

Abstract
Selective serotonin reuptake inhibitors (SSRIs) are commonly prescribed for depression, but sexual side effects often compromise compliance. These reproductive dysfunctions are likely mediated by elevations of the hormone prolactin. Yet, how serotonin (5-HT) and SSRIs cause changes in prolactin secretion is not known. Here, using in vitro whole-cell patch-clamp recordings, we show that 5-HT hyperpolarizes and abolishes phasic discharge in rat neuroendocrine tuberoinfundibular dopamine (TIDA) neurons, the main inhibitor of prolactin secretion. This process is underpinned by 5-HT1A receptor-mediated activation of G-protein-coupled inwardly rectifying K+-like currents. We further demonstrate that the SSRIs, fluoxetine and sertraline, directly suppress TIDA neuron activity through parallel effects, independent of 5-HT transmission. This inhibition involves decreased intrinsic excitability and a slowing of TIDA network rhythms. These findings indicate that SSRIs may inhibit neuroendocrine dopamine release through both 5-HT-dependent and -independent actions, providing a mechanistic explanation for, and potential molecular targets for the amelioration of, the hyperprolactinemia and sexual dysfunction associated with these drugs.
SIGNIFICANCE STATEMENT Depression affects approximately one-tenth of the population and is commonly treated with selective serotonin reuptake inhibitors (SSRIs; e.g., Prozac). Yet, many patients withdraw from SSRI therapy due to sexual side effects (e.g., infertility, menstrual disturbances, and impotence). Although it is generally accepted that sexual side effects are due to the ability of these drugs to elevate blood levels of the hormone prolactin, the mechanism for this hormonal imbalance is not known. Here, we show that SSRIs can inhibit hypothalamic dopamine neurons that normally suppress the secretion of prolactin. Intriguingly this inhibition can be explained both by increased serotonin activity and also by parallel serotonin-independent actions.
Keywords: depression, dopamine, fluoxetine, neuroendocrine, prolactin, tuberoinfundibular
Introduction
Selective serotonin reuptake inhibitors (SSRIs) were originally developed for the treatment of mood disorders but have also been approved to treat a widening spectrum of conditions, including anxiety, obsessive compulsive disorder, and bulimia nervosa. Depressive disorders, with a global prevalence exceeding 4% (Vos et al., 2012), are projected to be the leading cause of disease burden by 2030 (World Health Organisation, 2008). SSRIs are recommended as a front-line therapy for moderate to severe depression (American Psychiatric Association, 2000), and population prevalence of SSRI use as high as 3.5% has been described (Olié et al., 2002). Unfortunately, SSRI compliance is compromised by the high incidence of adverse effects. Sexual side effects, including amenorrhea, infertility, anorgasmia, impotence, and impaired libido, are often under-reported (Montejo-González et al., 1997) but, when systematically assessed, are experienced by ∼40% of all SSRI users (Clayton et al., 2002), often leading to cessation of treatment (van Geffen et al., 2007).
The adverse sexual effects associated with SSRIs are commonly attributed to elevation of the pituitary hormone, prolactin (Safarinejad, 2008; Madhusoodanan et al., 2010). Prolactin exerts a range of physiological effects that decrease fertility and libido (Bole-Feysot et al., 1998), and hyperprolactinemia, caused, for example, by pituitary adenomas or induced iatrogenically, results in reproductive dysfunction, such as menstrual disturbances and impotence (e.g., Schlechte et al., 1980; Buvat et al., 1985). Hyperprolactinemia is common with SSRIs, such as fluoxetine (Prozac) and sertraline (Urban and Veldhuis, 1991; Ekselius et al., 1997; Gregorian et al., 2002; Petit et al., 2003), and was originally described in animal studies (e.g., Morgan and Herbert, 1978). Importantly, serotonin (5-HT) itself was first shown to be a potent prolactin-releasing agent >4 decades ago (Kamberi et al., 1971; Lu and Meites, 1973).
It is not well understood how 5-HT and SSRIs promote prolactin release (Emiliano and Fudge, 2004). Direct stimulation of prolactin-producing lactotrophs is unlikely because administration of 5-HT to isolated pituitary preparations does not promote prolactin secretion (Birge et al., 1970). The release of prolactin is, however, under powerful control by neuroendocrine tuberoinfundibular dopamine (TIDA) neurons located in the hypothalamic arcuate nucleus (Lyons and Broberger, 2014). Dopamine released from TIDA neurons reaches the pituitary through the portal capillary system and tonically suppresses prolactin release via inhibitory dopamine D2-type receptors. Recent work suggests that neuromodulators and hormones that influence prolactin release and lactation may do this by altering the passive and active membrane properties of TIDA neurons (Lyons et al., 2010, 2012; Briffaud et al., 2015; Zhang and van den Pol, 2015) Here, we have investigated whether 5-HT and three SSRIs (fluoxetine, sertraline, and fenfluramine) can alter the electrophysiology of TIDA cells in a way that may explain the prolactin-releasing actions of these compounds.
Materials and Methods
Animals
Male Sprague Dawley rats (Charles River), 22- to 30-d-old for electrophysiology experiments and 4- to 6-weeks-old for immunofluorescence, were housed with free access to standard chow and tap water in a temperature-controlled environment under 12 h/12 h light/dark conditions. All animal experiments had received prior approval by the local ethical board, Stockholms Norra Djurförsöksetiska Nämnd, and were performed in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC).
Electrophysiology
Electrophysiological experiments were performed as previously described (Lyons et al., 2012). Rats (n = 83) were anesthetized with sodium pentobarbital and decapitated. The brain was removed and placed in cold, oxygenated (95%O2/5%CO2) “slicing” solution containing the following (in mm): 214 sucrose, 2.0 KCl, 1.2 NaH2PO4, 26 NaHCO3, 1.3 MgSO4, 2.4 CaCl2, and 10 d-glucose. The brain was glued to a vibratome (Leica), and 250 μm thick coronal sections of the hypothalamus, including the arcuate nucleus, were prepared. Slices were immediately transferred to a “recording” solution containing the following (in mm): 127 NaCl, 2.0 KCl, 1.2 NaH2PO4, 26 NaHCO3, 1.3 MgCl2, 2.4 CaCl2, and 10 d-glucose, in a continuously oxygenated holding chamber at 35°C for a period of 25 min. Subsequently, slices were allowed to recover in “recording” solution at room temperature for a minimum of 1 h before recording. For whole-cell recordings, slices were transferred to a submerged chamber and placed on an elevated grid that allows perfusion both above and below the slice. An Axioskop 2 FS Plus upright microscope (Carl Zeiss) was used for infrared-differential interference contrast visualization of cells. Recordings were performed at room temperature (22°C), and slices were continuously perfused with oxygenated “recording” solution (as above) unless otherwise described at a rate of ∼5 ml/min. All pharmacological compounds were bath applied.
Whole-cell current- and voltage-clamp recordings were performed with pipettes (3–7 mΩ when filled with intracellular solution) made from borosilicate glass capillaries (World Precision Instruments) pulled on a P-97 Flaming/Brown micropipette puller (Sutter Instruments). The intracellular recording solution contained (in mm) the following: 140 K-gluconate, 10 KCl, 10 HEPES, 1 EGTA, 2 Na2ATP, pH 7.3 (with KOH). When investigating IPSCs, a High Cl− intracellular solution was used comprised of the following (in mm): 150 KCl, 10 HEPES, 1 EGTA, 2 Na2ATP), pH 7.3 (with KOH). The concentration used for TTX was 500 nm. In experiments that required the raising of K+ conductances, reversal potential high K+ extracellular recording solution containing the following (in mm): 127 NaCl, 10 KCl, 1.2 NaH2PO4, 26 NaHCO3, 1.3 MgCl2, 2.4 CaCl2, 10 d-glucose was used. Recordings were performed using a Multiclamp 700B amplifier and pClamp9 software (Molecular Devices). Access resistance was monitored throughout the experiments, and neurons in which the series resistance was >25 mΩ or changed >15% were excluded from the statistics. Liquid junction potential was 16.4 mV and not compensated. The recorded current was sampled at 10 kHz and filtered at 2 kHz unless otherwise stated. In voltage-clamp experiments, the holding potential was −60 mV unless otherwise stated.
To determine the 5-HT-induced current in the presence of TTX frequency distribution, histograms were plotted for 10 s of control, 5-HT, and wash traces with Gaussian fits performed on the individual and pooled distributions. The mean difference between the Gaussian peaks for control and 5-HT was used as the value for 5-HT-induced current. The spectral density plot of the underlying oscillation was produced by filtering the trace with a 1 Hz low pass filter and performing spectral analysis with a rectangular window function on 56.5 s segments giving a spectral resolution of 0.019 Hz. To quantify GABAergic synaptic events, 100 s segments in control and during response were selected for analysis and comparison.
Immunofluorescence
Rats (n = 4) were perfused with a fixative of 5% glutaraldehyde/1% sodium bisulfite in cacodylate buffer; 14-μm-thick coronal brain sections were processed for indirect immunofluorescence as previously described using mouse monoclonal anti-TH antibodies (1:2000; Millipore) and rabbit anti-serotonin (5-HT) antiserum (1:250; Millipore) (Foo et al., 2014).
Statistical analysis and reagents
Data analysis was performed with OriginPro8.5 (OriginLab), Clampfit 9 (Molecular Devices), and Mini Analysis 6.0.9 (Synaptosoft) software. Statistical significance was set at p < 0.05. All reagents were purchased from Sigma Pharmaceuticals, with the exception of TTX, which was purchased from Alomone Labs, and 8-hydroxy PIPAT oxalate (±)-8-hydroxy-2-dipropylaminotetralin hydrobromide (8-OH-DPAT), NAD299 hydrochloride, fluoxetine hydrochloride, sertraline hydrochloride, and dexfenfluramine hydrochloride, which were purchased from Tocris Bioscience.
Results
5-HT directly inhibits oscillating TIDA neurons
TIDA neurons display a synchronized network oscillation (Lyons et al., 2010, 2012), characterized by alternating periods of hyperpolarization and depolarization (Fig. 1A). This stereotyped oscillation provides a reliable electrical signature for rat TIDA neurons in vitro (Lyons et al., 2010, 2012). When 5-HT (10 μm) was applied to TIDA neurons (in current-clamp mode), a reversible hyperpolarization and cessation of both oscillation and phasic discharge were consistently observed (n = 27 of 27; 100%; Fig. 1A,B).
Figure 1.
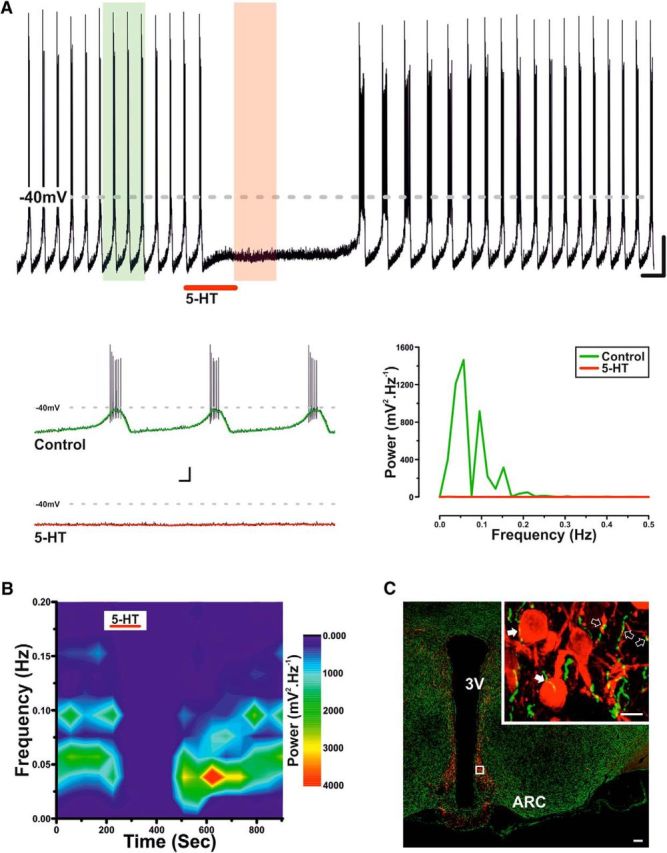
Serotonin inhibits TIDA neurons. A, Current-clamp, oscillating rat TIDA neuron. 5-HT (10 μm) application results in hyperpolarization and abolishment of phasic discharge. Calibration: 20 mV, 30 s. Bottom, Expanded sections highlighted in A with the color-coordinated 1 Hz filter superimposed and the corresponding spectral analysis opposite. Calibration: 20 mV, 2 s. B, Frequency spectrogram of the entire trace depicted in A. There is reversible abolishment of TIDA oscillation power by 5-HT. C, Confocal micrographs from rat hypothalamus stained by immunofluorescence for TH (red) and 5-HT (green). Boxed area enlarged in Inset. 5-HT-immunoreactive terminals form close appositions on somata (filled arrows) and dendrites (open arrows) of TH-expressing TIDA neurons. Scale bars: 100 μm; Inset, 10 μm. 3V, Third ventricle; ARC, arcuate nucleus.
Immunohistochemical staining for 5-HT and TH (the rate-limiting enzyme in catecholamine biosynthesis) revealed 5-HT-immunoreactive fibers in close apposition to TIDA somata and dendrites, thus providing an anatomical substrate for the direct 5-HT-ergic modulation of TIDA activity (Fig. 1C). This was confirmed pharmacologically, as in the presence of the Na+ channel blocker TTX (500 nm), which also abolishes the TIDA oscillation (Lyons et al., 2010, 2012), 5-HT (10 μm) induced a rapid and reversible 22.9 ± 1.6 mV hyperpolarization (n = 5; p < 0.005; ANOVA; F(2,12) = 163.4; Fig. 2A,B) (this value was used as a control 5-HT response for current-clamp tests where paired comparisons were not possible), and a 9.7 ± 2.1% reduction in input resistance (n = 5; p < 0.01; ANOVA; F(2,12) = 10.7; Fig. 2A,B). A second application of 5-HT resulted in a 19.3 ± 2.0 mV (n = 5; p < 0.01; ANOVA; F(2,12) = 21.8) hyperpolarization (88.0 ± 3.6% of the initial response; n = 5; Fig. 2C) indicative of small, yet significant, tachyphylaxis. The 5-HT response also demonstrated a degree of dose dependence, with 1 μm 5-HT inducing a significant −5.3 ± 1.2 mV (n = 5, p < 0.01; t test paired; t = 4.4) hyperpolarization, whereas 250 nm failed to elicit a response (−0.8 ± .1.1; n = 5; not significant; t test paired; t = 0.7; data not shown). These results demonstrate that 5-HT inhibits TIDA neurons via a direct, postsynaptic mechanism.
Figure 2.

Serotonin inhibits TIDA neurons via a postsynaptic mechanism. A, Current-clamp trace, oscillating TIDA neuron. The 5-HT (10 μm)-induced hyperpolarization endures in the presence of TTX (500 nm). Initial membrane potential was set at ≈−65 mV, a value achieved with the injection of negative DC current. Input resistance was assessed by square-form negative current injection (asterisks). For purposes of comparison at peak of response, membrane potential was returned to −65 mV with additional DC current (arrow). Inset, Superimposed membrane potential deflections in control (green) and 5-HT (red). Calibration: 10 mV, 20 s; Inset, 20 mV, 250 ms. B, Summary of 5-HT-induced effects on membrane potential (green) and input resistance (red) recorded as in A. Thick lines indicate mean ± SEM. Dashed lines indicate raw data (n = 5). C, Desensitization of the 5-HT response recorded as in A following repeated applications (after 15 min of wash). Thick lines indicate mean ± SEM. Dashed lines indicate raw data (n = 5). *p < 0.05. **p < 0.01. ***p < 0.005. ns, Not significant.
5-HT activates a G-protein-coupled inwardly rectifying potassium (GIRK)-like current
The ionic mechanisms underlying the 5-HT-induced hyperpolarization were investigated in voltage clamp. In the presence of TTX, application of 5-HT (10 μm) resulted in a reversible outward current of 17.3 ± 2.2 pA (n = 10; p < 0.005; ANOVA; F(2,27) = 52.4; Fig. 3A) (this value was used as a control 5-HT response for voltage-clamp tests where paired comparisons were not possible, “Control I5-HT”). Digital subtraction of voltage-clamp ramps performed in control from those at the peak of response revealed the 5-HT-induced current (I5-HT) to be a net outward current (Fig. 3Ai) with an estimated reversal potential of −128.1 mV (Fig. 3Aii), lower than that calculated for K+ (−110.6 mV).
Figure 3.

Serotonin activates GIRK-like channels. A, Voltage-clamp, oscillating TIDA neuron recorded in the presence of TTX. Application of 5-HT results in an outward current. Right, Gaussian fits of averaged (solid lines) holding current frequency distributions in control (green), 5-HT (red), and wash (black) (n = 10), sharing its y-axis with the raw trace. Dashed lines indicate raw data used to produce averages. Calibration: 5 s. Ai, Averaged 5-HT induced current ± SEM obtained by the digital subtraction of voltage-clamp ramps in control and at peak of 5-HT response (n = 10). Aii, Linear extrapolation of the 5-HT-induced current indicated by the box in Ai. There is projected reversal potential (Erev) of −128.1 mV. B, Voltage-clamp, oscillating TIDA neuron recorded as in Figure 3A, and with Cs+-based intracellular solution used to block K+-currents. Application of 5-HT induces a substantially reduced outward current (compare A). Right, Gaussian fits of holding current frequency distributions in control (green) and 5-HT (red). (n = 10). Calibration: 10 s. Bi, Averaged current-voltage relationship ± SEM of I5-HT in control (green, n = 10) and with Cs+-based intracellular solution (red, n = 10), obtained as in Ai. There is reduction of I5-HT when Cs+-based intracellular solution is used. Bii, Box plot of I5-HT recorded in voltage clamp with Cs+-based or control intracellular solution. Boxes represent 25th, 50th, and 75th percentile with superimposed mean ± SEM and maximum and minimum values (control n = 10, Cs+ n = 10). C, Voltage-clamp, oscillating TIDA neuron recorded as in A in the presence of high extracellular K+. Application of 5-HT fails to induce a significant current (n = 10). Calibration 5 s. Ci, Averaged current-voltage relationship ± SEM of I5-HT in the presence of High extracellular K+ (n = 10), obtained as shown in Ai. There is a shift of the reversal potential and presence of inward rectification (compared Ai). Cii, High [K+]O significantly reduces I5-HT. Box plot organized as Bii (control n = 10, High [K+]O n = 10). D, Voltage-clamp trace, oscillating TIDA neuron recorded as in A (there is abolishment of oscillation in response to TTX). 5-HT is applied before, during, and after Ba+ is present in the bath. There is Ba+-induced reduction of I5-HT. Calibration: 10 pA, 60 s. Di, Ba+ significantly and reversibly reduces I5-HT. Box plot organized as Bii (n = 5). Dii, TQ (n = 5) and the G-protein inhibitor NEM (control n = 10, NEM n = 5) significantly reduces I5-HT. Box plot organized as Bii. ***p < 0.005. ns, Not significant.
As this reversal potential was lower than that predicted for K+, possibly due to the role of gap junctions in the TIDA oscillation (Lyons et al., 2010) and their potential impact on space clamp, we sought to further investigate the role of K+ currents in the generation of I5-HT. Potassium channels were blocked through a Cs+-based internal solution. Here, 5-HT (10 μm) application induced only a 5.8 ± 0.7 pA outward current (n = 10; p < 0.005; t test paired; t = −8.9; Fig. 3B,Bi), a significant 66.5 ± 3.8% reduction of the control 5-HT response (n = 10; p < 0.005 vs control I5-HT; t test unpaired; t = 5.1; Fig. 3A,Bii).
Next, [K+]o was raised to 10 mm, shifting the calculated K+ reversal potential to −68 mV. At our holding potential of −60 mV, application of 5-HT (10 μm) failed to induce an outward current (1.0 ± 0.6 pA; n = 10; not significant; t test paired; t = 1.7; Fig. 3C), with I5-HT exhibiting a reversal potential of −58.9 mV and mild inward rectification (Fig. 3Ci).
Neuronal hyperpolarization by 5-HT has been associated with activation of GIRK channels (Penington et al., 1993; Lüscher et al., 1997). To investigate the potential role of GIRK channels, we sequentially applied 5-HT to TIDA neurons in the absence and presence of Ba2+ (300 μm), a nonspecific blocker of inwardly rectifying K+ channels. In the presence of Ba2+, 5-HT induced a 6.7 ± 1.5 pA outward current (n = 5; p < 0.05; t test paired; t = −4.4; Fig. 3D), a significant 75.1 ± 6.5% reduction compared with the initial, 29.0 ± 2.5 pA (n = 5), response to 5-HT (n = 5; p < 0.005; ANOVA; F(2,12) = 35.3; Fig. 3Di). This reduction was not due to desensitization as following washout of 5-HT and Ba2+, reapplication of 5-HT induced an outward current of 26.4 ± 2.4 pA (n = 5; p < 0.005; t test paired; t = −11.1), which proved indistinguishable from the first response (n = 5; not significant; ANOVA; F(2,12) = 35.3; Fig. 3D,Di). The selective GIRK channel blocker Tertiapin Q (TQ), a derivative of the honey bee venom tertiapin (Jin and Lu, 1999), was also tested. The application of 5-HT in the presence of TQ (300 nm) induced a 9.5 ± 2.2 pA outward current (n = 5; p < 0.01, t test paired; t = −4.4 Fig. 3Dii), a significant 48.5 ± 6.4% reduction compared with the initial, 18 ± 3.1 pA (n = 5), response to 5-HT (n = 5; p < 0.005, t test paired; t = 4.8; Fig. 3Dii). Furthermore, preincubation of slices (15–20 min) with the G-protein blocker N-ethylmaleimide (NEM; 250 μm) resulted in an 81.9% diminution of the 5-HT control response (3.0 ± 1.0 pA; n = 5; p < 0.005 vs control I5-HT; t test unpaired; t = 4.5; Fig. 3A,Dii).
Collectively these results indicate that I5-HT in TIDA neurons likely involves activation of GIRK-like channels.
5-HT inhibits TIDA neurons via the 5-HT1A receptor
In vivo studies have implicated 5-HT1A (Mulroney et al., 1994; Jørgensen et al., 2001), 5-HT2A (Liang and Pan, 2000), and 5-HT2C (Jørgensen et al., 1992) receptors in 5-HT's modulation of prolactin secretion. The broad-spectrum 5-HT2 receptor antagonist methiothepin (5 μm) failed to block the 5-HT-induced hyperpolarization of TIDA neurons (Control 5-HT −23.6 ± 1.8 mV vs methiothepin 5-HT −19.8 ± 2.4 mV; n = 3; not significant; t test paired; t = −2.4; data not shown). In contrast, the selective 5-HT1A antagonist, NAD 299 (1 μm) completely blocked the 5-HT-induced effect (0.3 ± 0.7 pA; n = 12; not significant; t test paired; t = −0.4; Fig. 4A), a response statistically different from initial response to 5-HT (20.9 ± 5.2 pA; n = 12; p < 0.005; ANOVA; F(2,33) = 25.6; Fig. 4Ai). Incubation with NAD 299 alone did not alter baseline current (Control −15.1 ± 4.1 pA vs NAD299 −15.0 ± 4.2 pA; n = 12; not significant; t test paired; t = −0.17; data not shown).
Figure 4.
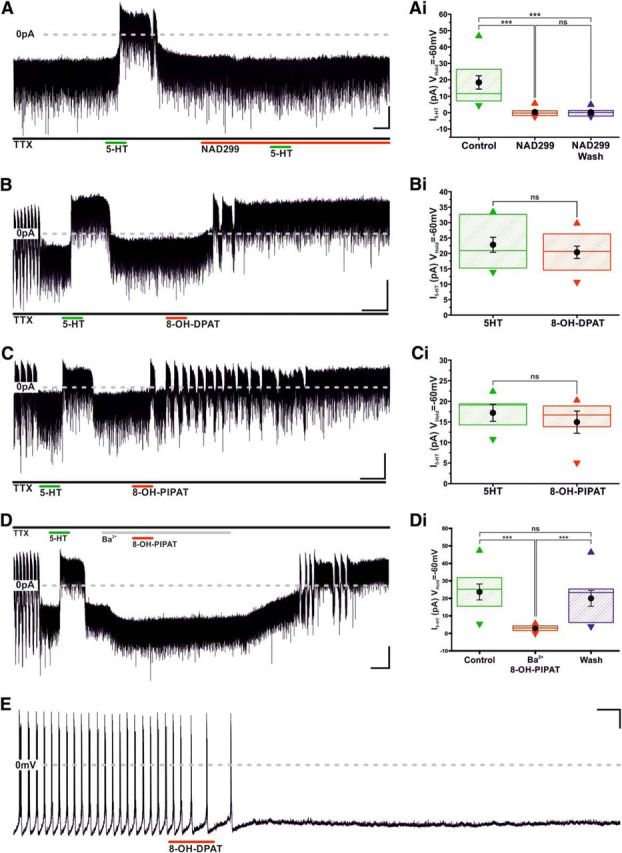
Serotonin inhibits TIDA neurons via activation of the 5-HT 1A receptor. A, Voltage-clamp, oscillating TIDA neuron recorded as in Figure 3A. 5-HT is applied before, during, and after the 5-HT 1A antagonist, NAD299, is present in the bath. Note the complete abolishment of 5-HT response by NAD299. Calibration: 20 pA, 60 s. Ai, Box plot organized as Figure 3Bii, illustrating irreversible I5-HT blockade by NAD299 (n = 12). B, Voltage-clamp trace, oscillating TIDA neuron recorded as in Figure 3A. Sequential application of 5-HT and the 5-HT1A agonist, 8-OH-DPAT; note outward current induced by both compounds. Calibration: 20 pA, 60 s. Bi, 5-HT and 8-OH-DPAT induce statistically indistinguishable outward currents in TIDA neurons. Box plot organized as Figure 3Bii (n = 10). C, Voltage-clamp trace, oscillating TIDA neuron recorded as in Figure 3A. Sequential application of 5-HT and the 5-HT1A agonist, 8-OH-PIPAT; note outward current induced by both compounds. Calibration: 20 pA, 60 s. Ci, 5-HT and 8-OH-PIPAT induce statistically indistinguishable outward currents in TIDA neurons. Box plot organized as Figure 3Bii (n = 5). D, Voltage-clamp, oscillating TIDA neuron recorded as in Figure 3A (note abolishment of oscillation in response to TTX). Following a first exposure to 5-HT, eliciting a reversible outward current, Ba+ (300 μm) is bath applied to the cell. Note the absence of a response to 8-OH PIPAT (compare C) in the presence of Ba+ and how, as a consequence of the enduring nature of its interaction with the 5HT-1A receptor, the drug response is fully uncovered following wash of Ba+ from the recording chamber. Calibration: 20 pA, 60 s. Di, Ba+ significantly and reversibly reduces I8-OH-PIPAT. Box plot organized as Figure 3Bii (n = 9). E, Current-clamp, application of 8-OH-PIPAT induces cessation of oscillation and AP discharge. There is lack of recovery following wash of 8-OH-PIPAT. Calibration 20 mV 60 s. ***p < 0.005. ns, Not significant.
The 5-HT1A-selective agonists, 8-OH-PIPAT and 8-OH-DPAT, were also tested. Application of 8-OH-DPAT induced a 20.4 ± 2.0 pA (n = 10; p < 0.005; t test paired; t = −10.2; Fig. 4B) outward current, corresponding to 90.2 ± 3.7% of the initial response to 5-HT (22.8 ± 2.4 pA; n = 10; not significant; t test; t = 1.9; paired; Fig. 4Bi). 8-OH-PIPAT also induced an outward current that was statistically indistinguishable from the initial response to 5-HT (Initial 5-HT 17.2 ± 2.1 pA vs 8-OH-PIPAT 14.9 ± 2.7 pA; n = 5; not significant; t test paired; t = 1.6; Fig. 4C,Ci). As peak response for 8-OH-PIPAT and 8-OH-DPAT often took time to stabilize and to ensure that it involved opening of GIRK-like channels, 8-OH-PIPAT was coapplied with Ba2+, under which conditions it induced a significantly reduced outward current compared with initial response to 5-HT (Initial 5-HT 23.7 ± 4.6 vs Ba2+ PIPAT 2.8 ± 0.6 pA; n = 9; p < 0.005; ANOVA; F(2,24) = 20.2; Fig. 4D,Di). However, subsequent removal of Ba2+ revealed the 5-HT1A-mediated outward current, which proved indistinguishable from the initial response to 5-HT (20.0 ± 4.6 pA; n = 9; not significant; ANOVA; F(2,24) = 20.2; Fig. 4D,Di). Unlike the effects of 5-HT, which were readily reversible, the response of TIDA neurons to 8-OH-PIPAT and 8-OH-DPAT did not recover in either voltage (n = 24 of 24) or current clamp (n = 6 of 6; Fig. 4E), with the 5-HT1A agonists having been washed from the recording chamber for a minimum of 15 min (Fig. 4E).
5-HT decreases the frequency of spontaneous and miniature IPSCs
Having established postsynaptic effects, we investigated whether 5-HT could modulate TIDA spontaneous IPSCs (sIPSCs). Using high Cl− pipette solution and in the presence of CNQX and AP-V to block glutamatergic events, we analyzed and compared sIPSCs from a 100 s segment before application of 5-HT with sIPSCs from a 100 s segment during response. Application of 5-HT induced a reversible increase in sIPSC interevent interval (IEI; control 460.8 ± 94.2 ms, 5-HT 1935.7 ± 358.9 ms, washout 604.1 ± 111.4 ms; n = 11; p < 0.005; ANOVA; F(2,30) = 25.6; Fig. 5A–D). To determine the effect of 5-HT on miniature IPSCs (mIPSCs), TIDA neurons were recorded as above but in the presence of TTX. Under these conditions, 5-HT did not affect mIPSC amplitude (control 60.6 ± 5.9 pA vs 5-HT 61.2 ± 5.4 pA; n = 5; not significant; t test paired; t = −0.14; Fig. 6A–D) but did increase IEI (Control 563.5 ± 92.6 ms vs 5-HT 1593.3 ± 197.3 ms; n = 5; p < 0.005; t test paired; t = −6.8; Fig. 6A–D). These results suggest that 5-HT reduces GABA release onto TIDA neurons, without affecting postsynaptic responsiveness to GABA.
Figure 5.
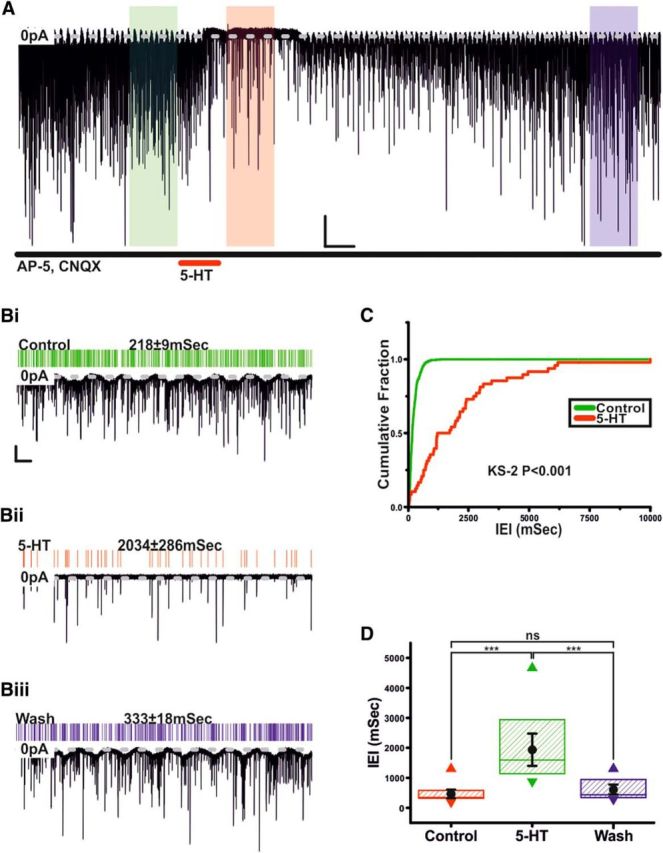
Serotonin inhibits sIPSCs in TIDA neurons. A, Voltage-clamp trace showing sIPSCs from an oscillating TIDA neuron, isolated pharmacologically by administration of CNQX and d-AP5 and recorded with High Cl− intracellular solution. Application of 5-HT reversibly induces an outward current and a decrease in sIPSC frequency. Calibration: 50 pA, 60 s. Bi–Biii, Enlarged views of the 100 s sections boxed in A with corresponding raster plots above. Green represents Control. Red represents 5-HT. Blue represents Wash. Calibration: 50 pA, 5 s. C, Cumulative probability curves of sIPSC IEIs generated from the control and 5-HT 100 s sections boxed in A. The distribution shifts to significantly greater IEIs. D, Application of 5-HT significantly and reversibly increases the IEI of sIPSCs. Box plot organized as Figure 3Bii (n = 12). ***p < 0.005. ns, Not significant.
Figure 6.
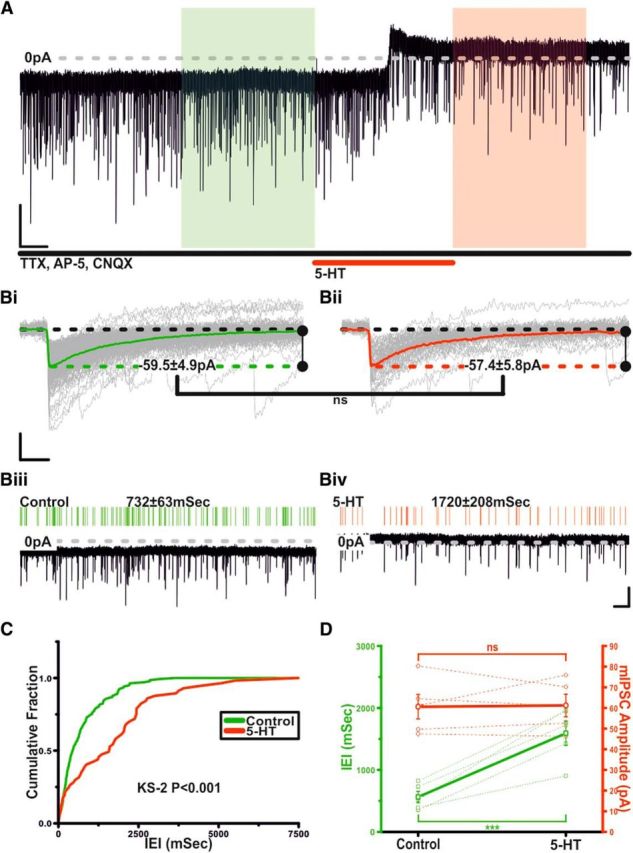
Serotonin inhibits mIPSCs in TIDA neurons via a presynaptic mechanism. A, Voltage-clamp trace showing mIPSCs from an oscillating TIDA neuron, isolated as in Fig. 5A, but with TTX added to abolish AP discharge. Application of 5-HT reversibly induces an outward current and a decrease in mIPSC frequency. Calibration: 40 pA, 20 s. Bi, Bii, Rise aligned mIPSCs (gray) with averaged event superimposed from boxed areas in A. Green represents Control. Red represents 5-HT. 5-HT application fails to alter mean mIPSC amplitude. Calibration: 30 pA, 10 ms. Biii, Biv, Enlarged views of the 100 s sections boxed in A with corresponding raster plots above. Green represents Control. Red represents 5-HT. Calibration: 40 pA, 5 s. C, Cumulative probability curves of mIPSCIEIs generated from the control (green) and 5-HT (red) sections boxed in A. The distribution shifts to significantly greater IEIs (K-S2; p < 0.001). D, 5-HT reversibly and significantly increases mIPSC IEI (green), whereas amplitude remains unchanged (red). Thick lines indicate mean ± SEM. Dashed lines indicate raw data (n = 5). ns, Not significant.
Clinically relevant doses of fluoxetine inhibit TIDA neurons' capacity for repetitive discharge and reduce action potential (AP) amplitude
Like 5-HT, SSRIs, such as fluoxetine (Prozac; Flx), increase plasma levels of prolactin in animals (e.g., Morgan and Herbert, 1978) and humans (e.g., Meltzer et al., 1979). Although SSRIs would, by enhancing ambient 5-HT, modulate TIDA output indirectly via the 5-HT mechanism described above, we sought to determine whether SSRIs could further modulate TIDA activity via a direct, 5-HT-independent mechanism. To this end, we again used our hypothalamic slice preparation, which, being rapidly perfused and having neither 5-HT cell bodies nor a means to stimulate deafferented 5-HT fibers, will have reduced 5-HT reuptake for SSRIs to block. As therapeutic concentrations of Flx in the brain are as high as ∼30 μm (Karson et al., 1993), we tested TIDA neurons with prolonged (60 min) exposure to Flx at a concentration of 25 μm. In contrast to 5-HT, under this challenge, TIDA oscillations endured, undergoing only slight changes in amplitude (Control 29.20 ± 2.39 mV vs Flx 60 min 23.84 ± 1.74 mV; n = 7; p < 0.01; t test paired; t = 5.9; Fig. 7A,B) and frequency (Control 0.059 ± 0.007 Hz vs Flx 60 min 0.072 ± 0.005 Hz; n = 7; p < 0.05; t test paired; t = −3.3; Fig. 7A,B). Intrinsic excitability was, however, severely diminished, as assessed by square-form current injection (20 pA, 500 ms) triggered from a membrane potential of −50 mV. Application of Flx reduced the depolarization-triggered AP number from 9.2 ± 1.2 to 1 (n = 6; p < 0.005; t test paired; t = 7; Fig 7C–E), most prominently after 60 min of drug exposure. AP waveform was also affected, with amplitude of the initial AP being reduced from 96.3 ± 2.3 mV in control to 31.7 ± 3.8 mV at t = 60 min (n = 6; p < 0.005; t test paired; t = 12.6; Fig. 7C,D). Threshold (Control −39.2 ± 0.7 mV; Flx 60 min −33.2 ± 1.3 mV; n = 6; p < 0.05; t test paired; t = −4.3; Fig. 7C) and half-width (Control 1.5 ± 0.1 ms; Flx 60 min 4.6 ± 0.5ms; n = 6; p < 0.005; t test paired; t = −5.6; Fig. 7C) were also affected.
Figure 7.
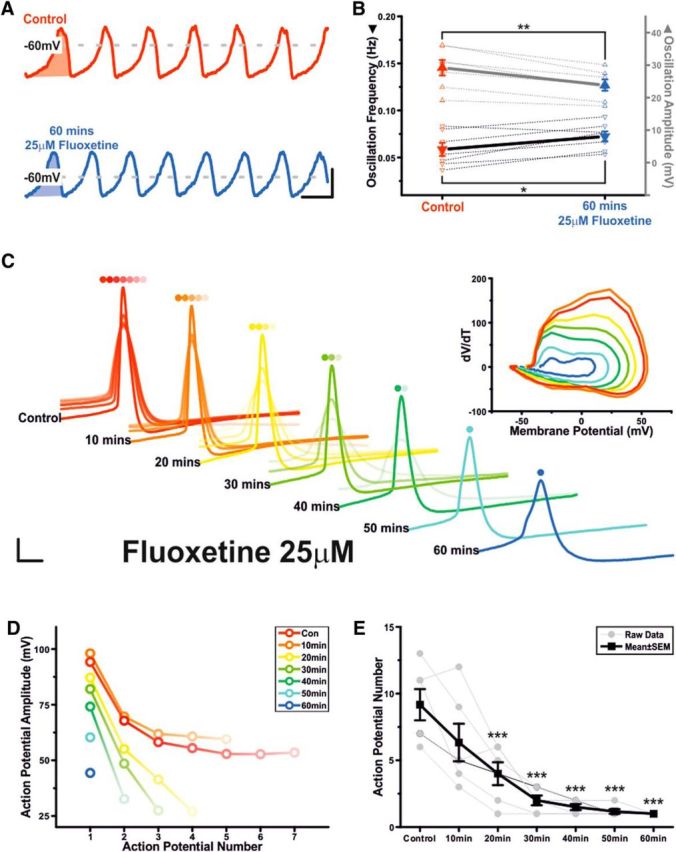
Clinically relevant concentrations of fluoxetine do not abolish the underlying TIDA oscillation but do inhibit TIDA neurons' capacity for repetitive discharge and reduce AP amplitude. A, Current-clamp trace of an oscillating TIDA neuron filtered at 1 Hz in control and following 60 min of 25 μm fluoxetine. Calibration 20 mV 20 s. B, Summary of fluoxetine-induced effects on oscillation amplitude (gray) and frequency (black). Thick lines indicate mean ± SEM. Dashed lines indicate raw data (n = 7). C, Peak aligned APs elicited in response to a square-form–positive current pulse during 60 min application of 25 μm fluoxetine. There is progressive reduction in AP number and amplitude. Calibration: 16 mV, 4 ms. Inset, Orbital plots of the first AP highlight change in threshold, amplitude, and rise and decay time. D, Plot showing AP number and amplitude data from the cell depicted in A. E, Fluoxetine progressively inhibits TIDA neurons' capacity for repetitive discharge. Thick lines indicate mean ± SEM. Thin lines indicate raw data (n = 6). *p < 0.05. **p < 0.01. ***p < 0.005.
With higher doses of Flx (100 μm), voltage-clamp recordings of TIDA neurons revealed a gradual reduction in cycle amplitude and increase in cycle duration, with the oscillation being completely abolished after 28.2 ± 3.1 min of continuous exposure (n = 10/10; Fig. 8A–D). Despite our recording conditions and use of a reduced preparation, there still remained the possibility that, rather than a 5-HT-independent effect, Flx's ability to abolish the TIDA oscillation was the consequence of either, 5-HT accumulation, or the dialysis of intracellular contents associated with prolonged whole-cell recording. To control for 5-HT accumulation, Flx was applied in the presence of NAD299, blocking 5-HT's effect at the 1A receptor (identified above as the mediator of direct electrophysiological actions of 5-HT on TIDA neurons) and presumably the effect of any increase in ambient 5-HT. These voltage-clamp recordings showed that prolonged application of Flx (100 μm) still abolished the TIDA oscillation (n = 7 of 7; 18.4 ± 2.4 min; Fig. 9A). In current clamp, Flx at the lower dose of 25 μm, applied in the presence of NAD299, continued to inhibit intrinsic excitability, reducing depolarization-induced AP number from 7.4 ± 0.5 to 1.5 ± 0.7 (n = 8; p < 0.005; t test paired; t = 6.6; Fig. 9B). We took a number of different approaches to control for potential effects of intracellular dialysis. In the cell-attached recording configuration, thus preserving the neuron's internal milieu, Flx (100 μm) also abolished TIDA AP discharge (n = 7/7; 15.1 ± 2.1 min; Fig. 9C). Second, whole-cell recordings of oscillating TIDA neurons were made after a 30 min incubation with Flx (25 μm), and their intrinsic excitability was compared with TIDA neurons recorded without exposure to Flx. This challenge also saw a reduction in intrinsic excitability with depolarization-induced AP discharge decreasing from 7.0 ± 0.6 APs in control neurons (n = 6) to 1.7 ± 0.5 APs in Flx-incubated neurons (n = 6; p < 0.005; t test unpaired; t = 8; Fig. 9D). Furthermore, when performed in the whole-cell configuration, prolonged (>60 min) control voltage- and current-clamp recordings (i.e., in the absence of any drug application) demonstrated that TIDA neurons maintain their oscillations (n = 9 of 9; Fig. 9E,F) and capacity for repetitive discharge (n = 6 of 6; Fig 9F). Collectively, the results from these experiments support the contention that Flx's ability to disrupt TIDA neurons' network oscillation and discharge pattern is independent of 5-HT's effects at the 1A receptor and the cellular dialysis associated with whole-cell recording.
Figure 8.

Supraclinical concentrations of fluoxetine abolish the TIDA oscillation. A, Voltage-clamp trace of an oscillating TIDA neuron. Application of 100 μm fluoxetine gradually abolishes TIDA oscillation. Calibration: 20 pA, 2 min. B, Enlarged color-coded 100 s boxed sections in D filtered at 1 Hz. There is gradual reduction in oscillation amplitude and frequency. Calibration: 10 pA, 5 s. C, Average oscillation cycle amplitude and duration from the color-coded data depicted in B. D, Spectral density plots of the color-coded data depicted in B. There is successively reduced oscillation strength and peak frequency.
Figure 9.
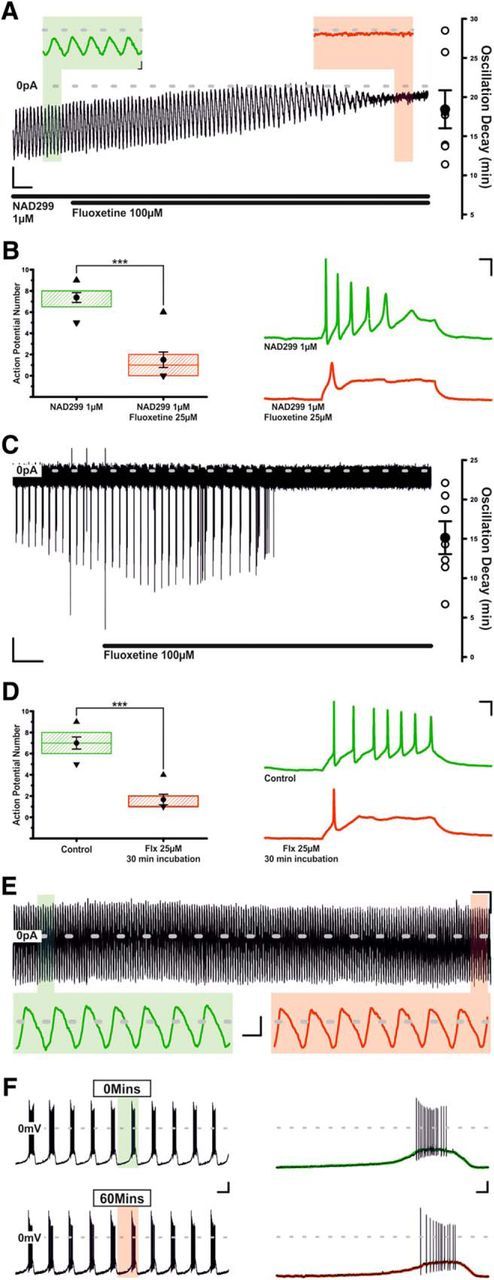
Fluoxetine's effects upon TIDA oscillation and repetitive discharge are independent of 5-HT1A receptor activation and intracellular dialysis. A, Voltage-clamp trace of an oscillating TIDA neuron filtered at 1 Hz. Application of 100 μm fluoxetine continues to abolish TIDA oscillation despite the continuous presence of the 5-HT1A receptor antagonist, NAD299. Calibration: 10 pA, 60 s. Inset, Enlarged areas of A denoted by color. Calibration: Inset, 10 pA, 2 s. B, Fluoxetine continues to inhibit TIDA neuron's capacity for repetitive discharge, reducing the number of APs in response to depolarizing current injection. Box plot organized as Figure 3Bii (n = 8). Right, Current-clamp traces showing the voltage response and AP discharge elicited by injection of a fixed-amplitude positive current pulse during control (green) and during application of fluoxetine (25 μm). Calibration: 20 mV, 50 ms. C, Cell-attached recording of an oscillating TIDA neuron. Fluoxetine (100 μm) continues to abolish the TIDA oscillation when intracellular contents remain undisturbed. Calibration: 10 pA, 2 min. D, Oscillating TIDA neurons patch clamped in whole-cell configuration following preincubation (30 min) with 25 μm fluoxetine (red) also exhibit reduced capacity for repetitive discharge, compared with TIDA neurons that have not been exposed to fluoxetine (green). Box plot organized as Figure 3Bii (n = 6). Right, Traces organized as in B. Calibration: 20 mV, 50 ms. E, Voltage-clamp trace of oscillating TIDA neuron. The 60 min of prolonged recording and intracellular dialysis fails to abolish the TIDA oscillation. Calibration: 20 pA, 2 min. Inset, Expanded areas of E denoted by color, filtered at 1 Hz. Calibration: Inset, 20 pA, 10 s. F, Current-clamp recording of an oscillating TIDA neuron at t = 0 and t = 60 min. The 60 min of prolonged recording fails to abolish TIDA neuron's capacity for repetitive discharge. Calibration: 20 mV, 10 s. Inset, Enlarged areas of F overlaid with trace filtered at 1 HZ according to color. Calibration: Inset, 20 mV, 1 s. ***p < 0.005.
SSRIs sertraline and fenfluramine also influence TIDA electrical properties
Hyperprolactinemia has been reported with the clinical use of other SSRIs, including sertraline (Petit et al., 2003) and fenfluramine (Siever et al., 1984). As with Flx, prolonged application of sertraline (50 μm) induced gradual reductions in AP amplitude, capacity for repetitive discharge, and oscillatory activity (Fig. 10A,B). AP number was reduced from 7.75 ± 0.75 to 1.75 ± 0.75 per test pulse (n = 4; p < 0.05; t test paired; t = 8.5; AP number fell to 1 in 3 of 4 neurons tested) and amplitude of the initial AP was reduced from 93.7 ± 2.1 mV to 41.8 ± 10.1 mV (n = 4; p < 0.05; t test paired; t = 4.4). Oscillatory activity was abolished in 4 of 4 neurons after 39.6 ± 6.1 min (Fig. 10B). Fenfluramine (20 μm), in contrast, abolished oscillation activity more rapidly than the other SSRIs tested (11.6 ± 2.6 min; n = 5; p < 0.05; ANOVA; F(2,11) = 11.0; Fig. 10C,D) but following 60 min of exposure did not significantly reduce either AP number (5.3 ± 0.7 in control, 3.3 ± 1.5 in fenfluramine; n = 3; not significant; t test paired, t = 1.3) or the amplitude of the initial AP (99.3 ± 3.8 mV in control, 87.0 ± 7.2 mV in fenfluramine; n = 3; not significant; t test paired; t = 3).
Figure 10.
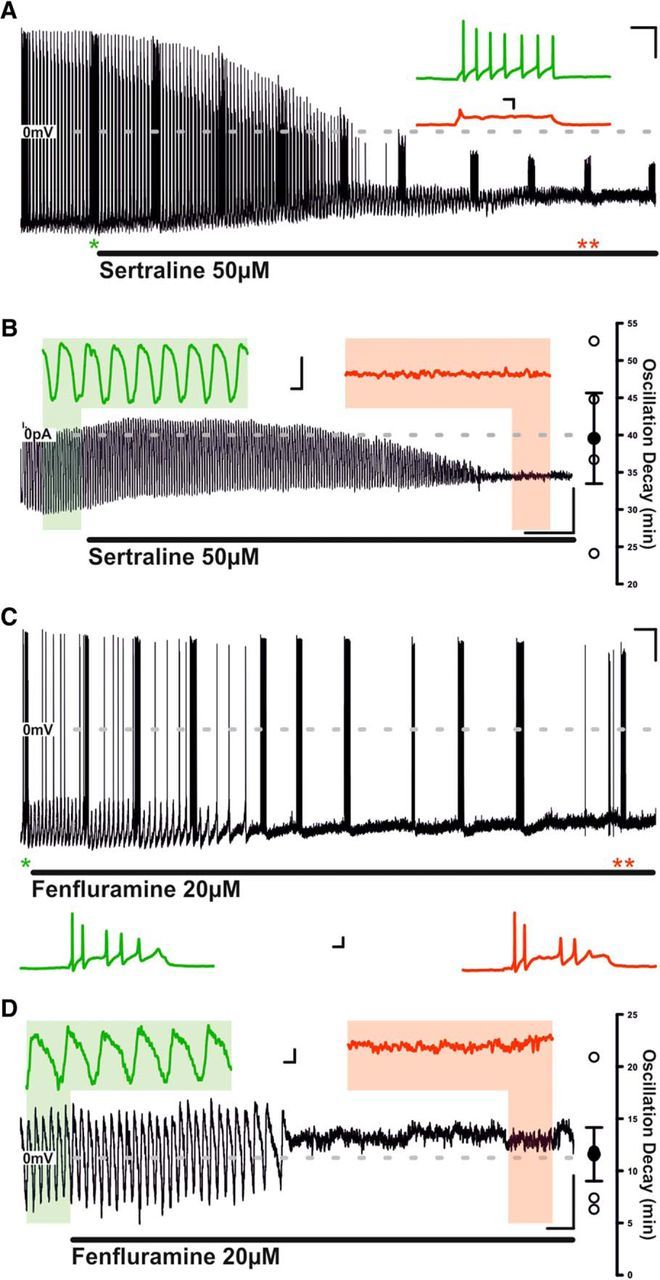
Sertraline abolishes both the TIDA oscillation and capacity for repetitive discharge, whereas fenlfuramine abolishes the TIDA oscillation but leaves the capacity for repetitive discharge intact. A, Current-clamp recording of an oscillating TIDA neuron. The response of the cell to injection of square-form–positive current pulses of constant size is tested at regular intervals (e.g., at asterisks). Prolonged bath application of sertraline results in gradual abolishment of oscillatory activity and reduction in AP amplitude and number, as well as of spontaneous discharge. Calibration: 20 mV, 2 min. Inset, Expanded sections from A denoted by asterisks. Calibration: Inset, 20 mV, 50 ms. B, Voltage-clamp recording of a TIDA neuron filtered at 1 Hz. Prolonged sertraline exposure results in a gradual amplitude reduction and eventual abolishment of oscillation. Calibration: 20 pA, 4 min. Inset, Expanded areas of B according to color. Calibration: Inset, 20 pA, 5 s. C, Current-clamp recording of an oscillating TIDA neuron. The response of the cell to injection of square-form–positive current pulses of constant size is tested at regular intervals (e.g., at asterisks). Prolonged bath application of fenfluramine results in gradual abolishment of oscillatory activity but fails to reduce capacity for repetitive discharge. Calibration: 20 mV, 2 min. Inset, Expanded sections from C denoted by asterisks. Calibration: Inset, 20 mV, 50 ms. D, Voltage-clamp recording of a TIDA neuron filtered at 1 Hz. Prolonged fenfluramine exposure results in a gradual amplitude reduction and eventual abolishment of oscillation. Calibration: 5 pA, 1 min. Inset, Expanded areas of D according to color. Calibration: Inset, 2 pA, 4 s. **p < 0.01.
Discussion
The ability of 5-HT and SSRIs to stimulate prolactin release was first described >40 years ago (Lu and Meites, 1973; Emiliano and Fudge, 2004). The hyperprolactinemia induced by many SSRIs has been linked to the sexual side effects associated with these drugs (Urban and Veldhuis, 1991; Amsterdam et al., 1997; Ekselius et al., 1997), which are common and sufficiently debilitating to cause drug withdrawal in many patients (Montejo et al., 2001; van Geffen et al., 2007). Yet, how 5-HT and SSRIs stimulate prolactin release is not well understood. Here we show that 5-HT activates a K+ conductance to directly hyperpolarize and inhibit the firing of rat TIDA neurons in vitro. We further show that fluoxetine and sertraline also decrease TIDA excitability, but that the cellular mechanisms, as revealed by recordings in a deafferented slice preparation lacking serotonergic neurons and in the continued presence of the 5-HT1A antagonist NAD299, are likely different from those through which 5-HT acts.
The outward current activated by 5-HT was mimicked by application of 8-OH-PIPAT and 8-OH-DPAT and was blocked by prior application of NAD299, indicating that it is mediated by 5-HT1A receptors. I5-HT displays characteristics of a GIRK current with its hyperpolarized reversal potential, inward rectification, sensitivity to [Cs+]i and [K+]o, and inhibition by Ba2+, the GIRK blocker, TQ, and the G-protein uncoupling agent, NEM. A GIRK-like current modulated by opioid and GABAB agonists has been described in TIDA neurons (Loose et al., 1990), and GIRK channel subunits are strongly expressed in the arcuate nucleus (Saenz del Burgo et al., 2008). Moreover, coupling of the 5-HT1A receptor to GIRK channels has previously been reported in hippocampal (Lüscher et al., 1997) and raphe (Penington et al., 1993) neurons, and mRNAs encoding these proteins are frequently coexpressed (Saenz del Burgo et al., 2008).
We thus propose that 5-HT-induced hyperpolarization, underpinned by a 5-HT1A receptor-mediated opening of GIRK-like channels, abolishes TIDA neuron discharge, which in turn reduces dopamine release in the portal capillaries. As such, the inhibitory D2R “brake” on lactotrophs is relieved, resulting in the secretion of prolactin into the general circulation. This scenario is in agreement with reports that 5-HT agonists, at concentrations that stimulate prolactin release, decrease the levels of dopamine in the TIDA-pituitary axis (Pilotte and Porter, 1981) and fail to increase prolactin when dopamine synthesis has been blocked (Quattrone et al., 1979). There is also evidence that 5-HT stimulates the release of a prolactin-releasing factor (Clemens et al., 1978; Pilotte and Porter, 1981), possibilities that are not mutually exclusive.
The persistence of the 5-HT-induced hyperpolarization in the presence of TTX and the presence of 5-HT terminals on TIDA cell bodies (confirming Bosler et al., 1984; Kiss and Halász, 1986) demonstrates direct, postsynaptic actions of 5-HT on TIDA cells. We also show, however, a decrease in presynaptic inhibitory inputs in response to 5-HT. This finding is perhaps best explained by anatomical evidence that TIDA neurons are GABAergic (Everitt et al., 1984) and may auto-innervate (Léránth et al., 1985), and also indicates the existence of 5-HT receptors on the axon terminal as well as the somatodendritic compartment of TIDA neurons. Thus, the loss of IPSCs may primarily reflect abolishment of TIDA discharge, further attesting to the powerful inhibition of the system by 5-HT. Modulation of non–TIDA-derived GABAergic input by 5-HT can, however, not be excluded.
Fluoxetine and other SSRIs were developed based on the hypothesis that hypoactivity of central serotonergic transmission underlies depression (see Wong et al., 1995). Given Flx's proven ability to increase ambient 5-HT, its prolactin-releasing effects in the whole animal would then be expected to depend on serotonin's ability to inhibit TIDA output. Accordingly, in a situation with little or no 5-HT release or accumulation (such as in our deafferented, rapidly perfused slice), or in the continued presence of the 5-HT1A antagonist NAD299, SSRIs would fail to inhibit TIDA neurons. However, this was clearly not the case, as during exposure to clinically relevant concentrations (25 μm) (Karson et al., 1993) of Flx, the intrinsic excitability of TIDA neurons was powerfully blunted with decreased AP amplitude and reduced capacity for repetitive discharge. Notably, however, the oscillation as such persisted, in sharp contrast to its abolishment in the presence of 5-HT. At 100 μm Flx, the oscillation disappeared, but with a strikingly different temporal profile to 5-HT, a response preserved despite 5-HT1A receptor blockade. Furthermore, similar effects were seen with sertraline and fenfluramine.
How can these apparently 5-HT-independent effects be explained? A solution can be found in the accumulating evidence for off-target (i.e., nonserotonergic) actions of SSRIs documented in the literature (see Bianchi, 2008). Studies in other systems have revealed that Flx can directly modulate particular membrane currents, including those carried by K+ (e.g., Tytgat et al., 1997), Ca2+ (e.g., Deák et al., 2000), and Cl− (e.g., Steele et al., 2005). Of particular relevance to the present results, Flx has been shown to attenuate both persistent (INaP) and transient sodium currents (INaT), inhibiting repetitive discharge and oscillatory activity in olfactory bulb neurons with a time course very similar to that shown here (Igelström and Heyward, 2012). In TIDA neurons, INaP is an important contributor to regenerative firing, as evidenced by the abolishment of oscillatory activity by the INaP blocker TTX, independent of this compound's effects on presynaptic input (Lyons et al., 2010; and unpublished observations). Thus, the decreased intrinsic excitability observed in TIDA neurons after application of Flx and sertraline may be due to inhibitory actions of these drugs on INaP. (It may be noted, in this context, that fenfluramine, which in addition to suppressing reuptake also stimulates nonexocytotic release of 5-HT [Kannengiesser et al., 1976], exerted an effect on a timescale more akin to 5-HT).
Thus, we propose that SSRIs promote prolactin release in vivo through two distinct actions on the TIDA network, both resulting in decreased DA output. First, by increasing 5-HT tone, SSRIs promote 5-HT1A/GIRK-mediated hyperpolarization. Second, in a 5-HT-independent manner, SSRIs reduce TIDA intrinsic excitability by inhibiting oscillatory activity and AP discharge, via reductions in INaT and INaP, respectively. Combined, these actions would result in a shutdown of the TIDA system, causing an increase in prolactin secretion. The present data thus highlight a novel mechanism for the hyperprolactinemia and sexual side effects of SSRIs. It should be noted that acute drug applications to slice preparations of TIDA from the young male rat offers far from a perfect model of the clinical situation where SSRIs are prescribed for long periods to a large and heterogeneous population for several diagnoses. The ability of these compounds to elevate circulating prolactin is also less robust when assessed in human subjects than in inbred experimental rodents. Yet, hyperprolactinemia, as well as adverse reproductive effects, are notably common when rigorously assessed in SSRI-treated patients (Montejo-González et al., 1997; Clayton et al., 2002). Thus, these results may be of importance for the development of antidepressant therapies with minimized reproductive side effects and consequently enhanced patient compliance.
Footnotes
This work was supported by European Research Council ENDOSWITCH 261286, Swedish Research Council 2014-3906, Strategic Research Programme in Diabetes at Karolinska Institutet, and Novo Nordisk Fonden to C.B. D.J.L. was supported by Wenner-Gren Foundations postdoctoral fellowship. The authors thank Dr. April Johnston for advice on serotonin pharmacology and Dr. Pradeep Atluri for discussions on clinical manifestations of SSRI treatment.
The authors declare no competing financial interests.
References
- American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision) Am J Psychiatry. 2000;157(Suppl):1–45. doi: 10.1176/ajp.157.1.1. [DOI] [PubMed] [Google Scholar]
- Amsterdam JD, Garcia-España F, Goodman D, Hooper M, Hornig-Rohan M. Breast enlargement during chronic antidepressant therapy. J Affect Disord. 1997;46:151–156. doi: 10.1016/S0165-0327(97)00086-4. [DOI] [PubMed] [Google Scholar]
- Bianchi MT. Non-serotonin anti-depressant actions: direct ion channel modulation by SSRIs and the concept of single agent poly-pharmacy. Med Hypotheses. 2008;70:951–956. doi: 10.1016/j.mehy.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Birge CA, Jacobs LS, Hammer CT, Daughaday WH. Catecholamine inhibition of prolactin secretion by isolated rat adenohypophyses. Endocrinology. 1970;86:120–130. doi: 10.1210/endo-86-1-120. [DOI] [PubMed] [Google Scholar]
- Bole-Feysot C, Goffin V, Edery M, Binart N, Kelly PA. Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr Rev. 1998;19:225–268. doi: 10.1210/edrv.19.3.0334. [DOI] [PubMed] [Google Scholar]
- Bosler O, Joh TH, Beaudet A. Ultrastructural relationships between serotonin and dopamine neurons in the rat arcuate nucleus and medial zona incerta: a combined radioautographic and immunocytochemical study. Neurosci Lett. 1984;48:279–285. doi: 10.1016/0304-3940(84)90051-X. [DOI] [PubMed] [Google Scholar]
- Briffaud V, Williams P, Courty J, Broberger C. Excitation of tuberoinfundibular dopamine neurons by oxytocin: crosstalk in the control of lactation. J Neurosci. 2015;35:4229–4237. doi: 10.1523/JNEUROSCI.2633-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buvat J, Lemaire A, Buvat-Herbaut M, Fourlinnie JC, Racadot A, Fossati P. Hyperprolactinemia and sexual function in men. Horm Res. 1985;22:196–203. doi: 10.1159/000180094. [DOI] [PubMed] [Google Scholar]
- Clayton AH, Pradko JF, Croft HA, Montano CB, Leadbetter RA, Bolden-Watson C, Bass KI, Donahue RM, Jamerson BD, Metz A. Prevalence of sexual dysfunction among newer antidepressants. J Clin Psychiatry. 2002;63:357–366. doi: 10.4088/JCP.v63n0414. [DOI] [PubMed] [Google Scholar]
- Clemens JA, Roush ME, Fuller RW. Evidence that serotonin neurons stimulate secretion of prolaction releasing factor. Life Sci. 1978;22:2209–2213. doi: 10.1016/0024-3205(78)90573-8. [DOI] [PubMed] [Google Scholar]
- Deák F, Lasztóczi B, Pacher P, Petheö GL, Valéria Kecskeméti, Spät A. Inhibition of voltage-gated calcium channels by fluoxetine in rat hippocampal pyramidal cells. Neuropharmacology. 2000;39:1029–1036. doi: 10.1016/S0028-3908(99)00206-3. [DOI] [PubMed] [Google Scholar]
- Ekselius L, von Knorring L, Eberhard G. A double-blind multicenter trial comparing sertraline and citalopram in patients with major depression treated in general practice. Int Clin Psychopharmacol. 1997;12:323–331. doi: 10.1097/00004850-199711000-00005. [DOI] [PubMed] [Google Scholar]
- Emiliano AB, Fudge JL. From galactorrhea to osteopenia: rethinking serotonin-prolactin interactions. Neuropsychopharmacology. 2004;29:833–846. doi: 10.1038/sj.npp.1300412. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Hökfelt T, Wu JY, Goldstein M. Coexistence of tyrosine hydroxylase-like and gamma-aminobutyric acid-like immunoreactivities in neurons of the arcuate nucleus. Neuroendocrinology. 1984;39:189–191. doi: 10.1159/000123977. [DOI] [PubMed] [Google Scholar]
- Foo KS, Hellysaz A, Broberger C. Expression and colocalization patterns of calbindin-D28K, calretinin and parvalbumin in the rat hypothalamic arcuate nucleus. J Chem Neuroanat. 2014;61–62:20–32. doi: 10.1016/j.jchemneu.2014.06.008. [DOI] [PubMed] [Google Scholar]
- Gregorian RS, Golden KA, Bahce A, Goodman C, Kwong WJ, Khan ZM. Antidepressant-induced sexual dysfunction. Ann Pharmacother. 2002;36:1577–1589. doi: 10.1345/aph.1A195. [DOI] [PubMed] [Google Scholar]
- Igelström KM, Heyward PM. The antidepressant drug fluoxetine inhibits persistent sodium currents and seizure-like events. Epilepsy Res. 2012;101:174–181. doi: 10.1016/j.eplepsyres.2012.03.019. [DOI] [PubMed] [Google Scholar]
- Jin W, Lu Z. Synthesis of a stable form of tertiapin: a high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry. 1999;38:14286–14293. doi: 10.1021/bi991205r. [DOI] [PubMed] [Google Scholar]
- Jørgensen H, Knigge U, Warberg J. Involvement of 5-HT1, 5-HT2, and 5-HT3 receptors in the mediation of the prolactin response to serotonin and 5-hydroxytryptophan. Neuroendocrinology. 1992;55:336–343. doi: 10.1159/000126134. [DOI] [PubMed] [Google Scholar]
- Jørgensen H, Kjaer A, Warberg J, Knigge U. Differential effect of serotonin 5-HT(1A) receptor antagonists on the secretion of corticotropin and prolactin. Neuroendocrinology. 2001;73:322–333. doi: 10.1159/000054649. [DOI] [PubMed] [Google Scholar]
- Kamberi IA, Mical RS, Porter JC. Effect of anterior pituitary perfusion and intraventricular injection of catecholamines on prolactin release. Endocrinology. 1971;88:1012–1020. doi: 10.1210/endo-88-4-1012. [DOI] [PubMed] [Google Scholar]
- Kannengiesser MH, Hunt PF, Raynaud JP. Comparative action of fenfluramine on the uptake and release of serotonin and dopamine. Eur J Pharmacol. 1976;35:35–43. doi: 10.1016/0014-2999(76)90298-3. [DOI] [PubMed] [Google Scholar]
- Karson CN, Newton JE, Livingston R, Jolly JB, Cooper TB, Sprigg J, Komoroski RA. Human brain fluoxetine concentrations. J Neuropsychiatry Clin Neurosci. 1993;5:322–329. doi: 10.1176/jnp.5.3.322. [DOI] [PubMed] [Google Scholar]
- Kiss J, Halász B. Synaptic connections between serotoninergic axon terminals and tyrosine hydroxylase-immunoreactive neurons in the arcuate nucleus of the rat hypothalamus: a combination of electron microscopic autoradiography and immunocytochemistry. Brain Res. 1986;364:284–294. doi: 10.1016/0006-8993(86)90841-3. [DOI] [PubMed] [Google Scholar]
- Léránth C, Sakamoto H, MacLusky NJ, Shanabrough M, Naftolin F. Intrinsic tyrosine hydroxylase (TH) immunoreactive axons synapse with TH immunopositive neurons in the rat arcuate nucleus. Brain Res. 1985;331:371–375. doi: 10.1016/0006-8993(85)91566-5. [DOI] [PubMed] [Google Scholar]
- Liang SL, Pan JT. An endogenous serotonergic rhythm acting on 5-HT(2A) receptors may be involved in the diurnal changes in tuberoinfundibular dopaminergic neuronal activity and prolactin secretion in female rats. Neuroendocrinology. 2000;72:11–19. doi: 10.1159/000054566. [DOI] [PubMed] [Google Scholar]
- Loose MD, Ronnekleiv OK, Kelly MJ. Membrane properties and response to opioids of identified dopamine neurons in the guinea pig hypothalamus. J Neurosci. 1990;10:3627–3634. doi: 10.1523/JNEUROSCI.10-11-03627.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KH, Meites J. Effects of serotonin precursors and melatonin on serum prolactin release in rats. Endocrinology. 1973;93:152–155. doi: 10.1210/endo-93-1-152. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron. 1997;19:687–695. doi: 10.1016/S0896-6273(00)80381-5. [DOI] [PubMed] [Google Scholar]
- Lyons DJ, Broberger C. TIDAL WAVES: network mechanisms in the neuroendocrine control of prolactin release. Front Neuroendocrinol. 2014;35:420–438. doi: 10.1016/j.yfrne.2014.02.001. [DOI] [PubMed] [Google Scholar]
- Lyons DJ, Horjales-Araujo E, Broberger C. Synchronized network oscillations in rat tuberoinfundibular dopamine neurons: switch to tonic discharge by thyrotropin-releasing hormone. Neuron. 2010;65:217–229. doi: 10.1016/j.neuron.2009.12.024. [DOI] [PubMed] [Google Scholar]
- Lyons DJ, Hellysaz A, Broberger C. Prolactin regulates tuberoinfundibular dopamine neuron discharge pattern: novel feedback control mechanisms in the lactotrophic axis. J Neurosci. 2012;32:8074–8083. doi: 10.1523/JNEUROSCI.0129-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhusoodanan S, Parida S, Jimenez C. Hyperprolactinemia associated with psychotropics: a review. Hum Psychopharmacol. 2010;25:281–297. doi: 10.1002/hup.1116. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, Young M, Metz J, Fang VS, Schyve PM, Arora RC. Extrapyramidal side effects and increased serum prolactin following fluoxetine, a new antidepressant. J Neural Transm. 1979;45:165–175. doi: 10.1007/BF01250091. [DOI] [PubMed] [Google Scholar]
- Montejo AL, Llorca G, Izquierdo JA, Rico-Villademoros F. Incidence of sexual dysfunction associated with antidepressant agents: a prospective multicenter study of 1022 outpatients. Spanish Working Group for the Study of Psychotropic-Related Sexual Dysfunction. J Clin Psychiatry. 2001;62(Suppl 3):10–21. [PubMed] [Google Scholar]
- Montejo-González AL, Llorca G, Izquierdo JA, Ledesma A, Bousoño M, Calcedo A, Carrasco JL, Ciudad J, Daniel E, De la Gandara J, Derecho J, Franco M, Gomez MJ, Macias JA, Martin T, Perez V, Sanchez JM, Sanchez S, Vicens E. SSRI-induced sexual dysfunction: fluoxetine, paroxetine, sertraline, and fluvoxamine in a prospective, multicenter, and descriptive clinical study of 344 patients. J Sex Marital Ther. 1997;23:176–194. doi: 10.1080/00926239708403923. [DOI] [PubMed] [Google Scholar]
- Morgan WW, Herbert DC. Elevation of serum prolactin levels after the inhibition of serotonin uptake. Endocrinology. 1978;103:1016–1022. doi: 10.1210/endo-103-4-1016. [DOI] [PubMed] [Google Scholar]
- Mulroney SE, Skudlarek C, Shemer A, Lumpkin MD, Kellar KJ. Tandospirone stimulates prolactin secretion in the rat by an action at serotonin-1A receptors. J Pharmacol Exp Ther. 1994;268:862–867. [PubMed] [Google Scholar]
- Olié JP, Elomari F, Spadone C, Lépine JP. Antidepressants consumption in the global population in France. L'Encéphale. 2002;28:411–417. [PubMed] [Google Scholar]
- Penington NJ, Kelly JS, Fox AP. Whole-cell recordings of inwardly rectifying K+ currents activated by 5-HT1A receptors on dorsal raphe neurones of the adult rat. J Physiol. 1993;469:387–405. doi: 10.1113/jphysiol.1993.sp019819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit A, Piednoir D, Germain ML, Trenque T. Drug-induced hyperprolactinemia: a case-non-case study from the national pharmacovigilance database. Thérapie. 2003;58:159–163. doi: 10.2515/therapie:2003023. [DOI] [PubMed] [Google Scholar]
- Pilotte NS, Porter JC. Dopamine in hypophysial portal plasma and prolactin in systemic plasma of rats treated with 5-hydroxytryptamine. Endocrinology. 1981;108:2137–2141. doi: 10.1210/endo-108-6-2137. [DOI] [PubMed] [Google Scholar]
- Quattrone A, Schettini G, Di Renzo GF, Preziosi P. Pharmacological evidence of an interaction between serotonergic and dopaminergic neurons in the control of prolactin secretion in male rats. Arch Int Pharmacodyn Ther. 1979;238:42–49. [PubMed] [Google Scholar]
- Saenz del Burgo L, Cortes R, Mengod G, Zarate J, Echevarria E, Salles J. Distribution and neurochemical characterization of neurons expressing GIRK channels in the rat brain. J Comp Neurol. 2008;510:581–606. doi: 10.1002/cne.21810. [DOI] [PubMed] [Google Scholar]
- Safarinejad MR. Evaluation of endocrine profile and hypothalamic-pituitary-testis axis in selective serotonin reuptake inhibitor-induced male sexual dysfunction. J Clin Psychopharmacol. 2008;28:418–423. doi: 10.1097/JCP.0b013e31817e6f80. [DOI] [PubMed] [Google Scholar]
- Schlechte J, Sherman B, Halmi N, VanGilder J, Chapler F, Dolan K, Granner D, Duello T, Harris C. Prolactin-secreting pituitary tumors in amenorrheic women: a comprehensive study. Endocr Rev. 1980;1:295–308. doi: 10.1210/edrv-1-3-295. [DOI] [PubMed] [Google Scholar]
- Siever LJ, Murphy DL, Slater S, de la Vega E, Lipper S. Plasma prolactin changes following fenfluramine in depressed patients compared to controls: an evaluation of central serotonergic responsivity in depression. Life Sci. 1984;34:1029–1039. doi: 10.1016/0024-3205(84)90016-X. [DOI] [PubMed] [Google Scholar]
- Steele EC, Jr, Chen X, MacLeish PR. Fluoxetine inhibits calcium-activated currents of salamander rod photoreceptor somata and presynaptic terminals via modulation of intracellular calcium dynamics. Mol Vis. 2005;11:1200–1210. [PubMed] [Google Scholar]
- Tytgat J, Maertens C, Daenens P. Effect of fluoxetine on a neuronal, voltage-dependent potassium channel (Kv1.1) Br J Pharmacol. 1997;122:1417–1424. doi: 10.1038/sj.bjp.0701545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban RJ, Veldhuis JD. A selective serotonin reuptake inhibitor, fluoxetine hydrochloride, modulates the pulsatile release of prolactin in postmenopausal women. Am J Obstet Gynecol. 1991;164:147–152. doi: 10.1016/0002-9378(91)90645-8. [DOI] [PubMed] [Google Scholar]
- van Geffen EC, van der Wal SW, van Hulten R, de Groot MC, Egberts AC, Heerdink ER. Evaluation of patients' experiences with antidepressants reported by means of a medicine reporting system. Eur J Clin Pharmacol. 2007;63:1193–1199. doi: 10.1007/s00228-007-0375-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S, Aboyans V, Abraham J, Ackerman I, Aggarwal R, Ahn SY, Ali MK, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2163–2196. doi: 10.1016/S0140-6736(12)61729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DT, Bymaster FP, Engleman EA. Prozac (fluoxetine, Lilly 110140), the first selective serotonin uptake inhibitor and an antidepressant drug: twenty years since its first publication. Life Sci. 1995;57:411–441. doi: 10.1016/0024-3205(95)00209-O. [DOI] [PubMed] [Google Scholar]
- World Health Organisation. The global burden of disease: 2004. Geneva: World Health Organisation; 2008. [Google Scholar]
- Zhang X, van den Pol AN. Dopamine/tyrosine hydroxylase neurons of the hypothalamic arcuate nucleus release GABA, communicate with dopaminergic and other arcuate neurons, and respond to dynorphin, Met-enkephalin, and oxytocin. J Neurosci. 2015;35:14966–14982. doi: 10.1523/JNEUROSCI.0293-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]