

Abstract
Ciliopathies are an emerging class of devastating disorders with pleiotropic symptoms affecting both the central and peripheral systems and commonly associated with hydrocephalus. Even though ciliary components and three master transcriptional regulators have been identified, little is known about the signaling molecules involved. We previously identified a novel gene, Unc51-like-kinase 4 (ULK4), as a risk factor of neurodevelopmental disorders. Here we took multidisciplinary approaches and uncovered essential roles of Ulk4 in ciliogenesis. We show that Ulk4 is predominantly expressed in the ventricular system, and Ulk4tm1a/tm1a ependymal cells display reduced/disorganized cilia with abnormal axonemes. Ulk4tm1a/tm1a mice exhibit dysfunctional subcommissural organs, obstructive aqueducts, and impaired CSF flow. Mechanistically, we performed whole-genome RNA sequencing and discovered that Ulk4 regulates the Foxj1 pathway specifically and an array of other ciliogenesis molecules. This is the first evidence demonstrating that ULK4 plays a vital role in ciliogenesis and that deficiency of ULK4 can cause hydrocephalus and ciliopathy-related disorders.
SIGNIFICANCE STATEMENT Ciliopathies are an emerging class of devastating disorders with pleiotropic symptoms affecting both the central and peripheral systems. Ciliopathies are commonly associated with hydrocephalus, and Unc51-like-kinase 4 (Ulk4) has been identified as one of 12 genes causing hydrocephalus in mutants. Here we uncover an essential role of Ulk4 in ciliogenesis. Ulk4 is predominantly expressed in the ventricles, and mutant ependymal cells display reduced/disorganized/nonfunctional motile cilia with abnormal axonemes and impaired CSF flow. Ulk4 modulates expression of the master regulator of ciliogenesis, Foxj1, and other ciliogenesis molecules. This is the first report demonstrating a vital role of Ulk4 in ciliogenesis. ULK4 deficiency may be implicated in human hydrocephalus and other ciliopathy-related disorders.
Keywords: ciliogenesis, CSF, hydrocephalus, hypomorph mouse, neurodevelopmental disorder, Ulk4
Introduction
Ciliopathies, which are associated with neurodevelopmental disorders of pleiotropic clinical symptoms, include Joubert syndrome, Bardet–Biedl syndrome, Meckel–Gruber syndrome, oral–facial–digital syndrome type 1, and nephronophthisis, all of which are commonly associated with hydrocephalus (Lee and Gleeson, 2011). Handedness, linked to dyslexia and schizophrenia, is also shown to involve ciliopathies (Brandler and Paracchini, 2014). MIPT3, an interactive protein of disrupted-in-schizophrenia 1 (DISC1), which is truncated in a large Scottish schizophrenic family (Millar et al., 2000), functions synergistically with the Bardet–Biedl syndrome protein Bbs4 and plays a critical role in assembling intraflagellar transport particle complexes (Li et al., 2008). Remarkably, when 41 candidate genes associated with schizophrenia, bipolar affective disorder, autism spectrum disorder, and intellectual disability are investigated, 23 are found to regulate cilium length in cultured cells (Marley and von Zastrow, 2012).
We recently demonstrated that the Unc51-like-kinase 4 (ULK4) gene is a rare risk factor for schizophrenia, autism, and bipolar disorder (Lang et al., 2014). Our unpublished data show that ULK4 is deleted in some patients with heterogeneous clinical features, including developmental delay, language delay, and severe intellectual disability (Liu et al., 2016, in press). Depleted ULK4 expression in neuroblastoma cells disrupts microtubular composition, compromises neuritogenesis and cell motility, and modulates multiple signaling pathways, which are associated with schizophrenia. Ulk4 is developmentally regulated by morphogens, and there is a switch in Ulk4 isoforms during mouse brain formation and neuronal maturation. Targeted Ulk4 deletion compromises corpus callosum integrity in mice (Lang et al., 2014), and agenesis of the corpus callosum is a frequent brain disorder found in >80 human congenital syndromes, including ciliopathies and neurodevelopmental disorders (Laclef et al., 2015).
To investigate the role of Ulk4 during brain formation, we recently characterized Ulk4 gene expression during Xenopus development, and found coexpression of Ulk4 mRNA with Sox3 (a neural progenitor cell marker) and Blbp (a radial glial marker) in the ventricular zone of the forebrain (Domínguez et al., 2015). To understand the consequence of genetic lesions, here we systematically investigated Ulk4tm1a/tm1a mice. We demonstrate that Ulk4 is predominantly expressed in the cells lining the ventricles and is essential for ciliogenesis. Ulk4tm1a/tm1a mice exhibit dysfunctional subcommissural organs (SCOs), obstructive aqueducts, and noncommunicating hydrocephalus. The CSF flow is impaired, and Ulk4tm1a/tm1a ependymal cells display reduced/disorganized/dysfunctional cilia with abnormal axonemes, which are required for coordinated beating of ependymal cilia and directional flow of CSF. Furthermore, we present the molecular mechanism that Ulk4 may regulate expression of Foxj1, a master switch of ciliogenesis, and numerous other ciliogenesis genes. Therefore, our findings indicate that Ulk4 may act as a scaffold protein regulating different processes of ciliogenesis and coordinated ciliary beating, and ULK4 may be associated with human diseases of heterogeneous clinical symptoms in relation to cilia dysfunction.
Materials and Methods
Ulk4 hypomorph mice.
The DNA sequence of the knock-out-first construct for creating the Ulk4tm1a(KOMP)Wtsi (shortened as Ulk4tm1a in this article) mutants was published online (http://www.ncbi.nlm.nih.gov/nuccore/JN950132.1). In the targeted allele, the intron 6 of sequence 5′-tctgtctgaagaaaggaggggccgccaggatcag-3′ was replaced by the FRT-En2SA-IRES-LacZ-PA-hBactP-Neo-PA-FRT-loxP cassette, so that a fusion mRNA containing Ulk4 exons 1–6 and En2SA-IRES-LacZ-PA could be transcribed. The intron 7 sequence of 5′-cagcctcctgcactgcgattacagatcaataccac-3′ was replaced by a second loxP site, to enable deletion of the critical exon 7 (76 bp) by the Cre-LoxP system if needed.
The Ulk4 mutant strain was created from ES cell clone EPD0182_4_E12 generated by the Wellcome Trust Sanger Institute and made into mice by the Knockout Mouse Project (KOMP) Repository (www.KOMP.org) and the Mouse Biology Program (www.mousebiology.org) at the University of California, Davis. Methods used to create the (CHORI/Sanger/UC Davis)-targeted alleles were published previously (Skarnes et al., 2011). Breeding pairs of Ulk4+/tm1a mice on C57BL/6N background were purchased from the KOMP Repository.
All experimental procedures were approved by both the Irish Department of Health and Children in accordance with Cruelty to Animals Act of 1876 with license number B100/4504; and by the institutional Animal Care and Research Ethics Committee with certificate number 12/SEP/02. Ulk4tm1a/tm1a and wild-type (WT) littermates were obtained from Ulk4+/tm1a × Ulk4+/tm1a mating. Each mouse was genotyped by genomic PCR with two pairs of primers at the same time. The WT allele was detected by a 271 bp DNA fragment using Ulk4EndE7For (5′-TAACTTGCTGGACGGATTGCTG-3′ in exon 7) and Ulk4EndIn7Rev (5′-TGATCTGTAATCGCAGTGCAGG-3′, nested within the sequence of 5′-CAGCCTCCTGCACTGCGATTACAGATCAATACCAC-3′ deleted in the Ulk4tm1a allele). The mutant allele was identified by a 621 bp PCR fragment using Ulk4KOMPKOFor (5′-GAGATGGCGCAACGCAATTAATG-3′, which was present only in the synthetic cassette) and Ulk4KOMPKORev (5′-CTGAGGAGACAATGTAACCAGC-3′ from intron 7).
Histology.
P12 Ulk4tm1a/tm1a (three females and one male) and littermate controls (one female and three males) were deeply anesthetized and intracardially perfused with 20 ml of 4% paraformaldehyde (PFA) in PBS. The brains were dissected, postfixed in 4% PFA for 24 h, and embedded into paraffin blocks. Serial coronal sections at 10 μm were produced with a microtome (SM2000R, Leica Instruments), stained with hematoxylin and eosin (H&E) and imaged under a bright-field microscope (IX41, Olympus) equipped with a camera.
CSF circulation assay.
Three Ulk4tm1a/tm1a (one female and two males) and four littermate controls (two females and two males) postnatal day (P) 12 mice [three were anesthetized by intraperitoneal injection of ketamine (150 mg/kg)/xylazine (15 mg/kg)]. The 30 gauge needle attached to a 10 μl glass syringe was positioned at 0.1 mm posterior and 1.0 mm lateral to the bregma on the head. Five microliters of Evans blue dye (4% in PBS) were slowly injected into the lateral ventricle (LV) of mice, which were killed 20 min later. Whole brains with a portion of the spinal cord were dissected and fixed in 4% PFA. Coronal sections at 1 mm thickness were generated by tissue chopper 48 h later. Images at 1× magnification were captured by a stereomicroscope with a digital camera.
X-gal staining.
Five P14 male mice were perfused, and brains were dissected, postfixed for 10 min in 4% PFA, and washed in PBS twice for 5 min. They were cryoprotected in 30% sucrose in PBS overnight at 4°C until sinking to the bottom of the tube, embedded in Optimal Cutting Temperature compound, and immersed in isopentane bath prechilled with liquid nitrogen. The samples were sectioned at 30 μm on a cryostat and mounted onto precoated slides. The sections were postfixed in 4% PFA for 10 min on ice, rinsed in PBS on ice for 2 × 10 min, and washed in PBS containing 2 mm MgCl2, 0.02% Nonidet P-40, 0.01% sodium deoxycholate for 10 min on ice. The sections were incubated in the dark with X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactoside) staining solution (1 mg/ml X-gal, 5 mm potassium ferricyanide, 5 mm potassium ferrocyanide, 2 mm MgCl2, 0.02% Nonidet P-40, 0.01% sodium deoxycholate in PBS) overnight at 37°C. The slides were then washed in PBS for 2 × 5 min at room temperature, quickly rinsed in distilled H2O, and counter-stained for 30 s in the eosin. They were subsequently washed in distilled H2O for 3 × 5 min, dehydrated through methanol (5 min each in 50, 70, and 100% methanol), cleared for 2 × 5 min in xylene, and mounted with mount medium (Sigma-Aldrich).
Immunohistochemistry.
P12 juvenile mutants (three females and one male) and WT littermates (one female and three males) were killed with overdoses of sodium pentobarbitone. Brains were postfixed in PBS containing 4% PFA at 4°C overnight, embedded in paraffin, and sectioned at 10 μm. The sections were immunostained using primary mouse monoclonal antibody against acetylated α-tubulin (1:1000; Sigma-Aldrich) and secondary goat anti-mouse antibody conjugated to Alexa Fluor 555 (1:500; Cell Signaling Technology), and mounted with mounting medium containing DAPI (Sigma-Aldrich).
Scanning electron microscopy.
P18 mice were perfused with 2.5% glutaraldehyde/2% PFA in PBS and brains postfixed in 2.5% glutaraldehyde/2% PFA in 0.1 m sodium cacodylate/HCl buffer, pH 7.2, overnight at 4°C. LVs were exposed by dissection under stereomicroscope. After dehydration through graded alcohols, tissue samples were transferred to hexamethyl disilizane for 2 × 15 min. The samples were then allowed to dry overnight in a fume hood, fixed to metal stubs using silver DAG paint, gold sputter coated, and viewed on a Hitachi S2600N variable-pressure scanning electron microscope.
Transmission electron microscopy.
LV walls were dissected from P18 brains fixed in 2.5% glutaraldehyde/2% paraformaldehyde/0.1 m sodium cacodylate/HCl buffer, pH 7.2, overnight at 4°C. Tissues were subsequently immersed in secondary fixative solution containing 1% osmium tetroxide in 0.1 m sodium cacodylate/HCl buffer, pH 7.2, for 4 h. They were then dehydrated through a series of graded alcohols (50, 70, 90, 95, and 100%), placed in propylene oxide, and transferred to a series of resin and propylene oxide mixtures (50:50, 75:25, pure resin). Finally, tissues were transferred to flat embedding molds, clearly labeled, and placed in 65°C oven for 48 h to polymerize. After polymerization, blocks were sectioned at 100 nm, lifted onto 3 mm copper grids, and stained for 30 min in 1.5% aqueous uranyl acetate for 10 min in lead citrate. Sections were dried and viewed on the Hitachi H7000 transmission electron microscope.
Whole-genome RNA sequencing.
Three pairs of P12 Ulk4tm1a/tm1a mice and WT controls were killed, and cortexes were quickly dissected and snap frozen in liquid nitrogen. RNA was extracted using RNeasy Kit (Qiagen) according to the manufacturer's instructions. RNA concentration was determined by the NanoDrop spectrophotometer. Equal amounts of RNA (∼6 μg/mouse, n = 3 each) were sent to BGI for whole-genome RNA sequencing. Libraries were constructed to convert RNA into cDNA, and quantitative RNA sequencing was performed by using the Illumina HiSeq2000 next-generation sequencer. Using bioinformatics tools and databases, 19,652 genes were identified from the P12 mouse cortex.
Relative gene expression was quantified and normalized in a format of FPKM (fragments per kilobase of transcript per million mapped reads). P values of multiple testing and false discovery rates (FDRs) were analyzed and supplied by BGI. We also used one-way ANOVA to statistically analyze the differential expression with two tails. p < 0.05 indicates statistical significance. The 414 cilia-associated genes were further analyzed statistically, and 34 Ulk4 targets were analyzed with the STRING database for pathway association and protein–protein interaction.
Quantitative RT-PCR.
Quantitative RT-PCR (qRT-PCR) was performed to validate expression of key Ulk4 targets detected by whole-genome RNA sequencing. Single-strand cDNA was synthesized from P14 cortical RNA of WT and Ulk4tm1a/tm1a mice. Triple qRT-PCRs were performed for each primer pair. GapdhFOR 5′-CTCATGACCACAGTCCATGC-3′ and GapdhREV 5′-CACATTGGGGGTAGGAACAC-3′ were used as cDNA loading control for each qRT-PCR plate. The Ulk4 RT-PCR primers were Ulk4E6FOR 5′-CCCCATTCTTCTCAGAAACG-3′ and Ulk4E8REV 5′-GTCTTTCCAGAATGGGTGCT-3′ (197 bp); Ulk4E21For 5′-TTGATAGTCCGTCCACTCCC-3′ and Ulk4E23Rev 5′-CATGGGGAGACACATCTTCA-3′ (189 bp); and Ulk4E34FOR 5′-CCCAGAGAACATGGTGACCT-3′ and Ulk4E36REV 5′-CGATAGTGTCGACGGGTGAG-3′ (197 bp). Other primers include Foxj1FOR 5′-GGCCACCAAGATCACTCTGT-3′ and Foxj1REV 5′-CTTCTTGAAGGCCCCACTGA-3′ (223 bp); Rfx3FOR 5′-TGCTAGCTTTGGCTCCTTTC-3′ and Rfx3REV 5′-GATTTCCGGGAGATACAGCA-3′ (152 bp); Vangl2FOR 5′-GCAGGAAGAGGAGCAGAAGA-3′ and Vangl2REV 5′-AATGCAGAACTCCAGGTGCT-3′ (166 bp); Pcm1For 5′-TCCTGTGGACATCCAGACTTC-3′ and Pcm1REV 5′-GCGAGTCCTTATGGGTAGCA-3′ (233 bp); Tubb4aFOR 5′-GGTCAATGCGGTAACCAGAT-3′ and Tubb4aREV 5′-CACGGCTCTGGGAACATAGT-3′ (165 bp); Rsph9FOR 5′-CCACTTCAGGGAGGCTATTG-3′ and Rsph9REV 5′-GAGCGTGGTAGAAGGTGAGG-3′ (194 bp); GsnFOR 5′-GACTCTTTGCCTGCTCCAAC-3′ and GsnREV 5′-TTGCTGGATCTGTCTCGATG-3′ (207 bp); Poc5FOR 5′-CAGGTCGGTCTGATTTTGGT-3′ and Poc5REV 5′-CTGCTTTCAGGATGGATGGT (172 bp); Cep120FOR 5′- AAACAGCAGGAGGAGTTGGA-3′ and Cep120REV 5′-GCTTCCCACTTGCAATCTCT-3′ (172 bp); Kif5aFOR 5′-TCCTACCAGAAGGCCAACAT-3′ and Kif5aREV 5′-CTGCAGCTACCTGAAAGTGC-3′ (178 bp); Spag6FOR 5′-ACATGTTGTTGGGCAGTTCAG-3′ and Spag6REV 5′-TTGGTAGCTGTCCACCCTCT-3′ (226 bp); and B9d2FOR 5′-TTCTCGGAAAGCAGCCTCTT-3′ and B9d2REV 5′-TAGCGAAGTGCAGGTCAATG-3′ (154 bp). Relative RNA abundance in the Ulk4tm1a/tm1a cortex was calculated using 2ΔΔCt, with average expression level of the corresponding gene in the WT littermates as 100%. The data were presented as mean ± SEM, n = 3 each (*p < 0.05, **p < 0.01).
Results
Ulk4 deficiency causes growth retardation and preweaning loss
The Ulk4 mutant strain (Ulk4+/tm1a) was created by the KOMP Repository using the knock-out-first strategy (Fig. 1A,B). A cassette of FRT-En2SA-IRES-LacZ-PA-hBactP-Neo-PA-FRT-loxP-Exon7-loxP was inserted into intron 6, so that Ulk4 exons 1–6 could be spliced with En2SA-IRES-LacZ-PA to generate a chimeric Ulk4-SA-IRES-LacZ-PA mRNA. Although a polyA signal was followed the lacZ reporter gene, the En2 splice acceptor site might not be fully efficient to knock-out Ulk4 gene completely. We evaluated Ulk4 mRNA expression in the Ulk4tm1a/tm1a mutants by qRT-PCR, using primers derived from the targeting region (exon 6–8), the middle (exon 21–23), and the 3′-end of the Ulk4 gene (exon 34–36). The Ulk4 mRNA expression was reduced to 4.0 ± 24.6% (p < 0.01, n = 3 each; Fig. 1D), 15.8 ± 23.5% (p = 0.01; Fig. 1E), and 22.8 ± 8.9% (p = 0.02; Fig. 1F) respectively in Ulk4tm1a/tm1a mice, showing an average 78–96% reduction of Ulk4 expression in the mutants. It is unknown whether the Ulk4 mRNA is generated from the leaky system of En2SA and/or from natural Ulk4 splicing variants using alternative promoter(s). It is also unclear whether they encode functional proteins. Nevertheless, these data demonstrate that the Ulk4tm1a/tm1a strain is a “hypomorph” model, not a “null” mutant. This further highlights the importance of the Ulk4 gene for brain development and function, given that 78–96% depletion of Ulk4 mRNA already resulted in striking phenotypes described in this article.
Figure 1.
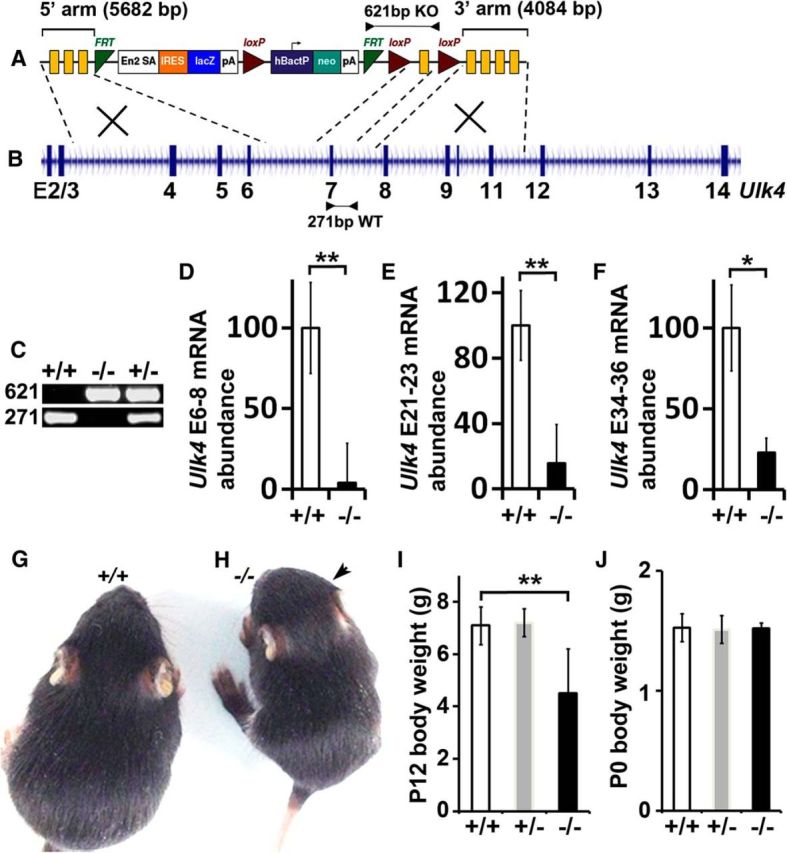
Ulk4 is essential for postnatal growth and survival. A, The Ulk4 knock-in construct harboring a cassette of FRT-En2SA-IRES-LacZ-PA-hBactP-Neo-PA-FRT-loxP-Exon7-loxP. The 5′ targeting arm was a 5682 bp fragment containing exons 4–6 of Ulk4, and the 3′ arm was a 4084 bp fragment containing exons 7–11. Exon 7 was considered a critical exon flanked by two loxP sites. B, The mouse Ulk4 genomic organization. Arrowheads indicate the primer sequence location. C, For mouse genotyping, the targeted allele was identified by a 621 bp PCR band with Ulk4KOMPKOFor and Ulk4KOMPKORev. The WT allele was detected by 271 bp PCR product with Ulk4E7For and Ulk4In7Rev. D–F, Ulk4 mRNA expression was assessed by qRT-PCR using primers derived from exon 6–8 (D; WT, 100.0 ± 28.2%; Ulk4tm1a/tm1a, 4.0 ± 24.6%, p < 0.01, n = 3 each), exon 21–23 (E; WT, 100.0 ± 21.4%; Ulk4tm1a/tm1a, 15.8 ± 23.5%, p = 0.01, n = 3 each), and exon 34–36 (F; WT, 100.0 ± 26.5%; Ulk4tm1a/tm1a, 22.8 ± 8.9%, p = 0.02, n = 3 each), showing a minimal 78% reduction of Ulk4 expression in Ulk4tm1a/tm1a mice. G, H, P12 WT (G; +/+) and Ulk4tm1a/tm1a (H) littermate mice with significantly reduced body size. The arrow in H highlights a domed-shaped head. I, The body weight of P12 (Ulk4+/+, n = 12; Ulk4+/tm1a, n = 12; Ulk4tm1a/tm1a, n = 5; p < 0.01) mice. J, The body weight of P0 mice (Ulk4+/+, n = 8; Ulk4+/tm1a, n = 9; Ulk4tm1a/tm1a, n = 5) were quantified statistically, showing no significant difference. +/+, WT Ulk4+/+; +/−, heterozygote Ulk4+/tm1a; −/−, homozygote Ulk4tm1a/tm1a; **p < 0.01.
To investigate Ulk4 gene function, we first bred Ulk4+/tm1a (Fig. 1A–C) males with WT (Ulk4+/+) females and observed the expected Mendelian ratio of Ulk4+/tm1a mice in offspring. For example, 117 pups were born from 19 litters with a standard litter size for C57BL/6N (6.2 ± 0.6, mean ± SEM). Of the 111 (95%) that survived to weaning, 52 were WT and 59 were heterozygous Ulk4+/tm1a. Therefore, deletion of one copy of Ulk4 gene did not affect mouse survival, sexual development, or reproduction.
However, Ulk4+/tm1a × Ulk4+/tm1a mating showed a significant preweaning loss. Among the 82 pups born from 12 litters (6.8 ± 0.5/litter), only 63 (76.8%) survived to weaning, with a Ulk4+/+-to-Ulk4+/tm1a-to-Ulk4tm1a/tm1a ratio of 16:37:10. To determine whether Ulk4tm1a/tm1a mice were embryonic lethal, we genotyped four litters of 28 pups (seven per litter) at birth (P0), and detected a Ulk4+/+:Ulk4+/tm1a:Ulk4tm1a/tm1a ratio of 9:11:8, showing that Ulk4tm1a/tm1a mice were not under-represented at birth. Subsequent genotyping further confirmed a Mendelian ratio of Ulk4tm1a/tm1a mice at birth, but they rarely survived to P28.
Ulk4tm1a/tm1a mice displayed marked growth retardation. At P12, their body weight was 5.10 ± 0.65 g (n = 5, p < 0.01), ∼30% lighter than that of Ulk4+/+ (7.10 ± 0.15 g, n = 12; Fig. 1E–G) or Ulk4+/tm1a littermates (7.20 ± 0.21 g, n = 12). However at P0, they (1.52 ± 0.02 g, n = 5) did not differ from WT (1.53 ± 0.04 g, n = 8; Fig. 1H) or Ulk4+/tm1a (1.51 ± 0.04 g, n = 9) mice. Therefore, the Ulk4 gene is essential for postnatal growth and survival.
Ulk4tm1a/tm1a mice develop hydrocephalus
All Ulk4tm1a/tm1a mice exhibited hydrocephalic appearance with domed-shaped heads 2 weeks after birth (Fig. 1F, arrow). To investigate the neuropathology, four P12 Ulk4tm1a/tm1a and four Ulk4+/+ littermates were histologically investigated. Ventriculomegaly was evident in Ulk4tm1a/tm1a mice, with an LV size of 7.00 ± 1.48 mm2 at the anterior commissural level, which was 17.5-fold larger (p < 0.01) than that in WT littermates (0.40 ± 0.11 mm2; Fig. 2R). The Ulk4tm1a/tm1a third ventricle (3V; 0.122 ± 0.070 mm2; Fig. 2H,S) was also 4.7-fold dilated (Fig. 2H; p = 0.03) compared with that of the controls (0.026 ± 0.003 mm2; Fig. 2G).
Figure 2.
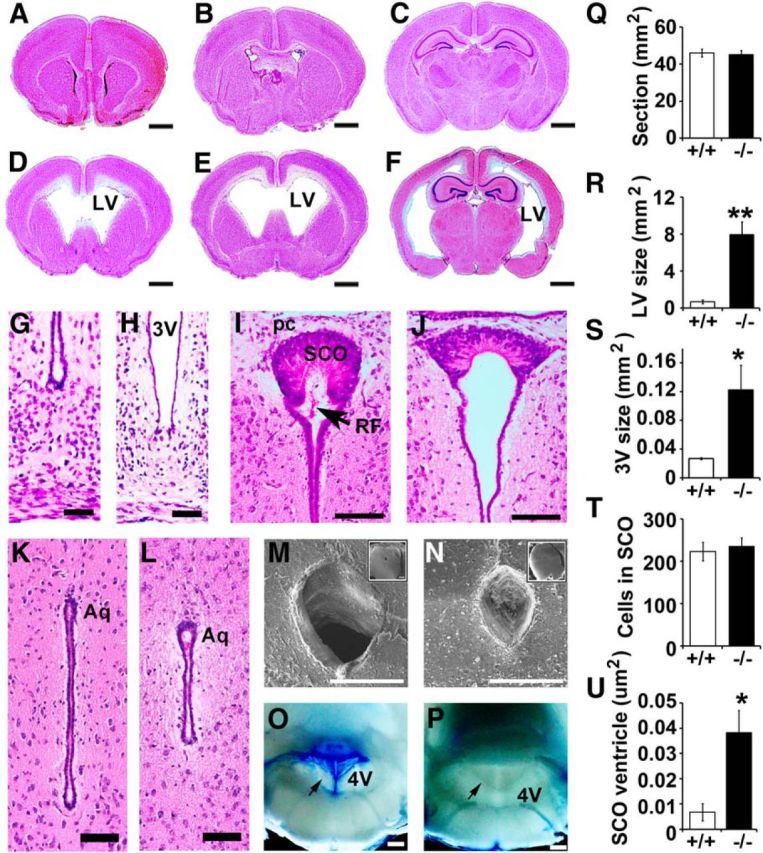
Ulk4tm1a/tm1a mice display hydrocephalus phenotype at P12. A–L, H&E-stained brain sections of WT littermate mice (A–C, G, I, K) and P12 Ulk4tm1a/tm1a brain sections (D–F, H, J, L). M, N, Scanning electron microscopy images of WT (M) and Ulk4tm1a/tm1a (N) aqueduct, showing aqueductal blockage in the mutant (N). O, P, Images of brain slice at the 4V (arrow) from WT (O) and mutant (P) mice. Q–U, Statistical quantifications show significantly enlarged LV (R), 3V (S), and SCO ventricles (U). Scale bars: A–F, 1 mm; G–J, 50 μm; K, L, 100 μm; M, N, insets, 500 μm; M–P, 200 μm. *p < 0.05; **p < 0.01. Aq, Aqueduct; LV, lateral ventricle; pc, posterior commissure; RF, Reissner's fiber; SCO, subcommissural organ; +/+, Ulk4+/+; −/−, Ulk4tm1a/tm1a.
The SCO is important for CSF flow, and the relationship between abnormal SCO and hydrocephalus has been well documented in animals (Lee et al., 2012). We quantified the number of SCO cells and found no significant difference between Ulk4tm1a/tm1a and WT mice (Fig. 2I,J,T; p = 0.67). However, the ventricle at the posterior commissural level was fivefold dilated in Ulk4tm1a/tm1a mice (Fig. 2I,J,U; p < 0.05). The SCO generates and secretes Reissner's fiber (RF) into the CSF, which extends caudally to the aqueduct, fourth ventricle (4V), and spinal cord. Impaired RF production/secretion can lead to aqueduct stenosis, causing obstructive hydrocephalus (McAllister, 2012). Whereas the RF was apparent in WT littermates (Fig. 2I, arrow), we failed to detect RF extension in Ulk4tm1a/tm1a mice (Fig. 2J), suggesting a dysfunction of Ulk4tm1a/tm1a SCO.
The aqueduct is a narrow channel connecting the 3V and 4V. Remarkably, in Ulk4tm1a/tm1a mice, the aqueduct was not overtly dilated (Fig. 2L,K). Scanning electron microscopy revealed aqueduct blockage in the mutants (Fig. 2N,P), which was not seen in the WT aqueduct (Fig. 2M,O). These data demonstrate that Ulk4tm1a/tm1a hydrocephalus was associated with SCO dysfunction and aqueduct stenosis.
The hydrocephalus phenotype is present in P0 Ulk4tm1a/tm1a mice
Congenital hydrocephalus accounts for ∼55% of all hydrocephalus (Carter et al., 2012). To determine the nature of Ulk4tm1a/tm1a hydrocephalus, we histologically analyzed Ulk4tm1a/tm1a brains and littermate controls at P0, when Ulk4tm1a/tm1a mice did not show gross abnormality. A significant dilation of the LV and 3V was detected (Fig. 3). However, the enlargement at P0 (Fig. 3E; 5.0-fold) was not as dramatic as at P12 (Fig. 2R; 17.5-fold). These results showed that Ulk4tm1a/tm1a hydrocephalus was congenital, but the phenotype was progressively worsened during the early postnatal period, which coincided with the development and maturation of ependymal cilia.
Figure 3.

The Ulk4tm1a/tm1a hydrocephalus is congenital. A, C, H&E-stained brain sections of P0 WT mice. B, D, H&E-stained brain sections of Ulk4tm1a/tm1a mice. E, F, The relative size of LV (E) and 3V (F) at the SCO position were quantified. *p < 0.05, **p < 0.01. Scale bars: A, B, 1 mm; C, D, 50 μm. 3V, third ventricle; LV, lateral ventricle; pc, Posterior commissure; SCO, subcommisural organ; +/+, Ulk4+/+; −/−, Ulk4 tm1a/tm1a.
CSF flow is obstructed in the Ulk4tm1a/tm1a aqueduct
CSF flows sequentially from the LV to the 3V, aqueduct, and 4V, before entering the subarachnoid space and draining into the blood primarily through arachnoid granulations. Ciliary beating and CSF flow may be visualized on brain slices in vitro (Sawamoto et al., 2006). Aqueduct obstruction is common in animal models. Evans blue is an azo dye that binds with high affinity to serum albumin, flows with the CSF, and is also used to trace CSF flow in animals (Kim et al., 2012).
We investigated CSF flow with Evans blue in three P12 Ulk4tm1a/tm1a mice and four WT littermate controls by injecting 5 μl of the dye (4% in PBS) into the LV (Fig. 4). The mice were kept in anesthetized status for 20 min to permit the dye to flow with CSF in vivo. Mice were then terminated and examined for dye distribution. The dye was detected throughout all ventricles in WT littermates (Fig. 4, top, W1–W4; n = 4), including the LV (Fig. 4, A), 3V (Fig. 4, B), SCO (Fig. 4, C), aqueduct (Fig. 4 D, Aq), 4V (Fig. 4, E), and spinal canal (Fig. 4, F, SC).
Figure 4.
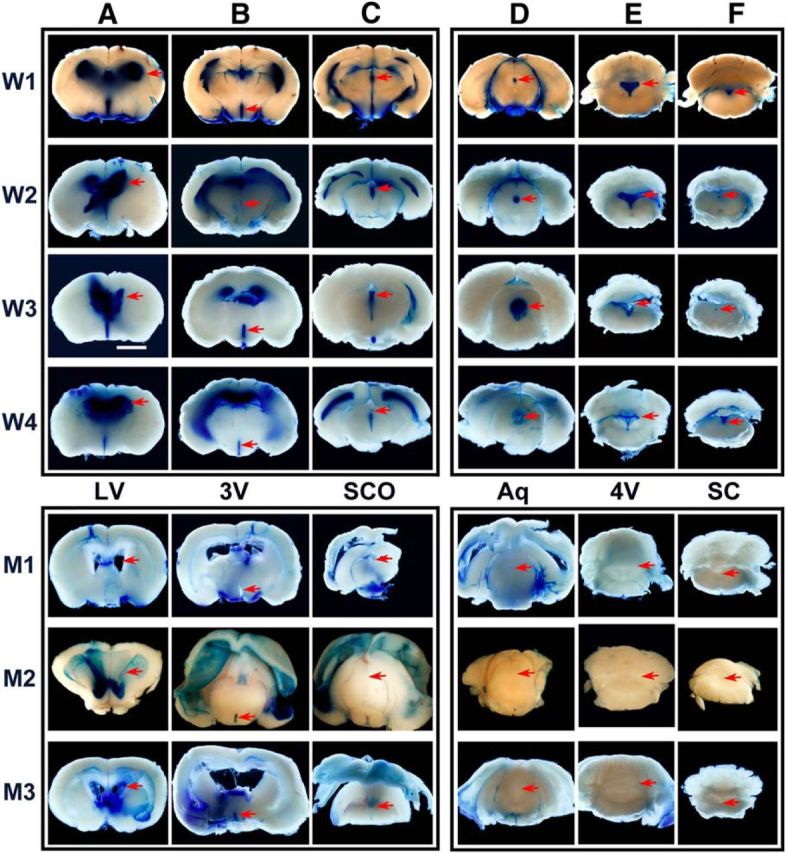
Impaired CSF circulation in Ulk4tm1a/tm1a mouse brain. Four P12 WT (W1–W4) and three mutant (M1–M3) mice were intraventricularly injected with 5 μl of Evans blue dye into the dorsal cortex (4% in PBS). The dye was allowed to flow with the CSF in vivo for 20 min. Mouse brains were subsequently fixed, sectioned, and imaged at the comparable levels of LV (A), 3V (B), SCO (C), aqueduct (Aq; D), 4V (E), and spinal canal (SC; F). Arrows indicate the anatomical positions of the LV, 3V, SCO, Aq, 4V, and SC of WT littermate and Ulk4tm1a/tm1a mutant brains. Note that in the WT brains, the dye flowed from the LV to SC with defined localization in the ventricles (W1–W4, A–F). In three mutants, the dye was largely diffused from LV to 3V passively, with little dye reaching the SCO and no dye reaching the Aq, 4V, or SC of the Ulk4tm1a/tm1a brain (D–F, bottom right), demonstrating an impaired CSF flow in the Ulk4tm1a/tm1a mice. Scale bar, 600 μm. +/+, WT Ulk4+/+; −/−, Ulk4tm1a/tm1a.
In contrast to well restrained dye distribution in the WT littermates from the LV to the spinal canal, there was little evidence of CSF flow in Ulk4tm1a/tm1a brains (n = 3; Fig. 4). The dye was detected in the LV (Fig. 4, A, M1–M3) and 3V (Fig. 4, B). However, little dye reached the SCO ventricle (Fig. 4, C), and no dye was seen in the Ulk4tm1a/tm1a aqueduct (Fig. 4, D), 4V (Fig. 4, E) or spinal canal (Fig. 4, F, SC). These data consistently demonstrated that the injected Evans blue was passively diffused from the LV to the 3V in Ulk4tm1a/tm1a mice. Ulk4 deficiency therefore functionally impairs the CSF circulation, leading to hydrocephalus in the mutants.
Ulk4 is predominantly expressed in the ventricular system
The impaired CSF flow suggested a failure of ependymal function in five Ulk4tm1a/tm1a male mice. To explore pathological mechanisms of Ulk4tm1a/tm1a hydrocephalus, we first investigated the expression pattern of Ulk4 in mice (Fig. 5). The Ulk4tm1a/tm1a mice were created by replacing exon 7 of Ulk4 with an IRES-lacZ reporter (Fig. 1A,B). Therefore an Ulk4-IRES-lacZ fusion mRNA was anticipated to be transcribed from the Ulk4 promoter. We stained P14 brain sections with X-gal and observed weak β-gal activity in the choroid plexus, where the CSF was produced (Fig. 5A, CP), and in the subfornical organ (Fig. 5C, SFO), which has been reported to be involved in hydrocephalus.
Figure 5.

Ulk4 is expressed in the ventricular system, and Ulk4tm1a/tm1a ependymal cilia are disorganized. A–F, X-gal staining (in blue) was performed to reveal the Ulk4 expression pattern. A moderate level of lacZ expression was observed in the choroid plexus (A, CP) and subfornical organ (C, SFO), and a high level of β-gal activity was detected in the ependymal cells along the LV (B), dorsal 3V (C, D3V), 3V (D), SCO (E), and aqueduct (F, Aq). Black arrow in F highlights no expression in the roof of aqueduct, which was occupied by nonciliated ependymal cells. Black arrowheads show strong expression at the lateral wall of the aqueduct with motile cilia. G, H, Immunohistochemical staining with an antiacetylated α-tubulin showed densely clustered cilia in the WT (G, white arrowhead), but fewer and disorganized cilia (H, white arrowhead) and absence of cilia (H, white arrow) in Ulk4tm1a/tm1a mice. I, J, Scanning electron microscopy confirmed highly organized cilia in Ulk4+/tm1a (I) mice, but disorganized/dysfunctional cilia, with the absence of cilia in large areas of Ulk4tm1a/tm1a (J) mice. +/+, Ulk4+/+; +/−, Ulk4+tm1a; −/−, Ulk4tm1a/tm1a. Scale bars: A–J, 100 μm.
The most intense lacZ staining was detected in the ventricular system, including ependymal cells in the LV, 3V, SCO, and aqueduct (Aq; Fig. 5B–F). Notably, Ulk4 was highly expressed in the lateral wall of the aqueduct with motile cilia (Fig. 5F, arrowheads), but not on the roof of the aqueduct, which was occupied by nonciliated cells (Collins, 1983). The predominant Ulk4 expression in mature ependymal cells suggests that Ulk4 plays a vital role in CSF circulation.
Ulk4tm1a/tm1a mice display impaired motile cilia
CSF circulation is largely considered unidirectional, and directional flow is driven by coordinated beating of ependymal cilia. To investigate ciliary development, we first immunohistochemically stained P12 brains with antiacetylated α-tubulin, a ciliary marker, when motile cilia were fully developed and matured. Highly organized cilium bundles were found to be arrayed in a linear fashion on the WT ependymal wall (Fig. 5G, arrowhead). In contrast, Ulk4tm1a/tm1a cells exhibited disorganized and less dense cilia (Fig. 5H, arrowhead), or absence of cilia (Fig. 5H, arrow). However, the cilia length appeared comparable (Fig. 5G–J).
We subsequently performed scanning electron microscopy to examine the LV surface (Fig. 5I,J). On the Ulk4+/tm1a LV wall, the ependymal cilia were organized into bundles and orientated in the same direction (Fig. 5I), indicative of coordinated/directional beating. In the Ulk4tm1a/tm1a mice, the number of cilia bundles was dramatically reduced (Fig. 5J), and a large proportion of cells did not have cilia arrays, suggestive of underdevelopment of motile cilia. The Ulk4tm1a/tm1a cilia were highly disorganized and randomly scattered on the surface of the ependymal wall, a sign of dysfunctional cilia with no directional beating (Fig. 5J).
Ulk4tm1a/tm1a mice display major defects in basal body orientation and axonemal organization
During ciliogenesis, the basel bodies (BBs) from centrioles migrate toward the surface of the cell. Along the way, the BBs attach to membrane vesicles and form a BB–vesicle complex. The vesicle membrane in the complex fuses with the plasma membrane, and axonemal microtubules then extend from the BB. To investigate the underlying mechanism of disorganized cilia in Ulk4tm1a/tm1a mice, we examined the BB position, which determines the cilium orientation. In WT ependymal cells, multicilia were fully developed, matured, clustered, and oriented in a same direction (Fig. 6A,D). In line with this, all BBs were aligned in parallel and leaned in the same direction (Fig. 6A,D, arrows).
Figure 6.

Ulk4 deficiency impairs axonemal structure and ciliary function. A–F, Transmission electron microscopic images of radial sections of ependymal cells. Clusters of multicilia with the same orientation appeared on P18 WT ependymal cells (A, D, arrows), but reduced (B, C, E) or absent (F) in Ulk4tm1a/tm1a cells (B, C, E, F). The black boxes in B and F show an immature cilium, namely a BB-vesicle fusion complex, beneath the Ulk4tm1a/tm1a cell membrane. Cilium length was not shortened in the Ulk4tm1a/tm1a mice (A, C–E), but the BB orientation was dispersed in the Ulk4tm1a/tm1a cells (A, D). Arrows show normally embedded BBs in WT (D), but differently oriented in the Ulk4tm1a/tm1a cells (E). The black box in E shows cross-sectional view of a BB foot in a vertical orientation. The black box in F shows a BB–vesicle fusion complex oriented parallel to the ependymal membrane, which varies by 90° from the normal orientation. G, WT littermates presented normal axonemes of 9 + 2. H–L, Some Ulk4tm1a/tm1a motile cilia exhibited 9 + 2 (H), but others either lost the central doublets without (9 + 0; I) or with disorganization of a peripheral doublet (also 9 + 0; J), or missed a peripheral doublet but had a supernumerary central doublet (8 + 4; K), or lost peripheral and central doublets (8 + 0; L). Scale bars: A–C, 1000 nm; D–F, 500 nm; G–L, 100 nm. Insets in B, E, and F were magnified three times from the respective images. MV, Microvilli; N, nucleus; V, vesicle; +/+, Ulk4+/+; −/−, Ulk4tm1a/tm1a.
The length of the Ulk4tm1a/tm1a cilia (Fig. 6A, ) was not markedly shortened. However, fewer cilia or the absence of cilia were observed on Ulk4tm1a/tm1a ependymal cells. Instead, microvilli of smaller and shorter projections abundantly occupied the Ulk4tm1a/tm1a ependymal cell surface (Fig. 6B,C,E,F, MV). The cilia development appeared delayed also in the Ulk4tm1a/tm1a mice, as the BB–vesicle complex was still present beneath the P18 Ulk4tm1a/tm1a ependymal membrane (Fig. 6B,F, black box). Dispersed and randomly orientated BBs were present in Ulk4tm1a/tm1a mice (Fig. 6B,C,E,F). Some adjacent BBs were oriented in completely different directions, and this was evident from the appearance of cross-sectional view of BB feet (Fig. 6E, black box). BB–vesicle fusion complexes with 90° difference to the normal orientation were also detected in Ulk4tm1a/tm1a ependymal cells (Fig. 6F). Collectively these data point to the delayed cilia development and misalignment of BBs in Ulk4tm1a/tm1a mice.
Motile cilia have a basic axonemal structure with nine peripheral doublets and a central pair of microtubules, termed 9 + 2, which is different from primary cilia lacking the central pair (9 + 0). To further investigate effects of Ulk4 on microtubular ultrastructure of ependymal cilia, we performed transmission electron microscopy on P18 mice. In WT mice, all axonemes exhibited typical 9 + 2 ultrastructure across sections (Fig. 6G). In Ulk4tm1a/tm1a mice, some exhibited normal structure (Fig. 6H), whereas others showed various defects in axonemes, i.e., missing the central doublets (9 + 0; Fig. 6I), with a disorganized peripheral doublet also lacking the central pair (also 9 + 0; Fig. 6J), losing a peripheral doublet but gaining a supernumerary central pair (8 + 4; Fig. 6K), or missing the central and a peripheral doublets (8 + 0; Fig. 6L). Together, these data showed that Ulk4tm1a/tm1a ependymal cilia were underdeveloped, disorganized, structurally disassembled, and functionally impaired.
Ulk4 deficiency dysregulates Foxj1 and other ciliogenesis molecules
Ulk4 is a relatively novel Ser/Thr kinase and nothing is known about it at the molecular level. To understand molecular mechanisms of cilium dysfunction in Ulk4tm1a/tm1a mice, we performed whole-genome RNA sequencing and obtained quantitative reads from 19,652 genes in three Ulk4tm1a/tm1a and three WT controls. Relative mRNA abundance was quantified, normalized, and expressed in FPKM. The data were analyzed by multiple testing, FDR, log2 ratio (knock-out/WT), mean, SEM, and one-way ANOVA, which identified 1824 genes significantly (p < 0.05) upregulated and 1005 genes significantly downregulated in P12 Ulk4tm1a/tm1a cortices.
We first compared expression of three master regulators of ciliogenesis, Foxj1, Rfx3, and Mcidas. None of the 8 Rfx family members (Rfx1–8, p > 0.05) nor Mcidas (30% reduced, p = 0.10) was significantly altered in Ulk4tm1a/tm1a mice (Fig. 7A,C). However, the expression of Foxj1 and Foxj3 increased by 1.59-fold (p = 0.03) and 1.33-fold (p = 0.04), respectively (Fig. 7A), suggesting a specific effect on the Foxj1 pathway in Ulk4tm1a/tm1a mice. Though no current literature suggests the involvement of Foxj3 in ciliogenesis, Foxj1 is overwhelmingly shown as a master switch that controls a series of ciliogenesis genes (Yu et al., 2008; Thomas et al., 2010; Choksi et al., 2014). We next pulled out 414 cilia-related genes from 19,652 transcripts and compared their expression levels, and identified 66 genes (15.94%, p < 0.05) that were significantly altered, with 18 downregulated and 48 upregulated. The bioinformatic analyses by the STRING database gave rise to a ciliary pathway with a p value of 1.949 × 10−35.
Figure 7.
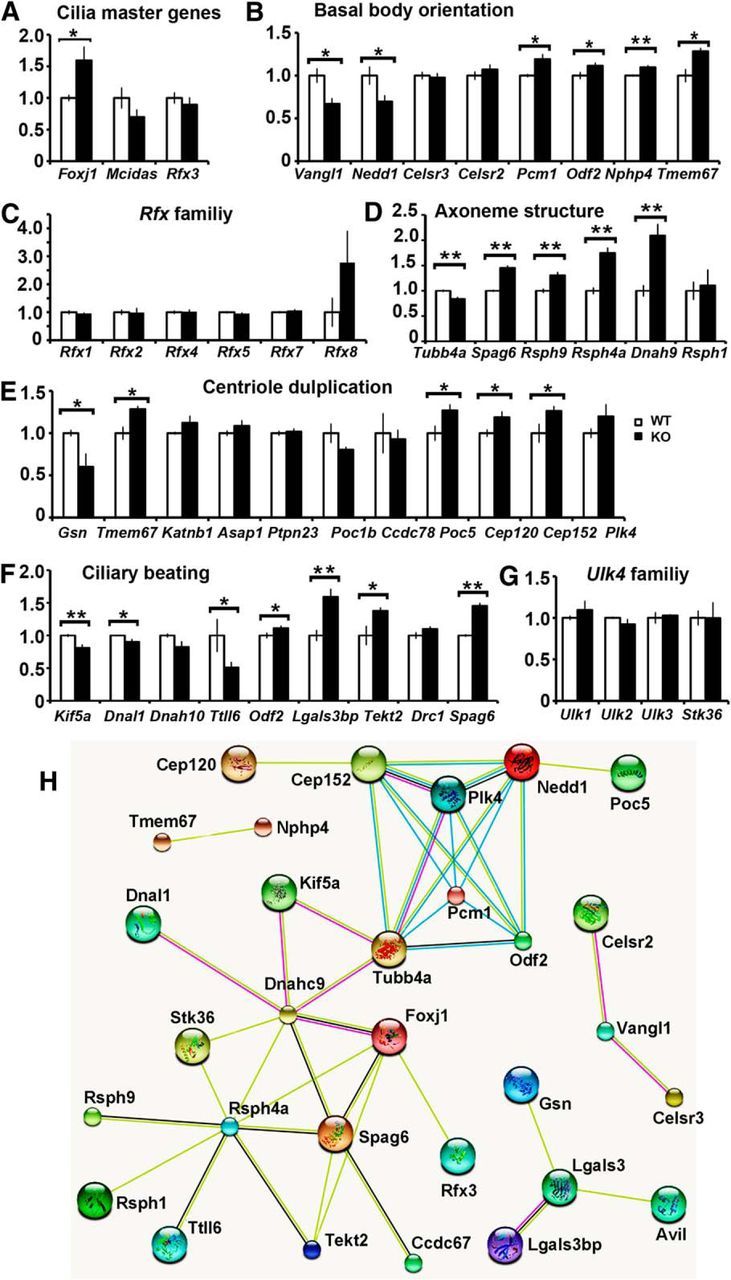
Changes in mRNA expression of genes associated with ciliary formation and function in Ulk4tm1a/tm1a mice. Whole-genome RNA sequencing was performed with P12 RNA from 3 WT and 3 knock-out (KO) cortices. Data were presented with folds of changes in the Ulk4tm1a/tm1a mice (KO/WT, mean ± SEM). A, Ulk4 regulates Foxj1 but not other (Rfx3, Mcidas) master ciliogenesis genes. C, None of the other Rfx3 family members was significantly altered. B, D–F, Key genes involved in (E) centriole amplification, (D) axoneme ultrastructure, (B) BB orientation, and (F) ciliary beating were statistically analyzed. G, The Ulk4 family members showed no compensation changes in expression. H, STRING GO analyses of the 38 Ulk4 dysregulated genes showed that 32 of them are interconnected. *p < 0.05; **p < 0.01.
Ciliogensis is a complicated and precisely controlled process, and amplification of tens to hundreds of centrioles is an initiation step. Gsn, Tmem67, Katnb1, Asap1, Ptpn23, Poc1b, Ccdc78, Poc5, Cep120, Cep152, and Plk4 have been implicated in centriole amplification (Arts et al., 2007; Lee and Gleeson, 2011). Among them, Gsn was 40% downregulated (p = 0.03; Fig. 7E), and Poc1b 20% reduced (p = 0.07). On the other hand, Tmem67 (1.28-fold, p = 0.01), Poc5 (1.27-fold, p = 0.037), Cep120 (1.19-fold, p = 0.04), and Cep152 (1.26-fold, p < 0.05) were significantly upregulated (Fig. 7E). The ependymal cilia should be fully matured at the time of analysis. Increased expression of procentriole components (Cep152, Cep120, and Poc5) in P12 Ulk4tm1a/tm1a cortex also suggested a delay of ciliogenesis in Ulk4tm1a/tm1a mice.
Transmission electron microscopy analyses showed misalignment of BBs in the Ulk4tm1a/tm1a ependymal cells. We next examined genes Odf2, Nphp4, Tmem67, Vangl1, Pcm1, Nedd1, Celsr2, and Celsr3, which were shown to regulate BB orientation (Ansley et al., 2003; Arts et al., 2007; Manning et al., 2008; Tissir et al., 2010; Kunimoto et al., 2012; Vladar et al., 2012; Leightner et al., 2013). Whereas expression of Odf2 (1.11-fold, p < 0.05), Nphp4 (1.43-fold, p = 0.02), and Tmem67 (1.28-fold, p = 0.01) increased, Vangl1 was significantly downregulated in Ulk4tm1a/tm1a mice (33%, p = 0.02; Fig. 7B), which is consistent with a previous report that cilia were randomly positioned in Vangl1−/−/Vangl2−/− mice (Song et al., 2010).
A centriolar satellite component, PCM1, was mutated in a family with ciliary defects and body-axis asymmetry (Ansley et al., 2003), and shown to colocalize and interact with BBS8 in centrosomes and BBs. Pcm1 increased 19% in Ulk4tm1a/tm1a mice (p = 0.01; Fig. 7B). On the other hand, Nedd1, a key component of the BB localized at the root of ciliated microtubules (Manning et al., 2008), was 30% reduced (p = 0.03; Fig. 7B). Gsn also regulates the cilia number by severing actin filaments (Kim et al., 2010), and the expression of Gsn was decreased by 40% in Ulk4tm1a/tm1a mice (p = 0.03, Fig. 7E).
The transmission electron microscopy data also revealed defects in axonemal assembly of microtubular doublets. Microtubules are heterodimers of α-tubulins and β-tubulins, and Tubb4a is the predominant subtype in the brain, which represents 46% of all β-tubulins (Leandro-Garcia et al., 2010). In Ulk4tm1a/tm1a mice, Tubb4a was 16% reduced (p < 0.01; Fig. 7D). Spag6, Rsph4a, Rsph9, and Rsph1 encode central pair components (Lee et al., 2012). RNA sequencing data showed that Foxj1 and three targets, Spag6 (1.46-fold, p < 0.001), Rsph4a (1.75-fold, p < 0.01), and Rsph9 (1.31-fold, p < 0.01), were significantly upregulated. Dnah9 and Dnal1 encode axonemal dynein heavy chain 9 and dynein light chain 1, respectively, as key components of the outer dyneins arm. Interestingly, the low-abundance gene Dnah9 was 2.1-fold increased (p < 0.01; Fig. 7D), whereas highly abundant Dnal1 was 10% reduced in the Ulk4tm1a/tm1a brain (p = 0.04, Fig. 7F).
CSF flow was impaired in Ulk4tm1a/tm1a mice and we next analyzed Odf2, Spag6, Lgals3, Tekt2, Dnah10, Dnal1, Ttll6, and Drc1, which were known to be involved in cilia beating (Fig. 7F; Mitchell et al., 2007; Suryavanshi et al., 2010; Mazor et al., 2011; Kunimoto et al., 2012; Clare et al., 2014). Four of them, Spag6 (1.46-fold, p < 0.01), Tekt2 (1.38-fold, p = 0.03), Odf2 (1.11-fold, p = 0.05), Lgals3 (7.48-fold, p < 0.01), and related Lgals3bp (1.59-fold, p < 0.01), were significantly upregulated. In contrast, Ttll6, encoding a tubulin tyrosine ligase, was 49% reduced (p = 0.06). Kif5a encodes a member of the kinesin family and functions as a microtubule motor. Clustering of kinesin-1 was shown to cause cilia-like beating of active microtubule bundles (31). In consistent with impaired CSF flow in Ulk4tm1a/tm1a mice, Kif5a expression was 19% reduced (p < 0.01; Fig. 7F).
Analyses of the RNA expression (FPKM) by log2 ratio, multiple testing (p < 0.05), and FDR (<0.01) confirmed significant changes of Foxj1, Pcm1, Tubb4a, Dnah9, Rsph4a, Gsn, Kif5a, Lgals3, Lgals3bp, and Dnal1 genes in Ulk4tm1a/tm1a mice. Together, these data showed that Ulk4 deficiency disturbed expression of the Foxj1 gene and a series of ciliogenesis genes, which consequently compromised ciliary development, axonema structure, coordinated beating, and CSF flow, which resulted in hydrocephalus in Ulk4tm1a/tm1a mice.
Discussion
Ciliopathies are pathogenic features of numerous human diseases, including hydrocephalus. Here we characterized the hypomorph Ulk4tm1a/tm1a mutant mouse strain with 4–22% of Ulk4 mRNA expressed. Ulk4tm1a/tm1a mice showed congenital hydrocephalus, which became severer postnatally with impaired CSF flow. The mutants displayed reduced/disorganized/nonfunctional ependymal cilia with defective axonemes. RNA sequencing analyses uncovered dysregulation of Foxj1 and other ciliogenesis genes, and the majority of them are interconnected by the STRING analysis (Fig. 7H). Therefore, Ulk4 is likely to act as an essential scaffold protein regulating ciliogenesis.
The Ulk4 family consists of five members. Ulk1 and Ulk2 are major regulators of autophagy (Egan et al., 2011; Lee and Tournier, 2011), whereas Ulk3 is involved in Shh signaling (Fuccillo et al., 2006). We previously reported the role of ULK4 in neuritogenesis, and ULK4 deficiency compromised neurite outgrowth in neuroblastoma cells and corpus callosum integrity in mice (Lang et al., 2014). Stk36 deficiency causes hydrocephalus in mice (Merchant et al., 2005), and this is confirmed by Lexicon Pharmaceuticals through screening of 4650 mouse models, which identify Stk36 and Ulk4 as risk factors among Ak7, Ak8, Celsr2, Dpcd, FZD3, Kif27, Mboat7, Nme5, Nme7, and RIKEN 4930444A02 genes. However, the pathological details or molecular mechanisms of these mutations remain elusive (Vogel et al., 2012). We show that neither Stk36 nor other family members is compensated for the Ulk4 hypomorph. Therefore both Ulk4 and Stk36 are indispensable for proper CSF circulation.
Ulk4 hydrocephaly is congenital as mild neuropathology appears at birth, which is worsened postnatally with aqueduct stenosis, dysfunctional SCO, and, more importantly, defective motile cilia. SCO cells secrete high-molecular-weight glycoproteins to maintain aqueduct patency. Impaired RF production/secretion can lead to obstructive hydrocephalus (McAllister, 2012), and abnormal RF and SCO dysplasia are associated with obstructive hydrocephalus in Sox3-overexpressing mice (Lee et al., 2012). Ulk4 protein colocalizes with Sox3 (Domínguez et al., 2015), but Sox3 expression (87%, p = 0.34) is not significantly altered in Ulk4tm1a/tm1a mice.
The characteristic feature of hydrocephalus is the ventricular dilation with excessive CSF. The Evans blue injection demonstrates no directional CSF flow, but passive diffusion in the Ulk4tm1a/tm1a brain. Directional CSF flow requires ependymal motile cilia, which are developed within the first postnatal week and coincide with progressive severity of the Ulk4tm1a/tm1a phenotype. Ulk4 is highly expressed in ependymal cells, SCO, and aqueducts consisting of multiciliated cells, but not on the roof of the aqueduct which is populated by nonciliated cells, highlighting the importance of Ulk4 for motile cilia.
The coordinated/directional ciliary beating from multiple ependymal cells produces stereotypical CSF flow (Breunig et al., 2010), and abnormal cilia are associated with hydrocephalus in patients (Fliegauf et al., 2007; Lee and Gleeson, 2011) and in mice with Foxj1, Ro1, Polaris/Ift188, Mdnah5, Hydin, or Spag6 mutation (Huh et al., 2009). We first observe abnormal and disorganized cilia in Ulk4 mice by anti-α-tubulin staining, which is supported by scanning and transmission electron microscopy data. Many Ulk4tm1a/tm1a ependymal cells lack motile cilia, and mutant cilia are highly disorganized and randomly scattered, display no consistent orientation, and are strikingly different from uniformly oriented and tightly clustered cilia, which are signs of coordinated beating, in WT littermates. The transmission electron microscopy reveals fewer cilia and misaligned BBs, subcellular mechanisms for disorganized cilia bundles. The 9 + 2 microtubular ultrastructure is the distinguishing feature of motile cilia. The central pair is crucial for beating. However, Ulk4tm1a/tm1a axonemes exhibit an array of 9 + 0, 8 + 4, and 8 + 0 ultrastructures. Together, these data show the fundamental importance of Ulk4 in ciliary development and function.
Ulk4 is a hypothetical kinase and nothing is known about its substrates or interactive partners. Other family members possess a conserved lysine at ULK1K46, ULK2K39, ULK3K26, and STK36K33, whereas the respective residue 33 in Ulk4 is leucine. Mutations of ULK1K46N or ULK2K39T dominantly inactivate the kinase activity (Chan et al., 2009). It would be interesting to determine ULK4 kinase activity.
In the absence of known Ulk4 substrates or binding partners, we performed whole-genome sequencing and identified dysregulated genes. While Foxj1 was specifically altered in the Ulk4tm1a/tm1a brain, Rfx3 and Mcidas, two other master genes of ciliogenesis, were not (Yu et al., 2008; El Zein et al., 2009; Thomas et al., 2010; Choksi et al., 2014). This is the first evidence suggesting that elevated Foxj1 signaling may also be pathogenic to the ciliogenesis and function. Consistent with Foxj1 upregulation, a whole array of ciliogenesis genes were modulated in Ulk4tm1a/tm1a mice. For example, among genes involved in centriole amplification (Keller et al., 2009; Al Jord et al., 2014), Tmem67, Poc5, Cep120, and Cep152 are upregulated, and Gsn is 40% downregulated, which is consistent with reduced ciliated cell numbers in Gsn knockdown cells (Kim et al., 2010).
The BBs are disorientated in Ulk4tm1a/tm1a mice. Odf2, Nphp4, Pcm1, and Tmem67 are involved in BB formation and orientation (Ansley et al., 2003; Arts et al., 2007; Kunimoto et al., 2012; Leightner et al., 2013), and their expression is significantly increased in Ulk4tm1a/tm1a mice. In contrast, Nedd1 is 30% downregulated, in line with its critical role as a BB component at the root of ciliated microtubules (Manning et al., 2008). Vangl1 is also 33% downregulated. Interestingly, mutations of VANGL1 have been identified in sporadic and familial neural tube defects (Kibar et al., 2007), and loss of Vangl2 causes neural tube defects in Lp/Lp mice (Kibar et al., 2001). Therefore decreased expression of Nedd1 and Vangl1 may contribute to misaligned BBs in Ulk4 mutants.
Ulk4tm1a/tm1a mice show defects in axoneme and motor function. Tubb4a, the predominant subtype of β-tubulins in the brain (Leandro-Garcia et al., 2010) is significantly reduced in Ulk4 mutants, and kinesin protein Kif5a as a microtubule motor is 19% decreased. Dnal1 and Ttll6 are downregulated by 10 and 49%, respectively, which is consistent with roles of Ttll6 in ciliary motility by modifying inner dynein arms (Suryavanshi et al., 2010), of DNAL1 as a component of outer dynein arms (Mazor et al., 2011), and identification of DNAL1 mutations in patients with CILD16 and motile cilia abnormalities (Mazor et al., 2011). Together, these data show that Ulk4 disruption results in disturbed balance of ciliogenesis genes, which consequently impairs cilia development, axoneme structure, and coordinated beating.
Ulk4tm1a/tm1a mice also die early postnatally. Hydrocephalus may partially contribute to this, but peripheral defects are likely. ULK4 is associated with hypertension (Levy et al., 2009) and multiple myeloma (Broderick et al., 2012), but we have no evidence of tumor formation, or gross pathology of heart, liver, lung, kidney, and spleen. The stomachs of Ulk4tm1a/tm1a mice are smaller than normal and colored white, suggestive of reduced food intake. Functional disturbance of lung and kidney is likely, as motile cilia are critical for these organs. Interestingly, Stumpy (B9D2) mutation also leads to preweaning loss, perinatal hydrocephaly, defective ciliogenesis, as well as polycystic kidney disease (Town et al., 2008). In addition, Stumpy mutation compromises hippocampal neurogenesis (Breunig et al., 2008), and we discover a reduced neural stem cell pool in the Ulk4 mutants (Liu et al., 2016). However, Ulk4 does not appear to directly regulate B9D2, and 109% (p = 0.16) of B9D2 mRNA is detected in Ulk4 mutants.
The RNA sequencing data suggest that Ulk4 regulates Foxj1 expression, modulates Foxj1 target genes, including Spag6, Rsph9, Rsph4a, Dnah9, Dnal1, Ttll6 and Tekt 2, and influences axoneme structure. Ulk4 may also modify expression of many other ciliogenesis genes (i.e., Vangl1, Nedd1, Odf2, Nphp4, Tmem67, Pcm1; Poc1b, Poc5, Cep120, Cep152; Tubb4a, Gsn; Kif5a, Lgals3, Lgals3b) and thereby affect BB alignment, ciliary number, maturation, and beating. In summary, the current study revealed defects in the structure, abundance, organization, and function of ependymal motile cilia in Ulk4tm1a/tm1a mice, together with aqueduct obstruction, SCO dysfunction, and impaired CSF flow. This report, the first to comprehensively demonstrate that Ulk4 is crucial for survival, postnatal growth, and ciliogenesis, supports broad implications for the ULK4 gene in human conditions.
Footnotes
This work was supported by Science Foundation Ireland (Grant 09/SRC/B1794s1 and 13/IA/1787 to S.S.) and the National University of Ireland Galway (Grant RSU002 to S.S.); We thank the staff of the Bio-Resources Unit, in particular Drs. Yolanda Garcia and Cathal O'Flatharta, for the support and assistance of experimental procedure. We would also like to thank the Knockout Mouse Project Repository (www.KOMP.org) and the Mouse Biology Program (www.mousebiology.org) at the University of California, Davis, for Ulk4+/tm1a breeding pairs. We also want to acknowledge the scientific and technical assistance of Dr. Kerry Thompson of the Centre for Microscopy & Imaging at the National University of Ireland. This facility is funded by the National University of Ireland Galway and the Irish Government's Programme for Research in Third Level Institutions, Cycles 4 and 5, National Development Plan.
The authors declare no competing financial interests.
References
- Al Jord A, Lemaître AI, Delgehyr N, Faucourt M, Spassky N, Meunier A. Centriole amplification by mother and daughter centrioles differs in multiciliated cells. Nature. 2014;516:104–107. doi: 10.1038/nature13770. [DOI] [PubMed] [Google Scholar]
- Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, Kim JC, Ross AJ, Eichers ER, Teslovich TM, Mah AK, Johnsen RC, Cavender JC, Lewis RA, Leroux MR, Beales PL, Katsanis N. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425:628–633. doi: 10.1038/nature02030. [DOI] [PubMed] [Google Scholar]
- Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT, Peters TA, Märker T, Voesenek K, Kartono A, Ozyurek H, Farin FM, Kroes HY, Wolfrum U, Brunner HG, Cremers FP, Glass IA, Knoers NV, Roepman R. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet. 2007;39:882–888. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- Brandler WM, Paracchini S. The genetic relationship between handedness and neurodevelopmental disorders. Trends Mol Med. 2014;20:83–90. doi: 10.1016/j.molmed.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breunig JJ, Sarkisian MR, Arellano JI, Morozov YM, Ayoub AE, Sojitra S, Wang B, Flavell RA, Rakic P, Town T. Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proc Natl Acad Sci U S A. 2008;105:13127–13132. doi: 10.1073/pnas.0804558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breunig JJ, Arellano JI, Rakic P. Cilia in the brain: going with the flow. Nat Neurosci. 2010;13:654–655. doi: 10.1038/nn0610-654. [DOI] [PubMed] [Google Scholar]
- Broderick P, Chubb D, Johnson DC, Weinhold N, Försti A, Lloyd A, Olver B, Ma YP, Dobbins SE, Walker BA, Davies FE, Gregory WA, Child JA, Ross FM, Jackson GH, Neben K, Jauch A, Hoffmann P, Mühleisen TW, Nöthen MM, et al. Common variation at 3p22.1 and 7p15.3 influences multiple myeloma risk. Nat Genet. 2012;44:58–61. doi: 10.1038/ng.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CS, Vogel TW, Zhang Q, Seo S, Swiderski RE, Moninger TO, Cassell MD, Thedens DR, Keppler-Noreuil KM, Nopoulos P, Nishimura DY, Searby CC, Bugge K, Sheffield VC. Abnormal development of NG2+PDGFR-alpha+ neural progenitor cells leads to neonatal hydrocephalus in a ciliopathy mouse model. Nat Med. 2012;18:1797–1804. doi: 10.1038/nm.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29:157–171. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choksi SP, Lauter G, Swoboda P, Roy S. Switching on cilia: transcriptional networks regulating ciliogenesis. Development. 2014;141:1427–1441. doi: 10.1242/dev.074666. [DOI] [PubMed] [Google Scholar]
- Clare DK, Magescas J, Piolot T, Dumoux M, Vesque C, Pichard E, Dang T, Duvauchelle B, Poirier F, Delacour D. Basal foot MTOC organizes pillar MTs required for coordination of beating cilia. Nat Commun. 2014;5:4888. doi: 10.1038/ncomms5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins P. Morphological features of the surface of the subcommissural organ and aqueduct in the red necked wallaby (Wallabia rufogrisea) J Anat. 1983;137:665–673. [PMC free article] [PubMed] [Google Scholar]
- Domínguez L, Schlosser G, Shen S. Expression of a novel serine/threonine kinase gene, Ulk4, in neural progenitors during Xenopus laevis forebrain development. Neuroscience. 2015;290:61–79. doi: 10.1016/j.neuroscience.2014.12.060. [DOI] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Zein L, Ait-Lounis A, Morlé L, Thomas J, Chhin B, Spassky N, Reith W, Durand B. RFX3 governs growth and beating efficiency of motile cilia in mouse and controls the expression of genes involved in human ciliopathies. J Cell Sci. 2009;122:3180–3189. doi: 10.1242/jcs.048348. [DOI] [PubMed] [Google Scholar]
- Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- Fuccillo M, Joyner AL, Fishell G. Morphogen to mitogen: the multiple roles of hedgehog signalling in vertebrate neural development. Nat Rev Neurosci. 2006;7:772–783. doi: 10.1038/nrn1990. [DOI] [PubMed] [Google Scholar]
- Huh MS, Todd MA, Picketts DJ. SCO-ping out the mechanisms underlying the etiology of hydrocephalus. Physiology (Bethesda) 2009;24:117–126. doi: 10.1152/physiol.00039.2008. [DOI] [PubMed] [Google Scholar]
- Keller LC, Geimer S, Romijn E, Yates J, 3rd, Zamora I, Marshall WF. Molecular architecture of the centriole proteome: the conserved WD40 domain protein POC1 is required for centriole duplication and length control. Mol Biol Cell. 2009;20:1150–1166. doi: 10.1091/mbc.E08-06-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibar Z, Vogan KJ, Groulx N, Justice MJ, Underhill DA, Gros P. Ltap, a mammalian homolog of Drosophila Strabismus/Van Gogh, is altered in the mouse neural tube mutant Loop-tail. Nat Genet. 2001;28:251–255. doi: 10.1038/90081. [DOI] [PubMed] [Google Scholar]
- Kibar Z, Torban E, McDearmid JR, Reynolds A, Berghout J, Mathieu M, Kirillova I, De Marco P, Merello E, Hayes JM, Wallingford JB, Drapeau P, Capra V, Gros P. Mutations in VANGL1 associated with neural-tube defects. N Engl J Med. 2007;356:1432–1437. doi: 10.1056/NEJMoa060651. [DOI] [PubMed] [Google Scholar]
- Kim IH, Carlson BR, Heindel CC, Kim H, Soderling SH. Disruption of wave-associated Rac GTPase-activating protein (Wrp) leads to abnormal adult neural progenitor migration associated with hydrocephalus. J Biol Chem. 2012;287:39263–39274. doi: 10.1074/jbc.M112.398834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee JE, Heynen-Genel S, Suyama E, Ono K, Lee K, Ideker T, Aza-Blanc P, Gleeson JG. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature. 2010;464:1048–1051. doi: 10.1038/nature08895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunimoto K, Yamazaki Y, Nishida T, Shinohara K, Ishikawa H, Hasegawa T, Okanoue T, Hamada H, Noda T, Tamura A, Tsukita S, Tsukita S. Coordinated ciliary beating requires Odf2-mediated polarization of basal bodies via basal feet. Cell. 2012;148:189–200. doi: 10.1016/j.cell.2011.10.052. [DOI] [PubMed] [Google Scholar]
- Laclef C, Anselme I, Besse L, Catala M, Palmyre A, Baas D, Paschaki M, Pedraza M, Métin C, Durand B, Schneider-Maunoury S. The role of primary cilia in corpus callosum formation is mediated by production of the Gli3 repressor. Hum Mol Genet. 2015;24:4997–5014. doi: 10.1093/hmg/ddv221. [DOI] [PubMed] [Google Scholar]
- Lang B, Pu J, Hunter I, Liu M, Martin-Granados C, Reilly TJ, Gao GD, Guan ZL, Li WD, Shi YY, He G, He L, Stefánsson H, St Clair D, Blackwood DH, McCaig CD, Shen S. Recurrent deletions of ULK4 in schizophrenia: a gene crucial for neuritogenesis and neuronal motility. J Cell Sci. 2014;127:630–640. doi: 10.1242/jcs.137604. [DOI] [PubMed] [Google Scholar]
- Leandro-Garcia LJ, Leskela S, Landa I, Montero-Conde C, Lopez-Jimenez E, Leton R, Cascon A, Robledo M, Rodriguez-Antona C. Tumoral and tissue-specific expression of the major human beta-tubulin isotypes. Cytoskeleton (Hoboken) 2010;67:214–223. doi: 10.1002/cm.20436. [DOI] [PubMed] [Google Scholar]
- Lee EJ, Tournier C. The requirement of uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy. 2011;7:689–695. doi: 10.4161/auto.7.7.15450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Gleeson JG. Cilia in the nervous system: linking cilia function and neurodevelopmental disorders. Curr Opin Neurol. 2011;24:98–105. doi: 10.1097/WCO.0b013e3283444d05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Tan J, Morris MB, Rizzoti K, Hughes J, Cheah PS, Felquer F, Liu X, Piltz S, Lovell-Badge R, Thomas PQ. Congenital hydrocephalus and abnormal subcommissural organ development in Sox3 transgenic mice. PLoS One. 2012;7:e29041. doi: 10.1371/journal.pone.0029041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leightner AC, Hommerding CJ, Peng Y, Salisbury JL, Gainullin VG, Czarnecki PG, Sussman CR, Harris PC. The Meckel syndrome protein meckelin (TMEM67) is a key regulator of cilia function but is not required for tissue planar polarity. Hum Mol Genet. 2013;22:2024–2040. doi: 10.1093/hmg/ddt054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, Glazer NL, Morrison AC, Johnson AD, Aspelund T, Aulchenko Y, Lumley T, Köttgen A, Vasan RS, Rivadeneira F, Eiriksdottir G, Guo X, Arking DE, Mitchell GF, Mattace-Raso FU, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41:677–687. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Inglis PN, Leitch CC, Efimenko E, Zaghloul NA, Mok CA, Davis EE, Bialas NJ, Healey MP, Héon E, Zhen M, Swoboda P, Katsanis N, Leroux MR. An essential role for DYF-11/MIP-T3 in assembling functional intraflagellar transport complexes. PLoS Genet. 2008;4:e1000044. doi: 10.1371/journal.pgen.1000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Guan Z, Shen Q, Flinter F, Domínguez L, Ahn J-W, Collier DA, O'Brien T, Shen S. Ulk4 regulates neural stem cell pool. Stem Cells. 2016 doi: 10.1002/stem.2423. in press. [DOI] [PubMed] [Google Scholar]
- Manning JA, Colussi PA, Koblar SA, Kumar S. Nedd1 expression as a marker of dynamic centrosomal localization during mouse embryonic development. Histochem Cell Biol. 2008;129:751–764. doi: 10.1007/s00418-008-0392-0. [DOI] [PubMed] [Google Scholar]
- Marley A, von Zastrow M. A simple cell-based assay reveals that diverse neuropsychiatric risk genes converge on primary cilia. PLoS One. 2012;7:e46647. doi: 10.1371/journal.pone.0046647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazor M, Alkrinawi S, Chalifa-Caspi V, Manor E, Sheffield VC, Aviram M, Parvari R. Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am J Hum Genet. 2011;88:599–607. doi: 10.1016/j.ajhg.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister JP., 2nd Pathophysiology of congenital and neonatal hydrocephalus. Semin Fetal Neonatal Med. 2012;17:285–294. doi: 10.1016/j.siny.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Merchant M, Evangelista M, Luoh SM, Frantz GD, Chalasani S, Carano RA, van Hoy M, Ramirez J, Ogasawara AK, McFarland LM, Filvaroff EH, French DM, de Sauvage FJ. Loss of the serine/threonine kinase fused results in postnatal growth defects and lethality due to progressive hydrocephalus. Mol Cell Biol. 2005;25:7054–7068. doi: 10.1128/MCB.25.16.7054-7068.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, Devon RS, St Clair DM, Muir WJ, Blackwood DH, Porteous DJ. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- Mitchell B, Jacobs R, Li J, Chien S, Kintner C. A positive feedback mechanism governs the polarity and motion of motile cilia. Nature. 2007;447:97–101. doi: 10.1038/nature05771. [DOI] [PubMed] [Google Scholar]
- Sawamoto K, Wichterle H, Gonzalez-Perez O, Cholfin JA, Yamada M, Spassky N, Murcia NS, Garcia-Verdugo JM, Marin O, Rubenstein JL, Tessier-Lavigne M, Okano H, Alvarez-Buylla A. New neurons follow the flow of cerebrospinal fluid in the adult brain. Science. 2006;311:629–632. doi: 10.1126/science.1119133. [DOI] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Hu J, Chen W, Elliott G, Andre P, Gao B, Yang Y. Planar cell polarity breaks bilateral symmetry by controlling ciliary positioning. Nature. 2010;466:378–382. doi: 10.1038/nature09129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryavanshi S, Eddé B, Fox LA, Guerrero S, Hard R, Hennessey T, Kabi A, Malison D, Pennock D, Sale WS, Wloga D, Gaertig J. Tubulin glutamylation regulates ciliary motility by altering inner dynein arm activity. Curr Biol. 2010;20:435–440. doi: 10.1016/j.cub.2009.12.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J, Morlé L, Soulavie F, Laurençon A, Sagnol S, Durand B. Transcriptional control of genes involved in ciliogenesis: a first step in making cilia. Biol Cell. 2010;102:499–513. doi: 10.1042/BC20100035. [DOI] [PubMed] [Google Scholar]
- Tissir F, Qu Y, Montcouquiol M, Zhou L, Komatsu K, Shi D, Fujimori T, Labeau J, Tyteca D, Courtoy P, Poumay Y, Uemura T, Goffinet AM. Lack of cadherins Celsr2 and Celsr3 impairs ependymal ciliogenesis, leading to fatal hydrocephalus. Nat Neurosci. 2010;13:700–707. doi: 10.1038/nn.2555. [DOI] [PubMed] [Google Scholar]
- Town T, Breunig JJ, Sarkisian MR, Spilianakis C, Ayoub AE, Liu X, Ferrandino AF, Gallagher AR, Li MO, Rakic P, Flavell RA. The stumpy gene is required for mammalian ciliogenesis. Proc Natl Acad Sci U S A. 2008;105:2853–2858. doi: 10.1073/pnas.0712385105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vladar EK, Bayly RD, Sangoram AM, Scott MP, Axelrod JD. Microtubules enable the planar cell polarity of airway cilia. Curr Biol. 2012;22:2203–2212. doi: 10.1016/j.cub.2012.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel P, Read RW, Hansen GM, Payne BJ, Small D, Sands AT, Zambrowicz BP. Congenital hydrocephalus in genetically engineered mice. Vet Pathol. 2012;49:166–181. doi: 10.1177/0300985811415708. [DOI] [PubMed] [Google Scholar]
- Yu X, Ng CP, Habacher H, Roy S. Foxj1 transcription factors are master regulators of the motile ciliogenic program. Nat Genet. 2008;40:1445–1453. doi: 10.1038/ng.263. [DOI] [PubMed] [Google Scholar]