

Abstract
Emerging evidence indicates that adipose stromal cells (ASC) are recruited to enhance cancer development. In this study, we examined the role these adipocyte progenitors play relating to intercellular communication in obesity-associated endometrial cancer. This is particularly relevant given that gap junctions have been implicated in tumor suppression. Examining the effects of ASCs on the transcriptome of endometrial epithelial cells (EEC) in an in vitro co-culture system revealed transcriptional repression of GJA1 (encoding the gap junction protein Cx43) and other genes related to intercellular communication. This repression was recapitulated in an obesity mouse model of endometrial cancer. Furthermore, inhibition of plasminogen activator inhibitor 1 (PAI-1), which was the most abundant ASC adipokine, led to reversal of cellular distribution associated with the GJA1 repression profile, suggesting that PAI-1 may mediate actions of ASC on transcriptional regulation in EEC. In an endometrial cancer cohort (n=141), DNA hypermethylation of GJA1 and related loci TJP2 and PRKCA was observed in primary endometrial endometrioid tumors and was associated with obesity. Pharmacologic reversal of DNA methylation enhanced gap junction intercellular communication and cell-cell interactions in vitro. Restoring Cx43 expression in endometrial cancer cells reduced cellular migration; conversely, depletion of Cx43 increased cell migration in immortalized normal EEC. Our data suggest that persistent repression by ASC adipokines leads to promoter hypermethylation of GJA1 and related genes in the endometrium, triggering long-term silencing of these loci in endometrial tumors of obese patients.
INTRODUCTION
The risk of developing endometrial cancer, the most common gynecologic malignancy in the United States, is associated with age, estrogen exposure, and obesity (1,2). The rise of obesity is expected to lead to further increase in the incidence of endometrial cancer. Recent studies show that while endometrial cancer is associated with old age (i.e., >60 years), this profile characteristic is changing with increasing incidence in obese women at a younger age (3). Obesity-associated endometrial tumors fall into the endometrioid endometrial cancer Type I, which is the most common histologic type of endometrial cancer (4). Type I lesions are characterized by well and moderately differentiated endometrioid histology, early stage, and favorable prognosis. Obesity is also a risk factor for other malignancies as well, including breast and colon cancers (5,6). Adipose stromal cells (ASCs), which are adipocyte progenitors within fatty tissue, have been implicated in the development of these cancers (7,8). While released factors from abdominal fat can be transported systemically, identification of circulating ASCs in obese subjects and recruitment of these cells by tumor-produced chemokines suggest that ASCs are trafficked to target tissue sites and embedded within the tumor microenvironment (9–12).
The tumor microenvironment is composed of complex cell types, including cancer-associated fibroblasts and leukocytes, which contribute to malignant development and progression (13,14). More recently, ASCs and adipocytes in the tumor microenvironment have been implicated as promoters of tumor progression (15,16). These cells produce hormones and cytokines that stimulate the growth of many tumor types, including endometrial cancer (7,17). Cancer-associated adipocytes may also contribute to sustenance of cancer cells by providing an energy source through lipolysis, supporting their growth (18). The interaction between the stromal compartment and cancer cells plays a role in regulating gene transcription during tumorigenesis (19). Persistent stimulation by tumor microenvironmental factors has long-term effect on gene repression through epigenetic mechanisms such as promoter CpG island hypermethylation (20,21).
Epigenetic repression frequently takes place in tumor-suppressor genes, including those that mediate or regulate the gap-junction intercellular communication (GJIC) (22,23). The gap junction (GJ) gene family, which encodes connexin (Cx) proteins that form the gap junction channels, and their structural and kinase modulators (herein collectively referred to as GJ-associated loci) control the transfer of metabolites and ions between adjacent cells (24,25). Gap junctions are also upregulated during normal invasive processes (e.g. oocyte implantation), as well as extravasation and distant metastatic lesion formation (26,27). This dichotomy in gap junction function reflects the complex cellular interactions occurring during tumor development and later in aggressive disease establishing metastatic growth.
In this study, we first examined the role of ASCs in repressing GJ-associated gene expression in immortalized endometrial cells. We then determined whether this repression can be recapitulated in a mouse model of obese endometrial cancer, and whether silencing is marked by DNA methylation in primary endometrial tumors of obese patients. Potential epigenetic regulation of cell-cell communication and interactions was first examined generally by DNA demethylation treatment of cancer cells. Then, cellular growth and motility effects by specific knockdown or reactivation of the major connexin Cx43 (encoded by methylation-target GJA1) in endometrial immortalized epithelial and cancer cells were examined, respectively. Our studies suggest that epigenetic repression of GJ-associated loci is involved in stunting gap junction intercellular communication and development of obesity-associated endometrial cancer.
Materials and Methods
Detailed description of the experimental procedures and reagents used in this study can also be found in Supplemental Experimental Procedures.
In Vitro Co-culture Exposure Model and Cell Lines
Adipose stromal cells (ASCs) were obtained from the Coriell Institute (Camden, NJ). The cells were isolated from fat tissue during subject’s abdomen and waist ante-mortem elective cosmetic tumescent liposuction. ASCs were cultured in non-differentiating media (DMEM supplemented with 0.5% FBS/0.2% BSA) and grown on fibronectin-coated culture dishes. For co-culture, ASCs were seeded on a fibronectin-layered insert of a Boyden chamber. In the bottom well, endometrial cells were seeded for co-culture experiment. Control wells contained the fibronectin-layered insert without ASCs. Co-cultures were carried out in duplicates for three weeks, followed by lysis of endometrial cells for RNA isolation. Ishikawa and HEC-1A cells were obtained from Millipore Sigma (St. Louis, MO) and ATCC (Manassas, VA), respectively. Ishikawa was derived from a well-differentiated human endometrial adenocarcinoma, whereas HEC-1A was derived from a Stage IA moderately differentiated adenocarcinoma (28,29). Both these cancer cell lines represent Type I endometrial cancer (28–30). Immortalized EM-E6/E7/TERT-1 cells, established and characterized by Kyo et al, are non-transformed, non-tumorigenic endometrial epithelial cell line (31). Cell line authentication was carried out by STR analysis at ATCC. Cells were used within 3 passages after thawing from frozen stock. Cells were routinely checked for mycoplasma while in use by the mycoplasma PCR detection kit (ABM, Richmond, BC).
RNA-seq analysis
RNA was subjected to cDNA conversion and sequencing library construction, and next generation sequencing was carried out using the HiSeq2000 platform (Illumina). Additional sequencing run information is reported in Supplementary Fig. 1.
Obesity Mouse Model for Endometrial Cancer
The studies were approved by the Institutional Animal Care and Use Committee at the Medical College of Wisconsin (AUA2927). Breeding pairs (B6.129-Ptentm1Rps, 01XH3) were obtained from Mouse Models of Human Cancers Consortium at National Cancer Institute (NCI, Frederick, MD). Mice were housed at the Medical College of Wisconsin Biomedical Resource Center facility at an appropriate temperature and a standard light-dark cycle. The mouse colony was established by breeding wild-type female mice with heterozygous Pten+/− male mice. Pten+/− female offspring were weaned at age 21 days. Ear or tail biopsies were obtained for genotyping using the primers specified by the website of Mouse Models of Human Cancers Consortium at the NCI. Female Pten+/− and wild-type mice at 3–4 weeks old were fed diets either with 16.4% energy from fat (standard AIN-93G diet) or with 63.7% fat to induce obesity (n=9, each group), prepared by TestDiet (St. Louis, MO). The mice were kept on these diets until they were sacrificed at 12, 20, or 28 weeks of age. All mice were weighed once per week to determine changes in body weight development from weaning to euthanization. One uterine horn was fresh frozen for molecular analyses, the other formalin fixed overnight. Changes in endometrial morphology were assessed from histopathologic sections processed in paraffin blocks, following the guidelines set by Fyles et al and examined by a board-certified pathologist blinded to treatment group and genotype (32).
Parachute Assay of Inter-cellular Communication
Gap junction intercellular communication (GJIC) was measured using intercellular transfer of the fluorescent dye calcein, which is permeable via gap junctions. Assays were performed in culture media supplemented with charcoal-stripped FBS (10%). Donor cells were incubated with the dye for 20 minutes at room temperature. Inside the cell, calcein is cleaved by non-specific esterases into calcein, making it impermeable to diffusion through the cell membrane. Recipient cells are grown to confluence. Calcein-labeled donor cells were then dropped (‘parachuted’) onto the recipient cell layer, and calcein transfer between donor and recipient cells was observed with fluorescent microscopic imaging. Data were expressed as # of fluorescent recipient cells/# of donor cells for each condition. Experimental conditions were performed in triplicates. Fluorescent images of 10–15 fields per well were captured on an Operetta automated microscope (Perkin Elmer). A program (Perkin Elmer) that allowed identification of all cells on the plate (from phase contrast image), as well as original donors (100±50 per well), and dye-filled recipients due to calcein transfer over time.
Atomic Force Microscope Analysis of Inter-cellular Communication
Nanomechanical properties of live adherent HEC-1A cells were analyzed in vitro with a Nanoscope Catalyst (Bruker) atomic force microscope mounted on a Nikon Ti inverted epifluorescent microscope. PeakForce Quantitative Nanomechanical Mapping (PF-QNM) mode of in-liquid imaging with SCANASYST-AIR probes (Bruker) was applied. Square fields from 30×30 µm to 50×50 µm corresponding to the electronic resolution from 0.19 to 0.47 µm/pixel were scanned. To calculate nanomechanical parameters of the cells, we analyzed the retrace images with Nanoscope Analysis software v.1.7 (Bruker). The Young’s modulus was calculated as a measure of cell elasticity. A force needed to separate a tip from a cell was determined to assess cell adhesion to a tip. The Sneddon model of tip-object interactions was applied in the calculations. Elasticity and adhesion mode values were calculated for each cell from corresponding distribution histograms of image pixel values and used in all the downstream statistical evaluations. Quantification of adhesion parameters governing HEC-1A cell-cell interactions was performed with the single cell force spectroscopy AFM as described (33) and detailed further in Supplementary Information.
DNA Methylation Pyrosequencing Analysis
All human tissue samples have been obtained under IRB approval. The studies were conducted with obtained written informed consent from the patients, and the studies were conducted in accordance with recognized ethical guidelines (e.g., Declaration of Helsinki, CIOMS, Belmont Report, U.S. Common Rule). Pyrosequencing of paraffin embedded tissue of endometrial tumors (n=141), adjacent normal tissue (n=43) and uninvolved normal uterine control (n=11) was performed as described previously (34). Clinical characteristics of this cohort are detailed under Supplementary Information. Briefly, tumor sites were macro-dissected from paraffin embedded tumor samples, and DNA was isolated. DNA (500 ng per sample) was processed for bisulfate conversion using the EZ DNA Methylation™ kit (Zymo Research, Irvine, CA). Primers used to amplify specific CpG island regions are listed in supplementary information. Methylated CpG sites were detected by the PyroMark Q96 MD system. The methylation percentage of each interrogated CpG site was calculated and visualized using MultiExperiment Viewer v4.8 (Dana-Farber Cancer Institute, Boston, MA) and GraphPad Prism® (GraphPad Software).
Statistical Analysis
The Shapiro-Wilk normality test was performed to assess parametric distribution of data. For statistical analysis t-test was used after normality of data was ensured. In cases were normality test failed, Mann-Whitney rank sum test was used as a nonparametric analysis method. p values <0.05 was considered statistically significant.
RESULTS
Repression of GJ-associated Loci via ASC Paracrine Actions in vitro
As an in vitro model of obesity for endometrial cancer, immortalized non-tumorigenic EM-E6/E7/hTERT-1 cells (referred to hereinafter as EME6/7t; (31)) were exposed to adiposity derived ASCs in a Boyden chamber for 21 days. Control cells were incubated in the Boyden chamber without ASCs. RNA-seq was used to determine the co-culture effects on the transcriptomes of ASC-exposed and control EME6/7t cells (Fig. 1A, Supplementary Fig. S1A). Robust differential expression was observed in EME6/7t cells exposed to ASCs compared to control, and high correlation in expression was observed between the biological replicates of each sample (Supplementary Figs. S1B and S1C). We identified 3811 differentially expressed genes between exposed and control cells, of which 2182 were induced and 1629 were repressed due to ASC exposure (p<0.05; Supplementary Fig. S1D and Table S1). On the other hand, ASC-exposed Ishikawa cancer cell line only showed 246 genes were differentially expressed in Ishikawa cells, of which 113 genes overlapped with EME6/7t (Supplementary Fig. S1D and S1E). The data indicate that EME6/7t cells may be more susceptible to ASC-mediated effects than the transformed Ishikawa cancer cells. Differentially expressed genes in EME6/7t cells (identified by a threshold of 1.9-fold difference in expression) encoding proteins involved in intercellular junctions or invasion/migration pathways (Fig. 1B and Supplementary Table S2). A number of genes suppressed in the ASC-exposed cells belonging to these pathways were significantly enriched by pathway analysis and formed a protein-protein network that is involved in mediating cellular interactions, including gap junction communication (p <0.001; Fig. 1C and D, respectively). These included gap junction (GJ) genes, encoding connexins (Cx), and GJ-associated loci, which encode structural scaffolding (e.g., tight junction proteins) and kinase modulators of gap junctions (see the model in Fig. 1E). For validation, RT-PCR of GJs and gap junction regulators, was performed using the EME6/7t cells co-cultured with and without ASCs. Among these genes tested, GJA1 (which encodes Cx43) showed the most prominent expression in EME6/7t cells, and its expression was reduced in ASC-exposed cells (Fig. 1F). In addition, the expression of kinases involved in gap junction modulation (i.e., MAPK1, MAPK3, PRKACA and PRKCA) and the tight-junction protein TJP1 were also suppressed in EME6/7t cells exposed to ASCs (Fig. 1F).
Figure 1. Gap junction genes are repressed in endometrial epithelial cells co-cultured with ASCs.
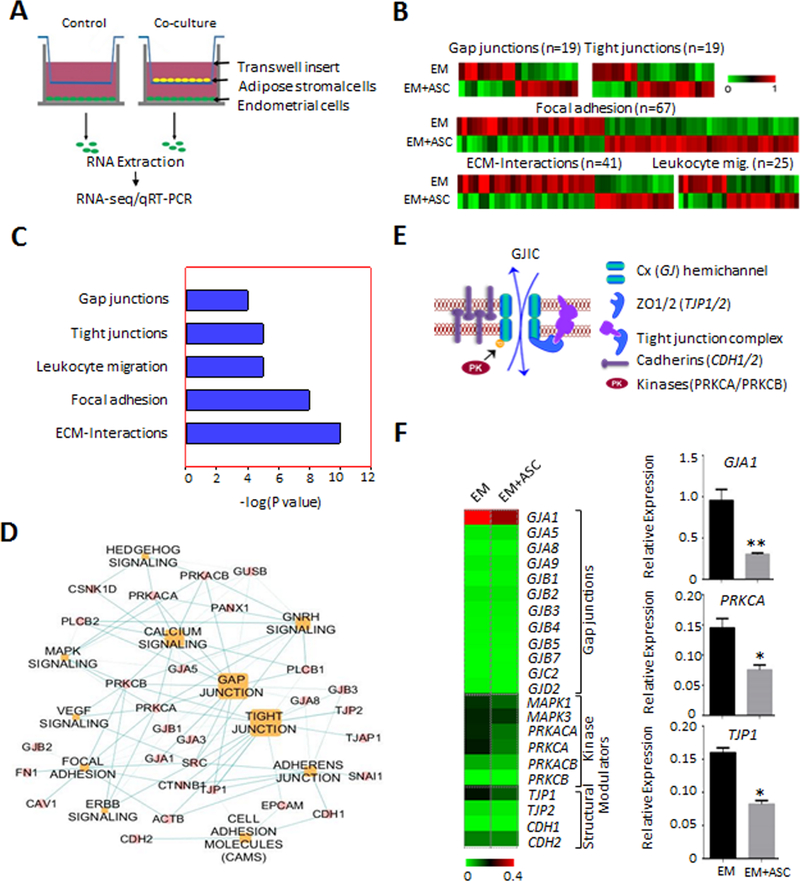
A, Diagram for the co-culture scheme of EME6/7t immortalized endometrial cells with ASCs, with ASCs plated in the transwell insert of a Boyden chamber and EME6/7t cells plated on the bottom of the well. Co-cultures and control (i.e., EME6/7t cells with no ASCs on the insert) were performed in duplicates. After three week co-culturing, EM-E6/7t cells were subjected to RNA extraction followed by RNA-seq analysis. B-D, Differential expression in cell communication and migration genes, including gap junction genes is observed (B). Pathway analysis reveal significant enrichment for gap junction, tight junctions, leukocyte endothelial migration, focal adhesion and ECM interaction genes, all of which play a role in cell-cell gap junctional coupling or are processes affected by this coupling (C). A gene network of the gap junction pathway is shown (D). E, A model for gap junction coupling is shown. It involves GJIC through formation of gap junction connexin channels between contiguous cells. Gap junction activities were regulated by kinases (such as PRKCB) as well as adhesion proteins (e.g., cadherins and tight junction proteins), facilitating formation of membrane gap junction plaques for efficient cell-cell communication. F, Validation of RNA-seq by RT-PCR reveals that these gap junction modulators are differentially repressed in EME6/7t cell exposed to ASCs. Repression of GJA1, encoding Cx43, the major connexin, is also repressed due to ASC exposure.
To identify the ASC paracrine factors acting on EME6/7t cells, we performed multiplexing assays for a panel of cytokines and adipokines. The major factors detected in conditioned media of ASCs were PAI-1, Adipsin, MCP-1, IL-6 and HGF (Fig. 2A). We chose PAI-1, the most abundant secreted factor determined in our assay, to evaluate its effect on the expression of GJ-associated genes in EME6/7t cells (Fig. 2A). These cells were treated with and without ASC conditioned media and with the ASC conditioned media in the presence of PAI-1 inhibitor tiplaxtinin for 24 hr and then subjected to single-cell expression analysis by microfluidic PCR. Expression profiling revealed 20 genes that were differentially expressed due to conditioned media and PAI-1 inhibition (Fig. 2B). t-distributed stochastic neighbor embedding (t-SNE) nonlinear dimensionality reduction analysis was used to cluster cells based on their expression profiles (Fig. 2B and 2C). Six subpopulations (a-f) were identified with unique expression profiles of these GJ-associated genes in treated and control cells (Fig. 2D). Subpopulations a-c showed higher coordinated expression levels of the abundant GJA1 and other GJ-associated genes, including GJB2, PRKACA and CDH2, while subpopulations d-f had lower expression levels of GJA1 and nine other genes GJA9, GJB1, GJB3, GJB5, GJB7, GJC2, TJP2, MAPK1, and PRKCB (Fig. 2D). The cell numbers of “GJA1-high” subpopulations a-c representing the majority (76%) of control cells were drastically reduced to 18% in cells exposed to conditioned media (Fig. 2E). In general, the cell composition shifted to “GJA1-low” subpopulations d-f after the exposure (Fig. 2E). This change of population dynamics was blocked by the addition of tiplaxtinin, partially restoring the two “GJA1-high” subpopulations a and b (Fig. 2E). This result suggests that the adipokine factor PAI-1 plays a role in suppressing the expression of GJA1 and other related loci in the majority of EME6/7t cell population. These single-cell data also corroborates the finding of bulk RNA-seq that obesity-derived ASCs have paracrine influences on suppressing expression of GJ-associated genes in endometrial epithelial cells.
Figure 2. PAI-1 secreted from adiposity derived ASCs modulates GJ-associated loci in EME6/7t cells.
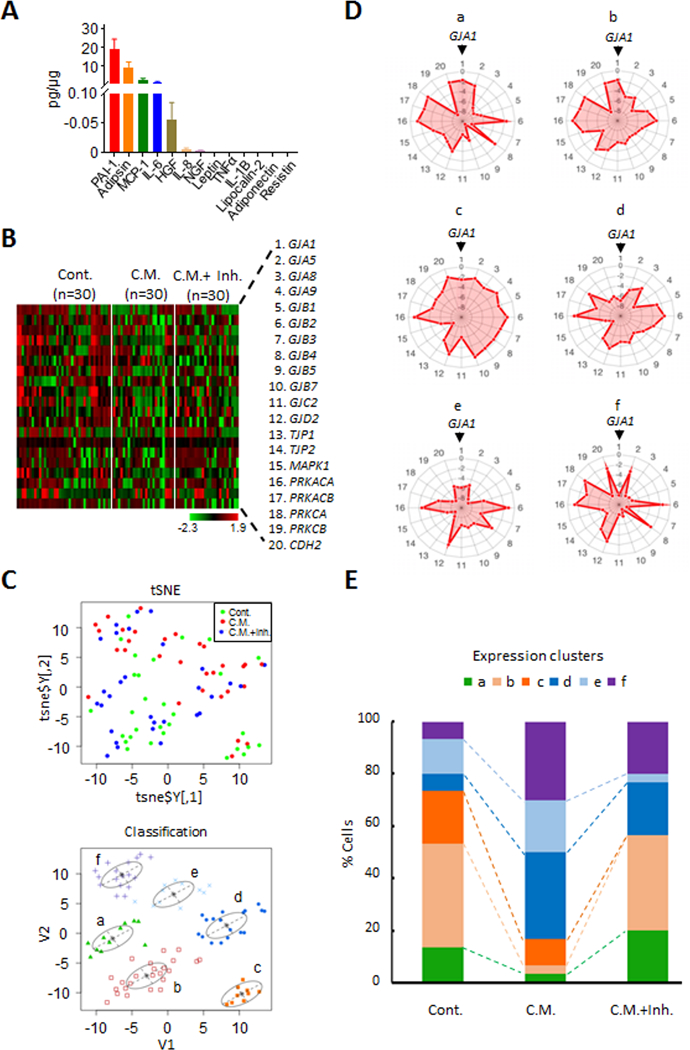
A, FlexMap multiplexing assays was used to determine secreted adipokine and cytokine factors from obesity derived ASCs after culturing for 48-hrs. B, Because PAI-1 was the most abundant factor in the 48-hr ASC conditioned media, EME6/7t cells were exposed to the conditioned media in the presence or absence of the PAI-1 inhibitor tiplaxtinin (50 μM). Treatments were carried out for 24-hrs. The cells were then subjected to single-cell isolation and single-cell microfluidic PCR expression analysis, and heat maps shown demonstrating the differential expression patterns of control cells, cells exposed to conditioned media alone, and cells exposed to conditioned media with tiplaxtinin. C-E, Single-cell expression analysis was performed to determine the effects of tiplaxtinin treatment on the expression of EME6/7t exposed to ASC conditioned media. tSNE analysis of expression profiles of control EME6/7t and cells exposed to conditioned media, with or without tiplaxtinin (C, top panel) reveals 6 clusters (C, tSNE lower panel, and D as shown by radar plots). In the radar plot, the genes from the expression heat map (numbered 1–20) in panel B are distributed around the circumference (starting with GJA1 on top), with the level of expression designated by the pink area emanating from the center of the circle (D). Histograms show these clusters corresponding to each treatment condition (E). Notably, clusters a and b present in control cells are drastically reduced in cells exposed to conditioned media (E). Treatment with tiplaxtinin restores these two clusters (E).
Repression of GJ-associated Loci in an Obesity Endometrial Cancer Animal Model
Since the repression of candidate GJ-associated genes was observed in this transient exposure system in vitro, we sought to solidify the role of obesity in transcription repression of GJ-associated loci in an in vivo model. Obesity was induced in an endometrial cancer mouse model with a Pten−/+ deletion fed with high-fat diet (32). Pten−/+ (n=9) and wildtype (n=9) mice fed with this diet exhibited an increase in body weight and associated phenotype such as serum estradiol and leptin levels (Supplementary Fig. S2A-E). Mutant mice fed high fat diet developed earlier endometrial atypia (as early as 12 weeks) and hyperplasia (20 weeks) than the control-fed mice (Fig. 3A & B). By 28 weeks, endometrial carcinomas were observed in both control- and high fat-fed Pten−/+ mice (Fig 3A). Furthermore, endometrium of mice fed with high-fat diet exhibited higher Ki67 proliferation levels than control-fed mice (Fig. 3C & D). qRT-PCR analysis of GJ-associated genes showed a significant decrease in PRKCB (p<0.01), PRKCA (p<0.05), TJP1 (p<0.01), TJP2 (p<0.05), GJA1 (p<0.01), and GJC2 (p<0.01) in lesions of the high-fat Pten−/+ group (n=9) compared to the wildtype group (n=9), but this difference was not observed in the control-fed control group (Fig. 3E). Interestingly, CDH1 was the only gene we studied that showed a significant decrease in Pten−/+ mice independent of the diet they were on. Collectively, these animal studies provide crucial evidence toward establishing a link between obesity and the silencing of key GJ-associated genes in endometrial cancer.
Figure 3. Gap junction modulators are repressed in an obesity mouse model of endometrial cancer.
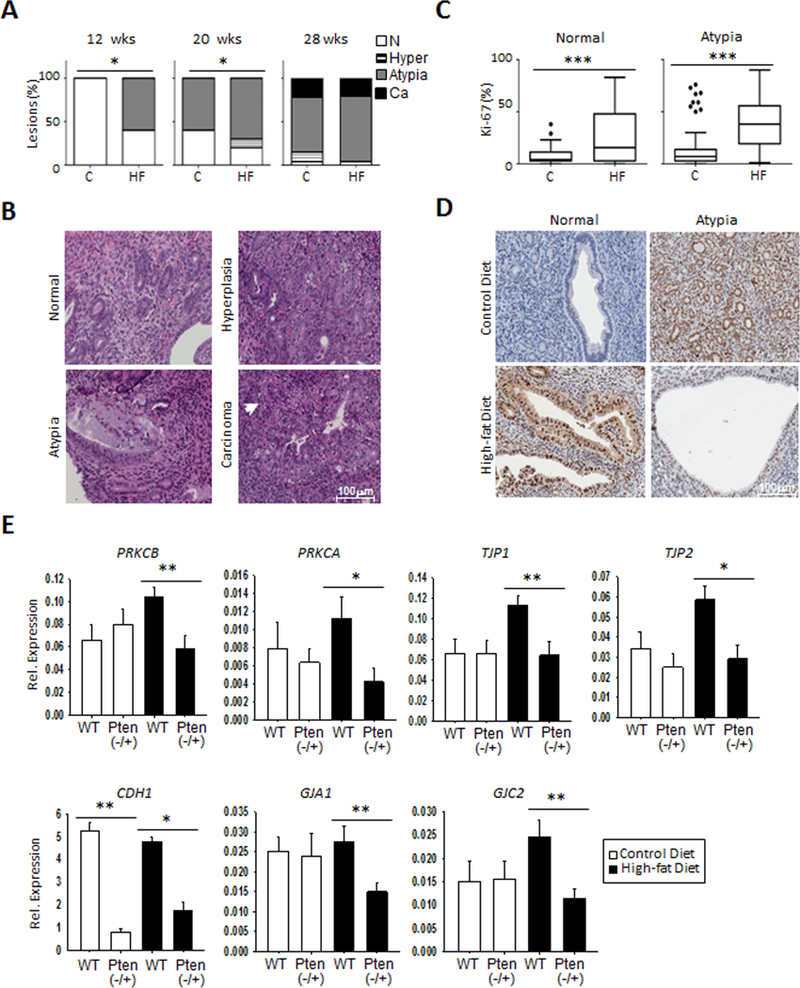
A-D, High fat diet leads to obesity, as shown in Figure S2A; increased initiation of preneoplastic lesions at 20 and 28 weeks in the Pten−/+ heterozygous mice, but not in mice fed control diet (A-B), and higher levels of the proliferative marker Ki67 are observed (C-D). The percentage of atypia is elevated as early as 12 weeks on the high fat diet (HF), and remains higher in the HF group at 28 weeks. Hyperplasia is also more evident on the HF mice at 20 weeks, but both groups show similar carcinoma development at that time point (A). Representative images of normal endometrial tissue and those with hyperplasia, atypia, and carcinoma is shown (B). Increased Ki67 is observed for the high fat diet group in normal and endometrial tissue with atypia (C). Representative images of Ki67 staining are shown (D). E, RNA expression analysis by qRT-PCR reveals suppression of GJ-associated genes in Pten−/+ mice compared to wildtype (n=9), but only in the HF group. Such differences are absent in the control diet. *p<0.05; **p<0.01. The notable exception was CDH1, which shows similar decreases in Pten−/+ mice in both control and HF diets.
Epigenetic Repression of Candidate GJ-associated Loci is Preferentially Observed in Endometrial Tumors Associated with Obesity
We then assessed profiles of promoter CpG island methylation in the GJ-associated loci in our endometrial methylome dataset from endometrioid endometrial carcinoma tumors obtained previously by MBDCap-seq analysis (n=67 primary tumors and 10 controls; Supplementary Fig. S3A; (35)). Methylation in CpG island cores, as well as in CpG island shores (defined as 2Kb upstream and downstream of the core island) has been associated with gene silencing (36). While hypermethylation was frequently observed in CpG island cores (e.g., PRKCB, GJC2 and TJP2), shore methylation also occurred in their flanking regions (e.g., left shores of PRKCA and MAPK3). Promoter CpG islands of PRKACA, TJP1 and CDH1 showed a slight increase in DNA methylation in some tumors compared to controls (Supplementary Fig. S3A). An increase in methylation was observed in non-CpG island (DNA with a G + C content <55%) promoters of GJA1, GJB4, GJB5 and GJC2 (Supplementary Fig. S3A). Non-CpG island promoter methylation had also been implicated in gene silencing (37). Analysis of The Cancer Genome Atlas (TCGA) endometrial cancer cohort confirmed that DNA hypermethylation of the six aforementioned promoter CpG islands (PRKCA, PRKCB, MAPK3, TJP1, TJP2 and CDH1) and three non-CpG island loci (GJA1, GJB5 and GJC2) was associated with reduced expression (Supplementary Figs. S3B and C).
Pyrosequencing analysis was then conducted to verify promoter methylation states in the major connexin-encoding gene GJA1 and three GJ-associated loci (PRKCA, PRKCB and TJP2) in primary endometrioid endometrial carcinoma (type I) tumors from a cohort of endometrial cancer patients (n=141; clinical characteristics in Supplementary Table S3). These loci exhibited high levels of DNA methylation in their promoter CpG islands in primary endometrial tumors relative to the control endometrium group (Fig. 4A; see also Supplementary Fig. S3A). Promoter hypermethylation was observed in the primary endometrial tumors compared to normal tissue adjacent to tumors and uninvolved noncancerous controls (Fig. 4A and B). GJA1 promoter methylation was significantly higher in tumor compared to normal tissue (Fig. 4B; p<0.05). This hypermethylation event was also observed for PRKCA, PRKCB and TJP2 (Fig. 4B; p<0.001, p<0.01, p<0.001, respectively), although in the former this was only on the right shore. After stratifying the cohort based on three groups - morbidly obese (BMI>35), obese (BMI 30–35), and non-obese (BMI<30), we found higher methylation levels of the PRKCA promoter in the obese group compared to those of the non-obese group (p<0.01; Fig. 4C). GJA1, PRKCB and TJP2 had no significant differences between the groups (Fig. 4C). To determine whether other factors may influence the methylation profile in the obesity groups, we further selected the patients that presented with comorbidity (e.g., diabetes) from each of the non-obese, obese and morbidly obese groups. In this selected cohort, we found higher promoter methylation levels in the morbidly obese group compared to non-obese patients for GJA1, PRKCA, and TJP2 (p<0.01, p<0.05, and p<0.01, respectively; Fig. 4D). For GJA1 and PRKCA, this increase in methylation level was also evident in the obese group in (p<0.01 and p<0.001, respectively; Fig. 4D, upper panel), whereas PRKCB showed no significant differences in any of the groups, even in this selected subset. These changes were starker between the non-obesity and the obesity groups when GJA1, PRKCA and TJP2 methylation profiles were compared in various combinations (p<0.001; Fig. 4D, lower panel). Taken together, these epigenetically repressed GJ-associated loci can be useful biomarkers to study pathophysiology of obesity-associated endometrial cancer.
Figure 4. Elevated promoter methylation of GJA1 and GJ-associated loci in primary endometrial tumors is associated with obesity.
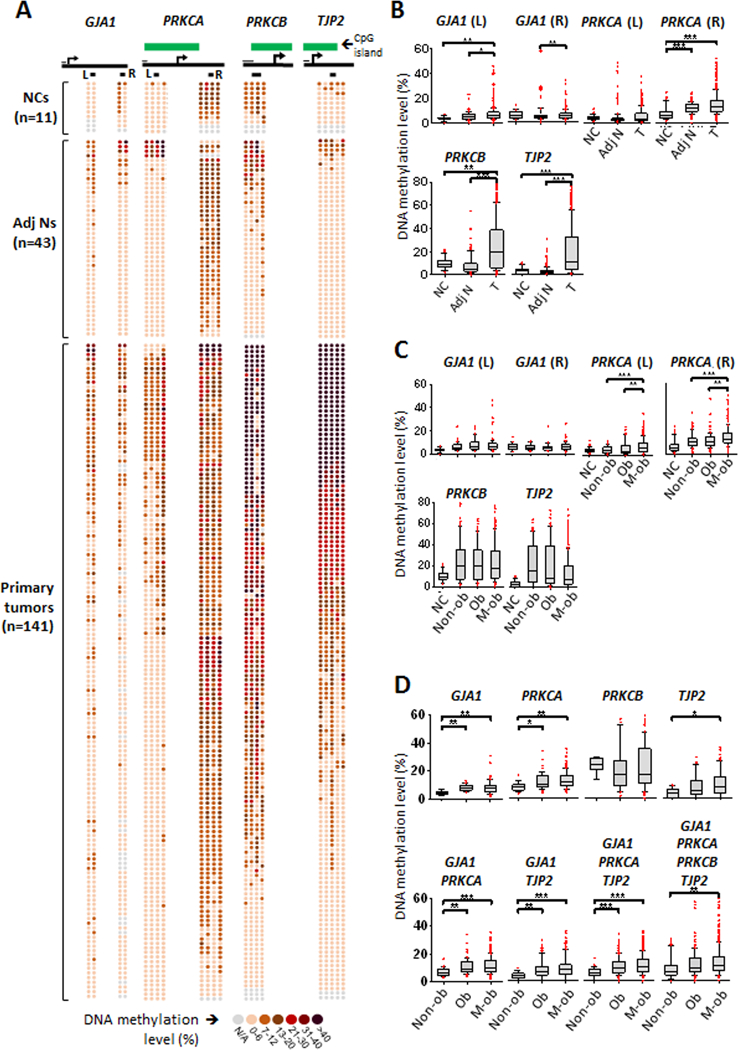
A-B, Hypermethylation of specific CpG sites for GJA1, PRKCA, PRKCB and TJP2 is observed in primary endometrial tumors from young, obese patients compared to normal endometrial tissue. Pyrosequecing of methylated CpG sites (circles) is shown for each sample (n=141); darker circles reflect higher methylation % (A). Green box designates CpG island. Under the CpG island box, gene body is represented as a thick solid black line with arrow indicating transcription start site (A). Short thin black line over the gene body represents scale of 100 bp, and thicker short black lines underneath the gene body indicates interrogated sites (A). For GJA1 and PRKCA, 2 sites were interrogated, whereas for PRKCB and TJP2 one site was interrogated based on the methylome patterns in Supplementary Fig. S3A. Box plots showing median expression of DNA methylation suggest increased DNA methylation in tumor compared for normal tissue (B). This pattern, uncovered by pyrosequencing, is consistent with the methylome shown in Supplementary Figure S3A. C, Promoter site methylation is interrogated for GJA1 (L) (non-cancerous n=16, non-obese n=48, obese n=42, morbidly obese n=118); GJA1 (R) (non-cancerous n=20, non-obese n=36, obese n=34, morbidly obese n=98); PRKCA (L) (non-cancerous n=4., non-obese n=88, obese n=64, morbidly obese n=236); PRKCA (R) (non-cancerous n=36, non-obese n=88, obese n=64, morbidly obese n=236); PRKCB (non-cancerous n=35, non-obese n=92, obese n=84, morbidly obese n=227); and TJP2 (non-cancerous n=45, non-obese n=118, obese n=105, morbidly obese n=292). Obese (BMI 30–35) and morbidly obese (BMI>35) patients exhibit significantly higher levels of methylation for PRKCA (p<0.01) compared to non-obese patients (BMI<30). Nonsignificant increases in median methylation are observed for GJA1, PRKCB, and TJP2. D, CpG Sites are interrogated for selected patients with co-morbidity for obesity (see Table S3) for GJA1 (L) (non-obese n=4, obese n=14; morbidly obese n=40); GJA1 (R) (non-obese n=4, obese n=8; morbidly obese n=26); PRKCA (L) (non-obese n=8, obese n=28, morbidly obese n=80); PRKCA (R) (non-obese n=8, obese n=28, morbidly obese n=80); PRKCB (non-obese n=8, obese n=28, morbidly obese n=80); and TJP2 (non-obese n=10; obese n=35; morbidly obese n=100). Significant methylation level increases in the morbidly obese group compared to non-obese group are observed for GJA1 (p<0.01), PRKCA (p<0.05) and TJP2 (p<0.01). Significant methylation level increases in the obese group compared to non-obese group are observed for GJA1 (p<0.01) and PRKCA (p<0.001). Increased methylation is observed in both the obese and morbidly obese (compared to non-obese) groups when different combinations of the profiles for GJA1, PRKCA and TJP2 are used (p<0.001).
DNA Demethylation Restores GJ-associated Loci expression, Gap-junction Intercellular Communication (GJIC) and Cell-cell Adhesiveness
To examine whether epigenetic actions are involved in deregulating gap junction activities, we initially determined the effect of demethylation on gene re-expression by treating endometrial cancer cell lines (HEC-1A and Ishikawa) and control EME6/7t cells with 2.5 and 5 µM 5-aza-2′-deoxycytidine (DAC), a general DNA demethylator (see an example in Supplementary Fig. S4A; (38)). Reactivation of GJB1, GJB3, GJB4, GJB5, GJB7, and GJC2, was observed mainly in HEC-1A cells (Supplementary Fig. S4B). Partial re-expression of kinase and structural modulators (PRKACA, PRKCB, TJP2, PRKACB, MAPK3, CDH1, and TJP1) with promoter CpG islands was also observed in these cells (Supplementary Fig. S4B). Re-expression of those loci without promoter CpG islands may be attributed to an effects of DAC on chromatin openness of inactive promoters (38), or secondary effects due to demethylation of regulatory genes. Consistent with this, The re-expression of some of these genes was affected to a lesser degree in Ishikawa cells (Supplementary Fig. S4B). This demethylating treatment had no obvious effect of gene reactivation in control EME6/7t cells.
To determine phenotypic effects of reactivated GJ-associated genes, we tested the homotypic gap junction intercellular communication (GJIC) of DAC treated and untreated cells using the ‘parachuting’ assay. Donor cells labeled with the gap junction permeable dye calcein were dropped (parachuted) on a confluent layer of recipient cells (Fig. 5A). GJIC was measured through the detection of calcein transfer from donor to acceptor cells (i.e., ratio of acceptor/donor cells). Without DAC treatment, the level of GJIC in EME6/7t cells was 3-fold higher than the endometrial cancer cells, consistent with the suppression of gap junction coupling in cancer (Fig. 5B). DAC treatment led to a significantly robust increase in GJIC in HEC-1A (p<0.001), and to a lesser, yet significant (p<0.01), increase in Ishikawa cells (Fig. 5B and C). We also determined whether DAC had an effect on the growth and motility in these cancer cells. Reduction in motility and growth, was observed in both DAC-treated HEC-1A and Ishikawa cells (p<0.001 and p<0.01, respectively; Supplementary Fig. S4C).
Figure 5. Treatment with demethylation agent DAC restores GJIC and enhances cell-cell adhesion.
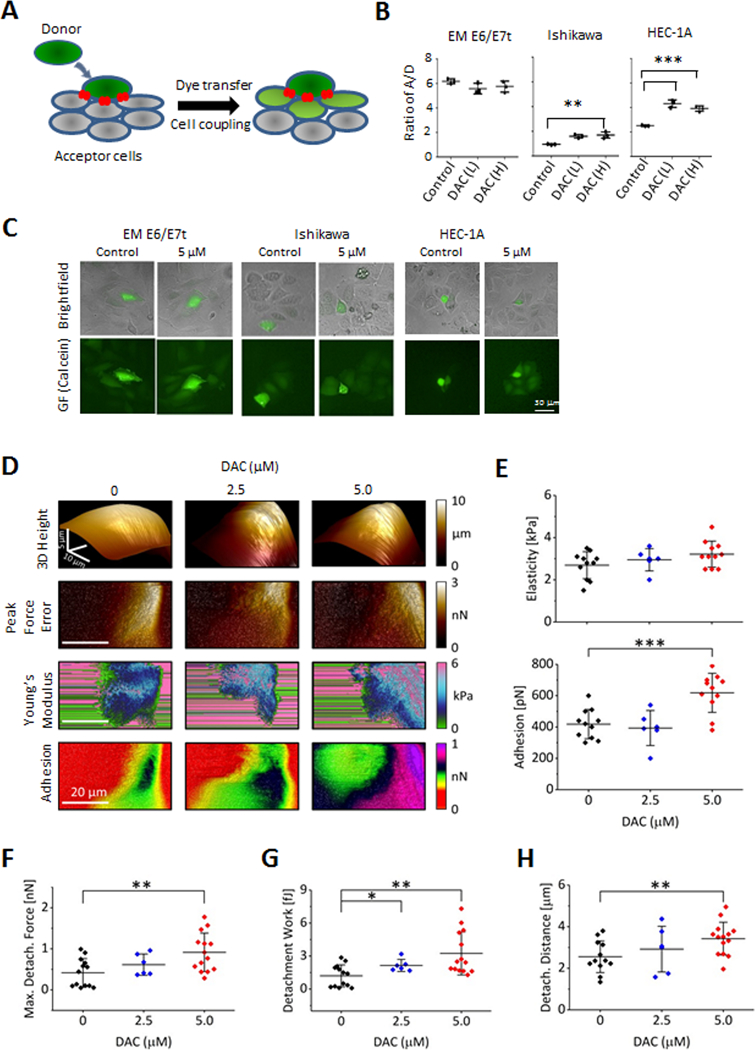
A, A scheme for the parachuting assay in which donor cells are labeled with gap junction permeable dye calcein. Transfer of dye (indicative of GJIC) is measured in recipient cells. B, In parachuting assays, DAC treatment restores GJIC in HEC-1A cells to the levels of the non-oncogenic immortalized line EME6/7t (B). A slight induction of GJIC is also observed in Ishikawa cells C, Representative images illustrative of GJIC induction of HEC-1A and Ishikawa cells are shown. D-E, DAC treatment increases cell to tip adhesion as shown with an array of AFM images of single cells scanned in the PF QNM mode. The first row shows a pseudo 3D height rendering of individual cells with different DAC treatments. Following rows represent 2D maps of surface topography (peak force error), elasticity (the Young’s modulus), and adhesion. An increase of the cell to AFM tip adhesion is noted only for HEC-1A cells treated with 5 μM DAC (D). Data extracted from the 2D AFM maps reveal that DAC treatments resulted in a significantly increased cellular adhesiveness (p<0.001) but had no effect on elasticity (E). F-H, Cell-cell adhesion was measured as described in Materials and Methods using AFM, by attaching a ‘tester’ living HEC-1A cell to the AFM tipless cantilever with PEI, which is then pressed with controlled force against a single HEC1–1A cell growing as part of a monolayer on the surface of a cell culture dish (illustrated in Supplementary Fig. S5). DAC treatment results in a significant systematic increase of the total detachment force (F; p<0.01), the maximum detachment work (G; p<0.01), and the distance required for a full separation of the interacting cells (H), confirming much stronger adhesion of the treated cells. Interestingly, the “snap point” distance – a distance at which a maximum pulling force is detected - significantly increased for the treated cells indicating involvement of substantial pull from cellular extensions (H; p<0.01). CTRL cells (n=13), and cells treated with 2.5 μM DAC (n=6) or 5.0 μM DAC (n=14) were tested over three separate experiments.
We then examined cellular nanomechanical characteristics that may contribute the restoration of GJIC and decreased cellular motility in DAC-treated HEC-1A cells using the Peak Force Quantitative Nanomechanical Mode of atomic force microscopy (AFM). This technique allows a direct assessment of several mechanical parameters of single cells by probing them with very low forces at a high spatial resolution (39). Here, we focused on cell elasticity, expressed as the Young’s modulus (39), and cell adhesion (to the AFM tip), since these parameters may reflect the strength of cell-cell interactions. DAC treatment had a little effect on cell shape or elasticity, but a significant increase in cell adhesiveness was detected (p<0.001, Figs. 5D and E).
While initially cellular adhesiveness was assayed as a function of non-specific cell stickiness to the AFM probe made with silicone nitride, we also examined whether DAC treatment affected cell-to-cell adhesiveness. To quantify adhesion between cells, we applied single cell force spectroscopy (33). In this AFM based method an AFM probe is constructed from a tipless cantilever and a single “tester” cell attached to it (Supplementary Fig. S5A). The tester cell is then brought to a close vicinity of a cell growing in a culture dish. After establishing contact, the tester cell is pressed on the target cell until the applied force reaches its predefined maximum value, and cellular interactions are measured by the force (Newton) and work (Joule) needed to separate the cells, as described in Supplemental Methods (Supplementary Fig. S5B and S5C). Here, a single tester HEC-1A cell attached to an AFM tipless probe was used it to test the interactions with target HEC-1A cells growing on a cell culture dish (Supplementary Fig. S5A). Representative data of detachment curves showing increased cell-cell adhesiveness in DAC treated HEC-1A cells (solid red trace) compared to control cells (solid black trace) are shown in Supplementary Fig. S5D. It is of note that the detachment of control cells passed through a sharp maximum, whereas the treated cells clutched together through a flat maximum. Importantly, tester cells interacted only weakly (dotted red and black traces) with the bottom of the dish allowing for a straightforward distinction between successful and false cell-cell adhesion (Supplementary Fig. S5D).
The maximal force required to separate the tester cell from target cell measured in nano-newtons (nN) was twice as large in HEC-1A cells treated with DAC (5 μM) than control cells (0.91 vs. 0.42 nN, p<0.001, Fig. 5F). In addition, the work required to separate the cells (defined as the force applied at a specific distance) in femto-joules (fJ) was about three-fold higher in the DAC-treated cells compared to untreated cells (3.2 vs. 1.2 fJ, p<0.001, Fig. 5G). Interestingly, the detachment distance during which separation takes place was ~30% longer for the treated cells, possibly indicating a larger and more complex contact area between the interacting cells (Fig. 5H). The observed differences in the mechanical parameters strongly point to the elevated cell-cell adhesiveness and the extensiveness of cell-cell contact in DAC-treated cells relative to untreated cells.
Epigenetically repressed Cx43 is involved in regulating endometrial cancer cell motility
To determine whether DAC treatment results in enhancing gap junction formation, we examined the localization of Cx43 within the cell, and the distribution of gap junction plaques at cell interfaces. More punctate Cx43 staining, indicative of gap junction formation, was observed in untreated EME6/7t cells compared to the two cancer cell lines, Ishikawa and HEC-1A. A striking redistribution of Cx43 to form gap junction plaques was observed in HEC-1A cells (Fig. 6A). Although Ishikawa expressed more Cx43 protein than HEC-1A (Fig. 6A and Supplementary Fig. S6A), and some redistribution of Cx43 to the cell surface was observed, the formation of gap junction plaques was not as organized along the cell surface as in the DAC treated HEC-1A cells (Fig. 6A). The coincident induction of GJIC and cellular adhesiveness, restoration of Cx43 gap junction plaque formation, and decreased motility in HEC-1A cells due to DAC treatment suggests epigenetic regulation of gap junction activity. Furthermore, ASC exposure led to significantly (p<0.01) inducing GJA1 promoter methylation in EME6/7t cells, confirming that this gene encoding Cx43 is subject to adiposity-mediated epigenetic suppression (Supplementary Fig. S6B). In this in vitro ASC exposure system, induction in PRKCA methylation was also observed but not TJP2 (Supplementary Fig. S6C and S6D). We then specifically examined whether restoration of Cx43, which is repressed in HEC-1A cells (Fig. 6A and Supplementary Fig. S6A), had an effect on cellular migration. Cx43 expression was exogenously induced by transfecting an expression vector in HEC-1A cells and selected several clones overexpressing Cx43 (Fig. 6B as an example). Two HEC-1A clones overexpressing Cx43 exhibited reduced migration compared to parental HEC-1A cells (p<0.001; Fig. 6C), with little effect on cellular proliferation (Fig. 6C). In complementary experiments, we examined whether Cx43 knockdown by specific siRNAs affected cellular motility and growth of EME6/7t endometrial epithelial cells, which expressed Cx43 (Supplementary Fig. S6A). Cx43 knockdown, as shown in Fig. 6D, resulted in a significant increase in migration (p<0.001) but had little effect on growth of these cells (Fig. 6E). Furthermore, treatment of EME6/7t cells with Gap27, a peptide mimicking the extracellular domain of Cx43 that has been shown to inhibit coupling (40), also diminished cellular motility in a dose-response fashion (p<0.001, Fig. 6F), but had a limited effect on cellular growth by Gap27 (Fig. 6F). Combined, the data suggest that endometrial cellular coupling by Cx43 gap junctions diminishes endometrial cancer cell motility, concomitant with enhanced cell-cell adhesion. Conversely, suppression of this Cx43 coupling serves to promote motility of these endometrial cancer cells.
Figure 6. Restoration of Cx43 gap junction suppresses motility in HEC-1A endometrial cancer cells.
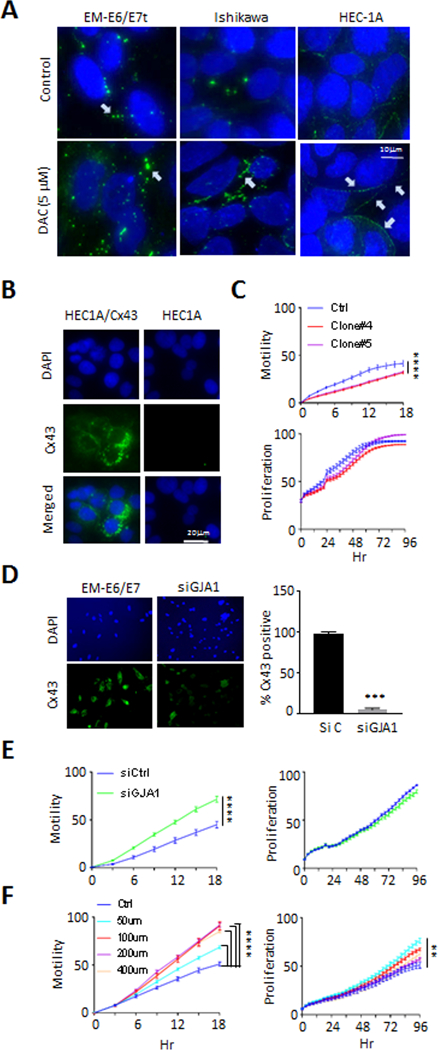
A, Labelling of Cx43 in intracellular pools was evident for EME6/7t and Ishikawa, and weekly observed in HEC-1A, with surface labeling of plaques between cells most evident in EME6/7t cells (white arrows). DAC treatment induces Cx43 gap junction plaque formation in Ishikawa and HEC-1A cells, although these were less organized in the Ishikawa cells. B-C, Restoration of Cx43 in HEC-1A cells by expression vector transduction was performed (B). This Cx43 induction resulted in decreased cellular migration of HEC-1A cells, with limited effects on proliferation (C). D-E, In complementary experiments, Knockdown of Cx43 by siRNA in EME6/7t cells, as demonstrated by immunofluorescence (D), led to enhanced cellular migration in scratch assays, with no effect on proliferation (E). Also, treating EME6/7t cells with the Cx43 gap junction specific peptide inhibitor, gap27, led to enhanced migration in a dose-dependent manner, with a limited effect on proliferation (F).
DISCUSSION
The present study demonstrates a strong link between obesity and epigenetic silencing of GJ-associated loci in endometrial cancer. While the development of obesity-mediated tumors is multifaceted, overgrowth of abdominal adipose tissue is highly associated with endometrial cancer (3,41). Abundant adiposity frequently undergoes tissue remodeling and releases ASCs into the circulation (11). These stromal cells can then home to endometrial tissue and contribute to their malignant growth via local secretion of tumor-promoting hormones, adipokines and cytokines (7,9,16,17,42). Our studies show that ASCs may act in part by PAI-1 to suppress GJ-associated genes in the immortalized EEC line EME6/7t, suggesting that cellular communication in EECs may be epigenetically targeted promoting type I endometrial cancer observed in obesity. While PAI-1 is known as a serine protease inhibitor, other functions of PAI-1 include regulation of gene expression via Stat-1 and enhancing motility in cancer cells (43,44). PAI-1 is elevated in visceral fat and may contribute to morbidity associated with obesity (45).
While transcriptional repression of candidate GJ-associated loci observed in vitro and in vivo may be transient, we further demonstrated that epigenetic mechanisms reinforce long-term repression in endometrial cancer development. Tumor DNA hypermethylation of these loci was observed in our endometrial cancer cohort, and ASC exposure induced GJA1 promoter methylation in vitro. Thus, while initially gene suppression may be reversible, de novo DNA methylation of the GJ-associated loci can spread to promoter CpG island shores or cores for permanent silencing.
The obesity-mediated epigenetic repression of GJ-associated loci has a profound effect on gap junction homeostasis. GJIC is regulated through multiple layers, which has been mainly learned from studies of the major connexin Cx43. Some kinase modulators (e.g. PKA and PKC) can affect connexin trafficking, while others (e.g. ERK and SRC) serve to directly modulate channel function. Scaffolding proteins (e.g., ZO1 and ZO2) can regulate the assembly of Cx43 gap junction plaques at the cell surface (46,47) by enhancing cell-cell adhesion as shown by the AFM studies (48,49). Disruption of the major connexin Cx43 gap junction activity plays a role in endometrial cancer cell migration potentially associated with tumor promotion. Reestablishment of gap junction activity, by re-expression of connexins and/or gap junction modulators, may be required for metastatic invasion during disease progression (50). This functional redundancy among members of gap junction proteins can be exploited by aggressive cancer cells to gain metastatic potential (50). Comprehensive analysis of these gap junction genes and their modulators is needed to provide a better understanding of their roles in endometrial cancer development and advanced disease.
The epigenetically repressed GJ-associated loci can be useful biomarkers to study pathophysiology of obesity-associated endometrial tumorigenesis, which is on the rise (3,41). There is an epidemic of obesity related endometrial cancers in Hispanics in our South Texas population which represents a health care disparity. These endometrial cancer patients tends to be considerably younger (~50 years) than the typical endometrial cancer patient (~60 years) (3,4). The association of endometrial cancer in obese women with aberrant DNA methylation of GJA1, a known tumor-suppressor, and at least two GJ-associated loci PRKCA and TJP2, provides mechanistic clues to these clinical observations. Similarly, endometrial lesions (atypia and hyperplasia) were observed at a younger age in the obese high-fat fed Pten mice compared to control-fat diet mice, whereas by 28 weeks of age both groups developed endometrial carcinomas, suggesting the contribution of obesity to early stages of tumor development, including initiation or promotion.
Our present study indicates that obesity, in part due to ASC paracrine actions, leads to downregulation of GJ-associated loci. We further confirmed the role of this epigenetic mechanism in the regulation of gap junction activity is in part mediated by effects on Cx43 assembly into functional gap junction plaques between cells. Future studies will examine the regulatory mechanisms leading to GJ-associated gene regulation and the functional changes in GJIC in obesity-associated endometrial cancer. These studies will improve our understanding of how obesity contributes to the development of endometrial cancer.
Supplementary Material
Significance: Studies reveal that adipose-derived stem cells in endometrial cancer pathogenesis influence epigenetic repression of gap junction loci, which suggests targeting of gap junction activity as a preventive strategy for obesity-associated endometrial cancer.
Acknowledgements
The authors acknowledge the assistance of the Next-Generation Sequencing Core for RNA-seq analysis, the Bioanalytics and Single-Cell Core for atomic force microscopy studies and single cell expression, and the High Throughput Screening Facility of the Center for Innovative Drug Discovery (partially supported by funds from the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1 TR001120) for the analyses of gap junction cell coupling studies, at the University of Texas Health Science Center, San Antonio, TX. This work was supported by NIH grants R01CA172279 and P30CA054174, the Cancer Prevention and Research Institute of Texas (CPRIT) grant RP150600, San Antonio Cancer Council, and the Max and Minnie Tomerlin Voelcker Fund.
Footnotes
The authors declare no potential conflicts of interest
References
- 1.Cramer DW. The epidemiology of endometrial and ovarian cancer. Hematol Oncol Clin North Am 2012;26(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen NE, Key TJ, Dossus L, Rinaldi S, Cust A, Lukanova A, et al. Endogenous sex hormones and endometrial cancer risk in women in the European Prospective Investigation into Cancer and Nutrition (EPIC). Endocr Relat Cancer 2008;15(2):485–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soliman PT, Oh JC, Schmeler KM, Sun CC, Slomovitz BM, Gershenson DM, et al. Risk factors for young premenopausal women with endometrial cancer. Obstet Gynecol 2005;105(3):575–80. [DOI] [PubMed] [Google Scholar]
- 4.Kost ER, Valente PT, Lynch BA, Krishnegowda NK, Hertz AM, Hall KL, et al. Clinical and Pathologic Features of Hispanic Endometrial Cancer Patients With Loss of Mismatch Repair Expression. Int J Gynecol Cancer 2016;26(6):1129–36. [DOI] [PubMed] [Google Scholar]
- 5.Gui Y, Pan Q, Chen X, Xu S, Luo X, Chen L. The association between obesity related adipokines and risk of breast cancer: a systematic review and meta-analysis. Oncotarget 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riondino S, Roselli M, Palmirotta R, Della-Morte D, Ferroni P, Guadagni F. Obesity and colorectal cancer: role of adipokines in tumor initiation and progression. World J Gastroenterol 2014;20(18):5177–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linkov F, Kokai L, Edwards R, Sheikh MA, Freese KE, Marra KG, et al. The role of adipose-derived stem cells in endometrial cancer proliferation. Scand J Clin Lab Invest Suppl 2014;244:54–8; discussion 57–8. [DOI] [PubMed] [Google Scholar]
- 8.Crop MJ, Baan CC, Korevaar SS, Ijzermans JN, Pescatori M, Stubbs AP, et al. Inflammatory conditions affect gene expression and function of human adipose tissue-derived mesenchymal stem cells. Clin Exp Immunol 2010;162(3):474–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang T, Tseng C, Zhang Y, Sirin O, Corn PG, Li-Ning-Tapia EM, et al. CXCL1 mediates obesity-associated adipose stromal cell trafficking and function in the tumour microenvironment. Nat Commun 2016;7:11674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma T, Luan SL, Huang H, Sun XK, Yang YM, Zhang H, et al. Upregulation of CC Chemokine Receptor 7 (CCR7) Enables Migration of Xenogeneic Human Adipose-Derived Mesenchymal Stem Cells to Rat Secondary Lymphoid Organs. Med Sci Monit 2016;22:5206–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghosh S, Hughes D, Parma DL, Ramirez A, Li R. Association of obesity and circulating adipose stromal cells among breast cancer survivors. Mol Biol Rep 2014;41(5):2907–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Kolonin MG. Cytokine signaling regulating adipose stromal cell trafficking. Adipocyte 2016;5(4):369–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erdogan B, Webb DJ. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem Soc Trans 2017;45(1):229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park J, Euhus DM, Scherer PE. Paracrine and endocrine effects of adipose tissue on cancer development and progression. Endocr Rev 2011;32(4):550–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Bellows CF, Kolonin MG. Adipose tissue-derived progenitor cells and cancer. World J Stem Cells 2010;2(5):103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saha A, Ahn S, Blando J, Su F, Kolonin MG, DiGiovanni J. Proinflammatory CXCL12-CXCR4/CXCR7 Signaling Axis Drives Myc-Induced Prostate Cancer in Obese Mice. Cancer Res 2017;77(18):5158–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 2011;17(11):1498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res 2013;52(4):585–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Studebaker AW, Storci G, Werbeck JL, Sansone P, Sasser AK, Tavolari S, et al. Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6-dependent manner. Cancer Res 2008;68(21):9087–95. [DOI] [PubMed] [Google Scholar]
- 20.Pistore C, Giannoni E, Colangelo T, Rizzo F, Magnani E, Muccillo L, et al. DNA methylation variations are required for epithelial-to-mesenchymal transition induced by cancer-associated fibroblasts in prostate cancer cells. Oncogene 2017. [DOI] [PubMed] [Google Scholar]
- 21.Lin HJ, Zuo T, Lin CH, Kuo CT, Liyanarachchi S, Sun S, et al. Breast cancer-associated fibroblasts confer AKT1-mediated epigenetic silencing of Cystatin M in epithelial cells. Cancer Res 2008;68(24):10257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yano T, Ito F, Yamasaki H, Hagiwara K, Ozasa H, Nakazawa H, et al. Epigenetic inactivation of connexin 32 in renal cell carcinoma from hemodialytic patients. Kidney Int 2004;65(4):1519. [DOI] [PubMed] [Google Scholar]
- 23.Aasen T, Mesnil M, Naus CC, Lampe PD, Laird DW. Gap junctions and cancer: communicating for 50 years. Nat Rev Cancer 2016;16(12):775–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans WH, Martin PE. Gap junctions: structure and function (Review). Mol Membr Biol 2002;19(2):121–36. [DOI] [PubMed] [Google Scholar]
- 25.Kanaporis G, Mese G, Valiuniene L, White TW, Brink PR, Valiunas V. Gap junction channels exhibit connexin-specific permeability to cyclic nucleotides. J Gen Physiol 2008;131(4):293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diao H, Xiao S, Howerth EW, Zhao F, Li R, Ard MB, et al. Broad gap junction blocker carbenoxolone disrupts uterine preparation for embryo implantation in mice. Biol Reprod 2013;89(2):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.el-Sabban ME, Pauli BU. Adhesion-mediated gap junctional communication between lung-metastatatic cancer cells and endothelium. Invasion Metastasis 1994;14(1–6):164–76. [PubMed] [Google Scholar]
- 28.Nishida M The Ishikawa cells from birth to the present. Hum Cell 2002;15(3):104–17. [DOI] [PubMed] [Google Scholar]
- 29.Kuramoto H, Tamura S, Notake Y. Establishment of a cell line of human endometrial adenocarcinoma in vitro. Am J Obstet Gynecol 1972;114(8):1012–9. [DOI] [PubMed] [Google Scholar]
- 30.Zhang W, Chen JH, Shan T, Aguilera-Barrantes I, Wang LS, Huang TH, et al. miR-137 is a tumor suppressor in endometrial cancer and is repressed by DNA hypermethylation. Lab Invest 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kyo S, Nakamura M, Kiyono T, Maida Y, Kanaya T, Tanaka M, et al. Successful immortalization of endometrial glandular cells with normal structural and functional characteristics. Am J Pathol 2003;163(6):2259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fyles A, Wood G, Li M, Manoukian AS, Gowing K, Khokha R, et al. Neither ovariectomy nor progestin treatment prevents endometrial neoplasia in pten+/− mice. Gynecol Oncol 2008;108(2):395–401. [DOI] [PubMed] [Google Scholar]
- 33.Friedrichs J, Legate KR, Schubert R, Bharadwaj M, Werner C, Muller DJ, et al. A practical guide to quantify cell adhesion using single-cell force spectroscopy. Methods 2013;60(2):169–78. [DOI] [PubMed] [Google Scholar]
- 34.Huang TT, Gonzales CB, Gu F, Hsu YT, Jadhav RR, Wang CM, et al. Epigenetic deregulation of the anaplastic lymphoma kinase gene modulates mesenchymal characteristics of oral squamous cell carcinomas. Carcinogenesis 2013;34(8):1717–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu F, Doderer MS, Huang YW, Roa JC, Goodfellow PJ, Kizer EL, et al. CMS: a web-based system for visualization and analysis of genome-wide methylation data of human cancers. PLoS One 2013;8(4):e60980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rao X, Evans J, Chae H, Pilrose J, Kim S, Yan P, et al. CpG island shore methylation regulates caveolin-1 expression in breast cancer. Oncogene 2013;32(38):4519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han H, Cortez CC, Yang X, Nichols PW, Jones PA, Liang G. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum Mol Genet 2011;20(22):4299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buchi F, Masala E, Rossi A, Valencia A, Spinelli E, Sanna A, et al. Redistribution of H3K27me3 and acetylated histone H4 upon exposure to azacitidine and decitabine results in de-repression of the AML1/ETO target gene IL3. Epigenetics 2014;9(3):387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsu YT, Osmulski P, Wang Y, Huang YW, Liu L, Ruan J, et al. EpCAM-Regulated Transcription Exerts Influences on Nanomechanical Properties of Endometrial Cancer Cells That Promote Epithelial-to-Mesenchymal Transition. Cancer Res 2016;76(21):6171–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wright CS, van Steensel MA, Hodgins MB, Martin PE. Connexin mimetic peptides improve cell migration rates of human epidermal keratinocytes and dermal fibroblasts in vitro. Wound Repair Regen 2009;17(2):240–9. [DOI] [PubMed] [Google Scholar]
- 41.Nevadunsky NS, Van Arsdale A, Strickler HD, Moadel A, Kaur G, Levitt J, et al. Obesity and age at diagnosis of endometrial cancer. Obstet Gynecol 2014;124(2 Pt 1):300–6. [DOI] [PubMed] [Google Scholar]
- 42.Freese KE, Kokai L, Edwards RP, Philips BJ, Sheikh MA, Kelley J, et al. Adipose-derived stems cells and their role in human cancer development, growth, progression, and metastasis: a systematic review. Cancer Res 2015;75(7):1161–8. [DOI] [PubMed] [Google Scholar]
- 43.Czekay RP, Wilkins-Port CE, Higgins SP, Freytag J, Overstreet JM, Klein RM, et al. PAI-1: An Integrator of Cell Signaling and Migration. Int J Cell Biol 2011;2011:562481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Degryse B, Neels JG, Czekay RP, Aertgeerts K, Kamikubo Y, Loskutoff DJ. The low density lipoprotein receptor-related protein is a motogenic receptor for plasminogen activator inhibitor-1. J Biol Chem 2004;279(21):22595–604. [DOI] [PubMed] [Google Scholar]
- 45.Shimomura I, Funahashi T, Takahashi M, Maeda K, Kotani K, Nakamura T, et al. Enhanced expression of PAI-1 in visceral fat: possible contributor to vascular disease in obesity. Nat Med 1996;2(7):800–3. [DOI] [PubMed] [Google Scholar]
- 46.Lampe PD. Analyzing phorbol ester effects on gap junctional communication: a dramatic inhibition of assembly. J Cell Biol 1994;127(6 Pt 2):1895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh D, Solan JL, Taffet SM, Javier R, Lampe PD. Connexin 43 interacts with zona occludens-1 and −2 proteins in a cell cycle stage-specific manner. J Biol Chem 2005;280(34):30416–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sariisik E, Popov C, Muller JP, Docheva D, Clausen-Schaumann H, Benoit M. Decoding Cytoskeleton-Anchored and Non-Anchored Receptors from Single-Cell Adhesion Force Data. Biophys J 2015;109(7):1330–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chakraborty S, Mitra S, Falk MM, Caplan SH, Wheelock MJ, Johnson KR, et al. E-cadherin differentially regulates the assembly of Connexin43 and Connexin32 into gap junctions in human squamous carcinoma cells. J Biol Chem 2010;285(14):10761–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016;533(7604):493–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.