

Abstract
Functional recovery of injured peripheral neurons often remains incomplete, but the clinical outcome can be improved by increasing the axonal growth rate. Adult transgenic GSK3αS/A/βS/A knock-in mice with sustained GSK3 activity show markedly accelerated sciatic nerve regeneration. Here, we unraveled the molecular mechanism underlying this phenomenon, which led to a novel pharmacological approach for the promotion of functional recovery after nerve injury. In vitro and in vivo analysis of GSK3 single knock-in mice revealed the unexpected contribution of GSK3α in addition to GSK3β, as both GSK3S/A knock-ins improved axon regeneration. Moreover, growth stimulation depended on overall GSK3 activity, correlating with increased phosphorylation of microtubule-associated protein 1B and reduced microtubule detyrosination in axonal tips. Pharmacological inhibition of detyrosination by parthenolide or cnicin mimicked this axon growth promotion in wild-type animals, although it had no effect in GSK3αS/A/βS/A mice. These results support the conclusion that sustained GSK3 activity primarily targets microtubules in growing axons, maintaining them in a more dynamic state to facilitate growth. Accordingly, further manipulation of microtubule stability using either paclitaxel or nocodazole compromised the effects of parthenolide. Strikingly, either local or systemic application of parthenolide in wild-type mice dose-dependently accelerated in vivo axon regeneration and functional recovery similar to GSK3αS/A/βS/A mice. Thus, reducing microtubule detyrosination in axonal tips may be a novel, clinically suitable strategy to treat nerve damage.
SIGNIFICANCE STATEMENT Peripheral nerve regeneration often remains incomplete, due to an insufficient growth rate of injured axons. Transgenic mice with sustained GSK3 activity showed markedly accelerated nerve regeneration upon injury. Here, we identified the molecular mechanism underlying this phenomenon and provide a novel therapeutic principle for promoting nerve repair. Analysis of transgenic mice revealed a dependence on overall GSK3 activity and reduction of microtubule detyrosination in axonal tips. Pharmacological inhibition of detyrosination by parthenolide fully mimicked this axon growth promotion in wild-type mice. Strikingly, local or systemic treatment with parthenolide in vivo markedly accelerated axon regeneration and functional recovery. Thus, pharmacological inhibition of microtubule detyrosination may be a novel, clinically suitable strategy for nerve repair with potential relevance for human patients.
Keywords: axon regeneration, DRG neuron, GSK3, microtubules, PNS, therapy
Introduction
Axon regeneration and optimal functional recovery upon nerve injury require a sufficient intrinsic ability of neurons to regrow axons, a growth-supportive environment and reconnection with the original target tissues before postsynaptic structures are lost. Although injured axons of the adult PNS show generally much greater regeneration potential than their counterparts of the CNS, functional recovery is nevertheless often incomplete. Poor regeneration is particularly observed after complete nerve transection or when axons need to travel long distances to reach their target. A decline in axonal growth rate as well as in neurotrophic support from Schwann cells over time combined with axonal misguidance have been made accountable for incomplete functional recovery that is, for example, observed after sciatic nerve or brachial plexus injuries (Nguyen et al., 2002; Witzel et al., 2005; Höke, 2006; Scheib and Höke, 2013). Although much progress has been made in recent years to understand the molecular mechanisms underlying regenerative failure, clinically relevant applications to improve the outcome upon peripheral nerve injuries have mostly been made in the area of surgical techniques. For this reason, the development of novel therapeutic approaches that significantly speed up the axonal growth rate is highly desirable from a clinical perspective (Ma et al., 2011).
We recently used GSK3αS21A/βS9A double knock-in (GSK3αS/A/βS/A) mice to investigate the role of protein kinase B (AKT)-modulated GSK3 activity in PNS axon regeneration (Gobrecht et al., 2014). In these mice, serine 21 of GSK3α and serine 9 of GSK3β, respectively, are substituted by alanine, preventing inhibitory GSK3 phosphorylation by AKT (McManus et al., 2005). These mutations render GSK3 constitutively active, without changing its physiological expression level or using pharmacological compounds. Unexpectedly, this global increase of GSK3 activity markedly facilitated axon growth of adult DRG neurons in culture. Moreover, in vivo axon regeneration upon sciatic nerve injury was strikingly accelerated, leading to improved motor and sensory recovery. These results strongly imply that translational approaches aiming to either maintain global GSK3 activity or to directly modulate GSK3 downstream targets in injured DRG neurons may be clinically applicable strategies to promote nerve regeneration.
GSK3 itself has a plethora of substrates and regulates many cellular processes. In particular, microtubule-associated proteins (MAPs), such as APC, CLASP, CRMP2, and MAP1B, have been implicated in axon growth regulation (Hur et al., 2012). Most of these MAPs (e.g., CRMP2) require priming by a distinct kinase before phosphorylation by GSK3 (Seira and Del Río, 2014) and are inhibited by GSK phosphorylation. MAP1B, on the other hand, is activated by GSK3-mediated phosphorylation at tyrosine 1265 without the need of prior priming (Lucas et al., 1998; Frame et al., 2001; Cole et al., 2004a; Trivedi et al., 2005) and can therefore be used as an indicator for GSK3 activity (Gobrecht et al., 2014). In non-neuronal cells, phosphorylated MAP1B binds to tyrosinated microtubules and thereby maintains a pool of dynamically unstable microtubules. Reduction of phospho-MAP1B levels upon GSK3 inhibition dramatically increases the numbers of stable (detyrosinated) microtubules in axonal growth cones (Owen and Gordon-Weeks, 2003). For this reason, it is generally assumed that GSK3-phosphorylated MAP1B promotes axonal growth in embryonic neurons by maintaining microtubules in a dynamically unstable state (Lucas et al., 1998; Goold et al., 1999; Owen and Gordon-Weeks, 2003). However, the detailed mechanism(s) underlying the regulation of MAP1B mediated microtubule detyrosination is not clearly understood. Moreover, it has not yet been directly shown whether inhibition of microtubule detyrosination would promote axon growth.
The current study investigated the molecular mechanism underlying improved axon regeneration in GSK3αS/A/βS/A mice. We found that growth stimulation depended on overall GSK3 activity in transgenic animals and led to reduced microtubule detyrosination in axonal tips. Pharmacological inhibition of detyrosination fully mimicked this axon growth promotion in wild-type (wt) animals, supporting the conclusion that sustained GSK3 activity primarily targets microtubules in growing axons and maintains them in a more dynamic state, thereby facilitating axon growth. Strikingly, either local or even systemic application of parthenolide in wt mice dose-dependently accelerated in vivo axon regeneration and recovery similar to GSK3αS/A/βS/A mice. Thus, reducing microtubule detyrosination in axonal tips may be a novel, clinically suitable strategy to promote nerve repair with potential relevance for human patients.
Materials and Methods
Surgical procedures.
All animal protocols adhered to respective animal care guidelines and were approved by the local authorities (LANUV Recklinghausen). Male and female adult (8–12 weeks) wt, GSK3αS21A, GSK3βS9A, and GSK3αS21A/GSK3βS9A mice of C57BL/6,129/Ola genetic background were maintained in cages with 1–5 animals on a 12 h light/dark cycle with ad libitum access to food and water. The expression levels of either naive or mutant GSK3α and GSK3β were similar in all genotypes (data not shown). Sciatic nerve crush (SNC) was performed as described previously (Gobrecht et al., 2014). In brief, animals were anesthetized by intraperitoneal injections of ketamine (80–100 mg/kg, Pfizer) and xylazine (10–15 mg/kg, Bayer), and a skin incision of ∼10 mm was made over the gluteal region. The ischiocrural musculature was carefully spread with minimal tissue damage to expose the right sciatic nerve from the sciatic notch to the point of trifurcation. The crush injury was performed for 30 s proximal to the tibial and peroneal divisions using carbon-tainted Dumont #5 forceps (Hermle) to mark the crush site. The skin was closed using 6–0 suture stitches. Injections into the injury site of the sciatic nerve were performed directly after nerve crush using a Nanoject II injector (Drummond Scientific) to apply five consecutive 50 nl injections with a speed of 23 nl/s and 30 s intervals between injections.
DRG neuron cultures and immunocytochemical stainings.
DRG neurons were harvested from adult wt, GSK3αS21A, GSK3βS9A, and GSK3αS21A/GSK3βS9A mice as described previously (Gobrecht et al., 2014). Isolated DRG (T8-L6) were incubated in 0.25% trypsin/EDTA (GE Healthcare) and 0.3% collagenase Type IA (Sigma) in DMEM (Invitrogen) at 37°C and 5% CO2 for 45 min and mechanically dissociated. Cells were resuspended in DMEM containing 10% FBS (GE Healthcare) and penicillin/streptomycin (500 U/ml; Merck, Millipore) and cultured at 37°C and 5% CO2 on poly-d-lysine (0.1 mg/ml, molecular weight <300,000 kDa; Sigma) and laminin (20 μg/ml; Sigma)-coated 96 well plates (Nunc). Cells were either treated with vehicle, 5 μm of the selective GSK3 inhibitor SB216763 (Coghlan et al., 2000; Eldar-Finkelman and Martinez, 2011), 0.1–100 nm parthenolide, 0.1–100 nm cnicin, 10 nm nocodazole, or 10 nm paclitaxel. Axonal growth was determined upon 48 h incubation by fixation in 4% PFA (Sigma) and immunocytochemical staining with antibodies against NeuN (1:2000; Abcam, ab177487) and βIII-tubulin (1:2000; Covance). Imaging and quantification of total axon length and neuron numbers per well were automatically performed with the Pathway 855 microscope system (BD Biosciences) and Attovision software, avoiding experimenter-induced quantification bias. Average axon length per neuron and neuron counts per experimental group were normalized to control groups. Data represent mean ± SEM of at least six replicate wells per experiment and three independent experiments. Significances of intergroup differences were evaluated using either one- or two-way ANOVA followed by the Holm–Sidak post hoc test.
The dynamics of microtubules in axon tips was evaluated after 3 d in culture using antibodies against βIII-tubulin (1:2000; Covance), pMAP1B (1:1000; Thermo Scientific), or detyrosinated tubulin (1:2000; Millipore). Axon tips were defined as the last 15 μm of βIII-tubulin-positive neurite extensions. Data represent mean ± SEM of three replicate wells with 30–60 tips per well from at least two independent experiments. Significances of intergroup differences were evaluated using either one- or two-way ANOVA followed by Holm–Sidak post hoc test.
Quantification of regenerating axons in the sciatic nerve and pMAP1B staining.
Sciatic nerves were isolated 3 d after SCN, postfixed in 4% PFA for 6 h, dehydrated in 30% sucrose overnight at 4°C, and embedded in Tissue-Tek (Sakura). Longitudinal and cross-sections (14 μm) were cut on a cryostat (Leica), thaw-mounted onto coated glass slides (Superfrost plus, Fisher), and stored at −20°C until further use. Cryosections were immunohistochemically stained with antibodies against the regeneration-associated protein SCG10 (1:1500; Novus Biologicals) (Shin et al., 2012) or phoshpo-MAP1B (1:500; Thermo Scientific). SCG10-positive axons were quantified at various points beyond the carbon-labeled injury site, as previously described (Gobrecht et al., 2014). Experimental groups comprise at least five animals and five different sections were analyzed per animal. Statistical significances of intergroup differences were evaluated using one-way ANOVA followed by the Holm–Sidak post hoc test.
Two-compartment cultures.
Two-compartment chambers (AXIS Axon Isolation Device, Millipore) were mounted on poly-d-lysine plus laminin-coated culture dishes according to the manufacturer's instructions. DRG neurons of adult mice were seeded in the somal compartment and cultured for 3 d to allow axon extension through the microchannels. Neurons were then axotomized by fast removal of the medium from the axonal compartment. Afterwards, medium containing either parthenolide (5 nm) or vehicle was applied to either the somal or the axonal compartment. Diffusion of parthenolide into the microchannels was prevented using a hydrostatic pressure difference between the somal and the axonal chambers. Regrowing neurons were fixed after 24 h with 4% PFA, and an average growth index was calculated by dividing the average length of βIII-tubulin-stained axons by the axon number.
Western blot assays.
Either L3/L4 DRG or ∼3 mm long pieces of sciatic nerves around the injury site from four mice per experimental group were combined in 100 μl lysis buffer (20 mm Tris-HCl, pH 7.5, 10 mm KCl, 250 mm sucrose, 10 mm NaF, 1 mm DTT, 0.1 mm Na3VO4, 1% Triton X-100, 0.1% SDS, and protease inhibitors; Merck, Millipore) and homogenized by sonification. To avoid a potential bias due to different numbers of regenerating axons in experimental groups we isolated tissue 5 d after injury. Lysates were centrifuged at 8000 × g for 10 min at 4°C, soluble proteins separated by SDS-PAGE and subsequently transferred to nitrocellulose membranes (Bio-Rad) according to standard protocols. Blots were blocked either in 5% dried milk or 5% BSA (Sigma) and incubated either with monoclonal antibodies against βIII-tubulin (1:2000; Covance), CRMP2 (1:1000; Abcam), phospho-CRMP2 (1:1000; Abcam), MAP1B (1:500; Abcam), or phospho-MAP1B (1:1000; Thermo Scientific) overnight at 4°C. Postnatal mouse brain lysate or HEK cells cotransfected with MAP1B and constitutively active GSK3 served as positive control for MAP1B and pMAP1B, whereas a lysate of HEK cells cotransfected with CRMP2 and constitutively active GSK3 was used as positive controls for CRMP2 and pCRMP2. All antibodies were detected with anti-rabbit or anti-mouse IgG secondary antibodies conjugated to HRP (1:80,000; Sigma). Antigen-antibody complexes were visualized using enhanced chemiluminescence substrate (Bio-Rad) on the Fluorchem E imaging system (ProteinSimple). Western blots were repeated at least twice.
RNA isolation and qRT-PCR.
DRG cultures were treated with DMSO, 100 nm NFκB inhibitor BMS 345541, 5 and 100 nm parthenolide, respectively, for 3 h at 37°C. Sciatic nerves were crushed and immediately injected either with vehicle (200 nl DMSO per injection), parthenolide (6.25 pg in 200 nl DMSO), or NFκB inhibitor BMS 345541 (50 ng in 200 nl DMSO). Four injections were performed into and 1, 2, and 3 mm proximal to the lesion site as longer sciatic nerve pieces were required for RNA isolation. At 3 h thereafter, 5 mm of proximal sciatic nerve was dissected from 3 mice per treatment. This time point was chosen to allow BMS 345541, which served as a positive control to inhibit NFκB activity. Total RNA was isolated using the RNeasy Mini kit (QIAGEN) according to the manufacturer's instructions. RNA was reverse transcribed using the SuperScript II reverse transcriptase kit (Invitrogen). NFκB inhibitor α (IκBα) and glyceraldehyde 3-phosphate dehydrogenase (gapdh) were amplified using respective QuantiTect primers (QIAGEN) and SYBR Green PCR Master Mix (Applied Biosystems) on an Applied Biosystems 7500 real-time PCR system (Thermo Fisher Scientific) using 45 amplification cycles. PCR specificity was verified with the dissociation curve analysis feature. IκBα mRNA expression levels in parthenolide- or BMS 345541-treated DRG cultures or sciatic nerves were quantified compared with vehicle controls using the ΔΔCt method. All reactions were performed in duplicate, and three independent samples were analyzed per experimental group.
α-Bungarotoxin (BTX) staining.
Mice were killed at 4 or 7 d after SNC, respectively, when first improvements of motor function were detected after parthenolide treatment and in GSK3 knock-in mice, respectively. The extensor hallucis longus muscle was dissected, fixed in 4% PFA for 1 h, and permeabilized in 2% Triton X/PBS overnight as previously described (Gobrecht et al., 2014). Axons were labeled with an antibody against heavy chain 200 kDa neurofilament (1:2000; Abcam) and Alexa488-conjugated secondary antibody. Reestablished synapses in neuromuscular endplates were visualized by incubation with Alexa594-conjugated α-BTX (1:1000; Invitrogen) for 1 h. Representative pictures of each experimental group were taken on an LSM510 confocal microscope (Zeiss).
Static sciatic index (SSI).
Motor recovery after SNC was determined by calculating the SSI of 10 GSK3αS21A, 10 GSK3βS9A, and 11 wt mice, as previously described (Baptista et al., 2007; Bobinski et al., 2011; Gobrecht et al., 2014). For experiments requiring local intraneural injections, 11 animals were treated with parthenolide and 11 mice with vehicle. At the same daytime (1:00–2:00 P.M.) of various days after SNC (0, 1, 4, 7, 9, 12, 14, and 21 d), mice were lifted from the ground to photograph the left and right hind feet, respectively. Toe spreading on contralateral (C, left) and ipsilateral (I, right) sides relative to the SNC was assessed by measuring paw length (PL) and distance between the first and fifth toe (FF). The SSI was then calculated using the previously described formula: SSI = 101.3 ((IFF − CFF)/CFF) − 54.03((IPL − CPL)/CPL) − 9.5 (Baptista et al., 2007). Data represent mean SSI ± SEM per experimental group. Statistical significances of intergroup differences were evaluated using two-way ANOVA followed by the Holm–Sidak post hoc test.
von Frey test.
Sensory recovery after SNC was determined at 0, 1, 4, 7, 14, and 21 d after SNC with the von Frey filament test in 10–11 animals per experimental group, as previously described (Gladman et al., 2012; Gobrecht et al., 2014). Tests were performed at the same time of day by the same experimenter. Mice were placed on an elevated metal grid (pore size: 2 mm) and allowed to acclimate for 15 min before testing. Starting with the smallest, differently sized, innocuous von Frey filaments (Muromachi Kikai) were consecutively poked into the ipsilateral hindpaw to elicit a positive response, which is indicated by sharp paw withdrawal. Statistical significances of intergroup differences were evaluated using two-way ANOVA followed by the Holm–Sidak post hoc test.
Results
Both isoforms contribute to GSK3S/A-mediated nerve regeneration
We previously showed that prevention of PI3K/AKT-triggered phosphorylation in GSK3αS/A/GSK3βS/A double knock-in mice markedly improves axonal growth of cultured adult DRG neurons and accelerates functional motor and sensory recovery after sciatic nerve injury (Gobrecht et al., 2014). To explore the molecular mechanism(s) underlying this phenomenon, we first addressed the question whether these observed effects were attributable to a specific GSK3 isoform. In DRG neuronal cultures from single knock-in mice, GSK3αS/A- as well as GSK3βS/A-expressing neurons showed ∼2-fold longer axon growth compared with wt controls (Fig. 1A,B). However, neither single knock-in reached the level of GSK3αS/A/GSK3βS/A neurons, indicating that both GSK3 isoforms contribute to the ∼3-fold enhanced axon regeneration of GSK3αS/A/GSK3βS/A neurons (Fig. 1A,B). Likewise, in vivo axon regeneration of the sciatic nerve 3 d after injury, visualized by SCG10 immunostaining on longitudinal sections (Fig. 1C), was significantly increased in both GSK3αS/A and GSK3βS/A mice relative to wt controls but remained below the level of GSK3αS/A/GSK3βS/A double knock-in animals (Fig. 1C,D). Similarly, neurofilament immunostaining on cross-sections revealed axons at 3 mm beyond the lesion site only in transgenic mice (Fig. 1E,F). Therefore, both GSK3α and GSK3β activities were required to achieve optimum axon regeneration.
Figure 1.
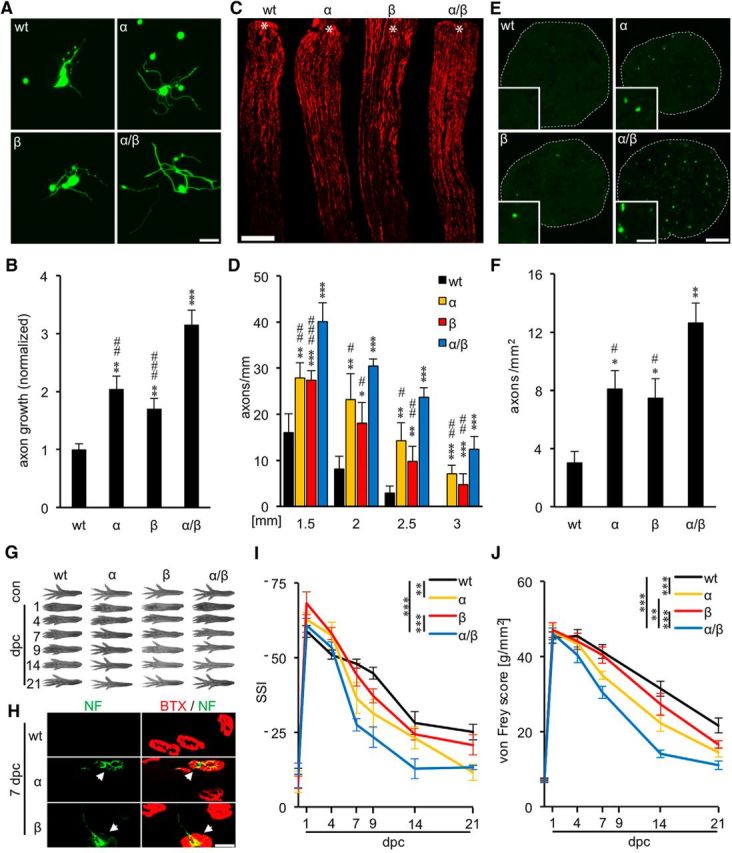
GSK3αS/A and GSK3βS/A each promote sciatic nerve regeneration. A, Representative pictures of DRG neurons isolated from adult wt, GSK3αS/A (α), GSK3βS/A (β), or GSK3αS/A/GSK3βS/A (α/β) mice, respectively, and stained for βIII-tubulin after 2 d in culture. Scale bar, 50 μm. B, Quantification of axon growth of cultures as depicted in A. Data from transgenic neurons were normalized to wt controls with an average axon length of 193 μm/neuron and represent mean ± SEM of three independent experiments. Treatment effects compared with wt: **p ≤ 0.01; ***p ≤ 0.001. Treatment effects compared with α/β: ##p ≤ 0.01; ###p ≤ 0.001. C, Longitudinal sections of sciatic nerves from adult wt, α, β, and α/β mice, respectively, stained for SCG10 3 d after SNC. Asterisks indicate the crush site. Scale bar, 500 μm. D, Quantification of regenerating axons at 1.5, 2, 2.5, and 3 mm beyond the injury site in wt and GSK3 mutant mice as described in C. Five different sections per animal were analyzed from 5 animals per genotype. Previously published, but concurrently evaluated data from α/β mice were included as reference (Gobrecht et al., 2014). Treatment effects compared with wt: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. Treatment effects compared with α/β: #p ≤ 0.05; ##p ≤ 0.01; ###p ≤ 0.001. Axon regeneration was not significantly different between α and β knock-in mice. E, Transverse sciatic nerve sections from adult wt, α, β, and α/β mice, respectively, 3 d after SNC at 3 mm distal to the lesion site. Insets, Single 200 kDa neurofilament (NF)-positive axons at higher magnification. Axons were only found in GSK3 transgenic animals. Scale bars: Whole nerve, 100 μm; Inset, 5 μm. F, Quantification of regenerating axons per mm2 in sciatic nerve sections of wt and GSK3 mutant mice as described in E. Three animals were analyzed per genotype. Treatment effects compared with wt: *p ≤ 0.05; **p ≤ 0.01. Treatment effects compared with α/β: #p ≤ 0.05. G, Representative pictures of right hindpaws of wt, α, β, and α/β mice before (con) and 1, 4, 7, 9, 14, and 21 d post SNC (dpc). H, Immunostaining of extensor hallucis longus muscle whole mounts with α-BTX (red) and neurofilament (NF, green) at 7 dpc. White arrows indicate reestablished synapses, which were only detected in α and β knock-in mice and not in wt controls at this time point (n = 3). Scale bar, 25 μm. I, Quantification of motor recovery in adult mice as depicted in G, using the SSI. Previously published, but concurrently evaluated data from α/β mice were included as reference (Gobrecht et al., 2014). At least 10 animals were analyzed per experimental group. Treatment effects: **p ≤ 0.01; ***p ≤ 0.001. J, Quantification of sensory recovery as determined by the von Frey test in wt, α, and β mice at 1, 4, 7, 14, and 21 dpc. Previously published, but concurrently evaluated data from α/β mice were included as reference (Gobrecht et al., 2014). Ten knock-in and 11 wt animals were analyzed per experimental group. Treatment effects: **p ≤ 0.01; ***p ≤ 0.001.
In addition to the histological analysis, we analyzed the separate impacts of GSK3αS/A and GSK3βS/A on motor and sensory recovery and compared them with previously published data of GSK3αS/A/GSK3βS/A mice (Gobrecht et al., 2014). Assessment of toe spreading before injury and 1 and 4 d after SNC showed no statistical differences in the SSI for mice of all four experimental groups (wt, GSK3αS/A, GSK3βS/A, GSK3αS/A/GSK3βS/A) (Fig. 1G,I). At 7–21 d after sciatic nerve injury, however, the SSIs of GSK3αS/A and GSK3βS/A mice were slightly higher compared with wt controls, with only GSK3αS/A reaching statistical significance (Fig. 1G,I). Consistent with this accelerated functional recovery, reestablishment of neuromuscular junctions in the extensor hallucis longus muscle was only detected in the GSK3 knock-in, but not in wt mice at 7 d after injury (Fig. 1H). Nevertheless, motor recovery in both single knock-in mice was slower than in GSK3αS/A/GSK3βS/A double knock-in mice (Fig. 1G,I). Comparable results were observed for sensory recovery using the von Frey test (Fig. 1J). Uninjured wt and all GSK3 mutant animals showed a similar increase in the force required to induce a hindpaw withdrawal response at 1 d after SNC, indicating dramatic injury-induced loss of touch sensation. Animals started to recover ∼7 d after injury, with GSK3αS/A/GSK3βS/A double knock-in animals recovering fastest, whereas wt animals showed the slowest improvement within the 21 d test period (Fig. 1J). Therefore, the mechanism underlying improved axon regeneration and functional recovery in mutant GSK3 animals is not isoform specific, and the rate of axon regeneration seems to depend on overall GSK3 activity.
Axon growth in GSK3 mutant mice correlates with axonal MAP1B phosphorylation
We previously showed that impaired GSK3 phosphorylation in GSK3αS/A/GSK3βS/A mice does not affect the induction of regeneration-associated genes upon SNC (Gobrecht et al., 2014). At the same time, GSK3-induced phosphorylation of MAPs, such as CRMP2 and MAP1B, reportedly affects axon growth by regulating microtubule assembly in axonal tips (Hur et al., 2011b, 2012; Liz et al., 2014). We therefore investigated whether phosphorylation of these substrates might be involved in the axon growth promoting effect in adult GSK3 mutant mice. To this end, we subjected wt and GSK3αS/A/GSK3βS/A mice to SNC and isolated respective DRG as well as lesioned sciatic nerves 5 d thereafter. As determined by Western blot analysis, phospho-CRMP2 levels were slightly increased in DRG lysates of GSK3αS/A/GSK3βS/A before as well as after injury compared with wt animals, reflecting the higher overall GSK3 activity in the transgenic mice (Fig. 2A–D). Surprisingly, despite abundant CRMP2 protein, almost no phospho-CRMP2 was detected in sciatic nerves of either GSK3αS/A/GSK3βS/A or wt mice (Fig. 2A–D). This observation suggests that CRMP2 phosphorylation in axons is regulated independently of GSK3 (potentially by priming kinases) and that CRMP2 activity is not significantly regulated in sciatic nerve regeneration. In comparison, phospho-MAP1B levels were increased precrush and postcrush in nerve lysates of GSK3αS/A/GSK3βS/A animals compared with wt controls, whereas little phospho-MAP1B was detectable in DRG (Fig. 2A–D). Neither total CRMP2 nor MAP1B levels were changed in GSK3αS/A/GSK3βS/A compared with wt mice, indicating that increased phosphorylation only depended on elevated GSK3 activity. Increased MAP1B phosphorylation in regenerating axons was verified by immunostainings of longitudinal nerve sections from single and double knock-in mice isolated 3 d after injury (Fig. 2E). Strongest pMAP1B staining was expectedly detected in regenerating axons of GSK3αS/A/GSK3βS/A mice, less in GSK3αS/A and GSK3βS/A animals and lowest in wt controls, confirming predicted GSK3 activity levels for the respective genotypes (Fig. 2E). To provide further evidence that increased MAP1B phosphorylation depends on elevated GSK3 activity, phospho-MAP1B was detected in axon tips of cultured sensory neurons from adult wt, GSK3αS/A, GSK3βS/A, and GSK3αS/A/GSK3βS/A knock-in animals that were exposed to the GSK3 inhibitor SB216763 (Coghlan et al., 2000). In wt neurons, 60% of axonal tips were positive for phospho-MAP1B (Fig. 2F,G). This percentage significantly increased in GSK3αS/A (79%) and GSK3βS/A (75.5%), whereas highest rates were quantified in GSK3αS/A/GSK3βS/A (95%) neurons (Fig. 2F,G). MAP1B phosphorylation returned to wt levels (65%) upon incubation with SB216763 (Fig. 2F,G), indicating that its phosphorylation depends on enzymatic GSK3 activity.
Figure 2.
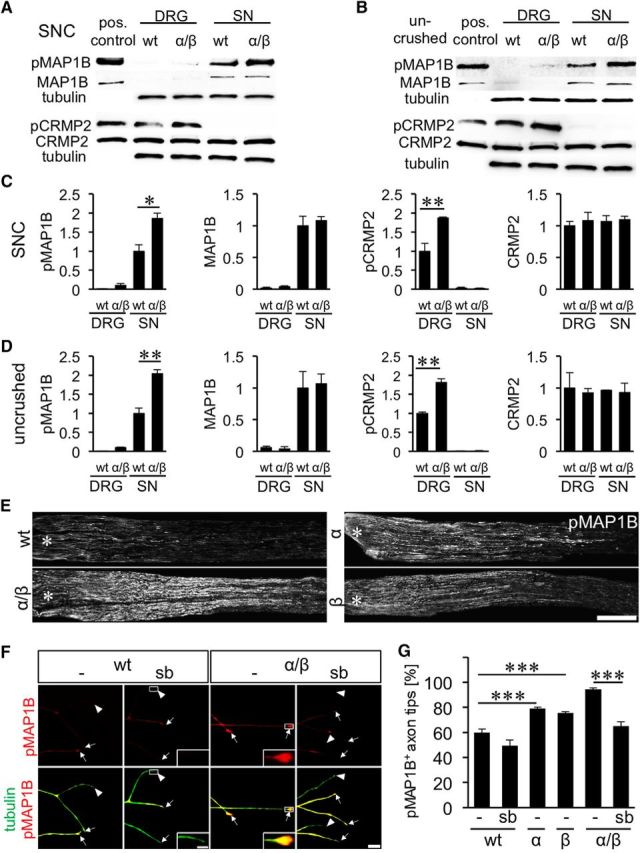
Effect of GSK3S/A on CRMP2 and MAP1B phosphorylation. A, B, Composition of Western blots analyzing levels of phosphorylated MAP1B (pMAP1B), total MAP1B, phosphorylated CRMP2 (pCRMP2), and total CRMP2 in DRG and sciatic nerves (SN), respectively, from wt and GSK3αS/A/GSK3βS/A (α/β) mice either 5 d after SNC (A) or in uninjured animals (B). Tubulin served as loading control. Lysates of HEK cells cotransfected with CRMP2 and constitutively active GSK3 served as positive controls for CRMP2 and pCRMP2, whereas HEK cells cotransfected with MAP1B and constitutively active GSK3 were used as positive control for pMAP1B and MAP1B. C, D, Quantification of Western blots as depicted in A and B. Consistent with increased GSK3 activity, MAP1B phosphorylation, but not total MAP1B, is increased in GSK3 knock-in mice. CRMP2 phosphorylation, however, is only elevated in DRG, despite abundant CRMP2 protein in sciatic nerves. Treatment effects: *p ≤ 0.05; **p ≤ 0.01. E, pMAP1B staining of longitudinal sciatic nerve sections of adult wt, α, β, and α/β mice 3 d after SNC verify MAP1B phosphorylation in regenerating axons. Scale bar, 500 μm. Asterisks indicate the crush site. F, Representative pictures of axons in DRG cultures of wt and α/β mice exposed to either vehicle (−) or 5 μm GSK3 inhibitor SB216763 (sb). Axons were stained for pMAP1B (red) and βIII-tubulin (tubulin, green). Arrows indicate pMAP1B-positive, arrowheads negative axon tips. Scale bar, 20 μm. Insets, Magnified examples of axon tips. Scale bar, 5, μm. G, Quantification of pMAP1B-positive axon tips of neurons from adult wt, α, β, and α/β mice exposed to either vehicle (−) or 5 μm GSK3 inhibitor SB216763 (sb). Data from transgenic neurons were normalized to wt controls and represent mean ± SEM of three independent experiments. Treatment effects: ***p ≤ 0.001. GSK3S/A increased the percentage of pMAP1B in axon tips, which was abrogated by GSK3 inhibition using SB216763.
Inhibition of microtubule detyrosination promotes axon growth
GSK3-mediated MAP1B phosphorylation is reportedly associated with reduced microtubule detyrosination in axonal tips of embryonic neurons (Goold et al., 1999; Owen and Gordon-Weeks, 2003; Trivedi et al., 2005), which suggestively increases microtubule dynamics in growth cones and thereby facilitates axon extension (Lucas et al., 1998; Trivedi et al., 2005). To test whether a similar mechanism could underlie the beneficial effects of GSK3S/A in adult DRG, we quantified the percentage of axonal tips with detyrosinated (stable) microtubules in wt and GSK3αS/A/GSK3βS/A neurons. Indeed, only ∼55% of the axonal tips of adult GSK3αS/A/GSK3βS/A neurons contained detyrosinated microtubules compared with 80% in wt controls (Fig. 3A,B). Reduced detyrosination depended on GSK3 activity as it was impaired in the presence of the SB216763 inhibitor (72%) (Fig. 3A,B). Thus, increased microtubule dynamics could be the underlying mechanism of GSK3S/A-mediated axonal growth promotion in adult sensory neurons.
Figure 3.
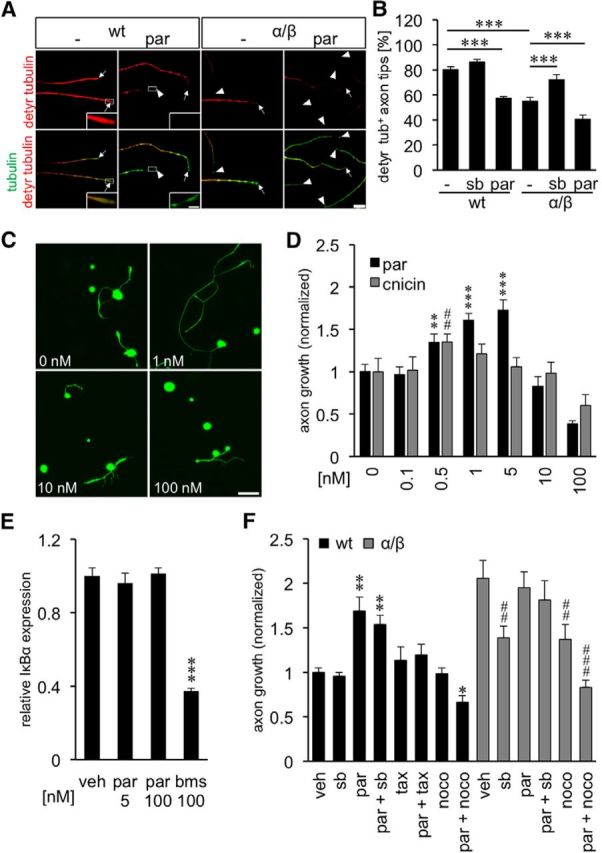
Inhibition of tubulin detyrosination promotes axon growth. A, Representative pictures of axons in DRG cultures from wt and GSK3αS/A/GSK3βS/A (α/β) mice exposed to either vehicle (−) or 5 nm parthenolide (par). Axons were stained for detyrosinated tubulin (detyr. tubulin, red) and βIII-tubulin (tubulin, green). Arrows indicate axon tips, arrowheads axon tips without detyrosinated tubulin. Scale bar, 20 μm. Insets, Magnified examples of detyrosinated tubulin-positive and -negative axon tips. Scale bar, 5 μm. B, Quantification of axon tips with detyrosinated tubulin in cultures from wt and α/β mice 3 d after exposure to vehicle (−), 5 μm SB216763 (sb), or 5 nm parthenolide (par). Data were normalized to wt controls and represent mean ± SEM of three independent experiments. Treatment effects: ***p ≤ 0.001. GSK3S/A as well as par treatment reduced the levels of detyrosinated tubulin in axonal tips. C, Representative pictures of dissociated wt DRG neurons treated with various par concentrations and stained for βIII-tubulin after 2 d in culture. Scale bar, 100 μm. D, Quantification of axon growth in wt DRG cultures after treatment with various cnicin and par concentrations. Data from treated neurons were normalized to vehicle controls with an average axon length of 933 μm/neuron for parthenolide and 546 μm/neuron for cnicin. Data represent mean ± SEM of three independent experiments. Treatment effects compared with vehicle control: **p ≤ 0.01; ***p ≤ 0.001; ##p ≤ 0.01; E, Quantitative PCR: relative expression of NFκB inhibitor α (IκBα) in dissociated DRG treated with DMSO (veh), 5 and 100 nm parthenolide (par), or 100 nm NFκB inhibitor BMS 345541 (bms), respectively. Data represent mean ΔΔCt ± SEM of two independent experiments. Treatment effect: ***p ≤ 0.001. NFκB activity was not affected at the concentrations of parthenolide used in our culture conditions. F, Quantification of axon growth in DRG cultures from wt or GSK3αS/A/GSK3βS/A mice (α/β), treated with vehicle (veh), 5 μm GSK3 inhibitor SB216763 (sb), 1 nm par, 10 nm paclitaxel (tax), 10 nm tax + 1 nm par, 5 μm sb + 1 nm par, 10 nm nocodazole (noco), or 1 nm par + 10 nm noco after 2 d in culture. Data from treated neurons were normalized to vehicle controls with an average axon length of 842 μm/neuron and represent mean ± SEM of three independent experiments. Treatment effects compared with vehicle-treated wt neurons: *p ≤ 0.05; **p ≤ 0.01. Treatment effects compared with vehicle-treated α/β neurons: ##p ≤ 0.01; ###p ≤ 0.001.
The exact mechanism how GSK3 might affect microtubule dynamics in axonal growth cones is still unclear, but phospho-MAP1B-mediated modulation of a tubulin-carboxypeptidase or a tubulin-tyrosine ligase has been discussed in this context (Gonzalez-Billault et al., 2004; Utreras et al., 2008). Downstream of GSK3 and MAP1B activities, detyrosination of microtubule plus ends can potently be compromised in non-neuronal cells by the sesquiterpene lactone parthenolide and to lesser extent by its derivate cnicin (Fonrose et al., 2007). We therefore used these compounds to test the hypothesis that decreased microtubule detyrosination could promote axon regeneration of mature DRG neurons. We first investigated whether parthenolide treatment would inhibit microtubule detyrosination also in axonal growth cones. Indeed, parthenolide decreased the percentage of axonal tips with detyrosinated microtubules in adult wt neurons at 5 nm to a similar extent as in GSK3αS/A/GSK3βS/A neurons (Fig. 3A,B). Moreover, parthenolide significantly and concentration-dependently increased axon growth of wt sensory neurons in culture (Fig. 3C,D). Strongest effects were measured at 1 and 5 nm, whereas concentrations ≥100 nm reduced axon growth. This bell-shaped efficacy profile suggests that only moderate reduction, but not strong inhibition, of microtubule detyrosination promotes axon growth. As cell numbers remained unaffected for all tested concentrations (data not shown), general toxicity was not an issue. Compared with parthenolide, the less efficacious derivate cnicin showed significant, but less pronounced, axon growth promotion with strongest effects at 0.5 nm (Fig. 3D).
Although parthenolide reportedly also inhibits NFκB, this effect has only been reported at much higher concentrations (1–4 μm) (Hehner et al., 1999; Yip-Schneider et al., 2005; Saadane et al., 2007). Nevertheless, to exclude a contribution of NFκB inhibition on parthenolide-induced axon growth, we analyzed the expression of the NFκB target IκBα in vehicle- and parthenolide-treated DRG cultures by qRT-PCR (Fig. 3E). As expected, IκBα expression was not changed by parthenolide treatment but significantly reduced by the positive control NFκB inhibitor BMS 345541 compared with vehicle-treated cultures. Therefore, we can exclude the possibility that NFκB inhibition considerably contributes to parthenolide-induced axon growth. Growth promotion by parthenolide in wt neurons was not significantly affected by the GSK3 inhibitor SB216763, providing further evidence that parthenolide acts downstream of GSK3 activities (Fig. 3F). In contrast, axon growth of GSK3αS/A/GSK3βS/A neurons was reduced to wt levels upon treatment with SB216763. These results indicate the dependence of GSK3αS/A/GSK3βS/A on enzymatic GSK3 activity, which is seemingly optimal, as parthenolide was unable to further increase axonal growth in these cultures.
To further demonstrate that the effects of GSK3αS/A/GSK3βS/A and parthenolide primarily rely on the modulation of microtubule dynamics, DRG cultures were treated with microtubule-stabilizing paclitaxel and microtubule-disrupting nocodazole. At concentrations that had no significant effect on axonal growth of wt neurons per se, both agents abrogated parthenolide-induced growth promotion (Fig. 3F). Compared with wt DRG, GSK3αS/A/GSK3βS/A neurons were more susceptible to nocodazole, in particular in combination with parthenolide (Fig. 3F). These data strongly suggest that the axon growth promoting effect of parthenolide and GSK3αS/A/GSK3βS/A is relayed by inhibition of microtubule detyrosination in axonal tips, which increases the dynamics of growth cones.
To verify that parthenolide targets axons/growth cones, we used two-compartment cultures that permit fluidic isolation of axons from their respective neuronal somas (Hur et al., 2011b). Cultured adult DRG neurons were allowed to enter a regenerative state for 3 d, after which they were axotomized. Although parthenolide in the somal chamber did not raise axon regrowth above vehicle-treated levels, parthenolide in the axonal chamber significantly increased it (Fig. 4A,B). These results confirm that parthenolide exerts its regenerative effect via interaction with axonal growth cones rather than neuronal cell bodies. Moreover, they indicate that parthenolide is able to further enhance axon growth of neurons already primed for regeneration.
Figure 4.

Parthenolide targets axonal growth cones. A, Representative pictures of cultures of adult DRG neurons in two-compartment chambers. Axons regrown beyond the microchannels 24 h after axotomy were visualized using βIII-tubulin immunostaining. The 5 nm par was applied either to the somal (blue) or the axonal (brown) compartments, as indicated in the schematic below the pictures. Scale bar, 250 μm. B, Quantification of axon regrowth beyond microchannels of two-compartment chambers 24 h after axotomy as depicted in A. Data are mean ± SEM from five independent experiments. Treatment effect: **p ≤ 0.01.
In vivo parthenolide application accelerates regeneration and functional recovery
As our cell culture experiments indicated that parthenolide targets axonal growth cones, we next investigated whether intraneural application of parthenolide would mimic the effects of GSK3S/A and promote sciatic nerve regeneration in vivo. To this end, we first applied increasing doses of parthenolide (1.25, 6.25, 12.5, 125, 1250, and 12,500 pg) into the crush site of sciatic nerves and evaluated axon regeneration on sciatic nerve sections to determine effective concentrations (Fig. 5A–C,E). Although intraneurally applied doses ranging from 1.25 to 12.5 pg markedly increased axon regeneration 3 d after SNC, 1000 times higher concentrations (12.5 ng) rather reduced it (Fig. 5A–C,E). Strongest growth promotion was determined at doses of 6.25 and 12.5 pg, which increased the number of axons >3-fold compared with vehicle-treated controls at a distance of 2.5 mm beyond the lesion site (Fig. 5A–C,E). We also tested whether systemic parthenolide administration would be able to promote sciatic nerve regeneration. To this end, increasing parthenolide doses (20 ng/kg; 200 ng/kg; 2 μg/kg; 20 μg/kg) were injected intraperitoneally after sciatic nerve injury. Strikingly, a single injection of 200 ng/kg significantly increased the number of regenerating axons ∼2.5-fold at 2.5 mm (Fig. 5A,D,F), which is slightly less pronounced compared with intraneural parthenolide application (Fig. 5E). Higher tested doses did not significantly affect sciatic nerve regeneration (Fig. 5F).
Figure 5.
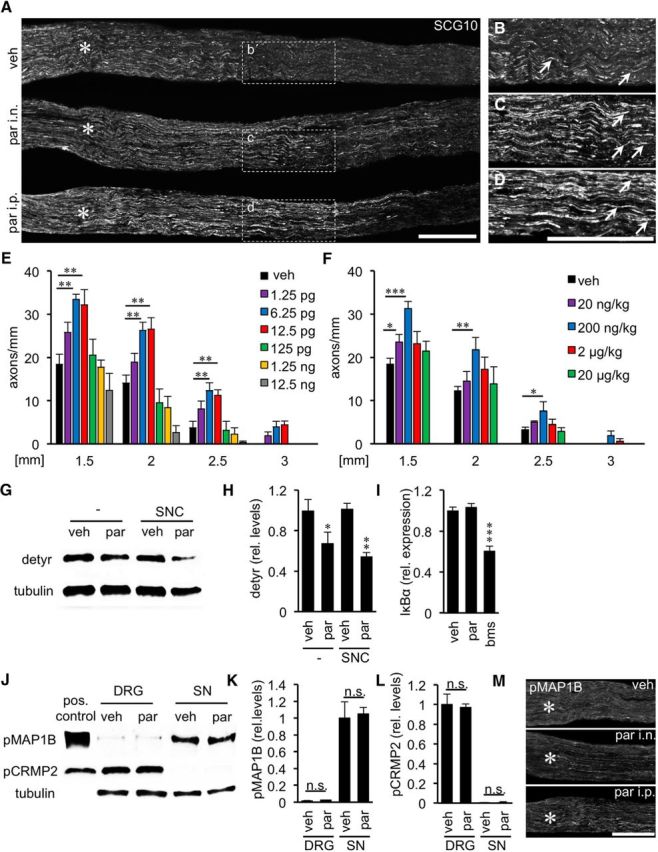
Parthenolide accelerates sciatic nerve regeneration and reduces tubulin detyrosination. A, Representative longitudinal sections of sciatic nerves 3 d after SNC and either single intraneural (i.n.) injection of vehicle (top) or parthenolide (6.25 pg par; middle) or intraperitoneal (i.p.) par injection (200 ng/kg; bottom). Regenerating axons were immunohistochemically stained for SCG10. Scale bar, 500 μm. Asterisks indicate the crush site. B–D, Magnifications of the respective areas indicated in A. Only a few axons (arrows) were detected at ∼2.5 mm past the lesion in vehicle-injected animals (B), whereas significantly more regenerating axons were present after intraneural (C) and intraperitoneal (D) par injection. E, F, Quantification of axons on longitudinal sections at 1.5, 2, 2.5, and 3 mm beyond the injury site of sciatic nerves from mice either intraneurally (E) or intraperitoneally (F) injected with vehicle (veh) or various doses of par as indicated. Data represent mean ± SEM of five sections from at least six individual mice per experimental group. Treatment effects compared with vehicle-injected animals: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. G, Western blot analysis of detyrosinated tubulin (detyr) in sciatic nerves 3 h after intraneural injection of either vehicle or 6.25 pg par without (−) or in combination with SNC. Tubulin served as loading control. H, Relative quantification of Western blots as depicted in G. Parthenolide injection decreased detyrosination of tubulin independent of nerve injury. Treatment effects compared with vehicle: *p ≤ 0.05; **p ≤ 0.01. I, Quantitative PCR: relative expression of NFκB inhibitor α (IκBα) in crushed sciatic nerves injected with DMSO (veh), 6.25 pg of parthenolide (par) or 50 ng of NFκB inhibitor BMS 345541 (bms), respectively. Data represent mean ΔΔCt ± SEM of three different mice per experimental group. Treatment effect: ***p ≤ 0.001. J, Western blot analysis of phosphorylated MAP1B (pMAP1B) and phosphorylated CRMP2 (pCRMP2) at 5 d after SNC in DRG and sciatic nerves (SN), respectively, from wt mice intraneurally injected either with vehicle (veh) or 6.25 pg of par. Tubulin served as loading control. K, L, Quantification of Western blots as depicted in J. Parthenolide application affects neither MAP1B nor CRMP2 phosphorylation. n.s., Not significant. M, Longitudinal sections of sciatic nerves treated as depicted in A. Regenerating axons were immunohistochemically stained for phospho-MAP1B (pMAP1B) and showed no signal differences. Scale bar, 500 μm. Asterisks indicate the crush site.
Expectedly, parthenolide treatment significantly reduced tubulin detyrosination in sciatic nerves before and after injury (Fig. 5G,H). Again, we tested the possibility that the observed increased axon regeneration might be caused by inhibition of NFκB rather than decreased tubulin detyrosination. To this end, we compared the expression of the NFκB target IκBα in vehicle- and parthenolide-injected sciatic nerves by qRT-PCR (Fig. 5I). As expected, IκBα expression was not changed by parthenolide treatment but significantly reduced by the NFκB inhibitor BMS 345541 compared with control animals. Therefore, we can exclude the possibility that NFκB inhibition considerably contributes to parthenolide-induced axon regeneration. Similarly, the phosphorylation status of MAP1B or CRMP2 in injured DRG and sciatic nerves was not affected by parthenolide, as determined by Western blot analysis and immunohistochemical staining of nerve sections (Fig. 5J–M), confirming that this drug acts downstream of GSK3 signaling.
To investigate whether in vivo parthenolide treatment might also be sufficient to accelerate functional recovery, we injected 6.25 pg of parthenolide into the lesion site and assessed functional recovery using the SSI and the von Frey tests. Strikingly, parthenolide-treated animals showed a significantly improved SSI score already at 4 d after injury compared with vehicle-treated control animals, and this difference was sustained over the total observation period of 3 weeks (Fig. 6A,B). To verify that regenerating axons already successfully reinnervated their targets 4 d after parthenolide treatment, neuromuscular endplates were detected using BTX and neurofilament staining of the extensor hallucis longus muscle. Consistently, neuromuscular junctions were found only in parthenolide-, but not vehicle-treated animals (Fig. 6C). Sensory recovery was likewise accelerated upon parthenolide treatment (Fig. 6D). Improvements of the touch response were first detectable in the von Frey test at 7 d after injury, and a significant difference between parthenolide- and vehicle-treated mice was observed over the total observation time of 21 d (Fig. 6D). The later onset of sensory versus motor function recovery thereby reflects the longer distance that sensory axons need to grow to reach their respective targets in the hindpaw. Together, these data demonstrate that in vivo treatment with parthenolide markedly promotes sciatic nerve regeneration and is sufficient to accelerate functional recovery.
Figure 6.

Parthenolide treatment accelerates functional recovery after sciatic nerve injury. A, Representative pictures of right hindpaws of wt mice intraneurally injected with either vehicle (veh) or 6.25 pg parthenolide (par) and tested before (con) and 1, 4, 9, 14, and 21 d after SNC (dpc). B, Quantification of motor function recovery in adult mice treated as depicted in A using the SSI. Eleven animals were analyzed per experimental group. Treatment effects: ***p ≤ 0.001. C, Extensor hallucis longus muscle whole mounts from veh- or par-treated mice stained with α-BTX (red) and 200 kDa neurofilament antibody (NF, green) 4 d after SNC. White arrows indicate reestablished synapses in par-treated mice. Scale bar, 50 μm. D, Quantification of sensory function recovery of adult wt mice intraneurally injected with either veh or par and tested at 1, 4, 7, 9, 12, 14, and 21 d after SNC using the von Frey test. Eleven animals were analyzed per experimental group. Treatment effects: ***p ≤ 0.001.
Discussion
We recently demonstrated that adult transgenic GSK3αS/A/GSK3βS/A mice with sustained GSK3 activity show markedly accelerated sciatic nerve regeneration (Gobrecht et al., 2014). Here, we investigated the molecular mechanism(s) underlying this phenomenon, which is seemingly caused by changed post-translational modifications of microtubules in axonal growth cones. Based on these results, we also devised a novel, clinically applicable approach to accelerate nerve regeneration using parthenolide to reduce the detyrosination of microtubules.
We initially tested whether the beneficial effects of GSK3αS/A/GSK3βS/A could be attributed to a GSK3 isoform-specific mechanism, as most previous studies only considered a role for GSK3β in axon regeneration. Interestingly, we found that single knock-in of either GSK3αS/A or GSK3βS/A similarly enhanced axon growth in culture and accelerated axon regeneration as well as functional recovery in vivo to comparable extent. These results provide strong evidence that both GSK3α and GSK3β activities are involved in regenerative processes of injured axons. However, axon growth promotion was less pronounced in each single knock-in mouse compared with double knock-in animals. The isoform-specific effects were not additive, indicating that GSK3α and GSK3β might act on the same target(s) and that the beneficial effects of GSK3αS/A/GSK3βS/A depend on overall GSK3 activity.
To further explore the mechanism(s) underlying GSK3αS/A/GSK3βS/A-promoted axon regeneration, we investigated the impact of elevated GSK3 activity on the two selected MAPs CRMP2 and MAP1B, which reportedly affect axon growth upon phosphorylation by GSK3 (Hur et al., 2011b). Whereas phosphorylation of CRMP2 rather compromises axon regeneration (Hur and Zhou, 2010; Liz et al., 2014), phosphorylation of MAP1B has been proposed to improve axon growth in embryonic neurons (Trivedi et al., 2005). Consistent with elevated GSK3 activity, phospho-CRMP2 levels were increased in DRG of GSK3αS/A/GSK3βS/A compared with wt mice before and after sciatic nerve injury. However, despite abundant CRMP2 protein, no phospho-CRMP2 was detected in normal and injured sciatic nerves of either wt or GSK3αS/A/GSK3βS/A animals. These results suggest differential regulation of CRMP2 phosphorylation in different neuronal compartments, potentially by the differential localization of the required priming kinase. This finding does not exclude an important role for CRMP2 in PNS regeneration (Liz et al., 2014) but rather indicates that it is not a key effector of GSK3 activity in this context. In conclusion, an expected compromising effect of increased CRMP2 phosphorylation upon increased GSK3 activity appears to be irrelevant for peripheral nerve regeneration. However, phospho-CRMP2 might show different subcellular distribution in other neurons (e.g., in the CNS), which could negatively impact axon regeneration, for example, in the optic nerve or spinal cord (Liz et al., 2014). Moreover, we cannot exclude the possibility that enhanced GSK3 activity inhibits the activity of other microtubule-binding proteins, such as APC or CLASP, which might still dampen the overall positive regenerative outcome in GSK3αS/A/GSK3βS/A mice (Zhou et al., 2004; Hur et al., 2011a). In contrast to CRMP2, phosphorylation of MAP1B at Thr1265 by GSK3 does not require prior priming (Frame et al., 2001; Cole et al., 2004b; Trivedi et al., 2005) and is reportedly restricted to axons (Avila et al., 1994; Ulloa et al., 1994; Goold et al., 1999; Trivedi et al., 2005; Scales et al., 2009). Thus, axonal phospho-MAP1B levels can be considered as a more reliable indicator for GSK3 activity. Consistently, phospho-MAP1B levels in axonal tips in culture and in precrushed and postcrushed sciatic nerves correlated with increased GSK3 activity in GSK3S/A knock-in mice and were reduced to wt levels by GSK3 inhibition. Notably, phosphorylation of MAP1B was enhanced in axons of each single knock-in mouse, indicating that MAP1B is a substrate for both GSK3α and GSK3β. This observation was rather unexpected as experiments in embryonic chick CNS tissue previously proposed MAP1B as a specific substrate of GSK3β (Lucas et al., 1998; Owen and Gordon-Weeks, 2003). Whether these differences are due to the different developmental stage, species, or specific experimental settings is currently unknown.
It has previously been proposed that increased MAP1B phosphorylation is associated with reduced microtubule detyrosination in embryonic neurons, which reportedly promotes axon extension (Lucas et al., 1998; Goold et al., 1999; Owen and Gordon-Weeks, 2003; Trivedi et al., 2005). Although the detailed enzymatic mechanism underlying this impaired microtubule detyrosination remains unclear, phospho-MAP1B-mediated modulations of a tubulin-carboxypeptidase or a tubulin-tyrosine ligase have been discussed in this context (Goold et al., 1999; Gonzalez-Billault et al., 2002; Owen and Gordon-Weeks, 2003; Gonzalez-Billault et al., 2004; Barnat et al., 2016). Interestingly, a recent publication shows that MAP1B is a key effector downstream of GSK3 and that GSK3-mediated phosphorylation of MAP1B directly impacts the stability of microtubules (Barnat et al., 2016). Regardless of the underlying mechanism, we found that microtubule detyrosination was indeed reduced in axonal tips of GSK3S/A knock-in mice. Others have previously shown that GSK3 inhibition rather than activation increases microtubule dynamics and axon growth via CRMP2 stimulation (Liz et al., 2014). Therefore, the overall outcome of GSK3 activation/inhibition seems to depend on the abundance of respective effectors and stresses the importance of context-specific analyses with respect to the role of GSK3 in axon regeneration (Diekmann and Fischer, 2015).
To investigate whether this modification of microtubules contributed to the observed enhanced axon regeneration in GSK3αS/A/GSK3βS/A mice, we inhibited microtubule detyrosination pharmacologically in wt animals using the sesquiterpene lactone parthenolide. Similar to non-neuronal cells (Fonrose et al., 2007), parthenolide reduced the detyrosination of tubulin in axonal tips of cultured DRG neurons. Consequently, parthenolide and to a lower extent also cnicin promoted axonal growth concentration-dependently in cell culture; and at optimal concentrations, parthenolide fully mimicked the beneficial effects of GSK3αS/A/GSK3βS/A on axon regeneration in wt mice. As parthenolide treatment did not further enhance axon growth of GSK3αS/A/GSK3βS/A neurons, reduced microtubule detyrosination is likely the main mechanism underlying GSK3αS/A/GSK3βS/A-facilitated growth. Moreover, these data provide the first direct evidence that inhibition of microtubule detyrosination can markedly promote axon growth. Inhibition of NFκB signaling is likely irrelevant for our results, as it would require ∼1000-fold higher parthenolide concentrations than used in our study (Hehner et al., 1999; Yip-Schneider et al., 2005; Saadane et al., 2007). Consistently, we found no effect of parthenolide on NFκB activity in vitro and in vivo using the expression of IκBα as sensitive readout. In addition, the dependence of growth promotion on axonal parthenolide exposure as indicated by the experiments with two-compartment chambers provided further evidence that parthenolide targets microtubules in axonal growth cones rather than gene expression in cell somata. The concentration dependency of the sesquiterpene lactones indicates that moderate reduction of microtubule detyrosination promotes, whereas strong inhibition at high concentrations rather reduces axon growth. General toxicity is no concern in this context, as the survival of DRG neurons was unaffected by any treatment and much higher parthenolide concentrations (1–4 μm) are usually applied to inhibit NFκB signaling (Hehner et al., 1999; Yip-Schneider et al., 2005; Saadane et al., 2007).
Conceptually, moderate inhibition of detyrosination at plus ends transforms microtubules in a more dynamic state, which is required for optimal axonal growth (Lucas et al., 1998). Consistent with increased microtubule dynamics, parthenolide-treated axons showed higher sensitivity toward microtuble-disrupting nocodazole, similar to GSK3αS/A/GSK3βS/A neurons (Gobrecht et al., 2014). Treatment with paclitaxel, which increases detyrosination and stabilizes microtubles (Ertürk et al., 2007; Hammond et al., 2010), counteracted the observed growth promotion. Our conclusion that increased microtubule dynamics promote axonal regeneration is consistent with a previous study showing that depletion of the injury-specific kinesin KIF3C, which destabilizes microtubules via binding to tyrosinated tubulin, compromises axon regeneration (Gumy et al., 2013). Similarly, knockdown of tubulin-tyrosin ligase reportedly increased the level of detyrosinated microtubules and impaired axon growth (Song et al., 2015). In this study, the observed effect was ascribed to impaired retrograd axonal transport of injury signals upon modification of the tubulin tyrosination cycle (Song et al., 2015). It remains to be investigated whether parthenolide treatment might additionally increase the regenerative response of neurons to axonal injury by accelerating this retrograde transport.
Strikingly, accelerated sciatic nerve regeneration in GSK3αS/A/GSK3βS/A mice could also be mimicked by in vivo parthenolide application in wt mice. Effective doses were identified for both systemic and intraneural applications. In agreement with our cell culture data, these in vivo effects of parthenolide were dose-dependent, with the highest tested doses having no or even adverse effects. Moreover, intraneural injection of effective parthenolide doses decreased microtubule detyrosination in the sciatic nerve in vivo, whereas the phosphorylation status of CRMP2 and MAP1B remained unaffected, thereby demonstrating that parthenolide promotes regeneration downstream of GSK3 signaling. Either one intraneural or one intraperitoneal injection of parthenolide was sufficient to significantly promote regeneration, including accelerated motor and sensory recoveries. However, it might still be possible to further improve the functional outcome by optimizing doses and application frequencies. Whereas intraneural application is quite an invasive intervention, it would be feasible to repeat intraperitoneal injections.
It is currently unknown whether parthenolide treatment would also promote axon regeneration in the CNS. In contrast to the PNS, inhibitory molecules in myelin and the gliotic scar limit axon regeneration in the CNS. We and others have previously demonstrated that axon regeneration in the CNS can be facilitated by paclitaxel-mediated stabilization of microtubules, which decreases the sensitivity of axonal growth cones toward inhibitory molecules (Ertürk et al., 2007; Hellal et al., 2011; Sengottuvel and Fischer, 2011; Sengottuvel et al., 2011). It will be interesting to see whether more dynamic microtubules, as induced by parthenolide, might also affect CNS regeneration. Experiments addressing these questions are currently in progress.
In conclusion, our study demonstrates that prevention of GSK3 phosphorylation markedly promotes axon regeneration in cell culture and in vivo by compromising detyrosination of microtubules in axonal tips. These effects can be mimicked by pharmacological inhibition of tubulin detyrosination using parthenolide. Therefore, treatments aiming to reduce microtubule detyrosination in the injured peripheral nerve may be a novel and clinically feasible approach to accelerate axon regeneration and to improve functional recovery.
Footnotes
This work was supported by the German Research Foundation. We thank Prof. Dr. Alessi (University of Dundee) for providing the GSK3S/A knock-in mice; and Marcel Kohlhaas for technical support.
The authors declare no competing financial interests.
References
- Avila J, Ulloa L, Diez-Guerra J, Diaz-Nido J. Role of phosphorylated MAPlB in neuritogenesis. Cell Biol Int. 1994;18:309–314. doi: 10.1006/cbir.1994.1079. [DOI] [PubMed] [Google Scholar]
- Baptista AF, Gomes JR, Oliveira JT, Santos SM, Vannier-Santos MA, Martinez AM. A new approach to assess function after sciatic nerve lesion in the mouse: adaptation of the sciatic static index. J Neurosci Methods. 2007;161:259–264. doi: 10.1016/j.jneumeth.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Barnat M, Benassy MN, Vincensini L, Soares S, Fassier C, Propst F, Andrieux A, von Boxberg Y, Nothias F. The GSK3-MAP1B pathway controls neurite branching and microtubule dynamics. Mol Cell Neurosci. 2016;72:9–21. doi: 10.1016/j.mcn.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Bobinski F, Martins DF, Bratti T, Mazzardo-Martins L, Winkelmann-Duarte EC, Guglielmo LG, Santos AR. Neuroprotective and neuroregenerative effects of low-intensity aerobic exercise on sciatic nerve crush injury in mice. Neuroscience. 2011;194:337–348. doi: 10.1016/j.neuroscience.2011.07.075. [DOI] [PubMed] [Google Scholar]
- Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee Cox L, Mills D, Brown MJ, Haigh D, Ward RW, Smith DG, Murray KJ, Reith AD, Holder JC. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol. 2000;7:793–803. doi: 10.1016/S1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- Cole A, Frame S, Cohen P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem J. 2004a;377:249–255. doi: 10.1042/bj20031259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ, Connolly CN, Sutherland C. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J Biol Chem. 2004b;279:50176–50180. doi: 10.1074/jbc.C400412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekmann H, Fischer D. Role of GSK3 in peripheral nerve regeneration. Neural Regen Res. 2015;10:1602–1603. doi: 10.4103/1673-5374.167753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Martinez A. GSK-3 inhibitors: preclinical and clinical focus on CNS. Front Mol Neurosci. 2011;4:32. doi: 10.3389/fnmol.2011.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertürk A, Hellal F, Enes J, Bradke F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci. 2007;27:9169–9180. doi: 10.1523/JNEUROSCI.0612-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonrose X, Ausseil F, Soleilhac E, Masson V, David B, Pouny I, Cintrat JC, Rousseau B, Barette C, Massiot G, Lafanechère L. Parthenolide inhibits tubulin carboxypeptidase activity. Cancer Res. 2007;67:3371–3378. doi: 10.1158/0008-5472.CAN-06-3732. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7:1321–1327. doi: 10.1016/S1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- Gladman SJ, Huang W, Lim SN, Dyall SC, Boddy S, Kang JX, Knight MM, Priestley JV, Michael-Titus AT. Improved outcome after peripheral nerve injury in mice with increased levels of endogenous omega-3 polyunsaturated fatty acids. J Neurosci. 2012;32:563–571. doi: 10.1523/JNEUROSCI.3371-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobrecht P, Leibinger M, Andreadaki A, Fischer D. Sustained GSK3 activity markedly facilitates nerve regeneration. Nat Commun. 2014;5:4561. doi: 10.1038/ncomms5561. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Billault C, Owen R, Gordon-Weeks PR, Avila J. Microtubule-associated protein 1B is involved in the initial stages of axonogenesis in peripheral nervous system cultured neurons. Brain Res. 2002;943:56–67. doi: 10.1016/S0006-8993(02)02534-9. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Billault C, Jimenez-Mateos EM, Caceres A, Diaz-Nido J, Wandosell F, Avila J. Microtubule-associated protein 1B function during normal development, regeneration, and pathological conditions in the nervous system. J Neurobiol. 2004;58:48–59. doi: 10.1002/neu.10283. [DOI] [PubMed] [Google Scholar]
- Goold RG, Owen R, Gordon-Weeks PR. Glycogen synthase kinase 3beta phosphorylation of microtubule-associated protein 1B regulates the stability of microtubules in growth cones. J Cell Sci. 1999;112:3373–3384. doi: 10.1242/jcs.112.19.3373. [DOI] [PubMed] [Google Scholar]
- Gumy LF, Chew DJ, Tortosa E, Katrukha EA, Kapitein LC, Tolkovsky AM, Hoogenraad CC, Fawcett JW. The kinesin-2 family member KIF3C regulates microtubule dynamics and is required for axon growth and regeneration. J Neurosci. 2013;33:11329–11345. doi: 10.1523/JNEUROSCI.5221-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond JW, Huang CF, Kaech S, Jacobson C, Banker G, Verhey KJ. Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol Biol Cell. 2010;21:572–583. doi: 10.1091/mbc.E09-01-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehner SP, Hofmann TG, Dröge W, Schmitz ML. The antiinflammatory sesquiterpene lactone parthenolide inhibits NFkappa B by targeting the I kappa B kinase complex. J Immunol. 1999;163:5617–5623. [PubMed] [Google Scholar]
- Hellal F, Hurtado A, Ruschel J, Flynn KC, Laskowski CJ, Umlauf M, Kapitein LC, Strikis D, Lemmon V, Bixby J, Hoogenraad CC, Bradke F. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science. 2011;331:928–931. doi: 10.1126/science.1201148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höke A. Mechanisms of disease: what factors limit the success of peripheral nerve regeneration in humans? Nat Clin Pract Neurol. 2006;2:448–454. doi: 10.1038/ncpneuro0262. [DOI] [PubMed] [Google Scholar]
- Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci. 2010;11:539–551. doi: 10.1038/nrn2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur EM, Saijilafu, Lee BD, Kim SJ, Xu WL, Zhou FQ. GSK3 controls axon growth via CLASP-mediated regulation of growth cone microtubules. Genes Dev. 2011a;25:1968–1981. doi: 10.1101/gad.17015911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur EM, Yang IH, Kim DH, Byun J, Saijilafu, Xu WL, Nicovich PR, Cheong R, Levchenko A, Thakor N, Zhou FQ. Engineering neuronal growth cones to promote axon regeneration over inhibitory molecules. Proc Natl Acad Sci U S A. 2011b;108:5057–5062. doi: 10.1073/pnas.1011258108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur EM, Saijilafu, Zhou FQ. Growing the growth cone: remodeling the cytoskeleton to promote axon regeneration. Trends Neurosci. 2012;35:164–174. doi: 10.1016/j.tins.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liz MA, Mar FM, Santos TE, Pimentel HI, Marques AM, Morgado MM, Vieira S, Sousa VF, Pemble H, Wittmann T, Sutherland C, Woodgett JR, Sousa MM. Neuronal deletion of GSK3beta increases microtubule speed in the growth cone and enhances axon regeneration via CRMP-2 and independently of MAP1B and CLASP2. BMC Biol. 2014;12:47. doi: 10.1186/1741-7007-12-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas FR, Goold RG, Gordon-Weeks PR, Salinas PC. Inhibition of GSK-3beta leading to the loss of phosphorylated MAP-1B is an early event in axonal remodelling induced by WNT-7a or lithium. J Cell Sci. 1998;111:1351–1361. doi: 10.1242/jcs.111.10.1351. [DOI] [PubMed] [Google Scholar]
- Ma CH, Omura T, Cobos EJ, Latrémoliere A, Ghasemlou N, Brenner GJ, van Veen E, Barrett L, Sawada T, Gao F, Coppola G, Gertler F, Costigan M, Geschwind D, Woolf CJ. Accelerating axonal growth promotes motor recovery after peripheral nerve injury in mice. J Clin Invest. 2011;121:4332–4347. doi: 10.1172/JCI58675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knock-in analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen QT, Sanes JR, Lichtman JW. Pre-existing pathways promote precise projection patterns. Nat Neurosci. 2002;5:861–867. doi: 10.1038/nn905. [DOI] [PubMed] [Google Scholar]
- Owen R, Gordon-Weeks PR. Inhibition of glycogen synthase kinase 3beta in sensory neurons in culture alters filopodia dynamics and microtubule distribution in growth cones. Mol Cell Neurosci. 2003;23:626–637. doi: 10.1016/S1044-7431(03)00095-2. [DOI] [PubMed] [Google Scholar]
- Saadane A, Masters S, DiDonato J, Li J, Berger M. Parthenolide inhibits IkappaB kinase, NFkappaB activation, and inflammatory response in cystic fibrosis cells and mice. Am J Respir Cell Mol Biol. 2007;36:728–736. doi: 10.1165/rcmb.2006-0323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scales TM, Lin S, Kraus M, Goold RG, Gordon-Weeks PR. Nonprimed and DYRK1A-primed GSK3 beta-phosphorylation sites on MAP1B regulate microtubule dynamics in growing axons. J Cell Sci. 2009;122:2424–2435. doi: 10.1242/jcs.040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheib J, Höke A. Advances in peripheral nerve regeneration. Nat Rev Neurol. 2013;9:668–676. doi: 10.1038/nrneurol.2013.227. [DOI] [PubMed] [Google Scholar]
- Seira O, Del Río JA. Glycogen synthase kinase 3 beta (GSK3beta) at the tip of neuronal development and regeneration. Mol Neurobiol. 2014;49:931–944. doi: 10.1007/s12035-013-8571-y. [DOI] [PubMed] [Google Scholar]
- Sengottuvel V, Fischer D. Facilitating axon regeneration in the injured CNS by microtubules stabilization. Commun Integr Biol. 2011;4:391–393. doi: 10.4161/cib.15552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengottuvel V, Leibinger M, Pfreimer M, Andreadaki A, Fischer D. Taxol facilitates axon regeneration in the mature CNS. J Neurosci. 2011;31:2688–2699. doi: 10.1523/JNEUROSCI.4885-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Miller BR, Babetto E, Cho Y, Sasaki Y, Qayum S, Russler EV, Cavalli V, Milbrandt J, DiAntonio A. SCG10 is a JNK target in the axonal degeneration pathway. Proc Natl Acad Sci U S A. 2012;109:E3696–E3705. doi: 10.1073/pnas.1216204109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Cho Y, Watt D, Cavalli V. Tubulin-tyrosine ligase (TTL)-mediated increase in tyrosinated alpha-tubulin in injured axons is required for retrograde injury signaling and axon Regeneration. J Biol Chem. 2015;290:14765–14775. doi: 10.1074/jbc.M114.622753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi N, Marsh P, Goold RG, Wood-Kaczmar A, Gordon-Weeks PR. Glycogen synthase kinase-3beta phosphorylation of MAP1B at Ser1260 and Thr1265 is spatially restricted to growing axons. J Cell Sci. 2005;118:993–1005. doi: 10.1242/jcs.01697. [DOI] [PubMed] [Google Scholar]
- Ulloa L, Díez-Guerra FJ, Avila J, Díaz-Nido J. Localization of differentially phosphorylated isoforms of microtubule-associated protein 1B in cultured rat hippocampal neurons. Neuroscience. 1994;61:211–223. doi: 10.1016/0306-4522(94)90225-9. [DOI] [PubMed] [Google Scholar]
- Utreras E, Jiménez-Mateos EM, Contreras-Vallejos E, Tortosa E, Pérez M, Rojas S, Saragoni L, Maccioni RB, Avila J, González-Billault C. Microtubule-associated protein 1B interaction with tubulin tyrosine ligase contributes to the control of microtubule tyrosination. Dev Neurosci. 2008;30:200–210. doi: 10.1159/000109863. [DOI] [PubMed] [Google Scholar]
- Witzel C, Rohde C, Brushart TM. Pathway sampling by regenerating peripheral axons. J Comp Neurol. 2005;485:183–190. doi: 10.1002/cne.20436. [DOI] [PubMed] [Google Scholar]
- Yip-Schneider MT, Nakshatri H, Sweeney CJ, Marshall MS, Wiebke EA, Schmidt CM. Parthenolide and sulindac cooperate to mediate growth suppression and inhibit the nuclear factor-kappa B pathway in pancreatic carcinoma cells. Mol Cancer Ther. 2005;4:587–594. doi: 10.1158/1535-7163.MCT-04-0215. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron. 2004;42:897–912. doi: 10.1016/j.neuron.2004.05.011. [DOI] [PubMed] [Google Scholar]