

Abstract
Patients with Type 6 episodic ataxia (EA6) have mutations of the excitatory amino acid transporter EAAT1 (also known as GLAST), but the underlying pathophysiological mechanism for EA6 is not known. EAAT1 is a glutamate transporter expressed by astrocytes and other glia, and it serves dual function as an anion channel. One EA6-associated mutation is a P>R substitution (EAAT1P>R) that in transfected cells has a reduced rate of glutamate transport and an abnormal anion conductance. We expressed this EAAT1P>R mutation in glial cells of Drosophila larvae and found that these larvae exhibit episodic paralysis, and their astrocytes poorly infiltrate the CNS neuropil. These defects are not seen in Eaat1-null mutants, and so they cannot be explained by loss of glutamate transport. We instead explored the role of the abnormal anion conductance of the EAAT1P>R mutation, and to do this we expressed chloride cotransporters in astrocytes. Like the EAAT1P>R mutation, the chloride-extruding K+-Cl− cotransporter KccB also caused astroglial malformation and paralysis, supporting the idea that the EAAT1P>R mutation causes abnormal chloride flow from CNS glia. In contrast, the Na+-K+-Cl− cotransporter Ncc69, which normally allows chloride into cells, rescued the effects of the EAAT1P>R mutation. Together, our results indicate that the cytopathology and episodic paralysis in our Drosophila EA6 model stem from a gain-of-function chloride channelopathy of glial cells.
SIGNIFICANCE STATEMENT We studied a mutation found in episodic ataxia of the dual-function glutamate transporter/anion channel EAAT1, and discovered it caused malformation of astrocytes and episodes of paralysis in a Drosophila model. These effects were mimicked by a chloride-extruding cotransporter and were rescued by restoring chloride homeostasis to glial cells with a Na+-K+-2Cl− cotransporter. Our findings reveal a new pathophysiological mechanism in which astrocyte cytopathology and neural circuit dysfunction arise via disruption of the ancillary function of EAAT1 as a chloride channel. In some cases, this mechanism might also be important for neurological diseases related to episodic ataxia, such as hemiplegia, migraine, and epilepsy.
Keywords: astrocyte, channelopathy, Drosophila, EAAT, episodic ataxia
Introduction
Episodic ataxias are characterized by occasional attacks of cerebellar incoordination or imbalance, and comprise several genetically and clinically distinct types (Jen et al., 2007). Like other paroxysmal diseases, episodic ataxias are caused by mutations in ion channels and have therefore been grouped among diseases known collectively as channelopathies (Russell et al., 2013). For example, episodic ataxia Type 1 and Type 2 are due to mutations in genes encoding a neuronal voltage-gated potassium channel (KCNA1) (Browne et al., 1994) or a P/Q-type calcium channel (CACNA1A) (Ophoff et al., 1996), respectively. However, patients with episodic ataxia Type 6 (EA6) have mutations in excitatory amino acid transporter 1 (EAAT1) (Jen et al., 2005; de Vries et al., 2009), a sodium-dependent glutamate transporter expressed in cerebellar astrocytes known as Bergmann glia. How EA6-associated mutations of EAAT1 affect astrocytes and impact function of neural circuits remains unknown.
One EA6-associated mutation is a proline-to-arginine (P>R) substitution (EAAT1P290R), at a residue that is conserved in other species, including Drosophila melanogaster (Jen et al., 2005) (see Fig. 1A). In transfected cells in culture, this P>R mutation in human EAAT1 impairs glutamate uptake and has reduced expression at the cell surface (Jen et al., 2005; Winter et al., 2012). However, EAAT glutamate transporters have dual function as anion channels (Fairman et al., 1995; Cater et al., 2016), and EAAT1P290R also causes a gain-of-function increase in EAAT1 anion currents (Winter et al., 2012). Activation of the EAAT anion channel is dependent upon both glutamate and sodium (Wadiche et al., 1995), and the homologous P>R mutation in human EAAT3 has been proposed to alter anion currents and reduce glutamate transport rates by decelerating a conformational change associated with sodium binding to the transporter (Hotzy et al., 2013). There has been recent progress in charting the anion permeation pathway through EAAT transporters (Cater et al., 2014; Machtens et al., 2015), but its importance for normal brain function is poorly understood, and its contribution to neurological diseases is not known. Here we used Drosophila as a model to examine in vivo the effects of the EAAT1 P>R mutation on astrocytes and neural circuit function, and to investigate whether pathogenesis involves unusual EAAT1-mediated conductance of chloride, a key physiological anion.
Figure 1.

Eaat1 function is conserved between humans and Drosophila, and the Eaat1P>R mutation is nonfunctional in vivo. A, Schematic of Venus-tagged transgenes, with sites of P>R mutations highlighted. B, Quantification in a box-and-whisker plot of full body contractions completed by L1 larvae in a 3 min period (one-way ANOVA, for trials using DmEaat1WT F(2,27) = 37.30, p < 0.0001, or HsEAAT1WT F(2,27) = 50.56, p < 0.0001, or DmEaat1P>R F(2,27) = 113.0, p < 0.0001, or HsEAAT1P>R F(2,27) = 134.7, p < 0.0001). ***p < 0.001. ns, Not significant.
Materials and Methods
Fly stocks and genetics.
Drosophila stocks were obtained from the Bloomington Stock Center (UAS-mCD8::GFP, UAS-CD4::tdTomato, Repo-Gal4) or published sources: UAS-KccB (Hekmat-Scafe et al., 2010), UAS-Ncc69 (Leiserson et al., 2011). Our laboratory produced Eaat1-null mutants (Eaat1SM2) and CG31235-Gal4 (Stacey et al., 2010), here renamed Nazgul-Gal4, which drives moderate levels of transgene expression in a restricted subset of CNS glia that includes astrocytes (Stacey et al., 2010; Peco et al., 2016).
To create UAS-DmEaat1WT::Venus and UAS-HsEAAT1WT::Venus transgenes, PCR was used to amplify the coding region from DmEaat1 cDNA RE20434 (Drosophila Genomic Research Centre) or from HsEaat1 cDNA 5264000 (Open Biosystems), respectively. Products were transferred into pTWV, a Gateway-amenable pUAST vector, which adds a C-terminal Venus epitope tag in-frame. Quikchange (Agilent Technologies) was used for site-directed mutagenesis of these constructs to generate the derivatives UAS-DmEaat1P>R::Venus and UAS-HsEAAT1P>R::Venus. Transgenic flies with random insertions on the second or third chromosome were generated via standard microinjection (BestGene). The transgenes produced Venus-tagged proteins of the correct size as determined by Western blotting (see Fig. 5C), and correct localization to the thin infiltrative processes of astrocytes within the Drosophila neuropil (see Fig. 2B). For experiments where we drove expression of these transgenes in a wild-type background, the number of transgene copies and their locations were as follows: DmEaat1WT (1 copy, second chromosome), DmEaat1P>R (1 copy, third chromosome), HsEAAT1WT (2copies, both on third chromosome), and HsEAAT1P>R (4 copies, 2 on second chromosome, 2 on third chromosome). For rescue experiments in Eaat1SM2-null mutants, they were as follows: DmEaat1WT (1 copy, third chromosome), DmEaat1P>R (1 copy, third chromosome), HsEAAT1WT (2copies, third chromosome), and HsEAAT1P>R (2 copies, third chromosome).
Figure 5.

Ncc69 rescues glial infiltration defect and paralysis caused by Eaat1P>R. A, Representative images of Eaat1 and Gat labeling of astrocyte processes within CNS neuropil. B, Astrocyte neuropil infiltration (mean ± SEM), normalized to controls (one-way ANOVA, for trials using DmEaat1P>R F(3,30) = 17.67, p < 0.0001, or HsEAAT1P>R F(2,25) = 112.3, p < 0.0001). C, Endogenous Eaat1 levels are reduced upon DmEaat1P>R expression in glia but restored with Ncc69 coexpression. Representative Western blot using anti-Eaat1, simultaneously detecting endogenous Eaat1, and ectopic Venus-tagged transgenes. Bottom, Bar graph showing quantification (mean ± SEM) of the expression level of endogenous Eaat1 expressed as a percentage of control Repo>DmEaat1WT. Three repeated Western blots were quantified (one-way ANOVA, F(3,8) = 47.07, p < 0.0001). D, Quantification of Naz+ glia per abdominal segment of L1 larvae (mean ± SEM) (Kruskal–Wallis, H = 5.718, p = 0.1262). E, Box-and-whisker plot for locomotor performance (one-way ANOVA, for trials using DmEaat1P>R F(3,44) = 7.742, p = 0.0003, or HsEAAT1P>R F(2,35) = 22.34, p < 0.0001). *p < 0.05. **p < 0.01. ***p < 0.001. ns, Not significant.
Figure 2.

Eaat1P>R reduces astrocyte infiltration. A, Representative images of infiltrative astrocyte processes labeled for the membrane-associated GABA transporter (Gat) (green) within the brain and ventral nerve cord (vnc) of a dissected, wild-type L1 larva. White box represents typical area selected for high-power views in other panels. B, Representative high-power images of astrocyte membranes, where each panel represents a single optical confocal section from the middle of the dorsal-ventral axis of the neuropil. Glial-specific expression of Venus-tagged transgenes (anti-GFP) with Repo-Gal4 (Repo>), with colabeling for Gat and Gal4-driven CD4-tdTomato (CD4-tdTom). Scale bar, 10 μm. C, Quantification of astrocyte neuropil infiltration (mean ± SEM), normalized to controls (two-tailed unpaired t test, for trials using Drosophila (Dm) transgenes t(16) = 6.529, p < 0.0001, or human (Hs) transgenes t(13) = 14.52, p < 0.0001). D, Quantification of Nazgul-positive (Naz+) glia per abdominal segment of L1 larvae (mean ± SEM) (two-tailed unpaired t test, for trials using Drosophila (Dm) transgenes t(14) = 0.8556, p = 0.4066, or human (Hs) transgenes t(13) = 0.9983, p = 0.3363). E, CD4-tdTom labeling at late embryonic stage 17 (E17), when astrocytes initiate ingrowth of CNS neuropil. F, Like heterozygous controls (Eaat1SM2/+), astrocytes of Eaat1-null mutants (Eaat1SM2/SM2) infiltrate the neuropil normally, as visualized with anti-Gat immunohistochemistry. Scale bar, 10 μm. Quantification of astrocyte neuropil infiltration (mean ± SEM), normalized to control (two-tailed unpaired t test, t(19) = 0.8674, p = 0.3965). ***p < 0.001. ns, Not significant.
Immunohistochemistry and Western blotting.
Larvae of either sex were dissected at first instar (L1) and prepared for immunohistochemistry according to standard procedures. PFA (4%) was routinely used for fixation, except for anti-Eaat1 immunohistochemistry, for which samples were incubated in Bouin's fixative for 10 min at room temperature. Primary polyclonal antibodies used were as follows: rabbit anti-Eaat1 (Peco et al., 2016) (1:4000), rabbit anti-Gat (Stork et al., 2014) (1:2000, C-terminal epitope), rabbit anti-GFP (Torrey Pines Biolabs, 1:1000), rabbit anti-Naz (von Hilchen et al., 2010) (1:500), and rat anti-HA (Roche, 1:1000). Secondary antibodies used were AlexaFluor-647-conjugated goat anti-rabbit (1:1000, Invitrogen, #A21244) and AlexaFluor-568-conjugated goat anti-rat (1:1000, Invitrogen, #A11077). Tissues were mounted in SlowFade Gold reagent (Invitrogen) and UltraPure Glycerol (Invitrogen, 1:1).
For Western blotting, lysates from 10 late-stage pupal heads of either sex were prepared in 25 μl extraction buffer (20 mm Tris-Cl, pH 7.4, 100 mm NaCl, 10 mm NaF, 1 mm Na3VO4, 0.3% Triton, 10% glycerol, 2 mm EDTA, 1× protease inhibitors, Roche), then mixed with 2× Laemmli buffer (1:1) (Bio-Rad) and incubated at 100°C for 1 min. The 15 μl of each sample was run on 10% SDS-PAGE gels, blotted to nitrocellulose membrane, and then probed sequentially with rabbit anti-Eaat1 (1:20000) and mouse anti-actin (1:5000, Sigma #A4700). For detection, HRP-conjugated secondary antibodies anti-rabbit (1:3000, Bio-Rad) or anti-mouse (1:20000, Promega #W4021) were used and revealed with chemiluminescence (HyGLO Chemiluminescent HRP Antibody Detection Reagent, Denville Scientific). To quantify the mean signal intensity for Eaat1 or GFP (Venus), normalized to that of actin, we analyzed three Western blots where each replicate was a lysate from a distinct pool of 10 pupae.
Microscopy, imaging, and quantification.
Confocal microscopy was performed with an Olympus Fluoview FV1000/BX63 confocal laser-scanning microscope using a 60× oil-immersion objective lens. Captured images were processed using Fiji software (Schindelin et al., 2012). To quantify infiltration of astrocyte processes immunostained with anti-Gat, we examined an area of 1400 μm2 in Z-stack images of the VNC neuropil, focused on abdominal segments starting from A1. Images were thresholded manually to distinguish astrocyte processes from background. Fiji's Tubeness plugin (http://fiji.sc/Tubeness) was first used to segment tubular processes; then the Skeletonize 2D/3D command was used to simplify the tubular processes to their centerlines. The percentage of area covered by processes was calculated for each of 9 sequential 1-μm-thick optical sections of the neuropil, situated in the z-axis between glial cell bodies flanking the dorsal and ventral surfaces of the neuropil. The mean neuropil infiltration for the control group was designated 100%. Seven to 10 larvae were examined in each group.
Locomotor behavior.
As described previously (Stacey et al., 2010), larval locomotor performance (see Figs. 1B, 4D, 5E) was quantified on 6-cm-diameter agar plates (after 5 min acclimatization) by counting full body peristaltic contractions in 3 sequential 3 min bouts for each animal. L1 larvae of either sex were examined in the afternoon, at least 1 h after hatching, and 10–14 larvae were examined for each genotype. For each experimental group, the control carries the same UAS-transgene but does not carry the Gal4 driver. To quantify episodic paralysis (see Fig. 3), movies of L1 larvae of either sex were recorded for 5.5 h, using a Sony Handycam where the plate was illuminated by ring-shaped LED source (Dioder, IKEA). Each video was imported into ImageJ (Schneider et al., 2012) as image sequence at 1/3 frames per second. Background signal from the arena was estimated by the time-series average and subtracted from each frame. Tracking of 20 animals per genotype was performed with TrackMate (Schindelin et al., 2012) upon median filtering. To ignore larval interactions with walls of each plate, tracking was limited to a circular ROI defined by 95% of the plate diameter. Larvae exiting the ROI terminated a track, and reentry initiated a new track. Tracks were divided into bouts defined as continuous time stretches in which animals were either crawling or motionless for at least 150 s. To do so, we used a custom Python script (Extract_Bouts_From_Tracks.py; http://dx.doi.org/10.5281/zenodo.49879) that analyzed a 150 s neighborhood centered around each time-point and processed the positioning of larvae as follows: (1) each tracked position was tagged with a binary flag based on an arbitrary cutoff for instantaneous speed (3 pixels/frame); (2) consecutive frames sharing the same binary flag were summed while (3) ignoring sporadic flag switches between consecutive frames.
Figure 4.
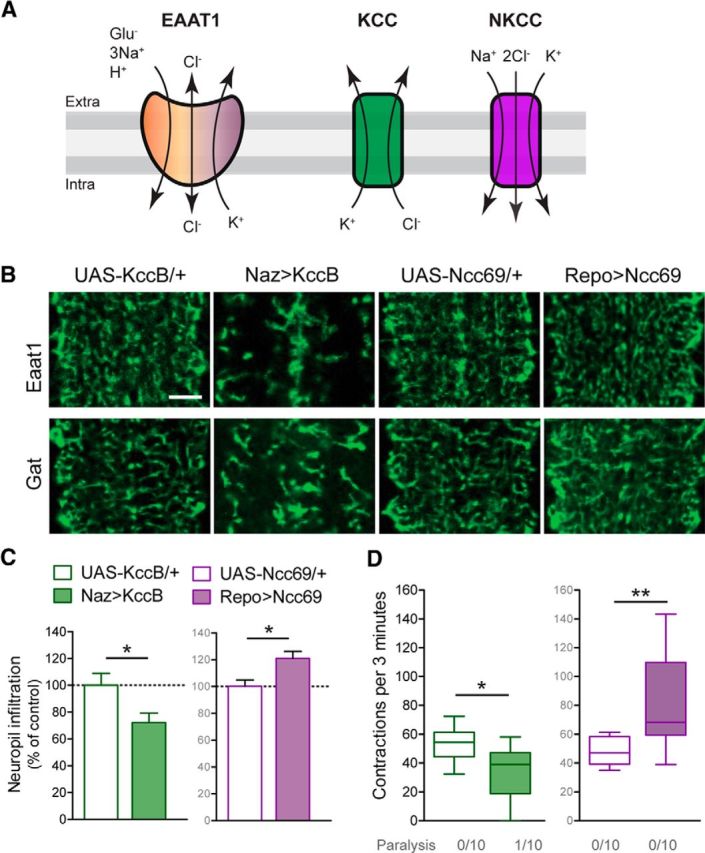
KccB and Ncc69 have opposing effects on astrocyte infiltration and larval locomotion. A, Schematic of ion exchange through EAAT1, KCC, and NKCC transporters. B, Representative images of astrocyte processes within larval neuropil labeled for Eaat1 or Gat. Animals expressing KccB with Repo-Gal4 did not hatch as larvae, and Naz-Gal4 was used instead. Scale bar, 10 μm. C, Quantification of astrocyte neuropil infiltration (mean ± SEM), normalized to controls (two-tailed unpaired t test, for trials using Kcc t(14) = 2.444, p = 0.0284, or Ncc69 t(12) = 2.973, p = 0.0116). D, Quantification of locomotor performance in a box-and-whisker plot (two-tailed unpaired t test, for trials using Kcc t(18) = 2.838, p = 0.0109, or Ncc69 t(18) = 3.044, p = 0.0070). *p < 0.05. **p < 0.01.
Figure 3.

Eaat1P>R causes episodic paralysis. A, B, The 5.5 h of video tracking of controls and DmEaat1P243R-expressing L1 larvae. A, Ten of the longest tracks are represented, showing bouts of crawling (gray) or motionlessness (black). B, Quantification (mean ± SEM, two-tailed unpaired t test) of motionless bouts/track (t(38) = 2.722, p = 0.0098), their average duration (t(38) = 2.034, p = 0.0489), and maximum instantaneous speed (t(38) = 0.2418, p = 0.8103). *p < 0.05. **p < 0.01. ns, Not significant.
Graphs and statistics.
Prism (GraphPad) software was used for graphing and statistical analyses. Graphs of larval crawling performance express data in box-and-whisker plots where boxes show the median (horizontal line) and the limits of the first and third quartiles, and whiskers depict the minimum and maximum of all data points. Data in each group were tested for normal distribution using the Shapiro–Wilk test. Data of two groups with normal distribution were analyzed using unpaired Student's t test (one-tailed), and data of more than two groups were compared using one-way ANOVA followed by Bonferroni's post hoc test if data were normally distributed, or with a Kruskal–Wallis test if not normally distributed in one or more groups.
Results
Drosophila larvae normally move via peristaltic crawling interrupted occasionally by brief pauses and turning. We showed previously that Eaat1-null larvae rarely make these peristaltic contractions but can be rescued by expressing Drosophila Eaat1 in CNS glial cells using the GAL4-UAS-system (Stacey et al., 2010). With transgenic flies carrying UAS-driven, Venus-tagged versions of wild-type Drosophila Eaat1 (DmEaat1WT) or wild-type human EAAT1 (HsEAAT1WT) (Fig. 1A), we used this in vivo rescue assay to demonstrate that EAAT function is conserved between Drosophila and humans (Fig. 1B). In contrast, neither the P>R mutation found in EA6 (HsEAAT1P>R) nor its Drosophila equivalent (DmEaat1P>R) was able to rescue Eaat1-nulls (Fig. 1A,B), indicating that this particular mutation renders EAAT1 nonfunctional in vivo.
We next examined the effects of expressing DmEaat1P>R or HsEAAT1P>R in wild-type larvae. Astrocytes expressing DmEaat1P>R or HsEAAT1P>R appeared to differentiate normally based on their expression of GABA transporter (Gat), but their mature morphology appeared abnormal relative to astrocytes expressing DmEaat1WT or HsEAAT1WT (Fig. 2A,B). We quantified their infiltration of CNS neuropil and found that it was dramatically reduced (Fig. 2C). This reduced infiltration was confirmed by expressing the plasma membrane-tethered reporter CD4-tdTom in astrocytes (Fig. 2B), and was not due simply to reduced astrocyte number as revealed by their expression of the neuropil-associated glial marker Nazgul (Naz) (Fig. 2D). Nor was it due to postembryonic deterioration of astrocytes because the defect was first evident when astrocytes normally initiate ingrowth (Stork et al., 2014) (Fig. 2E). The effects of the Eaat1P>R mutation contrast with Eaat1-null mutants, in which astrocytes infiltrate the CNS neuropil normally (Fig. 2F).
Unlike larvae expressing DmEaat1WT or HsEAAT1WT (data not shown), those expressing DmEaat1P>R or HsEAAT1P>R had episodes of complete paralysis during which they were flaccid and unable to respond to mechanical stimulation (for example of DmEaat1P>R, see Movies 1–3). With video tracking of DmEaat1P>R-expressing larvae, we quantified these episodes of prolonged motionlessness (Fig. 3A), and found that they were far more frequent and lengthy than in controls. Importantly, DmEaat1P>R-expressing larvae could achieve the same maximum speed as controls during phases when they were not paralyzed (Fig. 3B), suggesting that the underlying neuronal circuitry for locomotion is largely intact.
Movie 1.
Movies 1–3 in real-time demonstrating effects of expressing DmEaat1P>R (with Repo-Gal4) on L1 larval locomotion, and responses to mechanical stimulation. Movie 1, Crawling and responsive.
Movie 2.
Paralyzed and nonresponsive.
Movie 3.
A movie capturing the transition from paralysis to crawling.
The phenotypes caused by the P>R mutation are clearly distinct from those in Eaat1-null mutants where astrocytes infiltrate the CNS neuropil normally and where locomotor deficits include sustained and one-sided contractions, but these animals respond immediately to mechanical stimulation (Stacey et al., 2010). Therefore, the phenotypes caused by the P>R mutation cannot be explained by loss of glutamate transporter function alone and may instead arise via the ancillary function of EAAT1 as a chloride channel.
Chloride is important for cells to maintain membrane potential and osmotic balance, and chloride levels are influenced in large part by cation-chloride cotransporters expressed by neurons and glia (Kaila et al., 2014). These include the K+-Cl− cotransporters (KCCs), which extrude chloride, and the Na+-K+-2Cl− cotransporters (NKCCs), which drive chloride uptake (Fig. 4A). We tested whether these cotransporters might also influence astrocyte morphologies and locomotor function. We found that expression of the Drosophila KCC KccB reduced astrocyte infiltration of the neuropil in L1 larvae (Fig. 4B,C). It also led to reduced locomotor performance, with some larvae exhibiting paralysis and nonresponsiveness to mechanical stimuli (Fig. 4D). Thus, expression of a KCC mimicked the effects of the P>R mutation, suggesting that the P>R mutation causes increased chloride extrusion from Drosophila astrocytes. Conversely, expression of the Drosophila NKCC Ncc69 led to increased neuropil infiltration by astrocytes (Fig. 4B,C), and increased locomotor performance (Fig. 4D), showing that astrocyte morphology and wild-type behavioral output are influenced by chloride homeostasis via cotransporters.
If reduced astrocyte infiltration and paralysis caused by the P>R mutation were due to excessive chloride extrusion, they ought to be rescued by simultaneous expression of Ncc69 to counteract this extrusion with inward chloride flow. Indeed, Ncc69 rescued neuropil infiltration by astrocytes expressing DmEaat1P>R or HsEAAT1P>R (Fig. 5A,B). Consistent with the reduced astrocyte infiltration caused by the Eaat1P>R mutation, Western blots of pupal heads (largely CNS tissues) showed that DmEaat1P>R lowered endogenous levels of Eaat1 and, furthermore, that this too was at least partially restored by Ncc69 (Fig. 5C). Replacement of Ncc69 with a membrane marker (CD4-tdTomato) did not rescue these effects (Fig. 5A–C), demonstrating that these improvements were not due to titration of Gal4 from the DmEaat1P>R transgene. Nor were they due to increased astrocyte number (Fig. 5D). Remarkably, Ncc69 fully rescued locomotor performance of larvae expressing DmEaat1P>R, improved it for larvae expressing HsEAAT1P>R, and prevented paralysis for both (Fig. 5E).
Discussion
We developed an animal model in which an EA6-associated mutation was found to compromise astrocyte morphology and cause episodes of paralysis. Both of these effects could be specifically rescued by glial expression of an NKCC cotransporter to drive chloride uptake, and so we conclude that both are likely to stem from increased outward chloride flow from glia via the mutant transporter. Because of the importance of chloride for osmotic balance, we speculate this could lead to reduced volume of astrocytes and withdrawal of their thin infiltrative processes from synapses within neuropil, although additional experiments will be needed to test this model directly.
Reduced astrocyte infiltration could have manifold consequences for synapse function because it alters the proximity to synapses of transporters and channels for transmitters, including glutamate, ions, and other neuroactive substances. In one example of this from the supraoptic nucleus of the mammalian hypothalamus, alteration of the degree of synapse ensheathment by astrocytes during lactation leads to changes in the availability of extracellular d-serine, which has important effects on glutamate receptor activity at neuronal synapses nearby (Panatier et al., 2006). The paralysis seen in our Drosophila model could arise from altered availability or metabolism of neuroactive substances that are engaged during locomotor activity. During periods of paralysis, larval inactivity could allow these substances to return to levels that allow larvae to recover and resume crawling.
In animals expressing the Eaat1P>R mutation, endogenous Eaat1 levels are reduced in Western blots. Destabilization of endogenous Eaat1 following coassembly with Eaat1P>R might contribute to this reduction, but changes in astrocyte size and morphology likely play an indirect role because Ncc69 can partially rescue endogenous Eaat1 expression. Regardless of mechanism, one might imagine that lower endogenous Eaat1 levels causing reduced glutamate transport in some way contribute to the phenotypes we have observed. However, we note that loss-of-function nonsense mutations of EAAT1 have not been reported for EA6 patients, and that mice null for EAAT1 do not display ataxia (Watase et al., 1998). Here we found that Drosophila Eaat1-null animals do not have the defective astrocyte infiltration or episodic paralysis that we observed for animals expressing the Eaat1P>R mutation. Together, these observations suggest that the proximate cause of EA6 is not reduced glutamate transport. Rather, because the NKCC Ncc69 rescued astrocyte infiltration, endogenous Eaat1 levels, and paralysis in our model, we provide evidence that EA6 is principally a chloride channelopathy of glial cells, which may or may not be compounded by reduced glutamate recovery that accompanies astrocyte cytopathology and/or reduced endogenous Eaat1 protein levels. Because the EAAT1P290R mutation also led to hemiplegia, migraine, and seizures (Jen et al., 2005), these diseases might have a similar pathophysiological mechanism in some cases.
Footnotes
This work was supported by Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada, and Canada Foundation for Innovation grants to D.J.v.M.; and Research Institute of McGill University Health Center salary award to N.P., McGill Faculty of Medicine salary award to D.J.v.M., and McGill Integrated Program in Neuroscience salary award to N.P. We thank Marc Freeman, Ben Altenhein, Haig Keshishian, William Leiserson, Mark Tanouye, and the stock centers in Bloomington and Vienna for reagents; and Keith Murai, Brian Chen, Charles Bourque, Yong Rao, and members of the Murai, Rao, and D.J.v.M. laboratories for advice.
The authors declare no competing financial interests.
References
- Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, Litt M. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136–140. doi: 10.1038/ng1094-136. [DOI] [PubMed] [Google Scholar]
- Cater RJ, Vandenberg RJ, Ryan RM. The domain interface of the human glutamate transporter EAAT1 mediates chloride permeation. Biophys J. 2014;107:621–629. doi: 10.1016/j.bpj.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cater RJ, Ryan RM, Vandenberg RJ. The split personality of glutamate transporters: a chloride channel and a transporter. Neurochem Res. 2016;41:593–599. doi: 10.1007/s11064-015-1699-6. [DOI] [PubMed] [Google Scholar]
- de Vries B, Mamsa H, Stam AH, Wan J, Bakker SL, Vanmolkot KR, Haan J, Terwindt GM, Boon EM, Howard BD, Frants RR, Baloh RW, Ferrari MD, Jen JC, van den Maagdenberg AM. Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch Neurol. 2009;66:97–101. doi: 10.1001/archneurol.2008.535. [DOI] [PubMed] [Google Scholar]
- Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature. 1995;375:599–603. doi: 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Mercado A, Fajilan AA, Lee AW, Hsu R, Mount DB, Tanouye MA. Seizure sensitivity is ameliorated by targeted expression of K+-Cl− cotransporter function in the mushroom body of the Drosophila brain. Genetics. 2010;184:171–183. doi: 10.1534/genetics.109.109074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotzy J, Schneider N, Kovermann P, Fahlke C. Mutating a conserved proline residue within the trimerization domain modifies Na+ binding to excitatory amino acid transporters and associated conformational changes. J Biol Chem. 2013;288:36492–36501. doi: 10.1074/jbc.M113.489385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. 2005;65:529–534. doi: 10.1212/01.WNL.0000172638.58172.5a. [DOI] [PubMed] [Google Scholar]
- Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain. 2007;130:2484–2493. doi: 10.1093/brain/awm126. [DOI] [PubMed] [Google Scholar]
- Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci. 2014;15:637–654. doi: 10.1038/nrn3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiserson WM, Forbush B, Keshishian H. Drosophila glia use a conserved cotransporter mechanism to regulate extracellular volume. Glia. 2011;59:320–332. doi: 10.1002/glia.21103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machtens JP, Kortzak D, Lansche C, Leinenweber A, Kilian P, Begemann B, Zachariae U, Ewers D, de Groot BL, Briones R, Fahlke C. Mechanisms of anion conduction by coupled glutamate transporters. Cell. 2015;160:542–553. doi: 10.1016/j.cell.2014.12.035. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/S0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. Glia-derived d-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Peco E, Davla S, Camp D, Stacey MS, Landgraf M, van Meyel DJ. Drosophila astrocytes cover specific territories of the CNS neuropil and are instructed to differentiate by Prospero, a key effector of Notch. Development. 2016;143:1170–1181. doi: 10.1242/dev.133165. [DOI] [PubMed] [Google Scholar]
- Russell JF, Fu YH, Ptáèek LJ. Episodic neurologic disorders: syndromes, genes, and mechanisms. Annu Rev Neurosci. 2013;36:25–50. doi: 10.1146/annurev-neuro-062012-170300. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey SM, Muraro NI, Peco E, Labbé A, Thomas GB, Baines RA, van Meyel DJ. Drosophila glial glutamate transporter Eaat1 is regulated by fringe-mediated notch signaling and is essential for larval locomotion. J Neurosci. 2010;30:14446–14457. doi: 10.1523/JNEUROSCI.1021-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork T, Sheehan A, Tasdemir-Yilmaz OE, Freeman MR. Neuron-glia interactions through the Heartless FGF receptor signaling pathway mediate morphogenesis of Drosophila astrocytes. Neuron. 2014;83:388–403. doi: 10.1016/j.neuron.2014.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Hilchen CM, Hein I, Technau GM, Altenhein B. Netrins guide migration of distinct glial cells in the Drosophila embryo. Development. 2010;137:1251–1262. doi: 10.1242/dev.042853. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Amara SG, Kavanaugh MP. Ion fluxes associated with excitatory amino acid transport. Neuron. 1995;15:721–728. doi: 10.1016/0896-6273(95)90159-0. [DOI] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci. 1998;10:976–988. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- Winter N, Kovermann P, Fahlke C. A point mutation associated with episodic ataxia 6 increases glutamate transporter anion currents. Brain. 2012;135:3416–3425. doi: 10.1093/brain/aws255. [DOI] [PubMed] [Google Scholar]