

Abstract
Neuropathic pain, often caused by nerve injury, is a major clinical challenge. Mechanisms that underlie neuropathic pain remain elusive and effective medications are limited. Numerous investigations of pain mechanisms have focused on alterations and phenotypic switches of the nociceptive transmitters and modulators, as well as on their receptors and downstream signaling pathways that have already exerted roles in the pain processes of mature nervous systems. We have demonstrated recently that nerve injury may elicit neuronal alterations that recapitulate events occurring during development. Signaling of the representative activated molecule Wnt thus becomes a trigger for the development of neuropathic pain and is a potential therapeutic target. We report that the transcriptional regulators YAP and TAZ, which orchestrate Wnt response via incorporation in the β-catenin destruction complex, are key in the pathogenesis of neuropathic pain and may serve as an “ON–OFF” switch for neuropathic pain status in rats. Peripheral nerve injury causes rapid-onset and long-lasting nuclear accumulation of YAP/TAZ/β-catenin in the spinal dorsal horn. Spinal inhibition or knock-down of either YAP or TAZ suppresses mechanical allodynia induced by nerve injury or the pain initiators lysophosphatidic acid and Wnt3a. Promoting the nuclear accumulation of YAP/TAZ leads to mechanical hypersensitivity in naive animals. Further, we discovered a new small molecule, dCTB, which targets YAP/TAZ/β-catenin and can greatly suppress neuropathic pain and the associated neurochemical alterations. Our study reveals that YAP and TAZ are core mechanisms underlying the pathogenesis of neuropathic pain and are targets in the screening for potent analgesics for the treatment of neuropathic pain.
SIGNIFICANCE STATEMENT Mechanisms that underlie neuropathic pain remain elusive. We have demonstrated recently that nerve injury can activate Wnt signaling, which becomes a trigger for the development of neuropathic pain. We report that the transcriptional regulators YAP and TAZ, which orchestrate Wnt response via incorporation in the β-catenin destruction complex, are key in the pathogenesis of neuropathic pain and may serve as an “ON–OFF” switch for neuropathic pain status. Further, we discovered a new small molecule, dCTB, which targets YAP/TAZ/β-catenin and can greatly suppress neuropathic pain. Our study reveals that YAP and TAZ are core mechanisms underlying the pathogenesis of neuropathic pain and are targets in the screening of potent analgesics for the treatment of neuropathic pain.
Keywords: beta-catenin, drug discovery, nerve injury, neuropathic pain, TAZ, YAP
Introduction
Treatment of neuropathic pain, which is usually caused by direct nerve injury such as trauma, continues to be a major challenge. It is of clinical importance to determine the mechanisms underlying neuropathic pain and to develop medications that can treat it. Despite decades of investigation and numerous implicated processes, the specific cellular and molecular mechanisms underlying neuropathic pain remain elusive and clinical approaches are limited. In previous investigations, numerous hypotheses of basic pain mechanisms have focused on alterations and phenotypic switches of the nociceptive transmitters and modulators, as well as on their receptors and downstream signaling pathways that have already exerted roles in pain processes of mature nervous systems. The maladaptive plasticity has been the dominant model underlying neuropathic pain (Costigan et al., 2009). We have demonstrated recently that Wnt signaling, which is important in the regulation of cellular processes such as synapse formation and plasticity and axon guidance during the development of nervous systems and is not involved in pain processes in physiological conditions, becomes critical to the development of neuropathic pain after nerve injury (Zhang et al., 2013; Liu et al., 2015). This study may support the idea that neuropathic pain-inducing nerve injury may elicit neuronal alterations that recapitulate events occurring during development. This abnormally activated Wnt signaling becomes a trigger for the development of neuropathic pain. The core of the canonical Wnt signaling pathway is the regulation of β-catenin by a cytoplasmic destruction complex consisting of Axin, a central scaffold protein that interacts with adenomatous polyposis coli (APC), as well as glycogen synthase kinase-3 (GSK3), casein kinase 1 (CK1), and others (Clevers, 2006; Taelman et al., 2010; Li et al., 2012). A recent study demonstrated that YAP and TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cells without Wnt immediately sequester YAP and TAZ to the destruction complex and use them to recruit β-TrCP, which further degrades phospho-β-catenin (Azzolin et al., 2014). YAP and TAZ negatively regulate canonical Wnt/β-catenin signaling (Park et al., 2015). Wnt stimulation releases YAP and TAZ from the β-catenin destruction complex in the cytoplasm, stabilizing β-catenin, as well as YAP and TAZ nuclear translocation with the induction of their transcriptional responses (Azzolin et al., 2014).
We hypothesized that neuropathic-pain-inducing nerve injury might cause nuclear translocation of YAP/TAZ. YAP/TAZ activity then orchestrates the canonical Wnt/β-catenin signaling response through incorporation in the β-catenin destruction complex and thus plays a critical role in the pathogenesis of neuropathic pain. In this study, we have demonstrated that YAP and TAZ are core mechanisms underlying the pathogenesis of neuropathic pain and are targets for screening potent analgesics. We further discovered a new potent analgesic 4-((4-(3,4-dichlorophenyl)-1,2,5-thiadiazol-3-yl)oxy)butan-1-ol (dCTB), which greatly suppresses neuropathic pain by targeting YAP/TAZ/β-catenin.
Materials and Methods
Animals, anesthesia, drugs, and drug administration.
We purchased adult male Sprague Dawley rats (200–220 g) from the Department of Experimental Animal Sciences, Peking University Health Science Center. All protocols were approved by the Animal Care and Use Committee of Peking University. All surgeries were done under anesthesia with sodium pentobarbital (50 mg/kg, i.p.). Verteporfin was from the United States Pharmacopeial Convention, lysophosphatidic acid (LPA) and PEG400 from Sigma-Aldrich, and Wnt3a from R&D Systems. The drugs were delivered intrathecally (in a volume of 20 μl) into the CSF through lumbar puncture.
Chronic constriction injury of the sciatic nerve (CCI).
To produce peripheral nerve injury, a rat model of CCI was used in this study. The left common sciatic nerve of each rat was exposed at the midthigh level. Proximal to the sciatic nerve's trifurcation was freed of adhering tissue and four ligatures (4-0 surgical catgut) were tied loosely around it with ∼1 mm between ligatures. Animals in the sham group received surgery identical to that described in CCI but without nerve injury.
Assessment of mechanical allodynia and thermal hyperalgesia.
Mechanical allodynia was determined by measuring the incidence of foot withdrawal in response to mechanical indentation of the plantar surface of each hindpaw with a sharp, cylindrical probe with a uniform tip diameter of ∼0.2 mm provided by an Electro von Frey (ALMEMO 2390-5 Anesthesiometer; IITC Life Science). The probe was applied to six designated loci distributed over the plantar surface of the foot. The minimal force (in grams) that induced paw withdrawal was read off of the display. Threshold of mechanical withdrawal in each animal was calculated by averaging the six readings and the force was converted into milli-Newtons (mN). For the behavior test in the verteporfin (VP) administration experiment, the rats were fed in a dark environment from 3 d before VP administration to adapt the darkness. Further, the basic withdrawal threshold and the threshold of mechanical withdrawal after VP injection were measured under a dark environment. Thermal hyperalgesia was assessed by measuring foot withdrawal latency to heat stimulation. An analgesia meter (IITC Model 336 Analgesia Meter, Series 8; IITC Life Science) was used to provide a heat source. In brief, each animal was placed in a box containing a smooth, temperature-controlled glass floor. The heat source was focused on a portion of the hindpaw, which was flush against the glass, and a radiant thermal stimulus was delivered to that site. The stimulus shut off when the hindpaw moved (or after 30 s to prevent tissue damage). The intensity of the heat stimulus was maintained constant throughout all experiments. The elicited paw movement occurred at latency between 10 and 15 s in control animals. Thermal stimuli were delivered 3 times to each hindpaw at 5–6 min intervals. For the results expressing mechanical allodynia or thermal hyperalgesia, the values are mean of ipsilateral feet.
qRT-PCR.
Under deep anesthesia, the L4–L5 spinal cord segments of rats were quickly removed and analyzed. Total RNA was isolated with TRIzol reagent (Ambion) according to the manufacturer's instructions. cDNA was then synthesized using the Takara PrimeScript Master Mix (Perfect Real Time) kit. qRT-PCR was performed with the DyNAmo Flash SYBR Green qPCR Kit (Thermo Fisher Scientific). The standard conditions were as follows: 95°C for 7 min, then 40 cycles at 95°C for 10 s and 60°C for 30 s, then 95°C for 15 s, 60°C for 60 s and 95°C for 15 s for melt curve. Primers used for expression analysis of CTGF and BIRC5 were as follows: gene CTGF: forward (5′-3′)-GGCAGGGCCAACCACTGTGC; reverse (5′-3′)-CAGTGCACTTGCCTGGATGG; gene BIRC5: forward (5′-3′)-CTGATTTGGCCCAGTGTTTT; reverse (5′-3′)-TCATCTGACGTCCAGTTTCG; and gene GAPDH: forward (5′-3′)-AGAGAGAGGCCCTCAGTTGCT; reverse (5′-3′)-TTGTGAGGGAGATGCTCAGTGT. Relative mRNA levels were calculated using the 2-ΔΔCT method. Gene expression was first normalized to the housekeeping control gene GAPDH and, subsequently, the relative expression of genes of interest was compared with the respective experimental control.
Protein determinations.
To quantify temporal changes in protein levels, Western blotting analysis was used. The spinal cord at L4–L5 segments were quickly removed from deeply anesthetized rats and stored at −80°C. Sequential precipitation procedures were used on the tissue samples that were lysed in ice-cold (4°C) NP-40 lysis buffer containing a mixture of protease inhibitor, phosphatase inhibitors, and phenylmethylsulfonyl fluoride (Sigma-Aldrich). For the analyses of nuclear YAP, TAZ, and active-β-catenin, nuclear extractions were prepared using a NE-PER Nuclear and Cytoplasmic Extraction Kit (Pierce Biotechnology) according to the manufacturer's instructions. The protein concentrations of the lysates were estimated using the method of BCA (with reagents from Pierce) and the total protein content between samples was equalized. The total protein were separated by SDS-PAGE and transferred to PVDF membrane (both from Bio-Rad Laboratories). The following primary antibodies were used: anti-YAP1 (1:1000; Cell Signaling Technology), anti-TAZ (1:1000; Cell Signaling Technology), anti-active-β-catenin (1:1000; Millipore), anti-MST1/2 (1:1000; Cell Signaling Technology), anti-pMST1/2 (1:1000; Cell Signaling Technology), anti-LATS1 (1:1000; Sigma-Aldrich), anti-pLATS1 (1:1000; Cell Signaling Technology), anti-pNR2B (Tyr1472, 1:1000; Millipore), anti-pPKCγ (1:1000; Cell Signaling Technology), anti-laminB(1:10000; Abcam), and anti-GAPDH (1:8000; Sigma-Aldrich). The membranes were then developed by enhanced chemiluminescence reagents (PerkinElmer) with horseradish peroxidase-conjugated secondary antibodies (R&D Systems). Data were analyzed with the ImageJ.
Immunohistochemistry.
Deeply anesthetized rats were perfused transcardially with 0.9% saline followed by 4% paraformaldehyde. The L4–L5 spinal cord segments and DRGs were removed and postfixed in 4% paraformaldehyde 24–48 h. After being postfixed, the tissues were transferred into 40% sucrose (in 0.1 m PB) for 3 d for dehydration. The tissues were sectioned at 30 μm thickness for spinal cord and DRG sections. For immunofluorescence staining, free-floating sections were blocked in PBS containing 10% donkey serum with 0.3% Triton X-100 for 2 h and incubated in primary antibody at 4°C overnight. Sections were then washed in 0.1 m PBS with 0.05% Triton X-100, pH 7.6 (3 × 5 min) followed by incubating in the secondary antibody at room temperature for 2 h and washing. Sections were mounted on slides and covered with 90% glycerin for observation under a confocal microscope (FluoView FV1000; Olympus). The antibodies used included anti-YAP1 (1:800; Abcam), anti-TAZ (1:600; Abcam), anti-NeuN (1:4000; Millipore), anti-GFAP (1:200; Millipore), anti-Iba1 (1:200; Abcam), FITC conjugated Isolectin B4 (1:100; Sigma-Aldrich), anti-CGRP (1:100; Abcam), and anti-MAP2 (1:5000; Abcam). For nucleus counterstaining, sections were incubated with DAPI (1:3000; Sigma-Aldrich) for 3 min at room temperature. Data were analyzed with ImageJ. To obtain quantitative measurements of GFAP and IBA1 immunofluorescence in the dorsal horn, 15 fields covering the entire superficial dorsal horn in each group, which included 15 spinal cord sections from five rats (three sections from each rat), were evaluated and photographed at the same exposure time to generate the raw data. The average green fluorescence protein intensity of each pixel was normalized to the background intensity in the same image.
siRNA interference.
Stealth RNAi siRNA (set of three oligos) targeting rat Willin, YAP, TAZ, and Stealth RNAi Negative Control were purchased from Invitrogen. One from three siRNAs that target every molecule was selected by preliminary behavioral test and by immunoblotting. Sequences were as follows: siRNA for willin: ID RSS352387, 5′-AGUACUUGCUGGUUCUCUUCACGGG-3′ and 5′-CCCGUGAAGAGAACCAGCAAGUACU-3′; siRNA for YAP: ID RSS325772, 5′-UCUUCUGGUCAGAGAUACUUCUUAA-3′ and 5′-UUAAGAAGUAUCUCUGACCAGAAGA-3′; siRNA for TAZ: ID RSS355198, 5′-AGCAGACUCUACAUCAUUGAAGAGG-3′ and 5′-CCUCUUCAAUGAUGUAGAGUCUGCU-3′; and siRNA for NegativeControlLOGC_e0N: 5′-AGCUACACUAUCGAGCAAUUAACUU-3′ and 5′-AAGUUAAUUGCUCGAUAGUGUAGCU-3′. For administration, the siRNA was dissolved in RNase-free water at the concentration of 1 mg/ml as stock solution. The siRNA was mixed with branched polyethyleneimine (PEI; Sigma-Aldrich) for 10 min at room temperature before in vivo intrathecal injection to increase cell membrane penetration. PEI was dissolved in 5% glucose and 1 μg of siRNA was mixed with 0.18 μl of PEI. The negative siRNA, which was designed to minimize sequence homology to any known vertebrate transcript, was used as a control. In the naive rats, Willin siRNA was intrathecally injected in a single dose (10 μg) or 3 doses (10 μg each) daily for 3 consecutive days. In the rats that received CCI treatment, pretreatment of TAZ siRNA (10 μg/d) or YAP siRNA (5 μg/d) was administered 30 min before surgery. After surgery, the siRNA was administered continuously in the same dose daily for another 2 consecutive days during the early time course after CCI treatment. To determine the effect of YAP or TAZ targeting siRNA on the persistence of neuropathic pain, YAP siRNA (5 μg/d) and TAZ siRNA (10 μg/d), respectively, were administered continuously daily for 3 consecutive days during postoperative days 11–13.
Synthesis of dCTB.
(3,4-dichlorophenyl) boronic acid (27.40 g, 143.59 mmol, 1.00 equivalent), 3,4-dichloro-1,2,5-thiadiazole (21.14 g, 136.41 mmol, 0.95 equivalent), KF (25.03 g, 430.77 mmol, 3.00 equivalent), and Pd(PPh3)4 (3.03 g, 2.62 mmol) in a mixture of toluene (356 ml) and H2O (365 ml). The reaction mixture was turned red. The mixture was degassed and then heated to 90–100°C for 36 h under N2. TLC (PE, Rf = 0.8) showed that most of starting material was consumed completely. The organic layer was decanted off and the aqueous layer was re-extracted with ethyl acetate (40 ml). The organic phase was washed with saturated brine (40 ml), dried over anhydrous Na2SO4, and concentrated in a vacuum to give a residue that was purified by column chromatography (PE) to give the crude, then repurified by flash column (PE) to afford the pure intermediate product (3.20 g, 11.93 mmol, 8.31% yield, 95% purity) as a white solid.
To a suspension of NaH (2.89 g, 120.50 mmol, 8.00 equivalent) in tetrahydrofuran (THF) (60 ml) at 0°C was added butane-1,4-diol (5.43 g, 60.25 mmol, 4.00 equivalent) dropwise by syringe. The reaction mixture was heated to 70°C and let stir for 1 h. The reaction mixture was cooled to ∼20–25°C and a solution of compound the pure intermediate product (4.00 g, 15.06 mmol, 1.00 equivalent) in anhydrous THF (20 ml) was added dropwise by syringe. The reaction mixture was heated to70°C and stirred for 4 h. The suspension then turned light yellow. The reaction mixture was cooled to ∼20–25°C, treated with saturated aqueous sodium bicarbonate (300 ml), and concentrated under reduced pressure to remove the THF. The residual aqueous mixture was diluted with saturated aqueous sodium bicarbonate (200 ml) and extracted with EtOAc (3 × 80 ml). The combined organic phases were dried over Na2SO4, filtered, and concentrated. Purification was by flash column (PE:EtOAc = 0∼50%) to give the crude, which was triturated in hexane (20 ml) for 1 h, then filtered the suspension to give the product dCTB (1.29 g, 4.03 mmol, 27% yield) as a white solid.
Statistics.
Prism (GraphPad) was used to conduct all statistical analyses. Alterations of expression of the mRNA and proteins detected and the behavioral responses to mechanical and thermal stimuli over time among groups were tested with one-way or two-way ANOVA with repeated measures followed by Holm–Sidak tests, respectively. Individual t tests were used to test specific hypotheses about differences between each operated or drug-treated group and its corresponding control group between the naive or sham and the treatment for Western blot data. All data are presented as mean ± SEM. The criterion for statistical significance was p < 0.05.
Results
Nuclear accumulation of YAP/TAZ/β-catenin in the spinal dorsal horn after CCI
To verify the hypothesis that peripheral nerve injury might cause nuclear accumulation of YAP and TAZ with induction of their transcriptional responses, a process that may underlie pathogenesis of neuropathic pain, we began by examining the expression of YAP, TAZ, and active β-catenin protein in nerve-injured spinal cord and DRG cell nuclei at segments L4–L5. Western blot analyses showed that CCI treatment caused a rapid increase in the nuclear expression of YAP, TAZ, and active β-catenin in the spinal cord within a day, an increase that persisted for at least 14 d. YAP expression levels peaked at 7 d and then decreased significantly at 14 d, whereas TAZ and active β-catenin maintained high expression levels throughout the 1–14 d (Fig. 1A). Increases in YAP and active β-catenin were detectable at as early as 1 h, whereas no increase of TAZ was detected at either 1 or 6 h (Fig. 1B) after CCI treatment. Immunofluorescence validated the nuclear accumulation of YAP and TAZ in the dorsal horn (Fig. 1C), but not in the ventral horn of the spinal cord. Sham surgery did not alter the nuclear expression of YAP, TAZ, or active β-catenin. These results support our hypothesis that peripheral nerve injury triggers nuclear transportation of YAP/TAZ/β-catenin. However, in DRG cells, YAP and TAZ were mainly sequestrated in the cytoplasm, not in the nucleus. CCI treatment did not induce nuclear accumulation of YAP or TAZ. This is consistent with our recent findings that increased expression of β-catenin due to CCI, as well as its nuclear accumulation, was seen in the spinal dorsal horn, but not in the DRG (Zhang et al., 2013). This finding is also consistent with results of a study showing that canonical, β-catenin-dependent Wnt signaling in DRG neurons is not crucial in Wnt3a-induced pain hypersensitivity (Simonetti et al., 2014).
Figure 1.

Nuclear accumulation of YAP/TAZ/β-catenin and expression and cellular distribution of YAP/TAZ in the spinal dorsal horn after CCI. A, B, Western blot showing nuclear expression of YAP/TAZ/β-catenin in the long (A) and short (B) time course (n = 4 in each) after CCI. One-way ANOVA (A) YAP: p < 0.0001, F = 24.81; TAZ: p = 0.0111, F = 5.766; (B, C) p = 0.0072, F = 6.548; (B) YAP: p = 0.0116, F = 7.609; TAZ: p = 0.8791, F = 0.1397; (B, C) p = 0.0007, F = 18.12. (A, B) *p < 0.05, **p < 0.01 versus sham. C, Immunofluorescence showing nuclear (DAPI, blue) expression of YAP/TAZ (green) in the superficial dorsal horn. Arrows indicate nuclear expression of YAP or TAZ. D, Western blot showing time course of protein expression of YAP (n = 5) and TAZ (n = 4). One-way ANOVA followed by t test. YAP: p < 0.0014, F = 6.107; TAZ: p = 0.0061, F = 5.127. *p < 0.05, **p < 0.01 versus sham. A–D, Tissues were taken on the multiple time points as indicated. E, Overall distribution of YAP/TAZ. F, Tissues were taken on day 7 and cellular colocalizations of YAP/TAZ after CCI. Top, YAP (green) was predominantly colocalized with neuronal somata (NeuN, red), but not with dendrites (MAP2, red), astrocytes (GFAP, red), or microglial cells (IBA1, red). Bottom, TAZ (green) was mostly colocalized with NeuN and MAP2 and IBA1, but not with GFAP. Tissues were taken on day 7. Original magnifications: 1200× (C), 200× (E, F), and 800× (inserts in F). Scale bar, 50 μm.
Given that CCI can induce nuclear accumulation of YAP/TAZ, we continued to examine their protein expression and cellular distribution in the spinal cord. Western blot analysis showed that CCI treatment caused a rapid-onset (within 1 d) and long-lasting increase in the expression of YAP and TAZ in the spinal cord. YAP was increased starting from 1 d, peaked at 1–7 d, decreased at 14 d, and then returned to control levels at 21 d. TAZ increased gradually from 1 d, peaked at 3 d, and maintained a high level up to the last examination day after CCI, day 21 (Fig. 1D). Immunofluorescence showed that YAP and TAZ immunoreactivity was greatly increased and was distributed predominantly in the superficial layers of the dorsal horn, the critical regions in the nociceptive pathway ipsilateral to CCI (Fig. 1E). YAP immunoreactivity was specifically colocalized with neuronal somata (NeuN), but not with dendrites (MAP2), astrocytes (GFAP), or microglial cells (IBA1) (Fig. 1F, top row). In contrast, TAZ was additionally colocalized with neuronal dendrites and microglial cells (Fig. 1F, bottom row). Such distinct differences of cellular distributions of YAP and TAZ in the dorsal horn may suggest different functions of these cells and different roles in the pathophysiology of neuropathic pain. Immunofluorescence showed that neither YAP nor TAZ in the superficial dorsal horn was colocalized with CGRP- or IB4-positive primary afferent terminals (data not shown), indicating that the increased expression of YAP and TAZ in the dorsal horn originated from the dorsal horn of the spinal cord, but was not released from the central terminals of the DRG neurons.
Inhibition of the Hippo pathway after nerve injury
Mechanisms underlying the regulation of YAP/TAZ subcellular localization is not understood completely, but the Hippo pathway is well known as a crucial YAP/TAZ regulator (Azzolin et al., 2014). MST1/2 can phosphorylate and activate LATS1/2 kinases that in turn phosphorylate YAP/TAZ, leading to nuclear exclusion (Fig. 2A). Therefore, we examined the effect of CCI upon these core kinases of the Hippo pathway upstream of YAP/TAZ. Western blot results showed that CCI treatment caused persistent (1–14 d), significant downregulation of the phosphorylation of MST1/2 (p-MST1/2) and LATS1 (p-LATS1) (Fig. 2B), indicating an inhibition of the Hippo pathway after CCI treatment, which causes nuclear transportation of YAP/TAZ.
Figure 2.

Downregulation of phosphorylation of Hippo signaling components in the spinal cord after CCI. A, Schematic shows the upstream regulation of YAP/TAZ. MST1/2 can phosphorylate and activate LAST1/2 kinases that in turn phosphorylate YAP/TAZ, leading to nuclear exclusion. B, Western blot representative bands showing time course for expression of MST, LATS, and their phosphorylation after CCI. Bottom, Data summary. n = 4 in each group. One-way ANOVA followed by t test. LATS: p = 0.0148, F = 3.807; pLTAS: p = 0.0033, F = 5.405; MST: p = 0.0171, F = 3.732; pMST: p < 0.0001, F = 14.89. ratio/LATS: p < 0.0001, F = 38.61; ratio/MST: p < 0.0001 F = 30.79. *p < 0.05, **p < 0.01 versus sham in the corresponding group.
The expression of most of the p-MST and p-LATS Western blot bands were quite weak compared with those of MST and LATS, respectively. We adjusted brightness and contrast to make the bands of p-MST and p-LATS clear enough and to avoid MST and LATS overexposure. Density can thus be compared between each well on the same gel. Different gels run in the different conditions cannot be compared. This is consistent with previous findings (Yu et al., 2012; Chen et al., 2014). In addition to the Hippo signaling cascade, YAP/TAZ nuclear accumulation can also be regulated by other regulators such as mechanotransduction and tissue architectural cues (DuPont et al., 2011; Aragona et al., 2013), which were not discussed in this study.
Contributions of YAP and TAZ to the induction and maintenance of neuropathic pain
Given that CCI treatment causes nuclear accumulation and increased protein expression of YAP and TAZ, which are similar to each other in neuronal somata but dissimilar in neuronal dendrites and microglial cells and in the timing patterns of their expression after nerve injury, we investigated the roles of YAP/TAZ activity in neuropathic pain using pharmacological approaches and RNA interference. In the nucleus, TEAD families of transcriptional factors are known to mediate YAP/TAZ activities (Hansen et al., 2015). VP, a prescriptive medicine currently used in the clinical setting for treating macular degeneration (Brown et al., 2006), has the capability of impeding the YAP-TEAD association and causing inhibition of YAP activity (Zhao et al., 2008; Liu-Chittenden et al., 2012). We therefore tested whether VP could impede neuropathic pain. Our results showed that CCI treatment significantly increased expression of the endogenous YAP target genes Ctgf and Birc5 (Sorrentino et al., 2014), in addition to causing neuropathic pain manifested as mechanical hypersensitivity (allodynia). A single dose of VP (0.45 mg, i.t.) administered 30 min before CCI significantly inhibited the increased expression of Ctgf and Birc5 detected on 1 d after CCI (Fig. 3A). Repetitive intrathecal VP in the early phase (0.45 mg daily for 3 consecutive days starting 30 min before surgery) significantly delayed the onset of mechanical allodynia from day 3 to day 7 compared with the CCI-vehicle control (PEG400). Neither PEG400 nor VP changed the mechanical threshold in sham control animals (Fig. 3B). Furthermore, a single dose of VP (0.45 mg, i.t.) given transiently on postoperative day 14 significantly inhibited established mechanical allodynia. Such an inhibition started within 1 h and lasted for ∼6 h (Fig. 3C). Such repetitive VP treatments did not affect locomotor activities (data not shown) and no significant side effects were observed.
Figure 3.

YAP contributes to the early and later phases of neuropathic pain, whereas TAZ only contributes to the later phase in CCI rats. A, Effects of CCI and a single dose of VP (0.45 mg, i.t.) administered 30 min before CCI on the expression of Ctgf and Birc5. Tissues were taken on 1 d after CCI. B, C, Effects of intrathecal VP (0.45 mg each injection) on the production (B) and persistence (C) of mechanical allodynia. D, E, Effects of intrathecal YAP-targeting siRNA (10 μg each injection) on the production (D) and persistence (E) of mechanical allodynia. YAP siRNA (5 μg each injection). F, G, Effects of TAZ-targeting siRNA (10 μg each injection) on induction (F) and persistence (G) of mechanical allodynia. H, Effective knock-down of YAP and TAZ in vivo by their siRNA. Expression of the total (left) and nuclear (right) protein of YAP and TAZ was detected. Tissues were taken 24 h after a single (1i) or repetitive (daily for 3 consecutive days, 3i) spinal administration. PEG400 and negative siRNA were used as control. Each intrathecal administration is indicated by an arrow on the corresponding time point. Eight rats were included in each group. Solid triangle (▴) indicates CCI surgery. One-way ANOVA, (A) Ctgf: F = 12.93, p = 0.001; Birc5: F = 7.15, p = 0.009; (H) YAP: F = 44.59, p < 0.0001; TAZ: F = 54.73, p < 0.0001. Two-way ANOVA, (B) F(3, 252) = 347.8, p < 0.0001; (C) F(1, 207) = 31.08, p < 0.0001; (D) F(1, 207) = 31.08, p < 0.0001; (E) F(1, 182) = 30.56, p < 0.0001; (F) F(3, 252) = 409.7, p < 0.0001; (G) F(1, 210) = 160.9, p < 0.0001. *p < 0.05, **p < 0.01 versus sham (A), CCI + PEG400 (B, C), CCI + negative siRNA (D, E), CCI + Negative siRNA (F, G), or CCI control (H). #p < 0.05, ##p < 0.01 versus the corresponding group in CCI + PEG400 (A).
To validate the role of YAP in neuropathic pain, we further tested the effect of in vivo knock-down of YAP on mechanical allodynia. Spinal repetitive administration of YAP-targeting siRNA in the early phase after CCI (5 μg daily for 3 consecutive days starting 30 min before CCI) delayed the induction of mechanical allodynia for 2 d and significantly reduced the severity of mechanical allodynia by ∼50–70% for 11 d (Fig. 3D). Intrathecal YAP-targeting siRNA in the later phase after CCI (5 μg daily for 3 consecutive days starting 11 d after CCI) significantly suppressed the established mechanical allodynia (Fig. 3E). These results support the idea that nuclear activity of YAP is critical to the induction and persistence of neuropathic pain.
We continued to examine the possible role of TAZ in neuropathic pain by in vivo knock-down of TAZ. In the early phase of CCI, repetitive intrathecal TAZ-targeting siRNA (10 μg daily for 3 consecutive days starting 30 min before surgery) failed to change the induction and persistence of neuropathic pain (Fig. 3F). However, in the later phase of CCI, intrathecal TAZ-targeting siRNA (10 μg daily for 3 consecutive days starting 11 d after CCI) dramatically reversed the established mechanical allodynia (Fig. 3G). Compared with YAP-targeting siRNA, TAZ-targeting siRNA did not affect the induction of neuropathic pain, but had a strong analgesic effect on the established mechanical allodynia. The inhibition of TAZ-targeting siRNA on the established allodynia lasted for 10 d, whereas that of YAP-targeting siRNA lasted for 6 d (Fig. 3E,G). These results indicate that YAP is involved in both early induction and later persistence, whereas TAZ is involved only in later persistence and is unnecessary for the early induction of neuropathic pain after CCI.
In addition, we examined effective knock-down of YAP and TAZ in vivo in the nerve-injured spinal cord. Our results showed that CCI-induced expression of both total protein (Fig. 3H, left) and nuclear protein (Fig. 3H, right) levels of YAP and TAZ was significantly inhibited by either intrathecal YAP- or TAZ-targeting siRNA. A single dose (on postoperative day 11) or repetitive doses (daily for 3 consecutive days on postoperative days 11, 12, and 13) of YAP- or TAZ-targeting siRNA produced significant and similar knock-down of the total and nuclear protein of YAP and TAZ in the spinal cord. Meanwhile, a single injection of siRNA for YAP or TAZ resulted in significant inhibition of established mechanical allodynia (behavioral data not shown), which was measured 24 h after injection immediately before the surgery to collect the spinal cord tissues.
Promoting nuclear accumulation of YAP/TAZ leads to mechanical hypersensitivity in naive animals
After demonstrating that CCI treatment induces nuclear accumulation of YAP/TAZ and that blocking YAP/TAZ activity suppresses neuropathic pain, we inferred that promoting the nuclear accumulation of the transcriptional regulators YAP/TAZ could induce pain in naive animals. It is well known that the Hippo pathway is one of the crucial YAP/TAZ regulators. The Hippo pathway core MST1/2-LATS1/2 kinase cascade inhibits YAP/TAZ through direct phosphorylation, which results in cytoplasmic retention via 14-3-3 binding and further promotes β-TrCP-mediated YAP/TAZ ubiquitination and degradation (Azzolin et al., 2014; Hansen et al., 2015). This Hippo signaling pathway can be inhibited by knock-down of Willin/FRMD6, which decreases the phosphorylation of MST1/2 and LATS1 (Angus et al., 2012). Upon inhibition of the Hippo pathway, YAP/TAZ are activated and translocated into the nucleus to bind TEAD family transcription factors to stimulate target gene expression involved in cell proliferation, stem cell self-renewal, and tumorigenesis (Azzolin et al., 2014). We therefore performed knock-down of Willin/FRMD6 in vivo and monitored the subcellular localization of YAP, TAZ, and β-catenin. First, we examined the actual knock-down of Willin/FRMD6 protein and the subsequent alterations of phosphorylation of MST1/2 and LATS1 in vivo. Expression of Willin/FRMD6 was significantly inhibited after a single intrathecal injection or repetitive (once a day for 3 consecutive days) intrathecal injections of its targeting siRNA (10 μg each) (Fig. 4A). Phosphorylation of MST1/2 and LATS1 was also significantly downregulated after the same repetitive treatment of Willin/FRMD6-targeting siRNA (Fig. 4B). Then, we continued to show that knock-down of Willin/FRMD6 led to nuclear accumulation of all three transcriptional regulators, YAP/TAZ/β-catenin, in the spinal cord (Fig. 4C). Furthermore, Willin/FRMD6 knock-down induced significant mechanical hypersensitivity, which started 24 h after the first injection and persisted for up to 13 d (Fig. 4D). Such mechanical hypersensitivity is similar to that induced by CCI. These results show that promoting nuclear accumulation of YAP/TAZ/β-catenin can induce pain in naive animals, indicating that nuclear accumulation of YAP/TAZ/β-catenin is sufficient for inducing painful behavior.
Figure 4.

Promotion of nuclear accumulation of YAP/TAZ/β-catenin in the spinal cord leads to mechanical allodynia in naive animals. A, B, Western blot showing the actual knock-down of the Willin/FRMD6 protein in vivo by its siRNA (A) and a subsequent reduction of the p-MST and p-LATS protein (B). C, Western blot showing that intrathecal Willin siRNA induced nuclear accumulation of YAP/TAZ and β-catenin in the spinal cord. D, Intrathecal Willin siRNA induced a long-lasting mechanical allodynia. E, F, Western blot showing nuclear accumulation of YAP, TAZ, and β-catenin induced by intrathecal LPA (E) and Wnt3a (F), respectively. G, H, Intrathecal LPA or Wnt3a induced a rapid-onset and long-lasting mechanical allodynia. Coadministration of LPA with VP or Wnt3a with VP did not result in mechanical allodynia. Drug doses and administration: Willin siRNA, 10 μg, negative siRNA, 10 μg, a single injection (1i) or repetitive injection (daily for 3 consecutive days, 3i) (A–D); LPA, 40 nmol, daily for 3 consecutive days (E, G); Wnt3a, 60 ng, one single dose (F, H); VP, 0.45 mg, 2 h before each injection of LPA (G) or Wnt3a (H). Saline (E, G) and BSA (0.1% in PBS) (F, H) were used as control. Each injection is indicated by an arrow on the corresponding time point (D, G, H). The spinal cord tissues were collected 1 d (A), 3 d (A–C, E), or 4 h (F) after the last injection of Willin siRNA, LPA, or Wnt3a. n = 4 in each of the groups (A–C, E, F). Rats, n = 8 in each of the groups (D, G, H). One-way ANOVA, (A) F = 10.92, p = 0.001; (C) YAP: F = 10.14, p = 0.0050; TAZ: F = 9.268, p = 0.0065; (B, C): F = 16.56, p = 0.0010; (E) YAP: F = 4.894, p = 0.0365; TAZ: F = 39.53, p < 0.0001; (B, C): F = 8.807, p = 0.0076. F, YAP: F = 16.75, p = 0.0009; TAZ: F = 5.601, p = 0.0263; B-C: F = 9.967, p = 0.0052. *p < 0.05, **p < 0.01 versus saline (C, E) or BSA (G). Student's t test, (B) MST: p = 0.8066; p-MST: p = 0.0032; LATS: p = 0.7086; p-PATS: 0.0021. **p < 0.01 versus naive. Two-way ANOVA, (D) F(1, 154) = 322.9, p < 0.0001; (G) F(2, 297) = 231.8, p < 0.0001; (H) F(2, 243) = 51.84, p < 0.0001. *p < 0.05, **p < 0.01 versus negative siRNA (D), saline (G), or BSA (0.1% in PBS) (H).
We also investigated whether certain acknowledged neuropathic pain initiators could cause pain by inducing nuclear accumulation of YAP/TAZ. LPA and Wnt3a have been identified as typical neuropathic pain initiators (Inoue et al., 2004; Zhang et al., 2013). Our results showed that intrathecal LPA (40 nmol) daily for 3 consecutive days and a single injection of Wnt3a (60 ng), respectively, in naive rats resulted in a great increase of expression of nuclear YAP/TAZ/β-catenin in the spinal cord (Fig. 4E,F), in addition to the induction of a rapid-onset, long-lasting mechanical hypersensitivity (Fig. 4G,H). To further investigate whether YAP/TAZ activity would be necessary for LPA- and Wnt3a-induced mechanical allodynia, LPA or Wnt3a was injection with VP, which inhibits YAP/TAZ-TEAD function. The results showed that coadministration of LPA or Wnt3a with VP (0.45 mg, i.t., 2 h before LPA or Wnt3a) did not result in significant mechanical hypersensitivity (Fig. 4G,H). Together, these results support the idea that nuclear accumulation of YAP/TAZ is sufficient and necessary for pain induction.
Synthesis of the small molecule dCTB, which inhibits YAP/TAZ/β-catenin activity
The data presented so far strongly support the idea that the transcriptional response of YAP/TAZ may be the core mechanism that underlies the pathogenesis of neuropathic pain. Therefore, the corollary is that a reagent that causes nuclear accumulation of YAP/TAZ in the spinal dorsal horn may result in neuropathic pain and a reagent that inhibits YAP/TAZ activity may result in analgesia. dCTB is an existing antitumor small molecule and also an inhibitor of the Wnt and Hippo pathways. We tested the possible analgesic effect of dCTB in CCI rats in vivo. Because the chemical synthetic pathway described in the only existing report (Basu et al., 2014) did not lead to an effective synthesis of dCTB, we thus explored and developed a more efficient, new chemical synthetic pathway to produce such a molecule. The new chemical synthetic pathway of dCTB is shown in Figure 5A. We then validated the molecular effect of dCTB by Western blot and immunofluorescence staining. Repetitive intrathecal dCTB (1.6 mg daily for 3 consecutive days on postoperative days 11, 12, and 13) greatly inhibited CCI-induced nuclear accumulation of YAP, TAZ, and β-catenin (Fig. 5B,C) and led to a great decrease of protein expression of LATS and an increase of p-LATS, a core kinase of Hippo pathway. Expression of another core kinase of Hippo pathway MST and its phosphorylation were not significantly altered (Fig. 5D). These results indicate that dCTB may inhibit YAP/TAZ activity by activating the Hippo signaling pathway through increasing the ratio of p-LATS/LATS and may restore homeostasis under the condition of nerve injury.
Figure 5.

Synthesis of small molecule dCTB, which inhibits YAP/TAZ nuclear activity. A, Structure and chemical synthetic pathway of dCTB. B, Western blot showing the effects of intrathecal dCTB on the nuclear expression of YAP/TAZ/β-catenin after CCI. n = 4 in each group. One-way ANOVA, YAP: p = 0.0017, F = 13.97; TAZ: p = 0.0085, F = 8.481; (B, C): p = 0.0001, F = 27.35. **p < 0.01 versus sham; ##p < 0.01 versus CCI. C, Immunofluorescence staining showing effects of intrathecal dCTB on YAP/TAZ nuclear accumulation after CCI treatment. Original magnification: 800×. Scale bar, 50 μm. D, Western blot showing effects of intrathecal dCTB on the expression of MST and LATS and their phosphorylation. Left, Representative bands. Middle, Data summary. n = 4 in each group. One-way ANOVA, LATS: p = 0.0010, F = 16.30; pLATS: p = 0.0001, F = 29.13; MST: p = 0.0451, F = 4.461; p-MST: p < 0.0001, F = 72.56; ratio/LATS: p = 0.0131, F = 7.283; ratio/MST: p < 0.0001, F = 74.47. *p < 0.05, **p < 0.01 versus sham; #p < 0.05 versus CCI. Right, Schematic showing pharmacological mechanism of dCTB. In B–D, dCTB (1.6 mg, i.t.) was given on days 11, 12, and 13, respectively, after CCI. Tissues were collected 6 h after the last injection.
Spinal administration of dCTB inhibits induction and persistence of neuropathic pain
Given that dCTB has the potential to not only inhibit activity of YAP/TAZ/β-catenin, but also to reactivate CCI-induced inhibition of the Hippo signaling pathway, we continued to determine the pharmacological effect of dCTB on neuropathic pain. Rats that received CCI exhibited neuropathic pain manifested as thermal hyperalgesia and mechanical allodynia. Spinal administration of dCTB in the early phase (1.6 mg daily for 3 consecutive days starting 30 min before surgery) completely blocked the induction of thermal hyperalgesia for at least 27 d through the last examination (Fig. 6A). The onset of mechanical allodynia was greatly delayed for ∼3 weeks. The severity of the resumed mechanical allodynia was significantly less than that of the control animals without dCTB treatment (Fig. 6B). Administration of dCTB in the late phase (1.6 mg daily for 3 consecutive days on postoperative days 11, 12, and 13) completely abolished the established thermal hyperalgesia, which did not return until the last examination on postoperative day 27 (Fig. 6C). The established mechanical allodynia was also completely blocked for 10 d and then recovered ∼50% during the 10–14 d after termination of the last injection (Fig. 6D). We also tested the dose-dependent analgesic effects of intrathecal dCTB on the established neuropathic mechanical allodynia after CCI. dCTB exhibited positive, dose-dependent analgesic effects in both strength and duration of analgesia at the range of 0.08–0.32 mg. The inhibition reached peak within an hour and lasted for 2–12 h (≥6–12 h for 0.8 mg and 1.6 mg vs 2–3 h for 0.08–0.32 mg) (Fig. 6E). dCTB treatment at its highest dose used here (1.6 mg) did not affect locomotor activities and did not produce obvious side effects (Fig. 6F).
Figure 6.

Spinal administration of dCTB inhibits induction and persistence of neuropathic pain after CCI. A–D, Effects of repetitive intrathecal dCTB in the early phase (A, B) or late phase (C, D) on the induction and established thermal hyperalgesia (A, C) and mechanical allodynia (B, D). Solid triangle indicates surgery. Rats: n = 8 in each group. Each administration of dCTB (1.6 mg) is indicated by an arrow on the corresponding time point. Two-way ANOVA, F(3, 448) = 212.8, p < 0.0001 (A); F(3, 448) = 572.5, p < 0.0001 (B); F(3, 448) = 181.2, p < 0.0001 (C); F(3, 448) = 627.3, p < 0.0001 (D), *p < 0.05, **p < 0.01 versus sham+PEG400; #p < 0.05, ##p < 0.01 versus CCI+PEG400. E, Dose-dependent analgesic effect of dCTB. Six different doses dCTB was injected, respectively, on the 14th day (arrow) after CCI and its analgesic effect was tested in the following 1–24 h. F, dCTB, at the maximum dose used (1.6 mg), did not affect the locomotor activities evaluated by the running time and maximum speed in ROTA-ROD experiment in naive rats. Individual Student's t test was used to test the possible difference between the preinjection and postinjection of dCTB or PEG400 in each of the groups. In A–F, n = 8 rats were included in each of the groups.
In addition to the tremendous analgesic effects of dCTB, our results also showed that repetitive intrathecal dCTB (1.6 mg daily for 3 consecutive days) greatly inhibited the neurochemical signs accompanying neuropathic pain, including activation of the astrocytes and microglial cells (Fig. 7A), as well as phosphorylation of NR2B and PKCγ (Fig. 7B) in the spinal cord after CCI treatment.
Figure 7.

Spinal administration of dCTB inhibits activation of glial cells in the spinal cord after CCI. A, Immunofluorescence showing effects of dCTB on activation of microglial cells (IBA1) and astrocytes (GFAP) after CCI. dCTB (1.6 mg) was given daily for 3 consecutive days starting 30 min before surgery and on postoperative day 11 for detecting its effect on IBA1 and GFAP, respectively. Tissues were collected 6 h after termination of the last injection (20 spinal cord sections in each group). Original magnification: 200×. Scale bars, 50 μm. B, Western blot showing effects of dCTB on phosphorylation of NR2B and PKCγ. dCTB (1.6 mg) was given once a day on postoperative days 11, 12, and 13. Tissues were collected 6 h after termination of the last injection (n = 4). One-way ANOVA, (A) IBA1: p < 0.0001, F = 26.84; GFAP: p < 0.0001, F = 12.89; (B) NR2B: p = 0.0080, F = 11.97; PKC: p = 0.0025, F = 12.51. *p < 0.05 versus sham. #p < 0.05, ##p < 0.01 versus CCI.
Discussion
Our study reveals that YAP/TAZ are core mechanisms underlying the pathogenesis of neuropathic pain, making them targets in the screening for potent analgesics. We also discovered a potent analgesic dCTB for treating neuropathic pain after nerve injury. The principle findings are fourfold. First, nerve injury (CCI treatment) causes the rapid onset of long-lasting nuclear accumulation (activity) of YAP/TAZ/β-catenin in the spinal dorsal horn, but not in the DRG. Second, spinal inhibition or knock-down of YAP activity suppresses induction and persistence of CCI-induced mechanical allodynia, as well as allodynia induced by LPA and Wnt3a, whereas inhibition or knock-down of TAZ suppresses only established (persistent) mechanical allodynia. Third, promoting nuclear accumulation of YAP/TAZ leads to mechanical hypersensitivity in naive animals. Fourth, spinal administration of dCTB, a new small molecule that targets YAP/TAZ/β-catenin activity, inhibits the induction and persistence of neuropathic pain, as well as associated neurochemical alterations.
YAP/TAZ: an “ON–OFF” switch for neuropathic pain
We showed recently that canonical Wnt signaling, which is important for various developmental processes and shows dysregulation in certain diseases and disorders (Clevers and Nusse, 2012), is critical to the pathogenesis of neuropathic pain (Zhang et al., 2013). Canonical Wnt signaling activation is mediated by β-catenin, which is regulated by a cytoplasmic destruction complex (Clevers, 2006; Taelman et al., 2010; Li et al., 2012). In this study, we have demonstrated that the nuclear activity of the transcriptional regulators YAP and TAZ, which orchestrate the Wnt response by incorporating into the β-catenin destruction complex (Azzolin et al., 2014), is the core mechanism underlying the pathogenesis of neuropathic pain. Nerve injury causes rapid-onset and long-lasting nuclear accumulation of YAP/TAZ and β-catenin in the spinal dorsal horn neurons and of TAZ in microglial cells. There was a brief report showing alteration of YAP/TAZ in the spinal cord after nerve injury (Li et al., 2013). Spinal inhibition of YAP/TAZ activity suppresses neuropathic pain after nerve injury. Meanwhile, promotion of nuclear accumulation of YAP/TAZ leads to pain in naive animals. Nuclear activity of YAP/TAZ is sufficient and necessary for pain development precisely orchestrating pain status by flipping an ON–OFF switch after nerve injury and activation of certain pain initiators such as LPA and Wnt3a. A flowchart illustrating mechanisms underlying the contributions of YAP/TAZ and β-catenin to neuropathic pain is shown in Figure 8.
Figure 8.
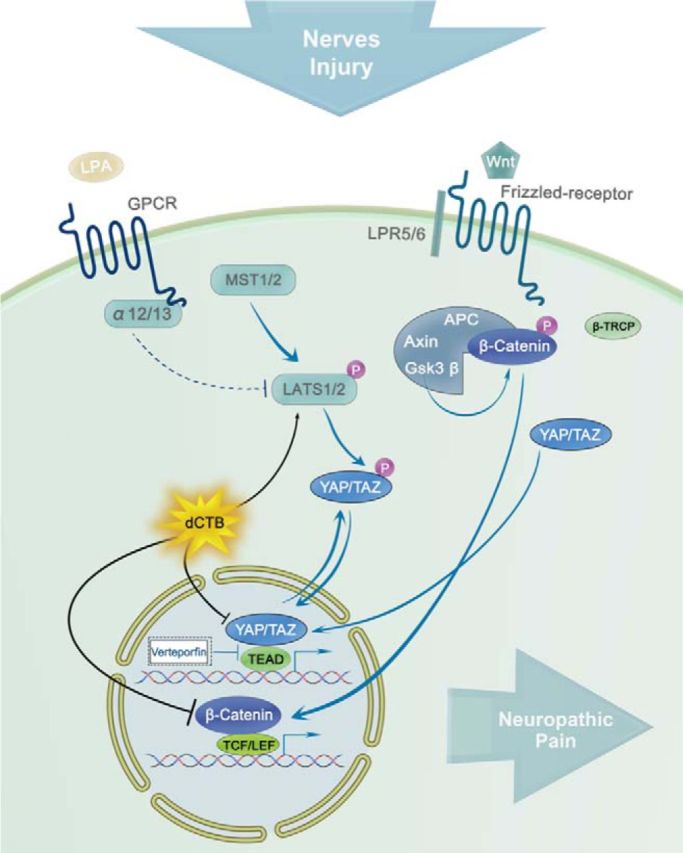
Schematic representation of the mechanisms underlying contributions of YAP/TAZ/β-catenin to the spinal central sensitization after nerve injury and regulation of dCTB on YAP/TAZ/β-catenin and Hippo signaling pathways. The flowchart illustrates the possible pathways for nuclear activity of YAP/TAZ. Inhibition of YAP/TAZ activity results in analgesia. The small molecule dCTB that we newly synthesized can effectively inhibit YAP/TAZ activity by activating the Hippo signaling pathway through increasing the phosphorylation of LATS.
We also found that YAP and TAZ may play different roles in neuropathic pain. YAP is involved in both early (induction) and later (persistence) phases of neuropathic pain, whereas TAZ is involved only in the later phase. Such different roles of YAP and TAZ are, in a certain way, correlated with the different timing patterns of their nuclear accumulation and distinct cellular distributions in the spinal dorsal horn neurons and suggest their different functions in these cells and different roles in the pathophysiology of neuropathic pain. These different roles of YAP and TAZ also suggest that YAP may contribute to the pathogenesis of neuropathic pain via TAZ-independent mechanisms, although they are two related transcriptional coactivators downstream of the Hippo pathway. A recent study reports that YAP and TAZ play different roles during cancer progression and their expression profiles differ depending on the cancer cell type. Hepatocellular carcinoma exhibited a TAZ-dominant expression profile rather than a YAP-dominant expression profile under normal conditions and a shift toward YAP-dominant expression may be a key step in conferring cancer stem-cell-like properties in hepatocellular carcinoma (Hayashi et al., 2015). Mechanisms underlying TAZ-independent YAP function need to be further investigated. Therefore, targeting YAP and TAZ may prevent the induction of neuropathic pain and greatly suppress established painful conditions.
YAP/TAZ: potent analgesic screening targets
In this study, we have shown that nuclear activity of the transcriptional factors YAP/TAZ are core factors orchestrating the pathogenesis of neuropathic pain, making them potent therapeutic targets. Therefore, a drug that inhibits YAP/TAZ may have a potent analgesic effect. We have proven that VP, which impedes YAP-TEAD association and inhibits YAP activity (Liu-Chittenden et al., 2012), is a potent analgesic, supporting the idea that VP, a prescriptive medicine currently used in clinic for treating macular degeneration (Brown et al., 2006), may also be used for treating neuropathic pain. Excitingly, we have further identified a small molecule, dCTB, and shown that it may be the most potent analgesic in treating neuropathic pain. dCTB is an existing antitumor small molecule and was reported as an inhibitor of the transcriptional responses of YAP/TAZ (Basu et al., 2014). A chemical synthetic pathway of dCTB described in the only report named compound 19 (C19) (Basu et al., 2014) did not lead to an effective synthesis. We have developed a new efficient chemical synthetic pathway (Fig. 5A) to produce the compound dCTB and have proven that dCTB is an activator of phosphorylation of LATS and a potent inhibitor of YAP/TAZ and β-catenin. The biological effects of YAP/TAZ and β-catenin do not fully overlap, but YAP/TAZ, as indicated in this study, and β-catenin, as indicated in our recent study (Zhang et al., 2013), are all critical to the development of neuropathic pain. Therefore, we believe that our newly synthesized dCTB is among the most potent analgesics, perhaps the strongest one so far, for treating neuropathic pain. This in turn firmly supports our hypothesis that YAP/TAZ and β-catenin are critical in orchestrating the development of neuropathic pain. The analgesic effect of dCTB via its regulation on Hippo and YAP/TAZ/β-catenin signaling pathways is illustrated in Figure 8.
dCTB can greatly reverse the downregulation of p-LATS1 without affecting the downregulation of p-MST1/2 after CCI treatment (Fig. 5D). These results suggest that dCTB may increase activities of LATS via an MST1/2-independent pathway. Studies have shown that the activity of LATS1/2 can be regulated by several inputs. NF2/Merlin and MAP4Ks activate LATS1/2 in parallel to MST1/2. LATS1/2 kinase activity can also be indirectly regulated by G-protein-coupled receptors and by the F-actin cytoskeleton (Piccolo et al., 2014). In addition, LATS activities can be increased by the adenosine monophosphate-activated protein kinase (AMPK) (DeRan et al., 2014; Wang et al., 2015). AMPK is a key cellular energy sensor that regulates energy homeostasis; therefore, energy stress can also influence the activity of LATS1/2. A recent study showed that a molecule compound C19, which is similar to dCTB in this study, can activate AMPK (Basu et al., 2014). Therefore, dCTB may increase activities of LATS via activation of AMPK, which is independent of MST1/2.
Recapitulation mechanism underlying neuropathic pain
The specific cellular and molecular mechanisms underlying neuropathic pain remain elusive. Numerous hypotheses have focused on alterations and phenotypic switches of the nociceptive transmitters and/or modulators and on their receptors and downstream signaling pathways that have already exerted roles in pain processes in mature nervous systems. They focus on the fact that the nociceptive system becomes sensitized after somatosensory nerve injury, which leads to neuropathic pain. This expression of maladaptive plasticity has been the dominant model underlying neuropathic pain (Costigan et al., 2009). We have recently provided evidence supporting the idea that neuropathic-pain-inducing nerve injury and other forms of severe stress such as bone cancer and withdrawal of chronic morphine treatment may elicit neuronal alterations that “recapitulate” events occurring during development. That is, after injury, the nerve may seek to repair itself and reconnect damaged nerve fibers by reawakening dormant developmental signaling pathways. The activated molecules may include Wnt signaling (Zhang et al., 2013; Liu et al., 2015) and its target gene EphB receptor signaling (Song et al., 2008; Liu et al., 2009; Liu et al., 2011; Liu et al., 2013), which become triggers for the development of neuropathic pain. These molecules therefore are potential therapeutic targets for treating neuropathic pain. In addition, the developmental signaling pathways Notch and Hedgehog are also known to contribute to the pathogenesis of neuropathic pain (Babcock et al., 2011; Sun et al., 2012). This contribution, due to nerve injury-induced activation of these developmental molecules, is a unique and fundamentally different concept from maladaptive plasticity. Through the present study, we have reinforced the “recapitulation” model of neuropathic pain, which is coherent with the “maladaptive plasticity” model in that both emphasize the hyperplastic state during neuropathic pain. We reveal that the transcriptional factors YAP/TAZ, which play a critical role as a nexus for Hippo signaling and an integrator of several other prominent signaling pathways, including Wnt, G-protein-coupled receptor, and Notch pathways (Hansen et al., 2015), are critical mechanisms underlying the pathogenesis of neuropathic pain and potent targets for treating neuropathic pain after nerve injury and probably similar disorders of the nervous systems.
Footnotes
This work was supported by the National Natural Science Foundation of China (Grants 81320108012, 81271241, and 81671086). We thank C.S. Zhao at Peking University for drawing the schematics and A.A. Song at University of Pennsylvania Perelman School of Medicine for help editing the manuscript.
The authors declare no competing financial interests.
References
- Angus L, Moleirinho S, Herron L, Sinha A, Zhang X, Niestrata M, Dholakia K, Prystowsky MB, Harvey KF, Reynolds PA, Gunn-Moore FJ. Willin/FRMD6 expression activates the Hippo signaling pathway kinases in mammals and antagonizes oncogenic YAP. Oncogene. 2012;31:238–250. doi: 10.1038/onc.2011.224. [DOI] [PubMed] [Google Scholar]
- Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013;154:1047–1059. doi: 10.1016/j.cell.2013.07.042. [DOI] [PubMed] [Google Scholar]
- Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, Bresolin S, Frasson C, Basso G, Guzzardo V, Fassina A, Cordenonsi M, Piccolo S. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell. 2014;158:157–170. doi: 10.1016/j.cell.2014.06.013. [DOI] [PubMed] [Google Scholar]
- Babcock DT, Shi S, Jo J, Shaw M, Gutstein HB, Galko MJ. Hedgehog signaling regulates nociceptive sensitization. Curr Biol. 2011;21:1525–1533. doi: 10.1016/j.cub.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu D, Lettan R, Damodaran K, Strellec S, Reyes-Mugica M, Rebbaa A. Identification, mechanism of action, and antitumor activity of a small molecule inhibitor of hippo, TGF-beta, and Wnt signaling pathways. Mol Cancer Ther. 2014;13:1457–1467. doi: 10.1158/1535-7163.MCT-13-0918. [DOI] [PubMed] [Google Scholar]
- Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS, Kim RY, Sy JP, Schneider S, ANCHOR Study Group Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1432–1444. doi: 10.1056/NEJMoa062655. [DOI] [PubMed] [Google Scholar]
- Chen SN, Gurha P, Lombardi R, Ruggiero A, Willerson JT, Marian AJ. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ Res. 2014;114:454–468. doi: 10.1161/CIRCRESAHA.114.302810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRan M, Yang J, Shen CH, Peters EC, Fitamant J, Chan P, Hsieh M, Zhu S, Asara JM, Zheng B, Bardeesy N, Liu J, Wu X. Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep. 2014;9:495–503. doi: 10.1016/j.celrep.2014.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Hansen CG, Moroishi T, Guan KL. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 2015;25:499–513. doi: 10.1016/j.tcb.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Higashi T, Yokoyama N, Kaida T, Sakamoto K, Fukushima Y, Ishimoto T, Kuroki H, Nitta H, Hashimoto D, Chikamoto A, Oki E, Beppu T, Baba H. An imbalance in TAZ and YAP expression in hepatocellular carcinoma confers cancer stem cell-like behaviors contributing to disease progression. Cancer Res. 2015;75:4985–4997. doi: 10.1158/0008-5472.CAN-15-0291. [DOI] [PubMed] [Google Scholar]
- Inoue M, Rashid MH, Fujita R, Contos JJ, Chun J, Ueda H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat Med. 2004;10:712–718. doi: 10.1038/nm1060. [DOI] [PubMed] [Google Scholar]
- Li N, Lim G, Chen L, McCabe MF, Kim H, Zhang S, Mao J. Spinal expression of Hippo signaling components YAP and TAZ following peripheral nerve injury in rats. Brain Res. 2013;1535:137–147. doi: 10.1016/j.brainres.2013.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, Mohammed S, Heck AJ, Maurice MM, Mahmoudi T, Clevers H. Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell. 2012;149:1245–1256. doi: 10.1016/j.cell.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Liu S, Liu WT, Liu YP, Dong HL, Henkemeyer M, Xiong LZ, Song XJ. Blocking EphB1 receptor forward signaling in spinal cord relieves bone cancer pain and rescues analgesic effect of morphine treatment in rodents. Cancer Res. 2011;71:4392–4402. doi: 10.1158/0008-5472.CAN-10-3870. [DOI] [PubMed] [Google Scholar]
- Liu S, Liu YP, Song WB, Song XJ. EphrinB-EphB receptor signaling contributes to bone cancer pain via Toll-like receptor and proinflammatory cytokines in rat spinal cord. Pain. 2013;154:2823–2835. doi: 10.1016/j.pain.2013.08.017. [DOI] [PubMed] [Google Scholar]
- Liu S, Liu YP, Huang ZJ, Zhang YK, Song AA, Ma PC, Song XJ. Wnt/Ryk signaling contributes to neuropathic pain by regulating sensory neuron excitability and spinal synaptic plasticity in rats. Pain. 2015;156:2572–2584. doi: 10.1097/j.pain.0000000000000366. [DOI] [PubMed] [Google Scholar]
- Liu WT, Li HC, Song XS, Huang ZJ, Song XJ. EphB receptor signaling in mouse spinal cord contributes to physical dependence on morphine. FASEB J. 2009;23:90–98. doi: 10.1096/fj.08-114462. [DOI] [PubMed] [Google Scholar]
- Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HW, Kim YC, Yu B, Moroishi T, Mo JS, Plouffe SW, Meng Z, Lin KC, Yu FX, Alexander CM, Wang CY, Guan KL. Alternative Wnt signaling activates YAP/TAZ. Cell. 2015;162:780–794. doi: 10.1016/j.cell.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94:1287–1312. doi: 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- Simonetti M, Agarwal N, Stösser S, Bali KK, Karaulanov E, Kamble R, Pospisilova B, Kurejova M, Birchmeier W, Niehrs C, Heppenstall P, Kuner R. Wnt-Fzd signaling sensitizes peripheral sensory neurons via distinct noncanonical pathways. Neuron. 2014;83:104–121. doi: 10.1016/j.neuron.2014.05.037. [DOI] [PubMed] [Google Scholar]
- Song XJ, Zheng JH, Cao JL, Liu WT, Song XS, Huang ZJ. EphrinB-EphB receptor signaling contributes to neuropathic pain by regulating neural excitability and spinal synaptic plasticity in rats. Pain. 2008;139:168–180. doi: 10.1016/j.pain.2008.03.019. [DOI] [PubMed] [Google Scholar]
- Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S, Rosato A, Piccolo S, Del Sal G. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol. 2014;16:357–366. doi: 10.1038/ncb2936. [DOI] [PubMed] [Google Scholar]
- Sun YY, Li L, Liu XH, Gu N, Dong HL, Xiong L. The spinal notch signaling pathway plays a pivotal role in the development of neuropathic pain. Mol Brain. 2012;5:23. doi: 10.1186/1756-6606-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I, Sabatini DD, De Robertis EM. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143:1136–1148. doi: 10.1016/j.cell.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xiao ZD, Li X, Aziz KE, Gan B, Johnson RL, Chen J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nature cell biology. 2015;17:490–499. doi: 10.1038/ncb3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, Fu XD, Mills GB, Guan KL. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780–791. doi: 10.1016/j.cell.2012.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YK, Huang ZJ, Liu S, Liu YP, Song AA, Song XJ. WNT signaling underlies the pathogenesis of neuropathic pain in rodents. J Clin Invest. 2013;123:2268–2286. doi: 10.1172/JCI65364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]