

Abstract
Neuroblastoma (NB) is a childhood tumor that arises from the sympathoadrenal lineage. MYCN amplification is the most reliable marker for poor prognosis and MYCN overexpression in embryonic mouse sympathetic ganglia results in NB-like tumors. MYCN cooperates with mutational activation of anaplastic lymphoma kinase (ALK), which promotes progression to NB, but the role of MYCN and ALK in tumorigenesis is still poorly understood. Here, we use chick sympathetic neuroblasts to examine the normal function of MYCN and MYC in the control of neuroblast proliferation, as well as effects of overexpression of MYCN, MYC, and activated ALK, alone and in combination. We demonstrate that MYC is more strongly expressed than MYCN during neurogenesis and is important for in vitro neuroblast proliferation. MYC and MYCN overexpression elicits increased proliferation but does not sustain neuroblast survival. Unexpectedly, long-term expression of activated ALKF1174L leads to cell-cycle arrest and promotes differentiation and survival of postmitotic neurons. ALKF1174L induces NEFM, RET, and VACHT and results in decreased expression of proapototic (BMF, BIM), adrenergic (TH), and cell-cycle genes (e.g., CDC25A, CDK1). In contrast, neuroblast proliferation is maintained when MYCN and ALKF1174L are coexpressed. Proliferating MYCN/ALKF1174L neuroblasts display a differentiated phenotype but differ from ALK-expressing neurons by the upregulation of SKP2, CCNA2, E2F8, and DKC1. Inhibition of the ubiquitin ligase SKP2 (S-phase kinase-associated protein 2), which targets the CDK inhibitor p27 for degradation, reduces neuroblast proliferation, implicating SKP2 in the maintained proliferation of MYCN/ALKF1174L neuroblasts. Together, our results characterize MYCN/ALK cooperation leading to neuroblast proliferation and survival that may represent initial steps toward NB development.
SIGNIFICANCE STATEMENT MYCN overexpression combined with activated anaplastic lymphoma kinase (ALK) is sufficient to induce neuroblastoma (NB) in mouse sympathoadrenal cells. To address cellular and molecular effects elicited by MYCN/ALK cooperation, we used cultures of chick sympathetic neuroblasts. We demonstrate that MYCN increases proliferation but not survival, whereas long-term expression of ALKF1174L elicits cell-cycle exit, differentiation, and survival of postmitotic neurons. Combined MYCN/ALKF1174L expression allows long-term proliferation and survival of neuroblasts with differentiated characteristics. In the presence of ALKF1174L signaling, MYCN induces the expression of the ubiquitin ligase SKP2 (S-phase kinase-associated protein 2), which targets p27 for degradation and is also upregulated in high-risk NB. SKP2 inhibition supports a function for SKP2 in the maintained neuroblast proliferation downstream of MYCN/ALK, which may represent an early step toward tumorigenesis.
Keywords: ALK, MYCN, neuroblast, neuroblastoma, neurogenesis, sympathetic
Introduction
Neuroblastoma (NB) is a childhood tumor that develops from neural crest-derived cells in sympathetic ganglia and adrenal medulla. NB displays a broad clinical spectrum that correlates with differentiation state. High-risk disease involving undifferentiated, metastatic tumors relapses in the majority of cases and is almost always fatal (Maris, 2010; Simon et al., 2011). A characteristic feature of high-risk NB is amplification of MYCN, a proto-oncogene encoding MYCN that belongs, together with c-MYC (hereafter called MYC) and MYCL, to the MYC-family of helix-loop-helix-Zip transcription factors (Brodeur et al., 1984). Targeted expression of MYCN in tyrosine hydroxylase (TH)-expressing cells of embryonic transgenic mice results in the postnatal generation of NB (Weiss et al., 1997; Hansford et al., 2004; Althoff et al., 2015).
Activating point mutations in the gene-encoding anaplastic lymphoma receptor tyrosine kinase (ALK) were discovered in the majority of familial and ∼8% of sporadic NB (Chen et al., 2008; George et al., 2008; Janoueix-Lerosey et al., 2008; Mosse et al., 2008). One of the most common somatic mutations results in a phenylalanine-to-leucine substitution at codon 1174 (F1174L). ALKF1174L is present in all NB subtypes, but in association with MYCN amplification it defines a subset of NB patients with poor outcome (De Brouwer et al., 2010). Expression of activated ALK in sympathoadrenal cells of transgenic and knock-in mice induced NB only when expressed transgenically using the strong β-actin promotor (Heukamp et al., 2012) but not when under control of the Th or endogenous Alk promotor (Berry et al., 2012; Cazes et al., 2014). Similarly, ALKF1174L expression was unable to induce NB in zebrafish (Zhu et al., 2012). In contrast, the combination of activated ALK and MYCN overexpression results in fully penetrant and rapid generation of NB without any additional genomic alterations (Berry et al., 2012; Heukamp et al., 2012; Cazes et al., 2014). Therefore, NB elicited by ALK/MYCN cooperation represents an interesting model to study cellular and molecular mechanisms of NB development. Comparing gene-expression profiles of ALK/MYCN with MYCN tumors identified (1) increased expression of components of the PI3K/AKT/mTor and MAPK pathway, which results in stabilization of MYCN protein; (2) increased MYCN transcription; and (3) reduced apoptosis (Berry et al., 2012; Heukamp et al., 2012). In addition, the tyrosine kinase receptor RET is induced in ALK/MYCN tumors and controls tumor growth (Cazes et al., 2014). Although the earlier onset and increased penetrance of tumor formation implies a function of these signaling conduits in tumor development, it remains unclear at which stage these mechanisms are effective. Neuroblasts may either be induced to expand from embryonic stages onwards or may depend for their postnatal survival on ALK/MYCN cooperation.
In embryonic sympathetic ganglia of both TH-MYCN and wild-type mice clusters of highly proliferating cells are present but selectively maintained postnatally in TH-MYCN ganglia (Hansford et al., 2004; Alam et al., 2009). In the Ki-Alk mouse, neuroblast proliferation is transiently increased in embryonic and early postnatal ganglia but terminated at postnatal day 18 (Cazes et al., 2014). The situation in sympathetic ganglia and adrenals coexpressing activated ALK and MYCN has not been investigated.
Here, we used chick sympathetic neuroblasts to investigate the effects and interactions of MYCN, MYC, and activated ALK on neuroblast proliferation and survival. We demonstrate that neuroblast proliferation depends mainly on MYC. Overexpression of MYCN or MYC supports continued high-level proliferation but not neuroblast survival. In contrast, ALKF1174L-expressing neuroblasts display only an initial proliferation increase and subsequently leave the cell cycle, acquire a mature neuron morphology, and display increased survival. Importantly, the combined expression of ALKF1174L and MYC proteins supports both neuroblast proliferation and survival. Compared with ALKF1174L-expressing neuroblasts, MYCN/ALKF1174L cells maintain elevated levels of neuronal differentiation markers and show increased expression of the cell-cycle-related MYCN target genes SKP2, DKC1, E2F8, and CCNA2. Cell-cycle exit observed upon SKP2 (S-phase kinase-associated protein 2) inhibition supports a function of SKP2 in the control of MYCN/ALKF1174L neuroblast proliferation.
Materials and Methods
Plasmid construction.
Expression vectors used were constructed by cloning restriction enzyme-digested or PCR-amplified DNA fragments with standard protocols. The PiggyBac (PB-CAG-eGFP) and PBase (mPB) expression vectors described previously (Cadiñanos and Bradley, 2007; Nagy et al., 2011), were obtained from the Wellcome Sanger Institute and from Andres Nagy at the Samuel Lunenfeld Research Institute, Mount Sinai Hospital, Toronto, Canada, respectively. PB-CAG-eGFP was used as parental plasmid replacing eGFP by either MYC, MYCN, or ALKF1174L or generating a control plasmid without eGFP [PiggyBac (PB) control]. Plasmids containing human MYCN and ALKF1174L were generously provided by Christian Beltinger (University Clinic Ulm, Ulm, Germany) and by Isabelle Janoueix-Lerosey (Institut Curie, Paris, France), respectively. The chicken MYC plasmid has been described previously (Zinin et al., 2014).
Cell culture, electroporation, pharmacological treatment, and immunostaining.
Paravertebral lumbosacral sympathetic chain ganglia were dissected from embryonic day (E) 7 chick embryos of either sex and dissociated to single cells as described previously (Rohrer and Thoenen, 1987; Zackenfels et al., 1995). Cells were either plated directly or transfected by electroporation using Amaxa Nucleofector II and the Basic Neuron Small Cell number (SCN) Nucleofector Kit (Program SCN2). For integration of MYC, MYCN, and ALKF1174L into the chick genome, the PB DNA-transposition method was applied (Ding et al., 2005), in which the genes of interest, cloned between the two PB arms of the PB vector pCYL50 are cotransfected with Pbase-expression vector (for a schematic representation of the PB system, see Woodard and Wilson, 2015). Specifically, cells were cotransfected with GFP-expression vector and PB (controls) or with GFP-expression vector, PB, and PB-expression vectors for MYC, MYCN, and ALKF1174L. For knockdown experiments, cells were also electroporated with siRNA against MYC, MYCN, or with control siN1. The following concentrations of DNA or RNA were used: PB-CAG-eGFP (0.25 μg/200,000 cells), PB-CAG-NMYC (0.25 μg/200,000 cells), PB-CAG-Cmyc (0.25 μg/200,000 cells), PB-CAG-ALKF1174L (0.2 μg/200,000 cells), mPBase (0.3 μg/200,000 cells), and empty PB-CAG vector (0.25 μg/200,000 cells) as control. iBONI siRNA Quattro siGgMYCN (50 pmol of each siRNA/200,000 cells; Riboxx), iBONI siRNA siGgMYCN(1) 5′-UAUUUCUUCUUCAUCAUCCCCC-3′ (Riboxx), siRNA siGgMYCN(2) 5′-CAACAGUUGCUAAAGAAAACCCCC-3′ (Riboxx), siRNA siGgMYCN(3) 5′-CGCCAAUUCACCAGCAGCAUAAUUA-3′ (Invitrogen Block-iT; stealth 905), siRNA siGgMYC 5′-AGAUCAGCAACAACCGAAA-3′ (Eurofins Genomics; all at 100 pmol siRNA/200,000 cells; Eurofins Genomics) and, as control, iBONI siRNA Negative Control-N1 and Invitrogen Block-iT control 3′-UAAUGUUGUAGUCGUCGUGUAAGCG-5′ (200 pmol/200,000 cells). After nucleoporation, cell debris was removed by density step-gradient centrifugation and 15,000 cells were plated per well on four-well culture dishes coated with 0.5 mg/ml poly-dl-ornithine (Sigma-Aldrich) in 0.15 m borate buffer, pH 8.3, and 10 μg/ml laminin (Invitrogen) in PBS, pH 7.3, and cultured in MEM, 10% (v/v) horse serum, 5% (v/v) fetal calf serum, 1% (w/v) penicillin/streptomycin, and 1% (w/v) glutamine at 5% CO2. Where indicated, 2% chick embryo extract (CEE) was added to the medium to support long-term survival (Haltmeier and Rohrer, 1990). BET bromodomain protein inhibitor JQ1 (ab141498, Abcam), RET inhibitor vandetanib (15706, Cayman Chemical), and SKP2 inhibitor SKPin C1 (4817, Tocris Bioscience) were dissolved in sterile DMSO at 10 mm (JQ1, C1) or 5 mm (vandetanib) and diluted to appropriate final concentrations in medium. Proliferating neuroblasts were detected by 5-ethynyl-2-deoxyuridine (EdU) labeling using the Click-iT EdU Alexa Fluor 647 imaging kit (Invitrogen) as described previously (Reiff et al., 2011; Holzmann et al., 2015) combined with immunostaining for TH and GFP or Alk and GFP. GFP expression was analyzed by staining with anti-GFP antibody (1:400, rabbit, polyclonal; A-11122, Invitrogen) followed by Alexa 488 anti-rabbit as secondary antibody. TH expression was detected by staining with anti-TH antibody (1:50, mouse, monoclonal; generated and characterized previously; Rohrer et al., 1986; Bonnefoy et al., 1988) followed by Alexa 546 anti-mouse secondary antibody. Alk expression was detected by anti-ALK antibody (1:1000, rabbit, monoclonal; #3633, Cell Signaling Technology) followed by Alexa 546 anti-rabbit secondary antibody. Cell nuclei were visualized with DAPI (Sanofi Aventis). Images were acquired at a magnification of 20× using Zeiss AxioImager.Z1 equipped with an AxioCam MRm camera.
The proportion of EdU-labeled, TH-positive, or Alk-positive and GFP-positive transfected neuroblasts was quantified in ≥3 independent experiments. For cell survival, the number of TH-positive and GFP-positive cells was determined for each experiment on 20 arbitrarily chosen visual fields per cultured dish and ≥3 independent experiments were evaluated. For apoptosis assays, the percentage of transfected GFP-positive cells with pyknotic nuclei was determined in five independent experiments. All results are provided as mean ± SEM of ≥3 independent experiments and statistically analyzed with unpaired two-tailed Student's t test or one-way ANOVA with the Tukey–Kramer multiple comparisons test.
In situ hybridization.
In situ hybridization (ISH) for MYC, MYCN, and SCG10 was performed in fixed tissue as described previously (Zinin et al., 2014; Ribeiro et al., 2016). Briefly, E7-derived sections were incubated with digoxigenin-labeled single-stranded RNA probes at 72°C for MYC and MYCN and 68°C for SCG10, followed by incubation with alkaline phosphatase-coupled antibody and nitroblue tetrazolium plus 5-bromo-4-chloro-3-indolyl phosphate (purple) substrates.
Chicken embryo electroporation.
In ovo electroporation was performed as previously described (Kaltezioti et al., 2010). Briefly, white Leghorn chicken eggs were incubated until Hamburger Hamilton stage 12–14 (E2). Plasmids were used at a concentration of 1–2 mg/ml with 0.025% Fast Green (Sigma-Aldrich), injected into the lumen of the neural tube (10 μl total volume). For PB transposition, a 2:1 ratio of transposon (PB-CAG-GFP alone or with PB-CAG-MYCN) and transposase (PBase) was used. Electrodes were spaced 4 mm part and positioned lateral to the neural tube. Embryos were pulsed 3× for 30 ms each at 28 V. Eggs were reincubated for 3 d followed by fixation for 4 h at 4°C in 4% paraformaldehyde in PBS. Embryos were washed in PBS, cryoprotected with 20% sucrose, mounted in Tissue Tec (Sakura Fintek), and sectioned at 12–14 μm. For EdU labeling, embryos received 500 μm (500 μl) EdU in PBS 4 h before fixation. Sections were triple-stained for GFP, TH, and EdU and analyzed for the percentage of EdU-labeled, transfected Th-expressing neuroblasts on ≥20 sections per embryo. Electroporation experiments were quantified from seven control and seven MYCN-transfected embryos.
qRT-PCR analysis.
For qRT-PCR analysis of cultured sympathetic ganglion cells, cultures were kept on poly-dl-ornithine/laminin-coated 3.5 cm culture dishes. After 2 or 8 d of cultivation, cells were harvested with a cell scraper and RNA was isolated using the RNeasy Kit (Qiagen) following the manufacturer's instructions. qRT-PCR was performed using the QuantiTect SYBR Green PCR Kit with the following QuantiTect primer assays (Qiagen): Gg_MAD2L1_1_SG; Gg_CCNA2_1_SG; Gg_PLK1_2_SG; Gg_SOX11_1_SG; Gg_SMC2_1_SG; Gg_CDK1_1_SG; Gg_SKP2-2-SG; Gg_E2F8_1_SG; Gg_CDC25A_1_SG; Gg-DKC1_1_SG; Gg_GAPDH_1_SG; Gg_SLC18A3_1_SG; Gg_BCL2L1_1_SG; Gg_BMF_1_SG; Gg_BOK_1_SG; Gg_VIP_1_SG; Gg_MYC_1_SG; Gg_NEFM_1_SG; Gg_DBH_1_SG; Gg_TH_1_SG; Gg_RET_1_SG; Gg_BIRC5_1_SG; Gg_BID_1_SG; Gg_BCL2_1_SG; Gg_MYCN_1_SG; Gg_TLX3_1_SG; Gg_LIN28B_1_SG; Gg_NTRK1_1_SG. Note that Gg-MYCN primers do not recognize human MYCN used for overexpression experiments. The temperature profile for all qPCR experiments was the following: 95°C for 15 min and 40 cycles (94°C for 15 s, 55°C for 30 s, 72°C for 30 s). At least triplets of every condition were performed in parallel and experiments were repeated independently at ≥3 times. Data are normalized to GAPDH as reference genes, expressed as a gene expression ratio, and statistically evaluated using the relative expression software tool (Pfaffl et al., 2002).
Results
MYCN and MYC are coexpressed in sympathetic neuroblasts during neurogenesis
In the embryonic chick spinal cord and dorsal root ganglia, MYC and MYCN mRNA are expressed in a mirror image pattern (Zinin et al., 2014). MYCN is restricted to SOX2-positive proliferating progenitors, whereas MYC is observed in differentiated Islet1-expressing neurons. In sympathetic ganglia, MYC is expressed at much higher levels compared with MYCN as judged from ISH and qRT-PCR analysis of sympathetic ganglia dissected at E7 (Fig. 1). Similar results were obtained for cultures of E7 sympathetic ganglion cells (15-fold higher by qRT-PCR; n = 3; p < 0.05). E7 represents the peak of neurogenesis in chick sympathetic ganglia, which are mainly composed of proliferating neuroblasts at this developmental stage (Holzmann et al., 2015). In view of the strong difference in their expression levels (Fig. 1), it was of interest to investigate to which extent neuroblast proliferation depends on MYC and/or MYCN.
Figure 1.

Expression of MYCN and MYC in developing chick sympathetic ganglia. A, ISH for MYC and MYCN on sections of E7 chick sympathetic ganglia reveals much lower signals for MYCN compared with the strong MYC expression. SCG10 expression is shown to illustrate the location of sympathetic ganglia (SYM), dorsal root ganglia (DRG), and spinal cord (SC). Arrowheads point to sympathetic ganglia. B, High MYC expression compared with MYCN is shown by qRT-PCR on dissected E7 sympathetic ganglia (mean ± SE; ***p < 0.001; statistical analysis by relative expression software tool).
Proliferation of sympathetic neuroblasts depends on MYC proteins
Neuroblast proliferation was analyzed in cultures of dissociated E7 chick sympathetic ganglia by EdU labeling as described previously (Zackenfels et al., 1995; Reiff et al., 2010, 2011). E7 is the earliest age sympathetic ganglia can be reliably dissected from the chick embryo. To investigate the role of MYC proteins in neuroblast proliferation, siRNA directed against MYC or MYCN was used (Zinin et al., 2014). siMYC led to a reduction in the proportion of proliferating EdU-labeled cells to 30 ± 0.5% of control siRNA cultures, whereas MYCN knockdown by siMYCN(2) had a much smaller effect (reduction to 67 ± 5%; Fig. 2A,B). Proliferation was reduced to a similar level in response to two additional siMYCN RNAs [siMYCN(1), siMYCN(3)] and an siMYCN RNA mixture (see Materials and Methods; data not shown).
Figure 2.
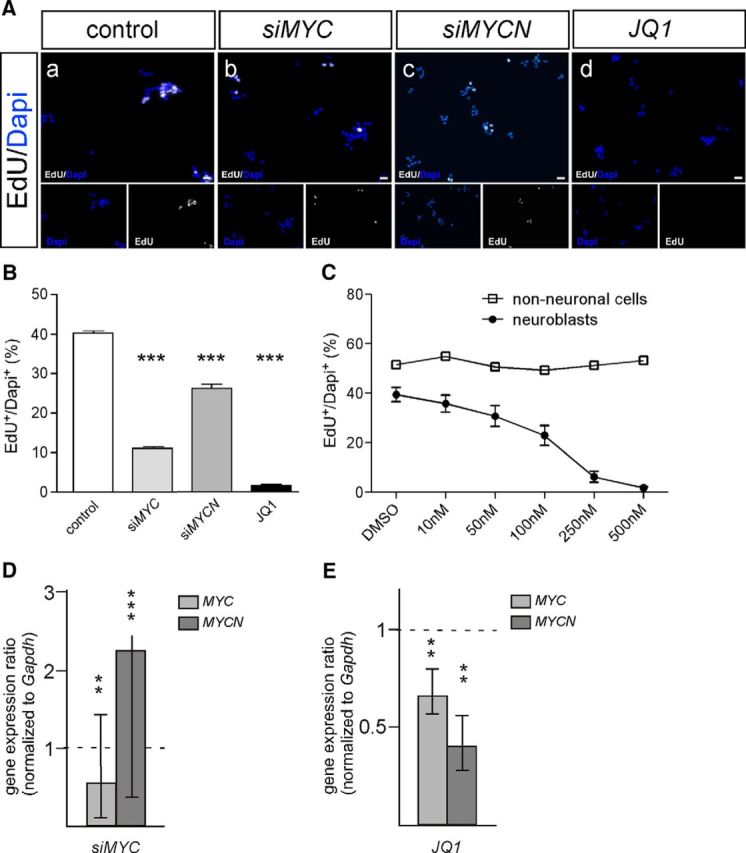
Proliferation of E7 sympathetic neuroblasts depends on MYC signaling. A, The number of proliferating neuroblasts, identified by EdU labeling (Aa–Ad) is reduced by siMYC (Ab), siMYCN(2) (Ac), and JQ1 (Ad). Scale bar, 20 μm. B, Quantification of neuroblast proliferation in cells transfected with control siN1, siMYC, siMYCN(2), or treated with JQ1 (500 nm; mean ± SEM; n ≥ 3; ***p < 0.001). C, Dose–response of the antiproliferation effect of JQ1. Whereas neuroblast proliferation is blocked at 500 nm, non-neuronal cell proliferation is not affected by JQ1. Non-neuronal cells were identified by the much larger size of their nucleus compared with neuroblasts. D, E, siMYC treatment leads to a significant reduction in MYC RNA and upregulation of MYCN RNA (D), whereas JQ1 treatment reduces both MYC and MYCN expression as shown by qRT-PCR (E; mean ± SE; **p < 0.01; ***p < 0.001; statistical analysis by relative expression software tool).
MYC mRNA level is decreased and MYCN mRNA is upregulated by siMYC as shown by qRT-PCR (Fig. 2D), which is consistent with previous evidence for inverse correlation between MYC and MYCN expression (Breit and Schwab, 1989; Henriksen et al., 2011; Zinin et al., 2014). Increased MYCN expression is unlikely to functionally compensate for the loss of MYC since MYCN is expressed at much lower levels than MYC in vivo (Fig. 1) and in cultured cells (see above). MYCN RNA levels were not affected by treatment with several different MYCN siRNAs. This may be explained by previous observations in NB cell lines that MYCN RNA, in contrast to MYCN protein, is affected only transiently by siRNA treatment (Bell et al., 2007; Henriksen et al., 2011). Together, expression and knockdown results suggest that MYC rather than MYCN is important for neuroblast proliferation during sympathetic neurogenesis.
As the BET bromodomain protein inhibitor JQ1 interferes with the expression of MYC and MYCN in tumor-cell lines and inhibits MYC-dependent tumor growth (Delmore et al., 2011; Heukamp et al., 2012; Puissant et al., 2013; Henssen et al., 2016), we also investigated the effects of JQ1 treatment in primary sympathetic neuroblast cultures. Proliferation was efficiently blocked by JQ1, with half-maximal effects at 100 nm and complete inhibition at 500 nm (Fig. 2B,C). MYC and MYCN levels were both reduced (Fig. 2E). The virtually complete block in neuroblast proliferation cannot be explained by the rather mild reduction in MYC and MYCN levels, which implies an action of JQ1 on MYC targets in addition to controlling MYC expression (Henssen et al., 2016). Notably, JQ1 did not interfere with the proliferation of ganglion non-neuronal cells present in the culture, excluding a nonspecific proliferation block (Fig. 2C).
Overexpression of MYCN and MYC leads to maintained neuroblast proliferation in vitro but does not sustain survival
In normal cells, expression of MYC family members is low and depends continuously on mitogenic signaling. Elevated levels are observed in many cancers, including NB where deregulated MYCN expression correlates with bad prognosis. To study effects of MYC protein overexpression, chick sympathetic neuroblasts were transfected with plasmids designed for genomic integration by Piggybac (Pb) transposase, allowing permanent expression (Ding et al., 2005; Lu et al., 2009; Nagy et al., 2011; Woodard and Wilson, 2015). As normal neuroblast proliferation depends on MYC, rather than MYCN, the effects of both MYC proteins were analyzed. Neuroblasts were cotransfected with Pb-flanked CAGGS-GFP, CAGGS-MYC, or CAGGS-MYCN and an episomally expressed Pb transposase plasmid. Transfected neuroblasts, identified by coexpression of GFP and TH, were analyzed for proliferation by EdU labeling after 2 and 8 d in culture (dic; Fig. 3). The proportion of EdU+/TH+-labeled transfected cells is significantly increased by both MYCN and MYC expression at 2 and 8 dic (Fig. 3B,C). MYCN overexpression increases cell numbers at 8 dic compared with GFP-transfected controls (Fig. 3E), but increased proliferation in response to MYC or MYCN does not result in a net increase in cell numbers compared with 2 dic (Fig. 3, compare D, E), which suggests that overexpression of MYC proteins is unable to maintain neuroblast survival. With increasing time in culture, survival of MYC-expressing and MYCN-expressing cells is further decreased, which may in part be due to tumor suppressor and apoptosis pathways induced by oncogenic levels of MYC proteins (Lowe et al., 2004). This notion is supported by the finding that the percentage of apoptotic cells is increased by MYCN expression (Fig. 3E).
Figure 3.
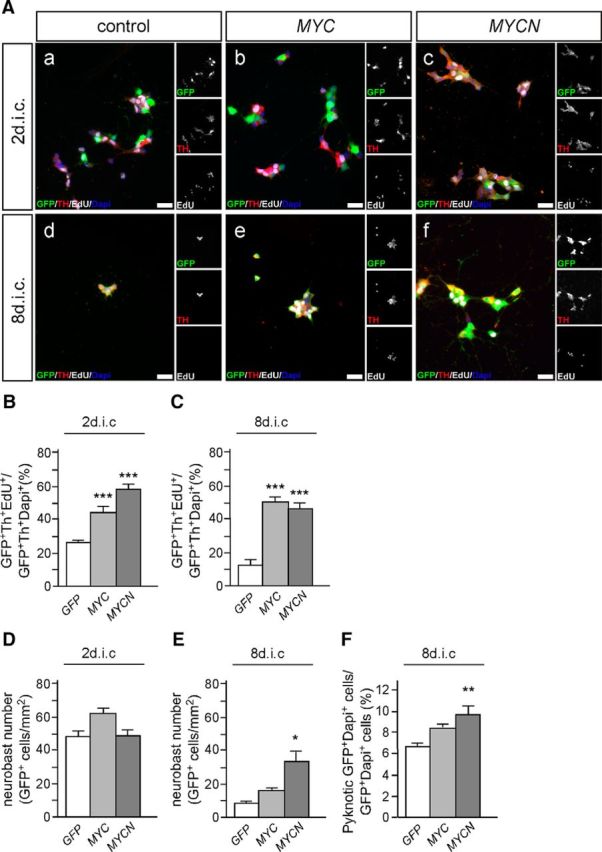
Sympathetic neuroblast proliferation is increased by MYC and MYCN overexpression. A, E7 sympathetic ganglion cells were transfected with expression vectors for GFP (Aa, Ad), GFP and MYC (Ab, Ae), or GFP and MYCN (Ac, Af) and analyzed by GFP/Th/EdU-triple immunostaining after 2 (Aa–Ac) and 8 dic (Ad–Af) for proliferating EdU-incorporating neuroblasts. Scale bar, 20 μm. B, C, Quantification reveals a significantly increased proliferation of MYC-expressing or MYCN-expressing neuroblasts compared with GFP controls at 2 (B) and 8 dic (C; mean ± SEM; n ≥ 3; ***p < 0.001). D, Neuroblast number is not significantly increased compared with controls at 2 dic. E, Neuroblast number in control and MYC cultures are strongly decreased at 8 dic compared with 2 dic. Overexpression of MYCN increases the number of surviving neuroblasts at 8 dic (mean ± SEM; n ≥ 3; *p < 0.05). F, The percentage of pyknotic cells is increased in MYC-expressing or MYCN-expressing neuroblasts, not quite reaching significance for MYC (mean ± SEM; n = 5; **p < 0.01).
MYCN overexpression increases neuroblast proliferation in vivo
MYCN is sufficient to elicit NB development, but in MYCN mouse NB models, neuroblast proliferation and sympathetic ganglion size are not increased during embryonic and early postnatal development (Hansford et al., 2004; Alam et al., 2009; Althoff et al., 2015). To address the in vivo effects of MYCN overexpression in chick sympathetic neuroblasts, we electroporated neural crest cells in 2-d-old (E2) chick embryos with Pb-flanked CAGGS-GFP, CAGGS-MYCN, and Pb plasmid and analyzed E5 embryos for the proportion of proliferating EdU+/TH+/GFP+ neuroblasts. MYCN overexpression leads to a significantly increased proliferation of sympathetic neuroblasts compared with control GFP-transfected embryos (Fig. 4). Thus, MYCN is sufficient to increase chick neuroblast proliferation both in vitro and in vivo. Only a small proportion of sympathetic ganglion cells was transfected and thus effects on ganglion size and survival could not be investigated.
Figure 4.

MYCN overexpression increases neuroblast proliferation in vivo. A, Migrating neural crest cells were transfected by electroporation with Pb plasmid and piggybac transposon vectors for GFP (control) or GFP and MYCN (MYCN). At E5, proliferating cells were labeled by an EdU pulse and, after fixation of the embryo, cross sections from the GFP-expressing transfected area were triple-immunostained for EdU, Th, and GFP. B, Quantification of the percentage of EdU-labeled GFP+/Th+ neuroblasts reveals a significant increase in MYCN-expressing cells (mean ± SEM; n = 7; **p < 0.01). Arrows point to GFP+/Th+ neuroblasts with increased EdU-labeling in MYCN transfected cells.
Expression of constitutively active ALKF1174L interferes with neuroblast proliferation and induces neuron differentiation and survival
As baseline for investigating the interaction between MYC proteins and activated ALK in cultured sympathetic neuroblasts, the long-term effects of activated ALK signaling were analyzed using the Piggybac transposon system. Constitutively active ALK increases neuroblast proliferation in short-term (2 dic) cultures (Fig. 5A,B; Reiff et al., 2011). Unexpectedly, proliferation is completely arrested after 8 dic (Fig. 5A,B). This seems not to be due to death of proliferating cells but rather to increased differentiation, reflected by mature morphology, including large, phase-bright cell bodies and a high number of long, branched neurites (Fig. 5Ad). Indeed, the number of ALKF1174L-transfected cells surviving up to 8 dic is significantly higher than the number of control transfected cells (Fig. 5C). ALKF1174L-induced differentiation generates sympathetic neurons that can be maintained for several weeks in culture (Fig. 5D). Increased neuronal differentiation of ALKF1174L-expressing sympathetic neuroblasts compared with MYCN-transfected cells is also reflected by augmented expression of differentiation genes, analyzed by qRT-PCR at 8 dic (Fig. 6). ALKF1174L-transfected cells were compared with MYCN-transfected rather than GFP-transfected control cells because it was not possible to get sufficient RNA due to the poor survival of the latter (Fig. 5Ac). As expected from the differentiated neuronal morphology of ALKF1174L cells, the pan-neuronal gene NEFM is strongly upregulated. The increase in RET and SLC18A3 (VACHT), together with decreased TH expression suggests a shift toward cholinergic differentiation observed also in ALK-induced NB (Cazes et al., 2014; Lambertz et al., 2015; Fig. 6). It should be noted, however, that TH expression is maintained in ALK-induced sympathetic neurons (Fig. 5D). Decreased MYCN, MYC, TLX3, LIN28B, and BIRC5 expression correlate with decreased proliferation and/or loss of properties characteristic for early neuroblasts in ALKF1174L-transfected versus MYCN-transfected cultures (Huang et al., 2013; Hennchen et al., 2015). The long-term survival of ALKF1174L-induced differentiated neurons may be explained by a decreased expression of the proapoptotic genes BMF and BID. As expected for cultures mainly composed of postmitotic sympathetic neurons, the expression of genes involved in cell-cycle regulation and mitosis (CDC25A, E2F8, SKP2, MAD2L1, DKC1, PLK1, CCNA2, CDK1, SMC2) and of a gene-controlling sympathetic neuroblast proliferation (SOX11; Potzner et al., 2010) are reduced in ALKF1174L cultures compared with proliferating MYCN-driven neuroblast cultures.
Figure 5.
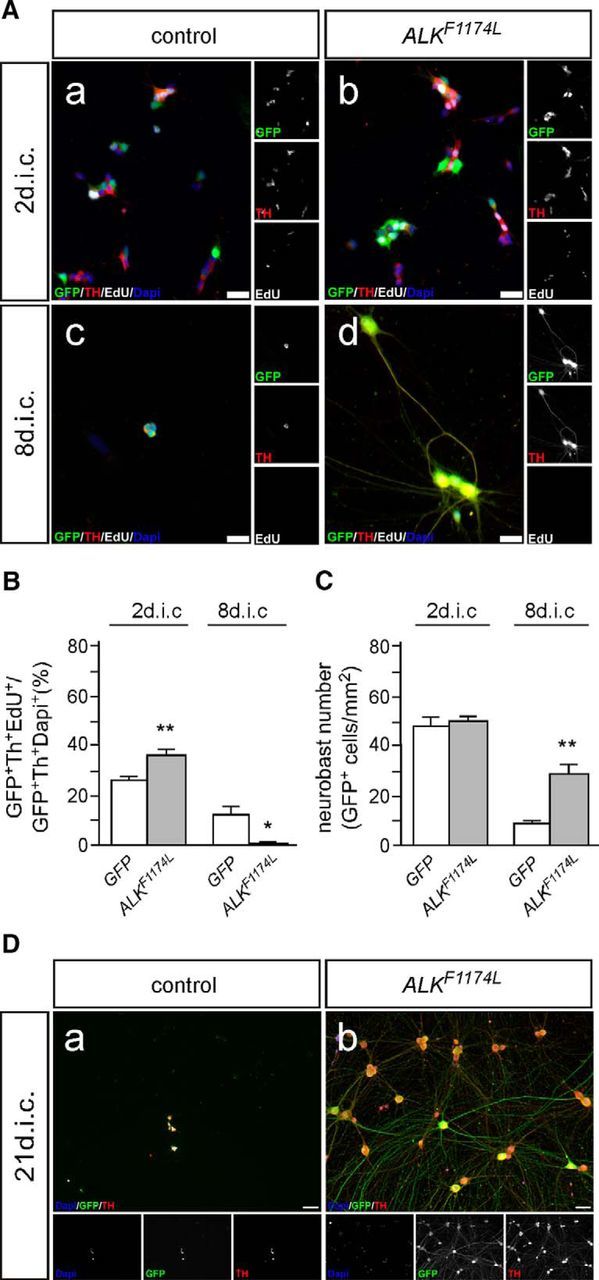
Effects of activated ALK on cultured chick sympathetic neuroblasts. A, E7 sympathetic ganglion cells were transfected with expression vectors for GFP (control; Aa, Ac) or GFP and ALKF1174 L (Ab, Ad) and analyzed by GFP/Th/EdU-triple immunostaining after 2 (Aa, Ab) and 8 dic (Ac, Ad) for proliferating EdU-incorporating neuroblasts. Note the neuronal morphology induced by activated ALK signaling at 8 dic (Ad). B, Quantification reveals a significantly increased proliferation of ALKF1174L-expressing neuroblasts compared with GFP controls at 2 dic (mean ± SEM; n ≥ 6; **p < 0.01). At 8 dic, in contrast, proliferating neuroblasts are absent (mean ± SEM; n ≥ 6; *p < 0.05; unpaired two-tailed t test). C, ALKF1174L expression does not affect neuroblast number at 2 dic but results in a strongly increased survival of Alk-induced neurons (mean ± SEM; n ≥ 4; **p < 0.01). D, Sympathetic neurons induced by activated ALK survive for extended culture periods (3 weeks; Db), in contrast to GFP-transfected control neuroblasts (Da). Please note the large neuron cell bodies and extensive neurite network of ALK-induced sympathetic neurons. Scale bar, 50 μm.
Figure 6.

Effects of ALK signaling on the expression of marker genes for apoptosis, neuron differentiation, early sympathetic neuroblasts, proliferation, and mitosis analyzed by qRT-PCR. Differential gene expression between ALKF1174L-transfected and MYCN-transfected 8 d cultures analyzed by qRT-PCR (mean ± SE; *p < 0.05; **p < 0.01; ***p < 0.001; statistical analysis by relative expression software tool). Note that the MYCN bar corresponds to endogenous chicken MYCN and not from the transfected gene.
ALKF1174L and MYC proteins cooperate to maintain long-term sympathetic proliferation and survival
To analyze whether cooperative interactions between activated ALK and MYCN described in mouse and zebrafish NB models (Berry et al., 2012; Heukamp et al., 2012; Zhu et al., 2012; Cazes et al., 2014) also take place in embryonic chick sympathetic neuroblasts, ALKF1174L and MYC or MYCN piggybac plasmids were cotransfected (Fig. 7). Interestingly, proliferating neuroblasts are maintained in these cultures, which is demonstrated by the presence of EdU-labeled neuroblasts not observed in ALKF1174L cultures at 8 dic (Fig. 7Ad–Af,B). Triple-immunostaining for ALK/GFP/EdU demonstrated that transfected ALK-expressing cells have incorporated the S-phase marker EdU (Fig. 7D,E). Whereas in ALKF1174L cultures, EdU-labeled cells are virtually absent at 8 dic (Figs. 5Ad, 7Ad,D,E), 24 ± 2.6% (mean ± SEM; n = 5) of ALK+/GFP+ cells are EdU-labeled in MYCN/ALKF1174L cultures (Fig. 7E). The higher percentage of GFP/Th/EdU cells compared with GFP/ALK/EdU cells (Fig. 7B,E) may reflect the presence of a small subpopulation of proliferating cells with low or absent ALK expression. The vast majority of neuroblasts in MYC/ALKF1174L and MYCN/ALKF1174L cells show a differentiated morphology, which is distinct from cells expressing either MYCN/MYC or ALKF1174L. They have increased cell-body size and neurite growth compared with MYCN/MYC cells, but remain smaller and display thinner neurites compared with ALKF1174L-expressing neurons (compare Figs. 3Ae,Af, 5Ad, 7Ad–Af). Comparative gene expression analysis of a number of marker genes by qRT-PCR revealed only relatively minor differences between MYCN/ALKF1174L and ALKF1174L cultures (Fig. 8A). The unchanged expression levels of TLX3, TRKA, TH, DBH, NEFM, LIN28B, and RET argues against major effects of MYCN on differentiated properties of ALKF1174L -transfected cells. The downregulation of endogenous chicken MYCN is explained by negative autoregulation in response to overexpressed human MYCN (Penn et al., 1990; Luo et al., 2004), which is not detected by the chicken-specific qRT-PCR primers. Although all analyzed genes controlling cell cycles and mitosis were previously shown to be direct or indirect MYCN target genes (Westermann et al., 2008; Otto et al., 2009; Bell et al., 2010; Valentijn et al., 2012; Puissant et al., 2013), only a subset (i.e., SKP2, CCNA2, DKC1, and E2F8) is induced when MYCN is acting in combination with ALK signaling. Notably, SKP2, CCNA2, DKC1, and E2F8 expression is also significantly increased in MYCN/ALKF1178L mouse NB compared with ALKF1178L sympathetic ganglia (Cazes et al., 2014). E2F8 is an atypical member of the E2F family of transcription factors (Maiti et al., 2005), but recent evidence demonstrates that E2F8 controls proliferation and tumorigenicity in hepatocellular carcinoma cell lines and is required for the growth of lung cancer cells (Deng et al., 2010; Park et al., 2015). The upregulation of the SKP2 oncogene is of particular interest as high SKP2 expression is observed in high-risk NB (Westermann et al., 2007). SKP2 is an essential component of the ubiquitin proteasome system and promotes proliferation largely by degradation of the cyclin-dependent kinase inhibitor p27 (Carrano et al., 1999; Nakayama et al., 2004; Hershko, 2008). The onset of p27 expression during sympathetic neurogenesis correlates with neuroblast cell-cycle exit and p27 overexpression blocks neuroblast proliferation, which together imply that p27 is involved in the termination of sympathetic neurogenesis (Reiff et al., 2010; Holzmann et al., 2015). Thus, it was of interest to test the function of SKP2 in MYCN/ALKF1174L cells using the SKP2-inhibitor C1, which specifically interferes with SKP2–p27 interaction and prevents p27 degradation (Wu et al., 2012). The complete block of neuroblast proliferation observed at 8 dic upon C1 addition between 5 and 8 dic (Fig. 8B) demonstrates an essential role of SKP2 in sympathetic neuroblast proliferation, downstream of MYCN and activated ALK.
Figure 7.
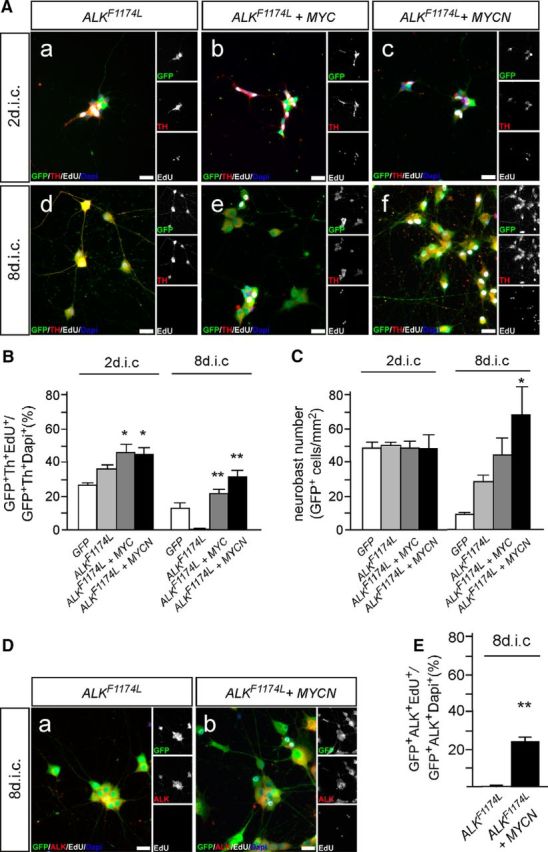
Coexpression of ALKF1174L and MYC proteins supports neuroblast proliferation and survival. A, E7 sympathetic ganglion cells were transfected with expression vectors for GFP and ALKF1174L (Aa, Ad); or GFP, ALKF1174L, and MYC (Ab, Ae); or GFP, ALKF1174L, and MYCN (Ac, Af); and analyzed by GFP/Th/EdU-triple immunostaining after 2 (Aa–Ac) and 8 dic (Ad–Af) for proliferating EdU-incorporating neuroblasts. Note that proliferating ALK/MYC cells (Ae, Af) are smaller and show a less mature neuronal morphology compared with neurons induced by activated ALK signaling at 8 dic (Ad). B, Quantification reveals a significantly increased proliferation of MYC/ALKF1174L-expressing and MYC/ALKF1174L-expressing neuroblasts compared with ALKF1174L neuroblasts at 2 dic (mean ± SEM; n ≥ 6; *p < 0.05). At 8 dic, proliferating neuroblasts are present in transfected cultures but absent in ALKF1174L neuroblasts (mean ± SEM; n ≥ 6; **p < 0.01; unpaired two-tailed t test). C, The combined MYCN/ALKF1174L expression does not affect neuroblast numbers at 2 dic, but results in a strongly increased neuron number at 8 dic compared with ALKF1174L (mean ± SEM; n ≥ 4; *p < 0.05). Data for GFP-transfected controls are included for comparison from Figure 5B,C. D, Proliferation of sympathetic neuroblasts transfected with expression vectors for GFP and ALKF1174L (Da); or for GFP, ALKF1174L, and MYCN (Db); and analyzed by GFP/ALK/EdU-triple immunostaining after 8 dic for proliferating EdU-incorporating ALK+ neuroblasts. E, Quantification reveals increased proliferation of ALK+ neuroblasts in MYCN/ALKF1174L compared with ALKF1174L cultures (mean ± SEM; n ≥ 3; **p < 0.01).
Figure 8.

Effects of combined MYCN/ALK signaling on the expression of marker genes for apoptosis, neuron differentiation, early sympathetic neuroblasts, proliferation, and mitosis analyzed by qRT-PCR; effects of SKP2 inhibition on the proliferation of MYCN/ALKF1174L neuroblasts. A, Differential gene expression between MYCN/ALKF1174L-transfected and ALKF1174L-transfected 8d cultures analyzed by qRT-PCR (mean ± SE; *p < 0.05; **p < 0.01; statistical analysis by relative expression software tool). B, E7 sympathetic ganglion cells were transfected with expression vectors for GFP, ALKF1174L, and MYCN. Cultures were supplemented at 5 dic with 10 μm SKP2 inhibitor SKPin C1 in DMSO or with DMSO alone and analyzed by GFP/Th/EdU triple immunostaining after 8 dic for the percentage of EdU-labeled proliferating MYCN/ALKF1174L neuroblasts. SKP2 inhibition resulted in a virtually complete proliferation block (mean ± SEM; n = 3; ***p < 0.001).
The specific functions of MYCN in the ALKF1174L context must also include pathways linking RET expression to sympathetic neuroblast proliferation. The tyrosine kinase receptor RET is upregulated in ALKF1178L/MYCN NB compared with TH-MYCN mouse NB and is important for tumor growth, as shown by the effects of the RET inhibitor vandetanib (Cazes et al., 2014). Interestingly, we now observed that RET levels are not increased in ALKF1174L/MYCN cultures compared with ALKF1174L neuron cultures (Fig. 8A) but in contrast are elevated in both ALKF1174L/MYCN and ALKF1174L compared with MYCN cultures (Fig. 6; data not shown). This implies that RET induced by activated ALK acquires a function in neuroblast proliferation only in combination with MYCN. Indeed, when ALKF1174L/MYCN cultures were treated with the RET-inhibitor vandetanib between 5 and 8 dic, the percentage of EdU-labeled neuroblasts was reduced from 43 ± 4% to 26 ± 6% (mean ± SEM; n = 3; *p < 0.05).
Long-term maintenance of ALK/MYCN-expressing neuroblasts
ALKF1174L-induced sympathetic neurons can be maintained for several weeks in culture (Fig. 5D), whereas the number of MYCN-expressing or MYC-expressing neuroblasts decreases despite continued cell proliferation (Fig. 3). The coexpression of ALKF1174L and MYCN increases survival compared with MYC-expressing or MYCN-expressing cells, but there is only a small net increase in neuroblast numbers in MYCN/ALKF1174L cultures at 8 dic (Fig. 7C). This is most likely due to MYC protein-induced apoptosis, which may be reflected by increased BID expression (Fig. 8A).
As the extent of MYC protein-induced apoptosis is strongly dependent on the presence of extracellular survival factors (Lowe et al., 2004), we took advantage of our previous observation that survival of avian sympathetic neuroblasts infected by the MYC-expressing myelocytomatosis virus (MC29) is improved by the addition of CEE, a classical component of avian neural crest culture media (Haltmeier and Rohrer, 1990). We now demonstrate a strong increase in the number of ALK/MYCN-neuroblasts at 8 dic in the presence of CEE (Fig. 9). As ganglion non-neuronal cells also proliferate under these conditions, it remains unclear whether increased survival is a direct effect of CEE components on sympathetic neuroblasts or caused by signals from non-neuronal cells. Irrespective of the underlying mechanism, this result demonstrates that survival of ALKF1178L/MYCN-expressing cells depends on environmental signals. Development of NB from ALKF1178L/MYCN-coexpressing neuroblasts may also depend on the presence of extrinsic survival signals.
Figure 9.

MYCN/ALKF1174L-transfected cells can be expanded in the presence of CEE. A, E7 sympathetic ganglion cells were transfected with expression vectors for GFP and ALKF1174L (Aa); or GFP, ALKF1174L, and MYC (Ab); or GFP, ALKF1174L, and MYCN (Ac); and analyzed by GFP/Th/EdU triple immunostaining after 8 dic in the presence or absence of 2% CEE for proliferating EdU-incorporating neuroblasts. Note the large proportion of EdU+/Th+ neuroblasts in MYC/ALKF1174L (Ab) and MYCN/ALKF1174L (Ac) cultures. Neuroblasts tend to grow in clusters in the presence of CEE that are, however, much sparser in MYC/ALKF1174L compared with MYCN/ALKF1174L cultures. Scale bar, 20 μm. B, Combined MYC/ALKF1174L and MYCN/ALKF1174L expression does not affect neuroblast number at 2 dic but MYCN/ALKF1174L increases the number of surviving cells compared with 2 d cultures (mean ± SEM; n ≥ 4; *p < 0.05). Data from cultures without CEE are included for comparison with Figure 7C.
Discussion
NB can be induced in mouse sympathetic ganglia and adrenals by forced expression of MYCN or activated ALK. MYCN/ALK coexpression results in earlier onset and increased penetrance of tumor formation, but the mechanisms that underlie this cooperation are incompletely understood. Using cultures of embryonic chick sympathetic neuroblasts, we now demonstrate that (1) MYC or MYCN overexpression alone increases proliferation but not long-term survival; (2) neuroblasts respond to ALKF1174L signaling by a transient increase in proliferation followed by cell-cycle withdrawal, differentiation, and long-term survival of ALKF1174L-induced sympathetic neurons, whereas (3) overexpression of both ALKF1174L and MYCN results in neuroblasts, which proliferate, are more differentiated, and show improved survival compared with neuroblasts expressing MYCN only; (4) E2F8, DKC1, CCNA2, and the ubiquitin ligase subunit SKP2, which targets p27 for degradation, are induced by MYCN under conditions of activated ALK signaling; and (5) SKP2 inhibition reduces proliferation of MYCN/ALKF1174L cells. Together, our data provide a novel view of how MYCN/ALK may cooperate and implicate SKP2 upregulation as an important early step preventing terminal cell-cycle withdrawal and thus promoting NB formation.
Expression and function of MYC genes during neurogenesis in sympathetic ganglia
The relative expression of MYC genes differs strongly between different parts of the nervous system (Stanton et al., 1992; Martins et al., 2008; Domínguez-Frutos et al., 2011). MYCN knock-out and targeted deletion in neuronal progenitors result in decreased brain size and reduced sensory ganglia (Charron et al., 1992; Knoepfler et al., 2002), whereas conditional deletion of MYC has only a minor effect (Hatton et al., 2006). The relative expression and function of MYC and MYCN in developing sympathetic ganglia has not been investigated, but the small reduction of ganglion size in MYCN knock-out mice (Sawai et al., 1993) suggests a function for other MYC family members. The present analysis shows that MYC is expressed at much higher levels than MYCN and is essential for sympathetic neuroblast proliferation, whereas MYCN knockdown has only a small effect. Together, these results indicate that endogenous proliferation of neuroblasts depends mainly on MYC. The importance of MYCN in NB generation does not reflect its developmental function but is related to MYCN gene amplification and redundant biological effects of MYCN and MYC (Malynn et al., 2000).
Interference with MYC gene expression by JQ1-mediated BET protein inhibition
Inhibition of BET bromodomain proteins by JQ1 treatment interferes with the transcription of MYC and MYCN genes (Delmore et al., 2011; Puissant et al., 2013). NB cell lines with MYCN amplification are very sensitive to JQ1, which was explained by BRD4-dependent MYCN transcription (Puissant et al., 2013). Recent studies suggest that JQ1 causes a preferential loss of BRD4 and transcription from super-enhancers controlling both MYC(N) and MYC(N) target genes (Lovén et al., 2013; Henssen et al., 2016). The complete inhibition of sympathetic neuroblast proliferation by JQ1 treatment, in contrast to findings with NB cell lines with normal MYCN expression (Puissant et al., 2013), demonstrates a strong difference between neuroblasts and NB cell lines with implications for clinical treatment. In contrast to other cells (Delmore et al., 2011), JQ1 treatment does not completely block MYC and MYCN expression in sympathetic neuroblasts. Thus, inhibition of sympathetic neuroblast proliferation may in addition reflect effects of JQ1 on MYCN targets (Henssen et al., 2016) and/or dependence on the activity of super-enhancers controlling master genes of sympathetic neuron identity (e.g., PHOX2B, HAND2; Whyte et al., 2013), all of which also affect neuroblast proliferation (Rohrer, 2011). Notably, the proliferation of ganglion non-neuronal cells is not affected by JQ1, indicating different BRD4-activated enhancers in neuroblasts and glia.
Sympathetic neuroblast propagation by forced MYCN and MYC expression
The initial steps in NB development are not well understood, as illustrated by the finding that targeted MYCN expression in E10.5 mouse sympathetic neuroblasts does not increase proliferation until postnatal stages (Hansford et al., 2004; Alam et al., 2009; Althoff et al., 2015). We now demonstrate that continued overexpression of MYC or MYCN in chick sympathetic neuroblasts results in a strong increase in the percentage of proliferating cells. MYCN overexpression also increases neuroblast proliferation in vivo. The difference between MYCN-transfected chick neuroblasts and embryonic sympathetic ganglia of MYCN-expressing mice may be due to higher MYCN expression levels in transfected neuroblasts using the strong CAG promotor. Increased proliferation of MYC(N)-expressing neuroblasts does not, however, lead to a net increase in neuroblast number, which is explained by increased apoptosis. The apoptotic loss of MYC(N)-expressing cells may either reflect the poor neuroblast survival also observed in controls or the induction of antioncogenic programs that lead to senescence and apoptosis (Lowe et al., 2004).
Proliferation and differentiation of sympathetic neuroblasts: alternative fates induced by activated ALK signaling
Although different tyrosine kinase receptors activate a common set of signal transduction pathways, distinct cellular effects are elicited by each receptor. For example, proliferation or differentiation is elicited in PC12 cells by EGF and NGF, respectively, correlating with transient and sustained activation of the MAPK signal cascade (Marshall, 1995). Here, we observed that ALKF1174L first increases proliferation and subsequently induces cell-cycle exit, differentiation, and neuron survival. These results may be explained by a maturation of sympathetic neuroblasts with time, leading to a different interpretation of ALK signaling in young versus more mature neuroblasts. This would be in agreement with the results in the Ki-Alk mouse where sympathetic neuroblasts show increased proliferation but eventually leave the cell cycle and differentiate (Cazes et al., 2014). An alternative explanation would be that an initially low ALK expression is unable to induce differentiation. This would be reminiscent to the finding that PC12 cells with low numbers of TrkA receptors do not differentiate in response to NGF (Schlessinger and Bar-Sagi, 1994). Proliferation effects may also be due to ALK located mainly in ER and Golgi (Mazot et al., 2011) in contrast to ALK located in the plasma membrane, where ALK elicits differentiation (Gouzi et al., 2005). Regardless of these explanations, our data indicate that cell-cycle exit and differentiation is the final result of activated ALK in cultured sympathetic neuroblasts. In contrast to our in vitro findings, expression of activated ALK in mouse sympathoadrenal cells leads to NB formation (Heukamp et al., 2012). The low penetrance and late onset of tumor formation suggests, however, that tumor initiation depends on additional genomic aberrations identified in these tumors (Heukamp et al., 2012).
ALK-induced sympathetic neuron differentiation and survival
Cell-cycle withdrawal and morphological differentiation in response to ALKF1174L is reflected by decreased expression of genes involved in cell-cycle progression and mitosis (e.g., CDC25A, CCNA2, CDK1, MAD2L1, and SMC2), upregulation of the generic neuron differentiation gene NEFM, and low expression of early neuroblast markers TLX3 and LIN28B (Huang et al., 2013; Hennchen et al., 2015). Upregulated RET has no proliferative function in ALKF1174L-induced sympathetic neurons, in contrast to MYCN/ALKF1174L neuroblasts and MYCN/ALK-induced mouse NB (Cazes et al., 2014; Lambertz et al., 2015). The long-term survival of ALKF1174L-induced neurons is surprising as sympathetic neuron death is tightly controlled by NGF/TRKA signaling with TRKA acting as an apoptosis-inducing dependence receptor (Nikoletopoulou et al., 2010). We propose that survival is enabled by the reduced expression of proapoptotic BH3-only genes BMF and BID leading to the net dominance of antiapoptotic proteins.
Cooperative effects of MYCN and activated ALK in sympathetic neuroblasts
Neuroblasts coexpressing MYC(N) and ALKF1174L continue to proliferate, whereas all ALKF1174L-expressing cells have left the cell cycle at 8 dic. Maintained proliferation results only in a small increase in MYCN/ALKF1174L cells, suggesting that ALK signaling may not be sufficient to interfere completely with the proapoptotic effects of forced MYC(N) expression as indicated by increased BID levels. Survival of MYCN/ALKF1174L-expressing cells is, however, maintained in CEE-supplemented cultures, leading to expansion and net increase of MYCN/ALKF1174L cells. Unexpectedly, this survival effect is selective for MYCN/ALKF1174L cells, raising the question whether MYC/ALK cooperation may be sufficient to induce NB.
Interestingly, the expression of the majority of genes analyzed by qRT-PCR does not differ between MYCN/ALKF1174L neuroblasts and ALKF1174L-expressing neurons, which may reflect their differentiated characteristics. From the analyzed genes involved in the cell cycle and mitosis, only SKP2, DKC1, CCNA2, and E2F8 are upregulated. Of particular interest is the MYCN-induced upregulation of the SKP2 oncogene (Hershko, 2008) and the reduction of MYCN/ALKF1174L neuroblast proliferation by SKP2 inhibition. SKP2 is a direct MYCN and MYC target and the main oncogenic mechanism of SKP2 is attributed to the degradation of the CDK inhibitor p27 (Carrano et al., 1999; Sutterlüty et al., 1999; Bretones et al., 2011; Evans et al., 2015). High SKP2 expression is characteristic for high-risk NB and is inversely correlated with p27 expression (Westermann et al., 2007; Chu et al., 2008). P27 is induced when sympathetic neuroblasts leave the cell cycle and p27 overexpression blocks cell cycle progression (Reiff et al., 2010; Holzmann et al., 2015). Together, these results suggest that SKP2 upregulation may be an essential early step induced by MYCN/ALKF1174L cooperation to maintain neuroblast proliferation during NB development.
Footnotes
This work was supported by grants from the Wilhelm-Sander-Stiftung to H.R and grants from the Swedish Cancer Society and Swedish Research Council to M.A.H. We thank Nikolay Zinin for technical support, Pascal Fries for support, Dorothea Schulte for discussion, and Uwe Ernsberger and Isabelle Janoueix-Lerosey for helpful comments on the manuscript.
The authors declare no competing financial interests.
References
- Alam G, Cui H, Shi H, Yang L, Ding J, Mao L, Maltese WA, Ding HF. MYCN promotes the expansion of Phox2B-positive neuronal progenitors to drive neuroblastoma development. Am J Pathol. 2009;175:856–866. doi: 10.2353/ajpath.2009.090019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althoff K, Beckers A, Bell E, Nortmeyer M, Thor T, Sprüssel A, Lindner S, De Preter K, Florin A, Heukamp LC, Klein-Hitpass L, Astrahantseff K, Kumps C, Speleman F, Eggert A, Westermann F, Schramm A, Schulte JH. A Cre-conditional MYCN-driven neuroblastoma mouse model as an improved tool for preclinical studies. Oncogene. 2015;34:3357–3368. doi: 10.1038/onc.2014.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell E, Lunec J, Tweddle DA. Cell cycle regulation targets of MYCN identified by gene expression microarrays. Cell Cycle. 2007;6:1249–1256. doi: 10.4161/cc.6.10.4222. [DOI] [PubMed] [Google Scholar]
- Bell E, Chen L, Liu T, Marshall GM, Lunec J, Tweddle DA. MYCN oncoprotein targets and their therapeutic potential. Cancer Lett. 2010;293:144–157. doi: 10.1016/j.canlet.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, Pei D, Sharma B, Vetharoy WR, Hallsworth A, Ahmad Z, Barker K, Moreau L, Webber H, Wang W, Liu Q, Perez-Atayde A, Rodig S, Cheung NK, Raynaud F, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell. 2012;22:117–130. doi: 10.1016/j.ccr.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefoy E, Ferrara P, Rohrer H, Gros F, Thibault J. Role of the N-terminus of rat pheochromocytoma tyrosine hydroxylase in the regulation of the enzyme's activity. Eur J Biochem. 1988;174:685–690. doi: 10.1111/j.1432-1033.1988.tb14152.x. [DOI] [PubMed] [Google Scholar]
- Breit S, Schwab M. Suppression of MYC by high expression of NMYC in human neuroblastoma cells. J Neurosci Res. 1989;24:21–28. doi: 10.1002/jnr.490240105. [DOI] [PubMed] [Google Scholar]
- Bretones G, Acosta JC, Caraballo JM, Ferrandiz N, Gómez-Casares MT, Albajar M, Blanco R, Ruiz P, Hung WC, Albero MP, Perez-Roger I, León J. SKP2 oncogene is a direct MYC target gene and MYC down-regulates p27(KIP1) through SKP2 in human leukemia cells. J Biol Chem. 2011;286:9815–9825. doi: 10.1074/jbc.M110.165977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121–1124. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- Cadiñanos J, Bradley A. Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res. 2007;35:e87. doi: 10.1093/nar/gkm446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- Cazes A, Lopez-Delisle L, Tsarovina K, Pierre-Eugène C, De Preter K, Peuchmaur M, Nicolas A, Provost C, Louis-Brennetot C, Daveau R, Kumps C, Cascone I, Schleiermacher G, Prignon A, Speleman F, Rohrer H, Delattre O, Janoueix-Lerosey I. Activated Alk triggers prolonged neurogenesis and Ret upregulation providing a therapeutic target in ALK-mutated neuroblastoma. Oncotarget. 2014;5:2688–2702. doi: 10.18632/oncotarget.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron J, Malynn BA, Fisher P, Stewart V, Jeannotte L, Goff SP, Robertson EJ, Alt FW. Embryonic lethality in mice homozygous for a targeted disruption of the N-myc gene. Genes Dev. 1992;6:2248–2257. doi: 10.1101/gad.6.12a.2248. [DOI] [PubMed] [Google Scholar]
- Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, Nakagawara A, Hayashi Y, Mano H, Ogawa S. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–974. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, Lakeman A, Vandesompele J, Hoebeeck J, Van Maerken T, De Paepe A, Laureys G, Schulte JH, Schramm A, Van Den Broecke C, Vermeulen J, Van Roy N, Beiske K, Renard M, Noguera R, et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res. 2010;16:4353–4362. doi: 10.1158/1078-0432.CCR-09-2660. [DOI] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, *akoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Wang Q, Zong WY, Zheng DL, Wen YX, Wang KS, Teng XM, Zhang X, Huang J, Han ZG. E2F8 contributes to human hepatocellular carcinoma via regulating cell proliferation. Cancer Res. 2010;70:782–791. doi: 10.1158/0008-5472.CAN-09-3082. [DOI] [PubMed] [Google Scholar]
- Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 2005;122:473–483. doi: 10.1016/j.cell.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Domínguez-Frutos E, López-Hernández I, Vendrell V, Neves J, Gallozzi M, Gutsche K, Quintana L, Sharpe J, Knoepfler PS, Eisenman RN, Trumpp A, Giráldez F, Schimmang T. N-myc controls proliferation, morphogenesis, and patterning of the inner ear. J Neurosci. 2011;31:7178–7189. doi: 10.1523/JNEUROSCI.0785-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans L, Chen L, Milazzo G, Gherardi S, Perini G, Willmore E, Newell DR, Tweddle DA. SKP2 is a direct transcriptional target of MYCN and a potential therapeutic target in neuroblastoma. Cancer Lett. 2015;363:37–45. doi: 10.1016/j.canlet.2015.03.044. [DOI] [PubMed] [Google Scholar]
- George RE, Sanda T, Hanna M, Fröhling S, Luther W, 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S, Gregor VE, Webb TR, Gray NS, Gilliland DG, Diller L, Greulich H, Morris SW, Meyerson M, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–978. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouzi JY, Moog-Lutz C, Vigny M, Brunet-de Carvalho N. Role of the subcellular localization of ALK tyrosine kinase domain in neuronal differentiation of PC12 cells. J Cell Sci. 2005;118:5811–5823. doi: 10.1242/jcs.02695. [DOI] [PubMed] [Google Scholar]
- Haltmeier H, Rohrer H. Distinct and different effects of the oncogenes v-myc and v-src on avian sympathetic neurons: retroviral transfer of v-myc stimulates neuronal proliferation whereas v-src transfer enhances neuronal differentiation. J Cell Biol. 1990;110:2087–2098. doi: 10.1083/jcb.110.6.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansford LM, Thomas WD, Keating JM, Burkhart CA, Peaston AE, Norris MD, Haber M, Armati PJ, Weiss WA, Marshall GM. Mechanisms of embryonal tumor initiation: distinct roles for MycN expression and MYCN amplification. Proc Natl Acad Sci U S A. 2004;101:12664–12669. doi: 10.1073/pnas.0401083101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton BA, Knoepfler PS, Kenney AM, Rowitch DH, de Alborán IM, Olson JM, Eisenman RN. N-myc is an essential downstream effector of Shh signaling during both normal and neoplastic cerebellar growth. Cancer Res. 2006;66:8655–8661. doi: 10.1158/0008-5472.CAN-06-1621. [DOI] [PubMed] [Google Scholar]
- Hennchen M, Stubbusch J, Abarchan-El Makhfi I, Kramer M, Deller T, Pierre-Eugene C, Janoueix-Lerosey I, Delattre O, Ernsberger U, Schulte JB, Rohrer H. Lin28B and Let-7 in the control of sympathetic neurogenesis and neurblastoma development. J Neurosci. 2015;35:16531–16544. doi: 10.1523/JNEUROSCI.2560-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen JR, Haug BH, Buechner J, Tømte E, Løkke C, Flaegstad T, Einvik C. Conditional expression of retrovirally delivered anti-MYCN shRNA as an in vitro model system to study neuronal differentiation in MYCN-amplified neuroblastoma. BMC Dev Biol. 2011;11:1. doi: 10.1186/1471-213X-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henssen AG, Althoff K, Odersky A, Beckers A, Koche R, Speleman F, Schäfers S, Bell E, Nortmeyer M, Westermann F, De Preter K, Florin A, Heukamp L, Spruessel A, Astrahanseff K, Lindner S, Sadowski N, Schramm A, Astorgues-Xerri L, Riveiro ME, et al. Targeting MYCN-driven transcription by BET-bromodomain inhibition. Clin Cancer Res. 2016;22:2470–2481. doi: 10.1158/1078-0432.CCR-15-1449. [DOI] [PubMed] [Google Scholar]
- Hershko DD. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer. 2008;112:1415–1424. doi: 10.1002/cncr.23317. [DOI] [PubMed] [Google Scholar]
- Heukamp LC, Thor T, Schramm A, De Preter K, Kumps C, De Wilde B, Odersky A, Peifer M, Lindner S, Spruessel A, Pattyn F, Mestdagh P, Menten B, Kuhfittig-Kulle S, Künkele A, König K, Meder L, Chatterjee S, Ullrich RT, Schulte S, et al. Targeted expression of mutated ALK induces neuroblastoma in transgenic mice. Sci Transl Med. 2012;4:141ra191. doi: 10.1126/scitranslmed.3003967. [DOI] [PubMed] [Google Scholar]
- Holzmann J, Hennchen M, Rohrer H. Prox1 identifies proliferating neuroblasts and nascent neurons during neurogenesis in sympathetic ganglia. Dev Neurobiol. 2015;75:1352–1367. doi: 10.1002/dneu.22289. [DOI] [PubMed] [Google Scholar]
- Huang T, Hu J, Wang B, Nie Y, Geng J, Cheng L. Tlx3 controls cholinergic transmitter and Peptide phenotypes in a subset of prenatal sympathetic neurons. J Neurosci. 2013;33:10667–10675. doi: 10.1523/JNEUROSCI.0192-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janoueix-Lerosey I, Lequin D, Brugières L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, Valteau-Couanet D, Frebourg T, Michon J, Lyonnet S, Amiel J, Delattre O. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–970. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- Kaltezioti V, Kouroupi G, Oikonomaki M, Mantouvalou E, Stergiopoulos A, Charonis A, Rohrer H, Matsas R, Politis PK. Prox1 regulates the notch1-mediated inhibition of neurogenesis. PLoS Biol. 2010;8:e1000565. doi: 10.1371/journal.pbio.1000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16:2699–2712. doi: 10.1101/gad.1021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertz I, Kumps C, Claeys S, Lindner S, Beckers A, Janssens E, Carter DR, Cazes A, Cheung BB, De Mariano M, De Bondt A, De Brouwer S, Delattre O, Gibbons J, Janoueix-Lerosey I, Laureys G, Liang C, Marchall GM, Porcu M, Takita J, et al. Upregulation of MAPK negative feedback regulators and RET in mutant ALK neuroblastoma: implications for targeted treatment. Clin Cancer Res. 2015;21:3327–3339. doi: 10.1158/1078-0432.CCR-14-2024. [DOI] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- Lu Y, Lin C, Wang X. PiggyBac transgenic strategies in the developing chicken spinal cord. Nucleic Acids Res. 2009;37:e141. doi: 10.1093/nar/gkp686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Q, Li J, Cenkci B, Kretzner L. Autorepression of c-myc requires both initiator and E2F-binding site elements and cooperation with the p107 gene product. Oncogene. 2004;23:1088–1097. doi: 10.1038/sj.onc.1207225. [DOI] [PubMed] [Google Scholar]
- Maiti B, Li J, de Bruin A, Gordon F, Timmers C, Opavsky R, Patil K, Tuttle J, Cleghorn W, Leone G. Cloning and characterization of mouse E2F8, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem. 2005;280:18211–18220. doi: 10.1074/jbc.M501410200. [DOI] [PubMed] [Google Scholar]
- Malynn BA, de Alboran IM, O'Hagan RC, Bronson R, Davidson L, DePinho RA, Alt FW. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000;14:1390–1399. [PMC free article] [PubMed] [Google Scholar]
- Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Martins RA, Zindy F, Donovan S, Zhang J, Pounds S, Wey A, Knoepfler PS, Eisenman RN, Roussel MF, Dyer MA. N-myc coordinates retinal growth with eye size during mouse development. Genes Dev. 2008;22:179–193. doi: 10.1101/gad.1608008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazot P, Cazes A, Boutterin MC, Figueiredo A, Raynal V, Combaret V, Hallberg B, Palmer RH, Delattre O, Janoueix-Lerosey I, Vigny M. The constitutive activity of the ALK mutated at positions F1174 or R1275 impairs receptor trafficking. Oncogene. 2011;30:2017–2025. doi: 10.1038/onc.2010.595. [DOI] [PubMed] [Google Scholar]
- Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, Kim C, Hou C, Hakonarson H, Torkamani A, Schork NJ, Brodeur GM, Tonini GP, Rappaport E, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–935. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy K, Sung HK, Zhang P, Laflamme S, Vincent P, Agha-Mohammadi S, Woltjen K, Monetti C, Michael IP, Smith LC, Nagy A. Induced pluripotent stem cell lines derived from equine fibroblasts. Stem Cell Rev. 2011;7:693–702. doi: 10.1007/s12015-011-9239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, Kitagawa M, Iemura S, Natsume T, Nakayama KI. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell. 2004;6:661–672. doi: 10.1016/S1534-5807(04)00131-5. [DOI] [PubMed] [Google Scholar]
- Nikoletopoulou V, Lickert H, Frade JM, Rencurel C, Giallonardo P, Zhang L, Bibel M, Barde YA. Neurotrophin receptors TrkA and TrkC cause neuronal death whereas TrkB does not. Nature. 2010;467:59–63. doi: 10.1038/nature09336. [DOI] [PubMed] [Google Scholar]
- Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R, Christiansen H, Berwanger B, Eilers M. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009;15:67–78. doi: 10.1016/j.ccr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Park SA, Platt J, Lee JW, Lopez-Giraldez F, Herbst RS, Koo JS. E2F8 as a novel therapeutic target for lung cancer. J Natl Cancer Inst. 2015;107:djr151. doi: 10.1093/jnci/djv151. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn LJ, Brooks MW, Laufer EM, Land H. Negative autoregulation of c-myc transcription. The EMBO J. 1990;9:1113–1121. doi: 10.1002/j.1460-2075.1990.tb08217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potzner MR, Tsarovina K, Binder E, Penzo-Méndez A, Lefebvre V, Rohrer H, Wegner M, Sock E. Sequential requirement of Sox4 and Sox11 during development of the sympathetic nervous system. Development. 2010;137:775–784. doi: 10.1242/dev.042101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, Garnett MJ, McDermott U, Benes CH, Kung AL, Weiss WA, Bradner JE, Stegmaier K. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3:308–323. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiff T, Tsarovina K, Majdazari A, Schmidt M, del Pino I, Rohrer H. Neuroblastoma phox2b variants stimulate proliferation and dedifferentiation of immature sympathetic neurons. J Neurosci. 2010;30:905–915. doi: 10.1523/JNEUROSCI.5368-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiff T, Huber L, Kramer M, Delattre O, Janoueix-Lerosey I, Rohrer H. Midkine and Alk signaling in sympathetic neuron proliferation and neuroblastoma predisposition. Development. 2011;138:4699–4708. doi: 10.1242/dev.072157. [DOI] [PubMed] [Google Scholar]
- Ribeiro D, Klarqvist MD, Westermark UK, Oliynyk G, Dzieran J, Kock A, Savatier Banares C, Hertwig F, Johnsen JI, Fischer M, Kogner P, Lovén J, Arsenian Henriksson M. Regulation of nuclear hormone receptors by MYCN-driven miRNAs impacts neural differentiation and survival in neuroblastoma patients. Cell Rep. 2016;16:979–993. doi: 10.1016/j.celrep.2016.06.052. [DOI] [PubMed] [Google Scholar]
- Rohrer H. Transcriptional control of differentiation and neurogenesis in autonomic ganglia. Eur J Neurosci. 2011;34:1563–1573. doi: 10.1111/j.1460-9568.2011.07860.x. [DOI] [PubMed] [Google Scholar]
- Rohrer H, Thoenen H. Relationship between differentiation and terminal mitosis: chick sensory and ciliary neurons differentiate after terminal mitosis of precursor cells whereas sympathetic neurons continue to divide after differentiation. J Neurosci. 1987;7:3739–3748. doi: 10.1523/JNEUROSCI.07-11-03739.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer H, Acheson AL, Thibault J, Thoenen H. Developmental potential of quail dorsal root ganglion cells analyzed in vitro and in vivo. J Neurosci. 1986;6:2616–2624. doi: 10.1523/JNEUROSCI.06-09-02616.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai S, Shimono A, Wakamatsu Y, Palmes C, Hanaoka K, Kondoh H. Defects of embryonic organogenesis resulting from targeted disruption of the N-myc gene in the mouse. Development. 1993;117:1445–1455. doi: 10.1242/dev.117.4.1445. [DOI] [PubMed] [Google Scholar]
- Schlessinger J, Bar-Sagi D. Activation of Ras and other signaling pathways by receptor tyrosine kinases. Cold Spring Harb Symp Quant Biol. 1994;59:173–179. doi: 10.1101/SQB.1994.059.01.021. [DOI] [PubMed] [Google Scholar]
- Simon T, Berthold F, Borkhardt A, Kremens B, De Carolis B, Hero B. Treatment and outcomes of patients with relapsed, high-risk neuroblastoma: results of German trials. Pediatr Blood Cancer. 2011;56:578–583. doi: 10.1002/pbc.22693. [DOI] [PubMed] [Google Scholar]
- Stanton BR, Perkins AS, Tessarollo L, Sassoon DA, Parada LF. Loss of N-myc function results in embryonic lethality and failure of the epithelial component of the embryo to develop. Genes Dev. 1992;6:2235–2247. doi: 10.1101/gad.6.12a.2235. [DOI] [PubMed] [Google Scholar]
- Sutterlüty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Müller U, Krek W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999;1:207–214. doi: 10.1038/12027. [DOI] [PubMed] [Google Scholar]
- Valentijn LJ, Koster J, Haneveld F, Aissa RA, van Sluis P, Broekmans ME, Molenaar JJ, van Nes J, Versteeg R. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc Natl Acad Sci U S A. 2012;109:19190–19195. doi: 10.1073/pnas.1208215109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16:2985–2995. doi: 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann F, Henrich KO, Wei JS, Lutz W, Fischer M, König R, Wiedemeyer R, Ehemann V, Brors B, Ernestus K, Leuschner I, Benner A, Khan J, Schwab M. High Skp2 expression characterizes high-risk neuroblastomas independent of MYCN status. Clin Cancer Res. 2007;13:4695–4703. doi: 10.1158/1078-0432.CCR-06-2818. [DOI] [PubMed] [Google Scholar]
- Westermann F, Muth D, Benner A, Bauer T, Henrich KO, Oberthuer A, Brors B, Beissbarth T, Vandesompele J, Pattyn F, Hero B, König R, Fischer M, Schwab M. Distinct transcriptional MYCN/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol. 2008;9:R150. doi: 10.1186/gb-2008-9-10-r150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodard LE, Wilson MH. piggyBac-ing models and new therapeutic strategies. Trends Biotech. 2015;33:525–533. doi: 10.1016/j.tibtech.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Grigoryan AV, Li Y, Hao B, Pagano M, Cardozo TJ. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem Biol. 2012;19:1515–1524. doi: 10.1016/j.chembiol.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zackenfels K, Oppenheim RW, Rohrer H. Evidence for an important role of IGF-I and IGF-II for the early development of chick sympathetic neurons. Neuron. 1995;14:731–741. doi: 10.1016/0896-6273(95)90217-1. [DOI] [PubMed] [Google Scholar]
- Zhu S, Lee JS, Guo F, Shin J, Perez-Atayde AR, Kutok JL, Rodig SJ, Neuberg DS, Helman D, Feng H, Stewart RA, Wang W, George RE, Kanki JP, Look AT. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell. 2012;21:362–373. doi: 10.1016/j.ccr.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinin N, Adameyko I, Wilhelm M, Fritz N, Uhlén P, Ernfors P, Henriksson MA. MYC proteins promote neuronal differentiation by controlling the mode of progenitor cell division. EMBO Rep. 2014;15:383–391. doi: 10.1002/embr.201337424. [DOI] [PMC free article] [PubMed] [Google Scholar]