

Abstract
Motor neurons (MNs) are the neuronal class that is principally affected in amyotrophic lateral sclerosis (ALS), but it is widely known that individual motor pools do not succumb to degeneration simultaneously. Because >90% of ALS patients have an accumulation of cytoplasmic TDP-43 aggregates in postmortem brain and spinal cord (SC), it has been suggested that these inclusions in a given population may trigger its death. We investigated seven MN pools in our new inducible rNLS8 transgenic (Tg) mouse model of TDP-43 proteinopathy and found striking differences in MN responses to TDP-43 pathology. Despite widespread neuronal expression of cytoplasmic human TDP-43, only MNs in the hypoglossal nucleus and the SC are lost after 8 weeks of transgene expression, whereas those in the oculomotor, trigeminal, and facial nuclei are spared. Within the SC, slow MNs survive to end stage, whereas fast fatigable MNs are lost. Correspondingly, axonal dieback occurs first from fast-twitch muscle fibers, whereas slow-twitch fibers remain innervated. Individual pools show differences in the downregulation of endogenous nuclear TDP-43, but this does not fully account for vulnerability to degenerate. After transgene suppression, resistant MNs sprout collaterals to reinnervate previously denervated neuromuscular junctions concurrently with expression of matrix metalloproteinase 9 (MMP-9), a marker of fast MNs. Therefore, although pathological TDP-43 is linked to MN degeneration, the process is not stochastic and mirrors the highly selective patterns of MN degeneration observed in ALS patients.
SIGNIFICANCE STATEMENT Because TDP-43 is the major pathological hallmark of amyotrophic lateral sclerosis (ALS), we generated mice in which mutant human TDP-43 expression causes progressive neuron loss. We show that these rNLS8 mice have a pattern of axonal dieback and cell death that mirrors that often observed in human patients. This finding demonstrates the diversity of motor neuron (MN) populations in their response to pathological TDP-43. Furthermore, we demonstrate that resistant MNs are able to compensate for the loss of their more vulnerable counterparts and change their phenotype in the process. These findings are important because using a mouse model that closely models human ALS in both the disease pathology and the pattern of degeneration is critical to studying and eventually treating progressive paralysis in ALS patients.
Keywords: amyotrophic lateral sclerosis, motor neuron, reinnervation, rNLS mice, selective vulnerability, TDP-43
Introduction
Amyotrophic lateral sclerosis (ALS) patients typically present with motor deficits in one particular region, a result of the dieback of motor neuron (MN) axons innervating individual muscle fibers and subsequent MN death (Munsat et al., 1988). The motor symptoms then spread to contiguous anatomic regions until the patient becomes nearly completely paralyzed and this followed by death usually within 2–5 years of disease onset (Ravits and La Spada, 2009). The question of how ALS progresses to affect more and more MN pools remains unanswered, but at least two possibilities are worthy of consideration and they are not mutually exclusive: (1) a pathologic factor(s) spreads from cell to cell and initiates degeneration or (2) the same pathologic process or processes occurs in all MN pools and subtypes, but individual MNs have different intrinsic vulnerabilities to degenerate and only begin to do so when some distinct threshold is reached.
We have recently generated a new transgenic (Tg) mouse model of ALS called rNLS8 Tg that harbor a doxycycline (DOX)-suppressible transgene engineered to express human TDP-43 (hTDP-43) with a defective nuclear localization signal (hTDP-43ΔNLS) under the control of the human neurofilament heavy chain promoter NEFH (Walker et al., 2015). After the removal of DOX chow, these Tg mice have widespread neuronal expression of this mutant hTDP-43 protein, followed by the accumulation of phosphorylated, cytoplasmic TDP-43 inclusions in multiple different neuronal populations, especially MNs, with the subsequent onset of paralysis culminating in the death of these Tg mice ∼10 weeks after the initiation of transgene expression. Because this transgene is driven by NEFH, a neuron-specific promoter, we can use this mouse model to investigate whether there are differences in how individual neuronal subtypes respond to the accumulation of TDP-43. Therefore, in this study, we investigated how different MN pools are affected by the expression of the mutant hTDP-43ΔNLS protein. The studies described here also examined a potential mechanism underlying the onset and progression of ALS (TDP-43 accumulation and loss of endogenous function) and compared the selective loss of MN subtypes in the rNLS8 line of Tg mice with those reported in human ALS. Finally, because these mice show a functional recovery upon suppression of the transgene (Walker et al., 2015), this model allows us to investigate the identity of the MNs that underlie this recovery and to monitor potential reorganization and reinnervation of muscle.
Materials and Methods
Tg mice.
As described in Walker et al. (2015), rNLS8 mice were generated by crossing new Tg lines overexpressing the tetracycline transactivator (tTA) protein under the control of the human NEFH promoter with an existing Tg line that can be induced with DOX manipulation to express human TDP-43 with a defective nuclear localization signal (hTDP-43ΔNLS, hereafter referred to as hTDP-43). A DOX diet inhibits tTA from binding to the tetracycline promoter element, repressing hTDP-43 expression. When mice are taken off DOX, hTDP-43 expression is activated. These mice were killed at various time points, including at disease end stage, which was defined as 30% weight loss from peak. For all studies, both male and female mice were used.
Retrograde labeling.
Cholera toxin subunit B (CTB) conjugated to Alexa Fluor 488 or 594 was injected into the soleus (Sol) or tibialis anterior (TA) muscles of adult rNLS8 mice on the same day that either the transgene expression was initiated by DOX withdrawal (for the 1 week off DOX time point) or after 1 year of transgene suppression in animals that had been off DOX for 6 weeks (for the recovery time point). Animals were perfused and spinal cords (SCs) collected ∼96 h later. At that time, the injected muscle and surrounding muscles were checked for fluorescence to ensure the specificity of injections.
Electron microscopy.
Sol and TA muscles were fixed by perfusion and 2–4 d immersion in 2.5% glutaraldehyde + 1% paraformaldehyde in 0.1 m cacodylate buffer, pH 7.4 + 0.005% CaCl2, washed, dehydrated in ethanol, and embedded in epoxy resin (EMbed 812; Electron Microscopy Sciences). One micrometer cross-sections were stained with toluidine blue for light microscopy and areas with neuromuscular endings were selected for ultrathin sectioning, cut, and stained with 2% uranyl acetate in 50% ethanol and Reynold's lead citrate buffer (Daddow, 1983).
Electrophysiological measurements.
Similar to previous studies (Arnold et al., 2015), mice were anesthetized and two electrodes were placed on either side of the sciatic nerve at a paraspinal site. Bipolar electrodes were inserted into the gastrocnemius (GC) muscle. A controlled stimulation was applied to the nerve to evoke contractions from the GC muscle in 5 mA increments from 10 mA.
Immuofluorescence and quantification.
rNLS8 mice and nTg controls were perfused with ice-cold PBS, followed by 10% formalin, and then the brain, cervical and lumbar SC, tongue, masseter, and hindlimb muscles were surgically removed. Muscles were washed in PBS overnight and the CNS tissue was postfixed in 10% formalin overnight. All tissues were then washed in PBS and processed in a sucrose gradient for cryoprotective embedding.
To visualize neuromuscular junctions (NMJs), 30 μm longitudinal cryosections were incubated with α-bungarotoxin conjugated to Alexa Fluor 488 (raised in mouse; 1:500; Invitrogen) and an antibody to vesicular acetylcholine transporter (VAChT; raised in rabbit; 1:32,000; gift from Christopher Henderson, Columbia University) to label motor endplates and nerve terminals, respectively. A lack of colocalization indicated muscle denervation.
To analyze MN populations in the midbrain, brainstem, and cervical and lumbar SC, the following primary antibodies were used: guinea pig anti-VAChT (1:10,000); CNDR, mouse anti-human TDP-43 monoclonal antibody (mAb) (0.06 μg/ml, clone 5104, CNDR), rabbit anti-C-terminal TDP-43 (0.15 μg/ml, CNDR C1039), and goat anti-MMP-9 (1:2000; Sigma-Aldrich).
Nuclear and cytoplasmic TDP-43 inclusions were distinguished by immunostaining for hTDP-43, total TDP-43, and DAPI to label cell nuclei. Using merged images, the presence of nuclear TDP-43 inclusions was indicated by colocalization of TDP-43 and DAPI, whereas the presence of cytoplasmic inclusions of TDP-43 was indicated by a lack of colocalization of TDP-43 and DAPI.
Size analysis.
For MN cell size measurements, the largest cross-sectional areas were determined using NIS-Elements 3.0 software (Nikon) for cells with a VAChT-immunopositive cytoplasm and a visible nucleus. All MNs with a visible nucleus were imaged at 10× and outlined. Distribution histograms were constructed for each level of MMP-9 staining intensity by grouping cell body cross-sectional areas in 400 μm2 bins.
Statistics.
Statistical significance was determined using unpaired two-tailed t tests when comparing two groups or a one-way ANOVA to compare multiple groups, using SigmaPlot. A p-value of <0.05 was considered significant.
Study approval.
All procedures were performed in accordance with the National Institutes of Health's Guide for the Care and Use of Experimental Animals. Studies were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Results
Selective MN death is observed in hypoglossal and spinal MNs
In most MN pools investigated in the rNLS8 mice, the majority of MNs identified by positive immunostaining for VAChT, a marker of all MNs (Arvidsson et al., 1997), had persistent expression of hTDP-43 as assessed by immunostaining with 5104, a human-specific TDP-43 mAb (Kwong et al., 2014), beginning 1 week after DOX removal (Fig. 1A–C, summarized in Table 1). At the lumbar SC level, >80% of all L4–L5 MNs were hTDP-43+, including those that innervated the fast hindlimb muscle TA or the primarily slow muscle Sol, with those specific pools identified by separate retrograde labeling from each muscle with the fluorescent retrograde tracer CTB-594 (Conte et al., 2009) from three separate animals (Fig. 1D,E). Further, the proportion of hTDP-43+ MNs remained stable over time from 1 week off DOX for all pools investigated (Fig. 1F). However, despite moderate to very high levels of expression of the mutant hTDP-43 transgene in all investigated MN pools, there was a selective cell death of the MNs in only the lumbar SC (L4–L5) and hypoglossal nucleus (XII) (with ∼49% and 32% of total MNs lost, respectively) after 8 weeks of transgene expression (Fig. 1G, n = 4 mice examined per MN pool). There was a nonsignificant reduction in average MNs in the cervical SC after 8 weeks of chronic transgene expression (with only ∼3 MNs per ventral horn or ∼14% lost, p = 0.14). Therefore, chronic, cytoplasmic hTDP-43 expression was not sufficient to induce cell loss in oculomotor (III), trigeminal (V), or facial (VII) nuclei in rNLS8 mice. In fact, there was no correlation between the percentage of MNs expressing hTDP-43 at 1 week off DOX and the percentage MNs lost after 8 weeks of chronic hTDP-43 expression (R2 = 0.29, p = NS; data available in Table 1).
Figure 1.
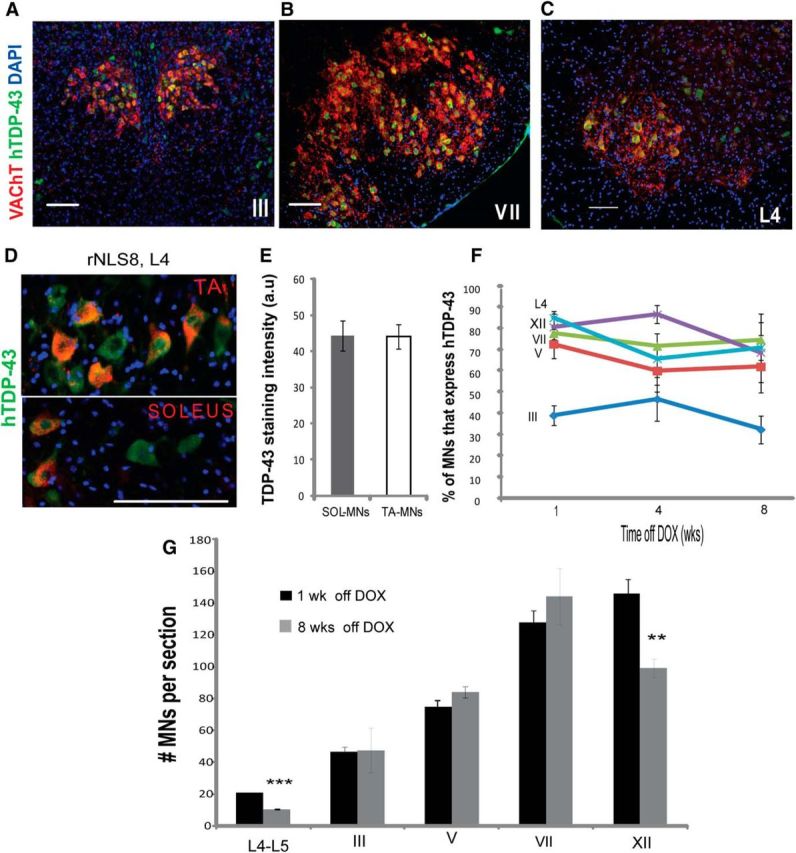
Selective MN death is observed in hypoglossal and spinal MNs despite uniformly high levels of hTDP-43 expression in all MN pools. A–C, Immunostaining for human hTDP-43 (green) and VAChT (MNs, red) in rNLS8 mice shows many MNs expressing high levels of hTDP-43 in oculomotor (A) and facial (B) nuclei, as well as in L4 SC (C) at 4 weeks off DOX. D, Almost all MNs of the fast-twitch TA pool (top) or slow-twitch Sol pool (bottom) identified by retrograde labeling with CTB-594 (red) are hTDP-43+ (green). E, There is no difference in hTDP-43 staining intensity (measured in arbitrary fluorescence units or a.u.) between these pools in the same mice at 1 week off DOX, 15–25 MNs per mouse from n = 3. t test, p = 0.97. F, After DOX removal, 65–80% of MNs in lumbar L4–L5 SC, the hypoglossal (XII), trigeminal (V), and facial (VII) nuclei, and ∼1/3 of those in the oculomotor nucleus (III) are hTDP-43+ in rNLS8 mice and these numbers remain stable over the disease course. Data are mean ± SD (n = 4 mice per MN pool). G, Eight weeks of cytoplasmic expression of hTDP-43 induces significant MN loss in lumbar L4–L5 SC and the hypoglossal nucleus (XII), but is not sufficient to induce MN loss in oculomotor, trochlear (III/IV), trigeminal (V), or facial (VII) nuclei in rNLS8 mice. Data are mean ± SD (n = 4 per motor pool), t test, **p < 0.01, ***p < 0.001. Scale bars, 100 μm.
Table 1.
Comparative MN vulnerability to degenerate: rNLS8 mouse model versus human
| Motor pool/subtype | rNLS8 mice |
Affected in ALS patients? | ||
|---|---|---|---|---|
| % MNs hTDP-43+ (mean ± SD)a | % lost by 8 weeks off DOX (mean ± SD) | % MNs w/o nuclear TDP-43 (mean ± SD)b | ||
| Oculomotor | 34.7 ± 4.7 | None | 7.3 ± 4 | Spared (Okamoto et al., 1993; Kanning et al., 2010) |
| Trigeminal | 64.6 ± 11.1 | None | 8.0 ± 2 | Mild degeneration (Iwanaga et al., 1997) |
| Facial | 71 ± 4.3 | None | 18.7 ± 4 | Mild to moderate degeneration (Iwanaga et al., 1997) |
| Hypoglossal | 76 ± 6.1 | 32.2 ± 5.7 | 24.7 ± 2 | ∼46% lost in ALS (Kiernan and Hudson, 1991); affected first in bulbar presentation |
| Lumbar MNs | 80.3 ± 3.3 | 49.5 ± 0.3 | 46 ± 3.2 | ∼50% lost in ALS (Kiernan and Hudson, 1991); affected early in the majority of patients |
| TA-innervating MNs | 100 [48, n = 3] | Not measured | Not measured | Affected early in disease (Dengler et al., 1990; Sharma et al., 1995; Kanning et al., 2010) |
| Sol-innervating MNs | 100 [23, n = 3] | Not measured | Not measured | Slow motor units are preferentially spared (Patten et al., 1979) |
aNumbers from rNLS8 mice at 1 week off DOX.
bNumbers from rNLS8 mice at 4 weeks off DOX.
Cell death and axonal dieback are not uniform from MNs of generally vulnerable pools
Next, we investigated in more detail vulnerable MN pools over time to define the timing and patterns of MN loss and axonal dieback. First, the hypoglossal nucleus was divided laterally using the natural functional grouping of MNs within this nucleus (dorsal and central groups, which are responsible for retraction, ridging, and furrowing of the tongue, compared with the ventral group, which underlie tongue protrusion; Fig. 2A) as described previously (Odutola, 1976). After 8 weeks of hTDP-43 expression, significantly more hypoglossal MNs were lost from the ventral portion than from the dorsal portion of the nucleus (38% vs 24%; Fig. 2B). Correspondingly, by 8 weeks off DOX, we found that 22 ± 4% of NMJs of the tongue muscle were denervated, whereas the motor axons that innervate the masseter muscle and originate from the resistant trigeminal nucleus remain intact (Fig. 2C,D).
Figure 2.
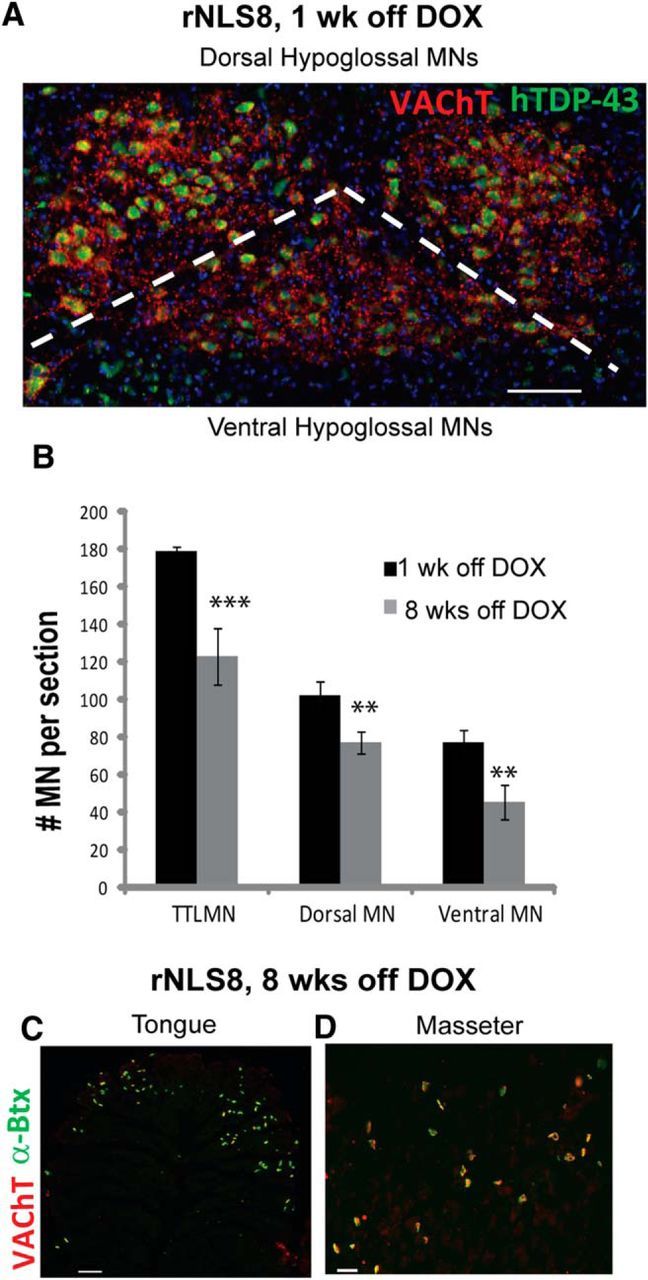
Within the generally vulnerable hypoglossal nucleus, certain MNs are more resistant to degeneration. A, Representative image of the hypoglossal nucleus from an rNLS8 mouse immunostained with VAChT (red) and hTDP-43 (green), with dorsal and ventral subdivisions separated by a dashed line. B, After 8 weeks of transgene expression, MNs are lost from both subdivisions, with a larger percentage lost from the ventral subdivision. Data are mean ± SEM, n = 3 animals, t test. C, D, Muscle denervation is observed in the tongue, but not the masseter muscles, of rNLS8 mice at late disease stages. Representative cryosections of muscles immunostained with VAChT (red) and labeled with α-Btx (green) to identify NMJs shows that, by 8 weeks off DOX, 22 ± 4% of NMJs of the tongue muscle are denervated (C), whereas the masseter muscle NMJs are still intact (D). **p < 0.01, ***p < 0.001. Scale bars, 100 μm.
At the level of MN subtype in the lumbar SC, the MNs that drive muscle contraction, α-MNs, can be divided into functional categories depending on the contractile properties of the muscle fiber that they innervate: fast-twitch fatigable (FF), fast-twitch fatigue-resistant (FR), and slow-twitch fatigue-resistant (S) (Burke et al., 1973). In rNLS8 mice, there is a selective loss of the fast MNs over time, as assessed by a fast MN marker, MMP-9 (Kaplan et al., 2014; Fig. 3A–D). At 1 week off DOX, 49 ± 2% of MNs at L4–L5 were MMP-9+. By 8 weeks off DOX, ∼44% of total MNs had been lost and, of those remaining, <2 MNs (∼11%) per ventral horn were MMP-9+, suggesting that the MMP-9+ MNs were those that were lost. In agreement with this assumption, the number of MMP-9−, putative slow MNs remained unchanged over time (Fig. 3D). Further, given that fast MNs have the largest perikaryal area among α-MN subtypes (Friese et al., 2009; Leroy et al., 2014), we also examined changes in the average soma size after 6 weeks off DOX, when significant neuron death had occurred. There was a dramatic increase in the proportion of small MNs by 6 weeks off DOX (Fig. 3E), consistent with a loss of the largest (likely FF) MNs. Moreover, the decrease in average MN size is unlikely due to tissue shrinkage because the average size of neurons containing the calcium-binding protein calbindin in the superficial dorsal horn did not change between 1 and 6 weeks off DOX (Fig. 3F,G).
Figure 3.
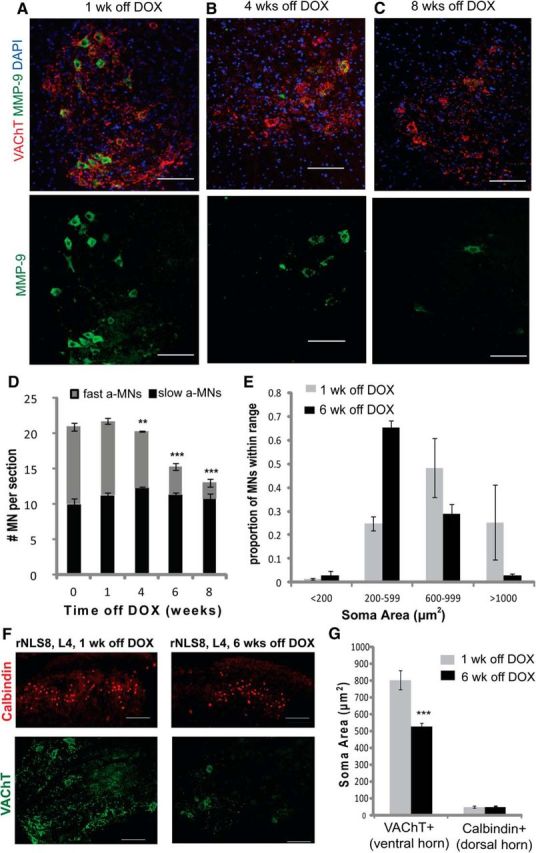
Fast MNs in the lumbar SC are those that are lost in rNLS8 mice over time. A–C, Immunostaining for VAChT (red) and MMP-9 (green) on SCs of rNLS8 mice 1 week (A), 4 weeks (B), and 8 weeks (C) off DOX shows a loss of fast (MMP-9+) MNs. D, At 1 week off DOX, 49 ± 2% of MNs at L4–L5 are MMP-9+. By 8 weeks off DOX, ∼44% of total MNs have been lost and only ∼11% of those remaining are MMP-9+, with no change in the number of MMP-9− MNs over time. E, MN size distributions of VAChT+ MNs show a decrease in size from 1 week off DOX (gray bars) to 6 weeks off DOX (black bars), consistent with a loss of the largest MNs. Data are mean ± SEM, n = 4 animals. F, Representative cryosection of an rNLS8 lumbar SC 1 week off DOX immunostained for VAChT (green) and calbindin (red). G, Average size of neurons containing calbindin in the superficial dorsal horn did not change between 1 week (gray bar) and 6 weeks off DOX (black bar), but there was a significant decrease in the average soma size of VAChT+ MNs in the ventral horn. This suggests that the reason for the decrease in average size of the MNs is that the largest cells have died, rather than that the tissue has shrunk. Data are mean ± SD (n = 3), ***p < 0.001, **p < 0.01.
To test whether there is a difference between axonal dieback for fast versus slow MNs, we used overlap of VAChT+ motor terminals (red) with acetylcholine receptors stained using α-bungarotoxin or BTX (green) as an indicator of innervated motor endplates in rNLS8 mice (Fig. 4A,B). There was symmetrical muscle denervation for all three hindlimb muscles investigated (data not shown). The fast-twitch TA muscle and the fast-twitch lateral portion of the gastrocnemius (LGC) (Pun et al., 2006) both exhibited significant denervation by 4 weeks off DOX, whereas the slow-twitch Sol remained almost completely innervated at that time point and did not have a significant decrease in the number of intact NMJs until 8 weeks off DOX (Fig. 4C). At 6 weeks off DOX, an intermediate time point in the disease course, we also observed ultrastructural differences between the majority of NMJs from the Sol and TA muscles (Fig. 4D). Whereas the Sol NMJs mostly look normal with the nerve's axonal terminal (Fig. 4D, arrow) contacting muscle fibers in a set of slit-like depressions (Fig. 4D, arrowhead) on the muscle fiber surface, the motor end plates of many TA muscle fibers are not supplied by axons and there is evident collagen deposition (Fig. 4D, #) in the place of a nerve. Finally, by disease end-stage, only 14 ± 2% and 15 ± 2% of NMJs of the lateral GC and TA muscles, respectively, remained intact, whereas 52 ± 5% of the Sol remained connected (Fig. 4C). This morphological change mirrors functional motor impairment because there was also a significant decrease in the evoked compound muscle action potential (CMAP) in the GC muscle by 4 weeks off DOX that persisted and worsened by 6 weeks off DOX (Fig. 4E,F). Therefore, consistent with human ALS patients (Fischer et al., 2004), muscles with more FF fibers lose innervation earliest (Table 1).
Figure 4.
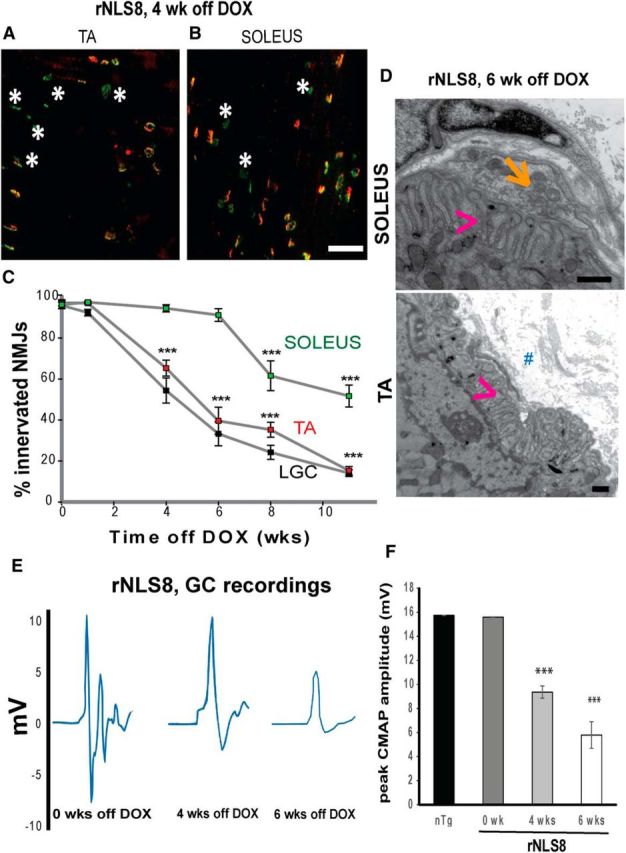
Axonal dieback is not uniform from MNs of vulnerable MN pools. A, Representative muscle cryosection from the TA of an end-stage rNLS8 mouse, with overlap of VAChT+ motor terminals (red) and acetylcholine receptors (BTX, green) as an indicator of innervated motor endplates. Denervated NMJs are marked with white asterisks. B, At the same time point, the slow Sol has far fewer denervated NMJs. Scale bar, 100 μm. C, Denervation of the Sol muscle (green) is later than for the fast TA (red) and LGC (black). Data are mean ± SEM, n = 4 per time point. D, At the electron microscopic level, NMJ structural differences are apparent between the Sol and TA muscle from an rNLS8 mouse after 6 weeks of transgene expression. Although the Sol still has a nerve ending (orange arrow) lying in the set of depressions on the muscle fiber surface (pink arrowhead) to form a NMJ, there is no nerve making contact with the motor terminal in the TA and there is evident collagen (marked with #) in the place of a nerve. Scale bar, 500 nm. E, F, Evoked CMAPs in the GC muscle after stimulation of the sciatic nerve in nTg and rNLS8 mice that are still on DOX and off DOX for 4 or 6 weeks. E, Individual traces showing the M-wave, the maximum peak-to-peak value of which is used to calculate the CMAP. F, CMAP measurements significantly decrease with hTDP-43 expression. Data are mean ± SD, n = 3–4 animals per genotype, *p < 0.05, **p < 0.01, ***p < 0.001.
As was the case with MN pools, differences in the timing of axonal dieback of different MN subtypes cannot be explained by the level of hTDP-43 expression given that all MNs of the fast-twitch TA MN pool or slow-twitch Sol MN pool identified by retrograde labeling with CTB-594 were hTDP-43+ (Fig. 1D, Table 1). Moreover, there was no difference in hTDP-43 staining intensity between resistant Sol-innervating MNs and susceptible TA-innervating MNs (Fig. 1E). Therefore, rather than widespread stochastic cell death, there was a specific ALS-relevant pattern of selective MN death in rNLS8 mice.
At 4 weeks off DOX, a time point before lower MN loss (Walker et al., 2015) but after initiation of axonal dieback from fast-twitch muscles (Fig. 4C), we investigated all MN pools using a polyclonal antibody that labels both endogenous mouse TDP-43 and hTDP-43 to examine potential differences in nuclear TDP-43 expression between resistant and vulnerable MNs (Fig. 5). We found that the percentage of neurons in a given MN pool with clearance of nuclear TDP-43 increased with greater vulnerability to degenerate in this ALS mouse model (Fig. 5A–E). For example, at 4 weeks off DOX, nearly half of all lumbar MNs had no nuclear TDP-43 (cf. TDP-43 localization in nontransgenic (nTg) to rNLS8 Tg CNS tissue samples in Fig. 5C,D and the quantitative data in Fig. 5E). However, this distinction does not hold up at the level of α-MN subtype because there was not a significant difference in nuclear TDP-43 signal between fast or slow subtypes using either size (<600 μm2 vs >600 μm2, data not shown) or MMP-9 status, a marker of fast MNs to distinguish subtypes (Fig. 5F,G), suggesting a lack of correlation between nuclear TDP-43 clearance and selective MN vulnerability.
Figure 5.
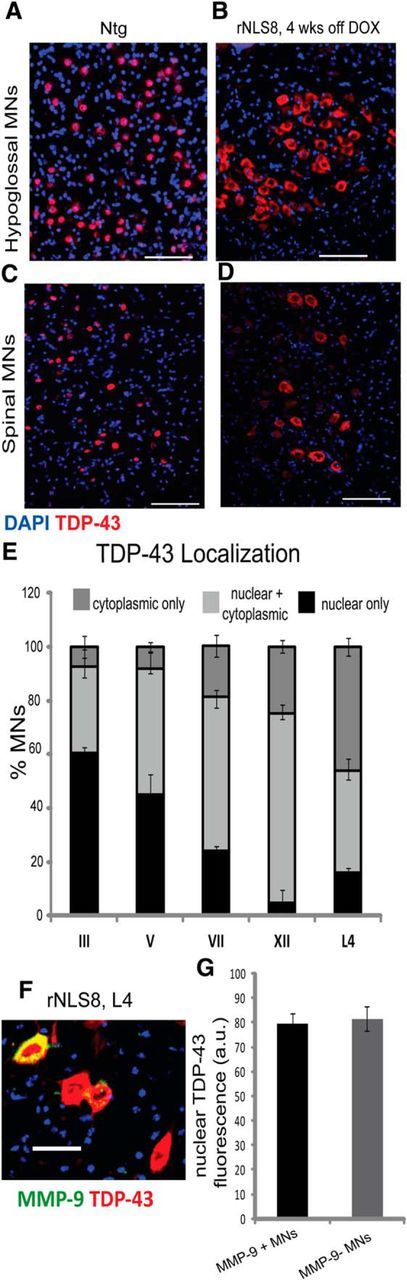
The percentage of MNs in a given MN pool without nuclear TDP-43 expression increases with increasing vulnerability to degenerate. A–D, Immunostaining for total TDP-43 (mouse + human, red) and DAPI on cryosections of the hypoglossal nucleus (A, B) and lumbar SC (C, D) shows the primarily nuclear localization of TDP-43 in the wild-type animals and the primarily cytoplasmic localization and loss of nuclear TDP-43 in rNLS8 mice. Scale bar, 100 μm. E, Percentage of MNs with nuclear TDP-43 at 4 weeks off DOX varies significantly between motor pools, with the two most vulnerable groups, the hypoglossal and L4 MNs, having the fewest MNs with nuclear TDP-43. Data are mean ± SEM, n = 3–4 animals, one-way ANOVA. F, There is no difference in the level of nuclear TDP-43 (red) between vulnerable MMP-9 (green) MNs and resistant MMP-9− MNs at L4. G, Fluorescence intensity (in arbitrary units) of the TDP-43 signal in the nucleus does not differ between MMP-9+ (black bar) and MMP-9− MNs (gray bar). Data are mean ± SEM of 280–300 MNs from 3 animals, p = 0.77.
Resistant FR and S MNs underlie motor recovery after hTDP-43ΔNLS suppression
In the rNLS mice, we reported previously that suppression of hTDP-43ΔNLS expression after disease progression allowed for recovery (Walker et al., 2015) and we hypothesize this is because the resistant, surviving MNs can reinnervate muscle. To investigate the nature of this recovery further, we examined both the muscle fibers and spinal MNs of rNLS8 mice that were off DOX treatment continuously for 6 weeks, followed by a reintroduction of DOX to suppress the hTDP-43ΔNLS transgene expression for 3 d to 1 year (Fig. 6A). Muscle fibers reinnervated by collateral sprouts are converted to the subtype of the reinnervating MN (Burke, 1982), a process leading to muscle fiber type grouping after disease. Therefore, this process allowed us to assess the pattern of MN axonal dieback and subsequent reinnervation by examining muscle fiber subtypes, identified by their different myosin isoform expression (FF: type-IIB; FR: type-IIA; S: type-I; Pun et al., 2006). We observed grouping of fast oxidative/glycolytic fibers, identified by immunohistochemistry with antibody to myosin heavy chain (MHC) IIA, suggesting reinnervation by the intermediate force, FR MNs, after transgene suppression (Fig. 6B–D).
Figure 6.

Resistant FR and S MNs reinnervate muscle and underlie motor recovery after hTDP-43ΔNLS suppression. A, Schematic of experimental timeline. B–D, Transverse cryosections of TA from a nTg mouse (B), an rNLS8 mouse after 6 weeks of transgene expression (C), and an rNLS8 mouse after 12 weeks of transgene suppression (D) immunostained with MHCIIA to label muscle fibers innervated by FR MNs. MHCIIA+ fibers are grouped after recovery, suggesting reinnervation by FR MNs after transgene suppression. Scale bars, 100 μm. E, Average fiber diameter is significantly decreased in the TA muscle after 6 weeks of transgene expression, reflecting muscle atrophy. Atrophy is reversed after 12 weeks of transgene suppression, when the average TA fiber diameter increases beyond nTg controls. Data are mean ± SD from n = 3–4 muscles per treatment, F(2, 8) = 67.8, p < 0.001. F, After 6 weeks, but not 1 week, of hTDP-43 expression, the largest TA-innervating FF MNs are lost and the remaining smaller FR/S MNs sprout to innervate unoccupied motor endplates during recovery from disease. Data are mean ± SEM from n = 3 animals, 20–30 MNs measured from each.
Further, there was clear muscle fiber atrophy at 6 weeks off DOX compared with nTg controls, but the TA muscle fibers regained their full average diameter size by 12 weeks after transgene suppression (28 ± 3 μm at 6 weeks off DOX vs 46 ± 2 μm after 12 weeks of recovery, Fig. 6B–E). Moreover, after 6 weeks of hTDP-43 expression, the largest TA-innervating FF MNs were lost and the remaining smaller FR/S MNs sprouted to innervate unoccupied motor endplates during recovery from ALS-like disease in this mouse model. This was assessed by backfilling MNs from Sol and TA with CTB-594 or CTB-488 after 1 year of recovery (i.e., 1 year back on DOX after having been off DOX for 6 weeks). Specifically, in nTg mice, the TA-innervating MNs were ∼300 μm2 larger than the Sol-innervating MNs (mean ± SEM for TA was 993 ± 51 μm2 vs 686 ± 64 μm2 for Sol, p = 0.02; Fig. 6F) because large FF MNs composed a large fraction of the TA, but not the Sol MN pool (Hegedus et al., 2007; Kanning et al., 2010). However, there was no difference between average MN size between these motor pools in rNLS8 mice after FF MN loss and 1 year of disease recovery and muscle reinnervation (TA vs Sol, 613 ± 47 μm2 vs 628 ± 19 μm2, p = 0.78; Fig. 6F).
Finally, after only 2 weeks of transgene suppression, there was a return of MMP-9 expression in ∼53% of lumbar (L4–L5) MNs (Fig. 7A). Surprisingly, this is nearly the same percentage of total L4–L5 MNs that expressed MMP-9 before axonal dieback and cell death (52% before transgene expression in rNLS8 mice, Fig. 3A,D, vs 50% after 12 weeks of recovery, Fig. 7D), suggesting that resistant FR and S MNs changed their phenotype to reach a fixed relative level of MMP-9 expression in a population. The reappearance of MMP-9 occurred as hTDP-43 was being cleared, but before muscle reinnervation, suggesting that contact with fast muscle fibers did not dictate MMP-9 expression. This was further confirmed in a model of sciatic nerve crush, which temporarily disconnects nerve and muscle (Baptista et al., 2007), because this did not change MMP-9 expression status at the L4–L5 level in nTg mice 7 d after nerve crush (Fig. 7B). In fact, MMP-9 reaches its full lumbar expression level after 2 weeks of transgene suppression (Fig. 7D), a time point when the TA is still nearly 70% denervated and motor dysfunction persists in the GC muscle, as reflected in evoked CMAP measurements (Fig. 7C). Although the functional relevance of this MMP-9 reappearance awaits future studies, we know the phenotype switch was complete because, after 8 weeks of transgene suppression, but not yet at 6 weeks of suppression, the CMAP recorded from reinnervated GC returned to its full maximal amplitude (Fig. 7C). We conclude that certain MN subpopulations are more resistant to the chronic expression of cytoplasmic hTDP-43 and these MNs underlie recovery from ALS-like disease after this TDP-43 is cleared in this mouse model.
Figure 7.
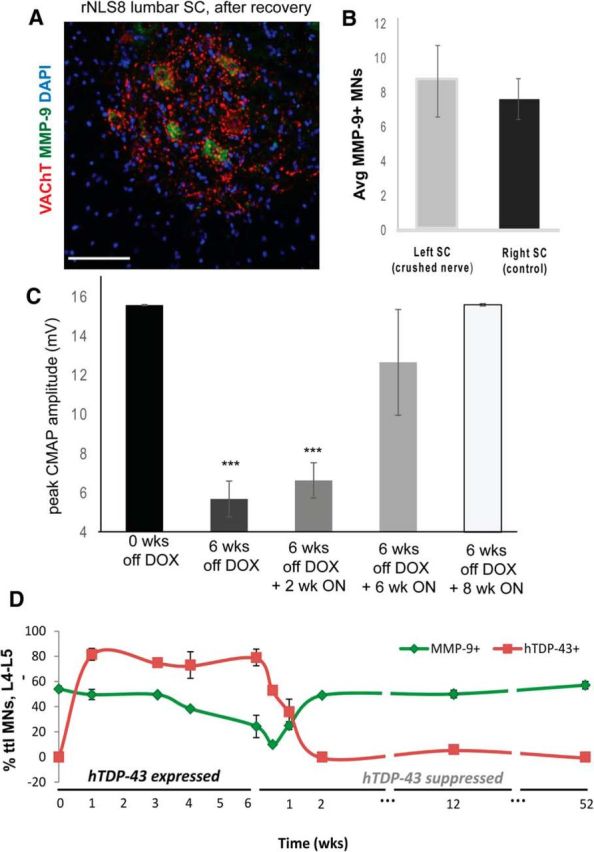
Remaining MNs in the lumbar SC change phenotype after hTDP-43 clearance. A, Immunostaining for MMP-9 (green), VAChT (to label MNs, red), and DAPI on cryosections of the lumbar SC of an rNLS8 mouse after only 2 weeks of transgene suppression showing a return of MMP-9 expression, which is lost during the disease course (Fig. 3A–C). Scale bar, 100 μm. B, Average (Avg) MMP-9 perikaryal MN expression at the L4–L5 level did not differ between the crushed side ventral horn and the control (noncrushed) side. Data are mean ± SD, n = 3, p = 0.49. C, Despite previous axonal dieback and lumbar FF MN loss, evoked CMAPs return to predenervation maxima after 8 weeks of transgene suppression, a result of muscle reinnervation and a phenotypic switch of former FR/S MNs. Data are mean ± SD, n = 3–6. D, Timeline of MMP-9 (red) and hTDP-43 (green) expression in rNLS8 mice shown as percentage of the total (ttl) MNs. Data are mean ± SD, n = 2–7 animals per time point, *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Using our new rNLS8 mouse model, which is based on a common point in the degenerative pathway (i.e., the mislocalization of TDP-43, an important RNA-binding protein from the nucleus to the cytoplasm) rather than a genetic mutation that only represents a minority of patients, the selective vulnerability to MN degeneration is recapitulated as described previously for this hallmark pathological signature of ALS (Saxena and Caroni, 2011; Brockington et al., 2013). We showed that, when moderate to very high levels of the mutant hTDP-43 transgene are expressed in all investigated MN pools, only MNs in the lumbar SC and hypoglossal nucleus are lost after 8 weeks of transgene expression. This demonstrates that pathological TDP-43 is linked to the initiation of ALS-like MN degeneration, but it is not sufficient to kill all MN subtypes in a given time frame. Therefore, the degenerative process that we observed in rNLS8 mice is not stochastic, but is characterized by temporally defined, selective vulnerabilities of distinct neurons and axons to degenerate in this mouse model of ALS.
Findings from autopsies of ALS patients had suggested that TDP-43 pathology spreads from an initial starting point throughout interconnected regions of the brain and SC in a stereotypical sequential pattern (Brettschneider et al., 2013). If the spread of pathological TDP-43 is the only factor that underlies the progression of ALS, then it is reasonable to assume that MN pools such as the oculomotor nucleus or Onuf's nucleus only appear resistant because patients usually die before pathological TDP-43 has spread to them and initiated degeneration. However, we see that these and other MN pools remain disease resistant even when cytoplasmic hTDP-43 expression is chronically induced. Further, the identity of the MNs that are vulnerable to degenerate after the accumulation of pathological hTDP-43 in the rNLS8 mice are those in the hypoglossal nucleus and fast lumbar MN pools. Because these are often the earliest MN populations affected in human patients (Kiernan and Hudson, 1991), our findings suggest that subtype-specific elements may exist in these MNs that permit toxic effects of accumulations of pathological TDP-43 cytoplasmic inclusions to emerge.
It is also worth noting that these MN pools are those with the greatest loss in the most commonly used murine model of familial ALS, Tg mice expressing mutant forms of human SOD1 (mSOD1) (Gurney, 1994). Specifically, the earliest motor defects detected in this model are in muscles innervated by the lumbar-level MNs (Pun et al., 2006; Kaplan et al., 2014), followed by those innervated by hypoglossal MNs (Smittkamp et al., 2008), whereas the oculomotor nucleus and pelvic MNs are resistant through to terminal stages of ALS-like disease in the mSOD1 model system (Ferrucci et al., 2010; Kaplan et al., 2014). Further, in both of these mouse models of familial and now sporadic ALS, low-threshold S MNs are resistant and still innervate muscle at end stage (Kanning et al., 2010; Saxena and Caroni, 2011; Kaplan et al., 2014). In addition, a recent study describing newly generated Tg mouse lines with ALS-associated mutations in another RNA-processing gene, FUS, shows selective degeneration of fast MNs in the SC, as well as oculomotor resistance (Sharma et al., 2016).
Therefore, the same MN pools are the most vulnerable in human ALS patients as in mouse models discussed above based on either a downstream pathological protein (as in rNLS8 mice) or genetic mutations (as in mSOD1 mice) (Table 1). Further, this observation that rNLS8 mice have a pattern of neuron death that recapitulates that of sporadic ALS patients with disease onset in the lower limbs demonstrates their future utility for probing biologically relevant questions, rather than simply showing that massively altered protein localization can have general deleterious effects on neurons.
What mechanism(s) underlies this difference in vulnerability? Because the loss of normal nuclear TDP-43 function is detrimental to MNs and can cause ALS/frontotemporal lobe degeneration phenotypes (Yang et al., 2014), we hypothesized that vulnerable and resistant MN pools might have differences in the clearance of nuclear TDP-43. We previously reported that the rNLS8 mice show a general loss of nuclear TDP-43 concomitant with the accumulation of cytoplasmic TDP-43 (Walker et al., 2015). We hypothesized that perhaps the presence of hTDP-43 in the cytoplasm causes a downregulation of endogenous TDP-43 and a loss of function in vulnerable MNs, whereas resistant MNs could maintain both endogenous mouse TDP-43 and hTDP-43. We found that, though the more resistant MN pools did have a higher percentage of MNs with nuclear TDP-43 than the vulnerable pools, there was no difference in the TDP-43 localization in FF and FR versus S MNs at the lumbar level (Fig. 5). Therefore, nuclear clearance of endogenous TDP-43 is not sufficient to explain the differences in a MN′s ability to cope with the transgene-driven expression of hTDP-43. Future studies should examine potential differences in TDP-43-shuttling dynamics or other regulatory pathways that might underlie the differences in TDP-43 nuclear clearance at different levels and/or cellular responses to cope with nuclear loss within specific MN pools and subtypes.
We have also demonstrated that the removal of pathological TDP-43 is beneficial even in late-stage disease. Specifically, we observed that the resistant MNs were able to compensate functionally for the loss of their more vulnerable counterparts by developing the physiological characteristics of these lost MNs. MMP-9 expression returns to spinal MNs as hTDP-43 is being cleared from MNs (Fig. 7D), suggesting that resistant FR and S MNs change their phenotype as part of the recovery process before their collateral sprouting to innervate the motor endplates left vacated after FF MN axonal dieback and cell death. The return of MMP-9 leaves two pressing questions unanswered: (1) what determines the overall expression of MMP-9 in a given MN pool?, and (2) what impact does MMP-9 expression have on an individual MN′s functional properties? One hypothesis that we explored was that MMP-9 expression returned after recovery because of signaling from the muscle. However, when we examined the timeline of reexpression of MMP-9, it became clear that MMP-9 fully returned by 2 weeks of transgene suppression, before TA or GC muscle reinnervation. Further, disconnecting motor axons from their target muscle by sciatic nerve crush did not change MMP-9 expression levels. An interesting future direction will be to assess whether MMP-9 is necessary for fast muscle reinnervation.
It has been well documented that skeletal muscle can switch fiber type to meet physiological demands or respond to injury (LeBrasseur et al., 2011). Several factors can induce fiber-type switching, with the best-studied factor being changes in nerve activity or loading (Schiaffino and Reggiani, 2011). We speculate that MMP-9 critically affects the functional properties of the resistant MNs to help them adapt to handling a larger innervation field. MMP-9 may exert its effect by altering neuronal activity, as it has been widely reported to do in the hippocampus (Gorkiewicz et al., 2010; Michaluk et al., 2011). In the future, it will also be interesting to measure the excitability of resistant, smaller MNs without MMP-9 before the onset of ALS-like disease and with MMP-9 after recovery.
In this study, we identified the most vulnerable cells in rNLS8 mice and established the time frame of their degeneration. We note that the same MNs (hypoglossal MNs and fast-twitch fiber-innervating lumbar SC MNs) are vulnerable to TDP-43-dependent degeneration, suggesting these neurons are intrinsically vulnerable in human sporadic ALS and should be targeted specifically for future ALS disease-modifying therapy. Finally, we demonstrate that resistant MNs are able to compensate for the loss of their more vulnerable counterpart MNs and change their phenotype in the process. This result is particularly encouraging because it suggests that patients may someday respond to interventional therapies and we can focus our research on these and on preventative measures.
Footnotes
This work was supported by The Brody Family Medical Trust Fellowship (K.J.S.). We thank Adam Walker for thorough initial characterization of these mice and for helpful advice, Guanghui Ge for assistance with mouse husbandry, and Tahiyana Khan and Jeremy Wertkin for assistance with tissue sectioning and immunostaining.
The authors declare no competing financial interests.
References
- Arnold WD, Sheth KA, Wier CG, Kissel JT, Burghes AH, Kolb SJ. Electrophysiological motor unit number estimation (MUNE) measuring compound muscle action potential (CMAP) in mouse hindlimb muscles. J Vis Exp. 2015;103:52899. doi: 10.3791/52899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvidsson U, Riedl M, Elde R, Meister B. Vesicular acetylcholine transporter (VAChT) protein: a novel and unique marker for cholinergic neurons in the central and peripheral nervous systems. J Comp Neurol. 1997;378:454–467. [PubMed] [Google Scholar]
- Baptista AF, Gomes JR, Oliveira JT, Santos SM, Vannier-Santos MA, Martinez AM. A new approach to assess function after sciatic nerve lesion in the mouse: adaptation of the sciatic static index. J Neurosci Methods. 2007;161:259–264. doi: 10.1016/j.jneumeth.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, Suh E, Van Deerlin VM, Wood EM, Baek Y, Kwong L, Lee EB, Elman L, McCluskey L, Fang L, Feldengut S, Ludolph AC, Lee VM, Braak H, Trojanowski JQ. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. 2013;74:20–38. doi: 10.1002/ana.23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockington A, Ning K, Heath PR, Wood E, Kirby J, Fusi N, Lawrence N, Wharton SB, Ince PG, Shaw PJ. Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 2013;125:95–109. doi: 10.1007/s00401-012-1058-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE. Motor units in cat muscles: anatomical considerations in relation to motor unit types. Adv Neurol. 1982;36:31–45. [PubMed] [Google Scholar]
- Burke RE, Levine DN, Tsairis P, Zajac FE., 3rd Physiological types and histochemical profiles in motor units of the cat gastrocnemius. J Physiol. 1973;234:723–748. doi: 10.1113/jphysiol.1973.sp010369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte WL, Kamishina H, Reep RL. Multiple neuroanatomical tract-tracing using fluorescent Alexa Fluor conjugates of cholera toxin subunit B in rats. Nat Protoc. 2009;4:1157–1166. doi: 10.1038/nprot.2009.93. [DOI] [PubMed] [Google Scholar]
- Daddow LY. A double lead stain method for enhancing contrast of ultrathin sections in electron microscopy: a modified multiple staining technique. J Microsc. 1983;129:147–153. doi: 10.1111/j.1365-2818.1983.tb04169.x. [DOI] [PubMed] [Google Scholar]
- Dengler R, Konstanzer A, Küther G, Hesse S, Wolf W, Struppler A. Amyotrophic lateral sclerosis: macro-EMG and twitch forces of single motor units. Muscle Nerve. 1990;13:545–550. doi: 10.1002/mus.880130612. [DOI] [PubMed] [Google Scholar]
- Ferrucci M, Spalloni A, Bartalucci A, Cantafora E, Fulceri F, Nutini M, Longone P, Paparelli A, Fornai F. A systematic study of brainstem motor nuclei in a mouse model of ALS, the effects of lithium. Neurobiol Dis. 2010;37:370–383. doi: 10.1016/j.nbd.2009.10.017. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Friese A, Kaltschmidt JA, Ladle DR, Sigrist M, Jessell TM, Arber S. Gamma and alpha motor neurons distinguished by expression of transcription factor Err3. Proc Natl Acad Sci U S A. 2009;106:13588–13593. doi: 10.1073/pnas.0906809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorkiewicz T, Szczuraszek K, Wyrembek P, Michaluk P, Kaczmarek L, Mozrzymas JW. Matrix metalloproteinase-9 reversibly affects the time course of NMDA-induced currents in cultured rat hippocampal neurons. Hippocampus. 2010;20:1105–1108. doi: 10.1002/hipo.20736. [DOI] [PubMed] [Google Scholar]
- Gurney ME. Transgenic-mouse model of amyotrophic lateral sclerosis. N Engl J Med. 1994;331:1721–1722. doi: 10.1056/NEJM199412223312516. [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Gordon T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2007;28:154–164. doi: 10.1016/j.nbd.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Iwanaga K, Hayashi S, Oyake M, Horikawa Y, Hayashi T, Wakabayashi M, Kondo H, Tsuji S, Takahashi H. Neuropathology of sporadic amyotrophic lateral sclerosis of long duration. J Neurol Sci. 1997;146:139–143. doi: 10.1016/S0022-510X(96)00297-3. [DOI] [PubMed] [Google Scholar]
- Kanning KC, Kaplan A, Henderson CE. Motor neuron diversity in development and disease. Annu Rev Neurosci. 2010;33:409–440. doi: 10.1146/annurev.neuro.051508.135722. [DOI] [PubMed] [Google Scholar]
- Kaplan A, Spiller KJ, Towne C, Kanning KC, Choe GT, Geber A, Akay T, Aebischer P, Henderson CE. Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron. 2014;81:333–348. doi: 10.1016/j.neuron.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan JA, Hudson AJ. Changes in sizes of cortical and lower motor neurons in amyotrophic lateral sclerosis. Brain. 1991;114:843–853. doi: 10.1093/brain/114.2.843. [DOI] [PubMed] [Google Scholar]
- Kwong LK, Irwin DJ, Walker AK, Xu Y, Riddle DM, Trojanowski JQ, Lee VM. Novel monoclonal antibodies to normal and pathologically altered human TDP-43 proteins. Acta Neuropathol Commun. 2014;2:33. doi: 10.1186/2051-5960-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBrasseur NK, Walsh K, Arany Z. Metabolic benefits of resistance training and fast glycolytic skeletal muscle. Am J Physiol Endocrinol Metab. 2011;300:E3–E10. doi: 10.1152/ajpendo.00512.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy F, Lamotte d'Incamps B, Imhoff-Manuel RD, Zytnicki D. Early intrinsic hyperexcitability does not contribute to motoneuron degeneration in amyotrophic lateral sclerosis. Elife. 2014;3 doi: 10.7554/eLife.04046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaluk P, Wawrzyniak M, Alot P, Szczot M, Wyrembek P, Mercik K, Medvedev N, Wilczek E, De Roo M, Zuschratter W, Muller D, Wilczynski GM, Mozrzymas JW, Stewart MG, Kaczmarek L, Wlodarczyk J. Influence of matrix metalloproteinase MMP-9 on dendritic spine morphology. J Cell Sci. 2011;124:3369–3380. doi: 10.1242/jcs.090852. [DOI] [PubMed] [Google Scholar]
- Munsat TL, Andres PL, Finison L, Conlon T, Thibodeau L. The natural history of motoneuron loss in amyotrophic lateral sclerosis. Neurology. 1988;38:409–413. doi: 10.1212/WNL.38.3.409. [DOI] [PubMed] [Google Scholar]
- Odutola AB. Cell grouping and Golgi architecture of the hypoglossal nucleus of the rat. Exp Neurol. 1976;52:356–371. doi: 10.1016/0014-4886(76)90211-9. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Hirai S, Amari M, Iizuka T, Watanabe M, Murakami N, Takatama M. Oculomotor nuclear pathology in amyotrophic lateral sclerosis. Acta Neuropathol. 1993;85:458–462. doi: 10.1007/BF00230482. [DOI] [PubMed] [Google Scholar]
- Patten BM, Zito G, Harati Y. Histologic findings in motor neuron disease: relation to clinically determined activity, duration, and severity of disease. Arch Neurol. 1979;36:560–564. doi: 10.1001/archneur.1979.00500450054009. [DOI] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology. 2009;73:805–811. doi: 10.1212/WNL.0b013e3181b6bbbd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron. 2011;71:35–48. doi: 10.1016/j.neuron.2011.06.031. [DOI] [PubMed] [Google Scholar]
- Schiaffino S, Reggiani C. Fiber types in mammalian skeletal muscles. Physiol Rev. 2011;91:1447–1531. doi: 10.1152/physrev.00031.2010. [DOI] [PubMed] [Google Scholar]
- Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, Shneider NA. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun. 2016;7:10465. doi: 10.1038/ncomms10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma KR, Kent-Braun JA, Majumdar S, Huang Y, Mynhier M, Weiner MW, Miller RG. Physiology of fatigue in amyotrophic lateral sclerosis. Neurology. 1995;45:733–740. doi: 10.1212/WNL.45.4.733. [DOI] [PubMed] [Google Scholar]
- Smittkamp SE, Brown JW, Stanford JA. Time-course and characterization of orolingual motor deficits in B6SJL-Tg(SOD1–G93A)1Gur/J mice. Neuroscience. 2008;151:613–621. doi: 10.1016/j.neuroscience.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AK, Spiller KJ, Ge G, Zheng A, Xu Y, Zhou M, Tripathy K, Kwong LK, Trojanowski JQ, Lee VM-Y. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 2015;130:643–660. doi: 10.1007/s00401-015-1460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Wang H, Qiao T, Yang B, Aliaga L, Qiu L, Tan W, Salameh J, McKenna-Yasek DM, Smith T, Peng L, Moore MJ, Brown RH, Jr, Cai H, Xu Z. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2014;111:E1121–1129. doi: 10.1073/pnas.1322641111. [DOI] [PMC free article] [PubMed] [Google Scholar]