

Abstract
Distinct types of GABAergic interneurons target different subcellular domains of pyramidal cells, thereby shaping pyramidal cell activity patterns. Whether the presynaptic heterogeneity of GABAergic innervation is mirrored by specific postsynaptic factors is largely unexplored. Here we show that dystroglycan, a protein responsible for the majority of congenital muscular dystrophies when dysfunctional, has a function at postsynaptic sites restricted to a subset of GABAergic interneurons. Conditional deletion of Dag1, encoding dystroglycan, in pyramidal cells caused loss of CCK-positive basket cell terminals in hippocampus and neocortex. PV-positive basket cell terminals were unaffected in mutant mice, demonstrating interneuron subtype-specific function of dystroglycan. Loss of dystroglycan in pyramidal cells had little influence on clustering of other GABAergic postsynaptic proteins and of glutamatergic synaptic proteins. CCK-positive terminals were not established at P21 in the absence of dystroglycan and were markedly reduced when dystroglycan was ablated in adult mice, suggesting a role for dystroglycan in both formation and maintenance of CCK-positive terminals. The necessity of neuronal dystroglycan for functional innervation by CCK-positive basket cell axon terminals was confirmed by reduced frequency of inhibitory events in pyramidal cells of dystroglycan-deficient mice and further corroborated by the inefficiency of carbachol to increase IPSC frequency in these cells. Finally, neurexin binding seems dispensable for dystroglycan function because knock-in mice expressing binding-deficient T190M dystroglycan displayed normal CCK-positive terminals. Together, we describe a novel function of dystroglycan in interneuron subtype-specific trans-synaptic signaling, revealing correlation of presynaptic and postsynaptic molecular diversity.
SIGNIFICANCE STATEMENT Dystroglycan, an extracellular and transmembrane protein of the dystrophin-glycoprotein complex, is at the center of molecular studies of muscular dystrophies. Although its synaptic distribution in cortical brain regions is long established, function of dystroglycan in the synapse remained obscure. Using mice that selectively lack neuronal dystroglycan, we provide evidence that a subset of GABAergic interneurons requires dystroglycan for formation and maintenance of axonal terminals on pyramidal cells. As such, dystroglycan is the first postsynaptic GABAergic protein for which an interneuron terminal-specific function could be shown. Our findings also offer a new perspective on the mechanisms that lead to intellectual disability in muscular dystrophies without associated brain malformations.
Keywords: cholecystokinin, dystroglycanopathy, dystrophin-glycoprotein complex, GABA, interneurons, perisomatic
Introduction
GABAergic interneurons, which provide the main source of inhibitory drive in the adult mammalian brain, form several distinct classes according to morphological, molecular, and functional criteria (Fishell and Rudy, 2011). This specialization allows interneurons to adapt to different demands of postsynaptic targets and thereby control membrane excitability in a spatially and temporally precise manner (Klausberger and Somogyi, 2008). Most interneuron classes innervate only a specific subcellular domain of target cells, for example, the axon initial segment, the cell soma, or dendritic regions. Synaptic transmission from different interneuron subtypes has thus fundamentally different impact on the activity of postsynaptic cells. It might be advantageous to account for this diversity of GABAergic innervation with postsynaptic specializations matching the specific properties and plasticity mechanisms of synaptic terminals they are contacted from. Indeed, the GABAergic postsynaptic density (PSD) is characterized by a large molecular heterogeneity (Tyagarajan and Fritschy, 2014). However, little is known about such subtype-specific postsynaptic GABAergic adaptations.
Basket cells are GABAergic interneurons that specifically target the perisomatic region of principal neurons. In cerebral cortex and hippocampus, expression of parvalbumin (PV) or cholecystokinin (CCK) identifies basket cells as belonging to one of two nonoverlapping groups (Freund and Katona, 2007). Although these two interneuron subtypes innervate the same subcellular domain, they are distinguished by various traits (Bartos and Elgueta, 2012). Only CCK-positive basket cells express presynaptic cannabinoid receptors, enabling retrograde signaling of endocannabinoids to suppress GABA release. Different firing patterns, expression profiles, and developmental origins further set the two subtypes apart. Therefore, it is conceivable that the two types of basket cells use different mechanisms for synapse formation and require a different set of postsynaptic proteins to exert their vastly different functions.
Dystroglycan (DG) is the central component of the dystrophin-glycoprotein complex (DGC). The extracellular α-DG and transmembrane β-DG, generated by proteolytic cleavage of a single gene product, bind the large cytoplasmic protein dystrophin, which in turn can interact with actin filaments. α-DG, through its glycosyl side chains, can bind to extracellular matrix components. The crucial role of the DGC in muscle tissue was revealed by mutations affecting DGC components that lead to muscular dystrophies (McNally and Pytel, 2007). The DGC, albeit differing slightly in its molecular composition, is also expressed in the CNS by glial cells and neurons (Waite et al., 2012). Developmental brain malformations and intellectual disability, observed frequently in muscular dystrophies caused by DGC dysfunction, testify to the importance of this complex for brain function. The finding that the DGC is present in pyramidal cells as large, mostly perisomatic clusters postsynaptic to GABAergic terminals spurred interest in the synaptic function of the DGC (Lidov et al., 1990). Because reduced GABAAR immunoreactivity was found in a mouse model of Duchenne's muscular dystrophy (DMD), a function for the DGC in clustering of PSD components was posited (Knuesel et al., 1999; Vaillend et al., 2010). Despite the selective DGC subcellular distribution, biochemical interaction with presynaptic neurexins and the obligatory association of DG with GABAergic presynaptic terminals in neuronal cultures, the role of the DGC in trans-synaptic signaling was never systematically assessed (Sugita et al., 2001; Brünig et al., 2002).
We hypothesized that the diversity of GABAergic PSD composition is functionally related to the heterogeneity of GABAergic innervation. Because of its restricted distribution and known role as a transmembrane complex, DG seemed ideally suited to address this issue. Ablation of DG specifically in pyramidal neurons allowed us to study the synaptic function of the DGC without confounding deficits in neuronal migration associated with loss of DG in other tissues. Using this approach, we demonstrate that the neuronal DGC plays an essential role in trans-synaptic signaling necessary for formation and maintenance of functional axon terminals from CCK-positive basket cells. Because the neuronal circuits depending on this signaling have been shown to be involved in major cognitive functions, our findings open new avenues in identifying the causes of intellectual disability in muscular dystrophies.
Materials and Methods
Animals.
All mice were bred on C57BL/6 background at the Laboratory Animal Service Center (Schlieren, Zurich, Switzerland) and kept in standard housing with food and water provided ad libitum. Mice harboring loxP sites in exon 2 of Dag1 were obtained from The Jackson Laboratory. NEX-Cre transgenic mice were used to achieve selective ablation of neuronal DG (Goebbels et al., 2006) and were provided by Dr. Sandra Goebbels (Max-Planck-Institute of Experimental Medicine, Goettingen, Germany). Dag1 T190M knock-in mice were provided by Dr. Kevin P. Campbell (Howard Hughes Medical Institute, Iowa City, IA). Dag1 floxed mice were genotyped by PCR analysis using primers 5′-GGAGAGGATCAATCATGG-3′ and 5′-CAACTGCTGCATCTCTAC-3′. Genotyping of NEX-Cre transgenic mice was performed as described previously (Goebbels et al., 2006). To obtain DG cKO and control mice, NEX-Cretg/+/Dag1loxP/+ mice were bred to NEX-Cre+/+/Dag1loxP/loxP mice. All experiments were approved by the veterinary office of the Canton of Zurich.
Western blotting.
Adult DG cKO and control mice of both sexes were anesthetized with pentobarbital (Nembutal; 50 mg/kg i.p.) and killed by decapitation. Cheek muscle was dissected on ice and transferred to lysis buffer (50 mm Tris, pH 7.6, 150 mm NaCl, 1% Triton X-100, Complete Mini Protease Inhibitor Mixture, Roche). Tissue was Dounce homogenized, sonicated, and incubated on ice for 1 h. Lysates were centrifuged at 50,000 RPM for 1 h at 4°C, and supernatants were stored at −80°C. For anti-α-DG blots, glycosylated proteins were enriched by incubating lysates with wheat germ agglutinin agarose beads (Vector Labs) at 4°C overnight. Proteins were eluted with 300 mm N-acetyl-glucosamine and stored at −20°C. Laemmli buffer was added to wheat germ agglutinin-enriched and nonenriched lysates (for loading control) and samples were run on 8% Tris-glycine polyacrylamide gels. Proteins were transferred to PVDF membranes. Mouse anti-α-DG (11H6C4; Millipore; 1:1000) and rabbit anti-actin (Sigma; 1:5000) antibodies were incubated in Tris-buffered saline with 0.05% Tween 20 (TBST), including 5% Western Blocking Solution (Roche) overnight at 4°C. Membranes were washed 5 times in TBST. HRP-coupled donkey secondary antibodies (1:20,000) were incubated for 1 h at room temperature, and membranes were washed again 5 times in TBST. SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) was applied, and membranes were developed on x-ray film (Fujifilm).
Tissue preparation for immunohistochemistry.
DG cKO and control mice of both sexes at the age of 8–12 weeks were anesthetized by intraperitoneal pentobarbital injection (Nembutal; 50 mg/kg) and perfused transcardially with ice-cold oxygenated ACSF, pH 7.4, for 2 min, as described previously (Notter et al., 2014). Brains were immediately dissected and fixed in 4% PFA for 100 min on ice. After rinsing in PBS, brains were incubated in 30% sucrose (in PBS) at 4°C overnight. The 50-μm-thick coronal sections were cut from frozen blocks using a sliding microtome (HM400; Microm) and stored at −20°C in antifreeze solution. Tissue preparation from P21 mice followed the same protocol with the following modifications: Mice were perfused with 4% PFA (after brief perfusion with PBS to rinse blood), and brains were postfixed for 3 h.
Immunohistochemistry.
After rinsing once in PBS, sections were incubated in primary antibody solution (50 mm Tris, 150 mm NaCl, 0.2% Triton X-100, 2% normal goat serum, pH 7.4) with antibodies listed in Table 1. Primary antibodies were incubated at 4°C overnight, or for 3 d if DG or dystrophin was labeled. Sections were washed 3 times for 10 min in PBS and incubated in secondary antibody solution (50 mm Tris, 150 mm NaCl, 0.05% Triton X-100, 2% normal goat serum, pH 7.4) for 30 min at room temperature with secondary antibodies raised in goat. Antibodies conjugated to AlexaFluor-488 and AlexaFluor-647 (Invitrogen) were diluted 1:1000, whereas antibodies conjugated to Cy3 (Jackson ImmunoResearch Laboratories) were diluted 1:500. Sections were washed 3 times for 10 min in PBS and mounted on gelatin-coated slides using Fluorescence Mounting Medium (Dako). For immunoperoxidase stainings, biotinylated secondary antibodies were diluted 1:300. After washing 3 times for 10 min in PBS, sections were incubated with avidin-peroxidase-complex solution (Vector Labs) for 30 min at room temperature and washed again 3 times for 10 min in PBS. Sections were preincubated in DAB solution (50 mm Tris, 150 mm NaCl, 0.05% Triton X-100, 0.5 g/L DAB, pH 7.7) for 5 min under agitation and DAB solution containing 0.01% H2O2 was added to sections. The reaction was stopped by washing in ice-cold PBS several times. Sections were mounted on gelatinized slides and dried overnight. After dehydration by immersion in increasingly concentrated ethanol solutions and clearing in xylene, slides were coverslipped with Eukitt (Merck).
Table 1.
Antibodies used for immunohistochemical stainingsa
| Target | Host species | Dilution | Catalog # | Company/origin |
|---|---|---|---|---|
| α-Dystroglycan (VIA4–1) | Mouse | 1:100 | 05-298 | EMD Millipore |
| α-Dystroglycan (11H6C4) | Mouse | 1:100 | 05-593 | EMD Millipore |
| β-Dystroglycan | Mouse | 1:100 | ab49515 | Abcam |
| Bassoon | Mouse | 1:2000 | VAM-PS003 | StressGen |
| CB1 | Rabbit | 1:3000 | 258003 | Synaptic Systems |
| Cholecystokinin 8 | Mouse | 1:1000 | ab37274 | Abcam |
| Cre recombinase | Rabbit | 1:1000 | PRB-106C | Covance |
| Dystrophin (C-terminal) | Mouse | 1:100 | BT39-9050-05 | Biotrend |
| GABAAR α1 subunit | Guinea pig | 1:20,000 | — | Fritschy and Mohler, 1995 |
| GABAAR α2 subunit | Guinea pig | 1:6000 | — | Fritschy and Mohler, 1995 |
| GABAAR γ2 subunit | Guinea pig | 1:10,000 | — | Fritschy and Mohler, 1995 |
| GAD65/67 | Rabbit | 1:2000 | GC3008 | Biomol |
| Gephyrin | Mouse | 1:1000 | 147021 | Synaptic Systems |
| NeuN | Mouse | 1:1000 | MAB377 | Chemicon |
| NL2 | Rabbit | 1:10,000 | — | Gift from Dr. Peter Scheiffele |
| PV | Rabbit | 1:1000 | 24428 | ImmunoStar |
| PSD-95 | Mouse | 1:1000 | MA1-045 | ABR |
| Synaptotagmin 2 | Rabbit | 1:1000 | 105123 | Synaptic Systems |
| synArfGEF | Guinea pig | 1:3000 | — | Gift from Dr. Hiroyuki Sakagami (Fukaya et al., 2011) |
| VGAT | Rabbit | 1:3000 | 131003 | Synaptic Systems |
| VGluT1 | Guinea pig | 1:1000 | 135304 | Synaptic Systems |
| VGluT3 | Guinea pig | 1:4000 | AB5421 | Merck Millipore |
aIf not otherwise stated, antibody VIA4–1 was used to label α-dystroglycan. For secondary antibodies, see Materials and Methods.
Stereology.
Immunoreactive cells were counted and the size of the dorsal hippocampus estimated with the help of Mercator software (Explora Nova). Four equidistant coronal sections per mouse were used to count cells in all layers of the hippocampus proper. The volume was estimated using Mercator software, and cell density was calculated.
Image acquisition and statistical analysis.
z-stack images (3 optical sections, 0.5 μm step size) were recorded of all specimens using confocal laser scanning microscopy (LSM 700, Carl Zeiss). Images were taken using a 40× objective with a numerical aperture of 1.4 and had a pixel size of 112 × 112 nm2. To reduce variability, 3 or 4 sections were imaged per mouse and cluster density values were averaged from these sections. All imaging parameters were kept constant between genotypes. For cluster analysis, maximum intensity projections were created from z-stacks and analyzed using ImageJ (National Institutes of Health). Representative example images were processed with Imaris (Bitplane). Statistical tests were performed using Prism software (GraphPad). A minimum of 4 mice per group were used and statistical tests were performed using data points from individual mice for density values and using pooled cluster data from all mice per group for size (Tables 2, 3).
Table 2.
Results of statistical tests performed for immunohistochemical stainingsa
| Epitope | Condition | Unpaired t test (density) | Kolmogorov–Smirnov test (size) |
|---|---|---|---|
| GABAAR α1 subunit | DG cKO/adult/CA1 | t(6) = 0.920, p = 0.393 | n = 33385, D = 12.662, p < 0.001 |
| GABAAR α2 subunit | DG cKO/adult/CA1 | t(15) = 3.816, p = 0.002 | n = 18495, D = 3.484, p < 0.001 |
| GABAAR γ2 subunit | DG cKO/adult/CA1 | t(7) = 1.607, p = 0.152 | n = 31807, D = 3.692, p < 0.001 |
| Gephyrin | DG cKO/adult/CA1 | t(7) = 1.252, p = 0.251 | n = 39289, D = 0.720, p = 0.677 |
| NL2 | DG cKO/adult/CA1 | t(6) = 0.979, p = 0.366 | n = 32217, D = 5.476, p < 0.001 |
| synArfGEF | DG cKO/adult/CA1 | t(6) = 1.093, p = 0.316 | n = 22737, D = 3.850, p < 0.001 |
| VGAT | DG cKO/adult/CA1 | t(15) = 0.646, p = 0.528 | n = 20569, D = 0.528, p = 0.943 |
| DG cKO/adult/neocortex | t(9) = 1.309, p = 0.223 | n = 24713, D = 1.107, p = 0.172 | |
| PV | DG cKO/adult/CA1 | t(7) = 0.198, p = 0.849 | n = 24929, D = 1.068, p = 0.204 |
| DG cKO/adult/neocortex | t(9) = 0.454, p = 0.661 | n = 20534, D = 4.845, p < 0.001 | |
| DG cKO/P21/CA1 | t(8) = 0.869, p = 0.410 | n = 8010, D = 1.037, p = 0.233 | |
| CB1 | DG cKO/adult/CA1 | t(6) = 16.869, p < 0.001 | n = 4654, D = 4.325, p < 0.001 |
| DG cKO/adult/neocortex | t(11) = 7.117, p < 0.001 | n = 8067, D = 6.960, p < 0.001 | |
| T190M/adult/CA1 | t(8) = 0.681, p = 0.515 | n = 9152, D = 0.881, p = 0.420 | |
| Synaptotagmin 2 | DG cKO/adult/CA1 | t(9) = 1.456, p = 0.179 | n = 3397, D = 1.977, p = 0.001 |
| Cholecystokinin 8 | DG cKO/adult/CA1 | t(17) = 6.292, p < 0.001 | n = 5111, D = 4.133, p < 0.001 |
| DG cKO/adult/neocortex | t(6) = 4.475, p = 0.004 | n = 1387, D = 1.874, p = 0.002 | |
| PSD-95 | DG cKO/adult/CA1/SP | t(6) = 1.106, p = 0.311 | n = 16281, D = 1.554, p = 0.016 |
| DG cKO/adult/CA1/SR | t(6) = 0.243, p = 0.817 | — | |
| Bassoon | DG cKO/adult/CA1/SP | t(10) = 0.767, p = 0.461 | n = 32489, D = 2.275, p < 0.001 |
| DG cKO/adult/CA1/SR | t(10) = 0.871, p = 0.404 | — | |
| VGluT1 | DG cKO/adult/CA1 | t(6) = 0.094, p = 0.928 | n = 12776, D = 0.492, p = 0.969 |
| VGluT3 | DG cKO/adult/CA1 | t(21) = 13.213, p < 0.001 | n = 5975, D = 8.273, p < 0.001 |
| DG cKO/adult/neocortex | t(9) = 0.456, p = 0.659 | n = 6523, D = 2.054, p < 0.001 | |
| DG cKO/P21/CA1 | t(8) = 13.437, p < 0.001 | n = 1830, D = 8.270, p < 0.001 | |
| T190M/adult/CA1 | t(8) = 0.494, p = 0.634 | n = 4763, D = 0.702, p = 0.708 |
aNumbers in subscript in t test represent degrees of freedom. D represents test statistics for Kolmogorov–Smirnov tests.
Table 3.
Results of statistical tests performed for immunohistochemical stainings of injected tissuea
| Condition | t test (unpaired between genotypes, paired between hemispheres; density) | Kruskal–Wallis test (size) | Dunn's multiple comparison test (size) |
|---|---|---|---|
| 28 dpi/loxP/+ | t(3) = 0.520, p = 0.639 | n = 13610 | y = 283.498, p < 0.05 |
| 28 dpi/loxP/loxP | t(4) = 2.895, p = 0.044 | H = 55.796 | y = 539.294, p < 0.001 |
| 28 dpi/contralateral | t(7) = 1.032, p = 0.336 | p < 0.001 | y = 136.188, p > 0.05 |
| 28 dpi/ipsilateral | t(7) = 2.894, p = 0.023 | y = 391.984, p < 0.001 | |
| 42 dpi/loxP/+ | t(4) = 0.870, p = 0.434 | n = 9915 | y = 93.906, p > 0.05 |
| 42 dpi/loxP/loxP | t(4) = 3.478, p = 0.025 | H = 87.162 | y = 693.091, p < 0.001 |
| 42 dpi/contralateral | t(8) = 1.040, p = 0.329 | p < 0.001 | y = 82.049, p > 0.05 |
| 42 dpi/ipsilateral | t(8) = 4.059, p = 0.004 | y = 681.234, p < 0.001 | |
| 84 dpi/loxP/+ | t(5) = 1.843, p = 0.125 | n = 7378 | y = 16.550, p > 0.05 |
| 84 dpi/loxP/loxP | t(3) = 8.578, p = 0.003 | H = 52.415 | y = 466.868, p < 0.001 |
| 84 dpi/contralateral | t(8) = 0.682, p = 0.515 | p < 0.001 | y = 143.428, p > 0.05 |
| 84 dpi/ipsilateral | t(8) = 3.495, p = 0.008 | y = 593.745, p < 0.001 |
aNumbers in subscript in t test represent degrees of freedom. H and y represent test statistics for Kruskal–Wallis tests and Dunn's multiple comparison tests, respectively.
Stereotactic injections.
Eight- to 10-week-old mice transgenic for loxP in exon 2 of Dag1 were anesthetized with isoflurane (Attane; Piramal). After mice were head-fixed on a stereotactic frame (David Kopf Instruments), a small longitudinal incision was made under continuous administration of isoflurane to reveal the skull. Bregma was identified, and the skull was perforated unilaterally using a surgical drill at the following coordinates relative to bregma: x = −1.9 mm, y = 1.6 mm. A glass pipette filled with virus solution was inserted into the brain to z = 1.5 mm. A total of 1 μl virus solution was injected using an automated injection pump in increments of 70 nl over 10 min. The pipette was removed and the incision sutured. Mice were injected intraperitoneally with 1 mg/kg buprenorphine (Temgesic; Essex Chemicals) and placed on a warm pad for recovery before returning to the home cage.
Virus.
AAV8-CaMKIIa-mCherry-Cre (dot blot titer 4.7 × 1012 VG/ml) was purchased from the University of North Carolina Vector Core (Chapel Hill, NC).
Acute brain slice preparation.
Five- to 6-week-old DG cKO and control mice were briefly anesthetized with isoflurane and decapitated. The brain was quickly removed and transferred to ice-cold solution containing 65 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 25 mm NaHCO3, 7 mm MgCl2, 0.5 mm CaCl2, 25 mm glucose, and 105 mm sucrose saturated with 95% O2 and 5% CO2. The 350-μm-thick transverse slices containing the hippocampus were cut from the tissue block with a vibratome (Microm HM 650V, Thermo Scientific) and kept in oxygenated ACSF (315 mOsm) containing 125 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 25 mm NaHCO3, 1 mm MgCl2, 2 mm CaCl2, and 25 mm glucose at 34°C for 25 min and then at room temperature until use.
Electrophysiology and data analysis.
For recording, individual slices were transferred to a recording chamber perfused with oxygenated ACSF solution (same as above) at a flow rate of 1–2 ml/min. Whole-cell recordings were made from hippocampal CA1 pyramidal neurons. Cells were first selected using oblique IR illumination with a BX51 microscope (40× water-immersion objective; Olympus). Subsequently, neurons were anatomically identified using a fluorescent dye (Alexa-488, 10 μm) included in the intracellular solution. The dye was excited with wLS broad-band LED illumination (488 nm), and images were acquired with Retiga R1 camera using Ocular software (Qimaging). The cells were patched with borosilicate glass pipettes (2–5 mΩ) containing the following: 135 mm KCl, 10 mm HEPES, 10 mm sodium phosphocreatine, 4 mm Mg-ATP, 0.3 mm Na-GTP, pH 7.3 with KOH. Recordings were performed using Multiclamp 700B amplifier, and data were acquired with a Digidata 1550A 16-bit board (all from Molecular Devices). All experiments were performed at room temperature. Spontaneous IPSCs (sIPSC) were recorded from CA1 pyramidal cells clamped at a membrane voltage of −70 mV in the presence of 10 μm NBQX to block excitatory transmission. Recordings with unstable baseline or >−400 pA were rejected. Currents were filtered off-line using a Butterworth low-pass filter (2 kHz) and analyzed in 1 or 2 min bins using the Mini-Analysis Program 6.0.7 (Synaptosoft). For pharmacology, baseline was analyzed 2 min before the application of carbachol (CCh, 10 μm). To study the effect of CCh, 1–2 min bins were analyzed at least 8 min following the arrival of CCh into the bath. Recordings with leak increasing >100 pA and access resistance changing >30% between the beginning and the end of the recording were discarded. At least 100 events were analyzed for any condition in any experiment. Events were identified as sIPSC by setting the event detection threshold at least twofold the baseline noise level and by checking that events had (1) rise times faster than the decay time, (2) rise times >0.5 ms, and (3) decay times >1.5 ms. Events not fitting the above parameters were rejected. Event amplitudes, interevent intervals (IEIs), and rise and decay times were first averaged within each experiment and regrouped by condition. The frequencies were calculated from the IEIs, and the resulting means were averaged between experiments. Single-cell properties (access resistance, membrane capacitance, etc.) were analyzed with Clampfit 10.5 (Axon Instruments). Graphs were done using Igor 6.37 software (Wavemetrics) and Illustrator 15.1.0 (Adobe).
Results
Use of NEX-Cre driver line for pyramidal cell-specific DG ablation
To study the role of neuronal DGC in the brain without gross morphological alterations, it is necessary to target DG in neurons but spare glial DG. For this reason, mice harboring loxP sites in Dag1 were crossed to the NEX-Cre driver line, which exhibits an exclusively neuronal Cre recombinase expression pattern (Goebbels et al., 2006; Satz et al., 2010). In hippocampus and neocortex, NEX promoter-mediated Cre expression is restricted to pyramidal cells, the cell type displaying most prominent DG expression in the forebrain. DG conditional knock-out mice (cKO; NEX-CreTg/+, Dag1loxP/loxP) showed reduced size compared with control mice (NEX-CreTg/+, Dag1loxP/+; Fig. 1A). The smaller size was reflected in reduced body and brain weight (Fig. 1B,C; 25.3 ± 1.0 g [mean ± SEM] vs 18.7 ± 1.0 g, t(28) = 4.670, p < 0.001 and 473.1 ± 5.4 mg vs 436.4 ± 6.5 mg, t(28) = 4.381, p < 0.001, unpaired t tests). Although cKO mice were born in Mendelian proportions, in adulthood less than the expected 25% cKO were observed due to higher lethality of cKO mice (Fig. 1D). To exclude a contribution of muscular dystrophy to this phenotype because of Cre leakage in muscle cells, α-DG levels were examined by Western blotting of wheat germ agglutinin-enriched muscle proteins. cKO mice showed similar levels of muscle α-DG as control mice (Fig. 1E). As reported before, DG cKO mice retained proper lamination of hippocampus (Fig. 1F) and neocortex (Fig. 1G) according to NeuN and DAPI labeling (Satz et al., 2010). Cre expression was restricted to pyramidal cells in hippocampus and neocortex and was not detected in dentate gyrus granule cells of adult mice (Fig. 1F,G) (Goebbels et al., 2006).
Figure 1.
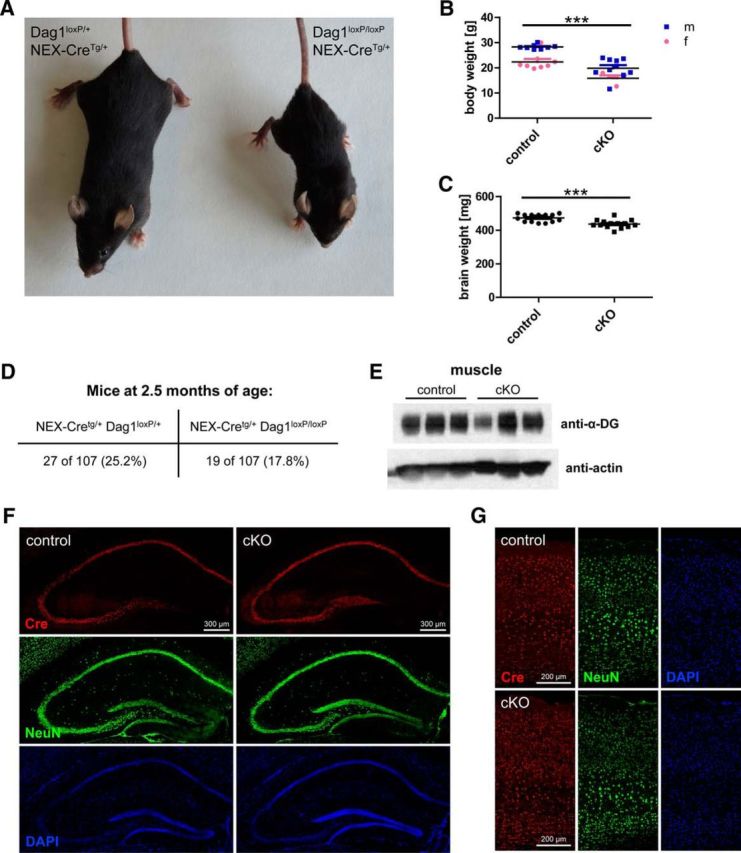
Characterization of NEX-Cre/Dag1 conditional KO mice. A, Representative examples of NEX-CreTg/+/Dag1loxP/+ (control) and NEX-CreTg/+/Dag1loxP/loxP (cKO) mice (4 months of age, siblings, both female). B, cKO mice exhibit reduced body weight compared with sibling control mice. C, Wet brain weight was lower in cKO mice than in controls. D, cKO mice exhibited a higher mortality rate than control mice, resulting in a frequency of cKO mice lower than the expected 25% at the age of 10 weeks. E, Similar levels of α-DG isolated from cheek muscle were found for cKO and control mice. F, Cre expression was restricted to pyramidal cells in the hippocampus of cKO and control mice. In adult mice, dentate gyrus granule cells were not immunoreactive for Cre recombinase. NeuN and DAPI labeling shows intact neuronal migration when NEX-Cre is used as driver line to ablate Dag1. G, In primary somatosensory cortex, Cre expression was also restricted to pyramidal cells. No migratory deficits were found in the neocortex in cKO mice. ***p < 0.001.
Efficiency and specificity of DG ablation were examined immunohistochemically in relevant brain regions. α-DG, β-DG, and dystrophin can be detected immunohistochemically in large perisomatic clusters in CA1 pyramidal cells (Fig. 2A) (Lidov et al., 1990; Knuesel et al., 1999). This characteristic immunolabeling was absent in DG cKO mice for DG and to the same extent for dystrophin, showing that dystrophin needs DG for synaptic clustering in vivo. In neocortex, perisomatic distribution of α-DG, β-DG, and dystrophin was replaced by diffuse unspecific staining in neuropil in DG cKO mice (Fig. 2B). Astrocyte endfeet are labeled prominently by antibodies to β-DG and dystrophin, and this labeling was preserved in DG cKO mice, as expected from neuron-specific Cre expression (Fig. 2A,B, asterisks). α-DG immunolabeling in dentate gyrus granule cells showed the same clustered distribution in both genotypes (Fig. 2C). The transient NEX promoter activity in these cells during early postnatal development might not be sufficient to achieve recombination (Goebbels et al., 2006). Alternatively, loss of DG during early development might be of little consequence in adulthood because of DG expression by granule cells that were born later. To further demonstrate specificity of NEX-induced DG ablation, striatum was selected as a control region. As expected, the characteristic sparse α-DG labeling persisted in striatum of DG cKO mice (Fig. 2D).
Figure 2.

NEX-Cre-mediated ablation of dystroglycan (DG) leads to specific loss of dystrophin-glycoprotein complex in pyramidal cells. A, Characteristic staining of α- and β-DG and dystrophin around CA1 pyramidal layer is lost in cKO mice. Labeling of β-DG and dystrophin in astrocyte endfeet is retained in cKO mice (asterisks). B, In primary somatosensory cortex layer 2/3, clustered labeling of DGC components around pyramidal cells is replaced by diffuse staining in the neuropil. Astrocyte endfeet labeling of β-DG and dystrophin is retained in cKO mice (asterisks). C, α-DG immunofluorescence in dentate gyrus is unaffected by NEX-Cre-mediated ablation of DG. D, α-DG expression in striatum is unaffected in cKO mice, confirming specificity of NEX-Cre expression to pyramidal cells. SR, Stratum radiatum; SP, stratum pyramidale; ML, molecular layer; GCL, granule cell layer.
Loss of neuronal DG results in minor alterations in GABAergic PSD protein clustering
Dependence of GABAergic PSD proteins on the DGC for synaptic clustering was suggested because of the subcellular localization of the DGC, its molecular interactions, and because a reduction in GABAAR clustering was observed in mice lacking full-length dystrophin (Knuesel et al., 1999; Sumita et al., 2007; Waite et al., 2012). However, a requirement of DG for clustering of GABAergic postsynaptic proteins was never systematically tested in vivo. We hypothesized that loss of neuronal DG affects neuroligin 2 (NL2) clustering, which might be important for clustering of GABAARs at perisomatic synapses through its interaction with gephyrin (Poulopoulos et al., 2009; Panzanelli et al., 2011). DG cKO and control mice were analyzed for changes in clustering of these markers in CA1 pyramidal layer. As previously reported (Knuesel et al., 1999; Brünig et al., 2002; Lévi et al., 2002), extensive colocalization of α-DG and dystrophin was observed with GABAergic markers, with a minority of DGC clusters showing no colocalization (Fig. 3A,B; arrowheads and arrows, respectively). Visual examination of GABAergic markers revealed no obvious differences between genotypes (Fig. 3A–C). However, quantification of cluster density and size showed a significant decrease of GABAAR α1 subunit size in cKO accompanied by an increase of GABAAR α2 subunit density (Fig. 3D,F). No changes were observed in GABAAR γ2 subunit and gephyrin clustering (Fig. 3E,H), indicating that total synaptic GABAAR content might be unchanged, whereas α subunit composition is altered by loss of DG. Surprisingly, NL2 clustering was barely affected in cKO mice, showing no difference in density and only a slight but significant reduction in cluster size (Fig. 3G). Neuronal DGC is also dispensable for normal colocalization of GABAAR γ2 subunit with gephyrin and of α1 subunit with NL2 (Fig. 3I). Furthermore, dystrophin was suggested to be important for anchoring synArfGEF at GABAergic PSDs (Fukaya et al., 2011). Although dystrophin clustering is lost in DG cKO mice, synArfGEF distribution remained almost unchanged in CA1 pyramidal layer of cKO mice (Fig. 3J).
Figure 3.
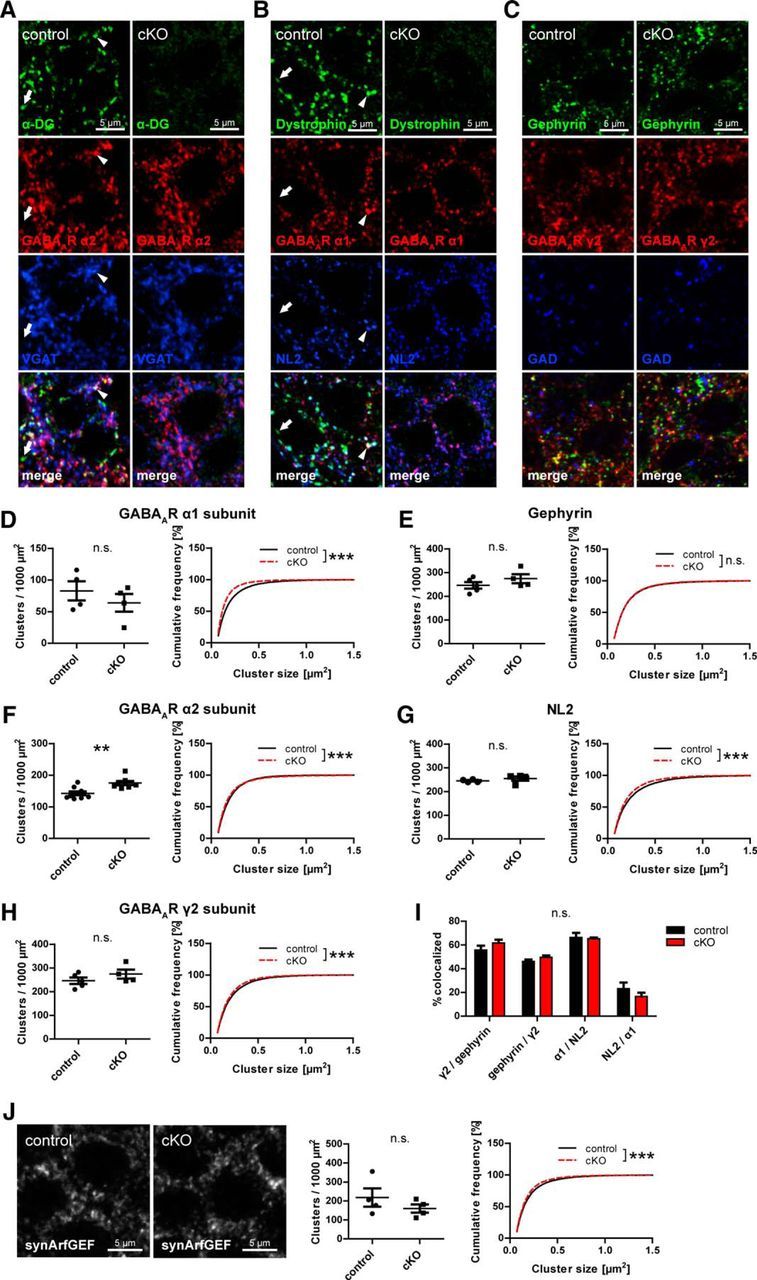
Loss of neuronal dystroglycan does not prohibit formation of GABAergic PSD but leads to minor changes in GABAAR subunit clustering. A–C, Triple immunofluorescence labeling of GABAergic postsynaptic markers in pyramidal layer of hippocampus CA1 area. The DGC is largely colocalized with α2 subunit and VGAT (A; arrowheads) but also with α1 subunit and NL2 (B; arrowheads). A minority of DGC clusters is not associated with GABAergic markers (A, B; arrows). D–H, Quantification of postsynaptic GABAergic markers in CA1 pyramidal cell layer. Cluster density and size are shown for GABAAR α1 (D), α2 (F), and γ2 (H) subunits and for gephyrin (E) and NL2 (G). A decrease of α1 subunit cluster size was accompanied by an increased α2 subunit cluster density. I, Colocalization of postsynaptic GABAergic markers was analyzed in cKO and control mice. Data are the number of colocalized clusters as percentage of first mentioned marker. No significant differences in colocalization were found between genotypes. J, Clustering of synArfGEF was analyzed in CA1 pyramidal layer of DG cKO and control mice. Data points represent individual mice (for statistical tests, see Table 2). **p < 0.01. ***p < 0.001.
Neuronal DG ablation leads to selective loss of markers of CCK-positive basket cell terminals
Many binding partners of α-DG have been identified, among them the presynaptic neurexins (Sugita et al., 2001). Together with the observation that DG is always apposed to GABAergic presynaptic terminals in primary neuronal culture, a trans-synaptic function for DG seemed probable (Brünig et al., 2002). We therefore probed DG cKO and control brain tissue with antibodies to presynaptic GABAergic markers. Perisomatic GABAergic terminals can be attributed to PV- or CCK-positive interneurons, which are labeled by PV/synaptotagmin 2 and CCK8/VGluT3/cannabinoid receptor 1 (CB1), respectively. We found that markers for presynaptic terminals from CCK-positive interneurons were virtually absent in CA1 pyramidal layer of DG cKO mice (Fig. 4A–D,G,I,K). Still, as in control mice, CCK-positive cell somata were occasionally observed in cKO CA1 pyramidal layer, and these were often covered with VGluT3-positive boutons (Fig. 4A, arrowhead). Synaptotagmin 2 and PV immunolabeling was still present in typical punctate distribution in the pyramidal layer of cKO mice (Fig. 4A–D,H,J), demonstrating the specific requirement of DG for formation of presynaptic terminals from CCK-positive interneurons. Preferential apposition of DG to CCK-positive interneuron terminals might be expected from this finding. However, apposition of DG to VGluT3 as well as to PV suggests no such distinction, at least at the resolution of conventional confocal laser scanning microscopy (Fig. 4C, arrowheads). Still, as a percentage of presynaptic immunofluorescence apposed to DG, VGluT3 showed more complete overlap with DG, indicating that PV apposition to DG might be caused by mere abundance of PV immunofluorescence in the pyramidal layer (data not shown). Loss of CCK-positive interneuron terminals extended from CA3 to CA1 (Fig. 4E). Surprisingly, no corresponding reduction in vesicular GABA transporter (VGAT) puncta was observed (Fig. 4F). To test whether sprouting of PV-positive axons compensates for the loss of CCK-positive terminals, the portion of VGAT-positive terminals containing PV was assessed. No significant difference was found in the fraction of VGAT puncta colocalized with PV between genotypes (31.05 ± 2.05% for control vs 33.24 ± 1.57% for cKO, t(8) = 0.847, p = 0.422, unpaired t test), suggesting that PV-positive axon terminal sprouting does not account for the persisting VGAT immunolabeling.
Figure 4.
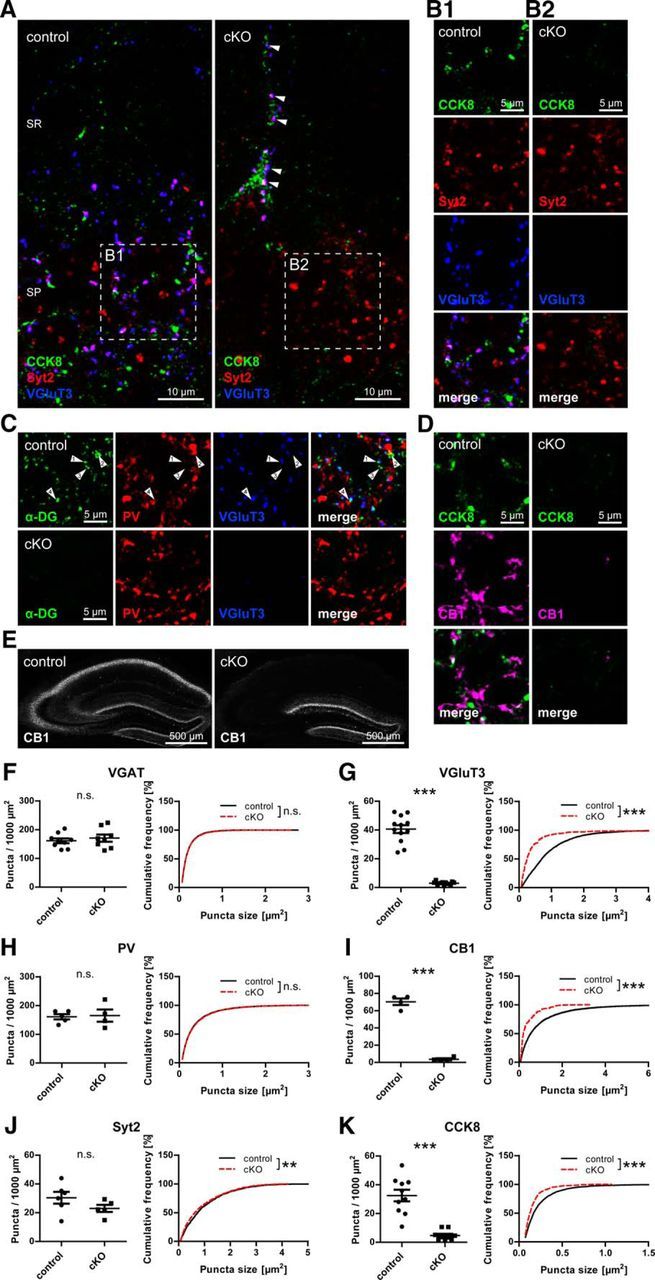
Neuronal dystroglycan ablation leads to specific loss of terminals from CCK-positive basket cells on hippocampal pyramidal cells. A–D, Triple immunofluorescence labeling of presynaptic GABAergic markers in hippocampus CA1 area. A, In pyramidal layer, markers labeling CCK-positive basket cell terminals (CCK8, VGluT3) are missing around pyramidal cell bodies but are still present on CCK-positive cell somata occasionally observed near the pyramidal layer (arrows). These VGluT3-positive boutons on CCK-positive somata were often immunopositive for synaptotagmin 2 (Syt2). In the pyramidal layer, immunostaining for Syt2 remained in cKO mice. B1, B2, Separated channels of A (insets). C, PV immunolabeling in CA1 pyramidal layer of DG cKO mice is indistinguishable from control. In CA1 pyramidal layer of control mice, the majority of α-DG clusters is either apposed to VGluT3 (arrow 1) or PV (arrow 2), but some clusters are not apposed to either marker (arrow 3). A minority of α-DG clusters showed apposition to both VGluT3 and PV (arrow 4). D, Along with CCK8 and VGluT3, CB1 staining is strongly reduced in CA1 pyramidal layer of DG cKO mice. E, Loss of CB1 immunofluorescence in DG cKO mice was observed from CA1 to CA3. F–K, Quantification of presynaptic GABAergic markers in CA1 pyramidal layer. No changes were found for VGAT (F) and PV-positive basket cell markers (H, J) between genotypes, but cluster density and size of markers of CCK-positive basket cell terminals (G, I, K) were strongly reduced in DG cKO mice. Data points represent individual mice (for statistical tests, see Table 2). **p < 0.01. ***p < 0.001. SR, Stratum radiatum; SP, stratum pyramidale.
Despite the drastic loss of CCK-positive terminals, somata exhibiting CCK immunofluorescence were observed in cKO hippocampi (Fig. 4A). To characterize the distribution and abundance of CCK-positive interneurons, sections of cKO and control dorsal hippocampus were immunoperoxidase-stained and CCK-positive cell density quantified using stereology. CCK-positive cell somata in cKO mice were reduced to less than half the density observed in control mice (816.77 ± 28.38 mm−3 vs 368.51 ± 4.88 mm−3, t(8) = 15.566, p < 0.001, unpaired t test). Along with cell density, immunoperoxidase staining intensity of CCK-positive cells was diminished in the CA1 to CA3 region in DG cKO mice.
Because the DGC is prominently expressed by pyramidal cells in the neocortex, it seemed likely that CCK-positive interneuron terminals in neocortex are also compromised by loss of neuronal DG. Indeed, CCK8 and CB1 immunolabeling was strongly reduced in primary somatosensory cortex (S1) of DG cKO mice, whereas PV staining was unchanged (apart from a minute difference in size; Fig. 5). VGluT3 puncta density was not decreased in neocortex of DG cKO mice, in agreement with histological studies showing that VGluT3 is present mostly in serotonergic fibers in this brain area (Fig. 5B,G) (Schäfer et al., 2002). As in hippocampus, reduction of CCK-positive terminals was not paralleled by a decrease of VGAT puncta (Fig. 5A,E). Markers for CCK-positive terminals were reduced uniformly across all cortical layers and in all regions of the neocortex that were examined (Fig. 5D).
Figure 5.
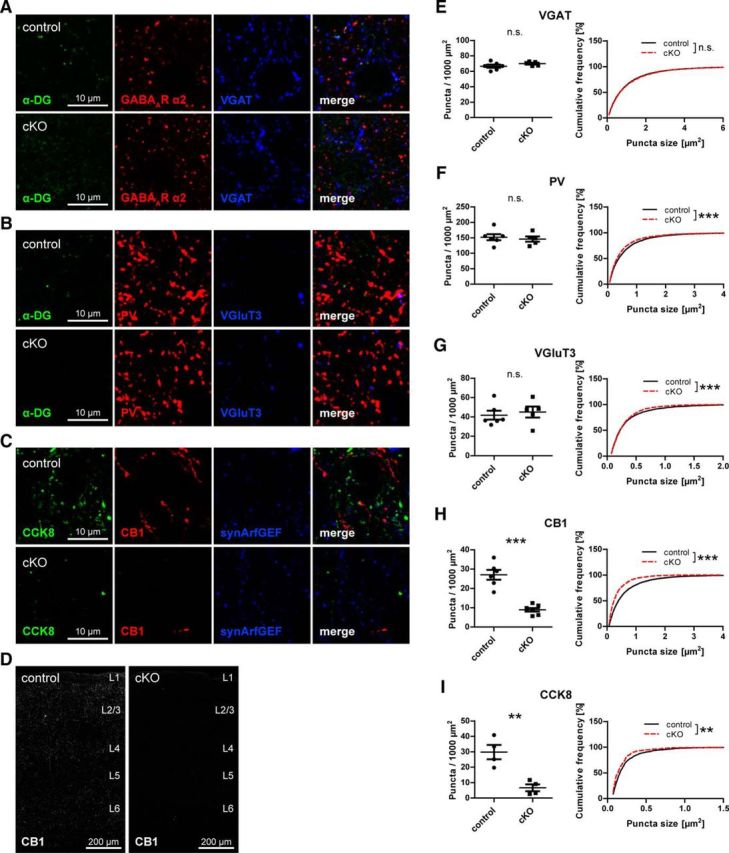
Neuronal dystroglycan ablation leads to specific loss of terminals from CCK-positive basket cells on pyramidal cells in neocortex. A–C, Triple immunofluorescence labeling of GABAergic markers in layer 2/3 of primary somatosensory cortex (S1) of DG cKO and control mice. A, As in hippocampus, the majority of DG clusters is colocalized with presynaptic and postsynaptic GABAergic markers in neocortex. B, Neocortical PV and VGluT3 immunolabeling is not affected by loss of neuronal DG. C, CCK8 and CB1 immunofluorescence is strongly reduced in neocortex of DG cKO mice. Immunolabeling of synArfGEF showed clustered distribution and did not differ between genotypes. D, Overview of S1 of DG cKO and control mice. Typical punctate CB1 immunofluorescence was lost across all layers of the cortex in DG cKO mice. E–I, Quantification of presynaptic GABAergic markers in S1 layer 2/3. VGAT and PV, and in contrast to hippocampus, also VGluT3 were not reduced in density in mice lacking neuronal DG (E–G). However, CB1 and CCK8 showed a similar reduction as in hippocampus in DG cKO mice compared with control mice (H, I). Data points represent individual mice (for statistical tests, see Table 2). **p < 0.01. ***p < 0.001.
Satz et al. (2010) have reported blunted long-term potentiation in CA1 pyramidal cells of mice with NEX-Cre-mediated DG ablation. To exclude that loss of CCK-positive interneuron terminals represents compensatory changes to large glutamatergic alterations, glutamatergic markers were examined as a proxy for integrity of glutamatergic synapses (Fig. 6). Clustering of the postsynaptic glutamatergic markers PSD-95 and bassoon did not differ significantly between genotypes (Fig. 6A,B), and neither did VGluT1 immunolabeling (Fig. 6C). Furthermore, the portion of PSD-95 clusters apposed to VGluT1 was similar in both genotypes (Fig. 6D).
Figure 6.

Neuronal dystroglycan is not necessary for clustering of glutamatergic synaptic proteins. A, B, To assess integrity of glutamatergic postsynaptic structures, antibodies to PSD-95 and bassoon were used and immunofluorescence quantified in stratum pyramidale and stratum radiatum. Cluster density and size were analyzed in stratum pyramidale and fluorescence intensity in stratum radiatum. All parameters analyzed did not differ substantially between genotypes. C, VGluT1 was used as a marker of glutamatergic presynaptic terminals, and puncta density and size in stratum pyramidale were quantified. No changes in VGluT1 puncta density and size were found between genotypes. D, PSD-95 apposition to VGluT1 was examined in stratum pyramidale and represented as percent PSD-95 clusters apposed to VGluT1 puncta. The apposition of PSD-95 to VGluT1 did not differ between genotypes. Data points represent individual mice (for statistical tests, see Table 2). *p < 0.05. ***p < 0.001.
Formation and maintenance of CCK-positive basket cell terminals require neuronal DG
DG expressed by pyramidal cells might have a function in synapse formation or in guidance of a subset of axons, similar to its role in the spinal cord (Wright et al., 2012). Alternatively, a function in maintenance of synapses through continuous trans-synaptic signaling is conceivable. If neuronal DG is crucial for synapse formation of CCK-positive terminals, these boutons should be reduced to the same degree as in adults at a time point right after initial synaptogenesis. Following this reasoning, we examined CCK-positive terminals of 21-day-old DG cKO and control mice in CA1 pyramidal layer. Indeed, VGluT3 puncta were largely missing also at this stage of development, whereas immunostaining of PV-positive terminals was not significantly different between genotypes (Fig. 7).
Figure 7.

CCK-positive terminals are not established in the absence of neuronal dystroglycan. A, Triple immunofluorescence labeling of DG cKO and control CA1 pyramidal layer at postnatal day 21. B, C, Quantification of puncta density and size reveals loss of VGluT3 puncta in DG cKO tissue to the same degree as in adult mice (B) but unchanged PV immunolabeling (C). Data points represent individual mice (for statistical tests, see Table 2). ***p < 0.001.
Although this finding indicates that synapse formation of functional CCK-positive terminals depends on DG, it does not rule out a role for DG in maintaining already formed connections. To assess this putative function of DG in synapse maintenance, we ablated DG long after developmental synapse formation, by viral delivery of Cre to adult mice carrying one or both floxed Dag1 alleles. AAV8-CaMKII-mCherry-Cre was stereotactically injected unilaterally into the CA1 region and mice killed at 14, 28, 42, or 84 d after injection (dpi; Fig. 8A,C). At 14 dpi, Cre and mCherry fluorescence was clearly visible (Fig. 8B). Loss of β-DG staining at 28 dpi in homozygously floxed mice indicated efficient recombination of loxP sites (Fig. 8D). In heterozygously floxed mice, only a moderate reduction of β-DG labeling was observed, suggesting that one wild-type allele is sufficient to sustain the bulk of DG expression. Because dystrophin immunostaining revealed a reduction that mirrored β-DG, and in addition showed lower background, dystrophin was used to assess DGC loss at subsequent time points (Fig. 8E–G). Examination of VGluT3-positive terminals at 28 dpi in Cre-expressing regions of CA1 pyramidal layer revealed a moderate but significant reduction of VGluT3 puncta density and size in homozygously floxed mice compared with contralateral side compared with the ipsilateral side of heterozygously floxed mice (Fig. 8H). VGluT3-positive terminals in heterozygous mice were not affected. Compromised VGluT3 immunolabeling was also found at later time points in homozygous mice, and the effect became more prominent with increased time after injection (Fig. 8I,J). Together, these results provide strong evidence for a role of DG both in synapse formation and in retrograde trans-synaptic signaling for maintenance of CCK-positive terminals.
Figure 8.
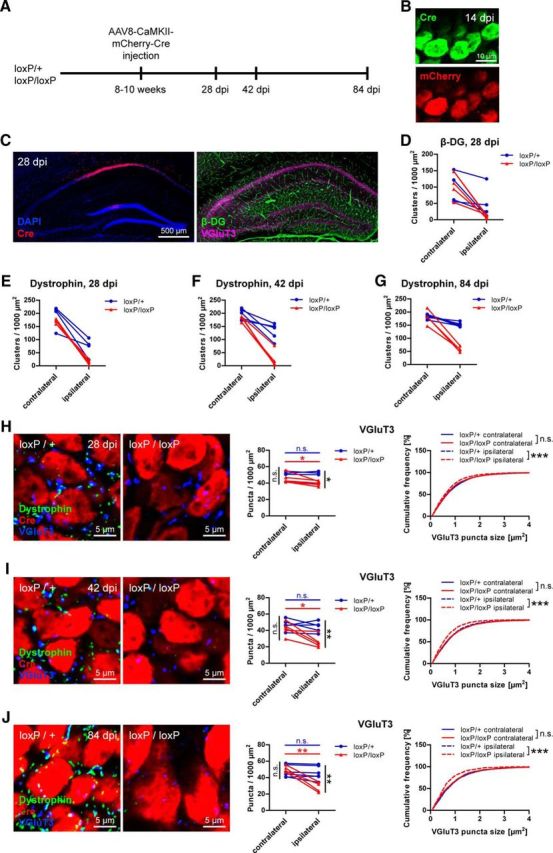
Maintenance of CCK-positive basket terminals requires dystroglycan. A, Overview of experimental design. Virus was stereotactically injected unilaterally into CA1 region in adult mice heterozygous or homozygous for loxP sites flanking the Dag1 gene. B, After 14 dpi, Cre recombinase immunolabeling and mCherry fluorescence were clearly visible in pyramidal cell somata. C, Example of the injection site at 28 dpi. Cre expression was mostly restricted to the CA1 pyramidal cell layer. VGluT3 and dystrophin or DG immunofluorescence was analyzed in the same sections. D, In mice containing homozygously floxed Dag1, β-DG immunostaining was markedly reduced in CA1 pyramidal layer at 28 dpi. Heterozygous mice showed a moderate reduction in β-DG immunofluorescence. E–G, As observed for β-DG, Cre expression led to loss of dystrophin in homozygously floxed mice, whereas only a slight decrease was observed in heterozygous mice. Reduction of dystrophin labeling was similar at 28 dpi (E), 42 dpi (F), and 84 dpi (G). H–J, Representative example images and quantifications of VGluT3 immunostaining in CA1 pyramidal layer at 28 dpi (H), 42 dpi (I), and 84 dpi (J). Ipsilateral VGluT3 size and density in homozygously floxed mice were significantly reduced compared with both contralateral side and ipsilateral side of heterozygously floxed mice. With increased time after injection, this reduction of VGluT3 puncta became more prominent. Data points represent individual mice (for statistical tests, see Table 3). *p < 0.05. **p < 0.01. ***p < 0.001.
Absence of CCK-positive basket cell terminals due to DG ablation impacts pyramidal cell inhibitory input and response to cholinergic activation
If axon terminals from CCK-positive basket cells are indeed lost in DG ablated mice, this should be reflected by functional changes of pyramidal cell inhibitory input. To test this hypothesis, acute slices were prepared from adult DG cKO and control brains and used for patch-clamp electrophysiological recordings from morphologically identified CA1 pyramidal cells (Fig. 9). With inhibitors of glutamatergic transmission present in the bath, occurrence of sIPSCs was probed in both genotypes. As anticipated from immunohistological changes, sIPSC frequency in DG cKO was reduced to approximately half of that in control slices (Fig. 9; 8.71 ± 1.52 Hz vs 4.46 ± 0.90 Hz, t(26) = 2.214, p = 0.036, unpaired t test). Furthermore, DG cKO pyramidal cells were marked by a significantly smaller sIPSC amplitude than that of control cells (61.49 ± 7.90 pA vs 39.18 ± 2.39 pA, t(26) = 2.374, p = 0.025, unpaired t test). No significant differences were found between genotypes in sIPSC rise and decay times (rise time 1.73 ± 0.09 ms vs 1.63 ± 0.12 ms, t(26) = 0.664, p = 0.512, unpaired t test; decay time 14.84 ± 0.64 ms vs 14.21 ± 0.53 ms, t(26) = 0.717, p = 0.480, unpaired t test).
Figure 9.

Frequency and amplitude of sIPSCs are reduced in dystroglycan cKO pyramidal cells. A, Position of the recording pipette in the hippocampal CA1 region (left, 4×), and an example of a typical CA1 pyramidal cell identified using LED illumination (AlexaFluor-488, right, 40×). B, Representative example traces of whole-cell sIPSC recordings from control mice (left) and DG cKO mice (right). Average sIPSCs are shown above the traces. C, Cumulative frequency plot of IEIs of sIPSCs from control (blue line) and DG cKO cell (red line) from the traces in B (left) and cumulative frequency plot of sIPSC amplitudes from the same cells (right). D, Comparison of average sIPSC frequency and amplitude between control and DG cKO slices. DG cKO mice exhibit significantly lower sIPSC frequency and amplitude than control mice. Data points represent individual cells. *p < 0.05.
The differences observed in baseline sIPSCs could be due to a general reduction of inhibitory transmission instead of interneuron subtype-specific loss of terminals. To gain insight into the origin of reduced inhibitory transmission in DG cKO pyramidal cells, we examined the effect of the acetylcholine receptor agonist CCh on inhibitory currents. In slices, CCh exposure leads to an increase of perisomatic inhibitory transmission in pyramidal cells, which is mediated by direct excitation of CCK-positive interneurons (Nagode et al., 2014). Given that CB1 receptor-containing terminals are required for increased inhibitory transmission after application of CCh, this effect should be absent in DG cKO mice if CCK-positive basket terminals are indeed nonfunctional in these mice. CCh was bath-applied to DG cKO and control acute slices from which sIPSCs were recorded in CA1 pyramidal cells. In control slices, CCh led to a robust increase in sIPSC frequency within minutes after application (Fig. 10A–C; frequency: 6.15 ± 1.45 Hz vs 10.53 ± 2.47 Hz, t(7) = 3.522, p = 0.010; amplitude: 63.36 ± 8.84 pA vs 70.35 ± 12.59 pA, t(7) = 0.943, p = 0.377; paired t tests). However, no statistically significant effect of CCh was observed in DG cKO pyramidal cells (Fig. 10D–F; frequency: 3.45 ± 0.75 Hz vs 4.47 ± 1.56 Hz, t(6) = 0.797, p = 0.456; amplitude: 45.70 ± 4.17 pA vs 53.03 ± 5.00 pA, t(6) = 1.239, p = 0.262; paired t tests). Together with the results from baseline recordings and immunohistochemical analysis, these findings strongly argue that functional connectivity between CCK-containing basket cells and pyramidal cells is lost in DG-ablated mice.
Figure 10.

Dystroglycan is necessary for CCh-induced increase of inhibitory currents in pyramidal cells. A, Representative example traces of sIPSC recordings before (baseline, left trace) and after the application of CCh (right) in control mice. Average sIPSCs are shown above the traces. B, Cumulative frequency plots of IEIs and amplitudes of sIPSCs from traces in A. C, Comparison of average sIPSC frequency and amplitude before and after application of CCh in control slices. Application of CCh resulted in typical increase of IPSC frequency in control pyramidal cells, but amplitude was not affected by CCh. D, Representative example traces of sIPSC recordings before (baseline, left trace) and after the application of CCh (right trace) in DG cKO mice. Average sIPSCs are shown above the traces. E, Cumulative frequency plots of IEIs and amplitudes of sIPSCs from traces in D. F, Comparison of average sIPSC frequency and amplitude before and after application of CCh in DG cKO slices. In contrast to control slices, application of CCh did not lead to a significant increase of sIPSC frequency in DG cKO pyramidal cells. Data points represent individual cells. **p < 0.01.
Persistence of CCK-positive terminals in DG T190M knock-in mice suggests trans-synaptic DG function is independent of neurexin binding
The intriguing finding that DG is required for formation and maintenance of CCK-positive terminals calls for an assessment of the clinical significance of this observation. In a subgroup of dystroglycanopathies, intellectual disability, although severe, is not accompanied by neuronal migration deficits (Godfrey et al., 2007). Dag1 T190M knock-in mice are a model of one such form of dystroglycanopathy and resemble the symptoms found in patients with the corresponding mutation (Dinçer et al., 2003; Hara et al., 2011). Interestingly, this mutation abolishes binding of DG to neurexin, a putative presynaptic DG binding partner. We compared markers of CCK-positive terminals in CA1 pyramidal layer of homozygous Dag1 T190M mice with wild-type mice (Fig. 11). Surprisingly, both VGluT3 and CB1 puncta were indistinguishable between Dag1 T190M and wild-type mice. Weaker and more diffuse labeling in Dag1 T190M mice using the α-DG glycosylation-specific antibody 11H6 confirmed that this mutation affects glycosylation of neuronal DG (Fig. 11B). Apposition of β-DG to PV or VGluT3-positive terminals was not changed by T190M mutation (Fig. 11E). Therefore, DG function for CCK-positive terminals is likely neurexin-independent, which suggests that a novel presynaptic receptor might be involved in this trans-synaptic connection.
Figure 11.

Neurexin and laminin binding of α-dystroglycan is not essential for formation of CCK-positive basket terminals on pyramidal cells. A, B, Triple immunofluorescence labeling of GABAergic markers in CA1 pyramidal layer of Dag1 T190M and wild-type mice. A, Antibody to β-DG revealed typical clustered distribution in Dag1 T190M mice. VGluT3 and PV immunofluorescence was indistinguishable between genotypes. B, Intensity of DG clusters was markedly reduced and background staining increased using the α-DG glycosylation-specific antibody 11H6, confirming glycosylation deficits of synaptic DG in T190M mice. CB1 and GABAAR γ2 subunit immunofluorescence was indistinguishable between genotypes. C, D, Quantification of density and size of VGluT3 (C) and CB1 (D) puncta in CA1 pyramidal layer of Dag1 T190M and wild-type mice. Density and size of puncta did not differ significantly between genotypes. E, Quantification of β-DG apposition to PV and/or VGluT3 in CA1 pyramidal layer. Data are number of β-DG clusters apposed to PV or VGluT3 or both (triple colocalized) as percentage of total β-DG clusters. Apposition of β-DG to presynaptic markers did not differ significantly between genotypes. Data points represent individual mice (for statistical tests, see Table 2).
Discussion
Our experiments have yielded five main findings about the synaptic function of DG. Ablation of neuronal DG, which also hindered synaptic clustering of dystrophin, led only to minor changes in clustering of GABAergic PSD proteins. These alterations might reflect compensatory changes to the massive presynaptic defects found in DG-deficient mice. Importantly, DG synaptic function is interneuron subtype-specific because loss of synaptic markers was restricted to CCK-expressing basket cell terminals. Formation and maintenance of these synapses required neuronal DG, indicating that trans-synaptic signaling is important both at the time of developmental synaptogenesis and continuously during adulthood. Function of CCK-positive basket cell terminals was likely compromised along with specific marker expression because loss of DG resulted in a reduced baseline spontaneous inhibitory activity in pyramidal cells that could not be increased by CCh. Finally, postphosphorylation glycosylation of DG is not necessary for CCK-positive synapse formation because Dag1 T190M knock-in mice showed normal CCK-positive terminals, suggesting that presynaptic receptors other than neurexins might be involved in DG trans-synaptic function.
Postsynaptic GABAergic alterations ascribed to DGC deficits may be secondary to innervation defects
Ablation of DG in primary hippocampal culture has revealed that DG is not necessary for GABAergic synapse formation and for clustering of main GABAergic PSD proteins, including GABAARs (Lévi et al., 2002). Yet, involvement of the DGC in clustering of GABAergic postsynaptic proteins was supported by several lines of evidence. Mdx mice, used as a DMD model because of their lack of full-length dystrophin, were shown to have reduced GABAAR (but not gephyrin) clustering in the hippocampus CA1 region (Knuesel et al., 1999). Overexpression of a shorter dystrophin construct in vivo rescued the decrease of GABAAR cluster density and size, adding to the notion that dystrophin loss directly caused GABAAR clustering defects (Vaillend et al., 2010). NL2 was shown to biochemically interact with dystrophin over the intracellular synaptic scaffolding molecule S-SCAM (Sumita et al., 2007). Furthermore, a functional connection between the DGC and NL2 is suggested by the observation that, in GABAAR α2 subunit KO mice, NL2 clustering is only compromised in dendritic but not in perisomatic areas (Panzanelli et al., 2011). The modest increase in GABAAR α2 subunit density and decrease in GABAAR α1 subunit size found in the present study do not correspond to the findings in mdx mice, in which both subunits cluster less efficiently than in wild-type mice (Knuesel et al., 1999; Vaillend et al., 2010). Rather, these alterations might reflect a subunit composition change because GABAAR γ2 subunit clusters were not affected by ablation of DG (except for a minute reduction in cluster size, which might be a reflection of reduced GABAAR α1 subunit cluster size). The finding that gephyrin clustering was unchanged in DG cKO mice further supports the conclusion that overall clustering of synaptic GABAAR subunits was not influenced by neuronal DG loss. The discrepancy between our results and published data from mdx mice might be explained by different roles of dystrophin isoforms at the GABAergic PSD. Short dystrophin isoforms still present in the mdx model might, by binding to DG, cause the reduction in synaptic GABAAR clustering. It is worth noting that the α2 subunit of GABAARs, which is localized preferentially at CCK-positive synapses (Nyíri et al., 2001), does not require the DGC or CCK-positive terminals for clustering. The DGC is thus likely involved in targeting the α2 subunit to synapses apposed to CCK-positive terminals, but clustering mechanisms seem to be DGC-independent. NL2 clustering was intact in DG cKO mice, apart from a slight decrease in cluster size. Therefore, the notion of the DGC as an obligatory stabilizer of postsynaptic NL2 clustering by mutual interaction with S-SCAM does not hold. Similarly, a role for the DGC in clustering the dystrophin-interacting protein synArfGEF at GABAergic synapses was suggested (Fukaya et al., 2011). Not excluding a contribution of the DGC to synArfGEF function by clustering additional signaling proteins, synArfGEF does not rely on the DGC to form clustered, presumably synaptic structures.
In the light of the dramatic changes in GABAergic innervation due to DG loss, an indirect presynaptic contribution to reduced postsynaptic clustering in dystrophin-deficient models should be considered. This hypothesis is supported also by the finding of reduced CCK-positive basket cell markers in mdx mice, suggesting that dystrophin plays a part in trans-synaptic signaling (Krasowska et al., 2014). The role of dystrophin in clustering signaling proteins at CCK-positive terminals is still unexplored but might include retrograde signaling by nitric oxide synthase. Resolution of conventional confocal laser scanning microscopy is not sufficient to conclusively answer whether the DGC is restricted to synapses from CCK-positive basket cells. Although apposition of DG to PV- and CCK-positive terminals was found with approximately equal frequency (Fig. 11E), the percentage of CCK-positive terminals apposed to DG was higher than that of PV-positive terminals (data not shown; Figs. 4C, 7A). Therefore, it seems likely that the DGC localizes preferentially postsynaptic to CCK-positive terminals to regulate synapse formation and function.
Basket cell type specificity of DG function implies specificity of trans-synaptic interaction with presynaptic binding partner
The selective dependence of the CCK-containing subtype of basket cells on neuronal DG for innervating target cells is a major finding of our study and has far-reaching implications. The DGC indeed acts as a trans-synaptic complex in central synapses, suggesting that presynaptic, rather than extracellular, binding partners enable DGC function in this context. Any such presynaptic adhesion molecule would have to be specifically localized at CCK-positive terminals. Interestingly, differential splicing of neurexins in PV- and CCK-expressing basket cells was recently reported (Fuccillo et al., 2015). Transcripts lacking neurexin1α alternative splice inserts 2 and 4, which prevent α-DG binding to LSM domains 2 and 6, respectively, were only found in CCK-positive basket cells (Sugita et al., 2001; Reissner et al., 2014; Fuccillo et al., 2015). This neurexin isoform specificity of basket cell subtypes would provide a mechanism for selective dependence of CCK-positive basket terminals on DG. However, we found terminals from CCK-positive basket cells to be intact in Dag1 T190M knock-in mice. DG containing the T190M mutation was found to lose neurexin binding capacity (Hara et al., 2011). This finding thus suggests that a novel presynaptic DG binding partner might be specifically localized at CCK-positive terminals. But because the diversity of neurexin isoforms was not considered in DG T190M binding assays, the possibility of a specific neurexin-DG trans-synaptic complex at CCK-positive terminals remains.
Continuous trans-synaptic signaling required for maintenance of CCK-positive terminals might reflect novel plasticity mechanism
Stopping trans-synaptic signaling mediated by the DGC by ablating DG in adulthood led to a decrease of CCK-positive terminals within weeks. This unexpected result implies that DG function goes beyond a potential role in validating newly formed synapses from CCK-positive basket cells. In addition to clustering signaling molecules at these synapses, our findings open the possibility that the DGC, by forming a trans-synaptic complex, is a direct target to regulate abundance of CCK-positive terminals. β-DG is a substrate of MMP-9 in a neuronal activity-dependent manner (Yamada et al., 2001; Kaczmarek et al., 2002; Michaluk et al., 2007). Cleavage of DG might therefore represent a physiological interneuron subtype-specific plasticity mechanism. In striking agreement with this hypothesis, CCK-positive terminals are selectively lost in a model of temporal lobe epilepsy (Wyeth et al., 2010).
Decreased inhibitory input to pyramidal cells in DG-ablated cells confirms functional significance of DG signaling for CCK-positive terminals
The possibility that loss of CCK-specific markers in DG cKO mice is only due to inability of terminals to differentiate was ruled out by the finding that DG-ablated pyramidal cells receive reduced inhibitory drive. Along with sIPSC frequency, amplitude was markedly reduced, possibly reflecting mistargeting of GABAARs in the absence of the DGC. Genesis of CCh-induced increase of inhibitory currents is not fully understood but involves Gad2-positive rather than PV-positive interneurons in the CA1 region (Nagode et al., 2014). Because the group of Gad2-expressing interneurons includes CCK-positive basket cells and CCh-induced currents are sensitive to depolarization-induced suppression of inhibition, our results add to the notion that CCK-positive basket cells play a crucial role in CCh-induced activity. Activity patterns elicited by CCh correlate with behaviorally relevant θ oscillations. Mechanisms of θ oscillation generation should thus be considered in future investigations of the etiology of intellectual disability associated with muscular dystrophies.
In conclusion, our investigation of the role of neuronal DG in GABAergic synapses has revealed a surprising interneuron type-specific function of DG in trans-synaptic signaling. It has shown that GABAergic postsynaptic diversity is functionally related to interneuron subtype heterogeneity and supports the emerging notion of a cell type-specific molecular code of synapse formation. Future studies will have to further characterize signaling and plasticity enabled by the DGC and delineate its behavioral consequences. Taking the interneuron-specific role of DG into consideration will help elucidate the mechanisms underlying intellectual disability observed in muscular dystrophies without developmental brain malformations.
Footnotes
This work was supported by University of Zurich Forschungskredit to S.F., Swiss National Science Foundation Grant 310030_146120 to J.-M.F., and Paul D. Wellstone Muscular Dystrophy Cooperative Research Center Grant 1U54NS053672 to K.P.C. K.P.C. is an investigator of the Howard Hughes Medical Institute. We thank Prof. Peter Scheiffele and Prof. Stephan Neuhauss for scientific discussion; and Dr. Tatjana Haenggi and Cornelia Schwerdel for genotyping.
The authors declare no competing financial interests.
References
- Bartos M, Elgueta C. Functional characteristics of parvalbumin- and cholecystokinin-expressing basket cells. J Physiol. 2012;590:669–681. doi: 10.1113/jphysiol.2011.226175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünig I, Suter A, Knuesel I, Lüscher B, Fritschy JM. GABAergic terminals are required for postsynaptic clustering of dystrophin but not of GABA(A) receptors and gephyrin. J Neurosci. 2002;22:4805–4813. doi: 10.1523/JNEUROSCI.22-12-04805.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinçer P, Balci B, Yuva Y, Talim B, Brockington M, Dinçel D, Torelli S, Brown S, Kale G, Haliloglu G, Gerçeker FO, Atalay RC, Yakicier C, Longman C, Muntoni F, Topaloglu H. A novel form of recessive limb girdle muscular dystrophy with mental retardation and abnormal expression of alpha-dystroglycan. Neuromuscul Disord. 2003;13:771–778. doi: 10.1016/S0960-8966(03)00161-5. [DOI] [PubMed] [Google Scholar]
- Fishell G, Rudy B. Mechanisms of inhibition within the telencephalon: “where the wild things are.”. Annu Rev Neurosci. 2011;34:535–567. doi: 10.1146/annurev-neuro-061010-113717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- Fuccillo MV, Földy C, Gökce Ö, Rothwell PE, Sun GL, Malenka RC, Südhof TC. Single-cell mRNA profiling reveals cell-type-specific expression of neurexin isoforms. Neuron. 2015;87:326–340. doi: 10.1016/j.neuron.2015.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukaya M, Kamata A, Hara Y, Tamaki H, Katsumata O, Ito N, Takeda S, Hata Y, Suzuki T, Watanabe M, Harvey RJ, Sakagami H. SynArfGEF is a guanine nucleotide exchange factor for Arf6 and localizes preferentially at post-synaptic specializations of inhibitory synapses. J Neurochem. 2011;116:1122–1137. doi: 10.1111/j.1471-4159.2010.07167.x. [DOI] [PubMed] [Google Scholar]
- Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B, Straub V, Robb S, Quinlivan R, Feng L, Jimenez-Mallebrera C, Mercuri E, Manzur AY, Kinali M, Torelli S, Brown SC, Sewry CA, Bushby K, Topaloglu H, North K, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130:2725–2735. doi: 10.1093/brain/awm212. [DOI] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 2006;44:611–621. doi: 10.1002/dvg.20256. [DOI] [PubMed] [Google Scholar]
- Hara Y, Balci-Hayta B, Yoshida-Moriguchi T, Kanagawa M, Beltrán-Valero de Bernabé D, Gündeşli H, Willer T, Satz JS, Crawford RW, Burden SJ, Kunz S, Oldstone MB, Accardi A, Talim B, Muntoni F, Topaloğlu H, Dinçer P, Campbell KP. A dystroglycan mutation associated with limb-girdle muscular dystrophy. N Engl J Med. 2011;364:939–946. doi: 10.1056/NEJMoa1006939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek L, Lapinska-Dzwonek J, Szymczak S. Matrix metalloproteinases in the adult brain physiology: a link between c-Fos, AP-1 and remodeling of neuronal connections? EMBO J. 2002;21:6643–6648. doi: 10.1093/emboj/cdf676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel I, Mastrocola M, Zuellig RA, Bornhauser B, Schaub MC, Fritschy JM. Short communication: altered synaptic clustering of GABAA receptors in mice lacking dystrophin (mdx mice) Eur J Neurosci. 1999;11:4457–4462. doi: 10.1046/j.1460-9568.1999.00887.x. [DOI] [PubMed] [Google Scholar]
- Krasowska E, Zablocki K, Górecki DC, Swinny JD. Aberrant location of inhibitory synaptic marker proteins in the hippocampus of dystrophin-deficient mice: implications for cognitive impairment in Duchenne muscular dystrophy. PLoS One. 2014;9:e108364. doi: 10.1371/journal.pone.0108364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévi S, Grady RM, Henry MD, Campbell KP, Sanes JR, Craig AM. Dystroglycan is selectively associated with inhibitory GABAergic synapses but is dispensable for their differentiation. J Neurosci. 2002;22:4274–4285. doi: 10.1523/JNEUROSCI.22-11-04274.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidov HG, Byers TJ, Watkins SC, Kunkel LM. Localization of dystrophin to postsynaptic regions of central nervous system cortical neurons. Nature. 1990;348:725–728. doi: 10.1038/348725a0. [DOI] [PubMed] [Google Scholar]
- McNally EM, Pytel P. Muscle diseases: the muscular dystrophies. Annu Rev Pathol. 2007;2:87–109. doi: 10.1146/annurev.pathol.2.010506.091936. [DOI] [PubMed] [Google Scholar]
- Michaluk P, Kolodziej L, Mioduszewska B, Wilczynski GM, Dzwonek J, Jaworski J, Gorecki DC, Ottersen OP, Kaczmarek L. Beta-dystroglycan as a target for MMP-9, in response to enhanced neuronal activity. J Biol Chem. 2007;282:16036–16041. doi: 10.1074/jbc.M700641200. [DOI] [PubMed] [Google Scholar]
- Nagode DA, Tang AH, Yang K, Alger BE. Optogenetic identification of an intrinsic cholinergically driven inhibitory oscillator sensitive to cannabinoids and opioids in hippocampal CA1. J Physiol. 2014;592:103–123. doi: 10.1113/jphysiol.2013.257428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notter T, Panzanelli P, Pfister S, Mircsof D, Fritschy JM. A protocol for concurrent high-quality immunohistochemical and biochemical analyses in adult mouse central nervous system. Eur J Neurosci. 2014;39:165–175. doi: 10.1111/ejn.12447. [DOI] [PubMed] [Google Scholar]
- Nyíri G, Freund TF, Somogyi P. Input-dependent synaptic targeting of alpha(2)-subunit-containing GABA(A) receptors in synapses of hippocampal pyramidal cells of the rat. Eur J Neurosci. 2001;13:428–442. doi: 10.1046/j.1460-9568.2001.01407.x. [DOI] [PubMed] [Google Scholar]
- Panzanelli P, Gunn BG, Schlatter MC, Benke D, Tyagarajan SK, Scheiffele P, Belelli D, Lambert JJ, Rudolph U, Fritschy JM. Distinct mechanisms regulate GABAA receptor and gephyrin clustering at perisomatic and axo-axonic synapses on CA1 pyramidal cells. J Physiol. 2011;589:4959–4980. doi: 10.1113/jphysiol.2011.216028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulopoulos A, Aramuni G, Meyer G, Soykan T, Hoon M, Papadopoulos T, Zhang M, Paarmann I, Fuchs C, Harvey K, Jedlicka P, Schwarzacher SW, Betz H, Harvey RJ, Brose N, Zhang W, Varoqueaux F. Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron. 2009;63:628–642. doi: 10.1016/j.neuron.2009.08.023. [DOI] [PubMed] [Google Scholar]
- Reissner C, Stahn J, Breuer D, Klose M, Pohlentz G, Mormann M, Missler M. Dystroglycan binding to alpha-neurexin competes with neurexophilin-1 and neuroligin in the brain. J Biol Chem. 2014;289:27585–27603. doi: 10.1074/jbc.M114.595413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satz JS, Ostendorf AP, Hou S, Turner A, Kusano H, Lee JC, Turk R, Nguyen H, Ross-Barta SE, Westra S, Hoshi T, Moore SA, Campbell KP. Distinct functions of glial and neuronal dystroglycan in the developing and adult mouse brain. J Neurosci. 2010;30:14560–14572. doi: 10.1523/JNEUROSCI.3247-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer MK, Varoqui H, Defamie N, Weihe E, Erickson JD. Molecular cloning and functional identification of mouse vesicular glutamate transporter 3 and its expression in subsets of novel excitatory neurons. J Biol Chem. 2002;277:50734–50748. doi: 10.1074/jbc.M206738200. [DOI] [PubMed] [Google Scholar]
- Sugita S, Saito F, Tang J, Satz J, Campbell K, Südhof TC. A stoichiometric complex of neurexins and dystroglycan in brain. J Cell Biol. 2001;154:435–445. doi: 10.1083/jcb.200105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumita K, Sato Y, Iida J, Kawata A, Hamano M, Hirabayashi S, Ohno K, Peles E, Hata Y. Synaptic scaffolding molecule (S-SCAM) membrane-associated guanylate kinase with inverted organization (MAGI)-2 is associated with cell adhesion molecules at inhibitory synapses in rat hippocampal neurons. J Neurochem. 2007;100:154–166. doi: 10.1111/j.1471-4159.2006.04170.x. [DOI] [PubMed] [Google Scholar]
- Tyagarajan SK, Fritschy JM. Gephyrin: a master regulator of neuronal function? Nat Rev Neurosci. 2014;15:141–156. doi: 10.1038/nrn3670. [DOI] [PubMed] [Google Scholar]
- Vaillend C, Perronnet C, Ros C, Gruszczynski C, Goyenvalle A, Laroche S, Danos O, Garcia L, Peltekian E. Rescue of a dystrophin-like protein by exon skipping in vivo restores GABAA-receptor clustering in the hippocampus of the mdx mouse. Mol Ther. 2010;18:1683–1688. doi: 10.1038/mt.2010.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite A, Brown SC, Blake DJ. The dystrophin-glycoprotein complex in brain development and disease. Trends Neurosci. 2012;35:487–496. doi: 10.1016/j.tins.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Wright KM, Lyon KA, Leung H, Leahy DJ, Ma L, Ginty DD. Dystroglycan organizes axon guidance cue localization and axonal pathfinding. Neuron. 2012;76:931–944. doi: 10.1016/j.neuron.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyeth MS, Zhang N, Mody I, Houser CR. Selective reduction of cholecystokinin-positive basket cell innervation in a model of temporal lobe epilepsy. J Neurosci. 2010;30:8993–9006. doi: 10.1523/JNEUROSCI.1183-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Saito F, Fukuta-Ohi H, Zhong D, Hase A, Arai K, Okuyama A, Maekawa R, Shimizu T, Matsumura K. Processing of beta-dystroglycan by matrix metalloproteinase disrupts the link between the extracellular matrix and cell membrane via the dystroglycan complex. Hum Mol Genet. 2001;10:1563–1569. doi: 10.1093/hmg/10.15.1563. [DOI] [PubMed] [Google Scholar]