

Abstract
A major challenge in drug delivery is to enhance the transport of drugs across biological barriers, such as the small intestine, the blood–brain barrier, and the blood–retinal/ocular barrier, and to effectively reach the site of action while minimizing the systemic impact. In recent years, piggybacking cell surface receptors have been considered a viable strategy for active drug delivery across the biological barriers. However, the ligands used to target drugs to plasma membrane receptors often have to compete against endogenous ligands, thereby limiting their binding to the cell surface and their transport across barriers. To address this problem, gambogic acid (GA) was identified as a noncompetitive ligand specific to the transferrin receptor (TfR), a receptor present on various barriers. However, the binding sites of the GA on TfR remain unknown, an essential step toward establishing structure–activity relationships. In silico binding site prediction tools, blind docking, and molecular docking simulation confirm that the GA binding site on the TfR is independent of the transferrin-bound iron binding sites. The GA-conjugated polyesters were processed into nanoparticles suitable for drug delivery applications that possess excellent storage stability under regulatory conditions. Traditionally, GA has been used as an anticancer compound that warrants safety assessment. The preliminary studies in healthy rodents on 10-repeated oral doses show no adverse effects. This work will generate paradigm shifting, new knowledge in the field of nanomedicines using unique noncompetitive nanosystems that do not compete with endogenous transferrin.
Keywords: active delivery, biodegradable, gambogic acid, nanoparticles, noncompetitive, oral delivery, polyesters, transferrin receptor
Graphical Abstract
INTRODUCTION
The recent use of receptor-mediated transport presents an exciting opportunity for enhancing the delivery of nanosystems across the barriers and bioavailability of the encapsulated drugs. In this approach, nanosystems are decorated with ligands that bind to active sites on cell surface receptors, subsequently facilitating transport across biological barriers.1–6 While promising, this drug-delivery approach is currently limited in its efficacy primarily due to the fact that all ligands used to date are outcompeted by physiological ligands present endogenously at high concentrations.7
To address this problem, we recently proposed the use of gambogic acid (GA), a small-molecule pigment, as a noncompetitive ligand useful for targeting nanosystems via the transferrin receptor (TfR), a target receptor expressed at high levels on the surface of cells of the small intestine barriers, the blood–brain barrier, and the blood–retinal/ocular barrier.6,8–11 Our approach, which uses GA-decorated nanosystems for delivering encapsulated drugs, takes advantage of noncompetitive (not competing with endogenous transferrin-bound iron [Fe(III)], subsequently referred to as transferrin, Tf) behavior of GA to navigate the TfR present on the intestinal barriers. Our recent results indicate that the reactive functional groups of the polyesters such as polylactide-co-glycolide (PLGA) can be coupled to GA to achieve noncompetitive active transport exploiting TfR, despite the receptor being blocked by a physiological ligand, transferrin.12 The GA-decorated nanosystems led to an increase in oral bioavailability of peptides and proteins such as cyclosporine12 and insulin13 that otherwise shows variable absorption12 or is not amenable by this route of delivery.13
Traditionally, GA has been extensively used as a pro-apoptotic anticancer compound, primarily exerting its activity via noncompetitive binding to TfR, a receptor that is abnormally expressed in various cancers, and these effects are dose dependent.14 In addition, GA is also used as an antioxidant and anti-inflammatory agent for the treatment of a plethora of chronic diseases.15 Because the contrasting toxic and nontoxic effects are dose-dependent, a comprehensive and detailed characterization of the in vivo toxicity of GA used a ligand in our delivery nanosystems is necessary to establish the utility of our approach. The safety of GA has been well established in rodents as well as humans; however, we do not know how GA-coupled nanosystems behave in vivo. The literature reports indicate that a 60 mg/kg GA administered by oral gavage for a total of 13 weeks at a frequency of one administration every other day is safe in rats, while in human patients, 45 mg/m2 (~75 mg for an average built human), 5 times a week over 2 weeks by intravenous administration is well tolerated.16,17 Finally, while there is a large body of work directed toward identifying the mechanisms of action of GA in various diseases, the binding sites of the GA on TfR remain unknown. It is however important to identify these sites as TfR is responsible for a wide variety of signaling responses in diverse cels types. In turn, such ligand binding may specifically elicit particular signaling responses that are deleterious. This foundational knowledge will allow us and other investigators to rationally design next-generation noncompetitive drug delivery agents that cross biological barriers with optimal efficiencies.
Here, we present In silico identification of GA–TfR binding sites, an essential step toward establishing structure–activity relationships, and contribute to new research directions of this novel concept. This study also establishes regulatory shelf-stability of the noncompetitive GA-decorated nanosystems that allows synthesis and storage of large quantities of dosage forms and their preliminary safety in healthy rodents, inching toward clinical translation.
RESULTS AND DISCUSSION
In silico Identification of Gambogic Acid–Transferrin Receptor Binding Sites.
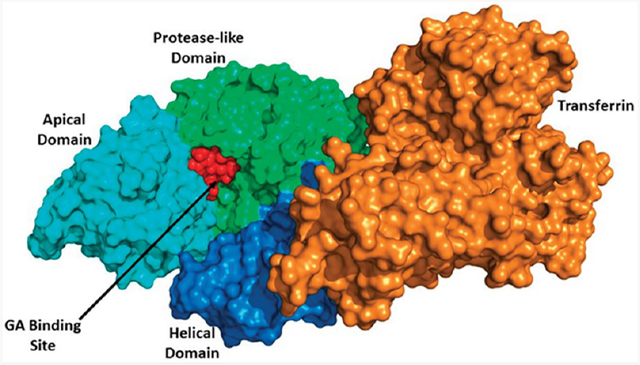
The structure of the human TfR–Tf complex, including the molecular details of the complex formation, has been extensively studied. The monomeric structure of TfR is composed of a protease-like domain proximal to the membrane, a helical domain accounting for all the dimer contacts, and a membrane-distal apical domain.18,19 The SiteMap20,21 module from the Schrödinger software package (Schrödinger, LLC, New York, NY) locates the potential binding sites on the entire surface of the TfR. Predicted potential binding sites by SiteMap were ranked based on SiteScore where a ligand can bind. Similarly, SiteHound22 and CASTp23 also identified the potential sites present on the TfR. GA was docked separately into the different sites predicted by all binding site prediction tools. All prediction tools identified a binding site for GA at the interface of the apical and protease-like domain of TfR that was subsequently confirmed by docking studies. Blind docking, which performs an exhaustive series of docking calculations across the whole protein surface, also finds that same site present at the interface of the apical and protease-like domain of TfR with best binding affinity for GA (Figure 1a, b). Results of different binding sites predicted by binding site prediction tools along with docking results are summarized in Tables S1–S4.
Figure 1.

(a) Summary of common binding sites of TfR for GA. (b) Image showing GA docked at the interface of the apical (cyan color) and protease-like domain (green color) of TfR.
A 50 ns molecular dynamics simulation was performed to validate and check the stability of the docked GA to the TfR at the common binding site concluded from different binding site prediction tools. Hydrogen bond occupancy analysis helps to identify the important TfR residues involved in the hydrogen bond interaction with GA (Figure 2a). Asp562 was the most predominant residue involved in hydrogen bonding followed by Lys261, Lys287, and Tyr563 during the whole 50 ns simulation. Lys495 has the least H-bond occupancy percentage, so it can be considered as the least critical residue among all. Per-residue decomposition analysis shows the amino acid residues which have a major contribution in the binding of GA to the TfR (Figure 2b), and these residues include: Lys287, Asp562, Gly260, Lys261, and Leu566. If we compare chain A which has ligand GA and chain B which is in apo form, there is a significant difference in energy decomposition. Per-residue decomposition energy was almost negligible in chain B for the same binding site residues.
Figure 2.

(a) Percentage hydrogen bond occupancy of different amino acid residues. (b) Per-residue decomposition analysis results for protein backbone (Chain A has ligand, and Chain B is without ligand).
Binding free energy (ΔGbind) contribution was calculated by MM-GBSA analysis. The binding energies (ΔGbind) for the GA (Table 1) with TfR were −48.13 kcal/mol at the end of the 20th ns simulation and −48.30 kcal/mol at the end of the 50th ns simulation. These binding free energy data suggest that there is a significant and stable interaction between TfR and GA during the whole 50 ns course of the simulation.
Table 1.
Average Binding Energies at 20, 30, 40, and 50 ns (Average over the Last 5 ns), along with Their Different Energy Components (Calculated Using MM-GBSA)a
| energy contribution (kcal/mol) |
Energy (TfR-GA) |
energy (TfR) | energy (GA) |
EVDW | EEL | EGB | EGSURF | ΔGgas | ΔGsol | ΔGbind |
|---|---|---|---|---|---|---|---|---|---|---|
| TfR_GA_20 ns | −116365.31 | −116320.48 | 3.3 | −57.67 | −104.53 | 123.29 | −9.22 | −162.20 | 114.07 | −48.13 |
| TfR_GA_30 ns | −116425.01 | −116382.19 | 3.19 | −55.86 | −103.01 | 121.91 | −9.05 | −155.87 | 112.86 | −46.01 |
| TfR_GA_40 ns | −116487.77 | −116450.56 | 6.16 | −58.32 | −91.58 | 115.62 | −9.09 | −149.90 | 106.53 | −43.37 |
| TfR_GA_50 ns | −116473.36 | −116427.81 | 2.76 | −56.97 | −94.68 | 112.62 | −9.26 | −151.66 | 103.35 | −48.30 |
EVDW = van der Waals contribution calculated by MM force field. EEL = electrostatic energy contribution calculated by MM force field. EGB = the electrostatic contribution to the solvation free energy, calculated by GB. ΔGgas = sum of VDW and ELE energy contribution. ΔGsol = solvation free energy contribution calculated by Generalized Born (GB). ESURF = nonpolar contribution to the solvation free energy, calculated by an empirical model. ΔGbind = final estimated binding free energy calculated from the terms above. All the values reported above are in kcal/mol.
The analysis of hydrogen bonding interactions over the 50 ns trajectory indicated the presence of two stable hydrogen bonds between GA and TfR that were present most of the time during the simulation (Figure S1). For most of the simulation time, the RMSD value of the TfR ranges near ~3 Å (Figure S2). For the first 12 ns, the backbone RMSD fluctuation for the TfR was under 3.0 Å, and after that it increased and went up to a maximum of 3.95 Å at 17.56 ns of simulation. During the last few ns of simulation, the RMSD value fluctuates around 3.1 Å. The overall RMSD of the TfR backbone and GA fluctuations indicate that the ligand was bound stably to the site of the protein predicted by various site prediction tools.
The identified GA binding site is present at the interface of the apical and protease like domain of the TfR (Figure S3). This predicted site is different from the active site where Tf binds, and this noncompetitive binding of GA to the TfR has been shown experimentally.12,13 So In silico identification of the GA binding site also satisfies the experimental finding.
The carboxyl group of GA connects with the linker during the formation of PLGA–GA conjugates. This carboxyl group, which interacts with the TfR and forms hydrogen bonds with Lys261 and Lys287, will not be available when PLGA–GA interacts with the TfR (Figure 3a). PLGA–GA was docked to the TfR using the PatchDock.24 In PLGA–GA, the carboxyl group of GA is linked to linker EDA, but the GA moiety was still able to interact with the same binding site. The binding free energy of the PLGA–GA and TfR complex was found to be −42.75 kcal/mol. Thr536, Lys261, and Asp562 present at the GA binding site of the TfR are the residues that interact with GA with and without PLGA–EDA (Figure 3b; GA portion shown in green color).
Figure 3.

(a) 2D interaction diagram for the GA:TfR complex (pink arrow depicts hydrogen bonding, and blue-red arrow depicts the salt bridge). (b) Interaction diagram for the PLGA–GA:TfR complex (blue line depicts hydrogen bonding; dotted gray line depicts hydrophobic interaction; and yellow dotted line depicts the salt bridge).
Asp562 is the same residue that has the highest hydrogen bond occupancy (Figure 2a), and Thr536 makes a significant contribution toward the binding of GA to TfR according to per-residue energy decompositions analysis (Figure 2b). These two residues are making hydrogen bonds with GA in PLGA–GA. Lys261 hydrogen bonds with the carboxyl group of free GA but forms a hydrophobic interaction of GA of PLGA-GA. Asn493, Phe494, Val496, Ser497, and Glu533 are the residues which interact with PLGA–EDA.
Synthesis and Characterization of PLGA–GA.
The PLGA–GA conjugation using EDC chemistry is robust and scalable. The association of GA to PLGA is confirmed by NMR, FTIR, and UV spectroscopy. The 1H NMR of PLGA–GA revealed the appearance of amide peaks (in the range 8–9 ppm) by replacing the −COOH peaks of PLGA and GA, at 13.14 and 12.80 ppm, respectively. FT-IR spectroscopy further corroborates the amide bond formation (1670–1630 cm−1 for C=O stretching; 1650–1560 cm−1 for N–H bending in amides; 1560–1530 cm−1 for C–N stretching). The characteristic peak of GA at 365 nm, upon conjugation to PLGA, shifts to 392 nm (bathochromic), which is confirmed by UV–vis spectroscopy.12,13
Preparation and Characterization of PLGA–GA Nano-systems.
The process for making PLGA nanosystems, including curcumin-encapsulated nanosystems, has been thoroughly optimized and is reproducibly made in large quantities.25–28 The PLGA–GA nanosystems are spherical in shape with the size range of 200–250 nm, as confirmed by dynamic light scattering (DLS) (Figure S4). The nano-formulations exhibit 75% entrapment efficiency with 15% CUR loading capacity. The use of a flexible hydrophilic linker between polymer and GA facilitates expression of GA on the surface of the nanosystems during the emulsification step and is confirmed by the presence of GA characteristic peaks in the UV–vis analysis.12,13 The freeze-drying process led to an intact cake formation with aesthetic appeal without exhibiting any significant difference between the fresh and freeze-dried particle size and zeta potential.
Shelf-Stability Testing of PLGA–GA Nanosystems.
Our lab has previously documented the shelf-stability of PLGA nanosystems.26 However, it is important to understand if polymer processing has any adverse effects on shelf-stability of the final dosage forms; therefore, we conducted the stability of nanosystems made of PLGA–GA. The PLGA–GA nano-systems demonstrate excellent stability by withstanding the ICH recommended accelerated room (Figure 4) and refrigerated test conditions (Figure S5). The particle size, morphology, and CUR content remained intact at the end of sixth months and are comparable to the fresh particles. Such stability will ensure preparation and storage of large quantities of drug-encapsulated PLGA–GA nanosystems for long-term efficacy testing as well as safe shipping of samples that can facilitate long distance collaborations.27,29
Figure 4.

PLGA–GA particle characteristics before and after subjecting to room-temperature conditions recommended by ICH at 40 ± 2 °C and 75% ± 5% RH. (a) Freeze-dried particles showing intact cake from 0-time point to post-6 month stability testing, (b) particle size, (c) CUR entrapment, and (d) SEM photomicrographs.
Safety Assessment of PLGA–GA Nanosystems.
The preliminary safety assessment of PLGA–GA nanosystems is essential before carrying out any efficacy studies considering their noncompetitive active transport mechanisms via the TfR present in the intestine.12,13 The role of the Tf–TfR-mediated endocytosis and its application in drug targeting have been widely studied; however, the majority of studies, if not all studies, uses endogenous mimicking ligands (e.g., Tf) that bind to the receptor’s active sites for transport. Therefore, the conventional TfR-targeted systems have to compete with endogenous Tf in order for them to bind to the TfR, and their safety studies in vivo if at all are pretty much assuming that the particles bind to the receptor, which may/may not be the case. In contrary, the PLGA–GA particles bind to nonactive sites on the TfR, facilitating active transport. To verify if such a noncompetitive active targeting strategy via nonactive sites causes any significant toxicities, a repeated dose toxicity study was performed in healthy rodents. The PLGA–GA nanosystems did not show any unusual variations in locomotor activity or signs of intoxication or ataxia. The hematological parameters for PLGA or PLGA–GA nanosystems are within the reference range, suggesting that 10 repeated administrations of GA-coupled PLGA particles are safe (Table 2).
Table 2.
Hematological Parameters at the End of the Study
| parameter | reference value | PLGA | PLGA-GA |
|---|---|---|---|
| RBC (M/μL) | 6.7–9.0 | 7.31 ± 0.13 | 6.73 ± 0.42 |
| WBC (K/μL) | 8.20–15.69 | 10.21 ± 0.94 | 10.68 ± 1.86 |
| HGB (g/dL) | 13.6–17.7 | 14.78 ± 0.17 | 13.98 ± 0.64 |
| HCT (%) | 37.9–52.2 | 51.88 ± 0.50 | 47.63 ± 2.08 |
| MCV (fL) | 51.8–63.6 | 71.03 ± 1.23 | 70.9 ± 2.24 |
| MCH (pg) | 17.9–20.9 | 20.31 ± 0.55 | 20.78 ± 0.68 |
| MCHC (g/dL) | 30.6–36.8 | 28.48 ± 0.34 | 29.38 ± 0.26 |
| rel. neutrophils (%) | 4.7–23.3 | 14.25 ± 2.75 | 15.75 ± 4.5 |
| rel. lymphocytes (%) | 71.1–89.4 | 80.25 ± 2.62 | 79.75 ± 3.30 |
| rel. monocytes (%) | 0.2–8.0 | 4.25 ± 1.25 | 4.00 ± 0.81 |
Similarly, the blood chemistry panel showed comparable results between PLGA and PLGA–GA nanosystems that are within the reference range and further demonstrate the safety of PLGA–GA nanosystems (Table 3). Blood chemistry panel and hematological assays give a good indication about the general wellbeing of the biological system. For example, albumin and globulins are proteins that are primarily made by the liver and, in the case of globulins, by the immune system to a lower extent. Albumin carries medicines and hormones and helps with tissue growth and healing, whereas globulins transport nutrients, bind antigens, and contribute to fighting infection. A high or low protein production indicates malfunctioning of various organs (e.g., liver, kidney), while the abnormal A/G ratios suggest the following: low suggests autoimmune diseases, cirrhosis, and nephrotic syndrome like features, and a high A/G indicates leukemia characteristics. A panel of markers such as alanine transaminase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), γ-glutamyl transpeptidase (GGT), glutamate dehydrogenase (GLDH), albumin, and bilirubin measure liver function, and again PLGA–GA nanosystems do not cause any abnormality. The increase in the ALP levels after dosing could be due to the post sacrifice terminal bleeding through a heart puncture.
Table 3.
Blood Chemistry Before (BD, before the Start of Dosing) and After (AD, at the End of the Study) PLGA and PLGA–GA Dosing over 10 Days
| parameter | reference value | PLGA-BD | PLGA-AD | PLGA-GA-BD | PLGA-GA-AD |
|---|---|---|---|---|---|
| total serum protein (g/dL) | 5.22–6.71 | 5.925 ± 0.28 | 6.65 ± 0.15 | 5.65 ± 0.17 | 6.35 ± 0.19 |
| albumin (g/dL) | 2.69–3.86 | 3.35 ± 0.17 | 3.675 ± 0.1 | 3.25 ± 0.1 | 3.47 ± 0.12 |
| calcium (mg/dL) | 9–11.3 | <4.0 ± 0 | 13.325 ± 0.51 | <4.0 | 12.62 ± 0.34 |
| phosphorus (mg/dL) | 7.5–10.8 | 7.75 ± 0.37 | 17.5 ± 1.53 | 7.65 ± 0.59 | 13.9 ± 0.48 |
| BUN (mg/dL) | 11.8–25.4 | 17.25 ± 1.5 | 17.75 ± 3.60 | 15.75 ± 1.70 | 17.25 ± 0.5 |
| creatinine (mg/d L) | 0.36–0.6 | 0.37 ± 0.05 | 0.5 ± 0 | 0.325 ± 0.05 | 0.4 ± 0 |
| total bilirubin (mg/dL) | 0.14–0.28 | 0.1 ± 0 | 0.125 ± 0.05 | 0.125 ± 0.05 | 0.1 ± 0 |
| ALP (U/L) | 91.0–250.1 | 66 ± 27.72 | 480.25 ± 113.93 | 155 ± 23.10 | 465.5 ± 26.81 |
| AST (SGOT) (U/L) | 63.9–228.8 | 81.25 ± 4.34 | 159.75 ± 133.67 | 79.75 ± 4.42 | 105.75 ± 8.73 |
| ALT (SGPT) (U/L) | 26.5–101.4 | 46.5 ± 7.41 | 48.75 ± 12.66 | 46.75 ± 6.84 | 50.75 ± 2.62 |
| globulins (g/dL) | 0–10.5 | 2.575 ± 0.15 | 2.975 ± 0.15 | 2.4 ± 0.14 | 2.87 ± 0.095 |
| A/G ratio | 0.93–1.72 | 1.325 ± 0.09 | 1.25 ± 0.05 | 1.35 ± 0.1 | 1.22 ± 0.05 |
| GGT (U/L) | 2.23–2.87 | <3 ± 0 | |||
| GLDH (U/L) | <10 | 6 ± 1.15 | 7 ± 3.05 | 8.5 ± 1.91 | 9 ± 0.81 |
| amylase (U/L) | 10–2000 | 627.25 ± 38.69 | 582.75 ± 46.55 | ||
| cholesterol (mg/dL) | 44.0–139.0 | 98.75 ± 2.87 | 81.5 ± 3.51 | 112.5 ± 5.06 | 92.5 ± 12.28 |
| sodium (mEq/L) | 141.0–149.0 | 141.75 ± 0.5 | 145.75 ± 1.52 | 142 ± 0.81 | 146.5 ± 1 |
| potassium (mEq/L) | 3.81–6.48 | >10.7 ± 2.34 | 9.325 ± 0.818 | >10.1 ± 0 | 8. ± 0.32 |
| chloride (mEq/L) | 103.0–110.0 | 95.75 ± 0.95 | 101.5 ± 1 | 97.25 ± 0.5 | 103.25 ± 0.5 |
| Na/K ratio | 9.33–27.5 | 14.7 ± 1.06 | 14.2 ± 0.08 |
The body and organ weight are comparable between PLGA and PLGA–GA groups (Table 4). This again suggests that receptor-mediated noncompetitive targeting across intestinal barriers did not affect food intake or body or organ weights, which is one of the most sensitive drug toxicity indicators, and the changes are often associated with morphological changes.
Table 4.
Body and Organ Weights of Rats after 10 Daily Doses of PLGA and PLGA–GA
| body/organ weight (g) | PLGA | PLGA-GA |
|---|---|---|
| body | 284.3 ± 9.7 | 277.5 ± 13.0 |
| liver | 14.9 ± 1.9 | 13.9 ± 0.05 |
| kidney | 2.2 ± 0.1 | 2.2 ± 0.1 |
| spleen | 0.6 ± 0.03 | 0.6 ± 0.07 |
| lungs | 1.5 ± 0.2 | 1.4 ± 0.1 |
| heart | 1.0 ± 0.09 | 1.0 ± 0.03 |
| brain | 1.9 ± 0.04 | 2.0 ± 0.04 |
| testis | 1.8 ± 0.04 | 1.9 ± 0.07 |
| pancreas | 0.7 ± 0.1 | 0.6 ± 0.06 |
The PLGA–GA particles are designed to facilitate active transport across the small intestine via the TfR upon oral administration, where the particles are expected to take portal or lymphatic or both routes.30–32 Therefore, it is important to understand the morphological changes that may arise in the small intestine due to the repeated administration, the ligand–receptor interactions, the longer residence of particles, or the bulk of the particle as such. The histological observations of jejunum sections suggest both PLGA and PLGA–GA nanoparticles did not cause any damage to the epithelium that is primarily made of absorptive cells and other cell types such as goblet cells, Paneth cells, and endocrine cells, playing important protective functions in the intestine. The sections demonstrate normal morphology with intact villi and dense brush border (Figure 5).
Figure 5.

Intestinal morphology in rats administered with PLGA or PLGA–GA nanosystems orally at ~33 mg of particles (polymer)/rat daily for 10 days. The histological sections did not show any difference between PLGA or PLGA–GA nanosystems, suggesting G-mediated active delivery across the intestine is safe.
The mucus layer is often considered the first line of defense against foreign particles, and this can be tampered by various factors including the particles themselves, leading to change in the expression of mucins. The histological examination of alcian blue-treated intestine sections reveals a similar expression of goblet positive cells between PLGA and PLGA–GA nanosystems, further suggesting the safety of PLGA–GA particles (Figure 6).
Figure 6.

Alcian blue positive staining reveals mucinous goblet cells in the jejunum of rats administered with PLGA or PLGA–GA nanosystems orally at ~33 mg of particles (polymer)/rat daily for 10 days. The alcian blue staining in goblet cells is primarily due to mucin glycoprotein (MUC2). The staining is carried out at pH 2.5.
The liver is the primary organ for drug metabolism and detoxification; therefore, it remains a challenge to contemporary hepatology. Many drugs, once deemed safe, including Aspirin, have recently been associated with the risk of hepatotoxicity.33 Since these particles are transcytosed and will eventually get to the liver, it is important to assess their safety. The histology studies suggest that there were no major areas of confluent tissue necrosis or inflammation (Figure 7), and these findings corroborate with the blood chemistry panel presented in Table 3.
Figure 7.

Hepatic histology of rats receiving 10 doses of ~33 mg of PLGA or PLGA–GA particles (polymer)/rat. Montage of liver sections stained with H&E. Both PLGA and PLGA–GA groups showed similar morphology suggesting safety of the PLGA–GA systems.(a) Branch of portal vein, (b) branch of the bile duct, (c) branch of the hepatic artery, and (d) hepatocytes.
The spleen is a vital and largest secondary immune organ in the body and is the site of direct and indirect toxicity. The spleen is responsible for initiating immune reactions to blood-borne antigens and for filtering the blood of foreign material and old or damaged red blood cells. These functions are carried out by the 2 main compartments of the spleen, the white pulp and the red pulp, which are vastly different in their architecture, vascular organization, and cellular composition. Therefore, it is important to diagnose pathological changes in the red pulp and white pulp to ensure the safety of the particles. The histology evaluations suggest that there were no major changes in the red and white pulp areas, and no areas of confluent tissue necrosis or inflammation were observed, suggesting the safety of PLGA–GA particles (Figure 8).
Figure 8.

Morphology of the spleen of rats administered with PLGA or PLGA–GA nanosystems orally at ~33 mg of particles (polymer)/rat daily for 10 days. Montage of entire spleen sections stained with H&E. The magnified photomicrographs 10× and 60× reveal normal architecture based on clear visibility of marginal sinus (MS) and its sinus lining cells, marginal zone metalophilic macrophages on the PALS side of the marginal sinus, and the outer ring of the marginal sinus composed of marginal zone macrophages, dendritic cells, B-cells, and reticular cells and blends with the red pulp (RP). Also seen are erythrocytes percolating through the marginal zone (MZ). OP = outer periarteriolar lymphoid sheaths (PALS).
Due to very high blood flow into the kidney, the particles in circulation are bound to be exposed to renal parenchyma leading to accumulation in parenchymal cells and tubular laminal fluid causing toxicity. The kidney histology analysis reveals the intact epithelium in glomeruli and tubules suggesting PLGA or PLGA–GA did not cause any deleterious effects to the kidney (Figure 9). These data corroborate the urine analysis where the protein–creatinine ratio is similar to PLGA nanosystems (Table S5).
Figure 9.

Histology of kidney tissue derived from rats treated with 10 doses of PLGA or PLGA–GA nanosystems. No major histological abnormalities were seen in both groups. The renal tubules and focal flattening of renal tubular epithelial cells and glomeruli are intact (PAS staining: Montage image at 4× (64 images)).
In recent years, increasing attention has been paid to safety assessment of the endocrine system that is traditionally considered as a system of glands capable of releasing chemical mediators that act on distant targets from the site of production, due to reports on the effects of nanomaterials as endocrine disruptors.34 In this study, we have analyzed the effects of PLGA or PLGA–GA nanosystems on the pancreas and testis. The histological examination of pancreas shows well-defined islets of Langerhans with intact exocrine of pancreatic tissues (Figure 10). On the other hand, the histological examination of testis revealed that the morphological features of seminiferous tubules, interstitial tissue, sperm flagella, basement membrane of tubule, and primary spermatocytes were normal in appearance (Figure 10).
Figure 10.

Histology of pancreas and testis tissues derived from rats treated with 10 doses of PLGA or PLGA–GA nanosystems. No major histological abnormalities were seen in both the tissues. In the pancreas, yellow arrows show intact pancreatic “islets”, and the number of islets is insignificant between both the treatment groups suggesting PLGA–GA is safe. In testis, the absence of obvious changes such as cytoplasmic vacuolation, necrosis, apoptosis, and atrophy indicates PLGA–GA particles are safe.
Overall, PLGA–GA showed a comparable safety profile to that of PLGA, while the former is superior concerning improving bioavailability as well as altering the pharmacokinetics and distribution profiles of drugs that are difficult to deliver by conventional dosage forms.12,13,35 This study is important in taking these next-generation nanosystems further as we have, for the first time, identified the nonactive binding sites of GA on the TfR as well as established the safety of the PLGA–GA, where GA is traditionally used as a cytotoxic agent in cancer therapy.14 It is important to note that the best dose is the dose level that maximizes efficacy while assuring safety, while a good dose is that in which efficacy is above a predefined boundary while maintaining safety. The amount of GA associated with 50 mg of particles is about 1 mg. While that dose is not sufficient to cause cell death, it is good enough to enable active transport of the particles across the barriers, improving the delivery of the cargo. It is interesting to note that GA is evolving as a poster child for its one-compound multiuses, such as cytotoxic properties, expanding number of antioxidant and anti-inflammatory drug targets in various chronic diseases, and importantly as a noncompetitive ligand for TfR-mediated active transport of nanosystems.
CONCLUSION
The In silico studies reveal potential sites for GA binding on the TfR, inching close to establishing a structure–activity relationship. We have shown that the noncompetitive active delivery systems employing the small-molecule ligand GA are stable at room and refrigerated storage conditions and are safe on 10 daily doses administered to healthy rats. Together, these findings suggest that these noncompetitive delivery strategies, for their ability to bind to the TfR, independent of Tf presence, to promote transport, will have a significant impact on the efficient use of existing molecules that are poorly bioavailable, as well as minimize the attrition rates, if used in the early stages of drug discovery programs.
MATERIALS AND METHODS
In silico Identification of Gambogic Acid–Transferrin Receptor Binding Sites.
We used a combined methodology in which different site prediction tools, molecular docking and molecular dynamics simulations, were used to predict the binding site of GA at the TfR. The 3D crystal structure of the human TfR in complex with transferrin (Tf) [TfR–Tf] (PDB ID: 3S9N) was retrieved from the Brookhaven Protein Data Bank.36 Apo-TfR was used for further study. Protein was prepared using the protein preparation wizard of the Schrödinger software package (Schrödinger, LLC, New York, NY). The probable GA binding sites at the TfR were identified using SiteMap,20,21 SiteHound,22 CASTp (Computed Atlas of Surface Topography of proteins),22,23 and Achilles Blind Docking server (http://bio-hpc.ucam.edu/achilles/) which is a customized version of Autodock Vina. After the identification of the binding sites from different site identification tools, all final docking studies of GA were performed by the Glide module of the Schrödinger software package (Schrödinger, LLC, New York, NY) using standard operating procedures with the extra precision (XP) protocol. PLGA–GA was docked to the TfR using the PatchDock,21 and the top ten docking solutions were obtained based on lowest energy. These docking solutions were clustered based on a 4 Å root-mean-square (RMS) deviation. Prime-MMGBSA37 calculations were performed to determine the binding free energy of the PLGA–GA–TfR docked complex.
Molecular Dynamic Simulations.
A 50 ns molecular dynamics simulation of the complex (GA and TfR) was carried out using AMBER 12 (Assisted Model Building with Energy Refinement) software.38 TfR is a homodimer, so both the chains were used for the MD study. The charges for ligand were defined by the Antechamber38 program of AMBER using a semiempirical (AM1) wave function with the bond charge correction (BCC) model.39 The necessary topology and coordinate files were prepared using General AMBER force field (GAFF)38 and ff99SB force field for GA and TfR, respectively. The complex was solvated using TIP3P40 water with a cubical box extended to 20 Å in each direction. Na+/Cl− ions were added to the system to make it neutral. The system was minimized in three steps in which the first and second steps involve minimization of solvent phase. 1000 and 500 minimization cycles using a force constant of 3 kcal/mol/Å2 and 1 kcal/mol/Å2 were used for the first and second step minimization, respectively. The third step minimization was an unrestrained minimization of the 25 000 cycles. Then the system was heated gradually from 0 to 300 K over a period of 50 PS under constant volume–constant temperature (NVT) conditions. A three-step density equilibration was performed in which the first 2 steps (50 PS each) had a restraint weight of 2 kcal/mol/Å2 and 1 kcal/mol/Å2, respectively, followed by the third step that included unrestrained density equilibration of 100 ps. The final equilibrated system was used for a 50 ns production run, and every 1 ns time interval was used to record the coordinates. The molecular mechanics-generalized born surface area (MM-GBSA) method was used to calculate the binding energies of the GA with the TfR at different time intervals. Hydrogen bond occupancy was calculated using the Visual Molecular Dynamics (VMD) package.41
Synthesis and Characterization of PLGA–GA.
The poly-(lactic-co-glycolic acid)–gambogic acid (PLGA–GA) conjugation is achieved according to protocols previously developed in our laboratory.12,13 Step 1: Preparation of PLGA–EDA: In brief, PLGA [50:50 Resomer R503H; MW 35–40 kDa: 500 mg; 0.016 mmol] was dissolved in 5 mL of dicholoromethane (CH2Cl2), and 1-ethyl-3-(dimethylaminopropyl) carbodiimide (EDC) [15.43 mg; 0.048 mmol] was added to stirring solution under nitrogen atmosphere and continued stirring for 30 min. After 30 min, n-boc-ethyelenediamine (12.75 μL; 0.048 mmol) and N,N-diisopropylethylamine (DIEA) (14.02 μL; 0.048 mmol) were added to the above reaction mixture and further continued stirring under inert conditions for 18 h at room temperature. Afterward, this reaction mixture was purified with cold diethyl ether washing (3 ×) which resulted in a white residue. The residue was dried under vacuum to get a dry powder (~400 mg). Further, this dry powder (~400 mg) was dissolved in 30% CH2Cl2:TFA solution and stirred under nitrogen atmosphere at 4 °C for 2 h. The solvent was evaporated on a rotary evaporator to get a clear viscous residue. The residue was dissolved in DCM and washed with diethyl ether washing (3 ×). The purified PLGA–EDA was dried under vacuum to a constant dry weight (~350 mg).
Step 2: Preparation of PLGA–GA: Gambogic acid (15.09 mg; 0.024 mmol) was dissolved in CH2Cl2 (1 mL) under nitrogen atmosphere. To the above solution, EDC (4.60 mg; 0.024 mmol) was added, and stirring was continued under nitrogen atmosphere for 30 min. To the GA–EDC mixture was added a solution of PLGA–EDA (~250 mg; 0.008 mmol) and DIEA (4.18 μL; 0.024 mmol) in CH2Cl2 (2 mL). The reaction mixture was further stirred for 18 h at room temperature. Further, the reaction mixture was precipitated in 50 mL of cold diethyl ether. The polymer was purified by dissolving in 3 mL of CH2Cl2 followed by precipitation in cold diethyl ether (3 ×). Lastly, the polymer was washed with distilled water and dried under vacuum to a constant dry weight of PLGA–GA (~150 mg). The PLGA–GA was characterized by various spectroscopic methods such as 1H NMR, FTIR, and UV spectroscopy.12,13
Preparation and Characterization of PLGA–GA Nano-systems.
The nanosystems were prepared by an oil-in-water emulsification and solvent evaporation method. In brief, the organic phases of PLGA–GA (50 mg) and CUR (7.5 mg, used as a model bioactive) were prepared separately in 2 and 0.5 mL of ethyl acetate, respectively, and mixed afterward, while the aqueous phase of poly(vinyl alcohol) (PVA, 60 mg) was prepared in 5 mL of water. The organic phase was emulsified into aqueous PVA solution under stirring for 7 min at 1500 rpm forming o/w emulsion followed by homogenization at 15 000 rpm for 7 min. The emulsion was added to 20 mL of water to facilitate the diffusion of ethyl acetate into the aqueous phase and subsequently evaporated. The curcumin-encapsulated nanoparticles were collected as pellets by centrifugation of the emulsion at 15 000g for 30 min at 4 °C. The pellet was resuspended in 5% sucrose solution and lyophilized (prefreezed for 3 h at −80 °C; ramp 1.0 °C/min hold at −50 °C for 48 h, later maintained at 20 °C for 12 h to ensure complete removal of water) for further use. The freeze-dried nanosystems upon resuspension were characterized for size and morphology using light-scattering technique (DLS) and scanning electron microscopy (SEM). The presence of GA on the nanosystem surface was confirmed by UV–vis analysis.12,13
Shelf-Stability Testing of PLGA–GA Nanosystems.
The freeze-dried CUR-encapsulated PLGA–GA nanosystems (~2.2 mg of CUR/vial, n = 24) were subjected to refrigerated (25 ± 2 °C/60 ± 5%RH) [Binder KBFP240 climate chamber] and room temperature (40 ± 2 °C/75 ± 5%RH) [Binder KBFP720 climate chamber] conditions over six months according to International Conference on Harmonization (ICH) guidelines. Each month, four vials were analyzed for particle size, morphology, and CUR content and were compared against zero time point freeze-dried samples.
Safety Assessment of PLGA–GA Nanosystems.
To assess the preliminary safety of PLGA–GA nanosystems, experiments were conducted using adult male Sprague–Dawley rats (220–250 g; ENVIGO) and PLGA nanosystems as control. A total of 12 rats (n = 6 per group) were used for the entire study. All procedures and protocols were approved by the Institutional Animal Care and Use Committee of Texas A&M University Institutional Animal Care and Use Committee and were performed in accordance with the National Institutes of Health Guide for the care and use of laboratory animals. The animals were housed in standard conditions and allowed free access to food and water throughout the experiment. The rats were administered with a daily single dose, a total of ten doses of PLGA–GA or PLGA (~33 mg of particles (polymer)/rat; equivalent to 20 mg/kg of CUR) nanosystems dispersed in water by oral gavage. During the study, we monitored the rats on a daily basis for general health as well as signs of acute toxicity, e.g., locomotor activity, prostration, rapid shallow breathing, pallor, and profound dullness. We terminated the study 2 h after the last dose, to collect blood and other organs for hematological and histological examination. Blood samples before the start of the experiment were collected via tail vein that served as a control group (before dosing). A complete blood picture (hematology and chemistry panel) was obtained using an automated analyzer. The intestine, kidney, liver, testis, spleen, and pancreas were isolated immediately on sacrifice. The tissues were washed with ice-cold saline and weighed before fixing in 10% neutral buffered formalin solution. The tissues were embedded in paraffin blocks for histopathological examination. Five micrometer thick sections were cut, deparaffinized, hydrated, and stained with hematoxylin and eosin, alcian, or PAS. The histological sections were examined under microscope [(Cytation 5 (biotech)] for any morphological changes.
Statistical Analysis.
The data were analyzed using multifactor ANOVA and unpaired t test. A value of p < 0.05 was considered statistically significant, and p > 0.05 was considered insignificant.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by National Institutes of Health (Grant No. R01EY028169). N. Kumar is thankful to NIPER S.A.S. Nagar for financial support in the form of a Fellowship.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsabm.9b00419.
Experimental and characterization data (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Petrus AK; Fairchild TJ; Doyle RP Traveling the vitamin B12 pathway: oral delivery of protein and peptide drugs. Angew. Chem., Int. Ed 2009, 48 (6), 1022–28. [DOI] [PubMed] [Google Scholar]
- (2).Fowler R; Vllasaliu D; Trillo FF; Garnett M; Alexander C; Horsley H; Smith B; Whitcombe I; Eaton M; Stolnik S Nanoparticle transport in epithelial cells: pathway switching through bioconjugation. Small 2013, 9 (19), 3282–3294. [DOI] [PubMed] [Google Scholar]
- (3).Anderson KE; Eliot LA; Stevenson BR; Rogers JA Formulation and evaluation of a folic acid receptor-targeted oral vancomycin liposomal dosage form. Pharm. Res 2001, 18 (3), 316–322. [DOI] [PubMed] [Google Scholar]
- (4).Roger E; Kalscheuer S; Kirtane A; Guru BR; Grill AE; Whittum-Hudson J; Panyam J Folic acid functionalized nanoparticles for enhanced oral drug delivery. Mol. Pharmaceutics 2012, 9 (7), 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pridgen EM; Alexis F; Kuo TT; Levy-Nissenbaum E; Karnik R; Blumberg RS; Langer R; Farokhzad OC Transepithelial transport of Fc-targeted nanoparticles by the neonatal fc receptor for oral delivery. Sci. Transl. Med 2013, 5 (213), 213ra167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Du W; Fan Y; Zheng N; He B; Yuan L; Zhang H; Wang X; Wang J; Zhang X; Zhang Q Transferrin receptor specific nanocarriers conjugated with functional 7peptide for oral drug delivery. Biomaterials 2013, 34 (3), 794–806. [DOI] [PubMed] [Google Scholar]
- (7).Lundquist P; Artursson P Oral absorption of peptides and nanoparticles across the human intestine: Opportunities, limitations and studies in human tissues. Adv. Drug Delivery Rev 2016, 106, 256–276. [DOI] [PubMed] [Google Scholar]
- (8).Bourassa P; Alata W; Tremblay C; Paris-Robidas S; Calon F Transferrin Receptor-Mediated Uptake at the Blood-Brain Barrier Is Not Impaired by Alzheimer’s Disease Neuropathology. Mol. Pharmaceutics 2019, 16 (2), 583–594. [DOI] [PubMed] [Google Scholar]
- (9).Johnsen KB; Bak M; Melander F; Thomsen MS; Burkhart A; Kempen PJ; Andresen TL; Moos T Modulating the antibody density changes the uptake and transport at the blood-brain barrier of both transferrin receptor-targeted gold nanoparticles and liposomal cargo. J. Controlled Release 2019, 295, 237–249. [DOI] [PubMed] [Google Scholar]
- (10).Wiley DT; Webster P; Gale A; Davis ME Transcytosis and brain uptake of transferrin-containing nanoparticles by tuning avidity to transferrin receptor. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (21), 8662–8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yefimova MG; Jeanny JC; Guillonneau X; Keller N; Nguyen-Legros J; Sergeant C; Guillou F; Courtois Y Iron, ferritin, transferrin, and transferrin receptor in the adult rat retina. Invest Ophthalmol Vis Sci. 2000, 41 (8), 2343–2351. [PubMed] [Google Scholar]
- (12).Saini P; Ganugula R; Arora M; Kumar MN The Next Generation Non-competitive Active Polyester Nanosystems for Transferrin Receptor-mediated Peroral Transport Utilizing Gambogic Acid as a Ligand. Sci. Rep 2016, 6, 29501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ganugula R; Arora M; Guada M; Saini P; Kumar MN V. R. Noncompetitive Active Transport Exploiting Intestinal Transferrin Receptors for Oral Delivery of Proteins by Tunable Nanoplatform. ACS Macro Lett. 2017, 6 (2), 161–164. [DOI] [PubMed] [Google Scholar]
- (14).Kasibhatla S; Jessen KA; Maliartchouk S; Wang JY; English NM; Drewe J; Qiu L; Archer SP; Ponce AE; Sirisoma N; Jiang S; Zhang HZ; Gehlsen KR; Cai SX; Green DR; Tseng B A role for transferrin receptor in triggering apoptosis when targeted with gambogic acid. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (34), 12095–12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Pandey MK; Karelia D; Amin SG Gambogic Acid and Its Role in Chronic Diseases. Adv. Exp. Med. Biol 2016, 928, 375–395. [DOI] [PubMed] [Google Scholar]
- (16).Qi Q; You Q; Gu H; Zhao L; Liu W; Lu N; Guo Q Studies on the toxicity of gambogic acid in rats. J. Ethnopharmacol 2008, 117 (3), 433–438. [DOI] [PubMed] [Google Scholar]
- (17).Chi Y; Zhan XK; Yu H; Xie GR; Wang ZZ; Xiao W; Wang YG; Xiong FX; Hu JF; Yang L; Cui CX; Wang JW An open-labeled, randomized, multicenter phase IIa study of gambogic acid injection for advanced malignant tumors. Chin Med. J. (Engl) 2013, 126 (9), 1642–1646. [PubMed] [Google Scholar]
- (18).Cheng Y; Zak O; Aisen P; Harrison SC; Walz T Structure of the human transferrin receptor-transferrin complex. Cell 2004, 116 (4), 565–576. [DOI] [PubMed] [Google Scholar]
- (19).Huebers HA; Finch CA The physiology of transferrin and transferrin receptors. Physiol. Rev 1987, 67 (2), 520–582. [DOI] [PubMed] [Google Scholar]
- (20).Halgren T New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des 2007, 69 (2), 146–148. [DOI] [PubMed] [Google Scholar]
- (21).Halgren TA Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model 2009, 49 (2), 377–389. [DOI] [PubMed] [Google Scholar]
- (22).Hernandez M; Ghersi D; Sanchez R SITEHOUND-web: a server for ligand binding site identification in protein structures. Nucleic Acids Res. 2009, 37 (Web Server), W413–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Dundas J; Ouyang Z; Tseng J; Binkowski A; Turpaz Y; Liang J CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34 (Web Server), W116–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Schneidman-Duhovny D; Inbar Y; Nussinov R; Wolfson HJ PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33 (WebServer), W363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Shaikh J; Ankola DD; Beniwal V; Singh D; Kumar MN Nanoparticle encapsulation improves oral bioavailability of curcumin by at least 9-fold when compared to curcumin administered with piperine as absorption enhancer. Eur. J. Pharm. Sci 2009, 37 (3–4), 223–230. [DOI] [PubMed] [Google Scholar]
- (26).Grama CN; Venkatpurwar VP; Lamprou DA; Ravi Kumar MN Towards scale-up and regulatory shelf-stability testing of curcumin encapsulated polyester nanoparticles. Drug Delivery Transl. Res 2013, 3 (3), 286–293. [DOI] [PubMed] [Google Scholar]
- (27).Grama CN; Suryanarayana P; Patil MA; Raghu G; Balakrishna N; Kumar MN; Reddy GB Efficacy of biodegradable curcumin nanoparticles in delaying cataract in diabetic rat model. PLoS One 2013, 8 (10), e78217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ganugula R; Arora M; Jaisamut P; Wiwattanapatapee R; Jorgensen HG; Venkatpurwar VP; Zhou B; Rodrigues Hoffmann A; Basu R; Guo S; Majeti N Nano-curcumin safely prevents streptozotocin-induced inflammation and apoptosis in pancreatic beta cells for effective management of Type 1 diabetes mellitus. Br. J. Pharmacol 2017, 174 (13), 2074–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Simon-Yarza T; Tamayo E; Benavides C; Lana H; Formiga FR; Grama CN; Ortiz-de-Solorzano C; Kumar MN; Prosper F; Blanco-Prieto MJ Functional benefits of PLGA particulates carrying VEGF and CoQ10 in an animal of myocardial ischemia. Int. J. Pharm 2013, 454 (2), 784–790. [DOI] [PubMed] [Google Scholar]
- (30).Kim KS; Suzuki K; Cho H; Youn YS; Bae YH Oral Nanoparticles Exhibit Specific High-Efficiency Intestinal Uptake and Lymphatic Transport. ACS Nano 2018, 12 (9), 8893–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zheng Y; Wu J; Shan W; Wu L; Zhou R; Liu M; Cui Y; Zhou M; Zhang Z; Huang Y Multifunctional Nanoparticles Enable Efficient Oral Delivery of Biomacromolecules via Improving Payload Stability and Regulating the Transcytosis Pathway. ACS Appl. Mater. Interfaces 2018, 10 (40), 34039–34049. [DOI] [PubMed] [Google Scholar]
- (32).Trevaskis NL; Kaminskas LM; Porter CJ From sewer to saviour - targeting the lymphatic system to promote drug exposure and activity. Nat. Rev. Drug Discovery 2015, 14 (11), 781–803. [DOI] [PubMed] [Google Scholar]
- (33).Cao Z; Liu S; Niu J; Wei B; Xu J Severe hepatoxicity caused by aspirin overdose: a case report. Front Med. 2015, 9 (3), 388–91. [DOI] [PubMed] [Google Scholar]
- (34).Iavicoli I; Fontana L; Leso V; Bergamaschi A The effects of nanomaterials as endocrine disruptors. Int. J. Mol. Sci 2013, 14 (8), 16732–16801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kaur G; Arora M; Ganugula R; Kumar M Double-headed nanosystems for oral drug delivery. Chem. Commun. (Cambridge, U. K.) 2019, 55, 4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res. 2000, 28 (1), 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Du J; Sun H; Xi L; Li J; Yang Y; Liu H; Yao X Molecular modeling study of checkpoint kinase 1 inhibitors by multiple docking strategies and prime/MM-GBSA calculation. J. Comput. Chem 2011, 32 (13), 2800–2809. [DOI] [PubMed] [Google Scholar]
- (38).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and testing of a general amber force field. J. Comput. Chem 2004, 25 (9), 1157–74. [DOI] [PubMed] [Google Scholar]
- (39).Jakalian A; Jack DB; Bayly CI Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem 2002, 23 (16), 1623–1641. [DOI] [PubMed] [Google Scholar]
- (40).Jorgensen WL; Chandrasekhar J; Buckner JK; Madura JD Computer simulations of organic reactions in solution. Ann. N. Y. Acad. Sci 1986, 482, 198–209. [DOI] [PubMed] [Google Scholar]
- (41).Humphrey W; Dalke A; Schulten K VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14 (1), 33–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.