

Abstract
Objective
To identify and characterize patients with autoantibodies against different neurofascin (NF) isoforms.
Methods
Screening of a large cohort of patient sera for anti-NF autoantibodies by ELISA and further characterization by cell-based assays, epitope mapping, and complement binding assays.
Results
Two different clinical phenotypes became apparent in this study: The well-known clinical picture of subacute-onset severe sensorimotor neuropathy with tremor that is known to be associated with IgG4 autoantibodies against the paranodal isoform NF-155 was found in 2 patients. The second phenotype with a dramatic course of disease with tetraplegia and almost locked-in syndrome was associated with IgG3 autoantibodies against nodal and paranodal isoforms of NF in 3 patients. The epitope against which these autoantibodies were directed in this second phenotype was the common Ig domain found in all 3 NF isoforms. In contrast, anti–NF-155 IgG4 were directed against the NF-155–specific Fn3Fn4 domain. The description of a second phenotype of anti–NF-associated neuropathy is in line with some case reports of similar patients that were published in the last year.
Conclusions
Our results indicate that anti–pan-NF-associated neuropathy differs from anti–NF-155-associated neuropathy, and epitope and subclass play a major role in the pathogenesis and severity of anti–NF-associated neuropathy and should be determined to correctly classify patients, also in respect to possible differences in therapeutic response.
The node of Ranvier has long been considered a potential site of attack in inflammatory neuropathies. With respect to the concept of complement-mediated reversible conduction block induced by antiganglioside autoantibodies, the term “paranodopathy/nodopathy” was introduced by Uncini et al.1,2 In the past few years, non–complement-fixing IgG4 autoantibodies against paranodal proteins were described in patients with immune neuropathies and were also comprised under that term.3,4 Patients with autoantibodies against paranodal proteins present with a distinct clinical phenotype of subacute-onset severe sensorimotor neuropathy with poor response to intravenous immunoglobulins but excellent response to rituximab.5–10 Autoantibodies are mostly of the IgG4 subtype, but IgG3 and IgG2 autoantibodies have also been described, possibly associated with early disease or monophasic course.9,10 In these cases, complement deposition could be demonstrated.11 Anti-neurofascin (NF)-155 appears to be the most prevalent paranodal autoantibody and is strongly associated with tremor.6,12,13 NF-155 is located at the paranodes, whereas NF-186 and -140 are nodal isoforms, the latter mainly expressed during embryonic development.14,15 Although all 3 isoforms share the Ig domain and some parts of the fibronectin domain,16 most autoantibodies described so far were selectively directed against NF-155.6,8 However, 2 recent studies could also detect anti-NF-186/140 autoantibodies, mostly of the IgG4 subclass, in patients with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP).17,18 Single cases of patients with autoantibodies against all isoforms and severe neuropathy have been described.17,19 In contrast to anti–NF-155-associated neuropathy, not much is known about neuropathy with anti–pan-NF autoantibodies.
In the present study, we aimed to identify and characterize patients with autoantibodies against NF to potentially establish anti–pan-NF-associated neuropathy as a distinct clinical phenotype.
Methods
Patients
Three different subcohorts of patients were included: sera of 182 patients with clinically suspected inflammatory neuropathy that were sent to our department for testing of paranodal autoantibodies, further referred to as “comprehensive diagnostic testing cohort,” sera of 306 patients with a differential diagnosis of inflammatory neuropathy who attended our department for diagnostic workup (32 finally diagnosed as Guillain-Barré syndrome (GBS), 81 as CIDP fulfilling the European Federation of Neurological Societies criteria,20 and 193 as noninflammatory neuropathy or suspected inflammatory neuropathy not fulfilling the EFNS criteria; “Würzburg cohort”), and 181 sera of a patient cohort from the University Hospitals of Kiel and Magdeburg that were collected between 2004 and 2016 (146 GBS, 21 CIDP, and 15 Miller-Fisher syndrome/Bickerstaff encephalitis; “Kiel/Magdeburg cohort”). Diagnosis of GBS was assessed by the Brighton criteria.21 CSF was obtained from diagnostic lumbar puncture. The study was approved by the Ethics Committees of the Universities of Würzburg and Kiel. Demographic data are summarized in table 1.
Table 1.
Demographic data of patients of all subcohorts

Enzyme-linked immunosorbent assay
All patients were screened for anti-NF-155 autoantibodies by ELISA, and 252 patients of the Würzburg cohort and all anti–NF-155-positive patients were also screened for anti-NF-186. All anti–NF-155-positive patients were tested for anti-NF-140. CSF was available from 3 seropositive patients and was tested at a dilution of 1:20. Five patients without a clinical history of neuropathy who had received lumbar puncture for other diagnostic purposes served as controls. NF-155/-186 ELISA was performed as previously described using the extracellular domains of human NF-155 and -186.8 For anti-NF-140 ELISA, wells were coated with recombinant human NF-140 protein (1 μg/mL, Sino Biological Inc, Beijing, China). Normal controls were run in each assay. The normal value was set at 5 SDs above the mean of all healthy controls that were tested in previous studies.22 Titers were determined by measuring serial dilutions of anti–NF-positive sera. Subclasses were detected using subclass-specific horseradish peroxidase-conjugated secondary antibodies (anti-IgG1: 1:1,000, anti-IgG2: 1:4,000, anti-IgG3: 1:3,000, and anti-IgG4: 1:2,000; Invitrogen, Carlsbad, CA).
Binding assays with murine teased fibers, sympathetic ganglia, cerebellar granular neurons, and rat cerebellum sections
All sera were screened for paranodal and nodal autoantibodies using binding assays with murine teased fibers as previously described.9 The preparations were incubated with patients' sera diluted 1:100 and 1:500 overnight at 4°C following incubation with appropriate anti–human Cy3-conjugated secondary antibodies (Dianova, Hamburg, Germany, 1:100). Cervical sympathetic ganglia were dissected from adult Lewis rats, and 10-μm cryosections were cut. Binding assays with patients' and controls' sera were performed with a dilution of 1:100 and 1:500 by incubating overnight. Cy3-conjugated anti-human secondary antibodies were added for 2 hours. All slides were assessed using a fluorescence microscope (Zeiss Ax10, Zeiss, Oberkochen, Germany). Cerebellar granular neurons were prepared from P5 mice: the cerebellum was dissected, and cells were dissociated and suspended using a papain dissociation system (Worthington Biochemical Corporation, Lakewood, NJ). Cells were plated on cover slips at a density of 200,000 cells per well and fixed with 4% paraformaldehyde after 3 days, and binding assays with sera of patients and controls were performed at a dilution of 1:500 using an anti-human Cy3-conjugated secondary antibody (Dianova, 1:300). Anti–pan-NF served as a positive control.
Binding assays on rat cerebellum sections were performed overnight at a serum dilution of 1:200 or CSF dilution of 1:4 on 6-μm sagittal adult rat brain cryosections optimized for detection of surface antigens as has been previously described.23
Binding assays with NF-transfected human embryonic kidney (HEK) 293 cells
Binding assays with HEK293 cells transiently transfected with plasmids of human NF-155 and -1868 were performed as previously described.22 Sera were used at a dilution of 1:200 and 1:500. Anti–pan-NF (1:1,000) served as a control antibody. Serum of 1 patient (patient 6) with immunoreactivity against NF-155 by ELISA but no binding to NF-transfected HEK293 cells at a dilution of 1:200 and 1:500 was additionally tested at a dilution of 1:40. Immunostaining was assessed using a fluorescence microscope (Zeiss Ax10).
Preabsorption experiments
Preabsorption was performed by serial incubation of patients' sera (1:500) 1 hour each in 3 wells of a 24-well plate with NF-155/-186–transfected HEK293 cells as described before.9 Sera incubated in wells with contactin-1–transfected cells served as controls. After incubation with transfected HEK293 cells, sera were used for binding assays on teased fibers as described above.
Epitope characterization
TE671 human rhabdomyosarcoma cells stably transfected with NF-155, NF-186, or 6 truncated forms of NF-155 and NF-186 were previously described.8 Cells were incubated with patients' and controls' sera (1:100). Detection of binding was performed as described,8 with some modifications: Serum IgG binding was detected using Biotin-SP-conjugated AffiniPure F(ab')2 antibodies (Jackson ImmunoResearch) and AF647-conjugated streptavidin (Jackson ImmunoResearch). Mean fluorescence intensity (MFI) of the AF647 channel was calculated after gating on enhanced green fluorescent protein (EGFP)high cells for NF truncations and empty EGFP vector–transfected cells, and delta MFI was calculated from these values. Patient 5 was only tested for Fn3Fn4, as the sample was obtained at a later time point and NF-155 isoform specificity had already been shown by ELISA.
Complement binding assay
ELISA plates were coated, blocked, and incubated with patients' sera as described above. The next day, the wells were incubated with C1q (10 μg/mL, Sigma-Aldrich, St. Louis, MO) for 2 hours at room temperature, following incubation with horseradish peroxidase–conjugated anti-C1q (LifeSpan Biosciences, Seattle, WA, 1:200) for 30 minutes. Measurement of optical density was made as described above.
Data availability statement
Any data not published within the article will be shared by request from any qualified investigator in anonymized form.
Results
Detection of anti-NF autoantibodies
Anti-NF-155 autoantibodies were detectable by ELISA in sera of 6 patients (figure 1). Titers ranged from 1:300 to 1:6,000 (table 2). Three of the 6 anti–NF-155-positive patients were also positively tested for anti-NF-186 and -140 (patients 1–3, table 2). Sera of 2 patients showed an optical density slightly above the cutoff value in the anti-NF-155 ELISA, but a second serum sample of these patients obtained during the course of disease was clearly negative. Of note, in none of the 252 neuropathy cases tested for anti-NF-186 independent of NF-155 positivity, did we observe isolated anti-NF-186 positivity. Anti-NF-155 and/or -186 autoantibodies were confirmed by binding assays with NF-155– and NF-186–transfected HEK293 cells in 5 of these patients (patients 1–5, figure 1). Binding assays on murine teased fibers revealed a distinct paranodal staining in the patients with anti-NF-155 autoantibodies only (patients 4–6; figure 2, F–H) and a nodal and paranodal staining in the 3 patients with anti–pan-NF autoantibodies (patients 1–3; figure 2, A, C, and D). In patient 1, paranodal staining was stronger than nodal staining, and in patients 2 and 3, nodal staining was stronger (Figure 2A, C-E). In patients 3 and 6, the fluorescence signal was weaker than in the other patients, most probably due to the low titer (Figure 2E, H). In patients 1 and 2, paranodal binding became stronger during the course of disease (Figure 2B, D).
Figure 1. Binding assays with NF-155– and NF-186–transfected HEK293 cells (A-G) and ELISA (H).

Patients 1, 2, and 3 show a clear binding to NF-155– and NF-186–transfected cells (A–C), whereas patients 4 and 5 only bind to NF-155–transfected and not to NF-186–transfected cells (F and G). Healthy control serum does not bind to the cells (E), and binding of commercial anti–pan-NF autoantibodies to NF-155– and NF-186–transfected cells is shown in (D). NF = neurofascin; OD = optical density.
Table 2.
Overview of assays and clinical data of anti–NF-positive patients

Figure 2. Microphotographs of binding assays of patients' sera to murine teased fibers.
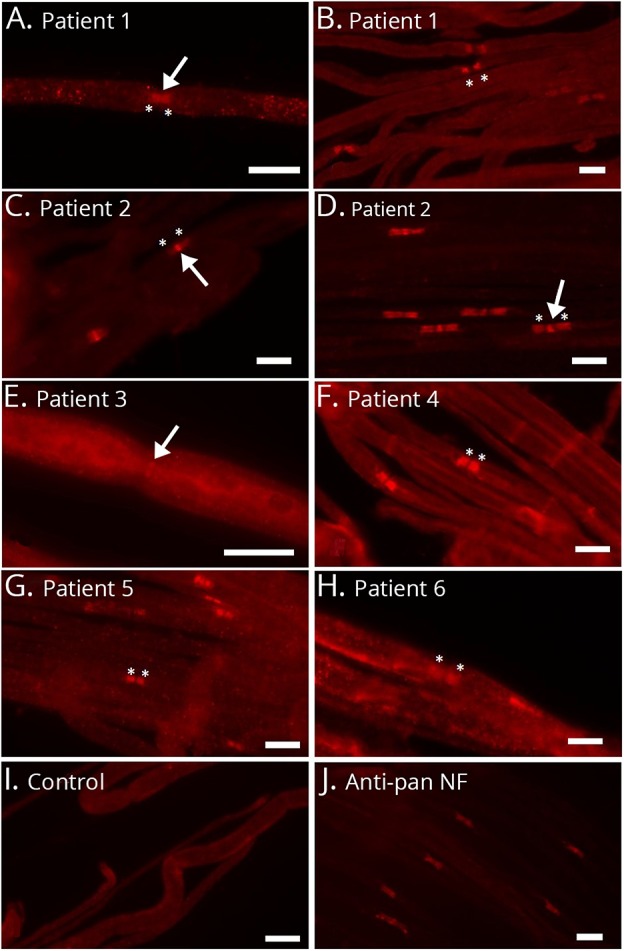
Nodal binding is marked with arrows, and paranodal binding with asterisks. Serum of patient 1 clearly binds to the nodal and paranodal regions at the onset of disease (A). Paranodal binding becomes more prominent during the course of disease (B). Patient 2 binds to the nodes stronger than to the paranodes (C and D), but paranodal binding becomes stronger during the course of disease (D). Patient 3 mainly binds to the nodal region (E), whereas patients 4 and 5 with anti-NF-155 autoantibodies bind to the paranodes only (F and G). Patient 6 weakly binds to the paranodes (H). A normal control does not show any binding (I), and the commercial anti–pan-NF control antibody clearly binds to the nodes and paranodes (J). Bar = 10 μm. NF = neurofascin.
Preincubation assays were performed to confirm paranodal/nodal binding as anti-NF specific. In the 2 patients who were only positive for anti-NF-155 (patients 4 and 5), paranodal binding was lost after preincubation with NF-155–transfected cells, not after incubation with NF-186–transfected cells. In 2 of the patients with autoantibodies against both isoforms (patients 1 and 2), paranodal and nodal binding were lost after incubation with NF-155-transfected cells, but weaker paranodal binding persisted after incubation with NF-186–transfected cells, suggesting the presence of at least 2 autoantibodies with different epitopes (supplemental figure 1, links.lww.com/NXI/A132, and figure 3).
Figure 3. Epitope mapping of anti-NF autoantibodies by binding assays to truncated NF mutants and flow cytometry.

The NF isoforms consist of a common Ig6 domain and different isoform specific fibronectin domains (A, right section). Reactivity of patient sera to NF-155 and NF-186 (A) and truncations of NF (B) by flow cytometry: patients 1, 2, and 3 show reactivity to both NF-155 (blue line) and NF-186 (orange line) compared with control (empty RSV vector, black, filled), whereas patients 4 and 5 only react to NF-155 (A). Epitope analysis using NF truncations (B) demonstrated that reactivity was against the 6Ig domain in 3 of the patients (patients 1–3) who were positive for both NF isoforms (green line). Patients 1 and 2 also reacted to the Fn3 domain (red line). The Fn3Fn4 domain (yellow line) was recognized by the sera of patients 4 and 5, who reacted only to NF-155. Other truncated NF variants (Fn1Fn2 = dark blue; Fn4-Mucin-Fn5 = brown; Mucin-Fn5 = purple; GFP-only control = gray filled) were not recognized by any of the sera tested (sera 1–4 and 6 tested for all mutants, serum 5 for Fn3Fn4 only). NF = neurofascin.
CSF was available from patients 2, 3, and 6. No anti-NF autoantibodies were detectable in CSF by ELISA in patients 3 and 6. In patient 2, no anti-NF autoantibodies were detectable in CSF by ELISA at the onset of disease (July 2014) or after 2 months (September 2014). However, 6 months after the onset of disease, anti-NF autoantibodies were detectable, most probably due to disruption of the blood-brain barrier (CSF/serum albumin index 37.2 × 10−3). There was no evidence of intrathecal IgG production at this time point, as no oligoclonal bands were detectable and the CSF IgG index was within the normal range (0.59).
Comparison of clinical phenotypes in patients with pan-NF or NF-155–specific antibodies
All 6 anti–NF-positive patients had a clinical diagnosis of CIDP except for patient 3 who had a monophasic course and was classified as GBS. Yet detailed analysis of clinical pictures showed that anti–NF-positive patients segregated into 2 distinct phenotypes.
Patients 1–3 who were seropositive for anti-NF-155, -NF-186, and –NF-140 had a fulminant course of disease. After GBS-like onset, patients 1–3 quickly developed tetraplegia and severe cranial nerve involvement. No antiganglioside antibodies were detectable. Patient 3 quickly recovered within a few months. Patients 1 and 2 needed mechanical ventilation for several months with complete tetraplegia and were almost locked-in. Patient 1 died due to sepsis, and patient 2 had a very protracted course with almost 2 years in tetraplegia and still recovering almost 5 years after onset of symptoms.
Patients 4 and 5 were young men with anti-NF-155 autoantibodies and presented with motor more than sensory neuropathy associated with tremor, as described in multiple reports.6,13 Remarkably, autonomic involvement was a major problem in patient 4. In patient 5, action and postural tremor and gait ataxia were the dominant symptoms with only minor neuropathic complaints and no autonomic symptoms.
In patient 6 anti-NF-155 autoantibodies could not be confirmed by cell-based assay. The patient had severe sensorimotor neuropathy without tremor.
For detailed clinical data, course of disease, and response to treatment, see Table 2 and Supplements (supplement course of disease, links.lww.com/NXI/A137).
Different patterns of reactivity of autoantibodies against truncations of NF
To find out whether the autoantibodies in patients 1–3 were directed against different epitopes or target a common epitope of all NF isoforms and were thus responsible for the positive reactivity to all 3 NF, binding assays with truncated NF mutants were performed. Patients 1–3 showed immunoreactivity against the Ig domains of NF that are part of all isoforms. In addition, patients 1 and 2 were found to be weakly immunoreactive to Fn3, a component of the NF-155 isoform. Immunoreactivity to Fn3 as a second NF-155–specific epitope is in line with the results of the preincubation assays, explaining persistence of weak paranodal binding after preincubation with NF-186–transfected HEK293 cells. Additional immunoreactivity to Fn3 as a NF-155–specific epitope also accounts for higher titers of anti-NF-155 compared with anti-NF-186. Patients 4 and 5 were immunoreactive against Fn3Fn4, a component of the fibronectin domain of NF-155 that is isoform specific (figure 3).
IgG subclasses and C1q binding
ELISA with subclass-specific secondary antibodies detected IgG4 and IgG2 as the predominant subclasses in patient 4, IgG4 in patient 5, and IgG3 in patients 1, 2, and 6. In patient 1, IgG2 was detectable at the onset of disease. No specific subclass was detectable in patient 3, most probably due to the low titer and lower sensitivity of the subclass-specific ELISA (figure 4). In patient 2, a minor titer of IgG4 was transiently detectable during the course of disease at a time when IgG3 titers peaked at extraordinarily high titers (supplemental table, links.lww.com/NXI/A133). Results of subclasses were confirmed by flow cytometry. Complement deposition induced by autoantibody binding was found in patients 1, 2, and 4 but not in patients 3 and 6 (both with low autoantibody titers and patient 6 without confirmation of anti-NF-155 by cell-based assays) and patient 5 (with IgG4 only) (figure 4). In patients 1 and 2, sera of different time points of disease with different autoantibody titers were measured, and in total, C1q deposition correlated with IgG3 immunoreactivity (p = 0.009, r = 0.72).
Figure 4. ELISA of total IgG and IgG subclasses (bars) and C1q deposition in the complement binding assay (line).

IgG3 can be identified as the dominant subclass in patients 1, 2, and 6, IgG 2 and 4 in patient 4, and IgG4 in patient 5. Complement deposition can be measured in patients 1 and 2 with IgG3 predominance and in patient 4 with IgG2 and IgG4 autoantibodies. No complement deposition was detectable in patient 5 with predominance of IgG4 and patients 3 and 6 with low titers of total IgG. IgG = immunoglobulin G.
Autonomic and cranial nerve involvement in anti–NF-positive patients and binding to the cerebellum
Autonomic involvement was present in 3 of the patients. Patients 1 and 2 developed severe autonomic dysfunction during intensive care treatment. Patient 1 was resuscitated after cardiac arrest. Patient 4 had disabling orthostatic dysfunction, increased transpiration, and pollakiuria. As there is no obvious link between NF and the autonomic nervous system and postganglionic autonomic fibers are unmyelinated, binding assays with patients' sera on sympathetic cervical ganglia were performed, but we did not detect any specific binding of patients' sera to autonomic neurons (data not shown). To assess binding of anti–NF-positive sera to preganglionic B-fibers, we performed binding assays with anterior root teased fibers of rats that contain preganglionic sympathetic B-fibers. B-fibers are distinguishable from A-alpha- and A-gamma-fibers by their diameter. Binding of patients' sera to ventral root fibers of all diameters (supplemental figure 3, links.lww.com/NXI/A135) and also to A-beta fibers of the dorsal root was detected. As former studies have proposed binding of anti-NF sera to the cerebellum as a possible correlate of tremor,6,13 we also performed binding assays with cerebellum sections that showed clear binding to the molecular and granular layer in patients 1–4 and to the molecular layer only in patient 5 and no cerebellar binding in patient 6. Of note, strong cerebellar binding was observed in patient 2 using CSF, which was not evident in patients 3 and 6 (figure 5). Positive binding to cerebellar granular neurons was also detectable in these patients (supplemental figure 3, links.lww.com/NXI/A135).
Figure 5. Binding assays with sera and CSF on rat cerebellum sections.

CSF is depicted as triangular inset on the right lower half of the panels. In contrast to a healthy control that only shows some unspecific staining to Purkinje cells due to tissue fixation optimized for detection of surface antigens (A), sera of patients 2 and 3 show distinct staining of the granular and molecular layers (B–D). Binding of CSF antibodies to the cerebellum can only be shown in patient 2 and increases during the course of disease (B and D), not in the healthy control (A) or patient 3 (C). Bar = 100 µm. CSF = cerebrospinal fluid; CWM = cerebral white matter; GCL = granular cell layer; ML = molecular layer; PCL = purkinje cell layer.
Cranial nerve involvement was detectable in the 3 patients with pan-NF autoantibodies. Patients 1 and 2 were almost locked-in with complete facial and oculomotor palsy and dysphagia. Patient 3 had dysphagia requiring a percutaneous endoscopic gastrostomy tube. As concomitant glomerulonephritis has been described in patients with paranodal autoantibodies,9,18,24 patients' records were checked for renal symptoms. There was no evidence of glomerulonephritis in patients 2–6, but patient 1 had acute-on-chronic renal failure with mild proteinuria and hematuria since the onset of disease, so concomitant glomerulonephritis is possible, although it was not formally diagnosed.
Discussion
In the present study, we identified 5 patients with definite anti–NF-associated neuropathy, 3 of them presenting with a distinct clinical phenotype of tetraplegia and cranial nerve involvement. Our data suggest that the clinical picture is associated with the IgG subclass and epitope—IgG4 autoantibodies against the Fn3Fn4 domain of NF-155 being associated with the well-known phenotype of severe sensorimotor neuropathy with tremor and IgG3 autoantibodies against the Ig domain of all isoforms leading to tetraplegia and cranial nerve involvement.
We describe multiple clinically uniform cases of this more severe form of anti–pan-NF-associated neuropathy, and the distinct clinical phenotype is strengthened by identification of a common pattern of reactivity to the 6Ig domain that is common to all NF isoforms. The notion of anti–pan-NF-associated neuropathy as a distinct subtype of anti-NF neuropathy is supported by reports of single cases similar to our 3 patients in the last years: In a cohort by Burnor et al., 1 patient with autoantibodies against NF-155, -186, and -140 clinically very much resembled our patients but had IgG4 subclass autoantibodies.17 Only recently, another patient with tetraplegia, respiratory failure, almost locked-in syndrome, and IgG3 autoantibodies against NF-186/-140 and also NF-155 was reported, and ultrastructural analysis of nodal architecture revealed occlusion of the nodal gap by myelin layers.19
In a recent study, 5 patients with anti-NF-186/-140 autoantibodies were compared with patients with anti-NF-155 IgG autoantibodies.18 Anti–NF-186/-140-positive patients were found to be clinically different from anti-NF-155 patients, and 2 anti–NF-186/-140-positive patients presented with cranial nerve involvement and respiratory failure, one of them with predominance of the IgG3 subclass.18 In contrast to our study, patients with pan-NF antibodies of the IgG4 subclass were also described in these studies, so possibly the epitope appears to be the major determinant of the clinical picture and not the IgG subclass. However, these subclasses may still contribute to the acuteness or other clinical aspects of the disease. This suggests that the unique clinical picture of our patients 1–3 may be mostly explained by the unique epitope—a common domain of nodal NF-186/-140 and paranodal NF-155—but may also be associated with the IgG3 subclass. In contrast to IgG4, IgG3 autoantibodies activate complement and are considered to have a strong proinflammatory effect.25 Indeed, C1q binding induced by anti–pan-NF autoantibodies could be shown in our patients by complement binding assays. Remarkably, complement deposition was also detectable in one of the patients with the typical anti-NF-155 IgG4 phenotype, most probably due to additional IgG2 autoantibodies that were detectable in this case. Complement deposition may contribute to the more severe acute course of disease in this patient compared with the other anti-NF-155 IgG patient in our cohort. Complement-mediated conduction block may either be reversible, as demonstrated by the case of patient 3 with GBS, or result in secondary axonal damage or nodal alterations (as demonstrated in an ultrastructural case description by Vallat et al.19) with a prolonged phase of recovery or lack of recovery as in patients 1 and 2. Pathophysiologic studies including passive transfer need to be conducted to further unravel the connection between clinical phenotype and autoantibodies in this subgroup of autoimmune neuropathy. CNS axonal damage and deterioration of clinical severity of experimental allergic neuritis induced by autoantibodies against the common domain of NF-155 and NF-186 were demonstrated by injection of pan-NF antibodies to rats indicating a pathogenic effect of autoantibodies against the common domain of NF.8,26
Patient 6 who was anti–NF-155 positive (IgG3) by ELISA did not present with the typical clinical phenotype of anti–NF-155-associated neuropathy. Lack of binding in the cell-based assay, cerebellar binding studies, and of complement deposition and only weak nodal/paranodal binding in teased fibers may reflect a low affinity of autoantibodies to the in vivo conformation of NF or even a protein associated with NF in this patient and may explain the mild phenotype.
Whereas all previous case reports of patients with pan-NF autoantibodies only described nodal but not paranodal binding on teased fibers,18,19 clear paranodal binding was found in all our anti–pan-NF patients. Stronger paranodal binding may be explained by additional binding to Fn3, part of the NF-155–specific fibronectin domain, in 2 of our patients. Remarkably, these 2 patients showed the most severe clinical phenotype and a very long course of disease. Thus, multiple epitopes may be a predictor of disease severity as also described in other autoimmune diseases.27,28 The underlying mechanism of multiple epitopes remains unclear: intramolecular epitope spreading could be an explanation and has also been described in other autoimmune diseases.27 Indeed, in patient 2, the Fn3 epitope was only detectable during the course of disease but not at disease onset. Paranodal binding in teased fiber assays increased respectively.
Tetraplegia was the dominant symptom in all of our patients with anti–pan-NF autoantibodies and in single cases described in the literature.17,29 We did not find any tremor in these patients, but tremor may be masked by tetraplegia. Tremor in anti-NF-155 neuropathy has been attributed to autoantibody binding to cerebellar neurons,6 and we could show cerebellar binding of anti-NF-155 and also anti–pan-NF serum. Our study is the first to detect paranodal autoantibodies in CSF: Anti–pan-NF autoantibodies were detectable in the CSF of patient 2 as a potential correlate of cerebellar involvement. In comparison to the 5 patients with anti-NF-186/-140 autoantibodies described by Delmont et al.,18 that also bound to the Ig domain, the course of disease was more severe in our patients and refractory to treatment. Lack of tremor, cranial nerve involvement, and older age compared with anti-NF-155 patients are features that were also found in the patients by Delmont et al.18 Differences may be explained by IgG3 subclass in our patients and mostly IgG4 in Delmont's patients.
Remarkably, 3 of our patients including a patient with only anti-NF-155 autoantibodies had severe autonomic involvement, a feature that has not yet been investigated in anti–NF-associated neuropathy. However, autonomic symptoms were also described in the single anti–pan-NF-positive patient in the study by Burnor et al.17 As postganglionic autonomic fibers are unmyelinated and we could not show any binding to sympathetic ganglia, this can most probably be explained by radiculitis involving preganglionic autonomic fibers.
The role of IgG subclasses in the pathogenicity of anti-NF autoantibodies and particularly association with therapeutic response needs to be further studied. Detection of complement binding in our study and by others argues for complement-mediated inflammation, which extends the concept of anti–NF-associated neuropathy as a pure IgG4-mediated disease. Our data indicate that testing of IgG subclasses should be performed when detecting autoantibodies against paranodal proteins, and testing of anti-NF autoantibodies should not be restricted to the NF-155 isoform.
Acknowledgment
The authors thank Barbara Reuter and Heike Rübsamen for expert technical assistance. The study was funded by a grant of the Interdisciplinary Center of Clinical Research to KD and CV. AV was supported by research fellowships from the European Academy of Neurology, The Scientific and Technological Research Council of Turkey (TUBITAK), and the Alexander von Humboldt Foundation. EM received support from SFBTR128 and the Clinical Competence Network for Multiple Sclerosis.
Glossary
- CIDP
chronic inflammatory demyelinating polyradiculoneuropathy
- GBS
Guillain-Barré syndrome
- HEK
human embryonic kidney
- MFI
mean fluorescence intensity
- MFS
Miller-Fisher syndrome
- NF
neurofascin
Appendix. Authors

Study funding
No targeted funding reported.
Disclosure
H. Stengel, A. Vural, A.M. Brunder, A. Heinius, L. Appeltshauser, B. Fiebig, F. Giese, C. Dresel, A. Papagianni, F. Birklein, J. Weis, T. Huchtemann, C. Schmidt, P. Körtvelyessy, and C. Villmann report no relevant disclosures. E. Meinl received personal fees from Roche, Novartis, and Genzyme and grants outside the submitted work from Novartis and Genzyme. C. Sommer received personal fees from Air Liquide, Alnylam, Astellas, Baxalta, CSL Behring, Genzyme, Grifols, Pfizer, UCB and Kedrion. F. Leypoldt reports no relevant disclosures. K. Doppler received personal fees from Baxter/Baxalta and Grifols and a grant from Kedrion (outside the submitted work). Go to Neurology.org/NN for full disclosures.
References
- 1.Uncini A, Susuki K, Yuki N. Nodo-paranodopathy: beyond the demyelinating and axonal classification in anti-ganglioside antibody-mediated neuropathies. Clin Neurophysiol 2013;124:1928–1934. [DOI] [PubMed] [Google Scholar]
- 2.Uncini A. A common mechanism and a new categorization for anti-ganglioside antibody-mediated neuropathies. Exp Neurol 2012;235:513–516. [DOI] [PubMed] [Google Scholar]
- 3.Uncini A, Vallat JM. Autoimmune nodo-paranodopathies of peripheral nerve: the concept is gaining ground. J Neurol Neurosurg Psychiatry 2018;89:627–635. [DOI] [PubMed] [Google Scholar]
- 4.Querol L, Illa I. Paranodal and other autoantibodies in chronic inflammatory neuropathies. Curr Opin Neurol 2015;28:474–479. [DOI] [PubMed] [Google Scholar]
- 5.Querol L, Nogales-Gadea G, Rojas-Garcia R, et al. . Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol 2013;73:370–380. [DOI] [PubMed] [Google Scholar]
- 6.Querol L, Nogales-Gadea G, Rojas-Garcia R, et al. . Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology 2014;82:879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Querol L, Rojas-Garcia R, Diaz-Manera J, et al. . Rituximab in treatment-resistant CIDP with antibodies against paranodal proteins. Neurol Neuroimmunol Neuroinflamm 2015;2:e149 doi:10.1212/NXI.0000000000000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng JK, Malotka J, Kawakami N, et al. . Neurofascin as a target for autoantibodies in peripheral neuropathies. Neurology 2012;79:2241–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doppler K, Appeltshauser L, Wilhelmi K, et al. . Destruction of paranodal architecture in inflammatory neuropathy with anti-contactin-1 autoantibodies. J Neurol Neurosurg Psychiatry 2015;86:720–728. [DOI] [PubMed] [Google Scholar]
- 10.Doppler K, Appeltshauser L, Villmann C, et al. . Auto-antibodies to contactin-associated protein 1 (Caspr) in two patients with painful inflammatory neuropathy. Brain 2016;139:2617–2630. [DOI] [PubMed] [Google Scholar]
- 11.Appeltshauser L, Weishaupt A, Sommer C, Doppler K. Complement deposition induced by binding of anti-contactin-1 auto-antibodies is modified by immunoglobulins. Exp Neurol 2016;287:84–90. [DOI] [PubMed] [Google Scholar]
- 12.Vural A, Doppler K, Meinl E. Autoantibodies against the node of Ranvier in seropositive chronic inflammatory demyelinating polyneuropathy: diagnostic, pathogenic, and therapeutic relevance. Front Immunol 2018;9:1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Devaux JJ, Miura Y, Fukami Y, et al. . Neurofascin-155 IgG4 in chronic inflammatory demyelinating polyneuropathy. Neurology 2016;86:800–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kriebel M, Wuchter J, Trinks S, Volkmer H. Neurofascin: a switch between neuronal plasticity and stability. Int J Biochem Cell Biol 2012;44:694–697. [DOI] [PubMed] [Google Scholar]
- 15.Zhang A, Desmazieres A, Zonta B, et al. . Neurofascin 140 is an embryonic neuronal neurofascin isoform that promotes the assembly of the node of Ranvier. J Neurosci 2015;35:2246–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Volkmer H, Hassel B, Wolff JM, Frank R, Rathjen FG. Structure of the axonal surface recognition molecule neurofascin and its relationship to a neural subgroup of the immunoglobulin superfamily. J Cell Biol 1992;118:149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burnor E, Yang L, Zhou H, et al. . Neurofascin antibodies in autoimmune, genetic, and idiopathic neuropathies. Neurology 2017;90:e31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delmont E, Manso C, Querol L, et al. . Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain 2017;140:1851–1858. [DOI] [PubMed] [Google Scholar]
- 19.Vallat JM, Mathis S, Magy L, et al. . Subacute nodopathy with conduction blocks and anti-neurofascin 140/186 antibodies: an ultrastructural study. Brain 2018;141:e56. [DOI] [PubMed] [Google Scholar]
- 20.Van den Bergh P, Hadden RD, Bouche P, et al. . European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society—First Revision. J Peripher Nerv Syst 2010;15:1–9. [DOI] [PubMed] [Google Scholar]
- 21.Sejvar JJ, Kohl KS, Gidudu J, et al. . Guillain-Barre syndrome and Fisher syndrome: case definitions and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine 2011;29:599–612. [DOI] [PubMed] [Google Scholar]
- 22.Doppler K, Appeltshauser L, Kramer HH, et al. . Contactin-1 and neurofascin-155/-186 are not targets of auto-antibodies in multifocal motor neuropathy. PLoS One 2015;10:e0134274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011;10:63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hashimoto Y, Ogata H, Yamasaki R, et al. . Chronic inflammatory demyelinating polyneuropathy with concurrent membranous Nephropathy: an anti-paranode and podocyte protein antibody study and literature Survey. Front Neurol 2018;9:997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol 2014;5:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mathey EK, Derfuss T, Storch MK, et al. . Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med 2007;204:2363–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sinmaz N, Nguyen T, Tea F, Dale RC, Brilot F. Mapping autoantigen epitopes: molecular insights into autoantibody-associated disorders of the nervous system. J Neuroinflammation 2016;13:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huijbers MG, Lipka AF, Plomp JJ, Niks EH, van der Maarel SM, Verschuuren JJ. Pathogenic immune mechanisms at the neuromuscular synapse: the role of specific antibody-binding epitopes in myasthenia gravis. J Intern Med 2014;275:12–26. [DOI] [PubMed] [Google Scholar]
- 29.Mathis S, Magy L, Diallo L, Boukhris S, Vallat JM. Amyloid neuropathy mimicking chronic inflammatory demyelinating polyneuropathy. Muscle Nerve 2012;45:26–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Any data not published within the article will be shared by request from any qualified investigator in anonymized form.