

Abstract
Objective
To evaluate the safety and efficacy of cholecalciferol in patients with relapsing-remitting MS (RRMS).
Methods
In this double-blind, placebo-controlled parallel-group, 2-year study, 181 patients with RRMS were randomized 1:1. Key inclusion criteria were a low serum 25-hydroxy vitamin D (25OHD) concentration (<75 nmol/L), a treatment with interferon beta-1a 44 μg (SC 3 times per week) 4 months ± 2 months before randomization, and at least one documented relapse during the previous 2 years. Patients received high-dose oral cholecalciferol 100,000 IU or placebo every other week for 96 weeks. Primary outcome measure was the change in the annualized relapse rate (ARR) at 96 weeks. Secondary objectives included safety and tolerability of cholecalciferol and efficacy assessments (ARR, MRI parameters, and Expanded Disability Status Scale [EDSS]).
Results
The primary end point was not met. In patients who completed the 2-year follow-up (45 with cholecalciferol and 45 with placebo), all efficacy parameters favored cholecalciferol with an ARR reduction (p = 0.012), less new hypointense T1-weighted lesions (p = 0.025), a lower volume of hypointense T1-weighted lesions (p = 0.031), and a lower progression of EDSS (p = 0.026). The overall rate of adverse events was well balanced between groups.
Conclusions
Although the primary end point was not met, these data suggest a potential treatment effect of cholecalciferol in patients with RRMS already treated with interferon beta-1a and low serum 25OHD concentration. Together with the good safety profile, these data support the exploration of cholecalciferol treatment in such patients with RRMS.
Clinicaltrials.gov identifier
Classification of evidence
This study provides Class II evidence that for patients with RRMS and low serum 25OHD, cholecalciferol did not significantly affect ARRs.
Optimizing treatment in relapsing-remitting MS (RRMS) is an unmet need. Immunomodulators or immunosuppressants are efficient to lower relapse rate, but they are not deprived of adverse effects, and poor response to treatment is still a concern.1,2
Vitamin D (VD) is a biological regulator of the immune system.3 VD deficiency is a risk factor for MS, and the risk of relapse and the risk of having new MRI lesions are inversely correlated with 25-hydroxy vitamin D (25OHD) blood levels, both in adults and children.4–8 VD is safe even at very high doses in RRMS.9 Double-blind, placebo-controlled trials gave controversial results and were characterized by a methodological heterogeneity.10,11 All studies with cholecalciferol, but one, gave positive results on biological parameters of inflammation, on MRI parameters of MS activity, or on quality of life.12–15 However, none of these studies showed a clinical benefit with VD treatment. The present study assessed the safety and efficacy of cholecalciferol in patients with RRMS, already treated with interferon beta-1a 44 μg subcutaneously 3 times per week (SCTIW), using 2 original parameters at inclusion, clinically active RRMS and VD insufficiency, combined with a 2-year duration.
Methods
Primary research question
This phase 2, multicenter, randomized, double-blind, placebo-controlled parallel-group study was designed to evaluate the efficacy of cholecalciferol (Crinex laboratories, Montrouge, France) in patients with RRMS that was conducted from January 2010 to June 2013 at 27 sites in France. Class II evidence is provided here as more than 20% of randomized patients did not complete the study.
Standard protocol approvals, registrations, and participant consents
Before initiating the study, the study protocol was approved by a national review board, the CPP Sud-Méditerranée IV ethics committee. Written informed consent was obtained from all the study participants before any study-related procedures were performed (ClinicalTrials.gov identifier NCT01198132). The study was conducted in accordance with the International Conference on Harmonization Guidelines for Good Clinical Practice and the Declaration of Helsinki.
Patients
Eligible patients were aged between 18 and 65 years, with a diagnosis of RRMS based on the 2005 revised McDonald criteria,16 low serum 25OHD concentration (VDC, < 75 nmol/L), a treatment with interferon beta-1a 44 μg (or 22 μg in case of intolerance to 44 μg) SCTIW for 4 months ± 2 months at the randomization visit, and an Expanded Disability Status Scale (EDSS) score from 0 to 5.0, with at least one documented relapse during the previous 2 years and stable disease (no episode) over the last 30 days. Main exclusion criteria were use of drugs affecting VD metabolism other than corticosteroids; previous or ongoing hypercalcemia; and abnormal renal function defined by eGFR < 60 mL/min. Eligible patients were identified in the clinic at each study site and recruited according to the protocol.
Study design and treatment
After enrollment, randomization was performed within an 8-week period, and patients were randomly assigned (1:1) to receive cholecalciferol or placebo at the second study visit. The composition, appearance, and packaging of placebo treatment were identical to those of active treatment. When starting medication, each subject was assigned a randomization number according to a chronological sequential order per site. A unique randomization number was assigned to each subject at the initial visit. This number was retained by the subject until the completion of the study. Treatment kits were labeled with batch number, expiry date, randomization number, storage/administration conditions, and a kit identification number.
Randomized patients were treated for 96 weeks. Treatment was an oral solution packaged in 2 mL yellow glass (type III) ampoules with 2 self-breakable tips, containing 2.5 mg of cholecalciferol corresponding to 100,000 IU or placebo. Concomitant medications included medications that had started before the first dose of cholecalciferol or placebo and continued after the first dose of blinded treatment. Study visits were V1 (screening, week W-8), V2 (baseline, W0), V3 (W24), V4 (W48), V5 (W72), and V6 (W96). At each visit from V2 to V5, patients received a kit to self-administering orally an ampoule every other week (equivalent to a daily dose of 7,143 IU of cholecalciferol) during the 96-week treatment period. Clinical (EDSS and Paced Auditory Serial Addition Task 3, second version [PASAT-3]) and quality of life (EQ-5D-3L) assessments were performed at all study visits since baseline. MRI scans were performed at baseline and V6 and were analyzed at a central MRI center by trained staff masked to study group assignment. Screening and baseline routine laboratory tests including blood 25OHD, renal function, and calcium levels were performed to comply with inclusion and exclusion criteria and repeated each month during the first 6 months and then quarterly to ensure safety.
Outcomes
The primary objective was the reduction of the annualized relapse rate (ARR), defined as the ratio of the number of relapses under treatment by the exposure duration, in years. Key secondary objectives were time to first relapse; EDSS progression; MRI parameters (number of new or enlarged T1-and T2-weighted lesions, T2-weighted MRI lesion load, number of T1-weighted Gd-enhancing lesions, volume change of hypointense T1-weighted MRI lesions, total brain volume, and brain gray and white matter volume); change in cognitive abilities; and change in quality of life and treatment safety.
Safety monitoring
Safety was assessed from reports of adverse events, serious adverse events, disease-related adverse events (DRAEs), routine laboratory tests, and vital signs. DRAEs were coded according to the Medical Dictionary for Regulatory Activities coding system. Values for clinical laboratory assessments were compared with both the appropriate normal ranges and ranges of potential clinical concern. Any abnormal test result or other safety assessment judged by the investigator to be clinically significant was recorded as an adverse event or a serious adverse event.
Population and statistical analysis
The Full Analysis Set was constituted by all the randomized patients (intention to treat [ITT] population). Supportive and post hoc analyses were conducted on the ITT set, after exclusion of subjects with major deviations, and a completers' population, which corresponded to the group of patients who completed the 96-week trial, as only those patients underwent EDSS and MRI examination at the final visit after 2 years (V6).
The primary efficacy analysis compared the cholecalciferol group with placebo using a generalized linear model featuring a Poisson regression adjusted for baseline values of annualized relapse rate (ARRb), sex, age, EDSS, controlled for overdispersion, and using the log-transformed exposure time as an offset variable. ARR values were descriptively reported by geometric means due to the positive skewness of the distribution.
The trial was powered according an assumed rate ratio (rR) between cholecalciferol and placebo of 0.75, a mean number of 1 relapse per year, and a correlation R(ARRb, ARR) of 0.7. Under these conditions, a sample size of 106 patients/group was needed for a power of at least 0.75 at a 95% 2-sided significance level.
Missing ARR data on the number of relapses after premature interruption were imputed by baseline ARR values.
Time to first relapse was compared using Cox regression adjusted on baseline ARR. Any dropout was considered as failure at trial interruption time. For sensitivity purposes, a supportive analysis was based in differentiating on drug-related dropout considered as failure, with drug-unrelated dropout considered as censored at dropout date, the differentiation established by an adjudication committee blind to treatment.
MRI values at V6, for the completers' population, were compared between treatment groups in adjusting for baseline, sex, and age. Log transformation was used for the number of new or enlarged T1- and T2-weighted lesions and the number of T1-weighted Gd lesions, and the ratio Final/Baseline was reported. Disability progression was defined as increase of at least 1 unit from baseline EDSS. EDSS at last visit was compared between the 2 groups by a linear model adjusting for baseline.
Supportive analyses were as follows: (1) for early drug-related dropouts (either for poor efficacy or safety), the short exposure duration inducing relapse-free profile (ARR = 0) may falsely conclude into therapy success: we corrected this bias in evaluating a corrected ARR in assuming post-dropout period ARR by baseline ARR following the Baseline Observed Carried Forward (BOCF) principle; and (2) final EDSS, for drug-related dropouts, was also assimilated to BOCF. Although ARR was the main end point of this study, progression of the disease now constitutes a primary concern for regulatory authorities. Therapy success may thus be considered on both end points: a patient characterized by relapse-free follow-up and without EDSS progression (EDSSb ≥ EDSSF). Response rate was compared between the 2 treatment groups in adjusting for baseline EDSS and ARRb. A response based on MRI was also defined in considering therapy success when radiologic disease-free status was observed (i.e., no Gd-enhancing lesions and no new or enlarging T2-weighted lesions on brain MRI). A last alternative combining the 2 first response definitions is the recently proposed No Evidence of Disease Activity (NEDA-3) binary end point defined as no relapses + no sustained disability progression + radiologic disease-free status.17
The effect of serum 25OHD concentration (VDC), measured at each visit, was also assessed. The summary mean of VDC (VDCM) was calculated during the follow-up period (V3 to V6). Poisson regression and linear model tested the VDCM effect on relapse count and EDSS, in adjusting for baseline values. The PASAT-3 and the EQ-5D-3L scores were compared between treatment groups using analysis of covariance adjusting for baseline value.
The safety analysis was performed on all randomized patients who received at least 1 dose of study drug during the study. The number of treatment emergent adverse events (TEAEs) of any type and the total number of subjects with at least one TEAE were compared between treatment groups. The same analysis was performed in comparing suspected DRAEs, serious DRAEs (SDRAEs), and deaths.
The study was analyzed in compliance with standard Good Clinical Practice regulations, database, and statistical analysis plan locked before unblinding. Software package SAS, version 9.4 (SAS Institute Inc) and R version 3.3.2 were used for analysis.
Data availability
We provide qualified researchers access to individual patient-level data through the Merck KGaA's data-sharing portal: merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html.
Results
Study population
Between January 2010 and June 2013, a total of 181 patients were screened, and 129 were randomized, constituting the ITT population (figure). The most common reason for failure to be randomized was a normal blood 25OHD level. Demographics and baseline characteristics of ITT population are shown in table 1. A total of 39 dropouts were recorded during the 2-year follow-up. Most reasons of dropout were related to the treatment of RRMS with either a lack of efficacy (relapse) or to patient willing or physician deciding to switch to another disease-modifying drug. A total of 90 patients (45 in each treated group) completed the trial, constituting the completers' population.
Figure. Trial flowchart.

Table 1.
Patient characteristics

Efficacy
A total of 85 patients (65.8%) were relapse-free, and the highest value of ARR = 1.5 was observed for 2 patients. Mean ARR in the cholecalciferol group was 0.094 compared with 0.110 for placebo; this difference was not statistically significant (ITT sample: rR = 0.799, 95% CI 0.481–1.32, p = 0.38, Poisson regression). Conversely, ARR was significantly lower on the completers' population: rR = 0.395, 95% CI 0.186–0.012, p = 0.01. A total of 19 relapses occurred in the cholecalciferol group compared with 25 with placebo. Time to first relapse was not significantly different between the 2 groups (hazard ratio [HR] = 0.801; 95% CI 0.403–1.454; p = 0.43).
For completers, at baseline, MRI measurements were in accordance with the protocol for 49 patients in the cholecalciferol group and for 47 patients in the placebo group, and final values at last visit were available for 44 and 41 of them. A significant mean reduction of new T1 lesions (rate ratio rR = 0.494, 95% CI 0.267–0.913; p = 0.03) and a decrease in the volume of hypointense T1-weighted MRI lesions (−312 mm3, 95% CI −596 to −29; p = 0.03) were recorded in the cholecalciferol group vs placebo. There were no significant changes in either of the other MRI parameters studied (number of Gd-enhancing T1 and new T2 lesions, as well as brain, T2 lesions, and gray matter and white matter volumes). Average progression of the EDSS score was significantly lower in the cholecalciferol group (−0.06 ± 0.78) compared with the placebo group (0.32 ± 0.87, 95% CI −0.614 to −0.043; p = 0.03). There was no difference in change of quality of life measured by the EQ-5D-3L or the PASAT-3 total score between the 2 experimental groups.
VDC at baseline was similar between treatment groups, whereas cholecalciferol administration roughly tripled, at V6, mean VDC compared with the placebo group: 156.92 mmol/L vs 57.23 mmol/L, p < 0.001. VDC showed a significant reduction effect on ARR (rR = 0.995 per nmol/L [95% CI 0.990–0.999], p = 0.04) and on EDSS progression with a mean decrease of EDSS of −0.003 per 1 nmol/L increase of VDC (95% CI −0.006 to −0.001; p = 0.006).
Post hoc analysis
There were 39 dropouts, almost 3 times higher than initially estimated. Drug-related dropouts were the most frequent cause. The corrected ARRc, used to avoid the potential bias of early dropout on ARR estimate, was significantly lower in the cholecalciferol group both in the ITT (rR = 0.502, 95% CI 0.326–0.764; p = 0.001) and in the completers' population (rR = 0.395, 95% CI 0.186–0.801; p = 0.01). When dropouts were considered as relapse, the observed effect of the cholecalciferol treatment was significant (HR = 0.508, 95% CI 0.286–0.901; p = 0.02). When considering drug-related dropout as a failure, 66% fewer relapses were found in the cholecalciferol group compared with placebo (HR = 0.333, 95% CI 0.164–0.675; p = 0.002). The proportion of relapse-free patients at last visit (V6) was not significantly higher in the cholecalciferol group (69.8%) vs placebo (62.1%, OR = 1.70, 95% CI 0.77–3.76; p = 0.19). When considering the ITT population and assimilating drug-related dropouts as relapsing, these proportions became 65.1% and 45.5% and were significantly different (OR = 3.22, 95% CI 1.45–7.19; p = 0.004). Similarly, in the completers' population, these proportions of relapse-free patients between cholecalciferol and placebo groups were significantly different (75.6% and 60%, respectively, OR = 3.24, 95% CI 1.14–9.22; p = 0.03). A bias being possible in case of an unbalance between completers and noncompleters, baseline demographics were compared between these 2 groups with the same set of parameters as for the ITT population in table 1. There was a female predominance in the noncompleters group, 82.1%, compared with completers, 63.3% (p = 0.03). No statistical difference could be noted between the 2 groups for any of the other demographic parameters.
When responders were defined by the absence of relapse (ARR = 0) and progression (EDSSf-EDSSb ≤ 1), 49.2% and 31.2% responders were found in the cholecalciferol group and the placebo group (OR = 2.18, 95% CI 1.05–4.53; p = 0.04). Similarly, the radiologic disease-free status was also in favor of the cholecalciferol treatment (OR = 2.172, 95% CI 1.016–4.640; p = 0.045). The NEDA-3 binary end point was met by 22.1% patients in the cholecalciferol group compared with 9.1% with placebo (OR = 3.44, 95% CI = 1.14–10.33; p = 0.03).
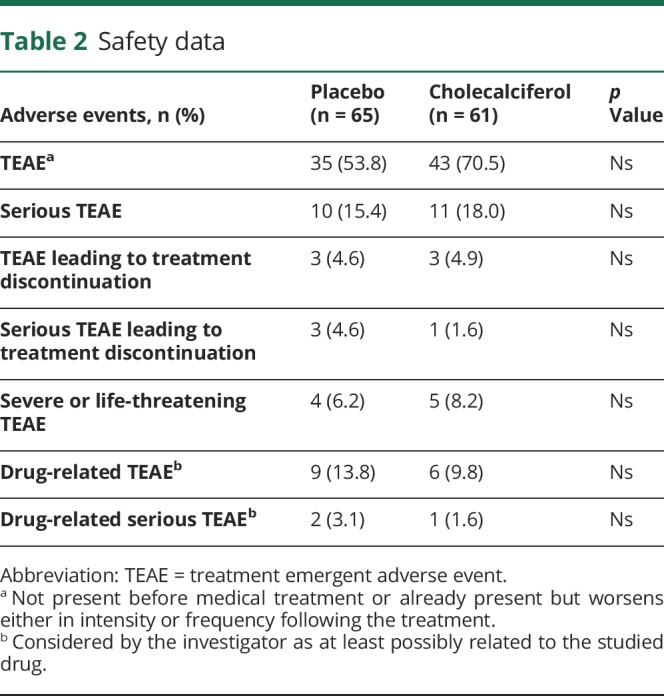
Safety
A total of 126 subjects (61 cholecalciferol, 65 placebo) were known to have been treated at least once during this study either by cholecalciferol or placebo (table 2). No patient died during the study. At least one TEAE was recorded in 78 subjects (61.9%): 43 in the cholecalciferol group (70.5%) and 35 in the placebo group (53.8%). Serious TEAEs were noted in 16.7% of the patients: 15.4% in the cholecalciferol group and 18% with placebo. A total of 6 TEAEs (4.8%) leading to treatment discontinuation were recorded: 3 (4.9%) in the cholecalciferol and 3 (4.6%) in the placebo group. Nine subjects (7.1%) discontinued treatment because of severe or life-threatening TEAEs: 5 patients (8.2%) in the cholecalciferol group and 4 (6.2%) for placebo. In the VD group, TEAEs were appendicitis, back pain, skin necrosis, fetal death, and breast cancer. The 4 severe or life-threatening TEAEs in the placebo group were abdominal abscess, lung malignant neoplasm, peripheral arterial occlusive disease, and nephrolithiasis. A total of 15 subjects (11.9%) had drug-related TEAEs, and 3 others (2.4%) had SDRAEs.
Table 2.
Safety data
Discussion
The present study, in line with previous works, showed that cholecalciferol at 100,000 IU every 2 weeks was well tolerated. The study did not demonstrate a reduction of the ARR, the primary efficacy measure, in the ITT population. However, in the completers' population, treatment with cholecalciferol was associated with a lower ARR, a reduction of the number of new T1 lesions, a lower volume of hypointense T1-weighted MRI lesions, and with a significantly lower progression of EDSS. Although post hoc analysis may be considered as “hypothesis generating,” it is in accordance with previous works showing that patients with low VDC were more likely to have MRI lesions, worse EDSS score, and more relapses.10 Of interest, in the present study, the number of relapses, adjusted for baseline, significantly decreased with increased mean 25OHD concentrations, and a benefit on EDSS progression was also noted.
To date, CHOLINE is the largest 2-year randomized controlled trial in RRMS using cholecalciferol. It was constructed with a cautious selection of patients to prevent some caveats from previous studies. Three selection criteria, at least, seemed key ones. The first one was to recruit clinically active RRMS patients, as ARR was the primary endpoint. Second, cholecalciferol was started in patients already treated with interferon beta-1a, as it has been proposed that those 2 molecules may act either synergistically or additively on blood monocytes and on MS parameters of activity.18–21 Third, a low 25OHD blood level was required for inclusion, a large literature showing deleterious effects of VD deficiency on human health with the lowest blood levels being associated with the worst conditions.
An important limitation of this study is its statistical power. One first reason for that was the difficulty to achieve inclusion goal (n = 210). This was likely due to the number of other alternatives for treating RRMS. It thus was decided to extend the inclusion period from 2 years to 3.5 years, but only 181 patients could be screened. Second, the sample size had been calculated on the basis of 1 relapse per year. This was an overestimation, all the more as interferon beta-1a was expected to reduce relapse rate. Third, dropouts, initially estimated at 10%, accounted in fact for 28.6% for the cholecalciferol group and 31.8% for placebo. Although unexpected and potentially explained by the numerous alternatives to the interferon treatment that led patients or physicians, or both, to discontinue the trial, we also considered this high dropout rate as potentially meaningful all the more as demographics of completers and noncompleters were similar. This suggests that dropouts were not biased by a difference in disease profile or disease severity at onset, and this was the basis for post hoc analyses.
Although the present study did not meet the primary end point of reducing the relapse rate in patients with RRMS, results in the completer's population are encouraging and support the use of the methodological parameters defined in this work. Further trials are warranted to refine the therapeutic interest of cholecalciferol in patients with RRMS.
Glossary
- 25OHD
25-hydroxy vitamin D
- ARR
annualized relapse rate
- BOCF
Baseline Observed Carried Forward
- DRAE
disease-related adverse event
- EDSS
Expanded Disability Status Scale
- HR
hazard ratio
- ITT
intention to treat
- NEDA
No Evidence of Disease Activity
- PASAT
Paced Auditory Serial Addition Task
- RRMS
relapsing-remitting MS
- SCTIW
subcutaneously 3 times per week
- SDRAE
serious DRAE
- TEAE
treatment emergent adverse event
- VD
vitamin D
- VDCM
mean of VDC
Appendix. Authors

Study funding
This study was funded by Merck France, a subsidiary of Merck KGaA, Darmstadt, Germany.
Disclosure
W. Camu reports personal fees from Actelion, Effik, Merck, MedDay Pharmaceuticals, Novartis Pharma, Roche, and Sanofi. P. Lehert reports personal fees from Merck, Biogen, Teva, and Genzyme and grants from Novartis. C. Pierrot-Deseilligny reports personal fees from Novartis. P. Hautecoeur reports no disclosures. A. Besserve, J. Deleglise, and M. Payet are employees of the sponsor, Merck. E. Thouvenot has received honoraria and travel grants or research grants from the following pharmaceutical companies: Biogen, Genzyme, Merck Serono, Novartis, Roche, and Teva Pharma. J.-C. Souberbielle reports no disclosures. Go to Neurology.org/NN for full disclosures.
References
- 1.Soelberg Sorensen P. Safety concerns and risk management of multiple sclerosis therapies. Acta Neurol Scand 2017;136:168–186. [DOI] [PubMed] [Google Scholar]
- 2.Trojano M, Tintore M, Montalban X, et al. Treatment decisions in multiple sclerosis—insights from real-world observational studies. Nat Rev Neurol 2017;13:105–118. [DOI] [PubMed] [Google Scholar]
- 3.Bscheider M, Butcher EC. Vitamin D immunoregulation through dendritic cells. Immunology 2016;148:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ascherio A, Munger KL, Simon KC. Vitamin D and multiple sclerosis. Lancet Neurol 2010;9:599–612. [DOI] [PubMed] [Google Scholar]
- 5.Mowry EM, Krupp LB, Milazzo M, et al. Vitamin D status is associated with relapse rate in pediatric-onset multiple sclerosis. Ann Neurol 2010;67:618–624. [DOI] [PubMed] [Google Scholar]
- 6.Mowry EM, Waubant E, McCulloch CE, et al. Vitamin D status predicts new brain magnetic resonance imaging activity in multiple sclerosis. Ann Neurol 2012;72:234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ascherio A, Munger KL, White R, et al. Vitamin D as an early predictor of multiple sclerosis activity and progression. JAMA Neurol 2014;71:306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fitzgerald KC, Munger KL, Köchert K, et al. Association of vitamin D levels with multiple sclerosis activity and progression in patients receiving interferon beta-1b. JAMA Neurol 2015;72:1458–1465. [DOI] [PubMed] [Google Scholar]
- 9.Burton JM, Kimball S, Vieth R, et al. A phase I/II dose-escalation trial of vitamin D3 and calcium in multiple sclerosis. Neurology 2010;74:1852–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pierrot-Deseilligny C, Souberbielle JC. Vitamin D and multiple sclerosis: an update. Mult Scler Relat Disord 2017;14:35–45. [DOI] [PubMed] [Google Scholar]
- 11.Pozuelo-Moyano B, Benito-León J, Mitchell AJ, Hernández-Gallego J. A systematic review of randomized, double-blind, placebo-controlled trials examining the clinical efficacy of vitamin D in multiple sclerosis. Neuroepidemiology 2013;40:147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mosayebi G, Ghazavi A, Ghasami K, Jand Y, Kokhaei P. Therapeutic effect of vitamin D3 in multiple sclerosis patients. Immunol Invest 2011;40:627–639. [DOI] [PubMed] [Google Scholar]
- 13.Kampman MT, Steffensen LH, Mellgren SI, Jørgensen L. Effect of vitamin D3 supplementation on relapses, disease progression, and measures of function in persons with multiple sclerosis: exploratory outcomes from a double-blind randomised controlled trial. Mult Scler 2012;18:1144–1151. [DOI] [PubMed] [Google Scholar]
- 14.Soilu-Hänninen M, Aivo J, Lindström BM, et al. A randomised, double blind, placebo controlled trial with vitamin D3 as an add on treatment to interferon β-1b in patients with multiple sclerosis. J Neurol Neurosurg Psychiatry 2012;83:565–571. [DOI] [PubMed] [Google Scholar]
- 15.Ashtari F, Toghianifar N, Zarkesh-Esfahani SH, Mansourian M. High dose vitamin D intake and quality of life in relapsing-remitting multiple sclerosis: a randomized, double-blind, placebo-controlled clinical trial. Neurol Res 2016;38:888–892. [DOI] [PubMed] [Google Scholar]
- 16.Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol 2005;58:840–846. [DOI] [PubMed] [Google Scholar]
- 17.Rotstein DL, Healy BC, Malik MT, Chitnis T, Weiner HL. Evaluation of no evidence of disease activity in a 7-year longitudinal multiple sclerosis cohort. JAMA Neurol 2015;72:152–158. [DOI] [PubMed] [Google Scholar]
- 18.Sintzel MB, Rametta M, Reder AT. Vitamin D and multiple sclerosis: a comprehensive review. Neurol Ther 2018;7:59–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stewart N, Simpson S Jr, van der Mei I, et al. Interferon-β and serum 25-hydroxyvitamin D interact to modulate relapse risk in MS. Neurology 2012;79:254–260. [DOI] [PubMed] [Google Scholar]
- 20.Munger KL, Köchert K, Simon KC, et al. Molecular mechanism underlying the impact of vitamin D on disease activity of MS. Ann Clin Transl Neurol 2014;1:605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waschbisch A, Sanderson N, Krumbholz M, et al. Interferon beta and vitamin D synergize to induce immunoregulatory receptors on peripheral blood monocytes of multiple sclerosis patients. PLoS One 2014;9:e115488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
We provide qualified researchers access to individual patient-level data through the Merck KGaA's data-sharing portal: merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html.