

Abstract
Motor cortical plasticity contributes to spontaneous recovery after incomplete spinal cord injury (SCI), but the pathways underlying this remain poorly understood. We performed optogenetic mapping of motor cortex in channelrhodopsin-2 expressing mice to assess the capacity of the cortex to re-establish motor output longitudinally after a C3/C4 dorsal column SCI that bilaterally ablated the dorsal corticospinal tract (CST) containing ∼96% of corticospinal fibers but spared ∼3% of CST fibers that project via the dorsolateral funiculus. Optogenetic mapping revealed extensive early deficits, but eventual reestablishment of motor cortical output maps to the limbs at the same latency as preoperatively by 4 weeks after injury. Analysis of skilled locomotion on the horizontal ladder revealed early deficits followed by partial spontaneous recovery by 6 weeks after injury. To dissociate between the contributions of injured dorsal projecting versus spared dorsolateral projecting corticospinal neurons, we established a transient silencing approach to inactivate spared dorsolaterally projecting corticospinal neurons specifically by injecting adeno-associated virus (AAV)-expressing Cre-dependent DREADD (designer receptor exclusively activated by designer drug) receptor hM4Di in sensorimotor cortex and AAV-expressing Cre in C7/C8 dorsolateral funiculus. Transient silencing uninjured dorsolaterally projecting corticospinal neurons via activation of the inhibitory DREADD receptor hM4Di abrogated spontaneous recovery and resulted in a greater change in skilled locomotion than in control uninjured mice using the same silencing approach. These data demonstrate the pivotal role of a minor dorsolateral corticospinal pathway in mediating spontaneous recovery after SCI and support a focus on spared corticospinal neurons as a target for therapy.
SIGNIFICANCE STATEMENT Spontaneous recovery can occur after incomplete spinal cord injury (SCI), but the pathways underlying this remain poorly understood. We performed optogenetic mapping of motor cortex after a cervical SCI that interrupts most corticospinal transmission but results in partial recovery on a horizontal ladder task of sensorimotor function. We demonstrate that the motor cortex can reestablish output to the limbs longitudinally. To dissociate the roles of injured and uninjured corticospinal neurons in mediating recovery, we transiently silenced the minor dorsolateral corticospinal pathway spared by our injury. This abrogated spontaneous recovery and resulted in a greater change in skilled locomotion than in uninjured mice using the same approach. Therefore, uninjured corticospinal neurons substantiate remarkable motor cortical plasticity and partial recovery after SCI.
Keywords: corticospinal, motor cortex, optogenetics, plasticity, recovery, spinal cord injury
Introduction
Spinal cord injury (SCI) typically results in sustained motor, sensory, and autonomic dysfunction as a result of the limited capacity of the adult mammalian CNS to regenerate lost connections (Ramer et al., 2014). However, some spontaneous functional improvement commonly occurs, even after cervical, functionally sensorimotor complete injuries (Roth et al., 1991; Steeves et al., 2011) by one year after injury. Such spontaneous recovery is well recapitulated by animal models of SCI, in which a detectable degree of functional recovery is observed without intervention in both cervical and thoracic injury models (Basso et al., 1995; Gensel et al., 2006; Hilton et al., 2013). The sensorimotor cortex is one substrate underlying spontaneous recovery after SCI and a wide range of other CNS disorders (Fouad et al., 2001; Raineteau and Schwab, 2001; Nudo, 2006; Nishimura and Isa, 2009). Corticospinal fibers exhibit anatomical plasticity that is correlated with spontaneous recovery based on some recovery observed when these fibers are spared in partial injury models (Weidner et al., 2001; Ghosh et al., 2009; Krajacic et al., 2010; Rosenzweig et al., 2010; Oudega and Perez, 2012). Corticospinal fibers also have the capacity to form relay circuits with propriospinal neurons after SCI in the absence of intervention (Bareyre et al., 2004; Vavrek et al., 2006; Ghosh et al., 2010). Nevertheless, the pathways through which the motor cortex mediates repair remain only partially understood and the extent to which corticospinal-mediated recovery is a result of injured versus uninjured pathways is unknown.
Here, we performed a series of optogenetic, pharmacogenetic, behavioral, and anatomical experiments to assess the role of the motor cortex in mediating spontaneous recovery after a cervical level C3/C4 dorsal column SCI that ablates the dorsal corticospinal tract (dCST) containing ∼96% of corticospinal fibers. This lesion also severs dorsal column sensory afferents bilaterally cranial to most spinal neurons subserving forelimb function (Asante and Martin, 2013) and all spinal neurons subserving hindlimb function. Using optogenetic mapping, which provides a noninvasive, longitudinal, and quantitative approach to assessing cortical motor output (Ayling et al., 2009), we find that the motor cortex is able to reestablish motor output to the forelimb and hindlimb at the same latencies as preoperatively by 4 weeks after injury despite massive corticospinal and sensory loss. In C57BL/6 background mice, there are ∼12,000 dorsal, ∼400 dorsolateral, and ∼100 ventral corticospinal axons at the cervical level, corresponding, respectively, to ∼96%, ∼3%, and ∼1% of the total (Fink et al., 2015). Hypothesizing that uninjured corticospinal neurons substantiated the reestablishment of cortical motor output and spontaneous recovery, we took a pharmacogenetic approach to transiently inactivate spared dorsolaterally projecting corticospinal neurons by injecting an adeno-associated virus (AAV)-expressing Cre-dependent hM4Di in sensorimotor cortex and an AAV-expressing Cre in the dorsolateral funiculus well caudal to the injury in both injured and uninjured mice (Rogan and Roth, 2011). After CNO administration in mice with C3/C4 dorsal column SCI, spontaneous recovery was abrogated and there was a greater change in skilled locomotion than in control uninjured mice using the same inactivation approach. Given that only a fraction of the corticospinal neurons in the dorsolateral funiculus are silenced effectively, these data indicate that small numbers of spared dorsolaterally projecting corticospinal neurons mediate remarkable motor cortical plasticity and recovery after SCI. Targeting the plasticity of a few spared corticospinal neurons may thus be sufficient to drive substantial recovery after SCI and perhaps other CNS disorders or diseases.
Materials and Methods
SCI.
All procedures were approved by the University of British Columbia Animal Care Committee in accordance with the guidelines of the Canadian Council on Animal Care. Female 3- to 4-month-old mice expressing Channelrhodopsin-2 under the Thy-1 promoter [line 18, stock 007612, strain B6.Cg-Tg(Thy1-COP4/EYFP)18Gfng/J] on the C57BL/6 background were used. For all surgeries and mapping procedures, body temperature was maintained at 37 ± 0.5°C with an electric heating pad regulated by a rectal thermometer. Animals were anesthetized with isoflurane (4% for induction and 1.5% for maintenance). The skin overlying the neck was shaved, disinfected with Betadine, wiped off with 70% ethanol, and cut with a scalpel blade. The muscles of the dorsal neck were split along the midline, a laminectomy was performed to expose the third cervical spinal cord segment, and the dura mater overlying the centers of the dorsal horns was pierced on each side of the spinal cord with a 26 Ga needle. Fine-tipped Dumont #5 forceps with blades ground to a width of ∼200 μm were inserted into the dorsal horns on each side of the dorsal column to a depth of ∼1 mm. The forceps were closed and held for 20 s to crush the dorsal column completely, including the main contingent of corticospinal axons; the forceps were removed and inserted again to repeat this crush once. Sham-operated mice received a laminectomy only (Hilton et al., 2013).
Horizontal ladder.
Error frequency was assessed for the forelimbs and hindlimbs and using a horizontal ladder modified as described previously (Cummings et al., 2007; Hilton et al., 2013). Briefly, mice were videotaped with a high-definition camera (Sony, HDR-Hc1) as they crossed a 5.2-cm-wide horizontal ladder with 31 rungs spaced 1.3 cm apart. Each mouse repeated the task five times. Frame-by-frame video analysis by a blinded observer was used to generate scores for the number of errors (rung misses, rung slips, or rung drags) and the number of steps for the FLs and HLs over each run and percentage error values were calculated from each (percentage error = number of errors/number of steps × 100). Mice were trained to perform the task by crossing the ladder 3 times each 2 and 3 d before injury. One day before injury, mice were tested preoperatively to obtain preinjury scores, 4 h postoperatively for acute scores, and on 3, 7, 14, 21, 28, 42, and 56 d postoperatively.
Chronic cranial window.
Under isofluorane anesthesia (4% for induction and 1.5% for maintenance mixed with 1% oxygen), C&B Metabond was applied generously on top of the skull and a 5 × 5 mm cover glass was placed on top of the right hemisphere over intact skull and pressed flat into the adhesive. If necessary, more adhesive was applied around the cover glass for reinforcement. Mice were permitted to recover under a heat lamp and preinjury mapping commenced 1 week after the chronic cranial window preparation.
Optogenetic motor mapping.
The methodology for optogenetic motor mapping has been described in detail previously (Ayling et al., 2009). Briefly, we used a scanning stage (ASAI, MS-2000) controlled by custom Igor Pro software (Wavemetrics) to direct a fixed 473 nm laser beam (Crystalaser, 10 ms pulses, 0.5–2 mW total or 63–252 mW/mm2) to a grid of cortical sites, typically 12 × 14, with 300 μm spacing. This process was repeated two to three times to obtain a mean value for each pixel of the map. Stimulation was delivered in a semirandom order with the requirement that sites stimulated in sequence be >750 μm away from each other. Optogenetic motor mapping occurred with identical stimulus intensity for all sites within a map and for each mouse at all time points before and after SCI. Movements were detected using laser range motion sensors (Keyence, LK-081) with submillimeter sensitivity targeted to the left FL and HL, which were fitted with bracelets made of rubber tubing (Etsy) glued to 12-mm-diameter round coverslips painted white, which provided large, flat targets for the motion sensors. The limbs were suspended slightly above the ground so that they could move freely along the axes of measurement of the motion sensors. Responses were considered to be genuine only if their amplitude exceeded three times the SD of the 500 ms prestimulus period within 100 ms after stimulus onset. A single injection of ketamine/xylazine (100/10 mg/kg) was administered at the beginning of each mapping session (duration 20–30 min). With this dose of anesthetic, mapping of the entire cortex could be performed in 20–30 min without requiring a supplementary dose, which might have introduced the possibility of anesthetic artifacts. We mapped each mouse 1 week before injury to establish baseline maps, acutely, and again at 3, 7, 14, 21, and 28 d postoperatively.
Dorsolateral corticospinal DREADD (designer receptor exclusively activated by designer drug) experiment.
Viral-mediated expression of a G-protein-coupled receptor (Gi/o-coupled human muscarinic M4 designer receptor exclusively activated by a designer drug, hM4Di) activated by the otherwise pharmacologically inert ligand clozapine-N-oxide (CNO) was used to pharmacogenetically and transiently (1–2 h; Rogan and Roth, 2011) silence dorsolateral corticospinal fibers that projected 3–4 segments caudal to the injury. Cre-dependent AAV1-hSyn-dio-hM4D(Gi)-mCherry (UNC Vector Core, University of North Carolina at Chapel Hill) was injected into the sensorimotor cortices of mice (1.5 μl total, 0.5 μl per injection site at 3 injection sites: rostral/caudal coordinates from bregma in mm: 0.6 mm, 0.1 mm, −0.4 mm; all 1.5 mm right of the midline and 0.7 mm depth from the surface of the brain) in n = 12 mice at 4 weeks after injury and n = 12 mice 4 weeks after sham operation. In n = 6 injured and n = 6 sham mice, cortical AAV injection was followed by spinal injection with AAV1-Cre (Vector Biolabs) into the left dorsolateral funiculus (0.6 μl total, 0.3 μl per injection site into 2 injection sites; into dorsolateral gray matter at 0.5 mm depth at C6/C7 and C7/C8 after hemilaminectomies at these sites). Control animals received cortical AAV injections only. At 56 d after injury, mice were recorded for 5 runs on the horizontal ladder task starting 10 min after injection of CNO (1 mg/kg, i.p.; Enzo Life Sciences). This was repeated at 58 d after injury; on days 55 and 57 after injury, mice were recorded on the horizontal ladder task after vehicle intraperitoneal administration. The number of errors and placements by the FLs were recorded in a blinded fashion by a naive rater scoring the videos. The results from days 56 and 58 after injury were averaged for each animal to generate a CNO error score and the results from days 55 and 57 were the vehicle error score.
Sensorimotor cortical immunohistochemistry.
After the DREADD experiment described above, at 59 or 60 d after injury, half of the injured and half of the sham-operated mice were administered CNO (1 mg/kg) and the other half received vehicle. Mice were then run repeatedly on the horizontal ladder for 15 min in an effort to induce c-fos gene expression. One hour after the conclusion of the horizontal ladder, mice were killed with an overdose of chloral hydrate (100 mg/kg, i.p.) and perfused transcardially with PBS, followed by phosphate-buffered freshly hydrolyzed 4% paraformaldehyde, pH 7.5. Motor cortices and cervical spinal cords were dissected, postfixed in 4% paraformaldehyde overnight, and cryoprotected in 12%, 18%, and 24% sucrose in 0.1 m phosphate buffer over 3 d before being snap frozen in isopentane over dry ice. Motor cortices (from ∼2 mm caudal to bregma to ∼2 mm rostral to bregma) were cut into 20 μm cross-sections on a cryostat and stored at −80°C. Frozen sections were thawed for 1 h, rehydrated in 10 mm PBS for 10 min, and incubated with 10% normal donkey serum (in 0.1% Triton X-100) for 30 min. Goat anti-c-Fos (1:500; Santa Cruz Biotechnology) and rabbit anti-dsRed (1:200; Clontech) were applied overnight at room temperature. Secondary antibody (1:200; Jackson Laboratories) raised in donkey and conjugated to Alexa Fluor 405 and 594 were applied for 2 h at room temperature in addition to fluorescent Nissl 640/660 (1:500, Neurotrace; Invitrogen). Sections were coverslipped in Fluoromount-G (Southern Biotech). In a blinded fashion, images were taken of sensorimotor cortex sections (∼2 mm caudal to bregma to ∼2 mm rostral to bregma; consistent with area that, when optically stimulated, led to limb movement) on which mCherry+ neurons could be identified with a Zeiss Axio Observer Z1 equipped with a Yokogawa X-1 Spinning Disk using Zen Blue software at 20× magnification. Counts of mCherry+ neuron numbers and c-fos+ mCherry+ neuron numbers were then made based on these images. The layer identification of mCherry+ neurons was made based on Nissl staining. Imaging and counts were made blinded to injury status and CNO/vehicle administration.
mCherry spinal cord analysis.
Sections of cervical spinal cord were incubated with rabbit anti-dsRed (1:200; Clontech) overnight at room temperature, followed by secondary antibody raised in donkey and conjugated to Alexa Fluor 594 (1:200; Jackson Laboratories) for 2 h at room temperature. Sections were coverslipped in Fluoromount-G (Southern Biotech). In a blinded fashion, images of three sections for each animal of the dorsal columns, left dorsolateral funiculus, and ventral funiculus were taken with a Zeiss Axio Observer Z1 equipped with a Yokogawa X-1 Spinning Disk using Zen Blue software at 40× magnification. Counts of mCherry+ fiber numbers in the dorsal and dorsolateral funiculi were then made based on these images and averaged. Imaging and counts were made blinded to injury.
dCST injury analysis.
Frozen cervical spinal cord sections were thawed for 1 h, rehydrated in 10 mm PBS for 10 min, and incubated with 10% normal donkey serum (in 0.1% Triton X-100) for 30 min. Rabbit anti-PKCγ (1:200; Millipore) and goat anti-green fluorescent protein (GFP, 1:1000; DAKO) were applied overnight at room temperature. Secondary antibody (1:200; Jackson Laboratories) raised in donkey and conjugated to Alexa Fluor 488 and 594 were applied for 2 h at room temperature. Sections were coverslipped in Fluoromount-G (Southern Biotech). To verify injury to the corticospinal tract, digital images of transverse sections of C1 and C8 spinal segments, in addition to transverse sections of the lesion site, were captured with an Axioplan 2 microscope (Zeiss) equipped with Northern Eclipse software (Empix Imaging) at 10×. Eriochrome Cyanine R staining was used to assess lesion epicenter area and PKCγ and YFP immunoreactivity were used to assess the completeness of dCST lesions.
Statistics.
All data are presented as mean ± SEM. Statistical significance was set at p < 0.05. Homogeneity of variance was tested for using Bartlett's test. Normality was tested for using the Shapiro–Wilk test. Horizontal Ladder data and optogenetic motor mapping data were analyzed using two-way repeated-measures ANOVAs (groups vs time). DREADD experimental data were analyzed using both a two-way ANOVA (groups vs time) and a one-way ANOVA comparing CNO versus vehicle scores across groups. mCherry fiber counts were analyzed using either the Mann–Whitney U test or a one-sample t test. mCherry and c-fos neuron counts/percentages were analyzed using Mann–Whitney U tests. Multiple-comparisons corrections and statistical significance were determined using the Holm–Sidak method. Statistical tests were performed using GraphPad Prism version 6.0 software.
Results
Longitudinal optogenetic and behavioral analysis of motor system deficits and recovery after SCI
We performed a series of optogenetic, pharmacogenetic, behavioral, and anatomical experiments in line-18 transgenic mice that express ChR-2 robustly in layer 5 pyramidal neurons driven by the Thy-1 promoter (Arenkiel et al., 2007; Ayling et al., 2009). In Thy1-ChR2 mice, we found high expression of ChR-2 within the dCST via immunostaining of sections of cervical spinal cord (Fig. 1A) and performed anterograde tracing of sensorimotor cortex with BDA to assess corticospinal anatomy. This revealed that the vast majority of corticospinal axons descended via the dCST (Fig. 1B) with a minor component descending via the dorsolateral funiculus (dlCST; Fig. 1C). Very little to no BDA was found within the ventral funiculus (Fig. 1D), although a new study with near complete labeling of the CST has found that there are ∼100 ventral corticospinal axons in C57BL/6 mice in addition to ∼12,000 dorsal and ∼400 dorsolateral (Fink et al., 2015), corresponding to ∼96% dCST, ∼3% dlCST, and ∼1% vCST, respectively.
Figure 1.

Optogenetic and pharmacogenetic dissection of motor cortical plasticity underlying recovery after SCI. A, Cross-section through cervical spinal cord in a Thy1-ChR2 mouse immunostained for PKCγ (red), YFP (green), and BDA (pink) with euthanasia 2 weeks after BDA injections into sensorimotor cortex. B, Close-up of the dorsal column showing BDA+ corticospinal axons comprising the dCST. C, Close-up of the dorsolateral funiculus showing BDA+ corticospinal axons comprising the dlCST and their branches into gray matter. D, Close-up of the ventral column showing sparse BDA+ immunoreactivity. E, Schematic of descending motor corticofugal pathways involved in distal limb control. The motor cortex projects directly to the spinal cord via the dCST (∼96%), dlCST (∼3%), and vCST (∼1%), but also provides major excitatory input to the red nucleus (RN) and reticular formation (RF), which descend the rubrospinal tract and reticulospinal tract, respectively. After C3/C4 SCI, the dCST is bilaterally interrupted, but other motor corticofugal pathways remain intact. F, Horizontal ladder task of sensorimotor dysfunction and recovery after C3/C4 dorsal column SCI. Arrows denote errors of the forelimb and/or hindlimb. G, Longitudinal optogenetic motor mapping through intact skull before and after SCI. Anesthetized, head-fixed mice are placed with their left FL and HL suspended from the ground to allow free movement. Laser motion sensors direct at targets on the limbs record movements evoked by optogenetic cortical stimulation. Mapping is repeated in the same animal at multiple time points after SCI. A chronic cranial window preparation allows optical access through completely intact skull. The window is directed over the right cortical hemisphere and centered around bregma (yellow square) rostro-caudally. A 12 × 14 array of cortical points with 300 μm spacing is stimulated in semirandom order by a fixed ∼100-μm-diameter 473 nm laser (10 ms pulse). After three repetitions of stimulation, a map of average evoked forelimb and hindlimb movements are assembled and scaled based on amplitude. Inset brain atlas was adapted from the Allen Brain Atlas and indicates approximate location of motor mapping area within the red outline. Bright green area is an approximate area of primary motor cortex. Scale bars: A, 500 μm; B–D, 50 μm.
The motor cortex projects to the spinal cord via the dCST, dlCST, and vCST, but also provides major excitatory input to brainstem motor nuclei which also descend spinal pathways involved in limb control, including the red nucleus (which projects the rubrospinal tract) and the reticular formation (which projects the reticulospinal tract) (Fig. 1E). After C3/C4 dorsal column SCI, the dCST is interrupted, but corticofugal pathways to these brainstem nuclei in addition to the dlCST and vCST are spared and might mediate recovery. We assessed sensorimotor behavioral dysfunction using a horizontal ladder task and quantified motor cortical output to the limbs from acute to chronic stages of injury using optogenetic motor mapping (Ayling et al., 2009; Harrison et al., 2012; Harrison et al., 2013; Anenberg et al., 2014), which provides repeated, fast, and relatively selective optical interrogation of motor corticofugal circuitry. To map as noninvasively as possible, we used a chronic cranial window preparation through the intact skull that provided optical access to the right cortical hemisphere (Fig. 1G; Silasi et al., 2013). Cranial windows were 5 × 5 mm in size, extending ∼1 mm across the midline and ∼2.5 mm rostral and ∼2.5 mm caudal from bregma. Mice were allowed at least 1 week to recover from cranial window surgery before motor mapping commenced. To generate cortical motor maps, laser motion sensors directed at targets worn by the left wrist and ankle of the mouse recorded movements evoked by optogenetic stimulation of a 12 × 14 grid of cortical points over the right hemisphere (Fig. 1G), with maps generated as the mean of responses from 2 to 3 repetitions with the minimum laser amplitude (0.5–2 mW) necessary to evoke movement for each animal before injury. To exclude artifactual movement, responses were only considered to be genuine if their amplitude exceeded three times the SD of the 500 ms prestimulus period within 100 ms after stimulus onset.
C3/C4 dorsal column SCI results in sustained deficits and partial recovery on the horizontal ladder task
We first used an SCI model to bilaterally ablate the dCST and dorsal column sensory afferents and assessed the effect of this injury on behavioral sensorimotor function using a horizontal ladder task (Hilton et al., 2013; Wang et al., 2015). To sever the dCST and sensory afferents, custom-made forceps with tips ground to ∼200 μm (Hilton et al., 2013) were used to lesion the dorsal column at the cervical C3/C4 level ventral to the level of at least the central canal, which disrupted the dorsal columns and dCST axons and partially compromised the gray matter of the dorsal horns and intermediate zone (Fig. 2A–C). Animals with lesions that did not sever the entire dCST by absence of either YFP or PKCy staining caudal to injury were excluded from analysis (n = 1). Sham-operated animals received a laminectomy at C3/C4 only. On the horizontal ladder task, although C3/C4 dorsal column injury resulted in a deficit in forelimb skilled locomotion to 56 d after injury, there was spontaneous yet incomplete recovery on days 42 and 56 after injury (Fig. 2D,E). At 7 d after injury, mice with C3/C4 dorsal column SCI made FL errors during 24.4 ± 2.6% of placement attempts made versus 3.78 ± 0.65% before injury. However, at 56 d after injury, the same mice made forelimb errors during 11.3 ± 1.02% of placement attempts, representing a 48.7 ± 5.17% recovery from the 7 d after injury time point (p < 0.01). Still, even at 56 d after injury, the error percentage for the forelimbs was higher than preoperatively, indicating that the recovery was only partial (p < 0.05). Hindlimb horizontal ladder function completely and spontaneously recovered by 14–21 d after injury relative to sham operation (Fig. 2F,G). Therefore, C3/C4 dorsal column SCI results in sustained deficits and partial spontaneous recovery in forelimb skilled locomotion by 56 d after injury.
Figure 2.

Partial spontaneous recovery and sustained deficits in skilled locomotion after C3/C4 dorsal column SCI. A, Left, Transverse (cross-) sections of cervical spinal cord rostral to a dorsal column lesion immunostained for YFP and PKCγ showing the presence of the dorsal corticospinal tract. YFP labels neurons diffusely in both gray and white matter and extensively labels the dCST in Thy1-ChR2/YFP mice. Middle, Transverse sections of cervical spinal cord at the lesion epicenter immunostained for PKCy and YFP. The lesion includes the dorsal column and the dCST bilaterally. Right, Transverse sections of cervical spinal cord caudal to the SCI immunostained for PKCy and YFP. B, C, Close-up on dorsal column for PKCy and YFP rostral (B) and caudal (C) to injury. Note the absence of the dCST caudal to injury. C–G, Horizontal ladder error percentage (errors/steps * 100) for the left forelimb (D), right forelimb (E), left hindlimb (F), and right hindlimb (G) analyzed using two-way ANOVA with a Holm–Sidak's multiple-comparisons test. The left and right FLs have significantly higher error percentages up to 56 d after injury versus preinjury. ***p < 0.001, **p < 0.01, *p < 0.05 versus preoperation. The left and right FLs have significantly lower error percentages at 42 and 56 d after injury versus 7 d after injury. ##p < 0.01, #p < 0.05 versus 7 d after injury. The left and right HLs completely recover horizontal ladder placement by 7–14 d after injury. ***p < 0.001, **p < 0.01, *p < 0.05 versus preoperation. n = 16 SCI mice and n = 8 sham-operated mice. Error bars indicate SEM. Scale bars: A, 500 μm; B, 50 μm.
Effect of acute SCI on cortical motor output
Because optogenetic motor mapping can generate representations in as few as 5 min, it is at least an order of magnitude faster than electrical-stimulation-based approaches and thus capable of assessing how the motor cortex responds immediately after injury (Anenberg et al., 2014). To assess how the motor cortex responds after acute SCI, we initiated mapping 10–30 min after injury. To aid map analysis, motor maps were upsampled to pixel sizes of 100 × 100 μm and aligned based on the position of bregma. Acutely, although optogenetic stimulation consistently evoked forelimb and hindlimb movement, forelimb motor cortex (FL) and hindlimb motor cortex (HL) were greatly diminished in area and output (Fig. 3). FL was reduced from an area of 7.69 ± 0.89 mm2 preoperatively to an area of 2.34 ± 0.17 mm2 acutely (p < 0.001). FL motor output was also reduced to 24.29 ± 12.25% that of baseline (p < 0.01). Notably, FL was displaced after acute SCI, with a caudal shift in center position from 0.30 ± 0.14 mm rostral to bregma preoperatively to 0.35 ± 0.07 mm caudal to bregma acutely, occupying a similar territory as HL (p < 0.01). HL was also reduced in area from 5.14 ± 0.49 mm2 to 1.95 ± 0.24 mm2 (p < 0.001) and output was reduced to 39.57 ± 22.81% that of baseline (p < 0.05). In sham-operated mice, no changes in map properties were observed (all p > 0.24).
Figure 3.
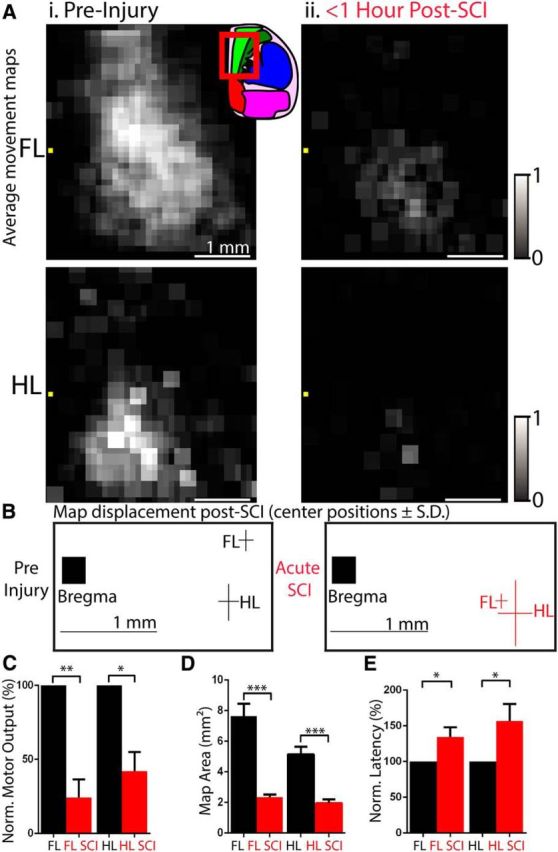
Effect of acute SCI on motor maps. A, Average upsampled ChR2-stimulated motor maps (100 × 100 μm pixels) normalized to their respective mean and aligned based on the location of bregma (yellow square). Average FL and HL motor maps before injury (Ai) and immediately after C3/C4 dorsal column SCI (Aii) (<1 h after injury; n = 8). Inset brain atlas was adapted from the Allen Brain Atlas and indicates approximate location of motor mapping area within the red outline. Bright green area is an approximate area of primary motor cortex. B, Position of center of gravity of FL and HL maps with respect to bregma. FL is displaced caudally from 0.30 ± 0.14 mm rostral to bregma preoperatively to 0.35 ± 0.07 mm caudal to bregma acutely. C, FL and HL motor amplitude are reduced after acute SCI. D, FL and HL map area defined by the number of cortical sites capable of generating FL and HL movement, respectively, are reduced after acute SCI. E, Latencies of optogenetic stimulation to FL and HL movement are increased after acute SCI. n = 8 injured mice. *p < 0.05, **p < 0.01, ***p < 0.001, preinjury versus acute injory. Error bars indicate SEM.
Longitudinal reestablishment of motor cortical output by 4 weeks after injury
Next, we investigated the capacity of FL and HL motor cortex to reestablish output after C3/C4 dorsal column injury by mapping the same mice at 3, 7, 14, 21, and 28 d postoperatively. We observed spontaneous reestablishment of both FL and HL motor maps longitudinally (Fig. 4). Although FL remained displaced caudally at 3 d after injury relative to preoperatively (p < 0.05), by 7 d after injury, it had reestablished the same position as preoperatively and remained in this position up to 28 d after injury. On a similar timeline, FL motor output was reestablished by 7 d after injury, whereas map area and latency to FL movement were reestablished by 21 d after injury. HL also reestablished output spontaneously after injury, with motor map area and output diminished at 3 d after injury but reestablished by 7 d after injury and latency to HL movement reestablished by 14 d after injury. Therefore, even in the absence of the vast majority of descending corticospinal transmission and substantial sensory loss, the adult mouse cortex has the capacity to reestablish FL and HL motor output (with properties such as latency similar to preinjury) by 4 weeks after injury.
Figure 4.

Spontaneous motor map reestablishment after C3/C4 dorsal column SCI. A, Baseline averaged ChR2-stimulated motor maps of the FL and HL at multiple time points before and after SCI. Bregma is denoted by the yellow square in each panel. B, FL motor map area, defined by the number of cortical sites from which movement could be generated is lower acutely and 3, 7, and 14 d after SCI. C, FL motor output, defined as the average maximal displacement at the 9 pixels encompassing the center of gravity, is significantly lower acutely and at 3 and 7 d after SCI. D, Latency to FL movement is longer acutely and at 3, 7, and 14 d after SCI. E, The FL map shifts caudally after SCI acutely and at 3 d after SCI. F, HL motor map area is lower acutely and at 3 d after SCI. G, HL motor output is lower acutely and at 3 and 7 d after SCI. H, Latency to HL movement is longer acutely and at 3 and 7 d after SCI. I, HL map does not shift in center position after SCI. n = 8 injured and n = 8 sham mice. *p < 0.05, **p < 0.01, ***p < 0.001 between groups. Error bars indicate SEM.
Specific DREADD receptor express ion in dorsolaterally projecting corticospinal neurons
Our optogenetic motor mapping experiments demonstrated that the motor cortex was able, over the course of 3–4 weeks, to reestablish output to forelimb and hindlimb longitudinally after C3/C4 dorsal column SCI at the same latency as preoperatively, suggesting that the corticospinal pathway underlies the partial recovery observed on the horizontal ladder. Axotomized corticospinal neurons harbor a capacity for relay formation with propriospinal neurons that may have mediated the recovery observed (Bareyre et al., 2004; Vavrek et al., 2006; Nishimura and Isa, 2012). Alternatively, recovery may have occurred due to rewiring of the minor dorsolateral corticospinal pathway that was spared from our injury paradigm. To dissociate between these two possibilities, we use a Cre-dependent pharmacogenetic strategy to specifically and transiently inactivate specifically and transiently spared dorsolaterally projecting corticospinal neurons (Fig. 5). Corticospinal neurons harbor a capacity for retrograde transduction by AAVs after spinal administration (Jara et al., 2012; Jara et al., 2014; Wahl et al., 2014). We took advantage of this characteristic by administering a Cre-dependent hM4Di expressing AAV to sensorimotor cortex and AAV-expressing Cre several (3–4) segments caudal to injury or sham operation at 4 weeks after injury (hM4Di Cre+). Therefore, only dorsolaterally projecting corticospinal neurons would be dual transduced in injured mice because the dCST would not be in proximity to the Cre virus at C6/C7 or C7/C8 after its ablation at C3/C4. In control mice, no Cre virus was administered (hM4Di Cre-). hM4Di is a Gi/o-coupled DREADD receptor that is activated specifically by the pharmacologically inert ligand CNO. After CNO activation, hM4Di activates G-protein-associated inwardly rectifying potassium channels, leading to membrane hyperpolarization and silencing of neurons for ∼1–2 h (Rogan and Roth, 2011). hM4Di was tagged with mCherry, allowing us to verify that expression was confined to dorsolaterally projecting corticospinal neurons in injured mice (Fig. 5D). No mCherry+ labeling was found within the ventral funiculus, excluding dual transduction of ventrally projecting corticospinal neurons (Fig. 5G). This was not a surprise because the vCST is an ipsilaterally projecting pathway (Weidner et al., 2001), so the left vCST descended from the left motor cortex, which was not injected with virus. In total, we counted 12.17 ± 0.79 mCherry+ dorsolateral corticospinal fibers in injured mice and 11.67 ± 0.67 dorsolateral corticospinal fibers in uninjured mice (Fig. 5E; p = 0.55). However, in sham-operated mice, our dual viral strategy also led to the appearance of 83.67 ± 22.23 mCherry+ dorsal corticospinal fibers, in contrast to the absence of any dCST fibers observed in SCI mice (Fig. 5F; p < 0.05), as a result of transduction of the uninjured dCST in sham-operated mice. Therefore, there was a significantly higher total number of mCherry+ corticospinal fibers in sham-operated mice versus SCI mice (p < 0.01) but, in injured mice, mCherry was confined to dorsolaterally projecting corticospinal neurons.
Figure 5.
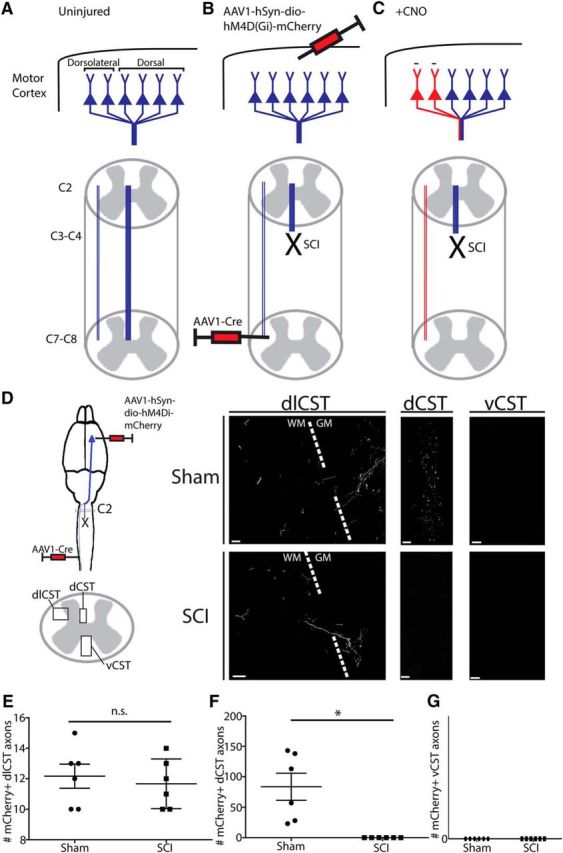
Specific targeting of DREADD receptor hM4Di to spared dorsolaterally projecting corticospinal neurons. A–C, Schematic of targeting uninjured dorsolaterally projecting corticospinal neurons. A, Corticospinal neurons originating in layer V motor cortex project their axons via the dCST and dlCST. B, At 4 weeks after injury, an AAV-expressing Cre-dependent hM4Di (AAV1-hSyn-dio-hM4D(Gi)-mCherry) was administered to sensorimotor cortex and an AAV-expressing Cre (AAV1-Cre) was administered to the dorsolateral funiculus at C7/C8, such that only spared dorsolaterally projecting corticospinal neurons are transduced by both viruses in injured animals. C, After CNO administration, only corticospinal neurons expressing both viruses are silenced for 1–2 h (red); dCST neurons axotomized by the dorsal column SCI are not silenced. D, Histological verification of mCherry+ dual-transduced corticospinal axons at C2 spinal cord rostral to the injury. After SCI, mCherry+ corticospinal axons can be observed in the dorsolateral funiculus, but not in the dorsal column. After sham operation, mCherry+ corticospinal axons can be observed in both the dorsolateral funiculus and in the dorsal column. E, F, Quantification of mCherry+ axon numbers in the dorsolateral funiculus and dorsal column in injured and sham-operated mice. E, There is no difference in the number of mCherry+ axons found in the dorsolateral funiculus between SCI and sham-operated mice. F, There are significantly more mCherry+ axons in the dorsal column in sham-operated mice versus SCI mice. G, There are no mCherry+ axons in the ventral funiculi. *p < 0.05. Scale bars in D are 20 μm. Error bars indicate SEM.
DREADD receptor activation in dorsolaterally projecting corticospinal neurons abrogates spontaneous recovery on the horizontal ladder
We assessed the necessity of dorsolaterally projecting corticospinal neuronal function in the spontaneous recovery observed on the horizontal ladder task with hM4Di activation by CNO at 56 and 58 d after injury and compared it with execution of the task at 55 and 57 d after injury after vehicle administration (Fig. 6). Although a lower left forelimb error percentage was observed after vehicle administration at 55 and 57 d versus 7 d after injury, no difference was observed versus 7 d after injury after CNO administration at 56 and 58 d (Fig. 6B; p < 0.01), indicating that the dorsolaterally projecting corticospinal neurons silenced were necessary for the spontaneous recovery observed. CNO administration led to a higher left forelimb error percentage in SCI and sham-operated hM4Di Cre+ mice (p < 0.001 for SCI and p < 0.05 for sham; Fig. 6C), but the loss of performance in CNO-treated versus vehicle-treated mice was higher in the SCI versus the sham-operated group. (p < 0.01; Fig. 6D) despite there being no difference in the number of mCherry+ dorsolateral corticospinal fibers between the groups and an overall higher number of mCherry+ corticospinal fibers in the sham-operated mice as a result of partial transduction of the uninjured dCST. Therefore, spared dorsolaterally projecting corticospinal neurons compensated for SCI in mediating partial behavioral recovery.
Figure 6.

Activation of hM4Di in spared dorsolaterally projecting corticospinal neurons abrogates spontaneous recovery on the horizontal ladder task after C3/C4 dorsal column SCI. A, Experimental schedule. AAV injections were made at 4 weeks after injury and DREADD receptor behavioral experiments at 55–58 d after injury by administering CNO on day 56 and 58. B, Timeline of left FL error percentage in injured and sham-operated mice with AAV1-hSyn-dio-hM4D(Gi)-mCherry + AAV1-iCre injections (hm4Di Cre+) or just AAV1-hSyn-dio-hM4D(Gi)-mCherry for control (hm4Di Cre−). HM4Di Cre− SCI mice have lower error percentage at 55–58 d after injury versus 7 d after injury. HM4Di Cre+ SCI mice have lower error percentage at 55 and 57 d after injury versus 7 d after injury, but not at 56 and 58 d after injury, the 2 d when CNO was administered. C, Left FL error percentages after vehicle administration and CNO administration for each group (calculated as an average of scores on 55 and 57 d and 56 and 58 d, respectively). HM4Di SCI Cre+ mice have a higher error percentage after CNO administration versus vehicle administration and versus sham hM4Di Cre+ mice after CNO administration. hM4DI sham-operated mice have a higher error percentage after CNO administration versus vehicle administration. D, Left FL delta error percentage calculated as the absolute difference between CNO error percentage and vehicle error percentage. hM4Di SCI Cre+ mice have a higher delta error percentage than hM4Di Sham Cre+ mice. E, Timeline of right FL error percentage in injured and sham-operated mice as in B. All four groups have a lower error percentage at 55–58 d after injury versus 7 d after injury. F, Right FL error percentages after vehicle administration and CNO administration for each group. hM4Di SCI Cre+ mice trend toward a higher error percentage after CNO administration versus vehicle administration (p = 0.06). G, Right FL delta error percentage. ***p < 0.001, **p < 0.01, *p < 0.05. Error bars indicate SEM.
Assessment of mCherry and c-fos in sensorimotor cortex after DREADD receptor activation
To validate our approach, we assessed mCherry labeling in sensorimotor cortices of both SCI and sham hM4Di Cre+ mice and counted the number of mCherry+ neurons to provide a more direct assessment of the number of hM4Di-mCherry+ neurons (Fig. 7). Consistent with specific corticospinal transduction, mCherry labeling was confined to layer V neurons (Fig. 7A). Counts of mCherry+ neurons revealed that there were significantly more in sham-operated mice versus SCI mice (p < 0.01). In total, we counted 30.83 ± 2.91 mCherry+ neurons in injured mice and 211.3 ± 55.49 mCherry+ neurons in uninjured mice (p < 0.01). Therefore, the behavioral effect observed, which included a greater loss of performance in SCI mice versus sham-operated mice (Fig. 6C), occurred despite there being substantially more mCherry+ corticospinal neurons in sham-operated mice than in SCI mice (Fig. 7B).
Figure 7.
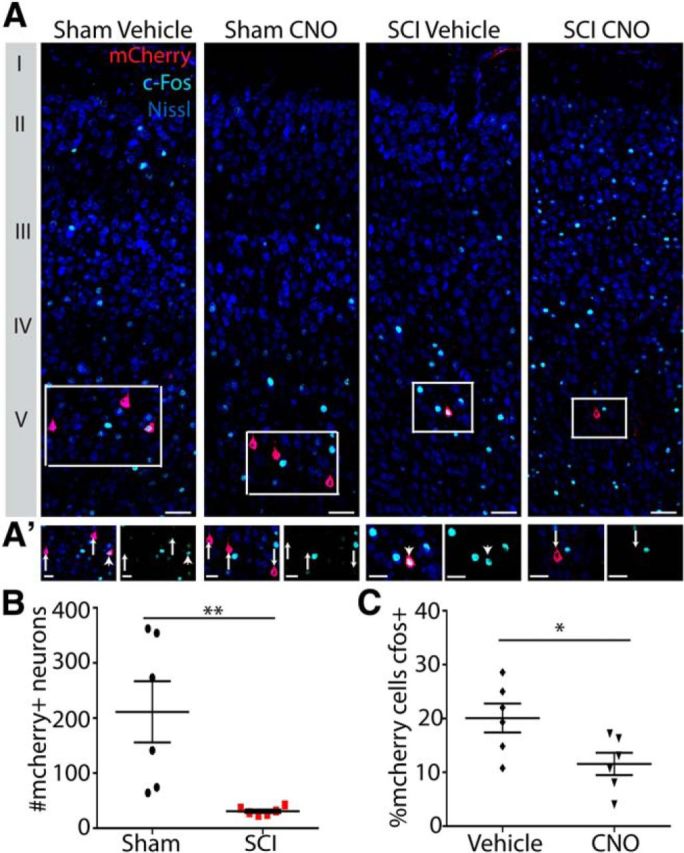
Sensorimotor cortical analysis of mCherry and c-fos expression. A, Photomicrographs of coronal sections of sensorimotor cortex with antibodies to mCherry (red) and c-fos (light blue) in addition to fluoro-Nissl staining (dark blue) to illuminate the cortical layers. MCherry is confined to layer V, consistent with specific transduction of corticospinal neurons. A′, Close-up images of mCherry+ neurons from A showing mCherry (red) and c-fos (light blue). Arrows denote c-fos negative mCherry+ corticospinal neurons, and arrowheads denote c-fos+ mCherry+ corticospinal neurons. B, Quantification of total mCherry+ neurons in sensorimotor cortex. There are significantly more mCherry+ neurons in sham-operated mice (with dual transduction of dlCST and dCST) than in C3/C4 dorsal column SCI mice (with transduction confined to dlCST). C, Quantification of percentage of c-fos+ nuclei in mCherry+ corticospinal neurons after administration of CNO to activate hM4Di or vehicle for control. **p < 0.01, *p < 0.05. Scale bars: A, 50 μm; A′, 25 μm. Error bars indicate SEM.
To provide a more direct demonstration that hM4Di activation silences CST neurons, we also assessed immunoreactivity for the marker of neuronal activity c-fos within mCherry+ neurons as a molecular indicator of hM4Di activation in mice that had been administered CNO or vehicle and subsequently ran on the horizontal ladder task repeatedly for 15 min with euthanasia ∼1.5 h later (Siegel et al., 2015; Fig. 7A,A′). In vehicle-administered mice, 20.10 ± 2.67% of mCherry+ neurons were c-fos+, whereas, in CNO-administered mice, significantly fewer (11.55 ± 2.054%) of mCherry+ neurons were c-fos+ (p < 0.05; Fig. 7C). Therefore, CNO administration significantly decreased the number of c-fos+ mCherry+ corticospinal neurons, providing direct evidence that hM4Di activation silences corticospinal neurons.
Discussion
We performed a series of optogenetic, pharmacogenetic, anatomical, and behavioral experiments to dissociate for the first time the role of spared dorsolaterally projecting corticospinal neurons in mediating spontaneous recovery after SCI. Using longitudinal mapping in Channelrhodopsin-2-expressing mice, we demonstrate that the motor cortex has the capacity to reestablish motor output to both forelimb and hindlimb after ablation of the dCST and substantial sensory loss. We then took a pharmacogenetic approach to discriminate between the contribution of plasticity in spared versus injured corticospinal neurons to recovery and show that transient silencing of uninjured dorsolaterally projecting corticospinal neurons is sufficient to result in the reappearance of deficits in skilled locomotion observed early after injury. This effect was only observed after CNO administration and only in SCI mice that were administered both Cre-dependent hM4Di and Cre-expressing viruses, highlighting the specificity of these neurons in mediating this behavioral effect. The dlCST represents ∼400 of 12,500 cervical CST axons (∼3%) in the adult mouse (Fink et al., 2015). Therefore, few spared corticospinal neurons can substantiate remarkable cortical plasticity in the injured adult mammalian CNS, which has important implications for treatment, rehabilitation, and recovery.
Early after injury, we found a substantial reduction in forelimb and hindlimb motor cortical output and map area after a C3/C4 dorsal column injury that ablated the dCST. Although optogenetic stimulation consistently evoked both forelimb and hindlimb movement despite our transection of the dCST, motor output to the forelimb and hindlimb was reduced to ∼25% and ∼40% that of baseline, respectively, acutely after injury. At this acute time point, the motor cortical output that does occur is likely substantiated by spared corticospinal neurons in addition to motor cortical circuits to brainstem targets such as the cortico-reticulospinal and cortico-rubrospinal pathways. This may be reflected by the increase in latency that occurs. Alternatively, the latency change might also reflect a challenge to fast signal propagation that occurs as a result of the injury itself. One strength of optogenetic mapping relative to traditional mapping techniques is its capacity to assess quantitatively motor cortical output in the same animals repeatedly from acute to chronic stages of injury. Doing this, we found longitudinal reestablishment of cortical motor output maps by 4 weeks after injury, which demonstrates that the adult mammalian motor cortex is able to reestablish limb output after interruption of >95% of the direct corticospinal transmission. This provides substantive evidence that, although the dCST is the major cortical output pathway to the forelimb and hindlimb in uninjured adult mice, the motor cortex spontaneously reroutes signals through other corticofugal pathways to reestablish cortical/limb connectivity after the dCST's ablation (Fig. 8). We have highlighted the dlCST, which represents ∼3% of corticospinal fibers in cervical spinal cord, as one pathway underlying this. However, multiple other corticofugal pathways might also exhibit such plasticity and underlie spontaneous recovery. The extent to which these pathways can be targeted for therapy will be an important avenue for further research. For example, a de novo rubral/raphe circuit after bilateral pyramidotomy was recently described (Siegel et al., 2015) and cortico-rubrospinal plasticity may be involved. One significant limitation of our motor mapping paradigm is the inability to dissociate movements along particular joints of the forelimb and hindlimb axes. It will be important in the future to assess whether motor cortical output reestablishment is limited to specific joints and motor tasks.
Figure 8.

Model of corticospinal plasticity after cervical SCI. A, In the uninjured adult mouse, ChR2 stimulation of layer V motor cortex evokes FL and HL movement principally via the dCST and there is baseline skilled locomotion on the horizontal ladder task. B, After C3/C4 dorsal column SCI, the dCST is interrupted (gray dashed line), resulting in depressed cortical motor output and a deficit in skilled locomotion. C, Chronically after C3/C4 SCI, cortical motor output is reestablished and there is partial recovery in skilled locomotion, likely in part mediated by dlCST neurons (green). D, When hM4Di is activated in dCST and dlCST neurons (red), there is a small but statistically significant change in skilled locomotion. E, When hM4DI is activated in dlCST neurons chronically in C3/C4 dorsal column SCI mice (red), there is an abrogation of spontaneous recovery and a greater deficit in skilled locomotion than in uninjured hM4Di activated mice as in IV despite similar dlCST silencing and no dCST silencing, representing a shift in function from the injured major dCST pathway to the uninjured minor dlCST pathway.
Our pharmacogenetic approach provides strong evidence that a small number of spared dorsolaterally projecting corticospinal neurons are necessary for spontaneous recovery on the horizontal ladder task after C3/C4 dorsal column SCI (Fig. 8D,E). Because we injected AAV-expressing Cre-dependent hM4Di into the sensorimotor cortex, only cortical neurons would be transduced by this virus. Because we injected AAV-expressing Cre 3–4 mm caudal to an SCI that resulted in complete ablation of the dCST, and thus the vast majority of corticospinal axons, only the small percentage of spared dorsolaterally projecting corticospinal neurons would be local to this virus administration. The administration of CNO led to a reappearance of the deficits observed early after injury in SCI mice administered both Cre-dependent hM4Di expressing and Cre-expressing viruses, but not in mice administered only the Cre-dependent virus. In addition, the difference observed after CNO administration versus vehicle administration in SCI hM4Di Cre+ mice was greater than in uninjured hM4Di Cre+ mice despite an equal level of mCherry+ dorsolateral corticospinal axons at C2, a higher level of total corticospinal mCherry+ fibers as a result of partial transduction of the dCST in uninjured mice, and a higher total number of mCherry+ neurons in uninjured mice. Nevertheless, although we were able to quantify the number of mCherry+ neurons in cortex, we cannot rule out the possibility that some corticospinal neurons may have been dual transduced by the required two viruses and silenced by the drug but not expressing the threshold level of mCherry necessary for our detection in cortex. Therefore, we cannot be certain of the minimum number of corticospinal neurons silenced necessary to exert the difference observed.
Numerous studies have demonstrated corticospinal axonal plasticity associated with recovery after CNS injury (Nudo, 2006; Nishimura and Isa, 2009), but, to the best of our knowledge, a causal demonstration of uninjured corticospinal neurons mediating spontaneous recovery has not been made. For example, in a seminal study, the sparing of ventral corticospinal axons was correlated with spontaneous recovery of skilled FL function in the adult rat (Weidner et al., 2001). We did not observe mCherry+ fibers within the ventral funiculus, however, suggesting that our approach was specific to dorsolaterally projecting corticospinal neurons in injured mice. Given that the vCST projects ipsilaterally (Weidner et al., 2001), we did not expect it to be targeted/silenced using our approach with Cre targeted to the contralateral side. More recently, substantial spontaneous plasticity of corticospinal axons associated with forelimb recovery was demonstrated in the adult primate (Rosenzweig et al., 2010; Friedli et al., 2015). Given the capacity of individuals with Brown–Séquard syndrome to exhibit a capacity to spontaneously recover volitionally guided locomotion (Roth et al., 1991) and the extent of corticospinal plasticity in nonhuman primates (Rosenzweig et al., 2010), it is likely that corticospinal plasticity occurs extensively in humans after incomplete SCI (Friedli et al., 2015). Our results demonstrate that spared dorsolateral corticospinal neurons are pivotal for spontaneous recovery of skilled locomotion in adult mice despite the limited reconstitution of corticospinal density after injury (Weidner et al., 2001; Brus-Ramer et al., 2007; Maier et al., 2008). Dorsolateral corticospinal fibers can increase their contact with lumbar motor neuron pools after midthoracic dorsal column injury (Bareyre et al., 2005) and are found in the vicinity of motor neuron pools after cervical dorsal column injury (Steward et al., 2004). We found that silencing of dorsolateral corticospinal neurons projecting caudal to the injury site was sufficient to result in the reappearance of deficits on the horizontal ladder task early after injury. However, whether dorsolaterally projecting corticospinal neurons contribute to spontaneous recovery in more skilled tasks such as pellet reaching remains to be determined.
Importantly, although our pharmacogenetic approach targeted dorsolaterally projecting corticospinal neurons after injury, our data do not demonstrate that compensation occurs specifically at the synapses of the dorsolateral corticospinal fibers at the spinal levels C6/C7 or C7/C8. Pharmacogenetic silencing via DREADD receptor activation would have silenced the entire neuron, so we cannot rule out the possibility (indeed, the probability) that these neurons project and synapse elsewhere between the sensorimotor cortex and the spinal cord (via collaterals) and that silencing these other connections between these neurons contributes to the deficits observed after CNO administration. Corticospinal neurons harbor a capacity for de novo connections with propriospinal neurons after injury (Bareyre et al., 2004; Vavrek et al., 2006) and corticofugal neurons project extensively throughout the brainstem to other motor nuclei, including the red nucleus and reticular formation, which harbor rubrospinal and reticulospinal tracts, respectively (Brown, 1974; Esposito et al., 2014).
Our pharmacogenetic approach to silencing a descending pathway after SCI is broadly applicable to the study of plasticity and regeneration after CNS injury. Such strategies have been used to decipher FL circuit function in the uninjured CNS (Esposito et al., 2014) and recovery after stroke (Wahl et al., 2014) and pyramidotomy (Siegel et al., 2015). We took advantage of the development of Cre-dependent versions of hM4Di to inactivate a specific descending pathway. Because some AAVs are capable of retrograde transduction of neurons, even after SCI (Klaw et al., 2013), this approach may yield valuable insight into the roles of other pathways in mediating spontaneous, regeneration-based, or rehabilitation-based recovery after SCI.
In conclusion, we demonstrate for the first time that small numbers of spared dorsolaterally projecting corticospinal neurons can substantiate remarkable motor cortical plasticity and partial recovery after SCI. Targeting spared corticospinal neurons represents a key substrate to promote repair after SCI and perhaps other CNS diseases/disorders.
Footnotes
This work was supported by the Wings for Life foundation (W.T.), the University of British Columbia (Djavad Mowafaghian Collaborative Grant to B.J.H., E.A., T.H.M., and W.T.), the Canadian Institutes of Health Research (CIHR Operating Grant MOP-12675; T.H.M. and CIHR Foundation Grant to T.H.M.). B.J.H. holds a CIHR Frederick Banting and Charles Best Canada Graduate Scholarship, a Zoology Graduate Fellowship, and receives funding from the CIHR Transplant Training Program. We thank Jeff LeDue for technical support; Cindy Jiang for animal husbandry; Rick White and Yi Huang from the Statistical Consulting and Research Laboratory (SCARL) at the University of British Columbia for help with statistical analysis; Annie Ren, Greg Duncan, Peggy Assinck, and Vrinda Ohri for help with analysis of behavioral data and microscopy; Matthieu Vanni for providing the mouse cortex atlas drawing; and Dr. Karim Fouad, University of Alberta, Edmonton for comments on an earlier version of the manuscript.
The authors declare no competing financial interests.
References
- Anenberg E, Arstikaitis P, Niitsu Y, Harrison TC, Boyd JD, Hilton BJ, Tetzlaff W, Murphy TH. Ministrokes in channelrhodopsin-2 transgenic mice reveal widespread deficits in motor output despite maintenance of cortical neuronal excitability. J Neurosci. 2014;34:1094–1104. doi: 10.1523/JNEUROSCI.1442-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenkiel BR, Peca J, Davison IG, Feliciano C, Deisseroth K, Augustine GJ, Ehlers MD, Feng G. In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron. 2007;54:205–218. doi: 10.1016/j.neuron.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asante CO, Martin JH. Differential joint-specific corticospinal tract projections within the cervical enlargement. PLoS One. 2013;8:e74454. doi: 10.1371/journal.pone.0074454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayling OG, Harrison TC, Boyd JD, Goroshkov A, Murphy TH. Automated light-based mapping of motor cortex by photoactivation of channelrhodopsin-2 transgenic mice. Nat Methods. 2009;6:219–224. doi: 10.1038/nmeth.1303. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Raineteau O, Mettenleiter TC, Weinmann O, Schwab ME. The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat Neurosci. 2004;7:269–277. doi: 10.1038/nn1195. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Misgeld T, Sanes JR. Transgenic labeling of the corticospinal tract for monitoring axonal responses to spinal cord injury. Nat Med. 2005;11:1355–1360. doi: 10.1038/nm1331. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Brown LT. Corticorubral projections in the rat. J Comp Neurol. 1974;154:149–167. doi: 10.1002/cne.901540204. [DOI] [PubMed] [Google Scholar]
- Brus-Ramer M, Carmel JB, Chakrabarty S, Martin JH. Electrical stimulation of spared corticospinal axons augments connections with ipsilateral spinal motor circuits after injury. J Neurosci. 2007;27:13793–13801. doi: 10.1523/JNEUROSCI.3489-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings BJ, Engesser-Cesar C, Cadena G, Anderson AJ. Adaptation of a ladder beam walking task to assess locomotor recovery in mice following spinal cord injury. Behav Brain Res. 2007;177:232–241. doi: 10.1016/j.bbr.2006.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito MS, Capelli P, Arber S. Brainstem nucleus MdV mediates skilled forelimb motor tasks. Nature. 2014;508:351–356. doi: 10.1038/nature13023. [DOI] [PubMed] [Google Scholar]
- Fink KL, Strittmatter SM, Cafferty WB. Comprehensive corticospinal labeling with mu-crystallin transgene reveals axon regeneration after spinal cord trauma in ngr1-/- mice. J Neurosci. 2015;35:15403–15418. doi: 10.1523/JNEUROSCI.3165-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouad K, Pedersen V, Schwab ME, Brösamle C. Cervical sprouting of corticospinal fibers after thoracic spinal cord injury accompanies shifts in evoked motor responses. Curr Biol. 2001;11:1766–1770. doi: 10.1016/S0960-9822(01)00535-8. [DOI] [PubMed] [Google Scholar]
- Friedli L, Rosenzweig ES, Barraud Q, Schubert M, Dominici N, Awai L, Nielson JL, Musienko P, Nout-Lomas Y, Zhong H, Zdunowski S, Roy RR, Strand SC, van den Brand R, Havton LA, Beattie MS, Bresnahan JC, Bézard E, Bloch J, Edgerton VR, et al. Pronounced species divergence in corticospinal tract reorganization and functional recovery after lateralized spinal cord injury favors primates. Sci Transl Med. 2015;7:302ra134–302ra134. doi: 10.1126/scitranslmed.aac5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensel JC, Tovar CA, Hamers FP, Deibert RJ, Beattie MS, Bresnahan JC. Behavioral and histological characterization of unilateral cervical spinal cord contusion injury in rats. J Neurotrauma. 2006;23:36–54. doi: 10.1089/neu.2006.23.36. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Sydekum E, Haiss F, Peduzzi S, Zörner B, Schneider R, Baltes C, Rudin M, Weber B, Schwab ME. Functional and anatomical reorganization of the sensory-motor cortex after incomplete spinal cord injury in adult rats. J Neurosci. 2009;29:12210–12219. doi: 10.1523/JNEUROSCI.1828-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Haiss F, Sydekum E, Schneider R, Gullo M, Wyss MT, Mueggler T, Baltes C, Rudin M, Weber B, Schwab ME. Rewiring of hindlimb corticospinal neurons after spinal cord injury. Nat Neurosci. 2010;13:97–104. doi: 10.1038/nn.2448. [DOI] [PubMed] [Google Scholar]
- Harrison TC, Ayling OG, Murphy TH. Distinct cortical circuit mechanisms for complex forelimb movement and motor map topography. Neuron. 2012;74:397–409. doi: 10.1016/j.neuron.2012.02.028. [DOI] [PubMed] [Google Scholar]
- Harrison TC, Silasi G, Boyd JD, Murphy TH. Displacement of sensory maps and disorganization of motor cortex after targeted stroke in mice. Stroke. 2013;44:2300–2306. doi: 10.1161/STROKEAHA.113.001272. [DOI] [PubMed] [Google Scholar]
- Hilton BJ, Assinck P, Duncan GJ, Lu D, Lo S, Tetzlaff W. Dorsolateral funiculus lesioning of the mouse cervical spinal cord at C4 but not at C6 results in sustained forelimb motor deficits. J Neurotrauma. 2013;30:1070–1083. doi: 10.1089/neu.2012.2734. [DOI] [PubMed] [Google Scholar]
- Jara JH, Villa SR, Khan NA, Bohn MC, Ozdinler PH. AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol Dis. 2012;47:174–183. doi: 10.1016/j.nbd.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jara JH, Genç B, Klessner JL, Özdinler PH. Retrograde labeling, transduction, and genetic targeting allow cellular analysis of corticospinal motor neurons: implications in health and disease. Front Neuroanat. 2014:8. doi: 10.3389/fnana.2014.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaw MC, Xu C, Tom VJ. Intraspinal AAV injections immediately rostral to a thoracic spinal cord injury site efficiently transduces neurons in spinal cord and brain. Mol Ther Nucleic Acids. 2013;2:e108. doi: 10.1038/mtna.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajacic A, Weishaupt N, Girgis J, Tetzlaff W, Fouad K. Training-induced plasticity in rats with cervical spinal cord injury: effects and side effects. Behav Brain Res. 2010;214:323–331. doi: 10.1016/j.bbr.2010.05.053. [DOI] [PubMed] [Google Scholar]
- Maier IC, Baumann K, Thallmair M, Weinmann O, Scholl J, Schwab ME. Constraint-induced movement therapy in the adult rat after unilateral corticospinal tract injury. J Neurosci. 2008;28:9386–9403. doi: 10.1523/JNEUROSCI.1697-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura Y, Isa T. Compensatory changes at the cerebral cortical level after spinal cord injury. The Neuroscientist. 2009;15:436–444. doi: 10.1177/1073858408331375. [DOI] [PubMed] [Google Scholar]
- Nishimura Y, Isa T. Cortical and subcortical compensatory mechanisms after spinal cord injury in monkeys. Exp Neurol. 2012;235:152–161. doi: 10.1016/j.expneurol.2011.08.013. [DOI] [PubMed] [Google Scholar]
- Nudo RJ. Mechanisms for recovery of motor function following cortical damage. Curr Opin Neurobiol. 2006;16:638–644. doi: 10.1016/j.conb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Oudega M, Perez MA. Corticospinal reorganization after spinal cord injury. J Physiol. 2012;590:3647–3663. doi: 10.1113/jphysiol.2012.233189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raineteau O, Schwab ME. Plasticity of motor systems after incomplete spinal cord injury. Nat Rev Neurosci. 2001;2:263–273. doi: 10.1038/35067570. [DOI] [PubMed] [Google Scholar]
- Ramer LM, Ramer MS, Bradbury EJ. Restoring function after spinal cord injury: towards clinical translation of experimental strategies. Lancet Neurol. 2014;13:1241–1256. doi: 10.1016/S1474-4422(14)70144-9. [DOI] [PubMed] [Google Scholar]
- Rogan SC, Roth BL. Remote control of neuronal signaling. Pharmacol Rev. 2011;63:291–315. doi: 10.1124/pr.110.003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig ES, Courtine G, Jindrich DL, Brock JH, Ferguson AR, Strand SC, Nout YS, Roy RR, Miller DM, Beattie MS, Havton LA, Bresnahan JC, Edgerton VR, Tuszynski MH. Extensive spontaneous plasticity of corticospinal projections after primate spinal cord injury. Nat Neurosci. 2010;13:1505–1510. doi: 10.1038/nn.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth EJ, Park T, Pang T, Yarkony GM, Lee MY. Traumatic cervical Brown-Sequard and Brown-Sequard-plus syndromes: the spectrum of presentations and outcomes. Paraplegia. 1991;29:582–589. doi: 10.1038/sc.1991.86. [DOI] [PubMed] [Google Scholar]
- Siegel CS, Fink KL, Strittmatter SM, Cafferty WB. Plasticity of intact rubral projections mediates spontaneous recovery of function after corticospinal tract injury. J Neurosci. 2015;35:1443–1457. doi: 10.1523/JNEUROSCI.3713-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silasi G, Boyd JD, Ledue J, Murphy TH. Improved methods for chronic light-based motor mapping in mice: automated movement tracking with accelerometers, and chronic EEG recording in a bilateral thin-skull preparation. Front Neural Circuits. 2013;7:123. doi: 10.3389/fncir.2013.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeves JD, Kramer JK, Fawcett JW, Cragg J, Lammertse DP, Blight AR, Marino RJ, Ditunno JF, Jr, Coleman WP, Geisler FH, Guest J, Jones L, Burns S, Schubert M, van Hedel HJ, Curt A EMSCI Study Group. Extent of spontaneous motor recovery after traumatic cervical sensorimotor complete spinal cord injury. Spinal Cord. 2011;49:257–265. doi: 10.1038/sc.2010.99. [DOI] [PubMed] [Google Scholar]
- Steward O, Zheng B, Ho C, Anderson K, Tessier-Lavigne M. The dorsolateral corticospinal tract in mice: an alternative route for corticospinal input to caudal segments following dorsal column lesions. J Comp Neurol. 2004;472:463–477. doi: 10.1002/cne.20090. [DOI] [PubMed] [Google Scholar]
- Vavrek R, Girgis J, Tetzlaff W, Hiebert GW, Fouad K. BDNF promotes connections of corticospinal neurons onto spared descending interneurons in spinal cord injured rats. Brain. 2006;129:1534–1545. doi: 10.1093/brain/awl087. [DOI] [PubMed] [Google Scholar]
- Wahl AS, Omlor W, Rubio JC, Chen JL, Zheng H, Schröter A, Gullo M, Weinmann O, Kobayashi K, Helmchen F, Ommer B, Schwab ME. Asynchronous therapy restores motor control by rewiring of the rat corticospinal tract after stroke. Science. 2014;344:1250–1255. doi: 10.1126/science.1253050. [DOI] [PubMed] [Google Scholar]
- Wang Z, Reynolds A, Kirry A, Nienhaus C, Blackmore MG. Overexpression of sox11 promotes corticospinal tract regeneration after spinal injury while interfering with functional recovery. J Neurosci. 2015;35:3139–3145. doi: 10.1523/JNEUROSCI.2832-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner N, Ner A, Salimi N, Tuszynski MH. Spontaneous corticospinal axonal plasticity and functional recovery after adult central nervous system injury. Proc Natl Acad Sci U S A. 2001;98:3513–3518. doi: 10.1073/pnas.051626798. [DOI] [PMC free article] [PubMed] [Google Scholar]