

Abstract
RNA binding proteins required for the maintenance of myelin and axoglial junctions are unknown. Herein, we report that deletion of the Quaking (QKI) RNA binding proteins in oligodendrocytes (OLs) using Olig2–Cre results in mice displaying rapid tremors at postnatal day 10, followed by death at postnatal week 3. Extensive CNS hypomyelination was observed as a result of OL differentiation defects during development. The QKI proteins were also required for adult myelin maintenance, because their ablation using PLP–CreERT resulted in hindlimb paralysis with immobility at ∼30 d after 4-hydroxytamoxifen injection. Moreover, deterioration of axoglial junctions of the spinal cord was observed and is consistent with a loss of Neurofascin 155 (Nfasc155) isoform that we confirmed as an alternative splice target of the QKI proteins. Our findings define roles for the QKI RNA binding proteins in myelin development and maintenance, as well as in the generation of Nfasc155 to maintain healthy axoglial junctions.
SIGNIFICANCE STATEMENT Neurofascin 155 is responsible for axoglial junction formation and maintenance. Using a genetic mouse model to delete Quaking (QKI) RNA-binding proteins in oligodendrocytes, we identify QKI as the long-sought regulator of Neurofascin alternative splicing, further establishing the role of QKI in oligodendrocyte development and myelination. We establish a new role for QKI in myelin and axoglial junction maintenance using an inducible genetic mouse model that deletes QKI in mature oligodendrocytes. Loss of QKI in adult oligodendrocytes leads to phenotypes reminiscent of the experimental autoimmune encephalomyelitis mouse model with complete hindlimb paralysis and death by 30 d after induction of QKI deletion.
Keywords: alternative splicing, axoglial junctions, myelin, neurofascin, Quaking, RNA-binding proteins
Introduction
Quaking (QKI) viable mice (qkv) display tremors by postnatal day 10 (P10). The genetic defect in qkv is a ∼1 Mb deletion encompassing the qkI gene promoter that reduces the expression of QKI proteins in myelinating glia (Ebersole et al., 1996). qkv mice exhibit severe dysmyelination in the CNS and PNS, with abnormalities in nodal, internodal, and paranodal regions with spotty disruption of paranodal junctions (Chénard and Richard, 2008; Rosenbluth and Bobrowski-Khoury, 2013). However, the exact cause of these defects remains unknown.
The qkI gene encodes KH-type RNA binding proteins that generates three major alternative splicing mRNAs (5, 6, and 7 kb) encoding QKI-5, QKI-6, and QKI-7 that differ in their C-terminal 30 aa (Ebersole et al., 1996). QKI-5 is exclusively nuclear, whereas QKI-6 is distributed throughout the cell, and QKI-7 is predominantly cytoplasmic (Wu et al., 1999). The QKI isoforms dimerize (Chen and Richard, 1998; Beuck et al., 2012; Teplova et al., 2013) and interact with RNA in a sequence-specific manner. The following bipartite sequence, ACUAAY-(N1–20)-UAAY, was identified and termed the QKI response element (QRE; Galarneau and Richard, 2005). Transcriptome-wide CLIP (crosslinking and immunoprecipitation) confirmed the QRE in vivo and also showed that QKI associates predominantly with intronic sequences (Hafner et al., 2010), implying a nuclear role for the QKI isoforms.
It was recognized that qkv mice harbor many RNA-associated defects. The QKI isoforms regulate the mRNA export (Larocque et al., 2002) and stability of the myelin basic protein (MBP) mRNAs (Li et al., 2000). The QKI isoforms also regulate the mRNA stability of Krox-20 (Egr-2; Nabel-Rosen et al., 2002), the cyclin-dependent kinase inhibitor p27KIP1 (Larocque et al., 2005), and the actin interacting protein 1 (Doukhanine et al., 2010). The qkv mice display alternative splicing defects of myelin components, including myelin associated glycoprotein (MAG), proteolipid protein (PLP; Wu et al., 2002), and Sirt2 (Zhu et al., 2012), and these have been shown to be regulated indirectly by QKI-6 that regulates the mRNA translational control of hnRNPA1 (Zhao et al., 2010; Zearfoss et al., 2011) and hnRNP F/H (Mandler et al., 2014). However, nuclear QKI-5 has been shown to regulate alternative splicing in vascular smooth muscle cells and skeletal muscle (Hall et al., 2013; van der Veer et al., 2013), but a role for QKI-5 in the regulation of alternative splicing in myelinating cells has yet to be demonstrated.
To circumvent the embryonic lethality of qkI mice (Li et al., 2003) and the large deletion in the qkv mice, we now report a qkI conditional allele designed to delete all three major isoforms. This new allele led to the identification of Neurofascin (Nfasc) as an alternative splicing target of QKI-5. Nfasc is extensively alternatively spliced with two major isoforms: Nfasc155 expressed in myelinating cells and Nfasc186 expressed in neurons (Ebel et al., 2014); however, the RNA binding proteins that regulate this alternative splicing event remain unknown. Nfasc155 is upregulated at the onset of myelination, in which it interacts with axonal Caspr and Contactin to form septate-like junctions also known as axoglial junctions (Collinson et al., 1998; Tait et al., 2000; Charles et al., 2002; Gollan et al., 2003). Genetic ablation of Caspr, Contactin-1, or Nfasc155 leads to disrupted paranodal axoglial junctions, failure to properly segregate ion channels, and impaired action potential conduction (Bhat et al., 2001; Boyle et al., 2001; Sherman et al., 2005; Pillai et al., 2009; Thaxton et al., 2010).
Herein, we show that deletion of the QKI isoforms in oligodendrocytes (OLs) using Olig2–Cre leads to severe CNS hypomyelination with tremors by P10 and death by postnatal week 3. Ablation of qkI in adult mice using PLP–CreERT resulted in hindlimb paralysis, thoracic kyphosis, and immobility ∼30 d after 4-hydroxytamoxifen (OHT) administration. Our work defines QKI-5 as a regulator of Nfasc alternative splicing required for axoglial junction development and maintenance.
Materials and Methods
qkI conditional allele.
All mouse procedures were performed in accordance with McGill University guidelines, which are set by the Canadian Council on Animal Care. The mouse qkI conditional allele was constructed using a previously described targeting vector (Yu et al., 2009). The targeting construct contained loxP sites flanking exon 2 of the qkI gene. Briefly, the sequenced plasmid was linearized and electroporated into 129/Sv ES cells. Three independent ES cell clones with homologous integration at the targeting site were injected into C57BL/6J blastocysts, and chimeras were obtained. These chimeras were subsequently crossed with C57BL/6J females, and those mice with successful germ-line transmission of the targeted allele were crossed with C57BL/6J mice expressing Flp recombinase to remove the neomycin resistance cassette resulting in a floxed allele (qkIFL/+). These mice were then intercrossed to obtain QKIFL/FL homozygote animals. Genomic DNA was isolated from ear biopsies, and a DNA fragment was amplified using the following primers: 5′-ACA GAG GCT TTT CCT GAC CA-3′ and 5′-TTC AGA ACC CCC ACA TTA CC-3′, resulting in a band of ∼191 bp in the wild-type allele and ∼309 bp in the floxed allele. To generate the embryonic or inducible qkI conditional knock-out mice, qkIFL/+ mice were crossed with transgenic mice expressing either Olig2–Cre or PLP–CreERT, respectively. To detect the Olig2–Cre (Ligon et al., 2007) and PLP–CreERT (Doerflinger et al., 2003) transgenes, generic Cre recombinase primers were used: 5′-GCG GTC TGG CAG TAA AAA CTA TC-3′ and 5′-GTG AAA CAG CAT TGC TGT CAC TT-3′. To verify recombination between the loxP sites, the following primers were used: 5′-CCT GGA ATG GTG CTT TCC TA-3′ and 5′-TTC AGA ACC CCC ACA TTA CC-3′. For tamoxifen treatment, a solution of 10 mg/ml tamoxifen (Sigma) was dissolved in corn oil, and 8-week-old mice were injected intraperitoneally once daily for 5 consecutive days with 1 mg/25 g.
Immunoblotting.
Mice were killed in accordance with a protocol approved by the Animal Care Committee at McGill University, and tissues were immediately flash frozen on dry ice and stored at −80°C. Tissues were lysed in cold RIPA buffer (Sigma-Aldrich) with protease inhibitor mixture (Roche). Samples were incubated on ice for 30 min, sonicated, and centrifuged at 10,000 rpm for 15 min at 4°C. Supernatants were collected, and protein concentration was measured using protein assay reagent (Bio-Rad). Samples were stored at −80°C or used immediately for immunoblotting. Forty micrograms of protein per well was separated by SDS-PAGE, transferred to nitrocellulose membranes using immunoblot TurboTransfer system (Bio-Rad), and immunoblotted with the indicated primary antibody: (QKI-5/QKI-6/QKI-7 (Millipore), β-tubulin (Sigma), glial fibrillary acidic protein (GFAP; Sigma), neurofilament light polypeptide (Nefl; Sigma), neuronal nuclei (NeuN; Millipore Bioscience Research Reagents), Nfasc155 and Nfasc186 (Millipore), pan-Nfasc (a kind gift from Dr. Peter J. Brophy, University of Edinburgh, Edinburgh, Scotland), MBP (Sternberger Monoclonals), Nav (Sigma), and Caspr (Abcam), followed by horseradish peroxidase-conjugated secondary antibody, and visualized by ECL Plus reagent (PerkinElmer Life and Analytical Sciences).
Reverse transcription, semiquantitative, and quantitative PCR.
Total RNA from tissues were isolated in appropriate amounts of TRIzol reagent according to the instructions of the manufacturer (Invitrogen). Thirty micrograms of RNA were DNase I (Promega) treated for 30 min at 37°C. One microgram of RNA was reverse transcribed using oligo-dT primer and MMLV reverse transcriptase according to the protocol of the manufacturer (Promega). cDNAs were then amplified by semiquantitative PCR, and the DNA fragments were separated by agarose gel electrophoresis. For real-time PCR, primers were designed and efficiency tested according to the MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) guidelines. Real-time PCR was performed in triplicates with a 1:4 dilution of cDNA using SyBR Green PCR Mastermix (Qiagen) on 7500Fast Real-Time PCR System (Applied Biosystems). All quantification data were normalized to GAPDH using the ΔΔCt method.
Primary mouse and rat OL cultures and siRNA transfection.
Primary oligodendrocyte progenitor cells (OPCs) were prepared from newborn Sprague Dawley rat brains as described previously (Almazan et al., 1993). The cells were plated on poly-d-lysine-coated dishes and grown in serum-free DMEM/F-12 mixture (1:1) medium supplemented with 10 mm HEPES, 0.1% bovine serum albumin, 25 μg/ml human transferrin, 30 nm triiodothyronine, 20 nm hydrocortisone, 20 nm progesterone, 10 nm biotin, 5 μg/ml insulin, 16 μg/ml putrescine, 30 nm selenium, and 10 ng/ml each of PDGF-AA and basic FGF. The progenitors are proliferative under these conditions, whereas removal of the mitogens initiates their differentiation. Culture medium was changed every 2 d under all experimental conditions. OPCs were induced to differentiate after withdrawal of growth factors for 4 d and transfected with an siRNA designed to target QKI (5′-GGA CUU ACA GCC AAA CAA C-3′) and luciferase control siRNA (5′-CGU ACG CGG AAU ACU UGA-3′) with 5′-tagged Alexa Fluor-488 fluorophore (Invitrogen). Primary OPCs were transfected using the Lipofectamine RNAiMax reagent (Invitrogen) according to the instructions of the manufacturer. The cells were harvested 48 h after transfection. Primary mouse OPCs were isolated from a pool of five to six wild-type or QKIFL/FL;PLP–CreERT P1 pups. The same isolation procedure as the rat OPCs was followed. Mouse OPCs were induced to differentiate for 4 d before treatment with tamoxifen.
Minigene assays.
Nfasc gene exons 20–23 with the intervening introns were amplified from mouse brain genomic DNA by PCR using the forward primer 5′-GGG GCTAGC CACC ATG GAC TAC AAA GAC GAT GAC GAC AAG CCT ATG AAT GCT ACC TCT GCC TTT GGC-3′, containing a FLAG epitope tag and an NheI site, and the reverse primer 5′-CCG GGC TCG AGT TAG GGC ACG TAT CTG AGG ATG TAT C-3, containing an XhoI site. The DNA fragment was then subcloned into the corresponding site of pVAX1. The mutation of the QREs within intron 21 of the minigene was performed in a two-step PCR using primers 5′-CCC AGC CCC ACG GAC CCA CCT TCT TGC GGG TCA GGG AAA GGG GTA GCA GCC-3′ (forward primer) and 5′-CTT TCC CTG ACC CGC AAG AAG GTG GGT CCG TGG GGC TGG GTG C-3′ (reverse primer) for QRE1 and PCR primers 5′-GTC ACT ACC ACC ACG GGT CAC GAG CCT GGC CCT GCT GCC TCG CCA C-3′ (forward primer) and 5′-AGG GCC AGG CTC GTG ACC CGT GGT GGT AGT GAC TGG TCC TCC CTT TGC-3′ (reverse primer) for QRE2 mutation. HEK293T cells were plated in six-well plates and transfected with 2 μg of the indicated minigene with 2 μg of pcDNA3.1 expression vectors encoding QKI-5, QKI-6, QKI-7, and QKI5:V157E. These plasmids have been described previously (Chen and Richard, 1998; Larocque et al., 2002). The common forward primer for RT-PCR detection of Nfasc minigene transcripts was 5′-TGA CGA CAA GCC TAT GAA TGC TAC-3′. The reverse primer for RT-PCR detection was 5′-CTA GAC TCG AGT TAG GGC ACG TAT C-′.
RNA binding assays.
Brains were lysed in cell lysis buffer (20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Triton X-100, and 40 U/ml RNase inhibitor, supplemented with Roche Complete Mini EDTA-free protease inhibitor) and incubated at 4°C with 4 μl of 100 μm biotinylated RNA (IDT) for 60 min, and subsequently, streptavidin beads were added for 30 min with end-over-end mixing. The beads were washed three times with lysis buffer with increasing salt concentrations (150, 300, or 500 mm) and once with 1× PBS. Protein samples were analyzed by SDS-PAGE and immunoblotting.
Layered and integrated system for splicing annotation.
Total RNA was extracted from brains of QKIFL/FL;− and QKIFL/FL;Olig2–Cre P14 mice (n = 3 for each genotype), reverse transcribed, and analyzed by high-throughput PCR amplification at the Sherbrooke University RNomics Platform as described previously (Klinck et al., 2008; Venables et al., 2009). A highly curated database has identified a genome-wide selection of 1328 alternative splicing events from the mouse RefSeq database whose isoform sizes differ by between 10 and 450 nt at a particular event. Spliced isoforms were characterized by endpoint PCR in an automated manner with the primers flanking the alternative splicing events, such that, after amplification and analysis by microcapillary electrophoresis on Caliper LC-90 instruments (Caliper Life Sciences), the relative ratio of the isoforms can be calculated. A percent splicing index (PSI) was assigned to each exon depending on its inclusion/exclusion.
Electron microscopy.
Mice were anesthetized and perfused with ice-cold PBS, followed by 2.5% glutaraldehyde in 0.1 m sodium cacodylate buffer. Tissues were dissected out and postfixed with 1% aqueous OsO4 (Mecalab) with 1.5% aqueous potassium ferrocyanide for 2 h. Tissues were embedded with Epon (Mecalab). Tissue sections were trimmed and cut into 90–100 nm sections with an UltraCut E ultramicrotome (Reichert-Jung) and put onto a 200 mesh copper grid (Electron Microscopy Sciences). Sections were then stained with uranyl acetate (Electron Microscopy Sciences), followed by Reynold's lead (Electron Microscopy Sciences). Images were captured using an FEI Tecnai 12 120 kV transmission electron microscope equipped with an AMT XR80C 8 megapixel CCD camera (McGill University, Department of Anatomy and Cell Biology). A total of two mice per genotype were analyzed. The g-ratio measurements were calculated using NIH ImageJ software. At least 100 axons per genotype were calculated.
Immunohistochemistry.
Mice were anesthetized with a mixture (ketamine–xylazine–acepromazine) and perfused with ice-cold PBS, followed by 4% paraformaldehyde (PFA). Brain tissues were postfixed in PFA for 3 d, cryoprotected in 30% sucrose, and embedded in OCT compound (Tissue-Tek) over dry ice in ethanol. Tissues were cryostat sectioned at a thickness of 10 μm and collected on Superfrost +/+ slides (Thermo Fisher Scientific). Tissue sections were blocked in 10% normal goat serum in PBS with 0.3% Triton X-100 for 1 h, followed by primary antibody: Olig2, MBP, PDGFRα, CC1, QKI-5, QKI-6, and QKI-7 (dilution at 1:200) incubation overnight at 4°C. Slides were then incubated with Alexa Fluor-488 or -546 IgG (Invitrogen) at a dilution of 1:200 for 2 h at room temperature. Pictures were taken using Zeiss LSM Pascal Confocal Imaging System with identical settings for wild-type and knock-out antibody pairs. For staining of spinal cords and sciatic nerves, mice were killed, and the tissues were fixed in 4% PFA in 0.1 m phosphate buffer (PB) for 30 min. Spinal cords were then washed extensively with PB buffer and placed in 30% sucrose overnight. Sciatic nerves were washed with PB buffer and teased. Spinal cords were sectioned on a cryostat into 20 μm sections. Spinal cord sections and teased sciatic nerves were blocked and permeabilized using 0.1 m PB buffer, 10% normal goat serum, and 0.3% Triton X-100 for 1 h, followed by primary antibodies diluted in blocking buffer: Nfasc155, Ankyrin G (AnkG; clone N106/36; Neuromab), and Caspr at 1:50 overnight at 4°C. Tissues were washed three times for 5 min each in the blocking buffers. Secondary antibodies were diluted in blocking buffer at 1:200, and slides were stained for 1 h. Pictures were taken using a Zeiss AxioImager 2 upright microscope. Images were processed with Photoshop. The intensity histogram was adjusted on grayscale images from each individual channel, and colored images were merged. Brightness/contrast adjustments were then applied across the entire set of images and equally to controls. For fluromyelin staining, brain sections were blocked with 10% normal goat serum in PBS for 1 h at room temperature, followed by 1 h fluoromyelin incubation in blocking solution (dilution at 1:300; Life Technologies). Images were acquired using an AxioImager 2 upright microscope (Carl Zeiss).
Statistical analysis.
All data presented were produced in at least three independent experiments with the exceptions indicated in the figure legends. Statistical analysis was performed using the Prism software (GraphPad Software) unless stated otherwise. Significance of differences was evaluated with either Student's t test, when only two groups were compared (g-ratio, axon diameter, nodal gap, and paranode length), or one-way ANOVA for more than two groups (RT-qPCR). One-way ANOVA was followed by Bonferroni's post hoc analysis. Significance was indicated as *p < 0.05, **p < 0.01 and ***p < 0.001.
Results
Ablation of qkI in OLs of mice leads to severe ataxia and death at postnatal week 3
Whole-body ablation of qkI leads to embryonic lethality at approximately E10 (Li et al., 2003), thus preventing the assessment of the role of QKI isoforms in mammalian myelination using a loss-of-function approach. We generated a conditional mouse knock-out allele of qkI in which exon 2 was flanked by loxP sites (Fig. 1A, QKIFL). Exon 2 encodes part of the KH RNA binding domain, common to all QKI isoforms, and its deletion should generate a frameshift, resulting in a null allele. QKIFL/FL mice were generated and crossed with mice expressing Cre recombinase under the control of the Olig2 promoter (Olig2–Cre), to induce recombination in the OL lineage from early developmental stages (Schüller et al., 2008). The deletion of exon 2 and the appearance of the one loxP allele was observed by genomic PCR using DNA extracted from the brain but not other organs of QKIFL/FL;Olig2-Cre mice (Fig. 1B,C). QKIFL/FL;Olig2-Cre mice were smaller in size (Fig. 1D), and, at P10, displayed progressive neurological defects with tremors, seizures, and severe ataxia, followed by death by postnatal week 3 (Fig. 1E). In the brain, the QKI isoforms are expressed in the glial lineage with high abundance in OLs (Hardy et al., 1996). We observed that, in P14 mice, the major QKI-expressing cell was indeed the OL, because brains of QKIFL/FL;Olig2–Cre mice were essentially devoid of the QKI-5, QKI-6, and QKI-7 isoforms at this time point, as visualized by immunoblotting and RT-qPCR (Fig. 2A,B). The loss of QKI isoform expression also led to a complete loss of the MBPs in brains of QKIFL/FL;Olig2–Cre mice but not QKIFL/FL;− (wild-type) or QKIFL/+;Olig2–Cre (heterozygous) mice (Fig. 2A). We detected the expected 50% reduction in QKI mRNA and protein levels in heterozygous mice (Fig. 2A,B), but no overt phenotype was observed and normal levels of MBPs were expressed. Immunohistochemical analysis of P14 brain sections of QKIFL/FL;Olig2–Cre mice confirmed the loss of MBP because the staining revealed only the occasional MBP-positive (MBP+) OL in the corpus callosum compared with the intense MBP staining in the QKIFL/FL;− mice (Fig. 2C). Similarly, QKI+ OLs were abundant in the corpus callosum of QKIFL/FL;− mice, whereas this staining pattern was severely diminished in the brains of QKIFL/FL;Olig2–Cre mice (Fig. 2C). These findings show that the loss of QKI in OLs leads to tremors and death by postnatal week 3.
Figure 1.

QKIFL/FL;Olig2–Cre mice display tremors and die at postnatal week 3. A, Schematic of the conditional construct engineered to target exon 2 of the qkI gene. QKIFL depicts the insertion of the loxP sequences flanking exon 2, and QKI1lox is the locus predicted after homologous recombination between the loxP sites. Primers F1/R1 are designed to detect the loxP site insertion. Primers F2/R1 are designed to detect the loss of exon 2 after recombination and the generation of the 1 loxP site. B, PCR amplification of genomic DNA from ear biopsies and brains to detect the 1 lox band confirming Cre-mediated recombination. C, PCR from genomic DNA from the respective tissues showing that the 1 lox PCR band is only detected in QKIFL/FL;Olig2–Cre brains. D, Photographs of QKIFL/FL;− and QKIFL/FL;Olig2–Cre mice at P14. E, Kaplan–Meier survival plot of QKIFL/FL;− (n = 12) and QKIFL/FL;Olig2–Cre (n = 12) mice. The statistical significance for survival was assessed by log-rank (p < 0.0001).
Figure 2.

Recombination in QKIFL/FL;Olig2–Cre mice leads to loss of QKI proteins expression in OLs. A, Total brain lysates from P14 wild-type (QKIFL/FL;−), heterozygotes (QKIFL/+;Olig2–Cre), and knock-out (QKIFL/FL;Olig2-Cre) mice were immunoblotted with anti-QKI-5, anti-QKI-6, anti-QKI-7, anti-MBP, and anti-β-tubulin antibodies. Molecular mass markers are shown on the left in kilodaltons. B, RT-qPCR analysis for QKI and MBP mRNAs in P14 whole brains for the indicated genotypes (n = 3 per genotype). The results are represented in terms of fold change after normalizing the mRNA levels to GAPDH mRNA. Each value represents the mean ± SEM. **p < 0.01. C, Representative images of coronal sections of P14 brains of wild-type (QKIFL/FL;−) and knock-out QKIFL/FL;Olig2–Cre mice stained with anti-QKI-5, anti-QKI-6, anti-QKI-7, and anti-MBP antibodies (n > 3). The area shown is the corpus callosum. Scale bar, 50 μm. Inset denotes higher magnification (scale bar, 25 μm).
Hypomyelination in the CNS of QKIFL/FL;Olig2–Cre mice
The brains of the QKIFL/FL;Olig2–Cre mice were smaller, but the gross anatomy appeared normal. The severe reduction in MBP staining in the brains of QKIFL/FL;Olig2–Cre mice implied a defect in CNS myelination. To examine this in detail, coronal brain sections were stained with fluoromyelin. A complete loss of myelin in the brains of QKIFL/FL;Olig2–Cre was visualized compared with QKIFL/FL;− mice (Fig. 3A,B). We also confirmed the absence of myelin by electron microscopy in the CNS using cross-sections of P14 optic nerves and spinal cords from QKIFL/FL;Olig2–Cre mice (Fig. 3C,D and E,F). There were normal levels of myelin lamellae in wild-type QKIFL/FL;− mice with a g-ratio of ∼0.7, whereas a g-ratio of ∼1.0 was observed in the optic nerve and spinal cord of QKIFL/FL;Olig2–Cre mice, confirming the absence of myelin lamellae (Fig. 3G,H). We noticed that axons in the optic nerves of QKIFL/FL;Olig2–Cre mice had a small diameter (Fig. 3C,D), whereas in the spinal cords, certain axons were larger and displayed axonal swelling (Fig. 3E,F), as often observed in the absence of myelin (Rosenfeld and Friedrich, 1986). These data show that the lack of QKI in developing OLs leads to hypomyelination.
Figure 3.

Ablation of QKI in OLs leads to hypomyelination. A, B, Representative images of coronal sections from brains of QKIFL/FL;− and QKIFL/FL;Olig2–Cre mice stained with fluoromyelin. CC denotes the corpus callosum. Scale bar, 500 μm. C, D, Representative cross-section electron micrograph images of optic nerves from (n = 2/genotype) P14 QKIFL/FL;− and QKIFL/FL;Olig2–Cre mice. Scale bar, 500 nm. E, F, Representative cross-section electron micrograph images of spinal cords from (n = 2/genotype) P14 QKIFL/FL;− and QKIFL/FL;Olig2–Cre mice. Scale bar, 500 nm. G, A scatter plot of the g-ratio determined from electron micrographs of cross-sections of P14 optic nerves from QKIFL/FL;− and QKIFL/FL;Olig2–Cre mice for n = 108 axons. g-ratio, p < 0.0001; axon diameter, p < 0.0001. H, A scatter plot of the g-ratio determined from electron micrographs of cross-sections of P14 spinal cords from QKIFL/FL;− and QKIFL/FL;Olig2–Cre mice for n = 108 axons. g-ratio, p < 0.0001; axon diameter, p < 0.05.
OL differentiation defects in QKIFL/FL;Olig2–Cre mice
To assess whether the lack of MBP in the brain of QKIFL/FL;Olig2–Cre mice was attributable to either a defect in differentiation of OPCs or a complete absence of the OL population, we evaluated the various stages of OL maturation by immunofluorescence. Brain immunostaining for the pan-OL marker Olig2 showed a slight reduction in Olig2+ cells in the corpus callosum of QKIFL/FL;Olig2–Cre mice (Fig. 4A,D). OPCs positive for PDGFRα appeared unaffected by the loss of QKI in OLs (Fig. 4B,D). However, CC1+ cells were absent in the brains of QKIFL/FL;Olig2–Cre mice, which is a marker for postmitotic mature OLs (Fig. 4C,D). In the QKIFL/FL;Olig2–Cre mice, the protein levels of GFAP, an astrocyte marker, Nefl, and NeuN, known neuronal markers, were comparable between genotypes as confirmed by immunoblotting (Fig. 4E). These findings show that the loss of QKI prevents OL maturation with minimal to no effect on the neuronal and astrocytic populations in the brain.
Figure 4.

Ablation of QKI in OLs leads to loss of mature OLs. A, Representative images of coronal sections of P14 brains of wild-type (QKIFL/FL;−) and knock-out QKIFL/FL;Olig2–Cre mice stained with anti-Olig2 and anti-MBP antibodies (n > 3). The area shown is the corpus callosum. Scale bar, 50 μm. Inset denotes higher magnification (scale bar, 25 μm). B, Representative images of coronal sections of P14 brains of wild-type (QKIFL/FL;−) and knock-out QKIFL/FL;Olig2–Cre mice stained with anti-PDGFRα and anti-MBP antibodies (n > 3). The area shown is the corpus callosum. Scale bar, 50 μm. Inset denotes higher magnification (scale bar, 25 μm). C, Representative images of coronal sections of P14 brains of wild-type (QKIFL/FL;−) and knock-out QKIFL/FL;Olig2–Cre mice stained with anti-Olig2 and anti-CC1 antibodies (n > 3). The area shown is the corpus callosum. Scale bar, 50 μm. Inset denotes higher magnification (scale bar, 25 μm). D, Total number of Olig2+, PDGRFα+, and CC1+ cells in the corpus callosum in QKIFL/FL;− and QKIFL/FL;Olig2–Cre. Data are shown as mean ± SEM (n = 3 brains for each phenotype, ***p < 0.001). E, Total brain lysates from P14 wild-type (QKIFL/FL;−), heterozygotes (QKIFL/+;Olig2–Cre), and knock-out (QKIFL/FL;Olig2–Cre) mice were immunoblotted with anti-GFAP, anti-Nefl, anti-NeuN, and anti-β-tubulin antibodies. Molecular mass markers are shown on the left in kilodaltons.
Alternative splicing defects in QKIFL/FL;Olig2–Cre brains
We used brain RNA from P14 wild-type and QKIFL/FL;Olig2–Cre mice to identify alternative splicing defects using layered and integrated system for splicing annotation (LISA), a high-throughput RT-PCR RNomics platform-based annotation of alternative splicing events (Venables et al., 2009). We performed the initial screen termed the “detection screen” using primers targeting 1328 characterized alternative splice events (ASEs), and the PSI was calculated for each event. From this list, 96 ASEs (with a ΔPSI of at least 0.10) were selected for the “validation screen,” and n = 3 mice per genotype were used and their PSI values were calculated. This screen yielded 31 ASEs that were significantly altered with the loss of QKI (Table 1). We searched for neighboring QREs (ACUAAY–N20–UAAY; Galarneau and Richard, 2005) adjacent to the alternatively spliced exons, and we identified at least one QRE near each splice event, suggesting that these splicing events are directly regulated by QKI (Table 1). The alternative splicing of MAG, PLP, and Sirt2 were shown previously to be deregulated in qkv mice and served as positive controls (Wu et al., 2002; Zhu et al., 2012; Mandler et al., 2014). We identified new splicing events of myelin components regulated by QKI, including Nfasc, MBP, MOBP, Mpzl1, and Magi1 (Table 1).
Table 1.
Top alternatively spliced events identified in QKIFL/FL:Olig2–Cre mice
| Gene | Exon number | PSI WT | PSI KO | ΔPSI | |ΔPSI| | QRE | WT versus KO p value |
|---|---|---|---|---|---|---|---|
| Nfasc | 2 | 0.233 | 0.945 | 0.712 | 0.712 | GCTAACCCACTGGAactaac | 5.412E-8 |
| Nfasc | 17 | 0.193 | 0.974 | 0.781 | 0.781 | TCTAACCCTGATAACCTGCTAAC | 1.407E-6 |
| Magi1 | 15 | 0.890 | 0.517 | −0.373 | 0.373 | ACGAATAAACTCTCTTTGCCCTATGTTAAC, CCTAACTAGTTGCAGTAAT | 2.775E-5 |
| Prom1 | 4 | 0.280 | 0.811 | 0.531 | 0.531 | GTCTTAGGTCAAT | 7.690E-5 |
| Lhfpl3 | 5 | 0.225 | 0.729 | 0.504 | 0.504 | ACTAACACATGAAAC | 1.627E-4 |
| Mpzl1 | 5, 6 | 0.270 | 0.099 | −0.171 | 0.171 | ACTAACATTTACTAAAAAC, CCTAACATATGTCTGTAAT | 2.064E-4 |
| Cadm1 | 9 | 0.684 | 0.323 | −0.361 | 0.361 | AGGAAGAACTTTTAACATCTGA | 2.456E-4 |
| Bin1 | 7, 8 | 0.642 | 0.884 | 0.242 | 0.242 | ACTAACCTGCTCCGAAAT, ACAAACCCTCTCTTTGTAAT | 2.772E-4 |
| Vldlr | 16 | 0.179 | 0.011 | −0.168 | 0.186 | ACTAACTGCCGCTTAAATAAT | 6.189E-4 |
| Plp1 | 3 | 0.502 | 0.012 | −0.490 | 0.490 | CAATAACAAGGGGTGGGGGACAATT | 6.791E-4 |
| Phka1 | 19 | 0.613 | 0.896 | 0.283 | 0.283 | TCTAACTCTCAATCTGTGAGGTTGTATAAT, ACTGATTATTTTAAAAATTATTTAAT | 7.481E-4 |
| Mbp | 2 | 0.964 | 0.409 | −0.555 | 0.555 | CAACAAAGAAGAAATTTCTAA | 9.313E-4 |
| Ctnnd1 | 18 | 0.834 | 0.678 | −0.156 | 0.156 | TCTAATGTTGCAGAATAGTGTTTAAT | 1.005E-3 |
| Pcdh15 | 4 | 0.589 | 0.897 | 0.308 | 0.308 | ACTACTAAATAGTTCTATGCTAAT | 2.111E-3 |
| Mon2 | 29 | 0.746 | 0.858 | 0.112 | 0.112 | CCTAACCCTGTCCTAAT | 2.331E-3 |
| Nfasc | 8 | 0.418 | 0.007 | −0.411 | 0.411 | CCTAATCCACTTTGTCTAAC | 2.606E-3 |
| Bnip2 | 10 | 0.392 | 0.127 | −0.265 | 0.265 | CAATAACTCTTCCCTACC | 2.613E-3 |
| Myo9b | 37 | 0.746 | 0.836 | 0.090 | 0.090 | ACTAACCTAGAGCTGCTGGTT | 4.063E-3 |
| Smarcb1 | 2 | 0.717 | 0.788 | 0.071 | 0.071 | CCTAACACTAAG | 5.568E-3 |
| Neo1 | 20 | 0.559 | 0.666 | 0.107 | 0.107 | ACTGACTCACCAGATTCAAGAGTTAAC | 6.321E-3 |
| Nfasc | 20, 21 | 0.197 | 0.000 | −0.197 | 0.197 | TAACCACCTTCTTGCTAAC, CACTAACCACTAAC | 6.660E-3 |
| Mobp | 4 | 0.975 | 0.885 | −0.090 | 0.090 | ACTAACATGGGGGATCTGTCCACAGTACCAAGCCTGTGTAAT, CAATCCATAGGACTGGAAGTCACTAAT | 8.288E-3 |
| Grm5 | 8 | 0.656 | 0.470 | −0.186 | 0.186 | ACTAAATAAATCTTTTCTGGTTAAT | 9.840E-3 |
| Pdlim5 | 5 | 0.234 | 0.127 | −0.107 | 0.107 | CAACTCATTGTTTTTCTTGAAGACATATTTGAGCTGCTAAC | 1.082E-2 |
| Nhsl1 | 5 | 0.048 | 0.448 | 0.400 | 0.400 | ACTAACCTCATGTGGTGCTGTCAAAT | 1.500E-2 |
| Sirt2 | 2 | 0.111 | 0.248 | 0.137 | 0.137 | ACTACCATCCCTGACTCTGAAATAAT | 1.919E-2 |
| Tnk2 | 12 | 0.850 | 0.715 | −0.135 | 0.135 | CTAAACCCTTCCCTCTGTGATCT | 2.143E-2 |
| Grik1 | 15 | 0.219 | 0.317 | 0.097 | 0.097 | CAATTTGCTCTTGGGGTGTCTTGTTTTATGCTACTAAT | 3.439E-2 |
| Sytl2 | 10 | 0.611 | 0.719 | 0.108 | 0.108 | GCTAATGAACTAATCCATTAAT,TCTAATGGTTGTGGATAAT | 4.256E-2 |
| Musk | 13 | 0.353 | 0.105 | −0.248 | 0.248 | CCTAATGCTCTCCTAAT, ATTAATGCTACACTTTATACATAGATAAC | 4.380E-2 |
| Oxr1 | 11 | 0.700 | 0.607 | −0.093 | 0.093 | AGTAACTTTAAT | 4.733E-2 |
The top identified genes in LISA screen that exhibit deregulated alternative splicing attributable to loss of QKI expression in QKIFL/FL;Olig2–Cre mice.
QKI regulates alternative splicing of Nfasc pre-mRNA
Nfasc was identified as a top hit with multiple exons showing splicing deregulation in QKIFL/FL;Olig2–Cre mice (Table 1). Alternative splicing of Nfasc produces cell adhesion proteins termed Nfasc155 and Nfasc186, which are the major isoforms (Sherman et al., 2005). The regulation of Nfasc alterative splicing is unknown. Nfasc155 is OL specific, and exons 21/22 encode a fibronectin III-like domain unique to this isoform (Collinson et al., 1998), whereas exon17 is unique to neuronal Nfasc186 (Ebel et al., 2014). In QKIFL/FL;Olig2–Cre mice, we observed a lack of Nfasc exons 21/22 inclusion and an increase in exon 17, as well as deregulation of exons 2 and 8 (Table 1). We confirmed the exclusion of Nfasc exons 21/22 and the inclusion of exon 17 by RT-PCR in the brains of QKIFL/FL;Olig2–Cre mice using primers flanking these exons (Fig. 5A). These findings were also confirmed by RT-qPCR (Fig. 5B), whereas the overall Nfasc transcript levels were unaffected (Fig. 5B). Nfasc155 protein was absent, whereas Nfasc186 remained unchanged by immunoblotting whole-brain lysates (Fig. 5C). The loss of Nfasc155 did not lead to reduced expression of the voltage-gated sodium channel (Nav) or Caspr (Fig. 5C), consistent with previous findings of Nfasc155−/− mice in OLs (Pillai et al., 2009).
Figure 5.
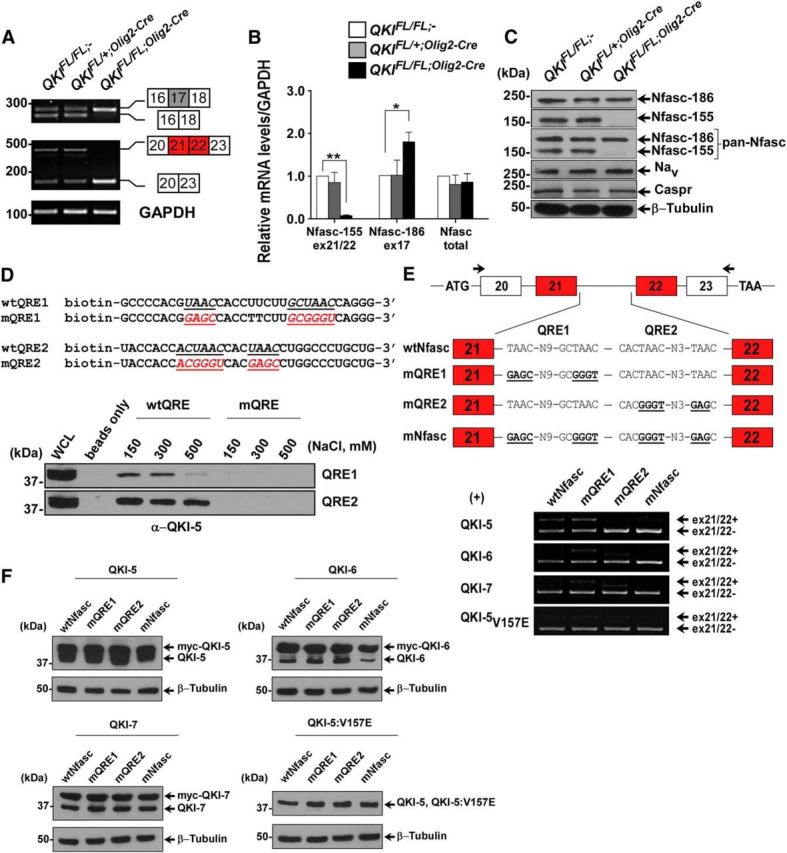
QKI regulates alternative splicing of Nfasc pre-mRNA. A, RT-PCR analysis from P14 mice brains with the indicated primers flanking exon 17 (inclusion denotes Nfasc186) and exons 21/22 (inclusion denotes Nfasc155). B, RT-qPCR using primers spanning the exon–exon junction for exon 17 of Nfasc186, exons 21/22 of Nfasc155, and primers that detect the total mRNA of Nfasc. The results are represented in terms of fold change after normalizing the mRNA levels to GAPDH mRNA from n = 3. Each value represents the mean ± SEM. *p < 0.05; **p < 0.01. C, Western blot analysis of total brain lysates immunoblotted with anti-Nfasc155, anti-Nfasc186, anti-pan-Nfasc, anti-Nav, anti-Caspr, and anti-β-tubulin. Molecular mass markers are shown on the left in kilodaltons. D, Immunoblot analysis of RNA pull-down assay from brains of wild-type P14 mice using biotinylated RNA harboring either wild-type or mutated QREs (mutated sequences indicated) using QKI-5 antibody. Increasing salt concentrations were used to compete with the binding. E, Schematics of the genomic DNA region used to generate the Nfasc155 minigene; the location of the primer set used for RT-PCR are marked by arrows. Wild-type sequences as well as sequences harboring mutant QRE1, QRE2, or both are shown. HEK293T cells were transfected with the Nfasc minigene plasmid and either expression vectors for QKI-5, QKI-6, QKI-7, or QKI-5:V157E. Total RNA was isolated 48 h later and analyzed by RT-PCR. F, Protein expression of transfected plasmids in HEK293T cells is determined by immunoblotting using the indicated antibodies. Myc-tagged QKI proteins migrate higher than endogenous QKI isoforms.
We analyzed the genomic sequence surrounding exons 21/22 of Nfasc155 for the presence of QREs. Two putative QREs were identified in intron 21 that could be responsible for the alternative splicing of the exons 21/22. QRE1 (UAAC–N9–GCUAAC) deviates by a single mismatch (underlined) from the consensus QRE, whereas QRE2 is a perfect binding site. To examine whether these QREs are indeed functional, biotinylated RNAs harboring wild-type and mutated binding sequences were synthesized for each Nfasc QRE. RNA pull-down assays were performed with brain cell lysates from wild-type mice, and the enrichment of QKI-5 was observed by immunoblotting in the presence of increasing concentrations of sodium chloride. Both QRE1 and QRE2 affinity purified QKI-5, although QRE2 bound with a higher relative affinity. Mutations within the sequences prevented binding (Fig. 5D). To show that QKI isoforms regulate Nfasc alternative splicing via these QREs, we next generated a minigene with a Nfasc genomic fragment spanning the exons 21/22 cassette (Fig. 5E). The minigene was cotransfected in HEK293T cells with expression vectors encoding QKI-5, QKI-6, QKI-7, or an RNA binding defective version of QKI-5 (QKI-5:V157E). QKI-5 expression led to inclusion of exons 21/22, but little to no inclusion was observed with QKI-6, QKI-7, or the RNA binding defective version of QKI-5 (QKI-5:V157E; Fig. 5E). The overexpression of the QKI-6 isoform led to a slight inclusion of exons 21/22 that could be explained by the endogenous expression of QKI-5 in HEK293T cells, which could dimerize with QKI-6 and lead to inclusion of these exons (Fig. 5E). Mutation of QRE2, but not QRE1, prevented this inclusion, suggesting that QRE2 is the major binding site used by QKI-5. Equivalent QKI protein expression was confirmed by immunoblotting (Fig. 5F). These observations demonstrate that Nfasc is a direct alternative splicing target of QKI.
Loss of QKI in the CNS and PNS of adult mice leads to paralysis
We subsequently investigated the role of QKI proteins in myelin maintenance in adult mice using the OHT inducible Cre line (PLP–CreERT; Doerflinger et al., 2003). Eight-week-old QKIFL/FL;PLP–CreERT mice and age-matched wild-type littermates were injected with OHT for 5 consecutive days. The presence of the qkI 1lox band was readily detected in brains of QKIFL/FL;PLP–CreERT mice as early as 5 d after OHT treatment (Fig. 6A). QKIFL/FL;PLP–CreERT mice progressively developed hindlimb paralysis with visible thoracic kyphosis at approximately post-OHT injection day 20 (Fig. 6B,C). Numerous QKIFL/FL;PLP–CreERT mice reached a clinical endpoint of accelerated breathing and immobility at approximately 30 d after OHT, so the animals were killed. As early as post-OHT day 15, the mice also displayed a characteristic shaking phenotype when held by the tail. These findings show that deletion of QKI in myelinating glia of adult mice results in paralysis and immobility.
Figure 6.

QKIFL/FL; PLP–CreERT mice display Nfasc pre-mRNA splicing defects. A, PCR amplification of genomic DNA from brains of 8-week-old wild-type and QKIFL/FL;PLP–CreERT mice injected with OHT for 5 consecutive days to confirm the Cre-mediated recombination and the presence of a 1 lox band. Mice were killed, and brains were collected at 30 d after OHT injection. B, Photographs of QKIFL/FL;− and QKIFL/FL;PLP–CreERT mice 30 d after OHT injection. C, Kaplan–Meier plot of onset of paralysis in QKIFL/FL;− (n = 11) and QKIFL/FL;PLP–CreERT (n = 11) mice. The statistical significance was assessed by log-rank (p < 0.0001). D, RT-PCR analysis of spinal cords, brains, and sciatic nerves from wild-type and QKIFL/FL;PLP–CreERT mice injected with OHT at 30 d after injection using primers flanking exon 17 (inclusion denotes Nfasc186) or exons 21/22 (inclusion denotes Nfasc155). E, Immunoblot analysis of lysates obtained from spinal cord and brain samples of 8-week-old wild-type and QKIFL/FL;PLP–CreERT mice injected with OHT for 5 consecutive days and harvested at 30 d after OHT injection. Blots were against anti-Nfasc155, anti-Nfasc186, and anti-β-tubulin. F, RT-qPCR analysis from spinal cord samples (n = 4) of 8-week-old wild-type and QKIFL/FL;PLP–CreERT mice injected with OHT for 5 consecutive days and collected at 30 d after OHT injection. Primers were specific for qkI, Nfasc155 ex21/22, and Nfasc186 ex17 mRNAs. The results are represented in terms of the mean of the fold change after normalizing the mRNA levels to GAPDH mRNA ± SEM. *p < 0.05; ***p < 0.001. G, RT-qPCR analysis from brain samples (n = 4) of 8-week-old wild-type and QKIFL/FL;PLP–CreERT mice injected with OHT for 5 consecutive days and collected 30 d after OHT injection. Primers were specific for qkI, Nfasc155 ex21/22, and Nfasc186 ex17 mRNAs. The results are represented in terms of the mean of the fold change after normalizing the mRNA levels to GAPDH mRNA ± SEM. **p < 0.01; ***p < 0.001.
QKI regulates Nfasc pre-mRNA alternative splicing in mature OLs
We next examined whether the loss of QKI in mature OLs could affect the Nfasc155 isoform expression. Indeed, we observed exclusion of Nfasc155 exons 21/22 in QKIFL/FL;PLP–CreERT mice as early as 5 d after OHT injection in the brains, spinal cords, and sciatic nerves with a concomitant inclusion of exon 17 (data not shown) and persisting at 30 d after OHT injection as shown by RT-PCR (Fig. 6D) and RT-qPCR (Fig. 6F,G). The exclusion of exons 21/22 was paralleled with an appreciable reduction in Nfasc155 protein observed at 30 d after OHT injection with no visible increase in Nfasc186 (Fig. 6E). We next isolated primary OL precursor (OPC) cells from P1 QKIFL/+;− and QKIFL/FL;PLP–CreERT mice and induced them to differentiate for 4 d before treating them with increasing concentrations of OHT. Genomic PCR confirmed recombination between the loxP sites in OLs with the generation of the 1 lox allele (Fig. 7A,B, top panels). The exclusion of exons 21/22 and the inclusion of exon 17 was observed in differentiated cultures treated with OHT of QKIFL/FL;PLP–CreERT OLs compared with QKIFL/+;− OLs using RT-PCR (Fig. 7A,B, bottom panels) and RT-qPCR (diff, Fig. 7C), although no change was observed in undifferentiated cultures without OHT treatment (Fig. 7A,C).
Figure 7.

Loss of QKI in differentiated primary mouse and rat OLs leads to defects in Nfasc pre-mRNA splicing. A, B, Top, PCR amplification of genomic DNA from primary mouse OPCs isolated from wild-type and QKIFL/FL;PLP–CreERT mice. Cultures were induced to differentiate for 4 d before OHT treatment with increasing concentrations (50, 100, 150, 200, and 250 nm) for 5 d to confirm the Cre-mediated recombination. The presence of a 1 lox band indicates recombination between LoxP sites. Bottom, RT-PCR analysis from wild-type and QKIFL/FL;PLP–CreERT differentiated cultures after 5 d treatment with OHT using primers flanking exon 17 (inclusion denotes Nfasc186) or exons 21/22 (inclusion denotes Nfasc155). C, RT-qPCR analysis for wild-type and QKIFL/FL;PLP–CreERT differentiated cultures after treatment with OHT using primers for QKI mRNA and Nfasc155 exons 21/22 and Nfasc186 exon 17. Cultures were isolated from pooled QKIFL/+ (n = 5) or QKIFL/FL;PLP–CreERT (n = 5) P1 mice. The results are represented as the average fold change obtained from the individual treatment after normalizing the mRNA levels to GAPDH mRNA. Each value represents the mean ± SEM. *p < 0.05; **p < 0.01. D, RT-PCR from differentiated rat OPCs transfected with either siLuc–GLO or increasing concentrations of siQKI–GLO using primers for exon 17 (Nfasc186-specific) and exons 21/22 (Nfasc155-specific). GAPDH is used as a control. E, Immunoblot analysis from undifferentiated and differentiated rat OPCs for 4 d and then transfected with increasing concentrations of siQKI–GLO (5, 10, 20, and 40 nm) blotted using anti-Nfasc186, anti-Nfasc155, and anti-β-tubulin. F, Immunoblot analysis from differentiated rat OPCs for 4 d and then transfected with increasing concentrations of siQKI–GLO (5, 10, 20, and 40 nm) blotted using anti-QKI-5, anti-QKI-6, anti-QKI-7, and anti-β-tubulin.
We investigated whether we could observe these Nfasc alternative splicing defects in primary rat OLs using fluorescently labeled (GLO) siRNAs targeting QKI. The transfection efficiency was >50% (data not shown). An increase in exons 21/22 exclusion (Fig. 7D; exon 20/23 fragment) and the inclusion of exon 17 was indeed observed (Fig. 7D; exon 16/17/18 fragment) even with the lowest siQKI–GLO concentration, whereas a decrease in the complementary isoforms was difficult to observe because of their elevated abundance after inducing differentiation (Fig. 7D). A significant decrease in Nfasc155 protein levels was observed with increasing concentrations of siQKI–GLO, but we were unable to detect the appearance of Nfasc186 in the primary rat OLs (Fig. 7E). We confirmed that the siQKI reduced the expression of the QKI isoforms, and this was evident at 40 nm, as assessed by immunoblotting (Fig. 7F). Together, our findings show that QKI regulates Nfasc alternative splicing to maintain Nfasc155 expression in mature OLs.
Paranodal defects in QKIFL/FL;Olig2–Cre and QKIFL/FL;PLP–CreERT mice
We next sought to assess the integrity of the nodes of Ranvier and the paranodes using immunofluorescence and electron microscopy. Ultrastructure analysis of optic nerves and spinal cords showed that wild-type mice had properly formed and spaced paranodal loops (Fig. 8A,C). Although in QKIFL/FL;Olig2–Cre mice, the optic nerves were translucent, a sign of hypomyelination (data not shown) and paranodal loops were difficult to observe being large with little periodicity (Fig. 8B,D).
Figure 8.

Ultrastructural paranodal defects in QKIFL/FL;Olig2–Cre mice. A–D, Representative electron micrograph images of longitudinal sections of optic nerves and spinal cords (n = 2/genotype) from wild-type QKIFL/FL;− and QKIFL/FL;Olig2–Cre mice. Scale bar, 100 nm.
Next, using spinal cords and sciatic nerves from adult QKIFL/FL;PLP–CreERT mice, we could assess the role of the QKI proteins in axoglial junction maintenance. In wild-type mice, immunostaining for paranodal Nfasc155 and Caspr along with the nodal AnkG at 30 d after OHT injection showed normal localization of Nfasc155 and Caspr at the paranodes, with AnkG being restricted to the nodes (Fig. 9A,B). The QKIFL/FL;PLP–CreERT mice harbored abnormal Nfasc155 and Caspr staining patterns at the paranodes. Staining for Nfasc155 in the spinal cord showed that loss of Nfasc155+ staining in one paranode was observed in ∼15% of the paranodes, whereas 69% displayed loss of Nfasc155+ staining in both paranodes and ∼16% had an increased distance in between the paranodes. Staining for Caspr showed that loss of Caspr+ staining in one paranode counted for ∼58% of the paranodes, whereas 9% displayed loss of Caspr+ staining in both paranodes and ∼32% had an increased distance in between the paranodes. We quantified the total number of each observed defect using both anti-Nfasc155 and anti-Caspr. The loss of both Nfasc155+ paranodes was the most observed abnormal staining pattern. In the sciatic nerves, a significant number of paranodes displaying increasing nodal gap and shorter paranode length compared with wild-type mice was observed (Fig. 9B, bottom panel). We also confirmed the paranodal defects in these mice by electron microscopy in spinal cord samples. Wild-type mice displayed regular paranodal loops that terminate at the axon with tight axoglial junctions and minimal spacing between the paranodal loop and the axolemma (Fig. 10A,B). In QKIFL/FL;PLP–CreERT mice, many terminal paranodal loops were found to be everting away from the axon and displayed irregular formations in which the loops abut each other instead of the axons (Fig. 10C). Absence of transverse bands is evident in these mice along with an increase in the paranodal junction gap width (Fig. 10D). These findings show that the loss of QKI expression in QKIFL/FL;PLP–CreERT mice leads to defective axoglial junctions and confirms the requirement for the ongoing expression of QKI to maintain expression of Nfasc155 and hence paranodal integrity.
Figure 9.

QKI-deficient mice exhibit altered distribution and expression of paranodal proteins. A, Immunostaining images of spinal cord sections from 8-week-old QKIFL/FL;− and QKIFL/FL;PLP–CreERT mice injected with OHT and harvested 30 d after injection and stained for Caspr, Nfasc155, and AnkG. Nfasc155 and Caspr colocalized to paranode structures and AnkG localized at the node. Number of defective paranodes observed in the spinal cord of QKIFL/FL;PLP–CreERT for each type of defect is shown on the right. B, Top, Immunostaining images of teased sciatic nerves from 8-week-old QKIFL/FL;− and QKIFL/FL;PLP–CreERT mice injected with OHT and harvested 30 d after injection and stained for Caspr, Nfasc155, and AnkG. Nfasc155 and Caspr colocalized to paranode structures and AnkG localized at the node. Bottom, Vertical scatter plot showing nodal gap size between Nfasc155+ paranodes. A total of 106 wild-type and 122 QKIFL/FL;PLP–CreERT nodal gaps were quantified (p < 0.001). Right, Vertical scatter plot displaying paranode length of QKIFL/FL mice versus QKIFL/FL;PLP–CreERT. Two hundred twelve wild-type and 244 QKIFL/FL;PLP–CreERT paranodes were quantified (p < 0.001).
Figure 10.
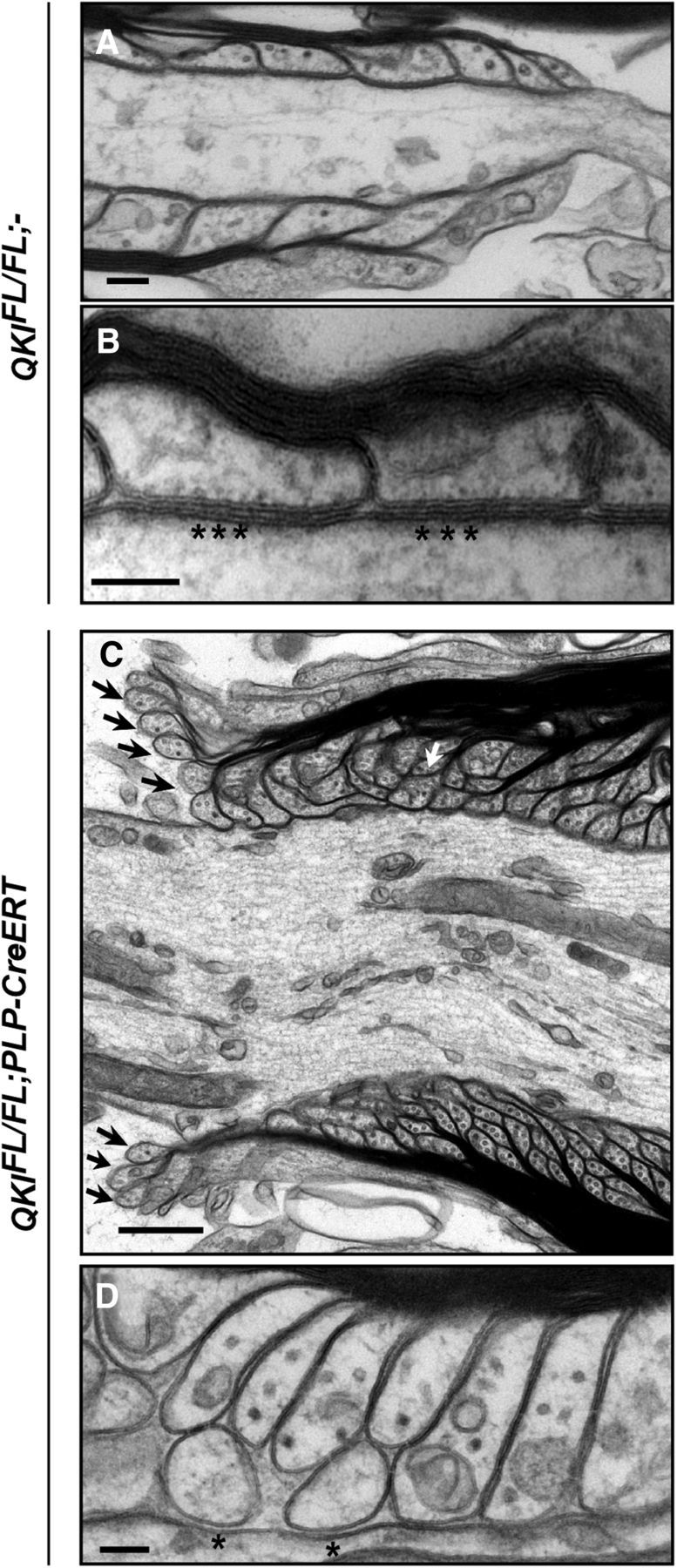
Ultrastructural paranodal defects in QKI-deficient mice. A, B, Representative electron micrograph images of longitudinal sections of spinal cords from wild-type mice at 30 d after OHT injection showing the attachment of paranodal loops to the axolemma and the formation of septate-like junctions (stars). Scale bar, 100 nm. C, Representative electron micrograph images of longitudinal sections of spinal cords from QKIFL/FL;PLP–CreERT mice displaying detached paranodal loops (black arrows) and the failure of several loops to terminate at the axolemma and instead abut each other (white arrow). Scale bar, 500 nm. D, Electron micrographs of QKIFL/FL;PLP–CreERT longitudinal spinal cord sections at 30 d after OHT injection showing the lack of septate-like junctions (stars) and increased space between the paranodal loops and the axolemma. Scale bar, 100 nm.
Discussion
Here, we report a mouse conditional null allele of the QKI RNA binding proteins. Deletion of QKI in OLs with Olig2–Cre caused the mice to develop rapid tremors at P10, followed by death at postnatal week 3. The mice had a severe hypomyelination phenotype, with OLs unable to fully mature beyond the PDGFRα+ stage. Removal of QKI expression in adult mice using PLP–CreERT resulted in hindlimb paralysis and immobility by ∼30 d after OHT injection. In these mice, we observed defects in axoglial junctions. High-throughput quantitative PCR identified several alternative splicing defects, including that of the axoglial protein Nfasc. Our findings define roles for QKI in myelin development and maintenance, as well as in the Nfasc isoform balance to maintain intact axoglial junctions.
The onset of tremors in the QKIFL/FL;Olig2–Cre mice at P10 and death at postnatal week 3 is similar to null alleles of Nfasc155 (Sherman et al., 2005; Pillai et al., 2009), Caspr (Bhat et al., 2001), and Contactin (Berglund et al., 1999) in that they die at postnatal week 3; however, they do differ in that QKIFL/FL;Olig2–Cre mice completely lack myelin, whereas the other mice have normal myelin. These findings suggest that a major function of the QKI proteins is to generate the Nfasc155 isoform for proper development of the axoglial junctions. Moreover, we identify a regulatory pathway through which QKI regulates Nfasc pre-mRNA splicing to generate Nfasc155. The binding of QKI to an RNA element (QRE2) in Nfasc intron 21 is required to promote inclusion of exons 21/22, which encode the third fibronectin-like domain unique to Nfasc155. A position-dependent asymmetric regulation of alternative splicing has been proposed for QKI with binding to QREs upstream of skipped exons and downstream of included exons in muscle (Hall et al., 2013). The fact that QRE2 is upstream of included exon 22 suggests that QKI does not exclusively function in a position-dependent asymmetric regulation, like nSR100 (Raj et al., 2014). The absence of QKI in primary OLs led to the production of Nfasc186 mRNA, but not protein. These observations suggest that the Nfasc neuronal isoforms may be the default mRNA produced in the absence of the QKI proteins. It is likely that QKI functions in collaboration with other splicing factors to regulate the Nfasc splicing events. Indeed RBFOX2, PTB, and SF1 have been shown to influence the alternative splicing activity of QKI in tumor microenvironment (Brosseau et al., 2014), muscle differentiation (Hall et al., 2013), and lung tumorigenesis (Zong et al., 2014), respectively. We also note the presence of eight QREs in the Nfasc155 mRNA. These observations suggest that the QKI isoforms may also regulate Nfasc mRNA export, transport, mRNA stability, and translation.
The qkI conditional allele provides a role for the QKI proteins in axoglial junction formation and maintenance. Although paranodal defects were observed in myelin mutants, such as Shiverer and qkv (Rosenbluth and Bobrowski-Khoury, 2013), these are likely a consequence of the severe dysmyelination and loss of mature OLs. We show that our observed paranodal defects are not solely an outcome of dysmyelination but are attributable to the lack of the generation of the Nfasc155 isoform. This was confirmed with primary rat and mouse QKIFL/FL;PLP–CreERT OLs. In vivo deletion of the QKI proteins in QKIFL/FL;PLP–CreERT mice further substantiated the requirement of QKI to maintain axoglial junctions. Despite adult mice possessing established axonal domains, removal of QKI resulted in the rapid loss of Nfasc155 mRNA and protein along with the observed paranodal axoglial junction defects. A similar defect is observed in adult OL-specific loss of Nfasc155−/− (Sherman et al., 2005; Pillai et al., 2009). This suggests that axoglial components are continuously being replenished to maintain the integrity of the nodes and paranodes and further demonstrate an ongoing requirement for glial QKI expression to maintain Nfasc155 protein. A complete absence of Nfasc155 at the paranodes in Nfasc155FL/FL;PLP–CreERT mice was only seen after 90 d after OHT injection, whereas by day 20 after OHT injection, only a 50% reduction in Nfasc155 levels was observed (Pillai et al., 2009). Consistent with this observation, QKIFL/FL;PLP–CreERT mice display the expected 50% reduction in Nfasc155 staining with a significant increase in nodal gap. However, unlike Nfasc155FL/FL;PLP–CreERT mice, the QKIFL/FL;PLP–CreERT mice exhibit progressive paralysis with immobility ∼30 d after OHT injection, preventing additional analysis of axoglial junction defects. Therefore, the death of the QKIFL/FL;PLP–CreERT mice cannot be explained solely by the loss of Nfasc155, indicating that deregulated control of RNA metabolism of other QKI targets also contribute to the phenotypes of these mice.
In humans, QKI has been associated with neurological disorders. Decreased expression of the QKI proteins is associated with schizophrenia along with other myelin abnormalities (Katsel et al., 2005; Haroutunian et al., 2006). Abnormally spliced Nfasc has been identified in schizophrenic brains (Wu et al., 2012), suggesting that reduced levels of QKI may be responsible for this event. Proper formation and maintenance of paranodal domains is critical, because disruption of Nfasc localization has been shown to lead to early changes that precede demyelination in multiple sclerosis (Howell et al., 2006). Thus, the regulation of Nfasc alternative splicing by QKI may be disrupted in certain demyelination disorders and schizophrenia. A breakpoint mutation in humans mapping to the QKI locus leads to a phenotype similar to 6q terminal deletion syndrome, with characteristics such as intellectual disabilities, hypotonia, seizures, brain anomalies, and specific dysmorphic features (Backx et al., 2010). Our findings suggest that the loss of QKI leads to paranodal defects, which may be associated with certain neurological disorders.
In conclusion, using a new conditional allele of qkI, we show that QKI not only plays a role in myelin development via the regulation of OL differentiation but also in myelin maintenance in adult mice. Our studies identify a novel role for the QKI proteins in axoglial junction maintenance by regulating the expression of the Nfasc155 splice isoform. These findings define the QKI RNA binding protein as a critical regulator of many RNA processes in myelinating glia, including alternative splicing.
Footnotes
This work was supported by a grant from the Multiple Sclerosis Society of Canada (S.R.). We thank Jun Fang and Jeannie Mui for technical assistance with the primary cultures and EM, respectively. We thank Roscoe Klinck, Durand Mathieu, and Philippe Thibault from the Université de Sherbrooke RNomics platform. We also acknowledge Kelly Sears and Hojatollah Vali for helpful discussions.
The authors declare no competing financial interests.
References
- Almazan G, Afar DE, Bell JC. Phosphorylation and disruption of intermediate filament protein in oligodendrocyte precursor cultures treated with calyculin A. J Neurosci Res. 1993;36:163–172. doi: 10.1002/jnr.490360206. [DOI] [PubMed] [Google Scholar]
- Backx L, Fryns JP, Marcelis C, Devriendt K, Vermeesch J, Van Esch H. Haploinsufficiency of the gene Quaking (QKI) is associated with the 6q terminal deletion syndrome. Am J Hum Genet. 2010;152A:319–326. doi: 10.1002/ajmg.a.33202. [DOI] [PubMed] [Google Scholar]
- Berglund EO, Murai KK, Fredette B, Sekerková G, Marturano B, Weber L, Mugnaini E, Ranscht B. Ataxia and abnormal cerebellar microorganization in mice with ablated contactin gene expression. Neuron. 1999;24:739–750. doi: 10.1016/S0896-6273(00)81126-5. [DOI] [PubMed] [Google Scholar]
- Beuck C, Qu S, Fagg WS, Ares M, Jr, Williamson JR. Structural analysis of the quaking homodimerization interface. J Mol Biol. 2012;423:766–781. doi: 10.1016/j.jmb.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St Martin M, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron. 2001;30:369–383. doi: 10.1016/S0896-6273(01)00294-X. [DOI] [PubMed] [Google Scholar]
- Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron. 2001;30:385–397. doi: 10.1016/S0896-6273(01)00296-3. [DOI] [PubMed] [Google Scholar]
- Brosseau JP, Lucier JF, Nwilati H, Thibault P, Garneau D, Gendron D, Durand M, Couture S, Lapointe E, Prinos P, Klinck R, Perreault JP, Chabot B, Abou-Elela S. Tumor microenvironment-associated modifications of alternative splicing. RNA. 2014;20:189–201. doi: 10.1261/rna.042168.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles P, Tait S, Faivre-Sarrailh C, Barbin G, Gunn-Moore F, Denisenko-Nehrbass N, Guennoc AM, Girault JA, Brophy PJ, Lubetzki C. Neurofascin is a glial receptor for the paranodin/Caspr-contactin axonal complex at the axoglial junction. Curr Biol. 2002;12:217–220. doi: 10.1016/S0960-9822(01)00680-7. [DOI] [PubMed] [Google Scholar]
- Chénard CA, Richard S. New implications for the QUAKING RNA binding protein in human disease. J Neurosci Res. 2008;86:233–242. doi: 10.1002/jnr.21485. [DOI] [PubMed] [Google Scholar]
- Chen T, Richard S. Structure-function analysis of Qk1: a lethal point mutation in mouse quaking prevents homodimerization. Mol Cell Biol. 1998;18:4863–4871. doi: 10.1128/MCB.18.8.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinson JM, Marshall D, Gillespie CS, Brophy PJ. Transient expression of neurofascin by oligodendrocytes at the onset of myelinogenesis: implications for mechanisms of axon-glial interaction. Glia. 1998;23:11–23. doi: 10.1002/(SICI)1098-1136(199805)23:1<11::AID-GLIA2>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Doerflinger NH, Macklin WB, Popko B. Inducible site-specific recombination in myelinating cells. Genesis. 2003;35:63–72. doi: 10.1002/gene.10154. [DOI] [PubMed] [Google Scholar]
- Doukhanine E, Gavino C, Haines JD, Almazan G, Richard S. The QKI-6 RNA binding protein regulates actin-interacting protein-1 mRNA stability during oligodendrocyte differentiation. Mol Biol Cell. 2010;21:3029–3040. doi: 10.1091/mbc.E10-04-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebel J, Beuter S, Wuchter J, Kriebel M, Volkmer H. Organisation and control of neuronal connectivity and myelinaition by cell adhesion molecule neurofascin. Adv Neurobiol. 2014;8:231–247. doi: 10.1007/978-1-4614-8090-7_10. [DOI] [PubMed] [Google Scholar]
- Ebersole TA, Chen Q, Justice MJ, Artzt K. The quaking gene product necessary in embryogenesis and myelination combines features of RNA binding and signal transduction proteins. Nat Genet. 1996;12:260–265. doi: 10.1038/ng0396-260. [DOI] [PubMed] [Google Scholar]
- Galarneau A, Richard S. Target RNA motif and target mRNAs of the Quaking STAR protein. Nat Struct Mol Biol. 2005;12:691–698. doi: 10.1038/nsmb963. [DOI] [PubMed] [Google Scholar]
- Gollan L, Salomon D, Salzer JL, Peles E. Caspr regulates the processing of contactin and inhibits its binding to neurofascin. J Cell Biol. 2003;163:1213–1218. doi: 10.1083/jcb.200309147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano MJ, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;14:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MP, Nagel RJ, Fagg WS, Shiue L, Cline MS, Perriman RJ, Donohue JP, Ares M., Jr Quaking and PTB control overlapping splicing regulatory networks during muscle cell differentiation. RNA. 2013;19:627–638. doi: 10.1261/rna.038422.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RJ, Loushin CL, Friedrich VL, Jr, Chen Q, Ebersole TA, Lazzarini RA, Artzt K. Neural cell type-specific expression of QKI proteins is altered in the quaking viable mutant mice. J Neurosci. 1996;16:7941–7949. doi: 10.1523/JNEUROSCI.16-24-07941.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haroutunian V, Katsel P, Dracheva S, Davis KL. The human homolog of the QKI gene affected in the severe dysmyelination “quaking” mouse phenotype: downregulated in multiple brain regions in schizophrenia. Am J Psychiatry. 2006;163:1834–1837. doi: 10.1176/ajp.2006.163.10.1834. [DOI] [PubMed] [Google Scholar]
- Howell OW, Palser A, Polito A, Melrose S, Zonta B, Scheiermann C, Vora AJ, Brophy PJ, Reynolds R. Disruption of neurofascin localization reveals early changes preceding demyelination and remyelination in multiple sclerosis. Brain. 2006;129:3173–3185. doi: 10.1093/brain/awl290. [DOI] [PubMed] [Google Scholar]
- Katsel P, Davis KL, Haroutunian V. Variations in myelin and oligodendrocyte-related gene expression across multiple brain regions in schizophrenia: a gene ontology study. Schizophr Res. 2005;79:157–173. doi: 10.1016/j.schres.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Klinck R, Bramard A, Inkel L, Dufresne-Martin G, Gervais-Bird J, Madden R, Paquet ER, Koh C, Venables JP, Prinos P, Jilaveanu-Pelmus M, Wellinger R, Rancourt C, Chabot B, Abou Elela S. Multiple alternative splicing markers for ovarian cancer. Cancer Res. 2008;68:657–663. doi: 10.1158/0008-5472.CAN-07-2580. [DOI] [PubMed] [Google Scholar]
- Larocque D, Pilotte J, Chen T, Cloutier F, Massie B, Pedraza L, Couture R, Lasko P, Almazan G, Richard S. Nuclear retention of MBP mRNAs in the Quaking viable mice. Neuron. 2002;36:815–829. doi: 10.1016/S0896-6273(02)01055-3. [DOI] [PubMed] [Google Scholar]
- Larocque D, Galarneau A, Liu HN, Scott M, Almazan G, Richard S. Protection of the p27KIP1 mRNA by quaking RNA binding proteins promotes oligodendrocyte differentiation. Nat Neurosci. 2005;8:27–33. doi: 10.1038/nn1359. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhang Y, Li D, Feng Y. Destabilization and mislocalization of the myelin basic protein mRNAs in quaking dysmyelination lacking the Qk1 RNA-binding proteins. J Neurosci. 2000;20:4944–4953. doi: 10.1523/JNEUROSCI.20-13-04944.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Takakura N, Oike Y, Imanaka T, Araki K, Suda T, Kaname T, Kondo T, Abe K, Yamamura K. Defective smooth muscle development in qkI-deficient mice. Dev Growth Differ. 2003;45:449–462. doi: 10.1111/j.1440-169X.2003.00712.x. [DOI] [PubMed] [Google Scholar]
- Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, Bachoo RM, Kane M, Louis DN, Depinho RA, Anderson DJ, Stiles CD, Rowitch DH. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503–517. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandler MD, Ku L, Feng Y. A cytoplasmic quaking I isoform regulates the hnRNP F/H-dependent alternative splicing pathway in myelinating glia. Nucleic Acids Res. 2014;42:7319–7329. doi: 10.1093/nar/gku353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel-Rosen H, Volohonsky G, Reuveny A, Zaidel-Bar R, Volk T. Two isoforms of the Drosophila RNA binding protein, How, act in opposing directions to regulate tendon cell differentiation. Dev Cell. 2002;2:183–193. doi: 10.1016/S1534-5807(01)00118-6. [DOI] [PubMed] [Google Scholar]
- Pillai AM, Thaxton C, Pribisko AL, Cheng JG, Dupree JL, Bhat MA. Spatiotemporal ablation of myelinating glia-specific neurofascin (Nfasc NF155) in mice reveals gradual loss of paranodal axoglial junctions and concomitant disorganization of axonal domains. J Neurosci Res. 2009;87:1773–1793. doi: 10.1002/jnr.22015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj B, Irimia M, Braunschweig U, Sterne-Weiler T, O'Hanlon D, Lin ZY, Chen GI, Easton LE, Ule J, Gingras AC, Eyras E, Blencowe BJ. A global regulatory mechanism for activating an exon network required for neurogenesis. Mol Cell. 2014;56:90–103. doi: 10.1016/j.molcel.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth J, Bobrowski-Khoury N. Structural bases for central nervous system malfunction in the quaking mouse: dysmyelination in a potential model of schizophrenia. J Neurosci Res. 2013;91:374–381. doi: 10.1002/jnr.23167. [DOI] [PubMed] [Google Scholar]
- Rosenfeld J, Friedrich VL., Jr Oligodendrocyte production and myelin recovery in heterozygous jimpy mice: an autoradiographic study. Int J Dev Neurosci. 1986;4:179–187. doi: 10.1016/0736-5748(86)90043-2. [DOI] [PubMed] [Google Scholar]
- Schüller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, Ma Q, Alvarez-Buylla A, McMahon AP, Rowitch DH, Ligon KL. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DL, Tait S, Melrose S, Johnson R, Zonta B, Court FA, Macklin WB, Meek S, Smith AJ, Cottrell DF, Brophy PJ. Neurofascins are required to establish axonal domains for saltatory conduction. Neuron. 2005;48:737–742. doi: 10.1016/j.neuron.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Tait S, Gunn-Moore F, Collinson JM, Huang J, Lubetzki C, Pedraza L, Sherman DL, Colman DR, Brophy PJ. An oligodendrocyte cell adhesion molecule at the site of assembly of the paranodal axo-glial junction. J Cell Biol. 2000;150:657–666. doi: 10.1083/jcb.150.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplova M, Hafner M, Teplov D, Essig K, Tuschl T, Patel DJ. Structure-function studies of STAR family Quaking proteins bound to their in vivo RNA target sites. Genes Dev. 2013;27:928–940. doi: 10.1101/gad.216531.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaxton C, Pillai AM, Pribisko AL, Labasque M, Dupree JL, Faivre-Sarrailh C, Bhat MA. In vivo deletion of immunoglobulin domains 5 and 6 in neurofascin (Nfasc) reveals domain-specific requirements in myelinated axons. J Neurosci. 2010;30:4868–4876. doi: 10.1523/JNEUROSCI.5951-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veer EP, de Bruin RG, Kraaijeveld AO, de Vries MR, Bot I, Pera T, Segers FM, Trompet S, van Gils JM, Roeten MK, Beckers CM, van Santbrink PJ, Janssen A, van Solingen C, Swildens J, de Boer HC, Peters EA, Bijkerk R, Rousch M, Doop M, et al. Quaking, an RNA-binding protein, is a critical regulator of vascular smooth muscle cell phenotype. Circ Res. 2013;113:1065–1075. doi: 10.1161/CIRCRESAHA.113.301302. [DOI] [PubMed] [Google Scholar]
- Venables JP, Klinck R, Koh C, Gervais-Bird J, Bramard A, Inkel L, Durand M, Couture S, Froehlich U, Lapointe E, Lucier JF, Thibault P, Rancourt C, Tremblay K, Prinos P, Chabot B, Elela SA. Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol. 2009;16:670–676. doi: 10.1038/nsmb.1608. [DOI] [PubMed] [Google Scholar]
- Wu JI, Reed RB, Grabowski PJ, Artzt K. Function of quaking in myelination: regulation of alternative splicing. Proc Natl Acad Sci U S A. 2002;99:4233–4238. doi: 10.1073/pnas.072090399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ, Wang X, Beveridge NJ, Tooney PA, Scott RJ, Carr VJ, Cairns MJ. Transcriptome sequencing revealed significant alteration of cortical promoter usage and splicing in schizophrenia. PLoS One. 2012;7:e36351. doi: 10.1371/journal.pone.0036351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Zhou L, Tonissen K, Tee R, Artzt K. The quaking I-5 protein (QKI-5) has a novel nuclear localization signal and shuttles between the nucleus and the cytoplasm. J Biol Chem. 1999;274:29202–29210. doi: 10.1074/jbc.274.41.29202. [DOI] [PubMed] [Google Scholar]
- Yu Z, Chen T, Hébert J, Li E, Richard S. A mouse PRMT1 null allele defines an essential role for arginine methylation in genome maintenance and cell proliferation. Mol Cell Biol. 2009;29:2982–2996. doi: 10.1128/MCB.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zearfoss NR, Clingman CC, Farley BM, McCoig LM, Ryder SP. Quaking regulates Hnrnpa1 expression through its 3′ UTR in oligodendrocyte precursor cells. PLoS Genet. 2011;7:e1001269. doi: 10.1371/journal.pgen.1001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Mandler MD, Yi H, Feng Y. Quaking I controls a unique cytoplasmic pathway that regulates alternative splicing of myelin-associated glycoprotein. Proc Natl Acad Sci U S A. 2010;107:19061–19066. doi: 10.1073/pnas.1007487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Zhao L, Wang E, Dimova N, Liu G, Feng Y, Cambi F. The QKI-PLP pathway controls SIRT2 abundance in CNS myelin. Glia. 2012;60:69–82. doi: 10.1002/glia.21248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong FY, Fu X, Wei WJ, Luo YG, Heiner M, Cao LJ, Fang Z, Fang R, Lu D, Ji H, Hui J. The RNA-binding protein QKI suppresses cancer-associated aberrant splicing. PLoS Genet. 2014;10:e1004289. doi: 10.1371/journal.pgen.1004289. [DOI] [PMC free article] [PubMed] [Google Scholar]