

Abstract
Glutamatergic input within the mesolimbic dopamine (DA) pathway plays a critical role in the development of addictive behavior. Although this is well established for some drugs of abuse, it is not known whether glutamate receptors within the mesolimbic system are involved in mediating the addictive properties of chronic alcohol use. Here we evaluated the contribution of mesolimbic NMDARs and AMPARs in mediating alcohol-seeking responses induced by environmental stimuli and relapse behavior using four inducible mutant mouse lines lacking the glutamate receptor genes Grin1 or Gria1 in either DA transporter (DAT) or D1R-expressing neurons. We first demonstrate the lack of GluN1 or GluA1 in either DAT- or D1R-expressing neurons in our mutant mouse lines by colocalization studies. We then show that GluN1 and GluA1 receptor subunits within these neuronal subpopulations mediate the alcohol deprivation effect, while having no impact on context- plus cue-induced reinstatement of alcohol-seeking behavior. We further validated these results pharmacologically by demonstrating similar reductions in the alcohol deprivation effect after infusion of the NMDAR antagonist memantine into the nucleus accumbens and ventral tegmental area of control mice, and a rescue of the mutant phenotype via pharmacological potentiation of AMPAR activity using aniracetam. In conclusion, dopamine neurons as well as D1R-expressing medium spiny neurons and their glutamatergic inputs via NMDARs and AMPARs act in concert to influence relapse responses. These results provide a neuroanatomical and molecular substrate for relapse behavior and emphasize the importance of glutamatergic drugs in modulating relapse behavior.
SIGNIFICANCE STATEMENT Here we provide genetic and pharmacological evidence that glutamate receptors within the mesolimbic dopamine system play an essential role in alcohol relapse. Using various inducible and site-specific transgenic mouse models and pharmacological validation experiments, we show that critical subunits of NMDARs and AMPARs expressed either in dopamine neurons or in dopamine receptor D1-containing neurons play an important role in the alcohol deprivation effect (the increase in alcohol intake after a period of abstinence) while having no impact on context- plus cue-induced reinstatement of alcohol-seeking responses. Medications targeting glutamatergic neurotransmission by selective inactivation of these glutamate receptors might have therapeutic efficacy.
Keywords: addiction, alcohol, dopamine, glutamate, relapse
Introduction
A major hypothesis in the addiction field is that glutamatergic neurotransmission and neuroadaptive changes in glutamate receptors play a central role in alcoholism (Tsai et al., 1995; Krystal et al., 2003; Gass and Olive, 2008; Holmes et al., 2013). In particular, glutamatergic input onto the mesolimbic dopamine (DA) system appears to mediate mechanisms of synaptic plasticity relevant for the development of addictive behavior. Thus, glutamatergic synapses on DA neurons in the ventral tegmental area (VTA) and medium spiny neurons (MSNs) of the nucleus accumbens (NAc) both modulate the reinforcing properties of drugs of abuse and reward-dependent learning processes (Lüscher and Malenka, 2011; Lüscher, 2013). Therefore, glutamate receptors within the mesolimbic system have been proposed to be important not only for synaptic plasticity mechanism, but also for the development of addictive behavior.
The contribution of glutamate receptors in alcohol-related behaviors has been studied in either knock-out mouse models or in pharmacological studies, and the results obtained point to a minor role of NMDARs and AMPARs in alcohol reinforcement and alcohol-seeking responses (Sanchis-Segura et al., 2006; Spanagel, 2009; Bilbao, 2013). However, relapse behavior, as measured by the alcohol deprivation effect (ADE), is attenuated or blocked by several NMDAR and AMPAR antagonists (Hölter et al., 1996; Vengeliene et al., 2005, 2008; Sanchis-Segura et al., 2006; Spanagel, 2009; Holmes et al., 2013). Thus, there is some indication that glutamate receptors are, at least in part, involved in mediating the addictive properties of alcohol, but the neuroanatomical substrates and the specific contributions of AMPARs and NMDARs are not well understood.
To study the contribution of NMDARs and AMPARs in mediating relapse-like behavior in the ADE model and in reinstatement of alcohol-seeking triggered by environmental stimuli (context and cue), we used site- and time-specific conditional mouse models lacking the glutamate receptor subunits GluN1 or GluA1 in the DAT- (a marker of DAergic neurons) and D1-expressing neurons (expressed on a subpopulation of MSNs). Although there is growing evidence that D2-expressing MSNs mediate several effects induced by alcohol (Spanagel, 2009), in a recent report by Wang et al. 2015)) it was demonstrated that alcohol consumption induces a long-lasting increase in AMPAR activity specifically in D1-MSNs, but not in D2-MSNs, and striatal blockade of D1R, but not D2R, activity attenuates alcohol consumption. Therefore, we studied the contribution of NMDARs and AMPARs onto D1-MSNs in mediating addictive-like behavior.
The ADE as a phenomenon in laboratory animals that models a relapse-like drinking situation was first described for rats (Sinclair and Senter, 1967) and later for mice (Salimov and Salimova, 1993). In this model, renewed access to alcohol solutions after a period of deprivation for several days leads to a pronounced, although temporary, increase in voluntary alcohol intake in animals (Vengeliene et al., 2014; Eisenhardt et al., 2015). In rats, the ADE was attenuated by repeated subacute treatment with memantine and other NMDAR antagonists (Hölter et al., 2000; Vengeliene et al., 2005, 2008; Spanagel, 2009).
The reinstatement model (de Wit and Stewart, 1981) examines the resumption of responding in an operant task of a previously extinguished drug-reinforced behavior in response to noncontingent drug delivery (i.e., priming), environmental stimuli previously associated with it (context or cues), or stressful stimuli (Sanchis-Segura and Spanagel, 2006; Bossert et al., 2013). A cue-induced increase in alcohol-seeking is attenuated by some NMDAR antagonists (Bäckström and Hyytiä, 2004; Spanagel, 2009), but these effects are usually accompanied by a generalized impairment in motor behavior (Bachteler et al., 2005; Spanagel, 2009; Holmes et al., 2013); therefore, the specific contribution of NMDARs to alcohol-seeking responses is unclear.
Here we show, with our selective and inducible mutant mouse models, that GluN1 and GluA1 receptor subunits in DAT- and D1-expressing neurons play a crucial role in relapse to alcohol while having no impact on environmental stimuli (context and cue) -induced reinstatement. This was further demonstrated by site-selective pharmacological manipulations with the drugs memantine and aniracetam.
Materials and Methods
Mice generation.
We generated mutant mice expressing GluN1 or GluA1 mutations under control of the DAT (Slc6a3) or D1 (Drd1a) promoter following the previously described procedure (Engblom et al., 2008; Mameli et al., 2009; Parkitna et al., 2009, 2010). In short, GluN1DATCreERT2, GluA1DATCreERT2, GluN1D1CreERT2, and GluA1D1CreERT2 mice were generated by crossing mice with an inducible Cre-recombinase under the DAT- or D1-promoter with mice carrying floxed alleles for GluN1or GluA1. The DATCreERT2 and D1CreERT2 mice were generated by recombining a construct containing an improved Cre-recombinase fused to a modified ligand binding domain of the estrogen receptor (CreERT2) into a bacterial artificial chromosome containing the gene encoding DAT (Slc6a3) or D1 (Drd1a) by recombineering. GluN1fl/fl and GluA1fl/fl mice, having exons 11–18 of the Grin1 or exon 11 of the Gria1 alleles, respectively, flanked with loxP sites were generated by gene targeting in embryonic stem cells (Zamanillo et al., 1999; Niewoehner et al., 2007). For induction of the mutation, mice were challenged with 1 mg of tamoxifen intraperitoneally twice a day for 5 consecutive days (Erdmann et al., 2007). For genotyping of the DATCreERT2 and D1CreERT2 transgene, we used the primers GGC TGG TGT GTC CAT CCC TGA A and GGT CAA ATC CAC AAA GCC TGG CA. The GluN1 and GluA1 flox variants were genotyped using the primers GGA CAG CCC CTG GAA GCA AAA T and GGA CCA GGA CTT GCA GTC CAA AT for GluN1, and CAC TCA CAG CAA TGA AGC AGG AC and CTG CCT GGG TAA AGT GAC TTG G for GluA1. For all experiments, adult male GluN1DATCreERT2, GluA1DATCreERT2, GluN1D1CreERT2, and GluA1D1CreERT2 and their wild-type littermate mice from at least six consecutive backcrosses with C57BL/6N were used (8–10 weeks at the beginning of the experiments). As controls, floxed littermates not carrying the Cre-recombinase were used.
Animals were single-housed in standard hanging cages at 21 ± 1°C and 50 ± 5% relative humidity on a reversed 12 h light/dark cycle, with lights on at 7:30 P.M. Animals were provided with standard rodent food and tap water ad libitum. All mice were offered two bottles of tap water to prevent habituation-induced side preference, except during the home cage drinking procedure where one of the bottles filled with tap water was replaced by an ethanol solution. Animals were handled on a daily basis before starting the experiments. Experiments were conducted in accordance with European Union guidelines on the care and use of laboratory animals.
Dual-labeling immunohistochemistry.
Four weeks after induction of the mutation, mice anesthetized by isoflurane inhalation were transcardially perfused with 1 m PBS for 1 min followed by perfusion with 4% PFA in 1 m phosphate buffer for 15 min. After perfusion, brains were removed and immersed in the same fixative for 2 h at 4°C. Afterward, brains were cryoprotected in 0.01 mm phosphate buffer with 15% sucrose and 0.05 mm sodium azide. Tissue blocks containing the area of DAergic (midbrain) or dopaminoceptive neurons were dissected and washed in 0.1 m phosphate buffer for several hours. Next, tissue blocks were embedded in 4% agarose and sectioned at 60 μm with a microtome. Sections were blocked in 10% normal goat serum and following incubation in a mixture of primary antibodies for GluA1 and TH, for GluN1 and TH, GluA1 and D1, or GluN1 and D1, in TBS containing 2% normal goat serum overnight at 4°C. For GluN1 labeling, the sections were pretreated with 2 mg/ml pepsin to improve accessibility to the epitopes. After washing with TBS, sections were incubated in a mixture of secondary antibodies coupled to Alexa-488 or to cyanine-derived fluorochrome Cy3, made up in TBS, for 2 h at room temperature. Subsequently, sections were washed with TBS, mounted on gelatin-coated slides, air-dried, and coverslipped with buffered glycerol. Fluorescent signals were determined using a confocal laser microscope (Zeiss, LSM 710).
Antibodies.
The primary antibodies (2 μg/ml) used were as follows: affinity-purified rabbit anti-GluA1 polyclonal (AB1504; lot #24040209; Millipore Bioscience Research Reagents, Merck), affinity-purified rabbit anti-GluN1 polyclonal (AB9864; lot #07-362; Millipore Bioscience Research Reagents, Merck), mouse anti-TH monoclonal (MAB5280, clone 2/40/15; Calbiochem, Merck, Millipore Bioscience Research Reagents), and affinity-purified guinea pig anti-D1 polyclonal (D1R-Gp-Af500; Frontiers Institute) antibodies. The characteristics and specificity of the anti-D1 antibody have been described previously(Narushima et al., 2006). Secondary antibodies conjugated to Alexa-488 or to cyanine-derived fluorochrome Cy3 (dilution 1:500) were purchased from Invitrogen, Thermo Fisher Scientific, and Jackson ImmunoResearch Laboratories, respectively.
Behavioral experiments.
All behavioral experiments were initiated 4 weeks after the tamoxifen treatment and were performed during the active, dark phase of the day, between 9:00 and 14:00 h.
Homecage activity.
Spontaneous locomotor activity was recorded in the home cage using the MOUSE-E-MOTION Data Logger (INFRA-E-MOTION). One data logger was placed above each cage (30 cm from the bottom) using a stainless steel mounting unit, so that the mouse could be detected at any position inside the cage. Locomotor activity was measured using a passive infrared sensor, which detects the emitted heat radiation of the mouse. The sensor can detect body movements of the mouse of ≥1.5 cm from one sample point to the next. The data logger was sampling every 4 min whether the mouse moved or not and lasted for 3 d.
Habituation to the activity boxes.
To assess reactivity to novel environments and habituation mechanisms, each mouse was placed in a clear Plexiglas box (40 × 40 cm), and locomotor activity was measured for a period of 30 min. Locomotor activity was recorded every 5 min using the TruScan photo beam activity system (Coulbourn Instruments).
Elevated-plus-maze.
Anxiety-like behavior was tested using the Elevated-plus-maze test. The Elevated-plus-maze consisted of a right-angled cross elevated 50 cm above the floor with two open and two enclosed arms. Each mouse was placed at the intersection of the 4 arms and allowed to explore all 4 arms freely for 5 min. Locomotion and spatial placement were recorded by EthoVision XT (Noldus Information Technology).
Taste preference test.
Taste preference over water for sweet 0.2% saccharin and bitter 0.25 mm quinine solutions was tested. Mice had the choice between one of the two solutions and water for 3 d. The preference of each solution was calculated as the percentage of the total fluid intake.
Home cage two-bottle free choice drinking and ADE.
Mice had access to increasing ethanol concentration in a two-bottle free choice paradigm in the home cage, having the choice between ethanol and water. First, mice had access to a 2% ethanol solution (v/v) and tap water for 3 d, followed by 4% ethanol solution and tap water for another 3 d. Next, mice had the choice between an 8% ethanol solution and tap water for 6 d; and finally, the concentration of ethanol was increased to 12%. Mice had access to 12% ethanol and tap water until they reached a stable intake (12–15 d). Following the establishment of a baseline, ethanol and water intake was recorded using the Drinkometer system (INFRA-E-MOTION) for 3 d. Mice were afterward deprived from ethanol for 12–15 d, during which they only had access to two bottles of tap water. After the deprivation period, the ADE was tested for 24 h by reintroducing the ethanol bottle.
For pharmacological experiments, drugs were administered intraperitoneally 30 min before the ADE test. For intra-VTA and intra-NAc administration, a guide cannula (see subsequent section) was implanted during the deprivation period (deprivation day 3–4), and the drugs were administered 60 min before the ADE test.
Ethanol (g/kg) and water (ml) intake as well as the ethanol preference (% of total fluid intake) were calculated for each ethanol concentration per day. During baseline and ADE measurement, ethanol and water intake was additionally calculated in 4 h time intervals. Baseline ethanol and water intake was calculated as the mean of the last 3 d of baseline recording.
Assessment of drinking patterns by a fully automated Drinkometer device.
It has very often been reported that mice do not show an ADE (Salimov et al., 1995; Bilbao, 2013; Vengeliene et al., 2014). By using a novel fully automated Drinkometer device, we have recently demonstrated a robust ADE with up to 10-fold increase in alcohol consumption compared with baseline drinking. This effect is, however, of short-lasting nature and restricted to the first 4–8 h of resumption of alcohol drinking. Therefore, a high time resolution monitoring is suggested to be essential to capture not only the ADE onset but also its duration (Eisenhardt et al., 2015).
Ethanol and water intake was measured during baseline and ADE with a fully automated, highly precise monitoring system. This Drinkometer system comprises the MOUSE-E-MOTION Universal Data Logger (INFRA-E-MOTION; http://www.infra-e-motion.de/en/) in connection with an extension module and is used to monitor simultaneously water and alcohol consumption. Additionally, the system enables the measurement of other variables, such as spontaneous locomotor activity and the lighting conditions. During recording, the standard lid of the mouse home cage was replaced with the Drinkometer lid containing two holes for special Drinkometer bottles and a spatially divided feeding dish to prevent hampered recording. Each cage was equipped with a metal holder to mount and connect the data logger. The metal holder contained two independent channels with weight sensors and an LCD display. Last, special Drinkometer bottles with a curved bottleneck and different tips for water (0.8 mm opening) and alcohol (0.5 mm opening) solutions were attached magnetically to the metal holder. The Drinkometer system was configured to sample every 4 min, the amount (g) of ethanol and water each mouse consumed. Ethanol and water intake was calculated every 4 h to assess the circadian drinking pattern and to obtain a temporal dissection of the ADE.
Operant self-administration, extinction, and context- plus cue-induced reinstatement of ethanol seeking.
Mice were trained to self-administer ethanol in eight operant chambers (TSE Systems). Each chamber was equipped with two ultrasensitive levers (required force, ≤1 g) on opponent sides: one functioning as the active and one as the inactive lever. Next to each lever, a front panel containing the visual stimulus was installed above a drinking microreservoir. When the programmed ratio requirements were met on the active lever, 10 μl of the ethanol solution was delivered into the microreservoir, and the visual stimulus was presented via a light located on the front panel. Responses on the inactive lever were recorded but had no programmed consequences. These responses were recorded as a measure of nonspecific behavioral activation. A microcomputer controlled the delivery of fluids, presentation of visual stimuli, and recording of the behavioral data.
Conditioning phase.
Mice were trained to self-administer 10% ethanol (v/v) in 30 min daily sessions on a fixed ratio 1 schedule of reinforcement, where each response resulted in delivery of 10 μl of fluid. A contextual stimulus predicting ethanol availability was presented during the ethanol self-administration sessions. The contextual stimulus consisted of a gray, smooth floor (S+). In addition, each lever press resulting in delivery of ethanol was paired with illumination of the chamber's cue light for 5 s (CS+). Concurrently with the presentation of these stimuli, a 5 s time-out period was in effect, during which responses were recorded but not reinforced. Criteria for the conditioning were met at stable baseline lever pressing for 3 consecutive days, with no significant differences in lever pressing.
Extinction phase.
After the last conditioning day, mice were subjected to daily 30 min extinction sessions. Responses at the active lever did not result in the delivery of liquids or the presentation of the response-contingent cue (light). During the extinction phase, a bar floor was used. The criteria for extinction were established at 50% of the baseline lever responses for 3 consecutive days.
Context- plus cue-induced reinstatement.
Reinstatement testing began the day after the last extinction session. Context- plus cue-induced reinstatement was tested under conditions identical to those during the conditioning phase, except that ethanol was not made available.
For pharmacological experiments, drugs were administered 30 min before the reinstatement test. Drug administrations were conducted every third day using a counterbalanced design.
Surgery and intra-NAc and VTA microinfusions.
Mice were anesthetized with 1.5%–1.8% of isoflurane (CP-Pharma) and placed in a stereotaxic frame (David Kopf Instruments). A 26-gauge stainless-steel guide cannula of 4.2 mm length (Plastics One) was unilaterally implanted to target the VTA or the NAc core. The stereotaxic coordinates for the VTA were as follows: from bregma, −3.16 mm anteroposterior, ±0.5 mm mediolateral, and −4.2 mm dorsoventral; and for the NAc core: from bregma, −1.65 mm anteroposterior, ±0.9 mm mediolateral, and −4.2 mm dorsoventral. The guide cannula was secured to the skull with dental cement (Super-Bond C&B, Sun Medical), and a 33-gauge dummy cannula was inserted into the guide cannula to prevent blockade and contamination. After the surgery, mice were allowed to recover for at least 7 d. Two days before the experiment, each mouse was habituated to the infusion procedure.
For microinfusion, the dummy cannula was removed and a 33-gauge injector cannula was slowly lowered into the guide cannula. The injection cannula projected 0.1 mm beyond the tip of the guide cannula. Drugs were microinfused into the target area with a flow rate of 0.25 μl/min over 2 min driven by a syringe pump (Harvard Apparatus). Injectors were left in place for an additional 1 min after the end of the injection, to ensure adequate diffusion from the tip and to reduce any possible backflow along the injection track. After completion of the injection, the injector cannula was removed and the dummy cannula inserted.
Histology.
Cannula placement was verified after the end of each experiment. Mice were therefore killed by decapitation. Brains were removed, snap frozen in −40°C isopentane for 1–2 min, and stored at −70°C until use. Brains were sliced in coronal sections of 50 μm. Sections were mounted on slides, stained with cresyl violet, and coverslipped. Cannula placement was examined under a light microscope, and the location of the guide cannula was verified using the atlas of Paxinos and Franklin (2001) as reference. Only mice with probe placement in the VTA or NAc (core or shell) were included in the statistical analysis. Although cannula placement was aimed at the NAc core, no differentiation between NAc core and shell was made. Because of the tight anatomical proximity, drug diffusion will comprise both subnuclei (Besheer et al., 2010).
Drugs.
Tamoxifen was dissolved in medium-chain triglycerides (Stadtklinik Frankenthal) to a final concentration of 10 mg/ml. Medium-chain triglycerides were used instead of conventional sunflower seed oil for better compatibility and made adding of alcohol dispensable. Sodium saccharin and quinine hydrochloride were added to water to achieve 0.2% or 0.25 mm (w/v), respectively. Ethanol dilutions (2%, 4%, 8%, 10%, and 12%; v/v) were made up with 96% ethanol and water. Memantine (Biotrend) was dissolved in saline and aniracetam (Cayman Chemical) suspended with 2–3 drops of Tween 80 in saline as vehicle. Memantine and aniracetam were administered as intraperitoneal injections at a dose of 25 and 5 mg/kg, respectively, and for intra-VTA and intra-NAc infusions at a dose of 5 μg/0.5 μl and 1.5 μg/0.5 μl, respectively.
We decided to use memantine for pharmacological validation experiments because of its translational potential. Thus, it has been suggested that memantine may be useful in the treatment of relapse behavior (Hölter et al., 1996, 2000; Spanagel and Vengeliene, 2013), and first clinical studies have already tested its therapeutic efficacy (Bisaga and Evans, 2004; Evans et al., 2007; Krupitsky et al., 2007). Another reason to choose memantine was that, because of its lower receptor affinity and faster off-rates, this drug displays some advantages over other NMDAR antagonists, especially that it produces a very low side effect profile compared, for example, with MK-801 (Parsons et al., 1999). The rationale for choosing the positive allosteric modulator aniracetam, which potentiates AMPAR activity (Tang et al., 1991) to rescue the abolished relapse-like behavior in our mutant mice, was the recent report showing that administration of this drug was able to increase alcohol-seeking in rats (Cannady et al., 2013). In addition, to our knowledge, there are no pharmacological agents available, which are selective for the AMPAR subunit GluA1.
Statistics.
The value n corresponds to the number of animals. Statistical analyses were performed by ANOVA with Newman–Keuls test for post hoc comparisons and Student's t test using Statistica 10 (StatSoft). All values are given as mean ± SEM, and statistical significance was set at p < 0.05.
Results
Basic characterization of mice with a specific lack of glutamate receptors within the mesolimbic DA system
In slices from control mice, GluN1 and GluA1 were expressed in both TH-negative and TH-positive as well as in D1-negative and D1-positive neurons. In GluN1DATCreERT2 and GluN1D1CreERT2 mutant mice, GluN1 was expressed only in TH-negative and in D1-negative cells, respectively (Fig. 1A). The same pattern was found in GluA1DATCreERT2 and GluA1D1CreERT2 mutant mice where GluA1 was expressed only in TH-negative and in D1-negative cells, respectively (Fig. 2). Together with our previous reports, this further confirms the ablation of individual glutamate receptor subunits within the mesolimbic DA system in our four mutant mouse lines.
Figure 1.

Lack of GluN1 in DAergic and D1-expressing cells in mutant mice. Representative coronal sections from the VTA showing immunofluorescence for GluN1 and TH in control (A, B) and GluN1DATCreERT2 (D, E) mice, respectively. C, F, Overlay of the images A, B and D, E. The deletion was efficient and specific in all cases. Hence, GluN1 is only expressed in TH-negative neurons in GluN1DATCreERT2 mice. Corresponding analysis for D1 and GluN1 in controls (G, H) and GluN1D1CreERT2 mice (J, K) in coronal sections from the NAc. I, L, Overlay of the images G, H and J, K. The deletion was efficient and specific in all cases. Hence, GluN1 is only expressed in D1-negative neurons in GluN1D1CreERT2 mice. Arrow indicates colocalization. *TH or D1 labeling. Triangle represents GluN1 labeling without colocalization.
Figure 2.
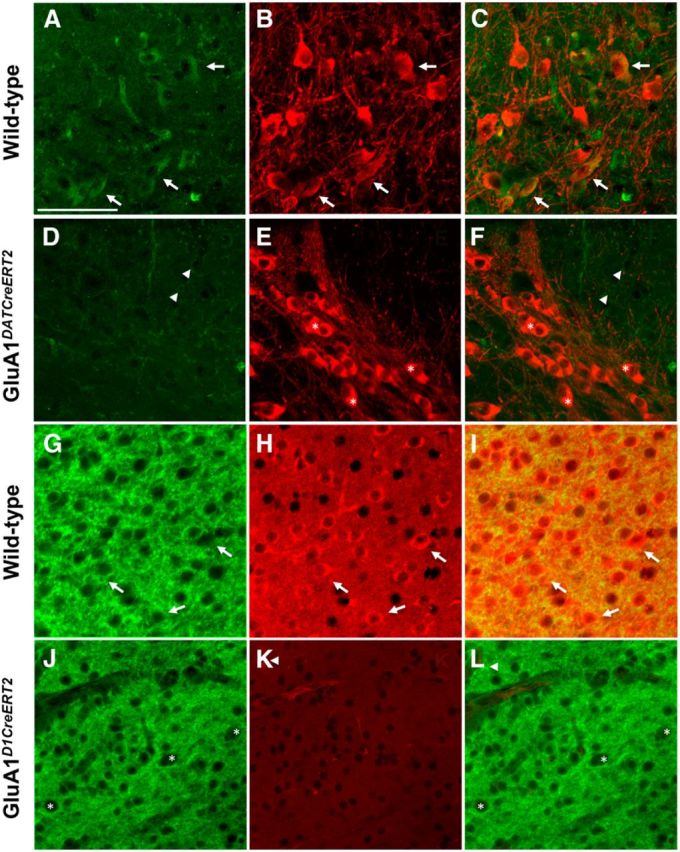
Lack of GluA1 in DAergic and D1-expressing cells in mutant mice. Representative coronal sections from the VTA showing immunofluorescence for GluA1 and TH in control (A, B) and GluA1DATCreERT2 (D, E) mice, respectively. C, F, Overlay of the images A, B and D, E. The deletion was efficient and specific in all cases. Hence, GluA1 is only expressed in TH-negative neurons in GluA1DATCreERT2 mice. Corresponding analysis for D1 and GluA1 in controls (G, H) and GluA1D1CreERT2 mice (J, K) in coronal sections from the NAc. I, L, Overlay of the images G, H and J, K. The deletion was efficient and specific in all cases. Hence, GluA1 is only expressed in D1-negative neurons in GluA1D1CreERT2 mice. Arrow indicates colocalization. *TH or D1 labeling. Triangle represents GluA1 labeling without colocalization.
Because motor and emotional components as well as alterations in taste sensitivity can potentially affect alcohol drinking and operant behavior, mice were tested for spontaneous locomotor activity, habituation to novelty, anxiety, and taste preference over water for a sweet (0.2% saccharin) and bitter (0.25 mm quinine) solution.
GluN1 mutations
All genotypes displayed typical diurnal rhythmicity, with higher activity levels during the dark, active phase compared with the light, inactive phase of the day (two-way ANOVA, Phase effect, (Fig. 3A) F(5,156) = 35.61, p < 0.001; (Fig. 3G) F(5,210) = 91.26, p < 0.001). However, both GluN1DATCreERT2 and GluN1D1CreERT2 mice displayed reduced locomotor activity during the active phase (Fig. 3A; two-way ANOVA, Genotype × Phase interaction effect, F(5,156) = 3.59, p < 0.01; Fig. 3G; Genotype effect, F(1,210) = 13.65, p < 0.001). We then tested habituation to novelty in an activity box. Although GluN1DATCreERT2 showed a similar habituation profile to novelty, they had reduced activity over the entire time course of testing (Fig. 3B; Genotype effect, F(1,26) = 37.5, p < 0.001), which resulted in a lower total locomotor activity (Fig. 3C; t(26) = 6.12; p < 0.001). GluN1D1CreERT2 showed slightly impaired habituation to novelty (Fig. 3H; Genotype × Habituation interaction effect, F(5,175) = 6.62, p < 0.001). In measurements for anxiety-like behavior (Fig. 3D,E,J,K) and taste preferences for saccharin and quinine (Fig. 3F,L), no genotype differences were observed.
Figure 3.

General behavioral characterization of GluN1DATCreERT2 and GluN1D1CreERT2 mice. A, G, Spontaneous home cage locomotor activity measured by the mouse-e-motion system in GluN1DATCreERT2 (n = 14), GluN1D1CreERT2 (n = 16), and respective wild-type (n = 14 or n = 16) control groups. All genotypes displayed typical diurnal rhythmicity with higher activity levels during the dark, active phase compared with the light, inactive phase of the day. However, both GluN1DATCreERT2 and GluN1D1CreERT2 mice showed lower locomotor activity during dark/active phase and during the first 4 h of the light/inactive phase. B, C, H, I, Habituation to novelty in the activity box. While total distance moved in the activity box was attenuated in GluN1DATCreERT2 mice, GluN1DATCreERT2 habituated faster to a novel environment compared with wild-type mice. D, E, J, K, Elevated-plus-maze test. Total time spent in the open and closed arms and visits to the open arm were similar in all genotypes. F, L, Preference over water for 0.2% saccharin and 0.25 mm quinine solution was not altered. Data are mean ± SEM. *p < 0.05 versus wild-type.
GluA1 mutations
GluA1DATCreERT2 and GluA1D1CreERT2 mice were almost indistinguishable from wild-type mice in all measurements (Fig. 4).
Figure 4.
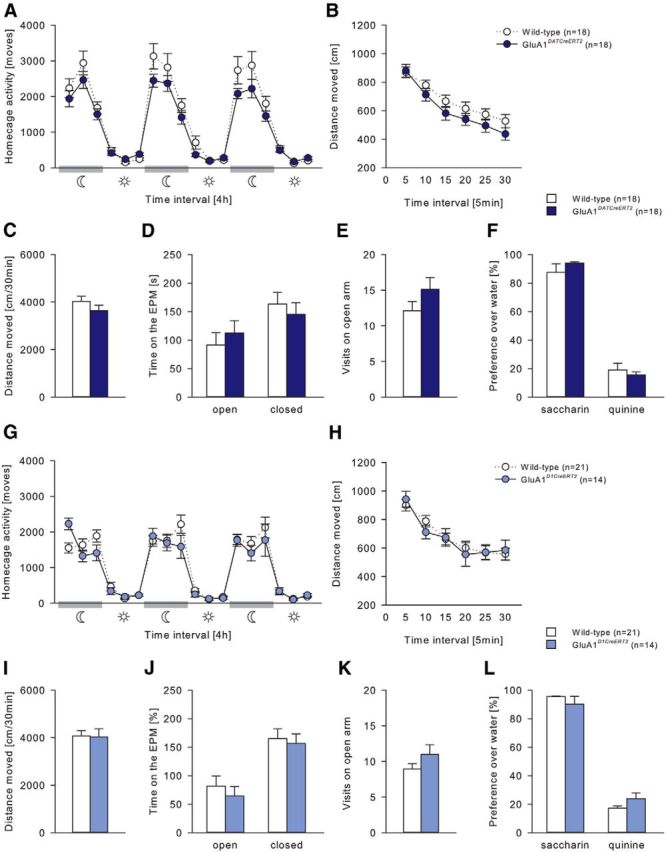
General behavioral characterization of GluA1DATCreERT2 and GluA1D1CreERT2 mice. A, G, Spontaneous home cage locomotor activity measured by the mouse-e-motion system in GluA1DATCreERT2 (n = 18), GluA1D1CreERT2 (n = 14), and respective wild-type (n = 18 or n = 14) control groups. All genotypes displayed similar diurnal rhythmicity with higher activity levels during the dark, active phase compared with the light, inactive phase of the day. B, C, H, I, Habituation to novelty in the activity box. No genotype difference in (C, I) total distance moved and (B, H) habituation to a novel environment was observed. D, E, J, K, Elevated-plus-maze test. Total time spent in the open and closed arms and visits to the open arm were not altered. F, L, Preference over water for 0.2% saccharin and 0.25 mm quinine solution was similar in all genotypes. Data are mean ± SEM.
Alcohol deprivation effect requires GluN1 and GluA1 containing receptors in the mesolimbic system
GluN1 mutations
During the acquisition of voluntary ethanol self-administration (Figs. 5A, 6A), GluN1DATCreERT2 and GluN1D1CreERT2 mice did not differ from their wild-type littermates at any ethanol concentration tested (Fig. 5A; two-way ANOVA, Genotype effect, F(1,27) = 0.16, p = 0.69; Genotype × Concentration interaction effect, F(3,81) = 0.14, p = 0.94; 6A, Genotype effect, F(1,36) = 0.02, p = 0.88; Genotype × Concentration interaction effect, F(3,108) = 0.64, p = 0.59), and all mice increased similarly the intake in a concentration-dependent manner (Fig. 5A; Concentration effect, F(3,81) = 26.23, p < 0.001; Fig. 6A; F(3,108) = 33.19, p < 0.001).
Figure 5.

Relapse-like behavior is mediated by GluN1 in DAergic neurons. A, Ethanol intake during the acquisition of voluntary ethanol self-administration (2%, 4%, 8%, and 12%) was not different in GluN1DATCreERT2 (n = 15) and wild-type (n = 14) mice. B–D, ADE. B, C, In the Drinkometer device, after 2 weeks of ethanol deprivation, GluN1DATCreERT2 mice showed a significant ADE during the first 4 h of reexposure compared with wild-type mice. B, Ethanol intake in g/kg per 4 h time blocks. C, Percentage change from baseline. D, The same significant effect was observable when wild-type mice received an intra-VTA (n = 7) administration of the NMDAR antagonist memantine (5 μg/0.5 μl) 60 min before the ADE test. Data are mean ± SEM. #p < 0.05 versus baseline. *p < 0.05 versus wild-type. Arrow indicates memantine infusion.
Figure 6.

Relapse-like behavior is mediated by GluN1 in D1-expressing neurons. A, Ethanol intake during the acquisition of voluntary ethanol self-administration (2%, 4%, 8%, and 12%) was not different in GluN1D1CreERT2 (n = 21) and wild-type (n = 17) mice. B–D, ADE. B, C, After 2 weeks of ethanol deprivation, the ADE in GluN1D1CreERT2 mice was reduced during the first 4 h of reexposure compared with wild-type mice. B, Ethanol intake in g/kg per 4 h time blocks. C, Percentage change from baseline. D, The same effect was observable when wild-type mice received an intra-NAc (n = 7) administration of the NMDAR antagonist memantine (5 μg/0.5 μl) 60 min before the ADE test. Data are mean ± SEM. #p < 0.05 versus baseline. *p < 0.05 versus wild-type. Arrow indicates memantine infusion.
A period of ethanol deprivation (Figs. 5B,C, 6B,C) significantly increased the ethanol intake in GluN1DATCreERT2, GluN1D1CreERT2, and the respective wild-type mice, indicative of an ADE, which was strongly pronounced during the first 4 h of reexposure and lasted not longer than 8 h in all genotypes (Fig. 5B; two-way ANOVA, Deprivation × Phase interaction effect, F(5,162) = 50.23, p < 0.001; Fig. 6B; Deprivation × Genotype interaction effect, F(5,216) = 9.9, p < 0.001). However, during the first 4 h period, GluN1DATCreERT2 and GluN1D1CreERT2 mice showed a reduced ADE as revealed by two-way ANOVA (Fig. 5B; Deprivation × Genotype × Phase interaction effect, F(5,162) = 4.27, p < 0.01; Fig. 6B; Deprivation × Genotype interaction effect, F(1,216) = 5.0, p < 0.05). When calculating the percentage change of ethanol intake during the first 4 h of reexposure compared with baseline, this effect was even more pronounced (Fig. 5C; two-way ANOVA, Deprivation × Genotype interaction effect, F(1,27) = 21.89, p < 0.001; Fig. 6C; F(1,36) = 7.22, p < 0.05).
If GluN1 in dopaminergic and dopaminoceptive neurons is critical for ethanol relapse behavior, site-specific pharmacological blockade of the NMDAR is expected to block this behavior. To test this, the effect of pharmacological blockade of the NMDAR in the VTA (Fig. 5D) and NAc (Fig. 6D) on the ADE was studied. Wild-type mice received either an intra-VTA or intra-NAc infusion of 5 μg/0.5 μl memantine or a vehicle infusion 60 min before the ADE test. Although all three treatment groups increased their ethanol intake after a period of deprivation during the first 4 h (Fig. 5D; two-way ANOVA, Deprivation × Phase interaction effect, F(5,84) = 3.47, p < 0.01; Fig. 6D; Deprivation effect, F(1,84) = 16.33, p < 0.001), memantine administration into the VTA and NAc significantly attenuated ethanol intake during the first 4 h compared with control mice, similarly to what was observed in the mutant mice (Fig. 5D, two-way ANOVA, Deprivation × Treatment interaction effect, F(1,84) = 5.32, p < 0.05; Fig. 6D, Treatment effect, F(1,84) = 12.05, p < 0.001).
We did not observe any side effects after memantine treatment. Water intake and locomotor activity were monitored with the Drinkometer system throughout all experiments, and microinjection of memantine did not affect water intake (two-way ANOVA, Treatment × Deprivation interaction effect, VTA: F(1,84) = 0.91, p = 0.34; NAc: F(1,84) = 0.20, p = 0.66) or locomotor activity (Treatment × Deprivation interaction effect, VTA: F(1,84) = 0.04, p = 0.85; NAc: F(1,84) = 1.80, p = 0.18) (data not shown).
GluA1 mutations
In GluA1DATCreERT2, GluA1D1CreERT2, and the respective wild-type mice, voluntary ethanol consumption during the acquisition phase was similar at any ethanol concentration offered (Fig. 7A; two-way ANOVA, Genotype effect, F(1,26) = 0.11, p = 0.75; Genotype × Concentration interaction effect, F(3,78) = 0.74, p = 0.53; Figure 8A; Genotype effect, F(1,28) = 0.81, p = 0.38; Genotype × Concentration interaction effect, F(3,84) = 0.12, p = 0.95), and all mice increased similarly the intake in a concentration-dependent manner (Fig. 7A; F(3,78) = 29.02, p < 0.001; Fig. 8A, F(3,84) = 8.37, p < 0.001). Resumption of ethanol drinking after 2 weeks of abstinence (Figs. 7B,C, 8B,C) induced a significant increase in ethanol consumption in all genotypes resulting in a characteristic ADE profile that was strongly pronounced during the first 4 h of exposure (Fig. 7B; two-way ANOVA, Deprivation × Phase interaction effect, F(5,156) = 10.35, p < 0.001; Fig. 8B; F(5,168) = 15.41, p < 0.001). However, the ADE was significantly attenuated in GluA1DATCreERT2 and GluA1D1CreERT2 during this 4 h period (Fig. 7B; Deprivation × Genotype interaction effect, F(1,156) = 4.36, p < 0.05; Fig. 8B; Deprivation × Genotype × Phase interaction effect, F(5,168) = 2.27, p < 0.05). When calculating the percentage change of ethanol intake during the first 4 h of ethanol resumption compared with baseline, the attenuated ADE of mutant mice during the first 4 h of reexposure was even more pronounced (Fig. 7D; Deprivation × Genotype interaction effect, F(1,24) = 4.74, p < 0.05; Fig. 8D; F(1,28) = 6.97, p < 0.05).
Figure 7.

Relapse-like behavior is mediated by GluA1 in DAergic neurons. A, GluA1DATCreERT2 (n = 13) and wild-type (n = 13) mice had similar ethanol intake during the acquisition of voluntary ethanol self-administration (2%, 4%, 8%, and 12%). B–D, ADE. B, C, A 2 week alcohol deprivation period induced an attenuated ADE in GluA1DATCreERT2 mice during the first 4 h of reexposure compared with wild-type mice. B, Ethanol intake in g/kg per 4 h time blocks. C, Percentage change from baseline. D, On the contrary, administration of the AMPAR potentiator aniracetam (1.5 μg/0.5 μl) into the VTA (n = 7) 60 min before the ADE test in wild-type mice significantly increased the ethanol intake during the first 4 h after reintroduction of the ethanol bottle. Data are mean ± SEM. #p < 0.05 versus baseline. *p < 0.01 versus wild-type. Arrow indicates aniracetam infusion.
Figure 8.

Relapse-like behavior is mediated by GluA1 in D1-expressing neurons. A, Ethanol intake during the acquisition of voluntary ethanol self-administration (2%, 4%, 8%, and 12%) was not different in GluA1D1CreERT2(n = 17) and wild-type (n = 13) mice. B–D, ADE. B, C, A 2 week alcohol deprivation period induced a reduced ADE in GluA1D1CreERT2 mice during the first 4 h of reexposure compared with wild-type mice. B, Ethanol intake in g/kg per 4 h time blocks. C, Percentage change from baseline. D, In contrast, administration of the AMPAR potentiator aniracetam (1.5 μg/0.5 μl) into the NAc (n = 7) 60 min before the ADE test in wild-type mice increased the ethanol intake during the first 4 h after reintroduction of the ethanol bottle. Data are mean ± SEM. #p < 0.01 versus baseline. *p < 0.01 versus wild-type. Arrow indicates aniracetam infusion.
Assuming that GluA1 in DAergic and D1-expressing neurons is similarly required for alcohol relapse behavior, acute potentiation of AMPAR activity is expected to increase relapse-like behavior. For this rescue experiment, we tested the effect of pharmacological potentiation of the AMPAR in the VTA (Fig. 7D) and NAc (Fig. 8D). Wild-type mice received either an intra-VTA or intra-NAc infusion of 1.5 μg/0.5 μl aniracetam or a vehicle infusion 60 min before the ADE test. In all treatment groups, a period of deprivation induced a significant increase in ethanol intake during the first 4 h of reexposure (Fig. 7D; two-way ANOVA, Deprivation × Phase interaction effect, F(5,72) = 23.83; p < 0.001; Fig. 8D; F(5,72) = 37.4; p < 0.001). However, aniracetam infusion into VTA and NAc led to a higher ethanol intake compared with control mice during this 4 h period (Fig. 7D; two-way ANOVA, Deprivation × Treatment interaction effect, F(1,72) = 6.82; p < 0.05; Fig. 8D; F(1,72) = 4.01; p < 0.05).
We did not observe any side effects after aniracetam treatment. Aniracetam microinfusion into VTA and NAc had no effect on water intake (two-way ANOVA, Treatment × Deprivation interaction effect, VTA: F(1,72) = 0.02, p = 0.88; NAc: F(1,72) = 0.38, p = 0.54) or locomotor activity (Treatment × Deprivation interaction effect, VTA: F(1,72) = 2.51, p = 0.12; NAc: F(1,72) = 0.41, p = 0.53) (data not shown).
An acute potentiation of AMPAR activity should also overcome the deficits in the relapse behavior seen in GluA1DATCreERT2 and GluA1D1CreERT2 mice. For this proof-of-concept experiment, mutant mice were challenged with a systemic injection of 0 or 5 mg/kg aniracetam 30 min before the ADE test (Fig. 9). In GluA1DATCreERT2 mice, aniracetam treatment induced a significant increase in ethanol intake after a period of abstinence compared with vehicle-treated mutant mice (Fig. 9A). This treatment effect was observable during the first 4 h after ethanol reexposure but was most pronounced during the following, second 4 h period (two-way ANOVA, Deprivation × Treatment interaction effect, F(1,60) = 8.41, p < 0.01). Similarly, in GluA1D1CreERT2 mice, aniracetam treatment induced a significant increase in ethanol intake during the first 4 h after a period of abstinence compared with vehicle-treated mutants (Fig. 9B; Deprivation × Treatment × Phase interaction effect, F(5,60) = 2.66, p < 0.05).
Figure 9.

Potentiation of AMPAR activity rescues the attenuated relapse behavior in GluA1DATCreERT2 and GluA1D1CreERT2 mice. A, Acute challenge of GluA1DATCreERT2mice with an intraperitoneal injection of the AMPAR potentiator aniracetam (5 mg/kg; n = 6/group) 30 min before the ADE test significantly increased the ethanol intake during the first 8 h after reintroducing the ethanol bottle compared with vehicle-treated mutant mice. B, In GluA1D1CreERT2 mice, an intraperitoneal injection of aniracetam (5 mg/kg; n = 6/group) induced elevated ethanol intake during the first 4 h after reexposure to ethanol compared with vehicle-treated mutant. Data are mean ± SEM. #p < 0.001 versus baseline. *p < 0.001 versus wild-type. Arrow indicates aniracetam administration.
Context- plus cue-induced reinstatement of ethanol-seeking behavior does not require GluN1 and GluA1 containing receptors in the mesolimbic system
GluN1 mutations
GluN1DATCreERT2, GluN1D1CreERT2, and corresponding wild-type littermates were trained across 22 daily 30 min sessions to lever press for ethanol with the presentation of specific cues predictive of ethanol availability (data not shown). During baseline responding (calculated as the mean of the last three conditioning sessions), there were no genotype differences (GluN1DATCreERT2: t(18) = −0.04; p = 0.97; GluN1D1CreERT2: t(23) = 1.61, p = 0.12) (Fig. 10A,B). Then, all mice underwent 9 daily extinction sessions of 30 min in a different context from alcohol-reinforced responding, where lever press responding for ethanol was extinguished by withholding further drug delivery and response-contingent cue presentation. One day after the last extinction session, mice were tested for context- plus cue-induced reinstatement of ethanol-seeking (Fig. 10A,B). The context and cue presentation significantly reinstated extinguished ethanol-seeking behavior almost identically in all groups (Fig. 10A; two-way ANOVA, Cue effect, F(2,36) = 21.93, p < 0.001; Genotype effect, F(1,18) = 0.06, p = 0.82; Figure 10B; Cue effect, F(2,46) = 24.29, p < 0.001; Genotype effect, F(1,23) = 3.93, p = 0.06). Because baseline responding of GluN1D1CreERT2 was lower, although not significant, compared with wild-type mice, the percentage change of lever responding during extinction and reinstatement compared with baseline was calculated (data not shown) and revealed that both genotypes reinstated ethanol-seeking behavior in a similar way (two-way ANOVA, Cue effect, F(1,23) = 30.22, p < 0.001; Genotype effect, F(1,23) = 0.02, p = 0.88).
Figure 10.

Context- plus cue-induced reinstatement of ethanol seeking-behavior is not mediated by GluN1-containing neurons within the mesolimbic system. A, B, In operant ethanol (10%) self-administration, GluN1DATCreERT2 (n = 10), GluN1D1CreERT2 (n = 10), and respective wild-type control (n = 10 or n = 15) groups did not differ in the lever pressing behavior during baseline or extinction sessions. Compared with extinction, the reinstatement of ethanol seeking behavior triggered by the presentation of the ethanol-associated cues was similar in all groups. C, Treatment of wild-type mice (n = 7/group) with the NMDAR antagonist memantine (25 mg/kg, i.p.) did not affect context- plus cue-induced reinstatement. Data are mean ± SEM. #p < 0.05 versus extinction.
Supporting these results, pharmacological blockade of the NMDAR did not alter context- plus cue-induced reinstatement. Thus, when wild-type mice were challenged with an intraperitoneal injection of 0 or 25 mg/kg memantine 30 min before the test (Fig. 10C), both treatment groups reinstated ethanol-seeking behavior in a similar way (two-way ANOVA, Cue effect, F(2,24) = 18.55, p < 0.001; Treatment effect, F(1,12) = 0.22, p = 0.65).
GluA1 mutations
GluA1DATCreERT2, GluA1D1CreERT2, and the respective wild-type mice were trained across 30 or 16, respectively, daily 30 min session to lever press for ethanol with the presentation of specific cues predictive of ethanol availability (data not shown). Statistical analysis of baseline responding (calculated as the mean of the last three conditioning sessions) did not reveal a Genotype effect (GluA1DATCreERT2: t(14) = 1.52, p = 0.15; GluA1D1CreERT2: t(15) = 0.32, p = 0.76) (Fig. 11A,B). During 9 daily 30 min extinction sessions, all groups reduced responding on the active lever and the presentation of the conditioned cues significantly reinstated extinguished ethanol-seeking behavior in all groups (11A; Cue effect, F(2,28) = 21.69, p < 0.001; 11B; Cue effect, F(2,30) = 16.94, p < 0.001). Ethanol-seeking behavior was not different between GluA1D1CreERT2 and wild-type mice (Fig. 11B; Genotype effect, F(1,15) = 0.01, p = 0.92). However, two-way ANOVA revealed a genotype differences between GluA1DATCreERT2 and wild-type mice (Fig. 11A; F(1,14) = 6.68, p < 0.05). Because GluA1DATCreERT2 showed a lower, although nonsignificant, baseline lever responding, the percentage change of lever responding during extinction and reinstatement compared with baseline was calculated and should in this case be considered as a better indicator for the ethanol-seeking behavior (data not shown). Two-way ANOVA revealed that both genotypes reinstated ethanol-seeking behavior in a similar way (two-way ANOVA, Cue effect, F(1,14) = 28.36, p < 0.001; Genotype effect, F(1,14) = 1.1, p = 0.31).
Figure 11.

Context- plus cue-induced reinstatement of ethanol seeking-behavior is not mediated by GluA1 containing neurons within the mesolimbic system. A, B, In operant ethanol (10%) self-administration, GluA1DATCreERT2 (n = 6), GluA1D1CreERT2 (n = 7), and respective wild-type control (n = 10) groups did not differ in the lever pressing behavior during baseline or extinction sessions. Compared with extinction, the reinstatement of ethanol seeking behavior triggered by the presentation of the ethanol-associated cues was similar in all groups. C, Treatment of wild-type (n = 7/group) mice with the AMPAR modulator aniracetam (5 mg/kg, i.p.) had no effect on context- plus cue-induced reinstatement. Data are mean ± SEM. #p < 0.05 versus extinction.
Furthermore, ethanol-seeking behavior was not affected by the pharmacological potentiation of the AMPAR activity during the context- plus cue-induced reinstatement test. Thus, when wild-type mice were challenged systemically with 0 or 5 mg/kg aniracetam 30 min before the test (Fig. 11C), both treatment groups reinstated extinguished ethanol-seeking behavior in a similar way (two-way ANOVA, Cue effect, F(2,24) = 20.66, p < 0.001; Treatment effect, F(1,12) = 0.17, p = 0.69).
Discussion
In this study, we demonstrate that DA neurons as well as D1-MSNs and their glutamatergic input act in concert to influence alcohol relapse responses. In particular, we show that GluN1 and GluA1 receptor subunits onto DAT- and D1R-expressing neurons play an important role in the ADE, the increase in alcohol intake after a period of abstinence. Thus, whereas initiation and maintenance of voluntary alcohol consumption in the home cage were normal in all four tested mutant lines, the ADE was attenuated in GluN1DATCreERT2, GluN1D1CreERT2, GluA1DATCreERT2, and GluA1D1CreERT2 mice. We further validated these results by showing similar reductions in the ADE after site-specific infusion of the NMDAR antagonist memantine into the NAc and VTA of control mice and an increase in the ADE using aniracetam to pharmacologically potentiate AMPAR activity in these brain regions. The ability of conditioned cues to reinstate alcohol-seeking behavior was normal in GluN1DATCreERT2, GluN1D1CreERT2, GluA1DATCreERT2, and GluA1D1CreERT2 mice. These negative findings were further validated by pharmacological manipulation of either NMDAR or AMPAR activity. In conclusion, relapse-like drinking as measured by the ADE is mediated by NMDARs and AMPARs within the mesolimbic system, whereas alcohol consumption, self-administration, and alcohol-seeking behavior, as measured by the context- plus cue-induced reinstatement procedure, are not influenced by these mesolimbic glutamate receptors. The possibility that DAT-expressing neurons from the substantia nigra and D1-expressing neurons from the dorsal striatum, respectively, contribute to the observed effects cannot be excluded. These observations suggest that NMDA- and AMPA-dependent mechanisms in cells other than DAT- or D1-expressing neurons may contribute to alcohol reinforcement.
To investigate the specific interaction between mesolimbic DA neurons and their glutamatergic input in behaviors relevant for alcohol addiction, we used genetically modified mice with an inducible knock-out of the NMDAR subunit GluN1 or AMPAR subunit GluA1 in DAT- or D1-expressing neurons. These four genetic mouse models have some advantages compared with other approaches for gene inactivation, allowing a more precise demonstration of the functional role regarding the gene of interest. First, these mutant models have high specificity of the deletion of GluN1 or GluA1 in DAT- and D1-expressing neurons, as shown here by our colocalization studies and previous work (Engblom et al., 2008). Second, the use of a temporally controlled gene deletion circumvents potential developmental compensatory mechanisms, which may offset the loss of the gene and consequently mask its functional role. Finally, most importantly, although global deletions of GluN1 or GluA1 result in neonatal death (Forrest et al., 1994) and hyperactivity (Cowen et al., 2003), respectively, the basic phenotype of our mutant mice was only slightly altered relative to wild-type controls.
Here we show that GluN1- and GluA1-dependent mechanisms in DAT- and D1-expressing neurons are important for relapse behavior, as demonstrated by the lack of ADE in GluN1DATCreERT2, GluN1D1CreERT2, GluA1DATCreERT2, and GluA1D1CreERT2 mice. In agreement with these findings, it was previously demonstrated that pharmacological blockade of NMDARs reduced the ADE in rats (Hölter et al., 1996; Vengeliene et al., 2005). However, evidence regarding AMPAR involvement in relapse is less clear. A global deletion of GluA1 failed to alter relapse-like drinking (Cowen et al., 2003), whereas a global deletion of GluA3 resulted in a lack of ADE in mice (Sanchis-Segura et al., 2006). Although these findings seem to contradict our results, an obvious difference between these previous studies and the present study is the use of noninducible conventional knock-outs. Furthermore, our results were confirmed by pharmacological experiments. We found a reduction in ADE in control mice infused with the NMDAR antagonist memantine into the VTA and NAc, a result previously demonstrated at the systemic level in rats (Hölter et al., 1996). In this regard, memantine has been suggested to be useful in the treatment of relapse behavior, and first clinical trials have already tested its therapeutic efficacy (Bisaga and Evans, 2004; Evans et al., 2007; Krupitsky et al., 2007). Consistent with these results, memantine administration has been shown to increase DA release within the mesolimbic system, suggesting interplay between memantine and the DAergic system (Spanagel et al., 1994). We further showed that treatment with the positive allosteric modulator aniracetam, which potentiates AMPAR activity and increases alcohol-seeking in rats (Tang et al., 1991; Cannady et al., 2013), but has not been used before in mice, potentiated the ADE in control mice during the first 4 h of alcohol resumption. The increased relapse-like drinking behavior likely depends on AMPAR activity in both the VTA and NAc, as local aniracetam administration in these brain areas similarly heightened the relapse-like drinking behavior. Pharmacological blockade on the systemic level of these receptors has been previously shown to reduce the ADE in rats (Sanchis-Segura et al., 2006). In further support of these findings, we demonstrated that an enhancement of AMPAR activity by aniracetam treatment rescued the attenuated relapse-like drinking behavior in GluA1DATCreERT2 and GluA1D1CreERT2 mice.
Interestingly, central treatments into the VTA, regardless of the target receptor, seem to be less effective in reducing the ADE compared with intra-NAc infusions. At least three possibilities may account for this differential effect in the magnitude of memantine and aniracetam in VTA versus NAc. First, the higher effect observed in the NAc may be due to higher receptor density in the NAc versus the VTA (http://mouse.brain-map.org/). Second, the VTA may have a less prominent role in driving the ADE than the NAc. However, this possibility is unlikely because such a difference in the ADE magnitude was not observed in mice with mutations in VTA compared with NAc. Finally, alcohol exposure and abstinence may induce differential long-term alterations in glutamate receptors or synaptic plasticity between the two regions. Unfortunately, our data cannot support or exclude one or another of these hypotheses. Further neuroanatomical and electrophysiological experiments are warranted to answer these questions.
Other key findings of our study are that mesolimbic GluN1 and GluA1 receptors (1) are not required for the initiation and maintenance of voluntary alcohol consumption in the home cage, (2) are not required for operant alcohol self-administration, and (3) do not influence alcohol-seeking responses in the context- plus cue-induced reinstatement procedure. In support of these findings are previous studies that have suggested a role of kainate, but not AMPA, receptors in mediating the reinforcing properties of alcohol (Stephens and Brown, 1999; Czachowski et al., 2012; Wang et al., 2012). However, our findings may be surprising in light of previous reports showing that pharmacological blockade of NMDARs decreases operant self-administration in rats (Sabino et al., 2013; Jeanblanc et al., 2014; Alaux-Cantin et al., 2015; but see also Piasecki et al., 1998). Furthermore, specific NMDAR blockade in the NAc has been found to reduce operant responding for alcohol (Rassnick et al., 1992). NMDAR antagonists might reduce alcohol operant self-administration by substituting for alcohol (Hölter et al., 2000), or impairing behavior by inducing dissociative states or sedation (Holmes et al., 2013). Nevertheless, our present study differs fundamentally from these previous reports via selective targeting of glutamate receptors in either DAT- or D1-expressing neurons.
The presentation of conditioned stimuli reinstated extinguished alcohol-seeking behavior in all four transgenic models. Consistent with this is the fact that memantine treatment had no effect on alcohol-seeking behavior, a finding supported by previous studies showing similar results with other NMDAR antagonists (Bäckström and Hyytiä, 2004; Alaux-Cantin et al., 2015). Furthermore, we also showed that pharmacological enhancement of AMPAR activity by aniracetam did not alter alcohol-seeking responses. In contrast to this finding, pharmacological blockade of AMPAR has been reported to reduce alcohol-seeking (Bäckström and Hyytiä, 2004; Sanchis-Segura et al., 2006), and pharmacological enhancement of AMPAR activity to increase alcohol-seeking in rats (Cannady et al., 2013). In addition to methodological differences, the apparent discrepancy in these findings may be due to the fact that pharmacological agents targeting AMPAR typically do not differentiate well between kainate receptors and AMPARs and show no subunit selectivity (Stephens and Brown, 1999; Natale et al., 2006), suggesting the potential involvement of other AMPAR subunits. Consistent with this explanation, mice with a global knock-out of Gria3 have been demonstrated to lack an alcohol-seeking response (Sanchis-Segura et al., 2006).
In conclusion, the present study demonstrates that glutamate receptors in DA and D1-MSNs are involved in the mechanism underlying the ADE, the increase in alcohol intake after a period of abstinence. The NMDAR subunit GluN1 and AMPAR subunit GluA1 in DAT- or D1-expressing neurons are proposed to influence alcohol relapse. Together, these findings support the assumption of a dysregulation of the glutamatergic system in the development of alcohol addiction (Tsai et al., 1995; Krystal et al., 2003; Gass and Olive, 2008; Holmes et al., 2013) and provide a neuroanatomical substrate for relapse behavior. Furthermore, it is noteworthy that relapse-like behavior, as measured by the ADE, can be clearly differentiated on the molecular level from alcohol-seeking responses triggered by environmental stimuli measured by the context- plus cue-induced reinstatement procedure. Medications targeting glutamatergic neurotransmission by selective inactivation of these glutamate receptors may have therapeutic efficacy. In this respect, more clinical trials using the NMDAR antagonist memantine (Evans et al., 2007; Spanagel and Vengeliene, 2013) are encouraged to fully evaluate its therapeutic efficacy. Further studies are warranted to specifically define how NMDARs and AMPARs interact within the mesolimbic system to mediate these responses.
Footnotes
This work was supported by the Bundesministerium für Bildung und Forschung (e:Med program; FKZ:01ZX1311A; Spanagel et al., 2013) to R.S., by a grant from the Deutsche Forschungsgemeinschaft (Grant ID SBF1158, B04) to R.S. and A.B, the Spanish Ministry of Education and Science Grant BFU-2012-38348, and Junta de Comunidades de Castilla-La Mancha Grant PPII-2014-005-P.
The authors declare no competing financial interests.
References
- Alaux-Cantin S, Buttolo R, Houchi H, Jeanblanc J, Naassila M. Memantine reduces alcohol drinking but not relapse in alcohol-dependent rats. Addict Biol. 2015;20:890–901. doi: 10.1111/adb.12177. [DOI] [PubMed] [Google Scholar]
- Bachteler D, Economidou D, Danysz W, Ciccocioppo R, Spanagel R. The effects of acamprosate and neramexane on cue-induced reinstatement of ethanol-seeking behavior in rat. Neuropsychopharmacology. 2005;30:1104–1110. doi: 10.1038/sj.npp.1300657. [DOI] [PubMed] [Google Scholar]
- Bäckström P, Hyytiä P. Ionotropic glutamate receptor antagonists modulate cue-induced reinstatement of ethanol-seeking behavior. Alcohol Clin Exp Res. 2004;28:558–565. doi: 10.1097/01.ALC.0000122101.13164.21. [DOI] [PubMed] [Google Scholar]
- Besheer J, Grondin JJ, Cannady R, Sharko AC, Faccidomo S, Hodge CW. Metabotropic glutamate receptor 5 activity in the nucleus accumbens is required for the maintenance of ethanol self-administration in a rat genetic model of high alcohol intake. Biol Psychiatry. 2010;67:812–822. doi: 10.1016/j.biopsych.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbao A. Advanced transgenic approaches to understand alcohol-related phenotypes in animals. In: Sommer W, Spanagel R, editors. Behavioral neurobiology of alcohol addiction. Berlin: Springer; 2013. pp. 271–311. [DOI] [PubMed] [Google Scholar]
- Bisaga A, Evans SM. Acute effects of memantine in combination with alcohol in moderate drinkers. Psychopharmacology. 2004;172:16–24. doi: 10.1007/s00213-003-1617-5. [DOI] [PubMed] [Google Scholar]
- Bossert JM, Marchant NJ, Calu DJ, Shaham Y. The reinstatement model of drug relapse: recent neurobiological findings, emerging research topics, and translational research. Psychopharmacology. 2013;229:453–476. doi: 10.1007/s00213-013-3120-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannady R, Fisher KR, Durant B, Besheer J, Hodge CW. Enhanced AMPA receptor activity increases operant alcohol self-administration and cue-induced reinstatement. Addict Biol. 2013;18:54–65. doi: 10.1111/adb.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen MS, Schroff KC, Gass P, Sprengel R, Spanagel R. Neurobehavioral effects of alcohol in AMPA receptor subunit (GluR1) deficient mice. Neuropharmacology. 2003;45:325–333. doi: 10.1016/S0028-3908(03)00174-6. [DOI] [PubMed] [Google Scholar]
- Czachowski CL, Delory MJ, Pope JD. Behavioral and neurotransmitter specific roles for the ventral tegmental area in reinforcer-seeking and intake. Alcohol Clin Exp Res. 2012;36:1659–1668. doi: 10.1111/j.1530-0277.2012.01774.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Stewart J. Reinstatement of cocaine-reinforced responding in the rat. Psychopharmacology. 1981;75:134–143. doi: 10.1007/BF00432175. [DOI] [PubMed] [Google Scholar]
- Eisenhardt M, Leixner S, Spanagel R, Bilbao A. Assessing alcohol drinking patterns in mice. Addict Biol. 2015 doi: 10.1111/adb.12325. in press. [DOI] [PubMed] [Google Scholar]
- Engblom D, Bilbao A, Sanchis-Segura C, Dahan L, Perreau-Lenz S, Balland B, Parkitna JR, Luján R, Halbout B, Mameli M, Parlato R, Sprengel R, Lüscher C, Schütz G, Spanagel R. Glutamate receptors on dopamine neurons control the persistence of cocaine seeking. Neuron. 2008;59:497–508. doi: 10.1016/j.neuron.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Erdmann G, Schütz G, Berger S. Inducible gene inactivation in neurons of the adult mouse forebrain. BMC Neurosci. 2007;8:63. doi: 10.1186/1471-2202-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans SM, Levin FR, Brooks DJ, Garawi F. A pilot double-blind treatment trial of memantine for alcohol dependence. Alcohol Clin Exp Res. 2007;31:775–782. doi: 10.1111/j.1530-0277.2007.00360.x. [DOI] [PubMed] [Google Scholar]
- Forrest D, Yuzaki M, Soares HD, Ng L, Luk DC, Sheng M, Stewart CL, Morgan JI, Connor JA, Curran T. Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron. 1994;13:325–338. doi: 10.1016/0896-6273(94)90350-6. [DOI] [PubMed] [Google Scholar]
- Gass JT, Olive MF. Glutamatergic substrates of drug addiction and alcoholism. Biochem Pharmacol. 2008;75:218–265. doi: 10.1016/j.bcp.2007.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A, Spanagel R, Krystal JH. Glutamatergic targets for new alcohol medications. Psychopharmacology. 2013;229:539–554. doi: 10.1007/s00213-013-3226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hölter SM, Danysz W, Spanagel R. Evidence for alcohol anti-craving properties of memantine. Eur J Pharmacol. 1996;314:R1–R2. doi: 10.1016/S0014-2999(96)00670-X. [DOI] [PubMed] [Google Scholar]
- Hölter SM, Danysz W, Spanagel R. Novel uncompetitive N-methyl-D-Aspartate (NMDA)-receptor antagonist MRZ 2/579 suppresses ethanol intake in long-term ethanol-experienced rats and generalizes to ethanol cue in drug discrimination procedure. J Pharmacol Exp Ther. 2000;292:545–552. [PubMed] [Google Scholar]
- Jeanblanc J, Coune F, Botia B, Naassila M. Brain-derived neurotrophic factor mediates the suppression of alcohol self-administration by memantine. Addict Biol. 2014;19:758–769. doi: 10.1111/adb.12039. [DOI] [PubMed] [Google Scholar]
- Krupitsky EM, NeznANOVA O, Masalov D, Burakov AM, Didenko T, RomANOVA T, Tsoy M, Bespalov A, Slavina TY, Grinenko AA. Effect of memantine on cue-induced alcohol craving in recovering alcohol-dependent patients. J Psychiatry. 2007;164:519–523. doi: 10.1176/ajp.2007.164.3.519. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Petrakis IL, Mason G, Trevisan L, D'Souza DC. N-methyl-d-aspartate glutamate receptors and alcoholism: reward, dependence, treatment, and vulnerability. Pharmacol Ther. 2003;99:79–94. doi: 10.1016/S0163-7258(03)00054-8. [DOI] [PubMed] [Google Scholar]
- Lüscher C. Drug-evoked synaptic plasticity causing addictive behavior. J Neurosci. 2013;33:17641–17646. doi: 10.1523/JNEUROSCI.3406-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, Lüscher C. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–1041. doi: 10.1038/nn.2367. [DOI] [PubMed] [Google Scholar]
- Narushima M, Uchigashima M, Hashimoto K, Watanabe M, Kano M. Depolarization-induced suppression of inhibition mediated by endocannabinoids at synapses from fast-spiking interneurons to medium spiny neurons in the striatum. Eur J Neurosci. 2006;24:2246–2252. doi: 10.1111/j.1460-9568.2006.05119.x. [DOI] [PubMed] [Google Scholar]
- Natale NR, Magnusson KR, Nelson JK. Can selective ligands for glutamate binding proteins be rationally designed? Curr Top Med Chem. 2006;6:823–847. doi: 10.2174/156802606777057535. [DOI] [PubMed] [Google Scholar]
- Niewoehner B, Single FN, Hvalby Ø, Jensen V, Meyer zum Alten Borgloh S, Seeburg PH, Rawlins JN, Sprengel R, Bannerman DM. Impaired spatial working memory but spared spatial reference memory following functional loss of NMDA receptors in the dentate gyrus. Eur J Neurosci. 2007;25:837–846. doi: 10.1111/j.1460-9568.2007.05312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkitna JR, Engblom D, Schütz G. Generation of Cre recombinase-expressing transgenic mice using bacterial artificial chromosomes. In: Kühn R, Wurst W, editors. Gene knockout protocols. New York: Humana; 2009. pp. 325–342. [DOI] [PubMed] [Google Scholar]
- Parkitna JR, Bilbao A, Rieker C, Engblom D, Piechota M, Nordheim A, Spanagel R, Schütz G. Loss of the serum response factor in the dopamine system leads to hyperactivity. FASEB J. 2010;24:2427–2435. doi: 10.1096/fj.09-151423. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Danysz W, Quack G. Memantine is a clinically well tolerated N-methyl-d-aspartate (NMDA) receptor antagonist: a review of preclinical data. Neuropharmacology. 1999;38:735–767. doi: 10.1016/S0028-3908(99)00019-2. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KB. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2001. [Google Scholar]
- Piasecki J, Koros E, Dyr W, Kostowski W, Danysz W, Bienkowski P. Ethanol-reinforced behaviour in the rat: effects of uncompetitive NMDA receptor antagonist, memantine. Eur J Pharmacol. 1998;354:135–143. doi: 10.1016/S0014-2999(98)00442-7. [DOI] [PubMed] [Google Scholar]
- Rassnick S, Pulvirenti L, Koob GF. Oral ethanol self-administration in rats is reduced by the administration of dopamine and glutamate receptor antagonists into the nucleus accumbens. Psychopharmacology. 1992;109:92–98. doi: 10.1007/BF02245485. [DOI] [PubMed] [Google Scholar]
- Sabino V, Narayan AR, Zeric T, Steardo L, Cottone P. mTOR activation is required for the anti-alcohol effect of ketamine, but not memantine, in alcohol-preferring rats. Behav Brain Res. 2013;247:9–16. doi: 10.1016/j.bbr.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salimov RM, Salimova NB. The alcohol-deprivation effect in hybrid mice. Drug Alcohol Depend. 1993;32:187–191. doi: 10.1016/0376-8716(93)80012-4. [DOI] [PubMed] [Google Scholar]
- Salimov R, Salimova N, Ratkin A, Shvets L, Maisky A. Genetic control of alcohol deprivation effect in congenic mice. Alcohol. 1995;12:469–474. doi: 10.1016/0741-8329(95)00033-N. [DOI] [PubMed] [Google Scholar]
- Sanchis-Segura C, Spanagel R. Behavioural assessment of drug reinforcement and addictive features in rodents: an overview. Addict Biol. 2006;11:2–38. doi: 10.1111/j.1369-1600.2006.00012.x. [DOI] [PubMed] [Google Scholar]
- Sanchis-Segura C, Borchardt T, Vengeliene V, Zghoul T, Bachteler D, Gass P, Sprengel R, Spanagel R. Involvement of the AMPA receptor GluR-C subunit in alcohol-seeking behavior and relapse. J Neurosci. 2006;26:1231–1238. doi: 10.1523/JNEUROSCI.4237-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair JD, Senter RJ. Increased preference for ethanol in rats following alcohol deprivation. Psychonom Sci. 1967;8:11–12. doi: 10.3758/BF03330642. [DOI] [Google Scholar]
- Spanagel R. Alcoholism: a systems approach from molecular physiology to addictive behavior. Physiol Rev. 2009;89:649–705. doi: 10.1152/physrev.00013.2008. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Vengeliene V. New pharmacological treatment strategies for relapse prevention. In: Sommer WH, Spanagel R, editors. Behavioral neurobiology of alcohol addiction. Berlin: Springer; 2013. pp. 583–609. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Eilbacher B, Wilke R. Memantine-induced dopamine release in the prefrontal cortex and striatum of the rat: a pharmacokinetic microdialysis study. Eur J Pharmacol. 1994;262:21–26. doi: 10.1016/0014-2999(94)90023-X. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Durstewitz D, Hansson A, Heinz A, Kiefer F, Köhr G, Matthäus F, Nöthen MM, Noori HR, Obermayer K, Rietschel M, Schloss P, Scholz H, Schumann G, Smolka M, Sommer W, Vengeliene V, Walter H, Wurst W, Zimmermann US, et al. A systems medicine research approach for studying alcohol addiction. Addict Biol. 2013;18:883–896. doi: 10.1111/adb.12109. [DOI] [PubMed] [Google Scholar]
- Stephens DN, Brown G. Disruption of operant oral self-administration of ethanol, sucrose, and saccharin by the AMP/Kainate antagonist, NBQX, but not the AMPA antagonist, GYKI 52466. Alcohol Clin Exp Res. 1999;23:1914–1920. doi: 10.1111/j.1530-0277.1999.tb04091.x. [DOI] [PubMed] [Google Scholar]
- Tang CM, Shi QY, Katchman A, Lynch G. Modulation of the time course of fast EPSCs and glutamate channel kinetics by aniracetam. Science. 1991;254:288–290. doi: 10.1126/science.1681589. [DOI] [PubMed] [Google Scholar]
- Tsai G, Gastfriend DR, Coyle JT. The glutamatergic basis of human alcoholism. J Psychiatry. 1995;152:332. doi: 10.1176/ajp.152.3.332. [DOI] [PubMed] [Google Scholar]
- Vengeliene V, Bachteler D, Danysz W, Spanagel R. The role of the NMDA receptor in alcohol relapse: a pharmacological mapping study using the alcohol deprivation effect. Neuropharmacology. 2005;48:822–829. doi: 10.1016/j.neuropharm.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Vengeliene V, Bilbao A, Molander A, Spanagel R. Neuropharmacology of alcohol addiction. Br J Pharmacol. 2008;154:299–315. doi: 10.1038/bjp.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vengeliene V, Bilbao A, Spanagel R. The alcohol deprivation effect model for studying relapse behavior: a comparison between rats and mice. Alcohol. 2014;48:313–320. doi: 10.1016/j.alcohol.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Wang J, Ben Hamida S, Darcq E, Zhu W, Gibb SL, Lanfranco MF, Carnicella S, Ron D. Ethanol-mediated facilitation of AMPA receptor function in the dorsomedial striatum: implications for alcohol drinking behavior. J Neurosci. 2012;32:15124–15132. doi: 10.1523/JNEUROSCI.2783-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Cheng Y, Wang X, Roltsch Hellard E, Ma T, Gil H, Ben Hamida S, Ron D. Alcohol elicits functional and structural plasticity selectively in dopamine D1 receptor-expressing neurons of the dorsomedial striatum. J Neurosci. 2015;35:11634–11643. doi: 10.1523/JNEUROSCI.0003-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, Kaiser KM, Köster HJ, Borchardt T, Worley P, Lübke J, Frotscher M, Kelly PH, Sommer B, Andersen P, Seeburg PH, Sakmann B. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284:1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]