

Abstract
Rab35 is a key protein for cargo loading in the recycling endosome. In neuronal immortalized cells, Rab35 promotes neurite differentiation. Here we describe that Rab35 favors axon elongation in rat primary neurons in an activity-dependent manner. In addition, we show that the p53-related protein kinase (PRPK) negatively regulates axonal elongation by reducing Rab35 protein levels through the ubiquitin-proteasome degradation pathway. PRPK-induced Rab35 degradation is regulated by its interaction with microtubule-associated protein 1B (MAP1B), a microtubule stabilizing binding protein essential for axon elongation. Consistently, axon defects found in MAP1B knock-out neurons were reversed by Rab35 overexpression or PRPK inactivation suggesting an epistatic relationship among these proteins. These results define a novel mechanism to support axonal elongation, by which MAP1B prevents PRPK-induced Rab35 degradation. Such a mechanism allows Rab35-mediated axonal elongation and connects the regulation of actin dynamics with membrane trafficking. In addition, our study reveals for the first time that the ubiquitin-proteasome degradation pathway regulates a Rab GTPase.
SIGNIFICANCE STATEMENT Rab35 is required for axonal outgrowth. We define that its protein levels are negatively regulated by p53-related protein kinase (PRPK). We show that microtubule-associated protein 1B (MAP1B) interacts with PRPK, preventing PRPK-dependent Rab35 proteasome degradation. We demonstrate that Rab35 regulates Cdc42 activity in neurons. This is the first evidence showing that a Rab protein is regulated by degradation dependent on the ubiquitin-proteasome system.
Keywords: axon development, MAP1B, p53-related protein kinase, Rab35, ubiquitin proteosome
Introduction
Neuronal function is highly dependent on the ability of neurons to acquire an asymmetric morphology during embryonic development, which is characterized by the presence of a single axon and several dendrites, in a process referred to as neuronal polarization. Neuronal asymmetry is initiated when neurons begin to elongate their axons, a process supported in part at the molecular level by the integrated regulation of cytoskeletal dynamics and membrane trafficking (Tahirovic and Bradke, 2009; Lalli, 2012; Villarroel-Campos et al., 2014). Neuronal polarized trafficking is an essential feature involved in the generation and maintenance of neuronal asymmetry, which is highly dependent on the supply of membrane needed for cell expansion (Futerman and Banker, 1996) and the differential distribution of proteins, such as receptors, adhesion molecules, and local mediators of polarity (Cheng and Poo, 2012; Namba et al., 2015). The family of Rab GTPase proteins regulates membrane trafficking by specific targeting of elements involved in membrane fission, transport, tethering, docking, and fusion (Zerial and McBride, 2001). Rab GTPases cycle between GDP/GTP loaded forms. GTP-loaded Rabs are the active forms of these small GTPases (Barr and Lambright, 2010). Upon activation, Rab proteins recruit effector proteins (Villarroel-Campos et al., 2014). Although there are >60 different Rabs in mammalian cells, the precise role of these proteins in neurons is only just beginning to be elucidated.
Among the many sources for the delivery of membranes, the recycling endosome has emerged in recent years as a prominent membrane-derived compartment that supports the development of axons in neurons. Rab4, a marker of fast endocytic recycling, is key to facilitating axon elongation in Xenopus laevis neurons. Both Rab4 knockdown and expression of dominant-negative forms of Rab4 inhibit axon elongation of Xenopus retinal ganglion cells (Falk et al., 2014). In rodent neurons, it was shown that Rab11, a marker for slow routes of recycling endosomes, is essential to support axon elongation. A constitutively active form of Rab11 (Rab11Q70L) promotes axon growth in cultured cortical neurons. In contrast, dominant-negative forms of Rab11 or genetic inactivation using shRNAs decrease axonal elongation (Takano et al., 2012). The small GTPase Rab35 also decorates the recycling endosome in many organisms ranging from fruit flies to nematodes and mammals (Kouranti et al., 2006; Sato et al., 2008; Zhang et al., 2009; Kobayashi and Fukuda, 2012). In neuronal cell lines, it has been proposed that Rab35 functions are important to support neurite extension (Kobayashi and Fukuda, 2012, Kobayashi et al., 2014). However, its role in axon specification in primary neurons still remains elusive. Here, we present evidence that Rab35 is essential to promote axon elongation in primary neurons. Its functions are regulated by a novel molecular mechanism involving Rab35 degradation by the ubiquitin proteasome system. Such degradation is dependent on the kinase activity of an atypical serine/threonine protein kinase that phosphorylates p53 at Ser15, its only known substrate in mammals, termed p53-related protein kinase (PRPK; Abe et al., 2001). PRPK is part of a complex known as KEOPS/EKC [kinase, putative endopeptidase and other proteins of small size (KEOPS)/endopeptidase-like kinase chromatin-associated], which participates in telomere uncapping and elongation (Downey et al., 2006), transcriptional regulation (Kisseleva-Romanova et al., 2006), and t6A tRNA modification (Srinivasan et al., 2011). Previously it was shown in yeast cells that Rab35 and PRPK interact (Abe et al., 2006); however, the functional consequence for this interaction is not known. Finally, we show here that the microtubule-associated protein 1B (MAP1B), which is the first MAP expressed in the nervous system during development (Tucker and Matus, 1988), protects Rab35 degradation by interacting with PRPK. MAP1B is necessary for proper axon outgrowth, as reduced MAP1B expression negatively affects axon length in cultured neurons (DiTella et al., 1996; González-Billault et al., 2001), as well as in brain (Villarroel-Campos and González-Billault, 2014).
Our results reveal a novel mechanism by which Rab35 functions are regulated by its degradation mediated by the ubiquitin proteosome system and the protein kinase PRPK. The neuronal cytoskeleton, through MAP1B function, regulates axonal elongation by buffering PRPK activity and favoring Rab35 protection from ubiquitin/proteasome degradation.
Materials and Methods
Antibodies and reagents.
The following commercially available primary antibodies were used: mouse anti-FLAG epitope (M2; Sigma-Aldrich), rabbit anti-PRPK (ab37606; Abcam), rabbit anti-PRPK (SAB1300461; Sigma-Aldrich), goat anti-MAP1B (N19; Santa Cruz Biotechnology), rabbit anti-Rab35 (9690; Cell Signaling Technology), rabbit anti-Rab11 (71-5300; Invitrogen), mouse anti-βIII-tubulin (G712A; Promega), mouse anti-α-tubulin (clone DM1A, T6199; Sigma-Aldrich), rabbit anti-Cdc42 (07-1466; Millipore), mouse anti-synaptotagmin 1 (105 011; Synaptic Systems), mouse anti-tau-1 (MAB3420; Millipore), rabbit anti-c-myc (A14; Santa Cruz Biotechnology), mouse anti-HA (F-7; Santa Cruz Biotechnology), mouse anti-ubiquitin (P4D1, #3936; Cell Signaling Technology), and mouse anti-β galactosidase (Z3781; Promega). Secondary antibodies included anti-mouse and anti-rabbit IgG conjugated to AlexaFluor 488, 543, 633, or 647 (Invitrogen), HRP-conjugated anti-rabbit and anti-mouse IgG (Jackson ImmunoResearch Laboratories), and HRP-conjugated anti-goat IgG (Santa Cruz Biotechnology). The ubiquitin proteasome inhibitor MG-132 was from Sigma-Aldrich (M7449), DMSO was from Calbiochem (317275), PR619 was from Sigma-Aldrich (SML0430), and Nocodazole was from Sigma-Aldrich (M1404).
Expression constructs and cloning.
Mouse PRPK WT cloned into the pCMV6-Entry expression vector was purchased from OriGene and then amplified by PCR (primers: forward, 5′-AATTGGATCCATGGCTGGTGTGTCCTCGGAG-3′ with a BamHI site added; reverse, 5′-AATTAAGCTTCTACCCGACCATGGACCGCTTTCG-3′ with a HindIII site added) and subcloned into the pCMV-Tag2 expression plasmid (Agilent Technologies). A D174N point mutation was introduced in pCMV6-Entry PRPK WT using the QuikChange Lightning Site-Directed Mutagenesis kit (Agilent Technologies). Additionally, PRPK WT and PRPK D174N were amplified by PCR and subcloned into the pmKate2-C vector (Evrogen). The generated constructs were sequenced by Macrogen.
Rab35 WT, Rab35 Q67L, and Rab35 S22N constructs in the pmStr-C1 vector, as well as GFP-tagged Rab11 Q70L, were prepared as described previously (Kanno et al., 2010; Ishida et al., 2012). GFP-tagged Rab11 WT was provided by F. Bronfman (Catholic University, Santiago, Chile). A Rab35(S5A) mutant carrying a swapping mutation between mouse Rab35 [amino acids (AA) 70–76] and Rab5A (AA 82–88) was prepared as described previously (Etoh and Fukuda, 2015). N- and C-terminal domain-swapping mutants between Rab35 and Rab5A [AA 1–8 of Rab35 and AA 1–20 of Rab5A, named Rab35(N5A)], and C-terminal region [AA 167–201 of Rab35 and Rab5A AA 180–215 of Rab5A, named Rab35(C5A)] were produced by conventional PCR techniques using the following pairs of oligonucleotides (restriction enzyme sites are underlined and stop codons are in bold) 5′-GGATCCATGGCTAATCGAGGAGCAACAAGACCCAACGGGCCAAA TACTGGAAATAAAATATGCCAGTTCAAGCTGCTCATCATGGCGACA-3′ [Rab35(N-Rab5A) primer, sense] and 5′-TTAGCAGCAGCTTTCTTTCTTTG-3′ (Rab35 stop primer, antisense); 5′-AAGCTTCCAAAGAATGAACCACA-3′ [Rab35(C-Rab5A) primer, sense] and 5′-TCAGTTACTACAACACTGGC-3′ (Rab5A stop primer, antisense); and 5′-CGGATCCATGGCCCGGGACTACGACCA-3′(Rab35 Met primer, sense) and 5′-AAGCTTCTCTGTGATACAGTTGAACA-3′ [Rab35(C-Rab35) primer, antisense]. Purified PCR products were directly inserted into the pGEM-T Easy vector (Promega). The cDNA inserts were sequenced and then subcloned into the BamHI/NotI site of pGEX4T-3 (GE Healthcare). pGEX-4T-3-Rab5A and -Rab35 were prepared as described previously (Itoh et al., 2008; Henríquez et al., 2012).
The Raichu-Cdc42 FRET biosensor probe was obtained from A. Cáceres (Instituto de Investigación Médica Mercedes y Martín Ferreyra, Córdoba, Argentina). pEGFP-N1-Lifeact was provided by R. Wedlich-Söldner (Max Planck Institute of Biochemistry, Martinsried, Germany). pGEX-Rab35 was provided by J. Presley (McGill University, Montreal, Quebec, Canada). HA-tagged ubiquitin (Ub-HA) was obtained from R. Felix (Centro de investigaciones y de estudios avanzados, CINVESTAV, DF, Mexico). shRNAs designed against the mouse PRPK coding sequence (#1 CTTTGTGGACTATGCGTCTAACTGCTTAT and #2 AGTCCAGTGCTGAAGAAGTTAGATGAGGT) and the corresponding scrambled shRNA (GCACTACCAGAGCTAACTCAGATAGTACT), which were cloned into the pGFP-V-RS vector, were purchased from OriGene. shRNA directed against MAP1B (CCCAGAGAGATGTCCTTATAT) was obtained from the Broad Institute. shControl and shRNA against Rab35 have been previously prepared and characterized (Kobayashi and Fukuda, 2012).
Cell culture.
COS7 and N1E-115 cell lines were cultured in DMEM (Invitrogen) containing 5% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin (Invitrogen) at 37°C and 5% CO2. Cells were plated on 100 mm, 60 mm culture dishes, or 6-well plates for biochemical experiments or on untreated glass coverslips in 24-well plates for immunofluorescence. Cells were transfected in serum-free DMEM with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The culture medium was replaced with fresh medium, and cells were processed 24–48 h later. MG-132 and nocodazole were added at 20 μm for 4 h, when indicated. PR619 was used at 50 μm for 2 h.
Culture of embryonic hippocampal neurons.
Neuronal hippocampal cultures were obtained as described previously (Kaech and Banker, 2006). Briefly, hippocampi were dissected from embryonic day (E)18 Sprague-Dawley rat embryos of either sex and treated with 0.25% trypsin in HBSS (Invitrogen) for 25 min at 37°C. The tissue was then mechanically dissociated, and neurons were seeded onto sterilized glass coverslips coated with poly-l-lysine (1 mg/ml, Sigma-Aldrich) in neurobasal medium (Invitrogen) containing 10% horse serum, 1% penicillin/streptomycin, and 1% glutamine (Invitrogen) for 1 h. The medium was then replaced with neurobasal medium containing 2% B27 (Invitrogen), 1% Glutamax (Invitrogen), and 1% penicillin/streptomycin (maintenance medium). For transfection experiments, the medium was replaced with serum-free neurobasal medium, and neurons were transfected using Lipofectamine 2000 according to the manufacturer's instructions. After 2 h, the medium was replaced with maintenance medium. Untagged constructs were cotransfected with a GFP-coding plasmid at a 4:1 ratio (construct of interest/GFP) to ensure a 98% rate of cotransfection, according to inner controls (data not shown). These same procedures were performed to obtain hippocampal neuronal cultures from MAP1B KO mice (González-Billault et al., 2000).
Expression of recombinant proteins.
Expression and purification of LC1, Rab35 WT, Rab35 swapping mutants (N5A, S5A, and C5A), and the CRIB domain (AA 67–150) of p21-activated kinase (Pak1) were performed as follows: BL21 (DE3) Escherichia coli strains carrying pGEX-LC1, pGEX-Rab35, pGEX-Swapping mutants, or pGEX-CRIB-GST were grown overnight at 37°C in LB-ampicillin. Cultures were diluted 1:100 and grown in fresh medium at 37°C to an OD600 of 0.7. Next, isopropyl β-D-1-thiogalactopyranoside was added to a final concentration of 1 mm, the cultures were grown for an additional 2 h and then samples were collected and sonicated in lysis buffer A (50 mm Tris-HCl, pH 8.0, 1% Triton X-100, 1 mm EDTA, 150 mm NaCl, 25 mm NaF, 0.5 mm PMSF, and 1× protease inhibitor complex; Roche). Cleared lysates were affinity purified with glutathione-Sepharose beads (GE Healthcare). Loaded beads were washed 10× with lysis buffer B (lysis buffer A with 300 mm NaCl) at 4°C. GST fusion proteins were quantified and visualized in SDS-polyacrylamide gels stained with Coomassie brilliant blue.
Pull-down assay.
Glutathione-sepharose beads (50 μg) loaded with GST-LC1 or GST-Rab35 were incubated overnight at 4°C with 1 mg E18 embryonic rat brain lysates. Beads were washed six times with lysis buffer B and then resuspended in SDS-PAGE sample buffer. Bound proteins were subjected to immunoblot analysis with the indicated antibodies. Beads loaded with GST alone were used as a control.
Cdc42 activity pull-down assay.
The CRIB domain was purified as described above. Loaded beads were incubated for 70 min at 4°C with 1 mg of either control or PRPK-expressing COS7 cell lysates using fishing buffer (50 mm Tris-HCl, pH 7.5, 10% glycerol, 1% Triton X-100, 200 mm NaCl, 10 mm MgCl2, 25 mm NaF, and protease inhibitor complex). The beads were washed three times with washing buffer (50 mm Tris-HCl, pH 7.5, 30 mm MgCl2, 40 mm NaCl), and then resuspended in SDS-PAGE sample buffer. Bound Cdc42-GTP was subjected to immunoblot analysis and quantified with respect to total Cdc42 using ImageJ.
Immunoprecipitation.
Rat E18 embryonic brains or cultured cell lines were lysed with radioimmunoprecipitation assay buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EGTA, 1 mm EDTA, 1% Nonidet P-40, 1% sodium deoxycholate, 5 mm NaF, 20 nm calyculin A, Cell Signaling Technology; and 1 mm PMSF and protease inhibitor complex), and the lysate was incubated with the indicated antibody overnight at 4°C. Protein A or G-sepharose beads (Sigma-Aldrich) were added and the mix was incubated for 4 h at 4°C. The beads were washed three times with lysis buffer A and resuspended in SDS-PAGE sample buffer. Immunoblots of the immunoprecipitates were probed with the indicated antibody. As a negative control for immunoprecipitation, we used anti-β galactosidase antibody.
Yeast two-hybrid screening.
AH109 yeast transformed with pGBKT7-LC1 were mated with Y187 yeast transformed with a mouse embryonic brain cDNA library (MatchMaker two-hybrid systems, Clontech) according to the manufacturer's protocols and the yeast protocol handbook (Clontech). Colonies grown for 10 d at 30°C were isolated and expanded, and then plasmid DNA was purified and sequenced.
Yeast two-hybrid binding assays.
The interaction between LC1 and PRPK was assessed using the two-hybrid assay. Protein interactions were detected with the MatchMaker two-hybrid systems according to the manufacturer's protocols and the yeast protocol handbook. The bait plasmid (pGBKT7, encoding the LC1 sequence) was analyzed for interaction with the prey plasmid (pGADT7, encoding the PRPK clone previously identified in the Y2H screen). AH109 yeast cells were cotransformed with both prey and bait plasmids. Positive interactions were assayed by growth on TDO (-Leu/-Trp/-His) and QDO (-Leu/-Trp/-His/-Ade) media after 3–7 d at 30°C. Media lacking amino acids were supplemented with X-α-Gal (20 mg/ml). The interaction between pGBKT7-p53 and pGADT7-T (SV40 large T-antigen) was used as a positive control (Clontech), and the empty pGADT7 vector cotransformed with pGBKT7-LC1 was used as a negative control. For selection, 8 μl of each suspension and three 10-fold serial dilutions were individually spotted on TDO/X-α-Gal and QDO/X-α-Gal plates and incubated at 30°C for 2 d.
Immunoblotting.
Protein extracts were obtained from cell lines, embryonic/adult brains, or primary cultures. Cells were lysed with radioimmunoprecipitation assay buffer, subjected to SDS-PAGE and electroblotted onto nitrocellulose or polyvinylidene difluoride membranes. Blots were probed with the appropriate primary and secondary antibodies, and immunoreactive signals were visualized with an enhanced chemiluminescent substrate (Thermo Scientific) and quantified by densitometry using ImageJ.
Immunocytochemistry and time-lapse imaging.
Cultures were fixed with 4% PFA/4% sucrose in PBS for 30 min at 37°C, washed three times with PBS and then permeabilized with 1% Triton X-100 in PBS. After cells were blocked with 5% BSA in PBS, primary antibodies were added in 1% BSA in PBS, and cells were incubated overnight at 4°C. Cells were washed three times with PBS, and the appropriate AlexaFluor-conjugated secondary antibodies were applied for 1 h at room temperature. Coverslips were mounted in FluorSave (Millipore) and analyzed using a confocal microscope (Zeiss LSM 510 Meta) with a 40×/1.3 NA oil-immersion objective. Axon length was measured using the microscope-associated software LSM Image Browser (Zeiss). Confocal images presented here were background substracted, and in most cases, color-inverted to improve morphological appreciation.
For live-cell imaging, N1E-115 cells were seeded onto 25 mm coverslips and imaged in HBSS-HEPES medium at 37°C with a 63×/1.4 NA oil-immersion objective. Images were acquired every 30 s for 10 min, and the number of filopodia per cell in each frame was determined manually, as described previously (Lim et al., 2008). For figures, the first, tenth, and last frames were selected from each time-lapse movies and processed as follows: a theoretical point spread function (PSF) was generated in Fiji ImageJ with the Diffraction PSF 3D plug-in, and the images were deconvoluted using the PSF and the Parallel Spectral Deconvolution plug-in.
Tubulin soluble fraction extraction.
The extraction of the soluble-tubulin fraction was performed based on previously described protocols (Brown et al., 1992; González-Billault et al., 2001). Briefly, hippocampal neurons were washed with PHEM buffer (60 mm PIPES, 25 mm HEPES, 10 mm EGTA, 2 mm MgCl2, pH 6.9) followed by extraction for 1 min with 0.2% saponin diluted in PHEM buffer containing 10 μm taxol and 0.1% DMSO (extraction buffer). Neurons were fixed with methanol for 5 min at 20°C and then rehydrated in PBS. Standard protocols for immunofluorescence were then followed. For the immunoprecipitation assay, neurons were rinsed with PHEM buffer and then treated with extraction buffer for 3 min at 4°C. The soluble fraction was then recovered and the insoluble fraction was obtained with a standard RIPA lysis protocol. The protein concentration of both fractions was determined using the Bradford method.
FRET experiments.
Cultured hippocampal neurons obtained as described before were transfected with the Raichu-Cdc42 FRET biosensor alone or with mKate2-C-PRPK WT at 1 d in vitro (DIV). After 18 h, neurons were fixed as described for immunocytochemistry, and then washed three times with PBS and immediately mounted. Image acquisition was performed using a 63×/1.4 NA oil-immersion objective, and images were recorded at 12 bits, 1024 × 1024 pixels per image. CFP and FRET channels were excited with a 458 nm laser, and the following filters were used for acquisition: 500/25 for CFP, 565/35 for the FRET channel, and LP650 for PRPK WT. Images were background subtracted and aligned using Fiji ImageJ software, then a binary mask was created using the FRET channel, which provided a higher signal-to-noise ratio than the CFP channel. The FRET channel was divided by the CFP channel, and the result was divided by the binary mask to obtain the FRET map. Pixel intensity was measured in the proximal third and distal third of each axon.
Statistical analysis.
Data were analyzed using GraphPad Prism 5 with the unpaired Student's t test or ANOVA test. Results are expressed as the mean ± SEM. Differences between values were considered significant if p < 0.05.
Results
Rab35 promotes axon elongation
Previous reports have shown that Rab35 promotes neuritogenesis in N1E-115 and PC12 cells (Chevallier et al., 2009; Kobayashi and Fukuda, 2012; Kobayashi et al., 2014). Nevertheless, its involvement in neuronal polarity still remains unexplored. First, to examine the effect of Rab35 on axon elongation, we transfected mStrawberry (mSt) fusion proteins for Rab35 WT, Rab35 Q67L (a constitutively active mutant), and Rab35 S22N (a dominant-negative mutant) into rat hippocampal neurons and evaluated their effect on axon elongation at 3 DIV. Control neurons were transfected with GFP. Neurons overexpressing Rab35 WT and Rab35 Q67L exhibited a significant increase in axon length (63% and 70%, respectively) compared with control (Fig. 1A–C,H). In contrast, Rab35 S22N significantly reduced axon length (to 61% of the mean control length; Fig. 1D,H). In a second approach, we used shRNA against Rab35 to discard possible undesired effects due to overexpression of the dominant-negative version of Rab35. Cultured neurons were transfected with shControl and shRab35 constructs, and axon elongation was assessed. Similarly to the effect of Rab35 S22N, Rab35 knockdown decreased axon elongation (Fig. 1E–G,I). These results clearly indicate that Rab35 is important in promoting axon elongation.
Figure 1.

Rab35 promotes axonal growth. A–D, Cultured hippocampal neurons expressing GFP (control; A), Rab35 WT (B), Rab35 Q67L (C) or Rab35 S22N (D). Cells were fixed at 3 DIV. Images were segmented and the color inverted. Scale bar, 50 μm. E–G, Cultured hippocampal neurons expressing GFP (control; E), shControl (F) or shRab35(g). Cells were fixed at 3 DIV. Images were segmented and the colors inverted. Scale bar, 50 μm. H, Quantification of axon length in neurons transfected as in A–D. n = 50 neurons from eight different cultures; one-way ANOVA with Dunnett's post-test (***p < 0.001). I, Quantification of axon length in neurons transfected as in E–G. n = 50 neurons from three different cultures; one-way ANOVA with Dunnett's post-test (***p < 0.001).
PRPK reduces axon elongation
Rab35 interacts with PRPK, an atypical protein kinase that is involved in p53 phosphorylation as its only described substrate (Abe et al., 2006). Nevertheless, an analysis of the role of this interaction has not yet been performed. First, and similar to experiments presented in the previous section, we evaluated the effect of PRPK overexpression upon axonal outgrowth. We cotransfected hippocampal neurons with GFP alone (control) or with GFP plus either PRPK wild-type (WT) or PRPK D174N. The latter carries a point mutation that affects the ATP-binding site, creating a variant described as kinase dead (KD) because it is unable to induce p53 Ser15 phosphorylation (Abe et al., 2001). We evaluated axon length in neurons transfected with the WT and the KD form of PRPK at 3 DIV (Fig. 2A,B), a stage when most neurons are fully polarized (Tahirovic and Bradke, 2009). Neurons that overexpressed PRPK WT had a significant reduction (51%) in axon length compared with control neurons that overexpressed GFP alone. Conversely, neurons overexpressing PRPK KD showed a mild but significant increase in axon length (13%; Fig. 2B). We also measured the length of minor neurites in neurons expressing either PRPK WT or PRPK KD. We did not find any changes in the mean minor neurite length (Fig. 2c). These results indicate that, unlike Rab35, PRPK is a negative regulator of axon elongation and that the effect is dependent on its kinase activity.
Figure 2.

PRPK negatively regulates axon elongation. A, Cultured hippocampal neurons transfected with GFP (control), GFP/PRPK WT, or GFP/PRPK KD. Cells were fixed and immunostained after 3 DIV. Tuj1 staining is also shown. Scale bar, 50 μm. B, Quantification of axon length in cells transfected as in A. n = 50 neurons from three different cultures; one-way ANOVA with Dunnett's post-test (**p < 0.01, ***p < 0.001). C, Quantification of minor neurite length in cells transfected as in A. n = 50 neurons from three different cultures; one-way ANOVA with Dunnett's post-test (no statistically significant differences were observed).
Rab35 counteracts PRPK-induced reduction in axon outgrowth
In the next set of experiments, we evaluated the potential epistatic relationship between PRPK and Rab35 during axon elongation by cotransfecting Rab35 variants (WT, Q67L, and S22N) with PRPK WT into hippocampal neurons and measuring axon length at 3 DIV. Cells were cotransfected with PRPK WT or GFP as a control. Both Rab35 WT and Q67L reversed the PRPK-induced deficiency in axon elongation (Fig. 3, compare C, D, to B). In contrast, Rab35 S22N was unable to rescue the inhibitory effect of PRPK (Fig. 3E). Quantitation experiments confirmed our visual inspection data, showing that active Rab35 overcomes axonal elongation defects induced by PRPK (Fig. 3F). Altogether, these results suggest that Rab35 rescue the inhibitory effect of PRPK on axonal outgrowth, which depends on Rab35 GTPase activity.
Figure 3.

Rab35 rescues PRPK-induced inhibition of axon growth. A–E, Cultured hippocampal neurons at 3 DIV expressing GFP (A) or PRPK WT (B) as controls, or coexpressing PRPK WT with either Rab35 WT (C), Rab35 Q67L (D), or Rab35 S22N (E). Images processed as in Figure 5. Scale bar, 50 μm. F, Quantification of axon length in neurons transfected as in A–E. n = 30 neurons from eight different cultures; one-way ANOVA with Dunnett's post-test (***p < 0.001).
PRPK antagonizes other functions of Rab35
Rab35 also regulates filopodial formation in Drosophila melanogaster S2 cells (Shim et al., 2010) via the effector protein fascin (Zhang et al., 2009) and in HeLa cells (Marat et al., 2012). Moreover, it has been suggested that Rab35 activates the small GTPase Cdc42 (Chevallier et al., 2009) by a still unknown mechanism. Therefore, we studied the possibility that PRPK by antagonizing Rab35, affected actin-based structures, such as filopodial formation in N1E-115 cells. We used EGFP-Lifeact to evaluate filopodial actin dynamics, and then coexpressed EGFP-Lifeact with either mKate2-PRPK WT, mKate2-PRPK KD or mSt-Rab35 WT to study filopodial growth and distribution (Fig. 4A,B). Strikingly, PRPK WT overexpression reduced the number of filopodia per cell by 72%. In the most severe cases, PRPK-expressing cells completely lacked filopodia (Fig. 4A,B). Conversely, expression of PRPK KD increased the number of filopodia by 90% (Fig. 4B), supporting the idea that PRPK kinase activity is essential to the inhibition of filopodial formation. Cells expressing Rab35 and EGFP-Lifeact showed a 132% increase in the number of filopodia per cell (Fig. 4B). Finally, we cotransfected EGFP-Lifeact, Rab35 WT and PRPK WT into N1E-115 cells to assess whether Rab35 rescues the filopodial phenotype induced by PRPK. Indeed, these cells exhibited a 104% increase in the number of filopodia per cell compared with control cells (Fig. 4B).
Figure 4.

Epistatic relationship of PRPK and Rab35 in actin-related processes. A, Still images taken from time-lapse recordings over a 10 min period, from undifferentiated N1E-115 cells transfected with EGFP-Lifeact (Control) or cotransfected with EGFP-Lifeact and either PRPK WT, PRPK KD, or Rab35 WT. Additionally, cells expressing EGFP-Lifeact, PRPK WT, and Rab35 were included. Movies were acquired at 2 frames/min. The first, middle, and last frames of each movie were deconvoluted, and are presented from left to right. Scale bar, 15 μm. B, Quantification of the mean number of filopodia per cell during the entire recording period. n = 20 cells, except for Rab35 WT, where n = 18; one-way ANOVA with Dunnett's post-test (***p < 0.001). C, Cdc42 activity pull-down assay in COS7 cells expressing PRPK WT or PRPK KD. Untransfected cells were used as a control. D, Cdc42 activity pull-down assay in COS7 cells expressing Rab35 WT, S22N, or the Q67L mutant. Untransfected cells were used as a control. E, Quantification of the Cdc42-GTP/Cdc42 ratio from C. n = 3; one-way ANOVA with Dunnett's post-test (*p < 0.05, **p < 0.01). F, Quantification of the Cdc42-GTP/Cdc42 ratio from D. n = 3; one-way ANOVA with Bonferroni's post-test (*p < 0.05, **p < 0.01; indicating differences with control condition. #p < 0.05, ##p < 0.01; indicating differences with S22N condition). G, Two DIV cultured hippocampal neurons expressing a FRET biosensor for Cdc42 alone (control condition) or coexpressing PRPK WT, Rab35 Q67L, or Rab35 S22N. FRET maps of representative neurons are depicted with a thermal scale. Scale bar, 40 μm.
The negative regulation exerted by PRPK on filopodial formation could be related to changes in the activity of Cdc42, a small GTPase proposed to be linked to Rab35 signaling by either activation of Cdc42 or tethering of vesicles containing polarity determinants in epithelial cells (Chevallier et al., 2009; Klinkert et al., 2016). Therefore, we overexpressed PRPK WT and PRPK KD in COS7 cells and performed GST-CRIB (Cdc42/Rac interactive binding, which binds specifically to Cdc42-GTP but not to its inactive form) pull-down assays to measure GTP-Cdc42 levels. Cells overexpressing PRPK WT had a 40% reduction in Cdc42 activity, whereas cells overexpressing PRPK KD showed an 80% increase (Fig. 4C,E). Changes in the activity of Cdc42 were indeed controlled by Rab35, as indicated by an increased proportion of Cdc42 active form in cells expressing WT or constitutively active versions of Rab35 (Fig. 4D,F). We then evaluated changes in Cdc42 activity in cultured primary neurons using a Förster resonance energy transfer (FRET)-based approach. We expressed a Cdc42 FRET biosensor alone (Control) or in conjunction with PRPK WT, Rab35 Q67L, or Rab35 S22N and measured pixel intensity in the resulting FRET maps in the distal axon. The relative pixel intensity in neurons overexpressing PRPK WT was decreased (Fig. 4G; PRPK WT). Similarly, overexpression of the dominant-negative form of Rab35 decreased active Cdc42 (Fig. 4G; Rab35S22N). Consistently, a constitutively active form of Rab35 enhanced Cdc42 activity (Fig. 4; Rab35 Q67L). These results clearly suggest that PRPK negatively regulates Cdc42 activity in both COS7 cells and cultured hippocampal neurons, and support the involvement of Rab35 in the regulation of Cdc42 activity.
PRPK is a novel MAP1B-interacting protein
To study potential regulatory mechanisms involving PRPK and Rab35 functions in neurons, we turned our attention to MAP1B, because we verified an interaction between MAP1B and PRPK in a Y2H screening using LC1 subunit as bait. MAP1B is a protein that was previously demonstrated to support axonal elongation by stabilizing microtubules (González-Billault et al., 2001) and regulating the activity of Cdc42 (Montenegro-Venegas et al., 2010). The interaction observed in our initial screening was confirmed in an Y2H binding assay, AH109 yeast cotransformed with pGBKT7-LC1 and pGADT7-PRPK (Fig. 5A) grew in both low-stringency TDO (-Leu/-Trp/-His) medium and high-stringency QDO (-Leu/-Trp/-His/-Ade) medium. The color of the colonies indicates they were able to hydrolyze the X-α-Gal present in the medium. Then, we used a pull-down approach to verify this novel interaction, and also to validate previous interactions described in literature for PRPK. Thus, GST-LC1 interacted with PRPK present in embryonic rat brain protein extracts (Fig. 5B, top). PRPK interacted with LC1 when the full-length protein was expressed. In contrast, N- or C-terminal deletions of LC1 did not interact with PRPK (Fig. 5C), suggesting that a likely region for the interaction may span the central domain of LC1. Although we did not detect an interaction between GST-LC1 and Rab35 (Fig. 5B, bottom), we observed that GST-Rab35 interacts with PRPK (Fig. 5D). In an attempt to define relevant domains in Rab35 that could mediate interactions with PRPK, we used a Rab35-Rab5 swapping assay (Etoh and Fukuda, 2015). In this approach, we swapped the N-terminal (Rab35N5A), the switch II (Rab35S5A), and C-terminal (Rab35C5A) domains of Rab35 with those derived from Rab5. Rab5 is a marker of early endosomes, which are vesicular precursors for the recycling endosomes (Gorvel et al., 1991). All Rab35 constructs evaluated (Rab35 WT, Rab35N5A, Rab35S5A, and Rab35C5A) retained the ability to interact with PRPK, suggesting that PRPK-Rab35 interaction proceeds through other domains in Rab35 (Fig. 5F). Interestingly, Rab5A itself did not interact with PRPK (Fig. 5F). Together, these experiments show that PRPK is a novel MAP1B binding partner that interacts with the LC1 subunit, and that PRPK also interacts specifically with Rab35.
Figure 5.

PRPK interacts with LC1 and Rab35. A, Y2H binding assay with AH109 yeast cotransformed with pGBKT7-LC1 and pGADT7-PRPK. Yeast carrying pGADT7-T and pGBKT7–p53 were used as a positive control (+); yeast cotransformed with pGBKT7-LC1 and empty pGADT7 were used as a negative control. Yeast from all three cotransformations were grown in low-stringency TDO (-Leu/-Trp/-His) and high-stringency QDO (-Leu/-Trp/-His/-Ade) media supplemented with X-α-Gal. Decreasing dilutions were seeded and allowed to grow for 4 d. B–D, GST pull-down assay from (E18) rat brain protein extracts. GST-LC1 and GST alone were incubated with brain supernatants, and the bound fraction was analyzed by immunoblotting for PRPK (B, top) and Rab35 (B, bottom). GST pull-down assays using full-length LC1, and N- and C-terminal domains were incubated with brain supernatants and the bound fraction analyzed by immunoblotting for PRPK (C). GST-Rab35 and GST alone were incubated with brain supernatants, and the bound fraction was analyzed by immunoblotting for PRPK (D). E, Schematic representation of Rab35 (swapping motif in light blue) and Rab5 (swapping motif in red) protein domains. Swapping mutants for Rab35 (N5A, S5A, and C5A). The numbers refer to the amino acid positions in mouse Rab35 and their substitution by the corresponding motif of Rab5. F, GST pull-down assays using proteins described in E were incubated with brain supernatants, and the bound fraction was analyzed by immunoblotting against PRPK (top). Purified proteins are indicated with an asterisk in the Ponceau red-stained membrane (bottom).
PRPK was originally described as a protein highly expressed in testis but undetected in brain (Abe et al., 2001). More recently, microarray experiments showed that all components of the KEOPS/EKC complex, including PRPK, are ubiquitously expressed, including the brain (Costessi et al., 2012). Therefore, we studied PRPK protein expression in rat brain by immunoblotting. PRPK was detected at E18 but was absent in the adult brain (Fig. 6A). Adult brain protein extracts were enriched in the synaptic marker synaptotagmin 1 (Syt1), which was used as a positive control for expression at adult stage. Because this is the first characterization of PRPK protein in the nervous system, we studied its expression at different developmental stages ranging from E18 through to postnatal day 21. PRPK is mostly expressed during embryonic and young postnatal brain (Fig. 6B). Its expression severely decreased 2 weeks after birth (Fig. 6C). In addition, we determined the relative expression levels of PRPK in cultured embryonic hippocampal neurons (Fig. 6D,E) from 18 h in vitro through 5 DIV. Axon elongation during this period was evidenced by increased levels of unphosphorylated tau protein, a canonical marker for growing axons (Fig. 6D). Compared with its expression at 18 h, PRPK levels were significantly reduced between 2 and 3 DIV, the phase during which axon determination and the beginning of axon elongation occurs. This suggests PRPK expression may be developmentally regulated during the time of neuron polarization and axon elongation.
Figure 6.

Characterization of PRPK expression. A, PRPK expression levels were assessed by immunoblotting of E18 embryonic and adult whole rat brain protein extracts. MAP1B was used as an embryonic marker and Syt1 as an adult marker. B, PRPK postnatal expression pattern was evaluated by immunoblotting of whole rat brain protein extracts obtained between P0 and P21. An additional E18 embryonic time-point was included. C, Quantification of relative PRPK postnatal expression from B, (n = 3). Data expressed as the normalized ratio of PRPK to α-tubulin; one-way ANOVA with Dunnett's post-test (**p < 0.01). D, PRPK expression in cultured hippocampal neurons was determined by immunoblotting, between 18 h in vitro and 5 DIV. tau-1 epitope expression was also assessed, as it is a widely used axonal marker, and its relative abundance increases following culture maturation. E, Quantification of relative PRPK expression from cultured neurons in D (n = 3). Data expressed as the normalized ratio of PRPK to α-tubulin; one-way ANOVA with Dunnett's post-test (*p < 0.05, ***p < 0.001). F, Two DIV cultured hippocampal neuron stained with anti-PRPK and anti-Tuj1. Scale bar, 50 μm. G, Quantification of anti-PRPK fluorescence intensity along the last 100 μm of the axon as in F, standardized against anti-Tuj1 fluorescence intensity. The axon tip is toward the right. H, I, Cultured hippocampal neurons from MAP1B WT (H) or KO (I) embryonic brains at 2 DIV stained with anti-PRPK and anti-Tuj1. Scale bar, 50 μm. J, Hippocampal neurons stained with anti-(Tuj1, anti-PRPK, and anti-MAP1B. Control neurons (without extraction of the soluble tubulin fraction) are shown in the top, and the axon is indicated with arrowheads. Neurons in the bottom were subjected to extraction of the soluble tubulin fraction, and the axon is indicated with arrows. The yellow strips indicate the axonal region where the fluorescence intensity profile was analyzed. Scale bar, 50 μm. K, Fluorescence intensity profile for each probe in control neurons (total fraction, red line) and after extraction of the soluble fraction (insoluble fraction, blue line). L, Immunoprecipitation from 4 DIV WT neurons using antibody anti-MAP1B and blotting against PRPK. Protein extracts correspond to both soluble and insoluble fractions. The red arrow highlights the PRPK band.
To characterize previously unknown function(s) of PRPK in neurons, we first studied its subcellular localization in cultured hippocampal neurons. In agreement with previous reports assessing PRPK localization in HeLa cells (Abe et al., 2006), endogenous PRPK exhibited a cytoplasmic distribution and was present in minor neurites and the axon (Fig. 6F). We measured PRPK fluorescence intensity, standardized with βIII-tubulin (Tuj1) intensity along the axon, to determine whether PRPK was enriched at the axonal tip. The fluorescence profile revealed a homogeneous distribution of PRPK in the last 100 μm of the axon (Fig. 6G). Because PRPK was isolated as a MAP1B-LC1 protein interactor, we evaluated PRPK distribution in neurons lacking MAP1B. The overall distribution in MAP1B deficient neurons was similar to WT controls, without any noticeable change in its sub cellular distribution (Fig. 6H,I). We then performed experiments to evaluate whether PRPK was associated with the microtubule cytoskeleton. We prepared neuronal cultures that were fixed after detergent extraction performed under microtubule-stabilizing conditions (Brown et al., 1992). This method removes unassembled tubulin from the cell, so that tubulin staining in immunofluorescence experiments is attributable to microtubules (Brown et al., 1992; González-Billault et al., 2001). PRPK immunofluorescence observed in the axonal compartment was barely detected when we extracted the soluble cytoskeleton (Fig. 6J, arrow in insoluble fraction). In contrast, the decrease in MAP1B immunofluorescence after detergent extraction was less severe than PRPK removal, as expected for a microtubule-binding protein (Fig. 6J, arrow in insoluble fraction). The immunofluorescence profile of PRPK and MAP1B after detergent extraction suggests that most of the PRPK was not associated to MAP1B bound to microtubules (Fig. 6K). Finally, we immunoprecipitated MAP1B from the soluble fraction after detergent treatments, and found PRPK was associated to MAP1B (Fig. 6L). These results suggest that the interaction between MAP1B and PRPK does not require that MAP1B is associated to microtubules.
PRPK, Rab35, and MAP1B functions are coupled to promote axon elongation
Having shown that Rab35 and PRPK function antagonistically during axon elongation, and that PRPK specifically interacts with MAP1B, we next sought to demonstrate a functional relationship linking these proteins. In first term, we set up a shRNA approach to inactivate PRPK. We used two different shRNA construct designed against the mouse PRPK coding sequence (shPRPK), along with its scrambled sequence as a control (shScr). Knockdown efficiency was evaluated in N1E-115 cells by immunoblotting (Fig. 7a) and by immunofluorescence in primary neurons (Fig. 7C). Knockdown in neuroblastoma cells showed a 64% decrease in PRPK expression (Fig. 7B). Similarly, a line scattering profile for PRPK in neurons expressing shPRPK#1 or shPRPK#2 showed a 60% and 53% decrease in PRPK fluorescence, respectively (Fig. 7E). Then, we transfected GFP alone or cotransfected it with PRPK WT, PRPK KD or Rab35 WT in cultured hippocampal neurons derived from MAP1B WT and KO mice. We measured axon length in WT and KO neurons as reported earlier (González-Billault et al., 2000, 2001). Consistent with these reports, KO neurons expressing GFP had a 49% reduction in axon length compared with WT neurons expressing GFP (Fig. 8A,B,G). Notably, PRPK WT did not exacerbate axon length defects found in MAP1B KO neurons (Fig. 8C,G). However, axonal length reduction in MAP1B neurons was rescued by overexpression of either PRPK KD or Rab35 WT, which increased axon elongation significantly compared with WT cells expressing GFP (41%; Fig. 8D, E,G). Having shown that PRPK KD mutant was able to reverse the MAP1B KO axon elongation deficiency, we used a knockdown approach to inactive PRPK and evaluate the effect on axon outgrowth. Each shRNA was cloned into a plasmid encoding GFP, and GFP alone was used as control for shRNA untransfected neurons. We used these shRNAs in cultured hippocampal neurons from MAP1B WT or KO mice and measured axon length (Fig. 8F,H). Neurons expressing the shScr construct did not exhibit changes in axon length. In contrast, axon length in KO neurons expressing shPRPK was rescued compared with KO neurons expressing GFP, indicating that PRPK knockdown restored axon outgrowth in neurons derived from MAP1B KO. Moreover, PRPK knockdown also restored axonal length in wild-type neurons treated with shRNA against MAP1B (Fig. 8I–K). Because we did not observe changes in the median axonal length of WT neurons treated with scrambled and shRNA against PRPK, we decided to evaluate axon length in neurons showing different levels of PRPK knockdown after shRNA treatment. We performed semiquantitative fluorescence analysis similarly to Figure 7E, and evaluated the relationship between axonal length and PRPK fluorescence intensity. Neurons displaying lower PRPK fluorescence extended longer axons compared with neurons where PRPK knockdown was less efficient (as evidenced by the increased remnant of PRPK fluorescence; Pearson r = −0.6382; p = 0.0346; Fig. 8L). These results suggest that even in the presence of MAP1B, PRPK loss-of-function modifies axonal length.
Figure 7.
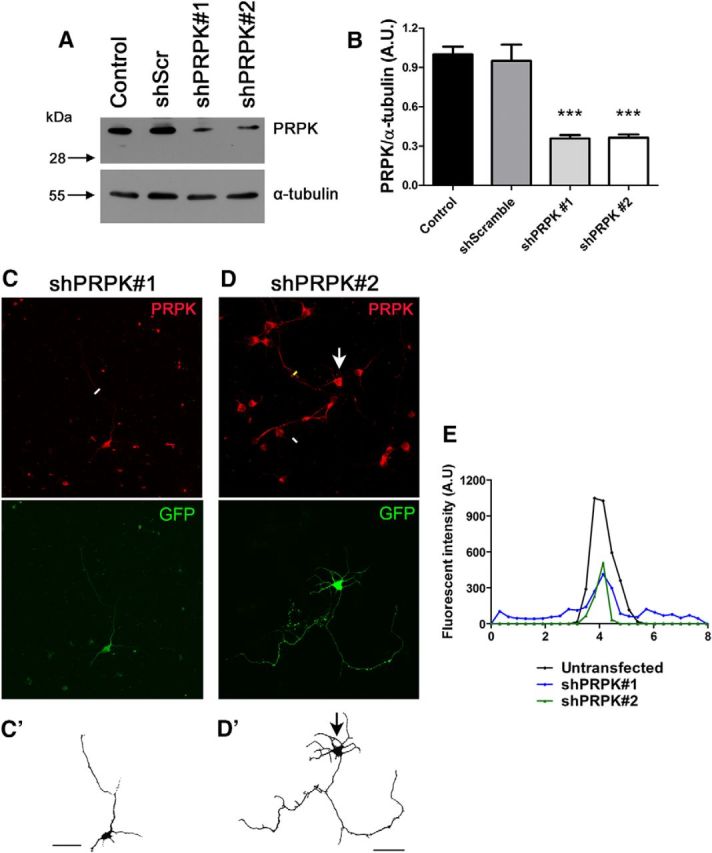
PRPK knockdown in neuroblastoma and primary neurons. A, N1E-115 cells were transfected with PRPK WT or were cotransfected with PRPK WT and either shScr, shPRPK#1, or shPRPK#2. Untransfected cells were included as an additional control. After 48 h, PRPK expression levels were assessed by immunoblotting using anti-PRPK and α-tubulin. B, Quantification of PRPK expression from A. n = 3; one-way ANOVA with Dunnett's post-test (***p < 0.001). C, D, Cultured hippocampal WT neurons at 2 DIV, expressing shPRPK#1 (C) or shPRPK#2 (D). Transfected neurons are shown in C′ and D′; a neuron in D′ is also indicated by an arrow. Scale bar, 50 μm. E, PRPK fluorescence intensity along the white lines in C and D (axonal sections of neurons expressing shPRPK#1 or #2) and yellow line (axonal sections of an untransfected neuron).
Figure 8.

Axon growth deficiencies in MAP1B KO and PRPK expressing neurons are rescue by Rab35 activity or PRPK inactivation. A, Cultured hippocampal neurons from WT brains transfected with GFP alone as control. B, Cultured hippocampal neurons from MAP1B KO brains transfected with GFP alone as control. C–E, Neurons from MAP1B KO brains cotransfected with GFP and PRPK WT (C), PRPK KD (D), or Rab35 WT (E). Cells were fixed at 2 DIV, images were segmented, and the color inverted. Scale bar, 40 μm. F, Cultured hippocampal neurons from MAP1B WT or KO embryonic brains transfected with GFP alone (control) or with shScr, shPRPK#1, or shPRPK#2. Cells were fixed at 2 DIV, images were segmented, and the color inverted. Scale bar, 50 μm. G, Quantification of axon length in neurons transfected from A to E. n = 30 neurons from seven independent cultures; one-way ANOVA with Dunnett's post-test (***p < 0.001). H, Quantification of axon length in neurons from F. n = 20 neurons from three independent cultures, except the shPRPK#1 condition where n = 9 for WT and 15 for KO; one-way ANOVA with Tukey's post-test (*p < 0.05, **p < 0,01, ***p < 0.001). I, J, Two DIV cultured WT neurons expressing shMAP1B and either shScr (F) or shPRPK#2 (G). Scale bar, 50 μm. K, Axonal length quantification from neurons in F and G. n = 8 neurons; unpaired Student's t test (*p < 0.05). L, Axonal length quantification in wild-type neurons expressing different levels of PRPK. Primary neurons were transfected with shRNA against PRPK (n = 11) and their axonal length plotted according to the PRPK decrease of fluorescence intensity induced by knockdown approach. (Pearson r = −0.6382; p = 0.0346).
PRPK induces Rab35 degradation
In the final part of this study we sought to elucidate the molecular mechanism linking PRPK and Rab35. First, we overexpressed PRPK WT and PRPK KD in COS7 cells and analyzed Rab35 expression levels. Strikingly, PRPK WT induced an 80% reduction in the amount of Rab35 protein (Fig. 9A,B). This effect was dependent on PRPK kinase activity, because expression of PRPK KD in COS7 cells did not modify Rab35 expression. Next, to explore the link between PRPK, Rab35 and MAP1B, we evaluated Rab35 protein levels in MAP1B KO mouse brains. Interestingly, Rab35 expression was reduced by 55% in MAP1B KO brain compared with WT littermates (Fig. 9C,D). Rab35 was originally described as a protein that regulates trafficking of recycling endosomes toward the plasma membrane (Kouranti et al., 2006). Recycling endosomes are also characterized by the presence of Rab11 (Ullrich et al., 1996). Therefore, we examined whether PRPK decreases Rab11 protein levels. We expressed PRPK WT and KD in COS7 cells and found that Rab11 expression remained unchanged in all experimental conditions (Fig. 9E,F), indicating that PRPK specifically targets Rab35.
Figure 9.

PRPK regulates Rab35 expression levels. A, Rab35 expression levels evaluated by immunoblotting in COS7 cells transfected with PRPK WT or PRPK KD. Untransfected cells were used as a control. B, Quantification of Rab35 expression shown in A. n = 3; one-way ANOVA with Dunnett's post-test (*p < 0.05). C, Rab35 expression levels in embryonic brain from MAP1B WT or KO mice. D, Quantification of Rab35 expression shown in B. n = 3; unpaired Student's t test (*p < 0.05). E, Rab11 expression in COS7 cells expressing PRPK WT or PRPK KD. Untransfected cells were used as a control. F, Quantification of Rab11 expression shown in E. n = 3; one-way ANOVA with Dunnett's post-test (no statistically significant differences were observed). G, Two DIV cultured neurons were treated with DMSO or nocodazole 20 μm for 4 h, and then processed for immunoblotting. Rab35 expression remains unaffected between conditions. H, Quantification of Rab35 expression shown in C. n = 3; unpaired Student's t test (no statistically significant differences were observed). I, Brain protein extracts derived from WT and tau KO mice were analyzed with anti tau-1 and anti Rab35. In the KO brain, tau-1 immunoreactivity was absent, and Rab35 levels were unmodified. J, Quantification of Rab35 expression from A. n = 3; unpaired Student's t test (no statistically significant differences were observed). K, COS7 cells expressing PRPK WT alone or cotransfected with PRPK WT and LC1-myc were used to assess Rab35 expression levels. L, Quantification of Rab35 expression. n = 3; unpaired Student's t test (*p < 0.05). M, COS7 cells expressing PRPK WT alone or cotransfected with PRPK WT and Tau were used to assess Tab35 expression levels. N, Quantification of Rab35 expression. n = 3; unpaired Student's t test (*p < 0.05).
Because MAP1B's main function is to stabilize microtubules we sought to address whether Rab35 protein reduction could be caused by decreased microtubule content in MAP1B KO neurons. We first treated neurons with 20 μm nocodazole for 4 h, to induce microtubule depolymerization. Of note, Rab35 levels were unaffected by microtubule loss induced by nocodazole (Fig. 9G,H). Additionally, we evaluated Rab35 in protein extracts derived from tau knock-out mice, another axonal microtubule associated protein. Null mutants for tau display Rab35 levels equivalent to control animals (Fig. 9I,J). These results suggest that Rab35 deficiency found in MAP1B KO neurons would not be primarily related to microtubule depolymerization.
We hypothesized that the LC1 subunit of MAP1B prevents Rab35 degradation due to its interaction with PRPK. To substantiate such hypothesis we overexpressed PRPK WT in COS7 cells that were either transfected with an empty vector or with a vector encoding LC1 (Fig. 9K,L). Coexpression of PRPK and LC1 rescue Rab35 protein degradation. We also evaluated whether heterologous expression of tau prevents Rab35 degradation induced by PRPK. Coexpression of tau and PRPK did not modify Rab35 degradation (Fig. 9M,N). These data strongly supports our hypothesis that the LC1 subunit of MAP1B buffers deleterious effects of PRPK on Rab35.
Finally, we studied Rab35 degradation by the ubiquitin-proteasome system, because it is a novel regulatory aspect for Rab GTPase proteins. First, we blocked proteasome degradation using MG132 in the presence of PR619, a cell-permeable broad spectrum deubiquitylating enzymes inhibitor, because such combined inhibition leads to the accumulation of polyubiquitinated conjugates (Lee et al., 2010; Silva et al., 2015). Combined MG132/PR619 treatment induced two changes: increased levels of Rab35 protein (Fig. 10A) but also the presence of high molecular weight species corresponding to ubiquitinated proteins recognized by Rab35 antibody (Fig. 10A; Rab35 2X and Rab35 poly-Ub). Such laddering indicates accumulation of poly-ubiquitinated proteins (Silva et al., 2015). Second, we coexpressed myc-tagged Rab35 and HA-ubiquitin constructs in cells incubated with MG132 and PR619. When we immunoprecipitated Rab35 using an anti-myc antibody, we observed higher molecular weight proteins recognized with the anti-HA antibody (as an indicator of ubiquitin presence in immunoprecipitated myc-Rab35; Fig. 10B). Third, we evaluated Rab35 laddering in brain protein extracts. Immunoprecipitated Rab35 also displays a protein laddering recognized by an anti-ubiquitin antibody, reinforcing the idea that Rab35 is ubiquitinated in brain (Fig. 10C). Finally, we showed that Rab35 protein reduction induced by PRPK was reversed by the ubiquitin proteasome inhibitor MG-132 (Fig. 10D,E). Indeed, PRPK expression levels are also increased after treatment with MG-132, as PRPK degradation is dependent on the ubiquitin-proteasome system and involves the E3 ubiquitin ligase Mdm2 (Guo et al., 2013). Thus, even though MG-132 treatment increases the amount of PRPK, its inhibitory effect on protein degradation blocks the PRPK-related decrease in Rab35.
Figure 10.

PRPK promotes Rab35 degradation. A, Rab35 ubiquitination levels evaluated by immunoblotting in COS7 cells treated with DMSO or PR619/MG132 (50 μm for 2 h and 20 μm for 4 h, respectively). B, The ubiquitination levels of Rab35 were analyzed in COS7 cells treated as (A) expressing Rab35-Myc and Ub-HA by immunoprecipitation with anti-myc antibody followed by immunoblotting against Ub-HA. Anti-β-galactosidase antibody was used as control. C, The endogenous ubiquitination levels of Rab35 were analyzed in E18 mice brain by immunoprecipitation with anti-Rab35 antibody followed by immunoblotting against ubiquitin. D, Effect of DMSO or MG-132 (20 μm for 4 h) on Rab35 expression in COS7 cells expressing PRPK WT. Untransfected cells were used as a control. E, Quantification of Rab35 expression shown in D. n = 3; one-way ANOVA with Dunnett's post-test (***p < 0.001).
Discussion
Rab35 is important in supporting axon elongation in primary neurons
Rab35 functions have been characterized in neuronal cell lines, where it promotes neurite outgrowth (Kobayashi and Fukuda, 2012; Kobayashi et al., 2014). In this paper, we extended those observations by showing that axonal elongation is dependent on the activity of Rab35, because dominant-negative forms or shRNA inactivation of Rab35 lead to shorter axons. The involvement of Rab35 in axon elongation highlights the importance of the recycling endosome for the acquisition of polarized morphology in primary neurons. Other Rabs present in recycling endosomes are also important during neuronal differentiation. For instance, active Rab11 promotes axon outgrowth downstream of lemur kinase 1 and Cdk5 in cultured hippocampal neurons (Takano et al., 2012) and promotes integrin-regulated neurite outgrowth in dorsal root ganglia (Eva et al., 2010), whereas Rab4 is necessary for axon elongation in Xenopus retinal ganglion cells (Falk et al., 2014). The hierarchy of recycling-related Rab GTPases during axon elongation remains unknown, and needs to be further studied.
Rab35 function is regulated through a proteolytic mechanism controlled by PRPK
The function of Rab35 in axon elongation is controlled by its proteolytic degradation in a mechanism that involves PRPK. Previous reports showed that Rab GTPases can be ubiquitinated. For example, the ubiquitination of Rab11 mediated by HACE1 is involved in the activation of Rab11 (Lachance et al., 2014). However, Rab GTPase ubiquitination and degradation is still relatively unexplored. Here, we demonstrated that Rab35 is ubiquitinated in brain, and its degradation is prevented by MG132. To our knowledge, our results on Rab35 proteolytic regulation are the first showing an ubiquitin-proteasome-dependent mechanism for Rab GTPase degradation.
We established an epistatic relationship suggesting that PRPK regulates Rab35 expression. This regulation is specific for Rab35, as the functionally related protein Rab11 was unaltered by PRPK overexpression. Proteolytic mechanisms are emerging as regulators of axon elongation, in which they act by selective degradation of components that are crucial for neuronal polarization (Cheng and Poo, 2012). Local degradation of a global axon growth inhibitor in one neurite favors elongation of that neurite to the detriment of others, as exemplified by RhoA degradation performed by the E3 ubiquitin ligase Smurf1 (Cheng et al., 2011).
PRPK inhibition of axon elongation
Negative regulation of axon outgrowth is the first role for PRPK reported in the nervous system and depends on its kinase activity. It is possible that additional modulatory roles for PRPK may emerge in the future, as PRPK homologs in both yeast and Drosophila melanogaster display functions that are independent of kinase activity (Lopreiato et al., 2004; Ibar et al., 2013). Moreover, it has yet to be determined whether the KEOPS/EKC complex is involved in axon outgrowth. The highest levels of PRPK found in embryonic brain may be counterintuitive for a suppressor of axonal growth. The high abundance at embryonic stages of a protein that negatively regulates axonal grown and/or extension is not uncommon. For example, RhoA a small GTPase involved in axon collapse (van Leeuwen et al., 1997) is robustly expressed during brain development (Olenik et al., 1999). In addition, Semaphorine, which are diffusible factors that regulate axonal guidance, are also highly expressed during nervous system development, even though their main function is to avoid axonal invasion of non-permissive territories (Culotti and Kolodkin, 1996). Other proteins negatively regulating axon elongation, such as BICDR1, reduce their expression during the main stage of axon outgrowth (Schlager et al., 2010). In the future, we need to improve our knowledge concerning the specific function of PRPK at different developmental stages and in different brain areas.
PRPK-dependent regulation of Rab35 is a suitable mechanism to explain the axon outgrowth phenotype, as Rab35 promotes neuritogenesis in neuronal cell lines (Chevallier et al., 2009; Kobayashi and Fukuda, 2012; Kobayashi et al., 2014). Indeed, in our experiments, Rab35 promoted axon elongation and rescued PRPK-induced axon shortening in a mechanism dependent on the GTP-active form. The precise localization and activity of Rab35 guanine exchange factors (GEFs) during neuronal polarization and axon elongation remain to be characterized.
Rab35 also rescued the PRPK-induced reduction in filopodial formation in N1E-115 cells. Rab35 enhances filopodial formation through its effector fascin (Zhang et al., 2009) and here we show that it enhances Cdc42 activity. Currently, it is unclear how Rab35 induces Cdc42 activation. One proposal is that Rab35 directs the transport of small GTPases toward the plasma membrane, allowing association with specific GEFs (Shim et al., 2010). An alternative explanation could be that PRPK-Rab35 regulates the activity of some Cdc42 GEFs, such as diffuse B-cell lymphoma (Dbl), dedicator of cytokinesis (DOCK) family proteins (Chua et al., 2010), or others. Recently, it has been proposed that vesicles carrying Rab35 also transport essential components involved in the development of polarized morphology in epithelial cells such as aPKC, Cdc42, and Crumbs3 (Klinkert et al., 2016). Whether such transport organization is mirrored in neurons still needs further evaluation.
PRPK is a novel MAP1B-binding protein
MAP1B is a microtubule-associated protein required to support axon elongation (González-Billault et al., 2001; Dajas-Bailador et al., 2012). The increasing number of MAP1B-interacting proteins suggests that it may regulate axon outgrowth through molecular mechanisms not strictly related to its MAP roles (Villarroel-Campos and González-Billault, 2014). In fact, MAP1B is involved in cellular functions regardless of its microtubule-stabilizing properties (Fuhrmann-Stroissnigg et al., 2012). Here, we identified PRPK as a novel MAP1B-LC1-interacting protein using Y2H and GST pull-down approaches.
We evaluated whether this novel mechanism involving PRPK, MAP1B, and Rab35 could be linked to deficiencies in axonal microtubule contents according to the canonical function of MAP1B. We believe this is not the case, because neurons depleted of microtubules by nocodazole, did not show Rab35 degradation. Rab35 levels were also unmodified in neurons derived from tau knock-out mice, another axonal MAP. Interestingly, although shorter than control, neurons derived from several knock-out lines targeting MAP1B (Takei et al., 1997; González-Billault et al., 2000, 2001; Meixner et al., 2000; Bouquet et al., 2004), tau (Harada et al., 1994; Dawson et al., 2001), or both (Takei et al., 2000) contain a distinguishable axon, an indication that in the absence of MAP1B or tau, compensatory mechanisms promote microtubule polymerization.
Axon defects in MAP1B-deficient neurons are rescued by Rab35 and PRPK KD
Previously, we showed that MAP1B KO neurons contain reduced levels of active Cdc42 (Montenegro-Venegas et al., 2010). We now propose that reduced levels of GTP-Cdc42 can be reverted by overexpressing PRPK KD or Rab35. Strikingly, PRPK KD promotes axon growth more efficiently in MAP1B KO neurons than in WT neurons, suggesting that in wild-type neurons PRPK cannot inactivate Rab35 over a threshold level needed to impair axon elongation, likely due to the presence of MAP1B. This idea is supported by three sets of experiments. First, treatment of control neurons with shRNAs against PRPK did not increase axon elongation. Second, WT neurons cotransfected with shRNAs against MAP1B and PRPK displayed longer axons than neurons transfected only with shRNAs against MAP1B. Finally, LC1 but not tau overexpression in cells lacking MAP1B prevented PRPK-dependent Rab35 degradation. Thus, the presence of MAP1B in WT neurons would be sufficient to buffer the deleterious effects of PRPK on axonal growth.
We are currently searching for PRPK substrates that may link PRPK kinase activity with Rab35 degradation. It is possible that PRPK phosphorylates an E3 ubiquitin ligase and then directs the ligase against Rab35. Indeed, KEOPS/EKC is recruited to Cullin2-based E3 ubiquitin ligases (Costessi et al., 2012), providing support for this hypothetical mechanism.
In conclusion, we demonstrate that Rab35 is important in supporting axon elongation, by regulating Cdc42 activity and actin dynamics. These results link actin and membrane dynamics, and highlight the role of the recycling endosomes during the acquisition of neuronal morphology. PRPK inhibits axon elongation by promoting Rab35 proteolysis, a mechanism that is regulated by the presence of MAP1B. To the best of our knowledge, this represents the first evidence for Rab GTPase proteasomal degradation, and places MAP1B as a scaffolding protein involved in the regulation of other cellular functions apart from microtubule stabilization.
Footnotes
This work was supported by grants from CONICYT: Fondecyt 1140325 and FONDAP 15150012 to C.G.-B., and Postdoctoral Fondecyt (3130316) to D.R.-H. We thank Michael Handford for English editing, Jesus Avila (CBMSO, Madrid, Spain) for providing samples derived from WT and tau knock-out mice, and Dr. Jose Wojnacki for FRET experimental setup.
The authors declare no competing financial interests.
References
- Abe Y, Matsumoto S, Wei S, Nezu K, Miyoshi A, Kito K, Ueda N, Shigemoto K, Hitsumoto Y, Nikawa J, Enomoto Y. Cloning and characterization of a p53-related protein kinase expressed in interleukin-2-activated cytotoxic T-cells, epithelial tumor cell lines, and the testes. J Biol Chem. 2001;276:44003–44011. doi: 10.1074/jbc.M105669200. [DOI] [PubMed] [Google Scholar]
- Abe Y, Takeuchi T, Imai Y, Murase R, Kamei Y, Fujibuchi T, Matsumoto S, Ueda N, Ogasawara M, Shigemoto K, Kito K. A small Ras-like protein Ray/Rab1c modulates the p53-regulating activity of PRPK. Biochem Biophys Res Commun. 2006;344:377–385. doi: 10.1016/j.bbrc.2006.03.071. [DOI] [PubMed] [Google Scholar]
- Barr F, Lambright DG. Rab GEFs and GAPs. Curr Opin Cell Biol. 2010;22:461–470. doi: 10.1016/j.ceb.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouquet C, Soares S, von Boxberg Y, Ravaille-Veron M, Propst F, Nothias F. Microtubule-associated protein 1B controls directionality of growth cone migration and axonal branching in regeneration of adult dorsal root ganglia neurons. J Neurosci. 2004;24:7204–7213. doi: 10.1523/JNEUROSCI.2254-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Slaughter T, Black MM. Newly assembled microtubules are concentrated in the proximal and distal regions of growing axons. J Cell Biol. 1992;119:867–882. doi: 10.1083/jcb.119.4.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng PL, Poo MM. Early events in axon/dendrite polarization. Annu Rev Neurosci. 2012;35:181–201. doi: 10.1146/annurev-neuro-061010-113618. [DOI] [PubMed] [Google Scholar]
- Cheng PL, Lu H, Shelly M, Gao H, Poo MM. Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron. 2011;69:231–243. doi: 10.1016/j.neuron.2010.12.021. [DOI] [PubMed] [Google Scholar]
- Chevallier J, Koop C, Srivastava A, Petrie RJ, Lamarche-Vane N, Presley JF. Rab35 regulates neurite outgrowth and cell shape. FEBS Lett. 2009;583:1096–1101. doi: 10.1016/j.febslet.2009.03.012. [DOI] [PubMed] [Google Scholar]
- Chua CE, Lim YS, Tang BL. Rab35: a vesicular traffic-regulating small GTPase with actin modulating roles. FEBS Lett. 2010;584:1–6. doi: 10.1016/j.febslet.2009.11.051. [DOI] [PubMed] [Google Scholar]
- Costessi A, Mahrour N, Sharma V, Stunnenberg R, Stoel MA, Tijchon E, Conaway JW, Conaway RC, Stunnenberg HG. The human EKC/KEOPS complex is recruited to Cullin2 ubiquitin ligases by the human tumour antigen PRAME. PLoS One. 2012;7:e42822. doi: 10.1371/journal.pone.0042822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culotti JG, Kolodkin AL. Functions of netrins and semaphorins in axon guidance. Curr Opin Neurobiol. 1996;6:81–88. doi: 10.1016/S0959-4388(96)80012-2. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador F, Bonev B, Garcez P, Stanley P, Guillemot F, Papalopulu N. microRNA-9 regulates axon extension and branching by targeting Map1b in mouse cortical neurons. Nat Neurosci. 2012;15:697–699. doi: 10.1038/nn.3082. [DOI] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114:1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]
- DiTella MC, Feiguin F, Carri N, Kosik KS, Cáceres A. MAP-1B/TAU functional redundancy during laminin-enhanced axonal growth. J Cell Sci. 1996;109:467–477. doi: 10.1242/jcs.109.2.467. [DOI] [PubMed] [Google Scholar]
- Downey M, Houlsworth R, Maringele L, Rollie A, Brehme M, Galicia S, Guillard S, Partington M, Zubko MK, Krogan NJ, Emili A, Greenblatt JF, Harrington L, Lydall D, Durocher D. A genome-wide screen identifies the evolutionarily conserved KEOPS complex as a telomere regulator. Cell. 2006;124:1155–1168. doi: 10.1016/j.cell.2005.12.044. [DOI] [PubMed] [Google Scholar]
- Etoh K, Fukuda M. Structure-function analyses of the small GTPase Rab35 and its effector protein centaurin-β/ACAP2 during neurite outgrowth of PC12 cells. J Biol Chem. 2015;290:9064–9074. doi: 10.1074/jbc.M114.611301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eva R, Dassie E, Caswell PT, Dick G, ffrench-Constant C, Norman JC, Fawcett JW. Rab11 and its effector Rab coupling protein contribute to the trafficking of beta 1 integrins during axon growth in adult dorsal root ganglion neurons and PC12 cells. J Neurosci. 2010;30:11654–11669. doi: 10.1523/JNEUROSCI.2425-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk J, Konopacki FA, Zivraj KH, Holt CE. Rab5 and Rab4 regulate axon elongation in the Xenopus visual system. J Neurosci. 2014;34:373–391. doi: 10.1523/JNEUROSCI.0876-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann-Stroissnigg H, Noiges R, Descovich L, Fischer I, Albrecht DE, Nothias F, Froehner SC, Propst F. The light chains of microtubule-associated proteins MAP1A and MAP1B interact with alpha1-syntrophin in the central and peripheral nervous system. PLoS One. 2012;7:e49722. doi: 10.1371/journal.pone.0049722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman AH, Banker GA. The economics of neurite outgrowth: the addition of new membrane to growing axons. Trends Neurosci. 1996;19:144–149. doi: 10.1016/S0166-2236(96)80025-7. [DOI] [PubMed] [Google Scholar]
- González-Billault C, Demandt E, Wandosell F, Torres M, Bonaldo P, Stoykova A, Chowdhury K, Gruss P, Avila J, Sánchez MP. Perinatal lethality of microtubule-associated protein 1B-deficient mice expressing alternative isoforms of the protein at low levels. Mol Cell Neurosci. 2000;16:408–421. doi: 10.1006/mcne.2000.0880. [DOI] [PubMed] [Google Scholar]
- González-Billault C, Avila J, Cáceres A. Evidence for the role of MAP1B in axon formation. Mol Biol Cell. 2001;12:2087–2098. doi: 10.1091/mbc.12.7.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorvel JP, Chavrier P, Zerial M, Gruenberg J. rab5 controls early endosome fusion in vitro. Cell. 1991;64:915–925. doi: 10.1016/0092-8674(91)90316-Q. [DOI] [PubMed] [Google Scholar]
- Guo Z, Wang X, Li H, Gao Y. Screening E3 substrates using a live phage display library. PLoS One. 2013;8:e76622. doi: 10.1371/journal.pone.0076622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- Henríquez DR, Bodaleo FJ, Montenegro-Venegas C, González-Billault C. The light chain 1 subunit of the microtubule-associated protein 1B (MAP1B) is responsible for Tiam1 binding and Rac activation in neuronal cells. Plos One. 2012;7:e53123. doi: 10.1371/journal.pone.0053123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibar C, Cataldo VF, Vásquez-Doorman C, Olguín P, Glavic A. Drosophila p53-related protein kinase is required for PI3K/TOR pathway-dependent growth. Development. 2013;140:1282–1291. doi: 10.1242/dev.086918. [DOI] [PubMed] [Google Scholar]
- Ishida M, Ohbayashi N, Maruta Y, Ebata Y, Fukuda M. Functional involvement of Rab1A in microtubule-dependent anterograde melanosome transport in melanocytes. J Cell Sci. 2012;125:5177–5187. doi: 10.1242/jcs.109314. [DOI] [PubMed] [Google Scholar]
- Itoh T, Fujita N, Kanno E, Yamamoto A, Yoshimori T, Fukuda M. Golgi-resident small GTPase Rab33B interacts with Atg16L and modulates autophagosome formation. Mol Biol Cell. 2008;19:2916–2925. doi: 10.1091/mbc.E07-12-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kanno E, Ishibashi K, Kobayashi H, Matsui T, Ohbayashi N, Fukuda M. Comprehensive screening for novel rab-binding proteins by GST pull-down assay using 60 different mammalian Rabs. Traffic. 2010;11:491–507. doi: 10.1111/j.1600-0854.2010.01038.x. [DOI] [PubMed] [Google Scholar]
- Kisseleva-Romanova E, Lopreiato R, Baudin-Baillieu A, Rousselle JC, Ilan L, Hofmann K, Namane A, Mann C, Libri D. Yeast homolog of a cancer-testis antigen defines a new transcription complex. EMBO J. 2006;25:3576–3585. doi: 10.1038/sj.emboj.7601235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinkert K, Rocancourt M, Houdusse A, Echard A. Rab35 GTPase couples cell division with initiation of epithelial apico-basal polarity and lumen opening. Nat Commun. 2016;7:11166. doi: 10.1038/ncomms11166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Fukuda M. Rab35 regulates Arf6 activity through centaurin-beta2 (ACAP2) during neurite outgrowth. J Cell Sci. 2012;125:2235–2243. doi: 10.1242/jcs.098657. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Etoh K, Fukuda M. Rab35 is translocated from Arf6-positive perinuclear recycling endosomes to neurite tips during neurite outgrowth. Small GTPases. 2014;5:e29290. doi: 10.4161/sgtp.29290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouranti I, Sachse M, Arouche N, Goud B, Echard A. Rab35 regulates an endocytic recycling pathway essential for the terminal steps of cytokinesis. Curr Biol. 2006;16:1719–1725. doi: 10.1016/j.cub.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Lachance V, Degrandmaison J, Marois S, Robitaille M, Génier S, Nadeau S, Angers S, Parent JL. Ubiquitylation and activation of a Rab GTPase is promoted by a beta(2)AR-HACE1 complex. J Cell Sci. 2014;127:111–123. doi: 10.1242/jcs.132944. [DOI] [PubMed] [Google Scholar]
- Lalli G. Crucial polarity regulators in axon specification. Essays Biochem. 2012;53:55–68. doi: 10.1042/bse0530055. [DOI] [PubMed] [Google Scholar]
- Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, Wilson SM, King RW, Finley D. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KB, Bu W, Goh WI, Koh E, Ong SH, Pawson T, Sudhaharan T, Ahmed S. The Cdc42 effector IRSp53 generates filopodia by coupling membrane protrusion with actin dynamics. J Biol Chem. 2008;283:20454–20472. doi: 10.1074/jbc.M710185200. [DOI] [PubMed] [Google Scholar]
- Lopreiato R, Facchin S, Sartori G, Arrigoni G, Casonato S, Ruzzene M, Pinna LA, Carignani G. Analysis of the interaction between piD261/Bud32, an evolutionarily conserved protein kinase of saccharomyces cerevisiae, and the Grx4 glutaredoxin. Biochem J. 2004;377:395–405. doi: 10.1042/bj20030638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marat AL, Ioannou MS, McPherson PS. Connecdenn 3/DENND1C binds actin linking Rab35 activation to the actin cytoskeleton. Mol Biol Cell. 2012;23:163–175. doi: 10.1091/mbc.E11-05-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meixner A, Haverkamp S, Wässle H, Führer S, Thalhammer J, Kropf N, Bittner RE, Lassmann H, Wiche G, Propst F. MAP1B is required for axon guidance and is involved in the development of the central and peripheral nervous system. J Cell Biol. 2000;151:1169–1178. doi: 10.1083/jcb.151.6.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montenegro-Venegas C, Tortosa E, Rosso S, Peretti D, Bollati F, Bisbal M, Jausoro I, Avila J, Cáceres A, González-Billault C. MAP1B regulates axonal development by modulating Rho-GTPase Rac1 activity. Mol Biol Cell. 2010;21:3518–3528. doi: 10.1091/mbc.E09-08-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba T, Funahashi Y, Nakamuta S, Xu C, Takano T, Kaibuchi K. Extracellular and intracellular signaling for neuronal polarity. Physiol Rev. 2015;95:995–1024. doi: 10.1152/physrev.00025.2014. [DOI] [PubMed] [Google Scholar]
- Olenik C, Aktories K, Meyer DK. Differential expression of the small GTP-binding proteins RhoA, RhoB, Cdc42u and Cdc42b in developing rat neocortex. Brain Res Mol Brain Res. 1999;70:9–17. doi: 10.1016/S0169-328X(99)00121-7. [DOI] [PubMed] [Google Scholar]
- Sato M, Sato K, Liou W, Pant S, Harada A, Grant BD. Regulation of endocytic recycling by C. elegans Rab35 and its regulator RME-4, a coated-pit protein. EMBO J. 2008;27:1183–1196. doi: 10.1038/emboj.2008.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlager MA, Kapitein LC, Grigoriev I, Burzynski GM, Wulf PS, Keijzer N, de Graaff E, Fukuda M, Shepherd IT, Akhmanova A, Hoogenraad CC. Pericentrosomal targeting of Rab6 secretory vesicles by Bicaudal-D-related protein 1 (BICDR-1) regulates neuritogenesis. EMBO J. 2010;29:1637–1651. doi: 10.1038/emboj.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J, Lee SM, Lee MS, Yoon J, Kweon HS, Kim YJ. Rab35 mediates transport of Cdc42 and Rac1 to the plasma membrane during phagocytosis. Mol Cell Biol. 2010;30:1421–1433. doi: 10.1128/MCB.01463-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva GM, Finley D, Vogel C. K63 polyubiquitination is a new modulator of the oxidative stress response. Nat Struct Mol Biol. 2015;22:116–123. doi: 10.1038/nsmb.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan M, Mehta P, Yu Y, Prugar E, Koonin EV, Karzai AW, Sternglanz R. The highly conserved KEOPS/EKC complex is essential for a universal tRNA modification, t6A. EMBO J. 2011;30:873–881. doi: 10.1038/emboj.2010.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahirovic S, Bradke F. Neuronal polarity. Cold Spring Harb Perspect Biol. 2009;1:a001644. doi: 10.1101/cshperspect.a001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tomomura M, Yoshioka N, Tsutsumi K, Terasawa Y, Saito T, Kawano H, Kamiguchi H, Fukuda M, Hisanaga S. LMTK1/AATYK1 is a novel regulator of axonal outgrowth that acts via Rab11 in a Cdk5-dependent manner. J Neurosci. 2012;32:6587–6599. doi: 10.1523/JNEUROSCI.5317-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei Y, Kondo S, Harada A, Inomata S, Noda T, Hirokawa N. Delayed development of nervous system in mice homozygous for disrupted microtubule-associated protein 1B (MAP1B) gene. J Cell Biol. 1997;137:1615–1626. doi: 10.1083/jcb.137.7.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei Y, Teng J, Harada A, Hirokawa N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol. 2000;150:989–1000. doi: 10.1083/jcb.150.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker RP, Matus AI. Microtubule-associated proteins characteristic of embryonic brain are found in the adult mammalian retina. Dev Biol. 1988;130:423–434. doi: 10.1016/0012-1606(88)90338-7. [DOI] [PubMed] [Google Scholar]
- Ullrich O, Reinsch S, Urbé S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol. 1996;135:913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen FN, Kain HE, Kammen RA, Michiels F, Kranenburg OW, Collard JG. The guanine nucleotide exchange factor Tiam1 affects neuronal morphology; opposing roles for the small GTPases Rac and Rho. J Cell Biol. 1997;139:797–807. doi: 10.1083/jcb.139.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarroel-Campos D, González-Billault C. The MAP1B case: an old MAP that is new again. Dev Neurobiol. 2014;74:953–971. doi: 10.1002/dneu.22178. [DOI] [PubMed] [Google Scholar]
- Villarroel-Campos D, Gastaldi L, Conde C, Caceres A, González-Billault C. Rab-mediated trafficking role in neurite formation. J Neurochem. 2014;129:240–248. doi: 10.1111/jnc.12676. [DOI] [PubMed] [Google Scholar]
- Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- Zhang J, Fonovic M, Suyama K, Bogyo M, Scott MP. Rab35 controls actin bundling by recruiting fascin as an effector protein. Science. 2009;325:1250–1254. doi: 10.1126/science.1174921. [DOI] [PubMed] [Google Scholar]