

Abstract
Fatty acid-binding protein 5 (FABP5) at the blood–brain barrier contributes to the brain uptake of docosahexaenoic acid (DHA), a blood-derived polyunsaturated fatty acid essential for maintenance of cognitive function. Given the importance of DHA in cognition, the aim of this study was to investigate whether deletion of FABP5 results in cognitive dysfunction and whether this is associated with reduced brain endothelial cell uptake of exogenous DHA and subsequent attenuation in the brain levels of endogenous DHA. Cognitive function was assessed in male and female FABP5+/+ and FABP5−/− mice using a battery of memory paradigms. FABP5−/− mice exhibited impaired working memory and short-term memory, and these cognitive deficits were associated with a 14.7 ± 5.7% reduction in endogenous brain DHA levels. The role of FABP5 in the blood–brain barrier transport of DHA was assessed by measuring 14C-DHA uptake into brain endothelial cells and capillaries isolated from FABP5+/+ and FABP5−/− mice. In line with a crucial role of FABP5 in the brain uptake of DHA, 14C-DHA uptake into brain endothelial cells and brain capillaries of FABP5−/− mice was reduced by 48.4 ± 14.5% and 14.0 ± 4.2%, respectively, relative to those of FABP5+/+ mice. These results strongly support the hypothesis that FABP5 is essential for maintaining brain endothelial cell uptake of DHA, and that cognitive deficits observed in FABP5−/− mice are associated with reduced CNS access of DHA.
SIGNIFICANCE STATEMENT Genetic deletion of fatty acid-binding protein 5 (FABP5) in mice reduces uptake of exogenous docosahexaenoic acid (DHA) into brain endothelial cells and brain capillaries and reduces brain parenchymal levels of endogenous DHA. Therefore, FABP5 in the brain endothelial cell is a crucial contributor to the brain levels of DHA. Critically, lowered brain DHA levels in FABP5−/− mice occurred in tandem with cognitive deficits in a battery of memory paradigms. This study provides evidence of a critical role for FABP5 in the maintenance of cognitive function via regulating the brain uptake of DHA, and suggests that upregulation of FABP5 in neurodegenerative diseases, where brain DHA levels are possibly diminished (e.g., Alzheimer's disease), may provide a novel therapeutic approach for restoring cognitive function.
Keywords: blood–brain barrier, brain microvascular endothelial cell, cognitive function, docosahexaenoic acid, fatty acid-binding protein
Introduction
Docosahexaenoic acid (DHA) is an ω-3 (n-3) polyunsaturated fatty acid with potent effects on synaptic transmission and long-term potentiation in the hippocampus (Itokazu et al., 2000; Young et al., 2000), and antiapoptotic effects in the CNS (Calviello et al., 1998). DHA promotes hippocampal neurite growth, synapsin puncta formation, and synaptic protein expression and is therefore important for cognitive function (Gómez-Pinilla, 2008; Cao et al., 2009). Epidemiological studies report that lower fish consumption or n-3 fatty acids/DHA dietary intake is associated with cognitive impairment (Kalmijn et al., 1997; Barberger-Gateau et al., 2002, 2007; Laitinen et al., 2006), although controversial findings exist (Engelhart et al., 2002). In Alzheimer's disease, lower plasma and brain DHA levels have been reported (Prasad et al., 1998; Lukiw et al., 2005; Cunnane et al., 2012; Yassine et al., 2016). DHA supplementation provides benefit in mild cognitive impairment and early Alzheimer's disease (Cole et al., 2009), however, people with late-stage Alzheimer's disease receive no benefit from DHA (Freund-Levi et al., 2009), likely due to the significant neuronal death at this stage of the disease.
The brain has a limited ability to synthesize its own DHA; therefore, DHA has to travel from the plasma across the blood–brain barrier (BBB) to reach the brain (Rapoport et al., 2001, 2007), although other factors may contribute to brain DHA levels, including CNS retention and metabolism of DHA, dietary fatty acid intake, and peripheral DHA pharmacokinetics (Arterburn et al., 2006; Dyall, 2015; Lavandera et al., 2016). Both passive and active transport have been shown to mediate the luminal uptake of DHA into brain endothelial cells (Chen et al., 2008; Hamilton et al., 2012; Nguyen et al., 2014); however, movement of DHA across the brain endothelial cytoplasm is likely to be limited by the aqueous intracellular environment; therefore, an intracellular protein would be required to facilitate this process. In support of this, we have demonstrated that fatty acid-binding protein 5 (FABP5) is involved in the brain uptake of exogenous DHA using an in situ transcardiac perfusion technique (Pan et al., 2015b).
Given the role of FABP5 in mediating brain uptake of DHA, and the role of DHA in cognition, it is likely that mice deficient in FABP5 would have lower endogenous brain DHA levels and cognitive deficit. Consistent with this suggestion, Yu et al. (2014) have demonstrated cognitive impairments in FABP5−/− mice, which they attributed to diminished peroxisome proliferator-activated receptor β/δ (PPARβ/δ) activation by arachidonic acid (which is transported to the nucleus by FABP5) (Kobilo et al., 2011) and a subsequent decrease in PPAR-mediated hippocampal neurogenesis (Kaczocha et al., 2009). Although others have shown FABP5 deficiency leads to cognitive deficits and have associated this with reduced PPAR-mediated hippocampal neurogenesis, we propose that an additional factor contributing to the cognitive deficit in FABP5−/− mice is a reduction in BBB transport of DHA, and therefore, diminished brain DHA levels.
In this study, we performed a broader range of cognitive assessments in FABP5−/− mice than those previously reported by Yu et al. (2014), to cover working, short-term, long-term, episodic, and fear memory. The effect of gender on cognitive function was also assessed, together with measurement of endogenous brain levels of DHA. Furthermore, we measured uptake of DHA into brain microvascular endothelial cells (BMECs) and isolated microvessels from FABP5−/− mice and their wild-type (WT) littermates to assess whether deletion of FABP5 correlates not only to reduced endogenous brain DHA levels and cognitive deficit, but decreased brain endothelial cell availability of DHA. To our knowledge, this is the first study associating cognitive deficits resulting from FABP5 deletion with reduced BBB transport and brain levels of DHA. These results may highlight FABP5 as a target that may be manipulated to modify brain DHA levels in disorders where brain DHA levels are reported to be lower.
Materials and Methods
Materials.
DNase I, collagenase, DMEM, FBS, TaqMan primers/probes, Hoechst 33342, and hydrogen peroxide (30% w/v) were purchased from Invitrogen. BSA, rat tail collagen Type I, PFA, PBS, Triton X, donkey serum, phloretin, Supelco 37-component FAME mix and polyethylenimine were obtained from Sigma-Aldrich. Chromatography-grade solvents hexane and dichloromethane were purchased from Merck. 14C-DHA, 14C-glucose, 14C-sucrose, and 3H-diazepam were sourced from American Radiolabeled Chemicals. DHA was purchased from Cayman Chemicals. The EBM-2 basal media was supplemented with EGM-2 Endothelial Cell Growth Medium-2 BulletKit as per the manufacturer's instructions (Lonza) to make the complete growth media for primary BMECs. pSG5-Fabp5 and pSG5 vector were obtained from Agilent Technologies. Mouse astrocytes and COS-7 cells were kindly provided by Professor Robert Medcalf (Monash University, Australia) and Professor Phillip Nagley (Monash University, Australia), respectively.
Cognitive assessment.
All animal experiments were approved by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee and performed in accordance with the National Health and Medical Research Council Guidelines for the care and use of animals for scientific purposes. FABP5−/− mice were generated according to a previously published method (Owada et al., 2002) and maintained on C57BL/6 background. FABP5−/− and littermate FABP5+/+ mice were bred in our laboratory and genotyped for the presence or absence of FABP5 gene by Transnetyx. Both male and female mice used in cognitive assessments were 12–15 weeks old. Before behavioral testing, mice were individually housed and acclimatized in a holding room on a 12:12 h reverse light-dark cycle (lights on 7:00 P.M.) with water and food ad libitum. All mice (including the breeders) were fed WEHI mouse breeder cubes (Ridley Agri Products), with ingredients and typical nutrients analysis listed in Table 1. Regular handling of the mice was performed by the experimenter for 3–5 min at a fixed time daily 1 week before cognitive assessments. Before experimentation, mice were allowed to acclimatize in the behavioral assessment room for 1 h. All mice received at least a 48 h break in between the cognitive assessments.
Table 1.
Nutrient information of BARASTOC WEHI Mouse Breeder Cubes Irradiateda
| Nutrient | % | Vitamin | Amount |
|---|---|---|---|
| Minimal crude protein | 20 | Vitamin A | 15 IU/g |
| Crude fat | 8.5 | Vitamin D3 | 2 IU/g |
| Arachidonic acid | 0.03b | Vitamin E | 260 mg/kg |
| DHA | 0.40b | Vitamin K3 | 55 mg/kg |
| Linolenic acid | 36.59b | Vitamin B1 | 64 mg/kg |
| Oleic acid | 39.83b | Vitamin B2 | 48 mg/kg |
| Palmitic acid | 13.69b | Vitamin B6 | 30 mg/kg |
| Stearic acid | 6.02b | Vitamin B12 | 0.08 mg/kg |
| Crude fiber | 3.2 | Niacin | 400 mg/kg |
| Acid detergent fiber | 4.4 | Panto | 220 mg/kg |
| Neutral detergent fiber | 10.4 | Biotin | 1.48 mg/kg |
| Digestible energy | 13.2 MJ/kg | Folic | 11 mg/kg |
| Calcium | 1.2 | Iron | 51 mg/kg |
| Phosphorous | 0.96 | Zinc | 60 mg/kg |
| Sodium | 0.35 | Manganese | 120 mg/kg |
| Potassium | 0.89 | Copper | 10 mg/kg |
| Chloride | 0.57 | Selenium | 0.1 mg/kg |
| Magnesium | 0.25 | Molybdenum | 0.4 mg/kg |
| Lysine | 1.22 | Cobalt | 0.6 mg/kg |
| Methionine | 0.38 | Iodine | 1.4 mg/kg |
| Starch | 25 |
a Ingredients include the following: wheat, wheat byproducts, groats, meat meal, canola oil, full fat soya bean meal, skim milk powder, molasses, salt, vitamins, trace minerals.
b Percentage of total fatty acids.
T-maze spontaneous alternation.
Spatial working memory was assessed using the T-maze spontaneous alternation task, similar to that previously described (Mishima et al., 2004). The T-maze apparatus has three equal arms in the shape of a capital T (30 cm L × 10 cm W × 20 cm H). The mice were placed at the base of the vertical arm and allowed to move freely through the maze during a 5 min test session. The sequence of arm entries was recorded manually (arm entry was counted if both the head and the base of the tail entered the arm). A correct alternation was defined as a nonrepeated entry into the arms for three consecutive entries. The percentage of spontaneous alternations was calculated as shown in Equation 1. Each arm of the T-maze was cleaned between sessions using ethanol to remove any olfactory cues, which may affect the behavior of the next mouse tested.
 |
Y-maze.
Short-term spatial memory was assessed using the Y-maze protocol as adapted from Dellu et al. (1997). The Y-maze consists of three arms (50 cm L × 16 cm W × 31 cm H) in the shape of a capital Y with spatial cues on the internal wall of each arm. In the training session, each mouse was allowed to explore the Y-maze for 5 min, with one of the arms (other than the start arm) closed. After a 5 h retention interval, the mice were placed into the same Y-maze and allowed to explore for 5 min, this time with all three arms accessible. The time the mouse spent in each arm was analyzed by Viewer tracking software (Biobserve). The time spent in the novel and familiar arms was compared for each genotype and gender combination. The distance traveled by the mice during the training session was used to measure locomotor activity.
Water maze.
Spatial learning was assessed using a water maze protocol, with minor modifications of the method previously described by Gulinello et al. (2009). The mice underwent a series of trials on 3 consecutive days in a pool (120 cm in diameter and 20 cm in depth) filled with opaque water (water containing white nontoxic paint), maintained at 24 ± 2°C. Consistent, distinct external spatial cues were placed in the room, and spatial cues were placed on the internal walls of the pool, dividing the pool into four quadrants. An escape platform (12 cm in diameter) was placed in a fixed position (the target zone) diagonally opposite the starting position. On day 1 (cued training), the mice were placed into the water at the starting position and allowed to find the visible platform (0.5 cm above water, with a flag attached) in four trials with an intertrial interval of 30 ± 10 min. Mice failing to find the platform by 60 s were manually guided to the platform. All mice remained on the platform for 10–20 s before being removed, dried, and placed in a holding cage. On day 2 (spatial trial), the mice underwent three hidden platform trials (the platform was submerged 0.5 cm below water), with an intertrial interval of 30 ± 10 min. On day 3 (probe test), the platform was removed, and the mice were placed into the pool and allowed to explore the maze for 60 s. Swimming behavior was recorded, and the position of the mouse was determined by video tracking. The platform/target zone visit latency (escape latency) and swimming speed were determined by Viewer tracking software.
Novel object recognition.
Episodic memory function was measured in a novel object recognition task (Antunes and Biala, 2012). Briefly, mice were allowed to acclimatize to the arena (40 cm L × 40 cm W × 30 cm H) for 5 min, twice on day 1. The distance traveled by mice in day 1 acclimatization sessions was used to assess locomotor activity. On day 2, two identical objects were placed into the arena, and mice were allowed to freely explore the arena for 5 min. After 3.5 h, the trained mice were tested in the arena with one of the familiar objects replaced with a novel object. The time that mice spent exploring each object (classified by nose sniffing the object) on day 2 was determined by Viewer tracking software. The ability of the mice to recognize the novel object was determined by the discrimination index, calculated as shown in Equation 2 as follows:
 |
Contextual fear conditioning.
Contextual fear conditioning was performed as previously reported (Kim et al., 1992; Do-Monte et al., 2015). Briefly, mice were subjected to a 5 min conditioning session, which consisted of two foot shocks (0.6 mA, 1 s) delivered at an interval of 60 s in an open top conditioning chamber (36 cm L × 18 cm W × 32 cm H) (Harvard Apparatus). The elevation of velocity of the mice after foot shocks was used to determine the sensorimotor activity, and the baseline velocity was used to monitor locomotor activity. Mice were reexposed for 5 min to the same context either 4 or 24 h after conditioning (referred to as 4 h reexposure and 24 h reexposure cohorts, respectively). Although 24 h retention interval is often used, the shorter 4 h retention interval was also included as it is a similar timeframe to the 3.5–5 h interval used in the novel object recognition (NOR) and Y-maze tasks. The duration of freezing during the second exposure was manually quantified from the recorded video clip by two experimenters independently, blinded from the mouse groups, and the average values were used for analysis.
Quantification of mouse brain DHA levels.
Following behavioral analysis, mice were anesthetized with isoflurane and decapitated to remove the brain. Fatty acids were extracted from the brain cortex of FABP5+/+ and FABP5−/− mice following cognitive assessment and were prepared for gas chromatography as detailed previously (Fraser et al., 2008). Briefly, 50 mg of brain cortex was added to 350 μl of ice-cold methanol and homogenized in a mechanical homogenizer (IKA T25 digital ULTRA-TURRAX, Thermo Fisher Scientific) at 6000 rpm for 2 × 30 s. The sample was cooled on ice after homogenization. Chloroform (350 μl) containing 100 mg/L of butylated hydroxytoluene (BHT) was added and the solution briefly vortexed. The blade of the homogenizer was rinsed with a further 150 μl of chloroform/BHT:methanol 2:1 v:v. The solutions were combined and centrifuged at 2000 × g for 5 min, and the supernatant was removed. The pellet was resuspended by vortexing in 200 μl of chloroform/BHT:methanol 1:1 v:v, and the solution was recentrifuged. The supernatant was removed and combined with the first supernatant; and after the addition of a further 400 μl of chloroform/BHT and 325 μl of 0.9% w/v potassium chloride, the solution was vortexed and centrifuged. The purified lipid fraction (obtained in the lower chloroform phase) was collected and dried under N2 before adding 250 μl of 1 m ethanolic potassium hydroxide. The solution was left overnight at room temperature in a sealed tube. A volume of 1250 μl of MilliQ water was added along with 400 μl of hexane:diethyl ether 1:1 v:v containing 100 mg/L BHT. The biphasic solution was vortexed, centrifuged as above, and the upper phase discarded. The addition of hexane:diethylether/BHT, vortexing, centrifugation, and removal of the upper phase was repeated three times. The pH of the lower phase was adjusted to pH 3 with concentrated HCl. A 400 μl volume of diethyl ether/BHT was added, before vortexing, centrifugation, and removal of the upper phase. This step was repeated twice, and the lower phase was discarded. The two upper phases (containing the free fatty acids derived from the lipid extract) were combined and dried under N2. Methylesters of the free fatty acids (FAME) were prepared in a sealed tube with a solution of 2.5% concentrated sulfuric acid in anhydrous methanol overnight at room temperature. The solution was mixed with 1.5 ml of MilliQ water and 500 μl of hexane and then vortexed. FAME were collected from the top hexane layer and diluted 10-fold before gas chromatography injection. Methyl nonadecanoate (C19:0ME) was used as an internal standard (IS) for quantitative analysis. Analysis of FAME was conducted using a 6850 Agilent gas chromatography with flame ionization detection (Agilent Technologies). A 30 m × 0.25 mm id column with a 0.25-m-thick film (df) of DB-5 ms phase was used for all sample analyses; 1 μl of FAME standard mixture or test sample was injected into the gas chromatography inlet at a split ratio of 20:1. The oven was temperature programmed from 60°C (0.1 min) to 280°C at 20°C/min (5 min hold). Hydrogen (99.999% pure) was used as carrier gas at a constant flow rate of 1.5 ml/min, and the detector temperature was 250°C. Data were processed using Agilent Chemstation software (Agilent Technologies). FAME peaks in the samples were identified according to the retention time and elution order of authentic FAME standards. The intensity of each major fatty acid peak was normalized to that of the IS, and the weight of cortex. Subsequently, all data were normalized by the average of FABP5+/+ mice, to represent the changes in FABP5−/− brain fatty acid levels relative to FABP5+/+ mice.
Isolation and characterization of mouse BMECs.
Before measuring the permeation of 14C-DHA across BMECs of FABP5+/+ and FABP5−/− mice, it was necessary to ensure the technique for endothelial cell isolation resulted in a pure endothelial cell population with expected permeability properties. The BMEC isolation procedure used was similar to that previously reported (Ruck et al., 2014). Brains of C57BL/6 mice (8–10 weeks) were removed under sterile conditions, and the cortex isolated and homogenized in DMEM (4:1 volume:tissue ratio) using a Dounce homogenizer (Tissue Grinder, Potter-ELV, Wheaton Industries) with 10 up-and-down strokes. The homogenate was centrifuged at 1000 × g, and the pellet was resuspended in DMEM, supplemented with 1% w/v collagenase Type II and DNase I (39 U/ml). The mixture was incubated at 37°C for 90 min before centrifugation at 2000 × g for 20 min. The pellet was mixed with 30% w/v BSA in DMEM, to achieve a final BSA concentration of 15% w/v, and centrifuged at 2000 × g for 30 min. The supernatant containing the myelin layer was carefully removed, and the capillary-enriched pellet was resuspended in DMEM and filtered through a 70 μm nylon mesh (BD Biosciences) to remove large blood vessels. The flow-through was centrifuged and resuspended with complete BMEC media (i.e., supplemented EBM-2 media) and seeded into either a 24-well plate (for immunocytochemistry studies) or a 24-well Transwell insert (for permeability studies). All surfaces were coated with 1% v/v rat tail collagen Type I in PBS. The cells were maintained at 37°C in a humidified incubator (5% CO2/95% O2) overnight, before being treated with 3 μg/ml puromycin in complete BMEC media for 72 h. Complete media changes were performed every second day, and cell growth was monitored using phase-contrast microscopy (Nikon). The purity of the primary BMEC culture was confirmed by positive immunohistochemical detection for the endothelial marker CD31 and RT-PCR to confirm lack of GFAP (a marker of astrocytes).
Immunocytochemistry for CD31 staining.
Primary C57BL/6 BMEC cultures (50%–60% confluent) were fixed with 4% w/v PFA for 15 min. After three PBS washes (5 min each), cells were incubated with 0.5% v/v Triton X-100 in PBS for 30 min and blocked with 5% v/v donkey serum in PBS for 1 h at room temperature. Cells were incubated with rat anti-mouse CD31 antibody (eBioscience) in PBS with 5% v/v donkey serum (1:300) overnight at 4°C. Cells were washed twice in PBS (5 min each) before incubation with the secondary AlexaFluor-594 goat anti-rat IgG (H+L) antibody (Invitrogen) diluted (1:200) in PBS with 5% v/v donkey serum at room temperature for 2 h. Cells were washed (3 × 5 min) with PBS and counterstained with Hoechst 33342 (2 μg/ml) for 15 min. The immunoreactivity was visualized using an Eclipse TE2000-E microscope (Nikon).
RT-PCR for GFAP expression.
The presence of GFAP mRNA in the primary cell culture isolated from C57BL/6 mice was screened, with RNA isolated from primary mouse astrocytes used as a positive control for GFAP detection. Total RNA was isolated using an RNeasy Plus Mini Kit (QIAGEN). iScript One-Step RT-PCR Kit (Bio-Rad) and TaqMan primers/probes were used for gene amplification, and detection of gene expression was performed in a CFX96 system (Bio-Rad).
In vitro permeability of probe compounds across C57BL/6 BMECs.
Cells grown on Transwell inserts (0.4 μm) were cultured for 7 d in EBM-2 basal media supplemented with growth factors, as detailed in Materials. The trans-endothelial electrical resistance (TEER) was monitored from days 5–7 using an EVOM-endohmeter (World Precision Instruments). Transwell inserts with TEER value >50 Ω cm2 on day 7 were selected for permeability studies. The probe compounds used for characterizing the permeability characteristics of the BMECs were 14C-sucrose (marker of passive paracellular diffusion), 3H-diazepam (marker of passive transcellular diffusion), and 14C-glucose (marker of active transport). On day 7, cells were incubated with blank EBM-2 basal media (transport medium) at 37°C in a humidified incubator (5% CO2/95% O2) 2 h before the commencement of the permeability study. In studies assessing the transport of 14C-glucose, BMECs were treated with or without phloretin 50 mm (an inhibitor of the glucose transporter) for 1 h before the commencement of the 14C-glucuose permeability study and during the permeability study. To commence permeability studies, 0.5 μCi of each radiolabeled probe was added to the apical chamber. A 10 μl aliquot of the apical chamber was taken at time 0, and 20 μl samples were taken from the receptor chamber for up to 30 min, with an equivalent volume of fresh receptor solution being replenished at each time point. All samples were measured for radioactivity in a Tri-Carb 2800TR liquid scintillation counter (PerkinElmer). An apparent permeability coefficient (Papp) was calculated based on the appearance of probe compound in the receptor chamber, using Equation 3 as follows:
 |
where dDPM/dt is the rate of change of the probe compound appearing in the receptor chamber with respect to time, A is the diffusion area (0.33 cm2), DPMdonor is the initial total radioactivity in the donor chamber, and Vdonor is the volume of the donor chamber.
DHA permeability across BMECs of FABP5+/+ and FABP5−/− mice.
BMECs from FABP5+/+ or FABP5−/− mice isolated using the validated technique used for isolation of BMECs from C57BL/6 mice were plated on 24-well Transwell permeable inserts (0.4 μm) and incubated at 37°C in a humidified incubator (5% CO2/95% O2) until confluent. The TEER value of the BMECs was measured, and only those with TEER values exceeding 50 Ω cm2 were used for permeability studies. The apical chamber was dosed with EBM-2 basal media containing 0.1 mm DHA (with 0.5 μCi 14C-DHA) and 0.1 mm BSA, and samples from the receptor chamber containing 0.1 mm BSA in EBM-2 basal media were removed over 10 min. At the completion of the permeability study, the cells on the Transwell insert were lysed using 1 m NaOH for 3 h at 37°C. The lysate was collected and neutralized using 2 m HCl. All donor and receptor samples and cell lysate were mixed with 2 ml of Ultima Gold scintillation mixture, and the radioactivity was quantified using the Tri-Carb 2800TR liquid scintillation counter. The Papp of 14C-DHA was calculated as per Equation 3, and the cellular concentration of 14C-DHA was normalized to total protein count determined by a BCA protein assay as per the manufacturer's protocol (Thermo Fisher Scientific).
Uptake of 14C-DHA into isolated brain capillaries of FABP5+/+ and FABP5−/− mice.
Capillary depletion was performed in a manner similar to that reported previously (Triguero et al., 1990). Briefly, the brain cortex of FABP5+/+ and FABP5−/− mice was removed and placed in an ice-cold Dounce homogenizer. The cortices were homogenized (10 strokes) in DMEM. BSA was added to the homogenate to achieve a final concentration of 15% w/v, and the homogenate was centrifuged at 2000 × g for 30 min at 4°C. The resulting supernatant (brain parenchymal fraction) and pellet (capillary fraction) were separated. The excessive BSA was removed by rinsing the capillary pellet with DMEM twice. The capillary pellet was resuspended in 100 μl of DMEM, transferred into a 96-well plate, and the optical density of the capillary suspension was estimated using EnVision 2101 multilabel reader (PerkinElmer) at a wavelength of 560 nm, to quantity capillary content in the sample. The plate was maintained at 37°C, and 0.3 μCi of 14C-DHA was dosed into each well at time 0. After 10 min, the microvessel suspension was collected and centrifuged at 2000 × g for 10 min at 4°C. A 10 μl aliquot was taken from the supernatant for quantification of 14C-DHA. The pellet was rinsed twice with 2 ml of DMEM and digested in 2 ml of Solvable at 50°C overnight and neutralized with 30% (v/v) hydrogen peroxide. Radioactivity of DHA was determined as described above. The percentage of 14C-DHA capillary uptake was calculated as per Equation 4 and normalized by the optical density of the capillary at 560 nm as follows:
 |
Uptake of 14C-DHA in COS-7 cells transiently transfected with FABP5.
To further confirm DHA uptake into BMECs and capillaries was dependent on FABP5, 14C-DHA uptake into COS-7 cells with and without FABP5 transfection was assessed. COS-7 cells (passage 15) were seeded into a 24-well plate (25,000 cells/cm2) and incubated in DMEM supplemented with 5% FBS at 37°C with 95%O2/5%CO2. One day after seeding, the cells were transfected drop-wise with 13 μl of NaCl (150 mm, pH 7) containing 0.4 μg polyethylenimine and either 0.1 μg of pSG5-Fabp5 DNA or blank pSG5, followed by the addition of 0.5 ml DMEM + 5% FBS. After 48 h, the vector control and FABP5 transfected cells were trypsinized and harvested by centrifugation (1000 × g, 10 min). The cell pellet underwent two freeze-thaw cycles followed by sonication for 30 s to release cellular proteins. The protein extract was loaded onto the ELISA plate, and FABP5 was detected as per the manufacturer's protocol (DL Sci and Tech Development, Wuxi). The uptake of 14C-DHA (0.5 μCi) into COS-7 cells over a 2 min period was assessed as previously reported (Pan et al., 2015b), and the cell-to-medium ratio was determined after normalizing 14C-DHA cellular uptake to total protein count as determined via a BCA protein assay.
Statistical analyses.
All data are expressed as mean ± SEM. Data analyses were performed using SPSS (IBM) or Prism software (GraphPad). The comparisons between experimental and control groups were undertaken by Student's t tests when only two groups were compared. ANOVA tests were performed for cognitive assessment analyses where genotype and gender effects were investigated, with repeated measures in the water-maze and Y-maze as appropriate. Statistical significance was set at a value of p < 0.05.
Results
Cognitive impairment in FABP5-deficient mice is associated with lower endogenous brain DHA levels
A previous study (Yu et al., 2014) has shown cognitive impairment in FABP5−/− mice using a limited number of behavioral paradigms, with a focus more on long-term memory. The current study used FABP5−/− mice and their WT littermates (FABP5+/+) of both genders, and a larger battery of cognitive assessments to assess the impact of FABP5 deletion on learning and memory, including working memory, short-term and long-term spatial memory, memory retrieval, episodic memory, and fear memory.
In the T-maze, analysis of the percentage of correct spontaneous alternations over 5 min revealed a significant effect of genotype (two-way ANOVA; F(1,35) = 16.2, p < 0.001), with no main effect due to gender and no interactions between the two factors. FABP5−/− mice demonstrated a significantly lower percentage of correct spontaneous alternations (Fig. 1A; 12.9 ± 3.3% lower than FABP5+/+ mice), indicating an impairment in working memory.
Figure 1.

A, The percentage of correct spontaneous alternations in the T-maze over 5 min, with reduced correct spontaneous alternations observed in FABP5−/− mice, regardless of gender (n = 18 or 19). ***p < 0.001 (two-way ANOVA). B, The time spent in the novel and familiar arm of the Y-maze paradigm over 5 min, with all mice, except female FABP5−/− mice spending significantly more time in the novel arm than familiar arm (n = 16 or 17). ***p < 0.001 (pairwise ANOVA comparison test). C, Performance of FABP5+/+ and FABP5−/− mice (n = 16–18) in the cued training trials and spatial trials in the water maze assessed as absolute escape latency, with FABP5−/− mice showing impaired learning ability for cued training, regardless of gender. ***p < 0.001 (two-way repeated-measures ANOVA). D, In the probe trial of the water maze, FABP5+/+ mice required significantly less time to reach the target zone relative to FABP5−/− mice, regardless of gender (n = 16–18). *p < 0.05 (two-way ANOVA). E, The discrimination index calculated from the novel object recognition, with FABP5−/− mice exhibiting a significantly lower ability to differentiate between the novel object and familiar object, regardless of gender. *p < 0.05 (two-way ANOVA). F, The percentage of contextual freezing reduces significantly in FABP5−/− mice, regardless of gender and retention interval (n = 18–20). ***p < 0.001 (two-way ANOVA). G, A more robust reduction in the percentage of contextual freezing time was observed in FABP5−/− mice in the 4 h reexposure cohort than the 24 h reexposure cohort, regardless of gender.
In the Y-maze, the time spent in the novel arm and previously explored arm were analyzed using a repeated-measures ANOVA, revealing an overall arm effect (F(1,32) = 86.5, p < 0.001), in combination with an arm × genotype × gender interaction (F(1,32) = 5.3, p < 0.05). Further post hoc analysis (pairwise ANOVA comparison) revealed that, although all groups were able to distinguish between the novel and familiar arm and spent more time in the newly available novel arm, there was no significant difference in the time spent in the two arms in female FABP5−/− mice (Fig. 1B). This indicated an impaired short-term spatial memory only in female FABP5−/− mice.
In the water maze, no significant gene effect on swimming speed of mice was identified. A significant effect of session was observed, with all mice exhibiting a decreased latency to obtain the platform over successive trials in the cued training stage on day 1 (Fig. 1C; two-way repeated-measures ANOVA; F(3,90) = 28.4, p < 0.001). A between-subject analysis also revealed a significant gene effect (F(1,30) = 9.7, p < 0.001), with FABP5−/− mice requiring more training to locate the escape platform than the FABP5+/+ mice. Once the task was successfully acquired, all mice (regardless of gender or genotype) were able to swim to the hidden platform on day 2. There was no difference in the platform escape latency for all three spatial trials, compared with that in the final session of cued training (day 1), suggesting unaffected spatial memory retention over the 24 h period (Fig. 1C; two-way repeated-measures ANOVA, comparing cued training session 4 and spatial trial session 1). Similarly, there were no significant differences in the latency to the hidden platform during the three spatial trials (Fig. 1C). However, in the day 3 probe trial, FABP5+/+ mice reached the target zone significantly quicker than FABP5−/− mice, regardless of gender (Fig. 1D; two-way ANOVA; F(1,33) = 5.3, p < 0.05), indicating a weakening in memory retrieval due to FABP5 deletion.
In the novel object recognition (NOR) paradigm, a two-way ANOVA of the discrimination index revealed a significant effect of genotype, but no effect of gender or an interaction between the factors (F(1,29) = 5.6, p < 0.05). In combining the gender data (Fig. 1E), FABP5−/− mice had a significantly lower discrimination index than FABP5+/+ mice, which was close to zero, demonstrating that FABP5−/− mice were unable to differentiate the novel object from the previously explored familiar object, spending an equivalent amount of time exploring both objects in the testing session.
In the contextual fear conditioning paradigm, a main genotype effect was observed, where FABP5−/− mice demonstrated significantly less freezing behavior than FABP5+/+ mice (three-way ANOVA; F(1,37) = 8.3, p < 0.001; Fig. 1F). The decrease in freezing duration was more robust in the 4 h reexposure cohort than in the 24 h reexposure group (Fig. 1G).
Overall, the cognitive assessment results suggested that working memory, short-term memory, episodic memory, and fear memory formation were affected due to FABP5 deficiency, whereas long-term memory remained intact. In addition, it has been confirmed that the different performance of FABP5+/+ and FABP5−/− mice was not due to alteration in sensorimotor or locomotor activity (Fig. 2). These memory deficits were not associated with an overall reduction in brain weight (Fig. 3A), but a 14.7 ± 5.7% reduction in total cortical DHA was observed with FABP5 deficiency (Fig. 3B). Of all the other major fatty acids quantified, only stearic acid (14.65 ± 5.73%, p < 0.05) and palmitic acid (14.50 ± 5.93%, p < 0.05) levels were reduced in the brains of FABP5−/− mice, with the composition of brain fatty acids in FABP5+/+ and FABP5−/− mice listed in Table 2.
Figure 2.

A, Sensorimotor activity of FABP5+/+ and FABP5−/− mice, with no main gene or gender effect identified (two-way ANOVA, n = 14) on the elevation in velocity (cm/s) immediately after foot shocks in fear conditioning, suggesting the absence of sensorimotor defects due to FABP5 genetic deletion. B, Locomotor activity measured by the baseline velocity of mice in the fear conditioning chamber during conditioning (n = 14), (C) the distance traveled in the Y-maze training session (n = 8 or 9), and (D) Novel object recognition (NOR) habituation sessions (n = 16–19), with no genotype or gender effect observed (two-way ANOVA), suggesting that FABP5 deletion does not affect locomotor activity. Data are mean ± SEM.
Figure 3.

A, Brain weight of FABP5+/+ and FABP5−/− mice, with no significant difference identified (n = 5 or 6), and (B) endogenous brain cortex DHA levels, with reduced DHA levels measured in FABP5−/− mice relative to FABP5+/+ mice (n = 6). *p < 0.05.
Table 2.
Composition of brain fatty acids in cortex of FABP5+/+ and FABP5−/− micea
| Fatty acid | Fatty acid | FABP5+/+ mice | FABP5−/− mice |
|---|---|---|---|
| C14:0 | Myristic acid | 0.13 ± 0.01 | 0.11 ± 0.01 |
| C16:1 | Palmitoleic acid | 0.30 ± 0.02 | 0.29 ± 0.02 |
| C16:0 | Palmitic acid | 9.05 ± 0.36 | 7.74 ± 0.39* |
| C18:0 | Stearic acid | 8.13 ± 0.29 | 6.94 ± 0.36* |
| C20:4n-6 | Arachidonic acid | 4.14 ± 0.12 | 3.69 ± 0.21 |
| C20:3n-6 | Eicosatrienoic acid | 0.23 ± 0.01 | 0.20 ± 0.01 |
| C20:1n-9 | Eicosenoic acid | 0.59 ± 0.01 | 0.53 ± 0.03 |
| C22:6n-3 | DHA | 7.60 ± 0.26 | 6.48 ± 0.35* |
a Data are mean ± SEM (n = 6), presented as intensity of fatty acid peak/IS peak normalized by weight of brain cortex.
*p < 0.05 (unpaired Student's t test).
Establishment of an in vitro model to assess 14C-DHA BBB transport
A previous in situ transcardiac perfusion study has shown a significant reduction in 14C-DHA transport into the brain of FABP5−/− mice, but whether FABP5 is responsible for brain endothelial cell uptake and transport of DHA across the brain endothelial cell is not fully characterized. Therefore, an in vitro BBB model based on mouse BMECs was developed and confirmed for appropriate barrier characteristics, which could then be translated to study the disposition of DHA across BMECs from both FABP5+/+ and FABP5−/− mice.
The current BMEC isolation protocol used 15% BSA density centrifugation to obtain microvessel-enriched fractions, which were seeded and treated with puromycin. The treatment with puromycin leads to a selection of murine BMECs as they express high levels of efflux transporters (e.g., P-glycoprotein), leading to a relative resistance to puromycin, whereas contaminating cells, such as pericytes and astrocytes, are much more susceptible to puromycin-mediated cytotoxicity (Perrière et al., 2005). Immediately after seeding, microvessels and contaminating cells formed conglomerates floating in the culture medium (Fig. 4A; day 0). During treatment with puromycin (3 μg/ml), the contaminating cells appeared to die, whereas the endothelial cells seeded well and started to proliferate (Fig. 4A; days 1–3). The cells that survived the puromycin treatment continued to grow (Fig. 4A; day 5) to become spindle-shaped endothelial cells (Fig. 4A, day 7). Furthermore, all of these cells stained positive for CD31 (an endothelial cell marker) (Fig. 4B), with the nuclei identified by Hoechst stain. In addition, the mRNA for GFAP (an astrocyte marker) was not detected in the final BMEC population by RT-PCR, unlike in primary astrocytes used as a positive control (Fig. 4C). Together, these results suggest that a highly purified mouse BMEC population was obtained using our isolation procedure.
Figure 4.
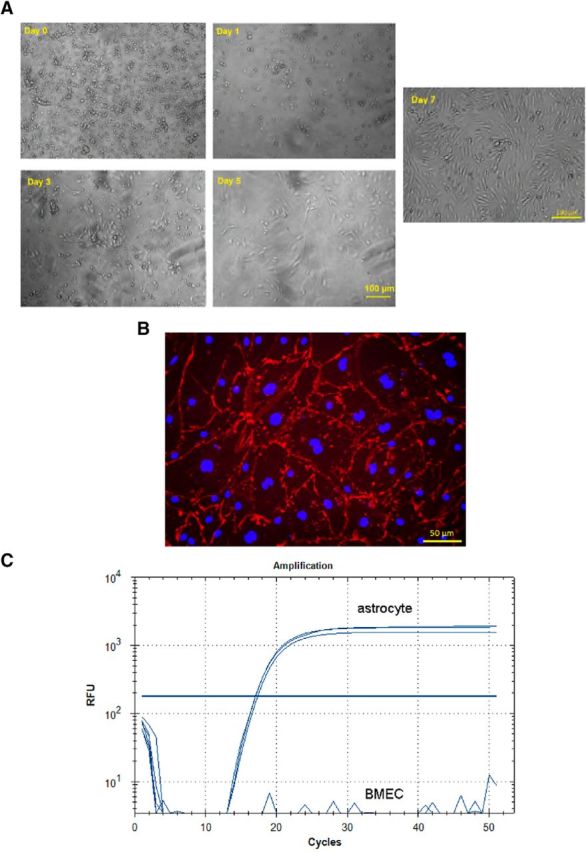
A, Morphology of BMECs from day of isolation (day 0) to day 7, with images taken by phase-contrast microscopy. On day 0, the population contains BMECs and contaminating cells (likely pericytes and astrocytes). From day 1 to day 3, the cells received puromycin treatment (3 μg/ml), which is toxic to contaminating cells but tolerated by BMECs. BMECs seeded well and started to proliferate (day 5). The cell morphology fully developed and reached 90% confluency 7 d after seeding. B, The expression of CD31 (red) on the cell surface with Hoechst staining the nucleus (blue) for cells surviving from the 72 h puromycin treatment (3 μg/ml) 5 d after seeding, imaged by phase-contrast microscopy. C, GFAP mRNA was expressed in primary mouse astrocytes but not in the isolated mouse BMEC population isolated using RT-PCR, indicating the cell population isolated was free from astrocytes.
After 7–8 d of culture on collagen-coated Transwell inserts, the functionality of tight junctions of the BMECs became apparent with TEER values of 50–170 Ω cm2 (any inserts with TEER values <50 Ωcm2 were not used for permeability studies). The Papp for 3H-diazepam (passive transcellular marker) was determined to be 3.09 ± 0.18 × 10−5 cm/s (Fig. 5A), in line with values reported for an established in vitro BBB model (Patabendige et al., 2013). The BMEC monolayer was relatively impermeable to 14C-sucrose over 10 min, with 0.39 ± 0.15% of the apical dose appearing in the basolateral chamber over 10 min (Fig. 5B), which was comparable with other validated in vitro BBB models (Wilhelm and Krizbai, 2014). After 10 min, the monolayer became significantly more permeable to 14C-sucrose (1.71 ± 0.34% of apical dose appearing in the basolateral chamber at 15 min, p < 0.05). Because of this observation, all further experiments were undertaken for 10 min only. The active transport of 14C-glucose was significantly (p < 0.05) reduced in the presence of phloretin (50 mm) treatment (Fig. 5C), suggesting that at least the GLUT1 active transport system was functional for the duration of the 10 min permeability study. The extent of inhibition was similar to previously obtained from mice transcardiac perfusion studies (Pan et al., 2015b).
Figure 5.
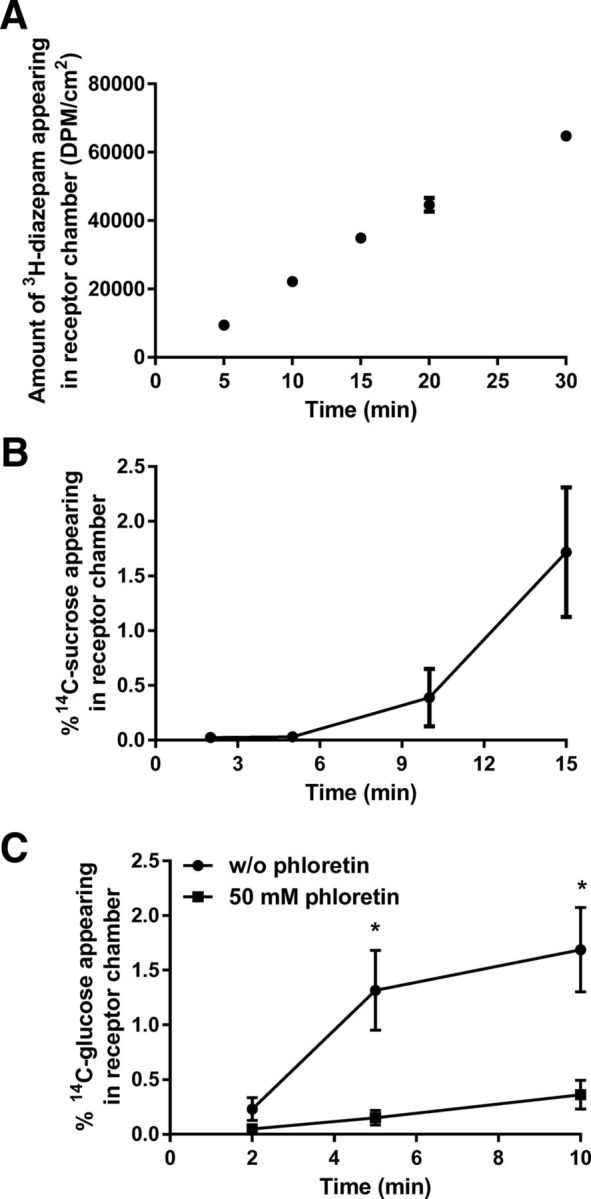
A, Amount of 3H-diazepam appearing in the receptor chamber (DPM/cm2) after application to primary mouse BMECs over a 30 min period. B, 14C-sucrose permeation across primary mouse BMECs, shown as percentage of 14C-sucrose appearing in receptor chamber relative to the dose applied in the apical chamber. C, The percentage of 14C-glucose transport across the primary mouse BMEC monolayer was significantly reduced in the presence of 50 mm phloretin (GLUT-1 inhibitor). Data are mean ± SEM (n = 3). *p < 0.05 (unpaired Student's t test).
14C-DHA BBB permeability study
Having validated the permeability characteristics of the BMECs from C57BL/6 mice, BMECs were isolated from FABP5+/+ and FABP5−/− mice using the same procedure and cultured on the Transwell inserts to assess 14C-DHA permeability. To confirm that the paracellular integrity of the BMECs from FABP5+/+ and FABP5−/− mice was similar and not different from that of C57BL/6 mice, the TEER values were measured. BMECs from FABP5+/+ and FABP5−/− mice were 78 ± 8 and 71 ± 10 Ω cm2, respectively. The accumulation of 14C-DHA in FABP5−/− mouse BMECs was reduced by 46.7 ± 5.0% compared with FABP5+/+ mouse BMECs over a 10 min period (Fig. 6A). The Papp values of 14C-DHA across BMECs from FABP5+/+ and FABP5−/− were 2.12 ± 0.13 × 10−6 and 1.93 ± 0.19 × 10−6 cm/s, respectively, with no statistically significant difference observed between the BMECs from each genotype (Fig. 6B).
Figure 6.
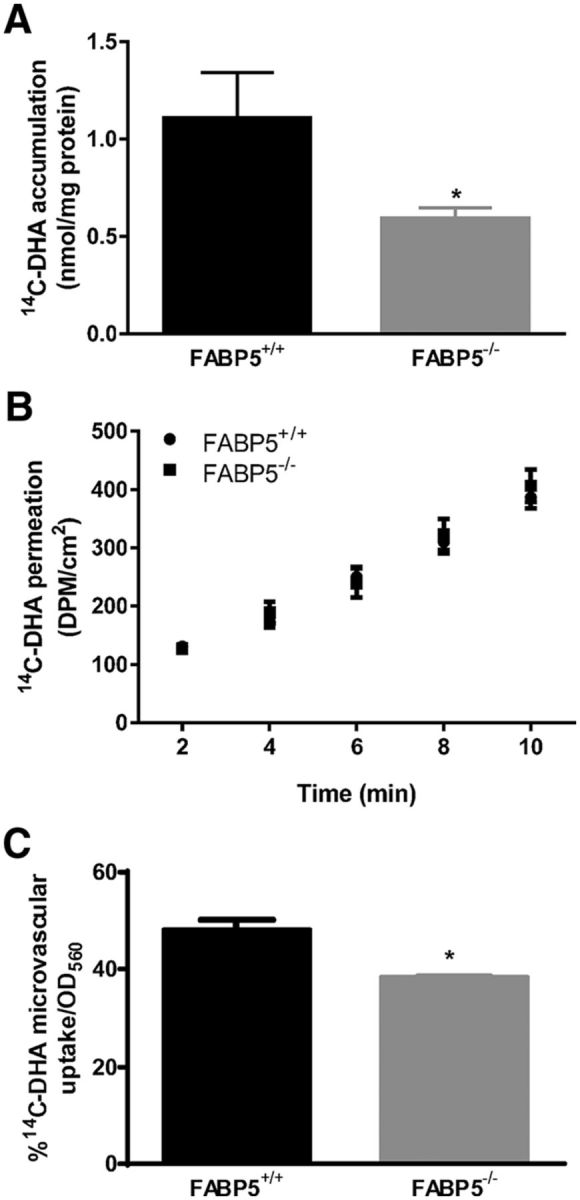
A, The amount (nmol) of 14C-DHA accumulated in FABP5−/− BMECs over 10 min was significantly lower than that in FABP5+/+ BMECs (n = 5 or 6). B, 14C-DHA permeation across BMECs isolated from FABP5+/+ and FABP5−/− mice, with Papp values determined to be 2.12 ± 0.13 × 10−6 and 1.93 ± 0.19 × 10−6 cm/s, respectively (n = 5 or 6). C, The percentage of 14C-DHA uptake (relative to amount in dosing solution; normalized by optical density at 560 nm) is significantly reduced into isolated brain capillaries of FAPB5−/− mice relative to FABP5+/+ mice (n = 3). Data are mean ± SEM. *p < 0.05.
Uptake of 14C-DHA is dependent on FABP5 expression
Mouse brain capillaries from FABP5+/+ and FABP5−/− mice were bathed in a 14C-DHA solution for 10 min, and a 14.0 ± 4.2% reduction in 14C-DHA uptake was observed in capillaries from FABP5−/− relative to those obtained from FABP5+/+ mice (Fig. 6C). To further demonstrate the role of FABP5 in general cellular uptake of 14C-DHA, COS-7 cells were transiently transfected to express FABP5. The protein expression of FABP5 in COS-7 cells was increased by 19.1 ± 6.7% following transfection (Fig. 7A), and this was associated with a 29.0 ± 10.4% increase in the uptake of 14C-DHA over a 2 min period (Fig. 7B).
Figure 7.

A, The concentration of FABP5 in COS-7 cells is significantly increased following transfection with FABP5 DNA, and this is associated with (B) increased cellular uptake of 14C-DHA compared with vector-transfected COS-7 cells (n = 3 or 4). Data are mean ± SEM. *p < 0.05.
Discussion
Although brain expression of FABP5 was reported decades ago, its role in the CNS has only been recently revealed, in concert with the roles of other FABP isoforms expressed in the brain, such as FABP3 and FABP7 (Shimamoto et al., 2014). FABP5 is important for neuron development with dynamic changes in the expression of this intracellular lipid binding protein observed during brain development, neuronal cell differentiation, and migration (Liu et al., 2010). Furthermore, FABP5−/− mice have been found to exhibit decreased hippocampal neurogenesis (Matsumata et al., 2012; Yu et al., 2012), which could result in cognitive dysfunction, as adult neurogenesis is associated with memory plasticity and capacity of memory formation (Shors et al., 2001; Leuner and Gould, 2010). Hippocampal-based cognitive dysfunction has been reported in FABP5−/− mice and has been attributed to diminished nuclear PPAR activation by arachidonic acid (carried to the nucleus by FABP5) and subsequent reduction in hippocampal neurogenesis (Yu et al., 2014). In addition to this postulated hypothesis, we propose that FABP5 deletion may impact on the brain levels of DHA, given our previous studies suggesting a role of FABP5 in mediating brain uptake of DHA (Pan et al., 2015b), and the role of DHA in maintaining cognitive function. Indeed, the present study revealed an association between cognitive impairment observed in FABP5−/− mice with reduced endogenous brain DHA levels in the brain cortex, in support of our hypothesis that FABP5 deletion attenuates CNS levels of the essential DHA.
In this study, we performed a comprehensive cognitive assessment in FABP5−/− mice and their WT littermates, including spontaneous alternation tasks and NOR tests to assess working and episodic memory, respectively. In addition, a modified water maze protocol was used to assess working memory, short-term spatial memory, long-term spatial memory, and memory retrieval ability, and two retention intervals (4 and 24 h) were used for contextual fear conditioning. Furthermore, we assessed the effect of gender on cognitive function in these mice. Genetic deletion of FABP5 resulted in a general cognitive deficiency in the extended battery of assessments used in this study, such as a reduced spontaneous alternation in the T-maze, indicating impaired working memory, and possible impairment in prefrontal cortex and hippocampus (Spellman et al., 2015). This is in agreement with the delayed acquisition of the visible platform location in the water maze (cued training), which requires working memory function. Such a delay is not due to the speed of swimming, as no difference between genotypes was observed. FABP5−/− mice also exhibited a deficiency in memory retrieval as demonstrated by an increased latency in the probe trial of the water maze, in line with previous observations (Yu et al., 2014), where a trend (p = 0.06) for memory retrieval deficiency in FABP5−/− mice was reported. A similar deficiency was also observed in the FABP5−/− mice for episodic memory tested in the NOR task, indicating that the perirhinal cortex could be affected due to FABP5 deletion (Abe and Iwasaki, 2001). Using the Y-maze, it was interesting that male FABP5−/− mice did not exhibit deficits in short-term spatial memory; however, this was observed in female FABP5−/− mice, indicating impaired hippocampal function (Shipton et al., 2014). A previous study has suggested that sex hormones may contribute to this gender difference, by interfering with the enzymatic synthesis of long-chain polyunsaturated fatty acids, such as DHA (Decsi and Kennedy, 2011). In addition, FABP5−/− mice showed impairment in fear memory formation, by exhibiting less freezing in the fear-conditioned context. It is worth noting that the difference in freezing duration between the FABP5+/+ and FABP5−/− mice was less 24 h after conditioning relative to 4 h, and this is likely due to the recruitment of the paraventricular nucleus of thalamus to consolidate fear memory after 24 h (Padilla-Coreano et al., 2012; Do-Monte et al., 2015). There may have also been delays in attaining/forming the required memory for FABP5−/− mice to respond to the stimuli appropriately, similar to that observed in the water maze, where FABP5−/− mice demonstrated a delay in obtaining the task in the cued training but were still able to retain the memory. Overall, the current study indicates that working memory, short-term memory, episodic memory, and fear memory formation are impaired due to FABP5 deletion, and apart from the hippocampus, other brain regions, such as the prefrontal cortex and perirhinal cortex, may also be affected.
Together with the cognitive deficits observed, we report a significant reduction in the endogenous brain levels of DHA in FABP5−/− mice. Unlike the Msfd2a-deficient mice, which also exhibit a substantial reduction in brain DHA levels due to deficiency in a membrane transporter involved in endothelial cell uptake of esterified DHA (Nguyen et al., 2014), the brain weight of FABP5−/− mice did not decrease significantly compared with FABP5+/+ mice. Despite this lack of difference in total brain weight with FABP5 deficiency, a significant 15% reduction in cortical DHA levels was observed. Given that unesterified DHA is the major plasma pool supplying the brain (Chen et al., 2015), the reduction in the observed brain DHA levels is likely a result of reduced BBB transport of unesterified DHA. Although phospholipids are the main repository of DHA in the brain, the beneficial effects of DHA on neurons are more dependent on unesterified DHA levels (Pan et al., 2015a); therefore, a reduction in unesterified DHA transport across the BBB would likely lead to cognitive impairment. The reduced brain DHA levels could be a cumulative effect of lower DHA transport across the BBB since embryonic phases and following birth. FABP5 is highly expressed in the embryonic brain (Pélerin et al., 2014); therefore, future studies could assess whether deletion of FABP5 also impacts on DHA brain levels during embryogenesis. Given the role of FABP in fatty acid storage and metabolism (Hotamisligil and Bernlohr, 2015), FABP5 deletion may affect DHA metabolism and/or retention within the CNS, leading to diminished brain DHA levels, although such alterations in the CNS have not yet been reported. Apart from DHA, reduced brain levels of stearic acid and palmitic acid were observed in the FABP5−/− mice relative to FABP5+/+ mice, which is in line with their high binding affinity to FABP5 (Liu et al., 2010). However, both fatty acids are not reported to have any cognitive-enhancing effects; therefore, it is likely that the cognitive deficits in FABP5−/− mice are associated with reductions in brain DHA levels. Indeed, this is consistent with observations of structural and functional changes in the hippocampal, hypothalamic, and cortical areas of the brain having been observed in animals with DHA (ω-3) deficiency (Moriguchi and Salem, 2003; Lim et al., 2005). Given the role of DHA in the CNS, the cognitive deficits associated with DHA deficiency could be as a result of impaired synaptic activity in the hippocampus, and altered neurite growth and synapsin puncta formation.
We have previously demonstrated that deletion of FABP5 results in reduced total brain homogenate concentrations of 14C-DHA following in situ perfusion. Although this is likely due to reduced BBB transport of 14C-DHA, this requires confirmation, as such changes in brain uptake of exogenous DHA could be a result of other factors, for example, altered brain binding of 14C-DHA with FABP5 deficiency or potential thickening of cerebral basement membranes (which has been suggested to reduce brain uptake of other lipophilic entities in a mouse model of Alzheimer's disease) (Mehta et al., 2013a, b). Therefore, the impact of FABP5 deletion on mouse brain endothelial cell uptake and trafficking was investigated in this study, using both BMECs and capillaries isolated from FABP5+/+ and FABP5−/− mice. It has to be acknowledged that in vitro models might not provide the architecture associated with the neurovascular unit, including presence of astrocytes, pericytes, basement membrane, and regulation by blood flow. However, assessing uptake of 14C-DHA in isolated brain endothelial cells allows for greater mechanistic insight to confirm that alterations to brain uptake observed in vivo (Pan et al., 2015b) are a direct result of reduced brain endothelial cell uptake and not due to potential confounders described above. We clearly demonstrated that FABP5 expression levels strongly affect the cellular accumulation of 14C-DHA, with a deletion of FABP5 resulting in a twofold reduction in accumulation of 14C-DHA in BMECs. This is concordant with studies we have previously undertaken in immortalized human BMECs where genetic silencing of FABP5 led to a 15% reduction in the cellular uptake of 14C-DHA (Pan et al., 2015b), and in the current study where transfection of COS-7 cells with FABP5 was associated with increased 14C-DHA uptake. Furthermore, this is in line with further experiments where we demonstrated that the brain capillary uptake of 14C-DHA was significantly reduced in capillaries from FABP5−/− mice relative to FABP5+/+ mice. This reduction in capillary uptake of DHA was not as dramatic as that seen in BMECs from FABP5−/− mice, possibly because the capillaries also contain nonendothelial cells that do not express FABP5, and this may have masked the overall effect of FABP5 deletion on 14C-DHA uptake. Overall, the in vitro data in the current study allow us to confirm that the reduced brain accumulation of endogenous DHA reported in this study and decreased brain uptake of exogenous DHA demonstrated in our previous study (Pan et al., 2015b) are truly a result of reduced brain endothelial cell uptake resulting from FABP5 deficiency.
Interestingly, the present study did not reveal a significant difference in the Papp of 14C-DHA across BMECs from FABP5+/+ and FABP5−/− mice over a 10 min period. A similar Transwell study performed by Mitchell et al. (2011) over a 30 min period demonstrated a reduction in the Papp of palmitic acid, oleic acid, and linoleic acid across human BMECs with FABP5 silenced by siRNA. Given the high affinity of DHA to FABP5 (Pan et al., 2015b), a reduction in DHA Papp across the FABP5−/− BMEC monolayer was expected in our study, particularly given that FABP5 was completely absent in our BMECs, and not just partially silenced. One possible reason for this lack of difference in DHA transport is the shorter duration used in the current permeability study. Being rather lipophilic, however, DHA is likely to be taken up into the BMECs quite rapidly, and release into the receptor chamber is possibly time-dependent and very much dependent on sink conditions provided in the receptor chamber. It is also possible that DHA at 0.1 mm may form micelles, and DHA-forming micelles would not interact with BMECs as would free unbound DHA; however, the addition of BSA might have removed this possibility. Finally, it is possible that sink conditions were not present in the in vitro receptor chamber, as would be present in vivo. The receptor chamber does not contain lipids and proteins that are present in brain parenchyma, which can assist in removal of DHA from BMECs, and this is a major driving force for DHA access into the brain parenchyma. Therefore, in vivo, reduced DHA uptake into BMECs (as a result of FABP5 deficiency) will ultimately lead to decreased DHA access into the brain parenchyma, which we have observed both by in situ brain perfusion (Pan et al., 2015b) and in the current study by measuring the endogenous brain levels of DHA in FABP5−/− mice. This reduction in endogenous brain levels of DHA is postulated to lead to cognitive dysfunction given the cognitive protective effects of DHA reported previously (Barberger-Gateau et al., 2002, 2007).
In conclusion, together with our previous study, we have demonstrated an association between reduced BBB transport of DHA, diminished brain DHA levels, and FABP5 deletion. Such a reduction in endogenous brain levels of DHA results from decreased brain endothelial cell uptake and trafficking of DHA, a process that could be biochemically modified to increase CNS access of DHA in diseases where brain levels of DHA are diminished. Given the widely accepted roles of DHA in cognitive function, the reduced endogenous brain DHA levels resulting from FABP5 deficiency is a likely factor contributing to the cognitive impairment observed in FABP5−/− mice.
Footnotes
This work was supported by Judith Jane Mason and Harold Stannett Williams Memorial Foundation and the William Buckland Foundation.
The authors declare no competing financial interests.
References
- Abe H, Iwasaki T. NMDA and muscarinic blockade in the perirhinal cortex impairs object discrimination in rats. Neuroreport. 2001;12:3375–3379. doi: 10.1097/00001756-200110290-00045. [DOI] [PubMed] [Google Scholar]
- Antunes M, Biala G. The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn Process. 2012;13:93–110. doi: 10.1007/s10339-011-0430-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arterburn LM, Hall EB, Oken H. Distribution, interconversion, and dose response of n-3 fatty acids in humans. Am J Clin Nutr. 2006;83:1467S–1476S. doi: 10.1093/ajcn/83.6.1467S. [DOI] [PubMed] [Google Scholar]
- Barberger-Gateau P, Letenneur L, Deschamps V, Pérès K, Dartigues JF, Renaud S. Fish, meat, and risk of dementia: cohort study. BMJ. 2002;325:932–933. doi: 10.1136/bmj.325.7370.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberger-Gateau P, Raffaitin C, Letenneur L, Berr C, Tzourio C, Dartigues JF, Alpérovitch A. Dietary patterns and risk of dementia: the Three-City cohort study. Neurology. 2007;69:1921–1930. doi: 10.1212/01.wnl.0000278116.37320.52. [DOI] [PubMed] [Google Scholar]
- Calviello G, Palozza P, Piccioni E, Maggiano N, Frattucci A, Franceschelli P, Bartoli GM. Dietary supplementation with eicosapentaenoic and docosahexaenoic acid inhibits growth of Morris hepatocarcinoma 3924A in rats: effects on proliferation and apoptosis. Int J Cancer. 1998;75:699–705. doi: 10.1002/(SICI)1097-0215(19980302)75:5<699::AID-IJC7>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Cao D, Kevala K, Kim J, Moon HS, Jun SB, Lovinger D, Kim HY. Docosahexaenoic acid promotes hippocampal neuronal development and synaptic function. J Neurochem. 2009;111:510–521. doi: 10.1111/j.1471-4159.2009.06335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CT, Green JT, Orr SK, Bazinet RP. Regulation of brain polyunsaturated fatty acid uptake and turnover. Prostaglandins Leukot Essent Fatty Acids. 2008;79:85–91. doi: 10.1016/j.plefa.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Chen CT, Kitson AP, Hopperton KE, Domenichiello AF, Trépanier MO, Lin LE, Ermini L, Post M, Thies F, Bazinet RP. Plasma non-esterified docosahexaenoic acid is the major pool supplying the brain. Sci Rep. 2015;5:15791. doi: 10.1038/srep15791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole GM, Ma QL, Frautschy SA. Omega-3 fatty acids and dementia. Prostaglandins Leukot Essent Fatty Acids. 2009;81:213–221. doi: 10.1016/j.plefa.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnane SC, Schneider JA, Tangney C, Tremblay-Mercier J, Fortier M, Bennett DA, Morris MC. Plasma and brain fatty acid profiles in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. 2012;29:691–697. doi: 10.3233/JAD-2012-110629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decsi T, Kennedy K. Sex-specific differences in essential fatty acid metabolism. Am J Clin Nutr. 2011;94:1914S–1919S. doi: 10.3945/ajcn.110.000893. [DOI] [PubMed] [Google Scholar]
- Dellu F, Fauchey V, Le Moal M, Simon H. Extension of a new two-trial memory task in the rat: influence of environmental context on recognition processes. Neurobiol Learn Mem. 1997;67:112–120. doi: 10.1006/nlme.1997.3746. [DOI] [PubMed] [Google Scholar]
- Do-Monte FH, Quiñones-Laracuente K, Quirk GJ. A temporal shift in the circuits mediating retrieval of fear memory. Nature. 2015;519:460–463. doi: 10.1038/nature14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyall SC. Long-chain omega-3 fatty acids and the brain: a review of the independent and shared effects of EPA, DPA and DHA. Front Aging Neurosci. 2015;7:52. doi: 10.3389/fnagi.2015.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhart MJ, Geerlings MI, Ruitenberg A, Van Swieten JC, Hofman A, Witteman JC, Breteler MM. Diet and risk of dementia: does fat matter? The Rotterdam Study. Neurology. 2002;59:1915–1921. doi: 10.1212/01.WNL.0000038345.77753.46. [DOI] [PubMed] [Google Scholar]
- Fraser T, Tayler H, Love S. Low-temperature improved-throughput method for analysis of brain fatty acids and assessment of their post-mortem stability. J Neurosci Methods. 2008;169:135–140. doi: 10.1016/j.jneumeth.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Freund-Levi Y, Hjorth E, Lindberg C, Cederholm T, Faxen-Irving G, Vedin I, Palmblad J, Wahlund LO, Schultzberg M, Basun H, Eriksdotter Jönhagen M. Effects of omega-3 fatty acids on inflammatory markers in cerebrospinal fluid and plasma in Alzheimer's disease: the OmegAD Study. Dement Geriatr Cogn Disord. 2009;27:481–490. doi: 10.1159/000218081. [DOI] [PubMed] [Google Scholar]
- Gómez-Pinilla F. Brain foods: the effects of nutrients on brain function. Nat Rev Neurosci. 2008;9:568–578. doi: 10.1038/nrn2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulinello M, Gertner M, Mendoza G, Schoenfeld BP, Oddo S, LaFerla F, Choi CH, McBride SM, Faber DS. Validation of a 2-day water maze protocol in mice. Behav Brain Res. 2009;196:220–227. doi: 10.1016/j.bbr.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J, Brunaldi K, Bazinet R, Watkins P. Brain fatty acid uptake. In: Choi IY, Gruetter R, editors. Neural metabolism in vivo. New York: Springer; 2012. pp. 793–817. [Google Scholar]
- Hotamisligil GS, Bernlohr DA. Metabolic functions of FABPs: mechanisms and therapeutic implications. Nat Rev Endocrinol. 2015;11:592–605. doi: 10.1038/nrendo.2015.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itokazu N, Ikegaya Y, Nishikawa M, Matsuki N. Bidirectional actions of docosahexaenoic acid on hippocampal neurotransmissions in vivo. Brain Res. 2000;862:211–216. doi: 10.1016/S0006-8993(00)02129-6. [DOI] [PubMed] [Google Scholar]
- Kaczocha M, Glaser ST, Deutsch DG. Identification of intracellular carriers for the endocannabinoid anandamide. Proc Natl Acad Sci U S A. 2009;106:6375–6380. doi: 10.1073/pnas.0901515106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmijn S, Launer LJ, Ott A, Witteman JC, Hofman A, Breteler MM. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann Neurol. 1997;42:776–782. doi: 10.1002/ana.410420514. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS, DeCola JP, Landeira-Fernandez J. Selective impairment of long-term but not short-term conditional fear by the N-methyl-d-aspartate antagonist APV. Behav Neurosci. 1992;106:591–596. doi: 10.1037/0735-7044.106.4.591. [DOI] [PubMed] [Google Scholar]
- Kobilo T, Yuan C, van Praag H. Endurance factors improve hippocampal neurogenesis and spatial memory in mice. Learn Mem. 2011;18:103–107. doi: 10.1101/lm.2001611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitinen MH, Ngandu T, Rovio S, Helkala EL, Uusitalo U, Viitanen M, Nissinen A, Tuomilehto J, Soininen H, Kivipelto M. Fat intake at midlife and risk of dementia and Alzheimer's disease: a population-based study. Dement Geriatr Cogn Disord. 2006;22:99–107. doi: 10.1159/000093478. [DOI] [PubMed] [Google Scholar]
- Lavandera JV, Sain J, Farina AC, Bernal CA, Gonzalez MA. N-3 fatty acids reduced trans fatty acids retention and increased docosahexaenoic acid levels in the brain. Nutr Neurosci. 2016;21:1–12. doi: 10.1080/1028415X.2016.1173343. [DOI] [PubMed] [Google Scholar]
- Leuner B, Gould E. Structural plasticity and hippocampal function. Annu Rev Psychol. 2010;61:C111–C113. doi: 10.1146/annurev.psych.093008.100359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SY, Hoshiba J, Salem N., Jr An extraordinary degree of structural specificity is required in neural phospholipids for optimal brain function: n-6 docosapentaenoic acid substitution for docosahexaenoic acid leads to a loss in spatial task performance. J Neurochem. 2005;95:848–857. doi: 10.1111/j.1471-4159.2005.03427.x. [DOI] [PubMed] [Google Scholar]
- Liu RZ, Mita R, Beaulieu M, Gao Z, Godbout R. Fatty acid binding proteins in brain development and disease. Int J Dev Biol. 2010;54:1229–1239. doi: 10.1387/ijdb.092976rl. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN, Bazan NG. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest. 2005;115:2774–2783. doi: 10.1172/JCI25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumata M, Sakayori N, Maekawa M, Owada Y, Yoshikawa T, Osumi N. The effects of Fabp7 and Fabp5 on postnatal hippocampal neurogenesis in the mouse. Stem Cell. 2012;30:1532–1543. doi: 10.1002/stem.1124. [DOI] [PubMed] [Google Scholar]
- Mehta DC, Short JL, Nicolazzo JA. Reduced CNS exposure of memantine in a triple transgenic mouse model of Alzheimer's disease assessed using a novel LC-MS technique. J Pharm Biomed Anal. 2013a;85:198–206. doi: 10.1016/j.jpba.2013.07.027. [DOI] [PubMed] [Google Scholar]
- Mehta DC, Short JL, Nicolazzo JA. Altered brain uptake of therapeutics in a triple transgenic mouse model of Alzheimer's disease. Pharm Res. 2013b;30:2868–2879. doi: 10.1007/s11095-013-1116-2. [DOI] [PubMed] [Google Scholar]
- Mishima K, Tanoue A, Tsuda M, Hasebe N, Fukue Y, Egashira N, Takano Y, Kamiya HO, Tsujimoto G, Iwasaki K, Fujiwara M. Characteristics of behavioral abnormalities in alpha1d-adrenoceptors deficient mice. Behav Brain Res. 2004;152:365–373. doi: 10.1016/j.bbr.2003.10.038. [DOI] [PubMed] [Google Scholar]
- Mitchell RW, On NH, Del Bigio MR, Miller DW, Hatch GM. Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J Neurochem. 2011;117:735–746. doi: 10.1111/j.1471-4159.2011.07245.x. [DOI] [PubMed] [Google Scholar]
- Moriguchi T, Salem N., Jr Recovery of brain docosahexaenoate leads to recovery of spatial task performance. J Neurochem. 2003;87:297–309. doi: 10.1046/j.1471-4159.2003.01966.x. [DOI] [PubMed] [Google Scholar]
- Nguyen LN, Ma D, Shui G, Wong P, Cazenave-Gassiot A, Zhang X, Wenk MR, Goh EL, Silver DL. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature. 2014;509:503–506. doi: 10.1038/nature13241. [DOI] [PubMed] [Google Scholar]
- Owada Y, Takano H, Yamanaka H, Kobayashi H, Sugitani Y, Tomioka Y, Suzuki I, Suzuki R, Terui T, Mizugaki M, Tagami H, Noda T, Kondo H. Altered water barrier function in epidermal-type fatty acid binding protein-deficient mice. J Invest Dermatol. 2002;118:430–435. doi: 10.1046/j.0022-202x.2001.01616.x. [DOI] [PubMed] [Google Scholar]
- Padilla-Coreano N, Do-Monte FH, Quirk GJ. A time-dependent role of midline thalamic nuclei in the retrieval of fear memory. Neuropharmacology. 2012;62:457–463. doi: 10.1016/j.neuropharm.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Khalil H, Nicolazzo JA. The impact of docosahexaenoic acid on Alzheimer's disease: is there a role of the blood–brain barrier. Curr Clin Pharmacol. 2015a;10:222–241. doi: 10.2174/157488471003150820151532. [DOI] [PubMed] [Google Scholar]
- Pan Y, Scanlon MJ, Owada Y, Yamamoto Y, Porter CJ, Nicolazzo JA. Fatty acid-binding protein 5 facilitates the blood-brain barrier transport of docosahexaenoic acid. Mol Pharm. 2015b;12:4375–4385. doi: 10.1021/acs.molpharmaceut.5b00580. [DOI] [PubMed] [Google Scholar]
- Patabendige A, Skinner RA, Morgan L, Abbott NJ. A detailed method for preparation of a functional and flexible blood–brain barrier model using porcine brain endothelial cells. Brain Res. 2013;1521:16–30. doi: 10.1016/j.brainres.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pélerin H, Jouin M, Lallemand MS, Alessandri JM, Cunnane SC, Langelier B, Guesnet P. Gene expression of fatty acid transport and binding proteins in the blood–brain barrier and the cerebral cortex of the rat: differences across development and with different DHA brain status. Prostaglandins Leukot Essent Fatty Acids. 2014;91:213–220. doi: 10.1016/j.plefa.2014.07.004. [DOI] [PubMed] [Google Scholar]
- Perrière N, Demeuse P, Garcia E, Regina A, Debray M, Andreux JP, Couvreur P, Scherrmann JM, Temsamani J, Couraud PO, Deli MA, Roux F. Puromycin-based purification of rat brain capillary endothelial cell cultures: effect on the expression of blood–brain barrier-specific properties. J Neurochem. 2005;93:279–289. doi: 10.1111/j.1471-4159.2004.03020.x. [DOI] [PubMed] [Google Scholar]
- Prasad MR, Lovell MA, Yatin M, Dhillon H, Markesbery WR. Regional membrane phospholipid alterations in Alzheimer's disease. Neurochem Res. 1998;23:81–88. doi: 10.1023/A:1022457605436. [DOI] [PubMed] [Google Scholar]
- Rapoport SI, Chang MC, Spector AA. Delivery and turnover of plasma-derived essential PUFAs in mammalian brain. J Lipid Res. 2001;42:678–685. [PubMed] [Google Scholar]
- Rapoport SI, Rao JS, Igarashi M. Brain metabolism of nutritionally essential polyunsaturated fatty acids depends on both the diet and the liver. Prostaglandins Leukot Essent Fatty Acids. 2007;77:251–261. doi: 10.1016/j.plefa.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruck T, Bittner S, Epping L, Herrmann AM, Meuth SG. Isolation of primary murine brain microvascular endothelial cells. J Vis Exp. 2014;93:e52204. doi: 10.3791/52204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto C, Ohnishi T, Maekawa M, Watanabe A, Ohba H, Arai R, Iwayama Y, Hisano Y, Toyota T, Toyoshima M, Suzuki K, Shirayama Y, Nakamura K, Mori N, Owada Y, Kobayashi T, Yoshikawa T. Functional characterization of FABP3, 5 and 7 gene variants identified in schizophrenia and autism spectrum disorder and mouse behavioral studies. Hum Mol Genet. 2014;23:6495–6511. doi: 10.1093/hmg/ddu369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipton OA, El-Gaby M, Apergis-Schoute J, Deisseroth K, Bannerman DM, Paulsen O, Kohl MM. Left-right dissociation of hippocampal memory processes in mice. Proc Natl Acad Sci U S A. 2014;111:15238–15243. doi: 10.1073/pnas.1405648111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372–376. doi: 10.1038/35066584. [DOI] [PubMed] [Google Scholar]
- Spellman T, Rigotti M, Ahmari SE, Fusi S, Gogos JA, Gordon JA. Hippocampal-prefrontal input supports spatial encoding in working memory. Nature. 2015;522:309–314. doi: 10.1038/nature14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triguero D, Buciak J, Pardridge WM. Capillary depletion method for quantification of blood–brain barrier transport of circulating peptides and plasma proteins. J Neurochem. 1990;54:1882–1888. doi: 10.1111/j.1471-4159.1990.tb04886.x. [DOI] [PubMed] [Google Scholar]
- Wilhelm I, Krizbai IA. In vitro models of the blood–brain barrier for the study of drug delivery to the brain. Mol Pharm. 2014;11:1949–1963. doi: 10.1021/mp500046f. [DOI] [PubMed] [Google Scholar]
- Yassine HN, Feng Q, Azizkhanian I, et al. Association of serum docosahexaenoic acid with cerebral amyloidosis. JAMA Neurol. 2016 doi: 10.1001/jamaneurol.2016.1924. Advance online publication. Retrieved Aug. 8, 2016. [DOI] [PubMed] [Google Scholar]
- Young C, Gean PW, Chiou LC, Shen YZ. Docosahexaenoic acid inhibits synaptic transmission and epileptiform activity in the rat hippocampus. Synapse. 2000;37:90–94. doi: 10.1002/1098-2396(200008)37:2<90::AID-SYN2>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Yu S, Levi L, Siegel R, Noy N. Retinoic acid induces neurogenesis by activating both retinoic acid receptors (RARs) and peroxisome proliferator-activated receptor β/δ (PPARβ/δ) J Biol Chem. 2012;287:42195–42205. doi: 10.1074/jbc.M112.410381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Levi L, Casadesus G, Kunos G, Noy N. Fatty acid-binding protein 5 (FABP5) regulates cognitive function both by decreasing anandamide levels and by activating the nuclear receptor peroxisome proliferator-activated receptor β/δ (PPARβ/δ) in the brain. J Biol Chem. 2014;289:12748–12758. doi: 10.1074/jbc.M114.559062. [DOI] [PMC free article] [PubMed] [Google Scholar]