice (A), Igf-1r+/− control mice (B), Igf-1r+/+ SMA mice (C), and Igf-1r+/− SMA mice (D) at 12 d of age. Scale bar, 25 μm. E, F, Total number (E) and cross-sectional area (F) of myofibers in the soleus, plantaris, and tibialis muscles from control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 4). G–J, Immunodetection of embryonic MyHC in the soleus of Igf-1r+/+ control mice (G), Igf-1r+/− control mice (H), Igf-1r+/+ SMA mice (I), and Igf-1r+/− SMA mice (J) at 12 d of age. Scale bar, 50 μm. K, L. Analysis of developmental (i.e., embryonic and neonatal; K) and adults (i.e., I, II, IIa and IIx/IIb; L) MyHC isoforms typology of the soleus, plantaris, and tibialis of control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 4). Data are represented as mean ± SEM and significance is reported versus Igf-1r+/+ control mice (*p < 0.05, **p < 0.01, ***p < 0.001).
Because a decrease of AKT activation has been reported in atrophic muscles of severe ALS patients (Yin et al., 2012), we analyzed the phosphorylation status of ERK and AKT in the tibialis of SMA, Igf-1r+/− SMA, control and Igf-1r+/− control mice at P12 (Fig. 7A–D). As in the spinal cord, reducing IGF-1R expression shifted the balance of ERK and AKT activation patterns, leading to a shift in the activation level of the transcription factors Elk-1 and CREB (Fig. 7E–H) and finally to a significant SMN expression increase in SMA muscles at the protein level (Fig. 7I,J). Interestingly, as found in the spinal cord, the IGF-1R reduction led to an increase of both the SMN transcript steady-state level and the proportion of exon-7-containing transcripts (Fig. 7K–M).
Figure 7.

Genetic reduction of IGF-1R expression restores the intracellular pathway activation profile in the skeletal muscle tibialis in SMA mice. A, B, Western blot analysis (A) and quantification (B) of ERK protein phosphorylation in the tibialis muscle of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 4). C, D, Western blot analysis (C) and quantification (D) of AKT protein phosphorylation in the tibialis muscle of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 4). E, F, Western blot analysis (E) and quantification (F) of Elk-1 protein phosphorylation in the tibialis muscle of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 4). G, H, Western blot analysis (G) and quantification (H) of CREB protein phosphorylation in the tibialis muscle of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 4). I, J, Western blot analysis (I) and quantification (J) of SMN protein expression in the tibialis muscle of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 4). K–M, Quantification by real-time qRT-PCR of the E7–E8 segment containing SMN2 transcripts normalized either by the E4–E5 segment containing SMN2 transcripts (K) or by 18S transcripts (L) and of total E4–E5 segment SMN2 transcripts normalized by 18S transcripts (M) in the tibialis muscle of Igf-1r+/+ and Igf-1r+/− control mice compared with Igf-1r+/+ and Igf-1r+/− SMA mice at 12 d of age (n = 8). Dotted lines on Western blot images symbolize some removed interspacing lanes for a side-by-side display of samples from all groups. Data are represented as mean ± SEM and significance is reported versus Igf-1r+/+ control mice (*p < 0.05, **p < 0.01, ***p < 0.001).
Discussion
This study provides the first lines of evidence that, regardless of the circulating IGF-1 level, which remained dramatically low in SMA compared with controls, the reduction of IGF-1R expression, be it at the physiological (through physical exercise) or genetic level, significantly protects motor neurons from SMA-induced cell death. Unexpectedly, reducing the first steps of IGF-1 axis activation, instead of resulting in an unselective depression of the three intracellular pathways known to lie downstream the receptor, exerted a specific effect on each pathway, leading to an alteration of the overall activation balance. These results suggest a crucial role of the surface receptor expression levels in the selective activation or depression of a given downstream intracellular pathway. In the specific case of SMA, IGF-1R reduction led to AKT/CREB pathway activation, which was associated with ERK and JAK inhibition in the spinal cord and skeletal muscle, both events contributing to the significant increase of SMN expression. This was most notable in in motor neurons, as revealed by Gem counts, neuroprotection, and subsequent benefits for SMA-like mice in terms of lifespan and motor behavior (Fig. 8).
Figure 8.

The modulation of IGF-1R expression levels is sufficient to restore the AKT/CREB and ERK/Elk-1 intracellular signaling pathway activation profile, resulting in SMN expression increase in SMA mice. A–C, Proposed mechanisms involved in the increase of SMN expression induced by the genetic reduction of IGF-1R expression in severe SMA-like mice. Modulation of the activation profile of ERK/Elk-1 and AKT/CREB signaling pathways in control (A) compared with Igf-1r+/+ (B) and Igf-1r+/− SMA (C) mice at 12 d of age. Large characters indicate activated molecules.
On the basis of accumulating in vitro evidence, it is well known that all types of cells in the CNS are targeted by IGF-1, including motor neurons (Kaspar et al., 2003; Rabinovsky et al., 2003; Dobrowolny et al., 2005; Ozdinler and Macklis, 2006). Whereas, for many years, canonical signaling through the IGF-1 receptors was considered to include the PI3K/AKT, Ras/ERK, and, more recently, the JAK/STAT pathways (Himpe and Kooijman, 2009), the signaling network elicited by IGF-1 is likely to be differentially activated depending on the cell context, making it difficult to predict the pathway that will ultimately be triggered and thus which biological effect will result. The present data suggest that there are some cell-specific events in which intracellular signaling routes triggered downstream of IGF-1R might depend on the expression levels of the receptor at the cell surface. Reducing IGF-1R expression results in the shift from both ERK and JAK to the AKT pathway and in a parallel alteration of their signaling cascade, leading to Elk-1, STAT, and CREB phosphorylation, respectively. This IGF-1R reduction-induced shift in intracellular pathways occurred only in SMA tissues and not in controls, regardless of tissue origin (e.g., human or murine, spinal cord or muscle). The reasons underlying this discrepancy remain unknown. However, in all of the conditions explored here, an IGF-1R reduction effect on the intracellular pathway activation profile was only found when these pathways were constitutively imbalanced (i.e., ERK overactivation and AKT underactivation). This imbalance could be essential for an IGF-1R reduction effect in restoring the selective pathway activation, ultimately resulting in SMN expression increase. In addition, and most unexpectedly, the IGF-1R reduction in control spinal cords and muscles did not result in half of the activation rate of ERK and AKT, as was reported previously in fibroblasts (Holzenberger et al., 2003), suggesting a cell-specific adaptation to the modulation of IGF-1R expression.
Whatever the molecular mechanism of the signal transduction switch, IGF-1R reduction results in the activation of the AKT/CREB pathway and the inhibition of the ERK/Elk-1 pathway in SMA tissues. In all likelihood, the resulting CREB activation led to its increased binding to the SMN2 promoter and, conversely, the Elk-1 inhibition to its binding decrease. This shift in transcription factor binding was followed by increased acetylation of H3 and H4 histones on sites that are involved in the control of SMN2 transcription (Majumder et al., 2004; Branchu et al., 2013), ultimately resulting in a significant increase in SMN expression. All of these molecular events resulting from the IGF-1R reduction occurred only in SMA and not in controls, in which the intracellular signaling pathways are not altered after IGF-1R expression decrease. Therefore, in SMA only, the IGF-1R reduction correlated with both an increase in SMN steady-state level, strongly suggesting an SMN2 transcriptional activation, and an increase in exon-7 inclusion in SMN2 transcripts, suggesting a modulation of the splicing process. This dual effect on SMN2 gene expression, both transcriptional and posttranscriptional, is unexpected in this mouse model at this age when one considers the effects of NMDA receptor activation in (SmnΔ7/Δ SMN2) mice at P12. In that case, the NMDA-induced activation of the AKT/CREB pathway specifically enhanced exon-7 inclusion in SMN transcripts without any observable effect on SMN steady-state levels (Biondi et al., 2010). A probable effect on the SMN2 gene transcription upon CREB phosphorylation induced either by NMDA receptor activation (Biondi et al., 2010) or ERK inhibition (Branchu et al., 2013) was only found during the first week (P6) in another severe mouse model of SMA. Clearly, identifying the mechanisms underlying IGF-1R reduction effects on the control of SMN2 expression may provide new tools aimed at modulating SMN2 gene expression trough transcriptional and posttranscriptional mechanisms.
Furthermore, whereas the ERK/Elk-1, JAK/STAT, and AKT/CREB pathways have all been correlated to neuroprotection, it is today widely admitted that IGF-1-mediated neuroprotection occurs through activation of the AKT/CREB pathway (D'Mello et al., 1997; Párrizas et al., 1997; Yadav et al., 2005). Furthermore, in the specific case of cerebellar granule neurons, IGF-1-induced neuroprotection results from both the activation of AKT and the inhibition of MEK-ERK1/2 through the AKT pathway (Subramaniam et al., 2005). Similarly, the neuroprotection induced by IGF-1R reduction in SMA spinal cords, as evidenced by the relative persistence of the motor neuron population and the reduced activation of caspase-3, might result from both an increased SMN protein expression and the presence of the two powerful anti-apoptotic molecules, AKT and CREB, at the activated state. Although no role has been reported to date for the JAK/STAT pathway in SMA pathogenesis, the fact that it parallels the activation profile of ERK, in contrast to AKT, suggests a detrimental role of this pathway in SMA.
The present results help to predict the consequences of the dramatic overexpression of IGF-1R in muscles of severe type 1 SMA patients compared with the IGF-1R down-expression in less severe type 3 SMA patients (Millino et al., 2009) in terms of intracellular signalization, SMN expression, cell survival, and muscle maintenance. Furthermore, it could be speculated considering the present results that the constitutive underactivation of AKT recently found in the muscles of ALS patients (Yin et al., 2012) might result from muscular IGF-1R overexpression (Lunetta et al., 2012). The fact that the expression of IGF-1R was found to be dramatically increased in these two genetic motor neuron diseases is puzzling and suggests that an alteration of the IGF-1/IGF-1R equilibrium might be directly involved in motor unit destabilization, ultimately resulting in specific motor neuron death. Over the past 20 years, compelling lines of evidence have accumulated throughout the literature designating IGF-1 as one of the most powerful anti-apoptotic agents for neurons. Several groups subsequently attempted to test the potential effects of IGF-1 in alleviating the symptoms in a variety of neurodegenerative diseases including ALS (Sakowski et al., 2009) and SMA (Bosch-Marcé et al., 2011; Murdocca et al., 2012; Tsai et al., 2012). When tested in different mouse models of SMA, treatments with IGF-1 (Bosch-Marcé et al., 2011; Tsai et al., 2012) or the IGF-1/IGF-BP3 complex (Murdocca et al., 2012) gave rise to disappointing results in terms of SMA mouse survival. Moreover, clinical trials using IGF-1 treatment failed to reduce ALS symptoms in humans (Sorenson et al., 2008; Howe et al., 2009). This paradoxical lack of IGF-1 effects might be due to an aberrant expression of IGF-1R in diseased tissues. More precisely, its overexpression might result in the activation of other, as yet unsuspected pathways that would restrict the beneficial effects of IGF-1. This newly emerging concept involving the modulation of the IGF-1R expression seems to be key for the selection of intracellular pathways and should be taken into account when designing IGF-1-based treatments for neurodegenerative diseases.
Footnotes
This work was supported by the Association Française contre les Myopathies. J.B. is the recipient of a fellowship from the Ministère de la Recherche et de la Technologie. J.C. is a Canada Research Chair (Tier 2) in RNA Metabolism funded through an operating grant from the Canadian Institutes for Health Research (Grant MOP-86746). G.S. is the recipient of a postdoctoral fellowship from Association Française contre les Myopathies. We thank S. Lefebvre for antibodies against SMN, K. Mamchaoui and V. Mouly (Human Cell Culture Platform of the Myology Institute, Paris, France) for providing the human cells from SMA patients, and C. Mader for animal care.
The authors declare no competing financial interests.
References
- Bigot A, Klein AF, Gasnier E, Jacquemin V, Ravassard P, Butler-Browne G, Mouly V, Furling D. Large CTG repeats trigger p16-dependent premature senescence in myotonic dystrophy type 1 muscle precursor cells. Am J Pathol. 2009;174:1435–1442. doi: 10.2353/ajpath.2009.080560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi O, Grondard C, Lécolle S, Deforges S, Pariset C, Lopes P, Cifuentes-Diaz C, Li H, della Gaspera B, Chanoine C, Charbonnier F. Exercise-induced activation of NMDA receptor promotes motor unit development and survival in a type 2 spinal muscular atrophy model mouse. J Neurosci. 2008;28:953–962. doi: 10.1523/JNEUROSCI.3237-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi O, Branchu J, Sanchez G, Lancelin C, Deforges S, Lopes P, Pariset C, Lécolle S, Côté J, Chanoine C, Charbonnier F. In vivo NMDA receptor activation accelerates motor unit maturation, protects spinal motor neurons, and enhances SMN2 gene expression in severe spinal muscular atrophy mice. J Neurosci. 2010;30:11288–11299. doi: 10.1523/JNEUROSCI.1764-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch-Marcé M, Wee CD, Martinez TL, Lipkes CE, Choe DW, Kong L, Van Meerbeke JP, Musarò A, Sumner CJ. Increased IGF-1 in muscle modulates the phenotype of severe SMA mice. Hum Mol Genet. 2011;20:1844–1853. doi: 10.1093/hmg/ddr067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchu J, Biondi O, Chali F, Collin T, Leroy F, Mamchaoui K, Makoukji J, Pariset C, Lopes P, Massaad C, Chanoine C, Charbonnier F. Shift from extracellular signal-regulated kinase to AKT/cAMP response element-binding protein pathway increases survival-motor-neuron expression in spinal-muscular-atrophy-like mice and patient cells. J Neurosci. 2013;33:4280–4294. doi: 10.1523/JNEUROSCI.2728-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, Masliah E, Dillin A. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell. 2009;139:1157–1169. doi: 10.1016/j.cell.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbo M, Lunetta C, Magni P, Dozio E, Ruscica M, Adobbati L, Silani V. Free insulin-like growth factor (IGF)-1 and IGF-binding proteins-2 and -3 in serum and cerebrospinal fluid of amyotrophic lateral sclerosis patients. Eur J Neurol. 2010;17:398–404. doi: 10.1111/j.1468-1331.2009.02815.x. [DOI] [PubMed] [Google Scholar]
- Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- D'Mello SR, Borodezt K, Soltoff SP. Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI 3-kinase in IGF-1 signaling. J Neurosci. 1997;17:1548–1560. doi: 10.1523/JNEUROSCI.17-05-01548.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolny G, Giacinti C, Pelosi L, Nicoletti C, Winn N, Barberi L, Molinaro M, Rosenthal N, Musarò A. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol. 2005;168:193–199. doi: 10.1083/jcb.200407021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U, Liu Q, Dreyfuss G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell. 1997;90:1023–1029. doi: 10.1016/S0092-8674(00)80368-2. [DOI] [PubMed] [Google Scholar]
- Fukaya M, Hayashi Y, Watanabe M. NR2 to NR3B subunit switchover of NMDA receptors in early postnatal motoneurons. Eur J Neurosci. 2005;21:1432–1436. doi: 10.1111/j.1460-9568.2005.03957.x. [DOI] [PubMed] [Google Scholar]
- Grondard C, Biondi O, Armand AS, Lécolle S, Della Gaspera B, Pariset C, Li H, Gallien CL, Vidal PP, Chanoine C, Charbonnier F. Regular exercise prolongs survival in a type 2 spinal muscular atrophy model mouse. J Neurosci. 2005;25:7615–7622. doi: 10.1523/JNEUROSCI.1245-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himpe E, Kooijman R. Insulin-like growth factor-I receptor signal transduction and the Janus Kinase/Signal Transducer and Activator of Transcription (JAK-STAT) pathway. BioFactors. 2009;35:76–81. doi: 10.1002/biof.20. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Géloën A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Howe CL, Bergstrom RA, Horazdovsky BF. Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology. 2009;73:1247. doi: 10.1212/WNL.0b013e3181b26ae6. author reply 1247–1248. [DOI] [PubMed] [Google Scholar]
- Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, Krainer AR. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya S, Park GH, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT, Monani UR. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:2552–2569. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar BK, Lladó J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- Kong L, Wang X, Choe DW, Polley M, Burnett BG, Bosch-Marcé M, Griffin JW, Rich MM, Sumner CJ. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kye MJ, Niederst ED, Wertz MH, Gonçalves Ido C, Akten B, Dover KZ, Peters M, Riessland M, Neveu P, Wirth B, Kosik KS, Sardi SP, Monani UR, Passini MA, Sahin M. SMN regulates axonal local translation via miR-183/mTOR pathway. Hum Mol Genet. 2014;23:6318–6331. doi: 10.1093/hmg/ddu350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Androphy EJ. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- Lunetta C, Serafini M, Prelle A, Magni P, Dozio E, Ruscica M, Sassone J, Colciago C, Moggio M, Corbo M, Silani V. Impaired expression of insulin-like growth factor-1 system in skeletal muscle of amyotrophic lateral sclerosis patients. Muscle Nerve. 2012;45:200–208. doi: 10.1002/mus.22288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder S, Varadharaj S, Ghoshal K, Monani U, Burghes AH, Jacob ST. Identification of a novel cAMP-response element (CRE-II) and the role of CREB-1 in the cAMP-induced expression of the survival motor neuron (SMN) gene. J Biol Chem. 2004;279:14803–14811. doi: 10.1074/jbc.M308225200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Millino C, Fanin M, Vettori A, Laveder P, Mostacciuolo ML, Angelini C, Lanfranchi G. Different atrophy-hypertrophy transcription pathways in muscles affected by severe and mild spinal muscular atrophy. BMC Med. 2009;7:14. doi: 10.1186/1741-7015-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdocca M, Malgieri A, Luchetti A, Saieva L, Dobrowolny G, de Leonibus E, Filareto A, Quitadamo MC, Novelli G, Musarò A, Sangiuolo F. IPLEX administration improves motor neuron survival and ameliorates motor functions in a severe mouse model of spinal muscular atrophy. Mol Med. 2012;18:1076–1085. doi: 10.2119/molmed.2012.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff NT, Prevette D, Houenou LJ, Lewis ME, Glicksman MA, Yin QW, Oppenheim RW. Insulin-like growth factors: putative muscle-derived trophic agents that promote motoneuron survival. J Neurobiol. 1993;24:1578–1588. doi: 10.1002/neu.480241203. [DOI] [PubMed] [Google Scholar]
- Oshima S, Fukaya M, Masabumi N, Shirakawa T, Oguchi H, Watanabe M. Early onset of NMDA receptor GluR epsilon 1 (NR2A) expression and its abundant postsynaptic localization in developing motoneurons of the mouse hypoglossal nucleus. Neurosci Res. 2002;43:239–250. doi: 10.1016/S0168-0102(02)00035-4. [DOI] [PubMed] [Google Scholar]
- Ozdinler PH, Macklis JD. IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat Neurosci. 2006;9:1371–1381. doi: 10.1038/nn1789. [DOI] [PubMed] [Google Scholar]
- Párrizas M, Saltiel AR, LeRoith D. Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathways. J Biol Chem. 1997;272:154–161. doi: 10.1074/jbc.272.1.154. [DOI] [PubMed] [Google Scholar]
- Pellizzoni L, Kataoka N, Charroux B, Dreyfuss G. A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell. 1998;95:615–624. doi: 10.1016/S0092-8674(00)81632-3. [DOI] [PubMed] [Google Scholar]
- Rabinovsky ED, Gelir E, Gelir S, Lui H, Kattash M, DeMayo FJ, Shenaq SM, Schwartz RJ. Targeted expression of IGF-1 transgene to skeletal muscle accelerates muscle and motor neuron regeneration. FASEB J. 2003;17:53–55. doi: 10.1096/fj.02-0183fje. [DOI] [PubMed] [Google Scholar]
- Sakowski SA, Schuyler AD, Feldman EL. Insulin-like growth factor-I for the treatment of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10:63–73. doi: 10.1080/17482960802160370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar-Grueso EF, Kim S, Kim H. Embryonic mouse spinal cord motor neuron hybrid cells. Neuroreport. 1991;2:505–508. doi: 10.1097/00001756-199109000-00002. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Sorenson EJ, Windbank AJ, Mandrekar JN, Bamlet WR, Appel SH, Armon C, Barkhaus PE, Bosch P, Boylan K, David WS, Feldman E, Glass J, Gutmann L, Katz J, King W, Luciano CA, McCluskey LF, Nash S, Newman DS, Pascuzzi RM, et al. Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology. 2008;71:1770–1775. doi: 10.1212/01.wnl.0000335970.78664.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Shahani N, Strelau J, Laliberté C, Brandt R, Kaplan D, Unsicker K. Insulin-like growth factor 1 inhibits extracellular signal-regulated kinase to promote neuronal survival via the phosphatidylinositol 3-kinase/protein kinase A/c-Raf pathway. J Neurosci. 2005;25:2838–2852. doi: 10.1523/JNEUROSCI.5060-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, Schoenenberger C, Ball R, Braun DG, Rosenfelder G. Glycosphingolipid-blotting: an immunological detection procedure after separation by thin layer chromatography. J Immunol Methods. 1984;72:471–479. doi: 10.1016/0022-1759(84)90015-2. [DOI] [PubMed] [Google Scholar]
- Tsai LK, Chen YC, Cheng WC, Ting CH, Dodge JC, Hwu WL, Cheng SH, Passini MA. IGF-1 delivery to CNS attenuates motor neuron cell death but does not improve motor function in type III SMA mice. Neurobiol Dis. 2012;45:272–279. doi: 10.1016/j.nbd.2011.06.021. [DOI] [PubMed] [Google Scholar]
- Wilczak N, de Vos RA, De Keyser J. Free insulin-like growth factor (IGF)-I and IGF binding proteins 2, 5, and 6 in spinal motor neurons in amyotrophic lateral sclerosis. Lancet. 2003;361:1007–1011. doi: 10.1016/S0140-6736(03)12828-0. [DOI] [PubMed] [Google Scholar]
- Yadav A, Kalita A, Dhillon S, Banerjee K. JAK/STAT3 pathway is involved in survival of neurons in response to insulin-like growth factor and negatively regulated by suppressor of cytokine signaling-3. J Biol Chem. 2005;280:31830–31840. doi: 10.1074/jbc.M501316200. [DOI] [PubMed] [Google Scholar]
- Yin F, Ye F, Tan L, Liu K, Xuan Z, Zhang J, Wang W, Zhang Y, Jiang X, Zhang DY. Alterations of signaling pathways in muscle tissues of patients with amyotrophic lateral sclerosis. Muscle Nerve. 2012;46:861–870. doi: 10.1002/mus.23426. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]