

Abstract
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by the pathological aggregation of the amyloid-β peptide (Aβ). Monomeric soluble Aβ can switch from helicoidal to β-sheet conformation, promoting its assembly into oligomers and subsequently to amyloid fibrils. Oligomers are highly toxic to neurons and have been reported to induce synaptic transmission impairments. The progression from oligomers to fibrils forming senile plaques is currently considered a protective mechanism to avoid the presence of the highly toxic oligomers. Protein nitration is a frequent post-translational modification under AD nitrative stress conditions. Aβ can be nitrated at tyrosine 10 (Y10) by peroxynitrite. Based on our analysis of ThT binding, Western blot and electron and atomic force microscopy, we report that Aβ nitration stabilizes soluble, highly toxic oligomers and impairs the formation of fibrils. We propose a mechanism by which fibril elongation is interrupted upon Y10 nitration: Nitration disrupts fibril-forming folds by preventing H14-mediated bridging, as shown with an Aβ analog containing a single residue (H to E) replacement that mimics the behavior of nitrated Aβ related to fibril formation and neuronal toxicity. The pathophysiological role of our findings in AD was highlighted by the study of these nitrated oligomers on mouse hippocampal neurons, where an increased NMDAR-dependent toxicity of nitrated Aβ oligomers was observed. Our results show that Aβ nitrotyrosination is a post-translational modification that increases Aβ synaptotoxicity.
SIGNIFICANCE STATEMENT We report that nitration (i.e., the irreversible addition of a nitro group) of the Alzheimer-related peptide amyloid-β (Aβ) favors the stabilization of highly toxic oligomers and inhibits the formation of Aβ fibrils. The nitrated Aβ oligomers are more toxic to neurons due to increased cytosolic calcium levels throughout their action on NMDA receptors. Sustained elevated calcium levels trigger excitotoxicity, a characteristic event in Alzheimer's disease.
Keywords: Alzheimer, amyloid, nitrotyrosination, NMDA Rc, oligomers, peroxynitrite
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder that leads to severe memory deficits, progressive cognitive decline, and neuronal death. Its pathophysiology involves oxidative and nitrative stress (Miranda et al., 2000) and the deposition of protein aggregates, that is, amyloid-β (Aβ) peptide in extracellular oligomers and senile plaques and tau in the intraneuronal neurofibrillary tangles (Masters et al., 1985; Grundke-Iqbal et al., 1986).
For >20 years, studies have pointed to Aβ accumulation as the primary event in AD pathogenesis. This constitutes the basis of the amyloid cascade hypothesis. Aβ is the product of the sequential cleavage of amyloid precursor protein (APP) by β-site APP cleaving enzyme 1 (BACE-1) (Vassar et al., 1999) and γ-secretase (Xia et al., 1998). It is widely accepted that an imbalance between Aβ production and clearance causes the pathogenic events that lead to AD (Hardy and Selkoe, 2002).
Soluble Aβ is able to aggregate in β-sheets forming primary structures called oligomers; these can assemble to form protofibrils as intermediate structures between aggregates and mature fibrils (Arimon et al., 2005). However, density of senile plaques does not correlate with cognitive deficits, and alterations in synaptic transmission take place before neuronal death (Murrell et al., 1991; Hsia et al., 1999). It has been proposed that Aβ effects on synaptic transmission are due to soluble forms of Aβ rather than mature fibrils (Selkoe, 2002). Oligomers have been shown to be the most neurotoxic form of Aβ and to impair synaptic plasticity (Klein, 2002; Selkoe, 2008).
The hippocampus, with its high density of glutamatergic neurons and NMDARs, has a crucial role in memory and learning. It is also one of the first and most severely affected structures in AD. Consistent with the involvement of NMDARs in memory and learning, alterations in the glutamatergic transmission by Aβ have been linked to the pathophysiological mechanisms underlying AD (Danysz and Parsons, 2012).
Chronic activation of NMDARs, which translates into higher cytoplasmic calcium (Ca2+) levels, leads to cell death and neurodegeneration through the process known as excitotoxicity (Esposito et al., 2013). In addition, elevated calcium levels trigger the activation of neuronal nitric oxide (NO) synthase (nNOS) through Ca2+-calmodulin, producing NO (Dawson and Dawson, 1996).
Aβ can increase the production of NO by promoting the activation of neuronal and inducible NOS (Meda et al., 1995; Rossi and Bianchini, 1996). NO reacts with the superoxide (O2·−) produced by the mitochondria and other free radical sources forming peroxynitrite anion (ONOO−), which nitrates proteins (Beckman and Koppenol, 1996). Protein nitration is an irreversible, post-translational modification that adds a nitro group to an amino acid, but mainly to a tyrosine, giving 3-nitrotyrosine residues (Guix et al., 2005); increased levels of protein nitration exist in AD (Smith et al., 1997). Numerous proteins have been reported to be affected by amyloid-dependent nitrotyrosination, thus severely affecting their function: presenilins (Guix et al., 2012), albumin (Ramos-Fernández et al., 2014), and the glycolytic enzymes triosephosphate isomerase (Guix et al., 2009; Ill-Raga et al., 2010; Tajes et al., 2013, 2014), α-enolase (Castegna et al., 2003), and glutamate dehydrogenase (Reed et al., 2009), among others.
Aβ has one tyrosine (Y10) in its sequence, which is surrounded by negatively charged amino acids, making protein nitration very likely (Guix et al., 2005). Accordingly, Kummer et al. (2011) reported increased levels of nitrotyrosinated Aβ and its presence in the core of amyloid plaques in AD brains. Study of the mechanisms underlying Aβ aggregation and the modulating factors is critical to the development of new therapeutic strategies targeting Aβ. We report the effect of Aβ nitrotyrosination in oligomer stability, fibrillation impairment, and increased excitotoxicity, and we propose and validate a bioinformatic model regarding the progression to fibrils of nitrated Aβ.
Materials and Methods
Immunofluorescence detection of nitrated amyloid in human hippocampal samples.
The 20 μm PFA-fixed and mounted slices of hippocampal human samples from an AD patient were supplied by the Banc de Teixits Neurològics (Neurological Tissue Bank of the Biobank-Hospital Clínic-IDIBAPS, Barcelona, Spain). Handling and investigation with the human samples have been performed according to the ethical standards, to the Declaration of Helsinki, and to the Ethics Committee of the Institut Municipal d'Investigacions Mèdiques-Universitat Pompeu Fabra (EC-IMIM-UPF). Slices were treated with freshly prepared 0.15 m glycine, 10 mg/ml NaBH4 for 30 min to reduce autofluorescence. Slices were immunostained with 1:500 polyclonal rabbit anti-nitrotyrosine (Cell Signaling Technology) and 1:1200 6E10 mouse monoclonal anti-Aβ (Covance) antibodies overnight at 4°C followed by 1:1000 Alexa-488-bound anti-rabbit and 1:1000 Alexa-555-bound anti-mouse as secondary antibodies (Dako) at room temperature (RT) for 1 h. Further reduction of autofluorescence was accomplished by a 10 min treatment of 0.3% Sudan black in ethanol. Samples were visualized using a Leica SPE confocal microscope.
Aβ peptide preparation.
Aβ42 WT (Anaspec) and mutants (Peptide Synthesis Facility, UPF) were solubilized as previously described (Bitan and Teplow, 2005). For the experiments to study the aggregation and structural analysis by atomic force microscopy (AFM), lyophilized Aβ42 was solubilized with 1 ml of 2 mm NaOH at 1 mg/ml, pH ≥ 10.5. The solution was then placed in a bath-type sonicator (Diagenode) for 1 min at medium intensity. The 25 μg aliquots were freeze-dried and stored at −20°C until used. When needed, aliquots were resuspended in MilliQ water immediately before thioflavin T (ThT; Sigma-Aldrich) experiments or PBS, pH 7.4, for the structural studies by AFM. For the experiments with cells and structural studies with the other techniques used in this work, 1 mg of the different Aβ was dissolved in 250 μl of MilliQ water and pH was adjusted to ≥ 10.5 with 1 m NaOH, to avoid the isoelectric point of Aβ, at which aggregation propensity is maximal and solubilization is minimal. The 250 μl of 20 mm phosphate buffer, pH 7.4, were added and the tubes were placed for 1 min in a bath-type sonicator (Bioruptor, Diagenode). Preparations were aliquoted and immediately used for subsequent experiments diluted in DMEM/F12 medium (Invitrogen).
Oligomer preparation.
The different Aβ were dissolved to 0.4 mg/ml in PBS (aggregation studies) or DMEM/F12 (cell and structural studies) and kept at 4°C at different times (from 0 to 72 h). For in vitro Aβ nitration, the peroxynitrite donor 3-morpholinosydnonimine hydrochloride (SIN-1; Sigma-Aldrich) was weighted, freshly dissolved in PBS, and immediately added to the Aβ preparation.
Study of Aβ nitrotyrosination by dot blot.
Aβ peptides (0.125 μg/μl) were incubated with or without the peroxynitrite donor SIN-1 in Nunc 96-well flat-bottom, black polystyrene microplates (Thermo Scientific) for 20 h (unless indicated otherwise) at RT; 2 μl drops of sample were spotted onto nitrocellulose membranes (Invitrogen) and left to dry at RT. Membranes were blocked with 5% BSA in 0.05% Tween TBS 1 h at RT and incubated with 1:1000 mouse monoclonal anti-nitrotyrosine (Millipore) and 1:1000 6E10 anti-Aβ antibodies in BSA/Tween TBS for 30 min at RT; 1:2500 HRP-linked goat anti-mouse secondary antibody (GE Healthcare) was applied for 30 min at RT, and then membranes were developed with ECL Substrate (Thermo Scientific) in the ChemiDoc MP imaging system.
ThT binding assay.
The 1 mm stock ThT solution was prepared dissolving the dye in PBS. The solution was then filtered through a 0.22 μm syringe filter. ThT solution was stored at −20°C in the dark. Aβ peptides (0.125 μg/μl) were incubated with or without the peroxynitrite donor SIN-1 and 10 μm ThT in Nunc 96-well flat-bottom, black polystyrene microplates (Thermo Scientific) for 20 h (unless indicated otherwise) at RT. ThT fluorescence was measured every 5 min using excitation and emission wavelengths of 430 and 470 nm, respectively, using a multiplate-reader fluorometer (FLUOstar optima, BMG Labtech).
Oligomer identification by Western blot.
Aβ42 samples incubated in the presence or absence of SIN-1 were set in aggregating conditions along 72 h. Samples were taken at different time points and resolved in Nu-PAGE 4%–12% Bis-Tris Protein Gels (Invitrogen) with 2-(N-morpholino)ethanesulfonic acid running buffer. Gels were transferred to PVDF (Immobilon-P, Millipore) using a dry blotting system (Invitrogen) and boiled in PBS for 5 min. After blocking the membranes with 5% nonfat milk 0.05% Tween PBS, they were incubated overnight with 1:1000 6E10 anti-Aβ antibody at 4°C. The 1:2500 HRP-linked goat anti-mouse secondary antibody (GE Healthcare) was used at RT for 1 h, and membranes were developed using the ECL Substrate in the ChemiDoc MP imaging system (Bio-Rad).
Structural study by AFM.
Aβ42 samples incubated in the presence or absence of SIN-1 were prepared as indicated above. Sample aliquots were frozen at different incubation times (0, 3, and 72 h). They were thawed at RT just before the AFM experiments. A multimode atomic force microscope (Veeco Instruments) equipped with a 12 μm scanner (E-scanner) was used. The images were taken in liquid in a cell without the O-ring seal, and 5–10 μl of sample was deposited on cleaved mica substrates. After an adsorption time of 5 min, 40 μl of double deionized water was added to form a drop suitable for the imaging procedure. The system was left for equilibration for at least 10 min before performing the experiments. NP-S (Veeco) probes were used to scan the samples in tapping mode at 0.5 Hz scan rate. Height and amplitude images were recorded simultaneously, although only the latter are presented in this paper.
Structural study by circular dichroism (CD) spectroscopy.
Aβ42 samples (wt, nitrated with SIN-1 and synthetically nitrated at Tyr10) were prepared as indicated above and kept for 24 h at 4°C. The spectral region was recorded from 200 to 260 nm, with a 0.5 nm step resolution, on a Jasco J-815 CD spectropolarimeter (Jasco) at RT using quartz cells of 1 mm optical path length. The scanning speed was 100 nm/min, and the spectra were collected and averaged over 6 scans. The DMEM/FC12 medium as background signal was subtracted from the CD spectra of the peptides.
Particle size distribution by dynamic light scattering (DLS).
Aβ42 samples (wt, nitrated with SIN-1 and synthetically nitrated at Tyr10) were prepared as indicated above and kept for 24 h at 4°C. DLS measurements were recorded on a Zetasizer instrument (Nano ZS; Malvern Instruments) at 20°C using a small-volume (40 μl) quartz cuvette of 1 cm path length. Scattering data were collected as an average of 5 measurements with 10 scans for each measurement. Refractive indices were 1.330 (water) and 1.45 (protein) for polydispersant and material, respectively. Data were processed with the Malvern Zetasizer Software (Malvern Instruments).
Structural study by attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectroscopy.
Aβ42 samples (wt, nitrated with SIN-1 and synthetically nitrated at Tyr10) were prepared as indicated above and kept for 24 h at 4°C. They were put in thin hydrated films through an N2 stream gentle drying. Spectra were obtained at RT by the average of 250 scans at 2 cm resolution in a VARIAN FTS-7000 infrared spectrometer. The second derivative was performed with 10 smoothing points.
Structural analysis by bioinformatic modeling.
The NMR structure 2LMN (Petkova et al., 2006) from the Protein Data Bank (Berman et al., 2000) was used as a template for the structural analysis of Aβ. A total of 500 models were built with MODELLER (Webb and Sali, 2014) for AβWT (wild-type form of the Aβ peptide) and the mutant AβE14. We evaluated with ZRANK (Pierce and Weng, 2007) the energies of all the interfaces involving the Aβ peptides. We observed a destabilization of AβE14 conformation with respect to AβWT, and we manually oriented the side-chains of H13 and E14 to regain the local stabilization (which was confirmed with ZRANK). There is a loss of conformational stability in AβWT when extending the multipeptide β-sheet due to the accumulation of identical charges. We postulate that an n-peptide complex (complex-1, forming a parallel β-sheet β1) packs with another n-peptide complex (complex-2, with an opposite parallel β-sheet β1′) and compensates for the charges. This observation agrees with the notion that phosphorylation of S26 has an effect over amyloid aggregation similar to that of the nitrotyrosination of Y10 (Rezaei-Ghaleh et al., 2014). Then, we suggest that the interaction between Y10 (in complex-1) and S26′ (in complex-2) fixes this new configuration. Through static-directed docking with PatchDock (Duhovny et al., 2002), we modeled the packed dimer of complex-1 and complex-2 and analyzed the potential conformations (poses) with ZRANK. The comparison between the wild-type (AβWT) and mutant form shows a putative alteration of affinity. Presumably, AβE14 is less stable, forcing a change of orientation between Y10 and S26′ that separates both residues and dissipates the effect of Y10 nitrotyrosination on fibril formation.
Mouse hippocampal primary cultures and transfection.
Mouse hippocampal neurons were isolated from 18-d-old CD1 embryos of either sex. Pregnant mice were killed by CO2 inhalation in accordance with the EC-IMIM-UPF and the directives of the Council of the European Communities 86/609/CEE. Embryos were rendered hypothermic and decapitated. Hippocampi were aseptically dissected in ice-cold HBSS (Invitrogen) supplemented with 4.5 g/L glucose and trypsinized for 17 min at 37°C. After 3 washes in HBSS + glucose and mechanical dissociation, cells were seeded in DMEM (Invitrogen) plus 10% horse serum (Invitrogen) onto 1% poly-L-lysine-coated plates. After 2 h, medium was removed and Neurobasal medium was added, containing 2% B27 supplement, 1% GlutaMAX, and 1% penicillin/streptomycin (Invitrogen). After 3 DIV, neurons were treated with 2 μm β-d-arabinofuranosylcytosine (Sigma-Aldrich) for 24 h to reduce the number of proliferating non-neuronal cells. Primary neuronal cultures were used at 10–14 DIV.
For the 3D rendering of dendrites, 14 DIV neurons were transfected with the pCAGLifeActTagGFP2 plasmid (Ibidi) using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions.
Cell viability study by methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay.
Hippocampal neurons were seeded in 48-well plates at a density of 5 × 105 cells/200 μl/well. The corresponding treatments were added to Neurobasal without phenol red supplemented with 1% GlutaMAX, and cells were incubated for 24 h at 37°C unless indicated otherwise. Cell viability was assessed by MTT reduction. Briefly, 22 μl of MTT (Sigma-Aldrich) stock solution (5 mg/ml) was added to the wells; and after 2 h, the media were replaced with 100 μl of DMSO to solubilize tetrazolium salts. MTT absorbance was determined in an Infinite 200 multiplate reader (Tecan) at A540 nm and corrected by A650 nm. Untreated cells were taken as 100%.
Structural analysis by transmission electron microscopy.
Aβ42 samples (wt, nitrated with SIN-1, synthetically nitrated at Tyr10 and H14 to E14 mutant; 0.125 μg/μl in PBS) were incubated for 72 h at RT without agitation. Silicon/Formvar-coated 300-mesh copper grids (Electron Microscopy Sciences) were rendered hydrophilic by being exposed for 20 min to UV light. Samples were gently agitated before pipetting 5 μl onto the grids. After 2 min adsorption time, the excess was drawn off using Whatman 541 filter paper (GE Healthcare). The grids were stained with filtered 2% uranyl acetate (Electron Microscopy Sciences) for 1 min. Excess was drawn off and grids were air-dried before examination. Samples were examined with a JEOL 1011 transmission electron microscope at an operating voltage of 80 kV.
Cytosolic calcium measurements.
Cells were loaded with 250 μl fura-2 AM (5 μm; Invitrogen) and 0.02% pluronic acid for 40 min at RT and washed thoroughly with isotonic solution containing 2.5 mm KCl, 140 mm NaCl, 1.2 mm CaCl2, 0.5 mm MgCl2, 5 mm glucose, and 10 mm HEPES (305 mOsm, pH 7.4). Experiments were performed at RT using a custom-made recording chamber on an inverted microscope. Excitation at 340 and 380 nm was supplied by a xenon arc lamp with an optical filter changer. Emitted fluorescence at 510 nm was recorded every 5 s using a digital camera controlled by AquaCosmos software. Cytosolic calcium levels are presented as the ratio of emitted fluorescence at 510 nm following excitation at 340 and 380 nm (340/380 ratio) and are corrected for the basal individual fluorescence measured before stimulation.
Cell viability study by TUNEL assay.
Hippocampal neurons were seeded in 48-well plates at a density of 5 × 105 cells/200 μl/well. Neurons were treated for 5 min with 10 μm Aβ42, prepared as indicated above, washed, and incubated with Neurobasal without phenol red supplemented with 1% GlutaMAX for 24 h at 37°C. Cell viability was assessed 24 h later with the Click-it TUNEL AlexaFluor-488 imaging assay (Invitrogen), following the manufacturer's instructions. Coverslips were visualized using a Leica TCS SP5 II confocal microscope and analyzed using Fiji (Schindelin et al., 2012) distribution of ImageJ (Schneider et al., 2012).
Study of Aβ binding to synaptic spines by immunofluorescence.
Hippocampal neurons were seeded in 48-well plates at a density of 5 × 105 cells/200 μl/well. Neurons were treated for 5 min with the different Aβ (wt, nitrated with SIN-1 and synthetically nitrated at Tyr10), prepared as indicated above, washed, and incubated with Neurobasal without phenol red supplemented with 1% GlutaMAX for 24 h at 37°C. Neurons were fixed with EM grade methanol-free 4% PFA in PBS for 15 min at RT. Samples were blocked with 0.3% Triton X-100 and 5% normal goat serum (Vector) for 1 h at RT. Then they were probed overnight with 1:200 6E10 anti-Aβ and 1:1000 rabbit polyclonal anti-GFP antibody (Sigma) at 4°C. Alexa-488-conjugated goat anti-rabbit and Alexa-555-conjugated goat anti-mouse secondary antibodies (1:1000, Invitrogen) were used at RT for 1 h. Coverslips were mounted using Fluoromount-G (eBioscience) and observed with a Leica TCS SP5 II confocal microscope. Deconvolution of the z-stacks was performed with Huygens deconvolution software (SVI), 3D rendering was performed with Imaris (Bitplane), and colocalization was analyzed using JaCoP plugin (Bolte and Cordelières, 2006) for ImageJ.
Study of Aβ binding to synaptic spine by Manders' coefficient measurement.
Manders' coefficients were calculated in the experiments of colocalization of spines and Aβ by immunofluorescence. They measure the overlap of two different immunofluorescence channels: green and magenta stainings for spines and Aβ, respectively. Therefore, we calculated the overlapping of the green channel (GFP) with the magenta channel (Aβ) as previously described (Ill-Raga et al., 2015). It is expressed in arbitrary units from 0 to 1, with 0 being a lack of overlap and 1 being the maximum overlap.
Study of Aβ binding to GluN1 by immunoprecipitation.
Hippocampal neurons were seeded in 48-well plates at a density of 5 × 105 cells/200 μl/well. Neurons were treated for 5 min with the different Aβ (wt, nitrated with SIN-1 and synthetically nitrated at Tyr10), prepared as indicated above, washed, and incubated with Neurobasal without phenol red supplemented with 1% GlutaMAX for 24 h at 37°C. Cells were washed three times in PBS and lysed with ice-cold 1% Triton X-100 PBS with protease inhibitor mixture (Roche). GluN1 was immunoprecipitated with the mouse monoclonal anti-GluN1 antibody and magnetic beads (Invitrogen), using a modified protocol of the manufacturer's instructions. Briefly, 2 μg of antibody was bound to 1.5 mg Dynabeads Protein G in 200 μl of Tween 20 PBS for 1 h at RT. The beads were washed and incubated overnight with 500 μl of lysate at 4°C. The supernatant was removed, the beads were washed thoroughly, and the antigen was eluted with a 67:30:3 mixture of 50 mm glycine, pH 2.8, LDS Sample Buffer and Reducing Agent (Invitrogen), and incubated at 70°C for 10 min before running the samples in a protein gel.
Statistical analysis.
Data are expressed as mean ± SEM of n experiments. Statistical analyses were performed with one-way ANOVA using GraphPad software, unless indicated otherwise.
Results
Aβ nitrotyrosination impairs fibril formation and stabilizes low molecular weight oligomers
Previous studies have shown the presence of nitrotyrosinated Aβ in AD brains and have nitrotyrosinated Aβ in vitro treated with pure peroxynitrite (Kummer et al., 2011). We were able to observe nitrotyrosine immunoreactivity in the plaques of AD patients (Fig. 1A). In addition, we wanted to determine whether the peroxynitrite donor SIN-1 also nitrotyrosinates Aβ in vitro, since pure peroxynitrite is more unstable (Szabó et al., 2007). Using a nitrotyrosine-specific antibody and dot blot analysis, we were able to detect Aβ nitrotyrosination using increasing concentrations of SIN-1 (Fig. 1B).
Figure 1.
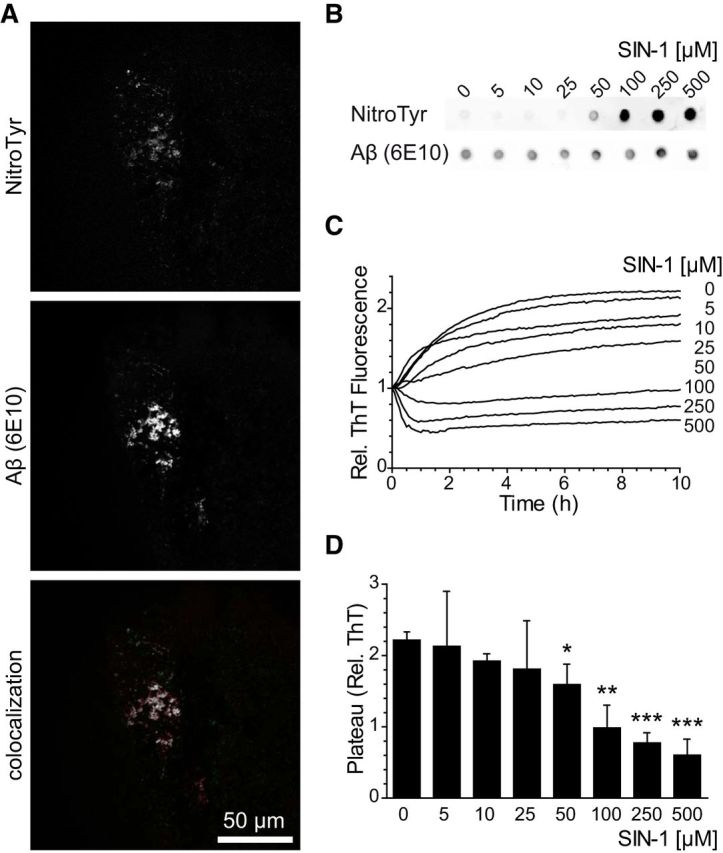
Aβ nitrotyrosination impairs fibril formation. A, Presence of nitrotyrosinated Aβ within amyloid plaques of the hippocampus from an AD patient. Colocalized pixels appear in white. B, Dot blot of synthetic Aβ42 treated with increasing concentrations of SIN-1 probed with an anti-nitrotyrosine and 6E10 anti-Aβ antibodies. C, ThT aggregation assay with synthetic Aβ42 treated with increasing concentrations of SIN-1. Graph represents fluorescence values relative to time 0. Mean of four independent experiments. D, Plateau phase ThT values of the data shown in C. Mean ± SEM of 3–6 independent experiments. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test).
Nitrotyrosination is well known to alter protein function; we hypothesized that peroxynitrite may alter Aβ aggregation. ThT measurements are useful to quantitatively assess the content of β-sheet-rich structures and are widely used to evaluate Aβ progression to fibrils. We assayed the aggregation of Aβ over time in the presence of increasing concentrations of SIN-1 and found that nitrotyrosination avoids fibrillation and favors oligomer formation (Fig. 1C). Untreated Aβ reached the plateau phase of the sigmoidal curve within 10 h of aggregation, which is consistent with the literature (Cohen et al., 2013), whereas peroxynitrite-treated Aβ showed remarkably decreased aggregation rates in a concentration-dependent manner. Moreover, fluorescence ThT values for treated Aβ remained similarly low after 1 week of aggregation (data not shown). Because we observed strong Aβ nitrotyrosination at 100 μm SIN-1 concentration (p < 0.01) and in parallel the aggregation rates at the plateau phase were significantly reduced (Fig. 1D), we chose this concentration for the detailed study of nitrotyrosinated Aβ (nitro-Aβ).
Our aggregation data with ThT pointed toward a clear enhancement of oligomerization and inhibition of Aβ aggregation upon nitrotyrosination. Focusing on soluble low molecular weight Aβ oligomeric species, we assayed Aβ and nitro-Aβ samples at different time points of aggregation and proceeded to blot them with the 6E10 monoclonal anti-Aβ antibody. Although dimers are not strictly oligomers, for the sake of simplicity, they will be referred to as such, together with trimers, tetramers, and other low molecular weight oligomeric species. Over time, the untreated Aβ low molecular weight oligomers tended to give way to the formation of larger aggregates, whereas the nitro-Aβ low molecular weight oligomers remained stable (Fig. 2A,B). This was further confirmed when these oligomers were visualized on freshly cleaved mica with AFM. In the case of untreated Aβ (Fig. 2C), the oligomers that remained stable after 72 h of aggregation for nitro-Aβ could not be seen. Overall, these results are in good agreement with our data regarding Aβ and nitro-Aβ aggregation (Fig. 1C,D). Particle size distribution by DLS shows that nytrotyrosinated and wild-type samples are very polydisperse (Fig. 3A), indicative of the existence of several size species. For wild-type samples, the presence of at least two populations (150 and 450 nm) contrasts with the nytrotyrosinated samples with a population more centered in the 150–250 nm region, in agreement with the NuPAGE data in Figure 1. Secondary structure analysis for nitrotyrosinated and wild-type Aβ42 shows a predominant β-sheet structure for all samples both by CD (Fig. 3B) and FTIR (Fig. 3C). CD-positive band at 200 nm and negative band at 225 nm representative of β-sheet secondary structure agree fairly well with the observed ATR-FTIR absorbance spectrum indicative of β-sheet structure with prominent peaks at 1625 and 1695 cm (Fig. 3C). For the sake of clarity, the ATR-FTIR spectra second derivative is shown for all samples (Fig. 3D). The 1625/1695 cm band splitting indicative of aggregation (Perálvarez-Marín et al., 2008) is highlighted showing a larger splitting (i.e., larger aggregation) for Aβ42 (1698 and 1624 cm). Aβ Ntyr and SIN1 treated band splitting are 1695 and 1628 cm and 1695 and 1629 cm, respectively, indicative of lesser aggregation. The ATR-FTIR data are in agreement with NuPAGE and DLS data in Figures 1 and 3A. Our next step was to explore the molecular basis for these observed behaviors.
Figure 2.
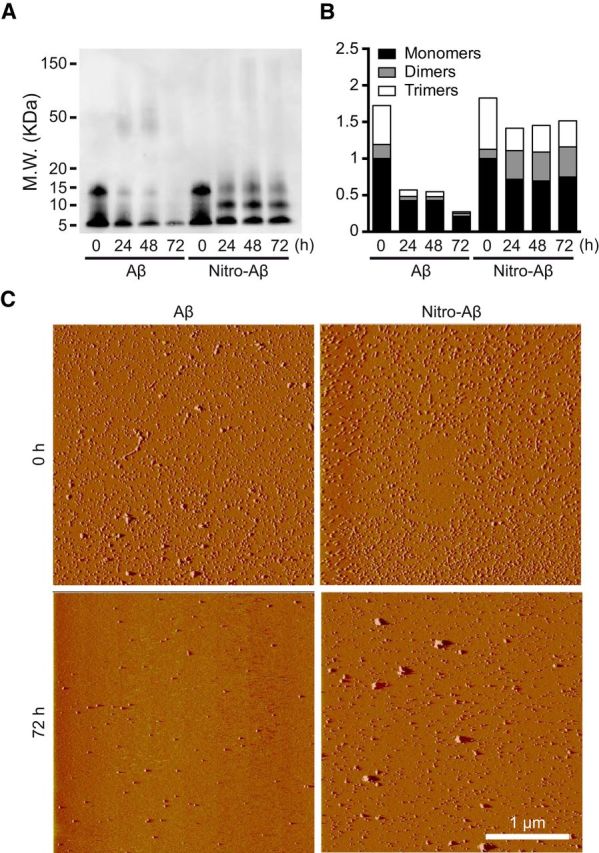
Aβ nitrotyrosination stabilizes low molecular weight oligomers. A, Representative Western blot of Aβ42 control and Aβ42 treated with 100 μm SIN-1 (nitro-Aβ) at different times of aggregation probed with 6E10 anti-Aβ antibody. B, Relative amounts of oligomeric species obtained by Western blot. Mean of three independent experiments. C, Oligomer visualization using AFM of Aβ42 and nitro-Aβ at 0 and 72 h of aggregation on a mica surface.
Figure 3.

Aβ nitrotyrosination generates low molecular weight oligomers with β-sheet structure. A, DLS. Size distribution measured by DLS as a function of number of particles. All the Aβ oligomers tested show a high and very similar polydispersity index (>0.6). B, CD spectroscopy. CD measurements of Aβ42 and nitrotyrosinated samples showing a minimum at ∼225 nm and a maximum at ∼200 nm, indicative of β-sheet structure. C, ATR-FTIR spectroscopy of the amide I region for different Aβ42 oligomers in a thin hydrated film. All spectra show the characteristic β-sheet secondary structure peak at ∼1630 cm, following the band assignments of Byler and Susi (1986). D, Second derivative of the ATR-FTIR is used here to illustrate better the different secondary structures due to band narrowing (Wahlstrom et al., 2008).
Disruption of Y10-S26 interaction impairs fibril formation
According to the experimentally determined Aβ42 fibril structure (PDB ID: 2BEG), there are two antiparallel β-sheets comprising residues L17-S26 (β1) and I31-I41 (β2). This complex is stabilized hydrophobically and by intermolecular salt bridges (between D23 and K28) and backbone H bonds (Lührs et al., 2005). However, protofibrils require further stabilization with interprotofilament interactions to conform the supramolecular architecture of the fibril. Indeed, the structurally heterogeneous fragment D1-H16 could possibly be involved in these interactions (Egnaczyk et al., 2001; Zhang et al., 2009). Here, we propose a model in which the interprotofilament interaction of Y10 and S26 is key to the elongation of the fibril (Fig. 4A). We hypothesized that, upon nitration of Y10, this interaction is lost, dramatically impairing fibril stability and thus fibril formation (Fig. 4B).
Figure 4.

Disruption of Y10–S26 interaction impairs fibril formation. A, Model of interprotofibrillar Aβ42. The interaction between Y10 and S26 stabilizes the fibril and contributes to its progression. B, Upon the nitration of Y10, because the nitro group is negatively charged, it is arrested by the side-chains of either R5 or H6, impeding its interaction with S26 and destabilizing the fibril. C, Our mutant H14 to E14 also would trigger a rearrangement of the side-chains of the N terminus, disrupting the interaction between Y10 and S26 as it happens upon nitration. D, ThT fluorescence after 72 h of aggregation of Aβ42, Aβ42 treated with 100 μm SIN-1, synthetically nitrated Aβ42 (AβNTyr), and AβE14 mutant. Values are expressed as a percentage compared with Aβ42. Mean ± SEM of four independent experiments. *p < 0.001 (one-way ANOVA using Dunnett's post test). E, Cell viability assay performed by the MTT method. Hippocampal neurons were treated with 10 μm of AβWT, AβNTyr, and the mutant AβE14 for 24 h. Values are expressed as the percentage of the control. Mean ± SEM of 13–16 independent experiments. *p < 0.001 (one-way ANOVA using Dunnett's post test).
Impaired fibrillation ability also has been reported in the S26 mutant (Rezaei-Ghaleh et al., 2014). Thus, we tried to mimic the effect of Y10 nitration by using a different strategy to avoid Y10-S26 interaction. By mutating H14 to E (AβE14), we expected the side-chains of the N terminus to rearrange, avoiding the interaction between Y10 and S26 (Fig. 4C). We synthesized AβE14 as well as an Aβ analog with nitrotyrosine in position 10 (AβNTyr) to check whether the effects on fibril formation impairment and oligomer stabilization were the same as those observed using in vitro nitration with SIN-1.
To evaluate the aggregation propensity of the mutants, we measured ThT fluorescence after 72 h of aggregation. We observed that fibril formation was reduced to half in Aβ treated with 100 μm SIN-1, compared with the control (Fig. 4D), which is consistent with our time course ThT data (Fig. 1). With AβNTyr, which is synthetically nitrotyrosinated and thus modified in 100% of the molecules, amyloid fibril formation was almost completely nonexistent, as hypothesized. AβE14 also had significantly reduced aggregation rates compared with untreated AβWT.
Regarding neuronal toxicity, we treated mouse hippocampal neurons with oligomer preparations from AβWT, AβNTyr, and AβE14 for 24 h and evaluated cell viability (Fig. 4E). In both cases, the mutants were significantly more toxic than untreated AβWT, suggesting the presence of a soluble, more toxic form of Aβ.
To explore their structure, we examined untreated AβWT, SIN-1 treated AβWT, AβNTyr, and AβE14 (Fig. 5) under a transmission electron microscope after 72 h of aggregation. With untreated AβWT (Fig. 5A), we could see mature, amyloid fibrils; but with AβNTyr (Fig. 5C) and AβE14 (Fig. 5D), we were unable to depict fibrils in any field. Nevertheless, the presence of oligomers was clear in both cases, as well as when AβWT was nitrotyrosinated (Fig. 5B).
Figure 5.

Nitrotyrosination stabilizes oligomers. Transmission electron microscopy of the following: A, AβWT; B, AβWT + 100 μm SIN-1; C, AβNTyr; D, AβE14 negatively stained with uranyl acetate after 72 h of aggregation.
Nitro-Aβ oligomers affect calcium homeostasis and exert NMDA-dependent toxicity
Oligomers are accepted to be the neuroactive, synaptically toxic form of Aβ. Dimers, trimers, tetramers, dodecamers, and Aβ-derived diffusible ligands have all been suggested as the molecular determinant of Aβ toxicity. As all coexist in microenvironments of the AD brain, it is difficult to make sharp distinctions between them. Aggregation is a dynamic process, and working with one only form of Aβ oligomer, even in vitro, can be misleading unless it is cross-linked and thoroughly purified. Given that a mixture of oligomeric species has greater physiological relevance and that finding the molecule responsible for Aβ toxicity is beyond the scope of this work, we assessed nitro-Aβ toxicity using high, acute doses of a heterogeneous oligomer preparation.
Considering that Aβ oligomers have been linked to calcium dyshomeostasis and that NMDAR plays a key role not only in synaptic plasticity but also in AD, we explored the effect of acute, high-concentration Aβ and nitro-Aβ oligomer treatment on hippocampal cultured cells. Ten DIV mouse hippocampal neurons were loaded with the cytosolic calcium dye fura-2 AM. A 10 μm (monomer equivalent) oligomer preparation of either Aβ or nitro-Aβ was applied extracellularly for 5 min. Surprisingly, we observed no cytosolic calcium changes upon the bath application of Aβ or nitro-Aβ (Fig. 6A,D). However, subsequent stimulation with the NMDAR synthetic agonist NMDA generated an increased response upon Aβ treatment and even larger responses in the case of nitro-Aβ treatment (Fig. 6A–C). Nevertheless, this effect was not observed when cells were stimulated with another physiological stimulus. Upon application of a high potassium solution, which activates voltage-dependent calcium channels, we did not observe differences in calcium responses (Fig. 6D–F). Thus, it appears that this effect is specific, although perhaps not exclusive, to NMDAR.
Figure 6.
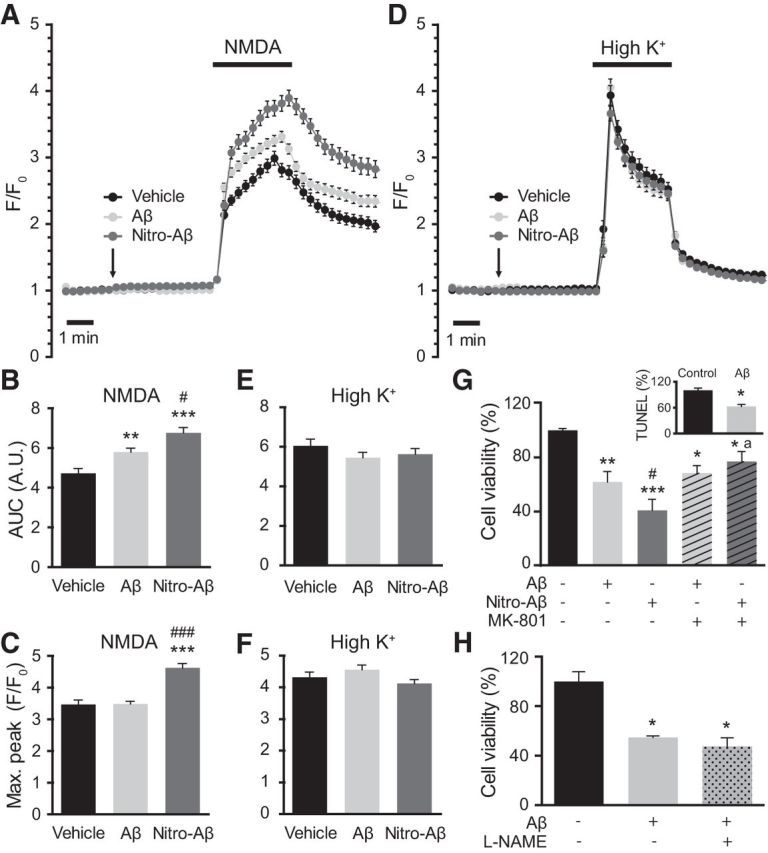
Nitro-Aβ oligomers affect calcium homeostasis and exert NMDA-dependent toxicity. Cells were treated for 5 min with 10 μm Aβ or nitro-Aβ and stimulated with bath application of either 100 μm NMDA (A) or 52.5 μm KCl (D). Mean ± SEM of 82–87 cells for NMDA and 98–104 for KCl. The area under the curve (AUC) (B, C) and the maximum response peak (E, F) were calculated for each cell response. G, Cell viability assay performed by the MTT method. Hippocampal neurons were treated with 10 μm Aβ and nitro-Aβ with or without 10 μm MK-801 for 5 min, and cell viability was assessed after 24 h. Untreated neurons were taken as 100% of viability. Inset, A control of Aβ toxicity studied by TUNEL assay. Mean ± SEM of three or four independent experiments. H, Cell viability assayed with the MTT method in hippocampal neurons treated with Aβ in the presence or absence of the NOS inhibitor l-NAME for 5 min. Cell viability was assessed after 24 h. Mean ± SEM of three independent experiments. *p < 0.05, compared with control (one-way ANOVA using Newman–Keuls post test). **p < 0.01, compared with control (one-way ANOVA using Newman–Keuls post test). ***p < 0.001, compared with control (one-way ANOVA using Newman–Keuls post test). #p < 0.05, compared with Aβ (one-way ANOVA using Newman–Keuls post test). ###p < 0.001, compared with Aβ (one-way ANOVA using Newman–Keuls post test). ap < 0.05 compared with nitro-Aβ by (one-way ANOVA using Newman–Keuls post test). Student's t test for the TUNEL study.
Having observed that differences in NMDA-induced calcium responses and extrasynaptic NMDARs can trigger excitotoxicity, we wanted to assess whether the nitro-Aβ insult made the neurons more susceptible to excitotoxicity using the NMDAR blocker MK-801. Similarly to calcium-imaging experiments, we treated the cells with Aβ and nitro-Aβ oligomers with or without MK-801, replaced the media after 5 min and evaluated cell viability after 24 h (Fig. 6G). Strikingly, 5 min of oligomer exposure was sufficient to trigger neuronal cell death. In addition, NMDA was partially responsible for Aβ oligomer toxicity, as toxicity was slightly reverted with MK-801. On the other hand, nitro-Aβ oligomers were significantly more toxic compared with Aβ, and cotreatment with MK-801 restored cell viability to the levels of Aβ plus MK-801. Therefore, it seems that the difference in cell viability between Aβ and nitro-Aβ oligomers was entirely NMDAR-dependent. A control on the accuracy of MTT method to measure cell death was performed by TUNEL assay (Fig. 6A, inset), resulting both in a drop of ∼40% in cell viability with Aβ. Furthermore, to address whether NO release by NOS is involved in the toxicity associated to Aβ, we performed a cell viability assay combining the treatment of Aβ with the NOS inhibitor l-NAME (Fig. 6H). However, we were not able to observe a prevention of Aβ toxicity. Hence, nitrotyrosination of Aβ inhibits fibrillation, stabilizing a soluble, highly toxic form of nitro-Aβ oligomers that make neurons more susceptible to excitotoxic insults (Fig. 7).
Figure 7.

Aβ oligomers and fibrils trigger the formation of nitrogen reactive species. In this context, peroxynitrite reacts with proteins, affecting their function. One of these modifications is protein nitrotyrosination, which is highly promoted in AD brains. Aβ has a potential nitration site in Y10 and is nitrated in the disease. Nitrotyrosination of Aβ impairs amyloid fibril formation and stabilizes soluble, highly toxic oligomers. Glutamatergic synapses are the most abundant within the hippocampus and the first to be affected in AD. Glutamate is released from the presynaptic neuron into the synaptic cleft; and when it is released in a large quantity, it spills to the perisynaptic space, partially activating extrasynaptic NMDARs. When glutamatergic stimulation is particularly high, and especially when extrasynaptic NMDARs are activated, the excitotoxic apoptotic pathway is triggered. Nitro-Aβ oligomers make the neurons more susceptible to excitotoxic insults.
We hypothesized that the differences we observed in excitotoxicity and in the NMDAR-dependent intracellular calcium increase could be due to an increased binding of nitro-Aβ oligomers to the synapses and, more specifically, to NMDAR. To address this hypothesis, we transfected hippocampal neurons with GFP and treated them with or without Aβ and nitro-Aβ for 5 min. We performed 3D rendering of confocal microscopy stacks of the dendritic spines to visualize the binding of Aβ and nitro-Aβ to the dendrites and spines (Fig. 8A). We observed an increased binding of oligomers when neurons were treated with nitro-Aβ (both SIN-1 treated Aβ and AβNTyr) compared with Aβ (Fig. 8B). Even for the neurons that were not transfected, the shape of the spines was visible because of the nitro-Aβ bound to the dendritic shaft. We obtained similar results when we immunoprecipitated the NMDAR subunit GluN1 from hippocampal neurons and checked the amount of bound Aβ and the nitrated forms of Aβ by Western blot (Fig. 8C,D).
Figure 8.

Increased nitro-Aβ oligomers binding to the synapses. Cells were treated for 5 min with 10 μm Aβ or nitro-Aβ (SIN-1-treated Aβ and AβNTyr). A, Three-dimensional rendering of dendritic spines. Green represents the neuron volume (GFP). Magenta represents Aβ and nitro-Aβ oligomers. Nitro-Aβ oligomers contact the dendritic spines (arrows). B, Colocalization analysis of A. M1 Manders' coefficient refers to the fraction of Aβ overlapping spines. Mean ± SEM of 5–8 neurons from three different experiments. C, Representative Western blot of Aβ and nitro-Aβ coimmunoprecipitated with the NMDAR subunit GluN1 from hippocampal neurons. D, Quantification of C. Mean ± SEM of 5–7 independent experiments. *p < 0.05, compared with control (one-way ANOVA using Newman–Keuls post test). **p < 0.01, compared with control (one-way ANOVA using Newman–Keuls post test). ***p < 0.001, compared with control (one-way ANOVA using Newman–Keuls post test). #p < 0.01, compared with Aβ (one-way ANOVA using Newman–Keuls post test).
Discussion
There are two main reasons to believe that nitro-oxidative stress is an etiopathogenic factor in AD (Dawson and Dawson, 1996; Smith et al., 1997; Miranda et al., 2000; Tamagno et al., 2002; Coma et al., 2008; Guix et al., 2009). First, aging is the major risk factor for AD and is associated with an unbalanced redox state due to the failure of antioxidant mechanisms (Hensley et al., 1994). Second, oxidative stress increases APP cleavage by raising the expression of BACE-1 and, as a consequence, enhances the production of both Aβ42 and Aβ40 (Tamagno et al., 2002; Coma et al., 2008), but nitro-oxidative stress increases the Aβ42/Aβ40 ratio (Guix et al., 2012).
Our findings revealed that Aβ nitrotyrosination impairs fibrillation dramatically, favoring the presence of toxic soluble oligomers. As described by Kummer et al. (2011), Aβ is nitrated at Y10 in AD brains; they observed nitrotyrosinated Aβ at the core of amyloid plaques in a triple transgenic mouse model. Our results are in good agreement with their findings regarding the increased toxicity, synaptic failure, and memory impairment caused by nitro-Aβ. However, in our study, nitro-Aβ did not progress to mature amyloid fibrils, producing low or virtually no signals in ThT assays. Furthermore, Zhao et al. (2015) have reported that nitration of Aβ40 seems to provoke impaired fibrillation by ThT assays and transmission electron microscopy. Therefore, given that the most toxic and neuroactive forms of Aβ are low molecular weight oligomeric species (Selkoe, 2008; Shankar et al., 2008; Li et al., 2009), our findings support the observed increase of soluble oligomers, rather than fibril formation.
In addition, our results show that nitro-Aβ oligomers exert higher toxicity in hippocampal neuronal primary cultures through an NMDAR-dependent mechanism. We hypothesize that the binding of Aβ oligomers, and specially of those of nitrated Aβ, depends directly on their hydrophobic nature (Serpel, 2000), allowing them to interact with the hydrophobic domains of the extracellular regions of GluN1 and GluN2B (Costa et al., 2012). This interaction would impair the proper and rapid closing of the NMDAR; an effect increased when Aβ oligomers are nitrated. NMDAR plays a key role in AD because glutamate is the most abundant neurotransmitter in the hippocampus; furthermore, one of the few treatments available for AD is the mild NMDAR inhibitor memantine. NMDAR overactivation leads to a particular apoptotic pathway known as excitotoxicity (Choi, 1985). Many researchers have explored calcium dyshomeostasis as one of the factors responsible for neurodegeneration in AD (Berridge, 2010).
Here, we propose a model by which the addition of a nitro group to Y10 would prevent its interaction with S26, leading to the destabilization of interprotofibrillar contacts and impairing the progression to amyloid fibrils. The phosphorylation of S26 or its replacement with another amino acid have been reported to impair the capacity to form amyloid fibrils (Rezaei-Ghaleh et al., 2014). We show how the disruption of Y10-S26 interaction by mutating H14 to E14 not only impairs fibril formation but also mimics nitro-Aβ in terms of neurotoxicity.
In terms of the structure of the Aβ fibrils, it has been demonstrated that oligomers are the most toxic amyloid aggregate forms. The formation of senile plaques by mature amyloid fibrils could be a mechanism to eliminate oligomers from the neuronal surroundings (Fernàndez-Busquets, 2013). We found that Aβ42 oligomers are stabilized by the presence of peroxynitrite, by using the peroxynitrite donor SIN-1, which delays the formation of mature Aβ fibrils. It resembles the pathological scenario leading to APP cleavage on the glutamatergic synapses in AD, where sustained NO production causes the oxidative stress. Thus, these nitrotyrosinated oligomers could be especially toxic to the brain because they exist in a more toxic conformation for a longer period than do non-nitrated oligomers that rapidly aggregate to form mature fibrils, a less toxic amyloid aggregate form.
In conclusion, our data support the role of peroxynitrite as a factor with an impact on AD pathophysiology at several levels. We propose a mechanism for Aβ toxicity that is relevant for AD pathophysiology because nitro-Aβ has been found in amyloid plaques. We also suggest a model for the assembly of Aβ fibrils, in which the aggregation is severely impaired by the disruption of the interprotofibrillar interaction between Y10 and S26. These nitrotyrosinated oligomers remain stable over time, thus preserving the higher toxicity of these shorter amyloid aggregates, compared with mature amyloid fibrils.
Footnotes
This work was supported by Spanish Plan Estatal de I+D+I 2013-2016 and the ISCIII-Subdirección General de Evaluación y Fomento de la Investigación Grants PI13/00408 and Red HERACLES RD12/0042/0014 and FEDER Funds, Spanish Ministerio de Economía y Competitividad BIO2014-57518-R, BIO2014-52872-R, and AGL2014-52395-C2-2-R and FEDER Funds, the Generalitat de Catalunya (Spain) 2014-SGR-938, and Chilean Funds for Science CONICYT-PFB 12/2007 and FONDECYT N° 1160724. A.P.-M. was the recipient of the Universitat Autònoma de Barcelona-Programa Banco de Santander Fellowship. We thank Salvador Bartolomé for generous access to the FTIR spectrometer; and Judit Fuentes for skillful DLS data acquisition.
The authors declare no competing financial interests.
References
- Arimon M, Díez-Pérez I, Kogan MJ, Durany N, Giralt E, Sanz F, Fernàndez-Busquets X. Fine structure study of Aβ1–42 fibrillogenesis with atomic force microscopy. FASEB J. 2005;19:1344–1346. doi: 10.1096/fj.04-3137fje. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Calcium hypothesis of Alzheimer's disease. Pflugers Arch. 2010;459:441–449. doi: 10.1007/s00424-009-0736-1. [DOI] [PubMed] [Google Scholar]
- Bitan G, Teplow DB. Preparation of aggregate-free, low molecular weight amyloid-beta for assembly and toxicity assays. Methods Mol Biol. 2005;299:3–9. doi: 10.1385/1-59259-874-9:003. [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Byler DM, Susi H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers. 1986;25:469–487. doi: 10.1002/bip.360250307. [DOI] [PubMed] [Google Scholar]
- Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer's disease brain. J Neurochem. 2003;85:1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett. 1985;58:293–297. doi: 10.1016/0304-3940(85)90069-2. [DOI] [PubMed] [Google Scholar]
- Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TP. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A. 2013;110:9758–9763. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coma M, Guix FX, Ill-Raga G, Uribesalgo I, Alameda F, Valverde MA, Muñoz FJ. Oxidative stress triggers the amyloidogenic pathway in human vascular smooth muscle cells. Neurobiol Aging. 2008;29:969–980. doi: 10.1016/j.neurobiolaging.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Costa RO, Lacor PN, Ferreira IL, Resende R, Auberson YP, Klein WL, Oliveira CR, Rego AC, Pereira CM. Endoplasmic reticulum stress occurs downstream of GluN2B subunit of N-methyl-D-aspartate receptor in mature hippocampal cultures treated with amyloid-β oligomers. Aging Cell. 2012;11:823–833. doi: 10.1111/j.1474-9726.2012.00848.x. [DOI] [PubMed] [Google Scholar]
- Danysz W, Parsons CG. Alzheimer's disease, β-amyloid, glutamate, NMDA receptors and memantine-searching for the connections. Br J Pharmacol. 2012;167:324–352. doi: 10.1111/j.1476-5381.2012.02057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM. Nitric oxide neurotoxicity. J Chem Neuroanat. 1996;10:179–190. doi: 10.1016/0891-0618(96)00148-2. [DOI] [PubMed] [Google Scholar]
- Duhovny D, Nussinov R, Wolfson H. Algorithms in bioinformatics SE-14. Berlin: Springer; 2002. Efficient unbound docking of rigid molecules; pp. 185–200. Lecture Notes in Computer Science. [Google Scholar]
- Egnaczyk GF, Greis KD, Stimson ER, Maggio JE. Photoaffinity cross-linking of Alzheimer's disease amyloid fibrils reveals interstrand contact regions between assembled β-amyloid peptide subunits. Biochemistry. 2001;40:11706–11714. doi: 10.1021/bi002852h. [DOI] [PubMed] [Google Scholar]
- Esposito Z, Belli L, Toniolo S, Sancesario G, Bianconi C, Martorana A. Amyloid β, glutamate, excitotoxicity in Alzheimer's disease: are we on the right track? CNS Neurosci Ther. 2013;19:549–555. doi: 10.1111/cns.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernàndez-Busquets X. Amyloid fibrils in neurodegenerative diseases: villains or heroes? Future Med Chem. 2013;5:1903–1906. doi: 10.4155/fmc.13.138. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guix FX, Uribesalgo I, Coma M, Muñoz FJ. The physiology and pathophysiology of nitric oxide in the brain. Prog Neurobiol. 2005;76:126–152. doi: 10.1016/j.pneurobio.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Guix FX, Ill-Raga G, Bravo R, Nakaya T, de Fabritiis G, Coma M, Miscione GP, Villà-Freixa J, Suzuki T, Fernàndez-Busquets X, Valverde MA, de Strooper B, Muñoz FJ. Amyloid-dependent triosephosphate isomerase nitrotyrosination induces glycation and tau fibrillation. Brain. 2009;132:1335–1345. doi: 10.1093/brain/awp023. [DOI] [PubMed] [Google Scholar]
- Guix FX, Wahle T, Vennekens K, Snellinx A, Chávez-Gutiérrez L, Ill-Raga G, Ramos-Fernandez E, Guardia-Laguarta C, Lleó A, Arimon M, Berezovska O, Muñoz FJ, Dotti CG, De Strooper B. Modification of γ-secretase by nitrosative stress links neuronal ageing to sporadic Alzheimer's disease. EMBO Mol Med. 2012;4:660–673. doi: 10.1002/emmm.201200243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ill-Raga G, Ramos-Fernández E, Guix FX, Tajes M, Bosch-Morató M, Palomer E, Godoy J, Belmar S, Cerpa W, Simpkins JW, Inestrosa NC, Muñoz FJ. Amyloid-β peptide fibrils induce nitro-oxidative stress in neuronal cells. J Alzheimers Dis. 2010;22:641–652. doi: 10.3233/JAD-2010-100474. [DOI] [PubMed] [Google Scholar]
- Ill-Raga G, Tajes M, Busquets-García A, Ramos-Fernández E, Vargas LM, Bosch-Morató M, Guivernau B, Valls-Comamala V, Eraso-Pichot A, Guix FX, Fandos C, Rosen MD, Rabinowitz MH, Maldonado R, Alvarez AR, Ozaita A, Muñoz FJ. Physiological control of nitric oxide in neuronal BACE1 translation by heme-regulated eIF2α kinase HRI induces synaptogenesis. Antioxid Redox Signal. 2015;22:1295–1307. doi: 10.1089/ars.2014.6080. [DOI] [PubMed] [Google Scholar]
- Klein WL. Abeta toxicity in Alzheimer's disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int. 2002;41:345–352. doi: 10.1016/S0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, Walter J, Pape HC, König S, Roeber S, Jessen F, Klockgether T, Korte M, Heneka MT. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron. 2011;71:833–844. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Döbeli H, Schubert D, Riek R. 3D structure of Alzheimer's amyloid-beta(1–42) fibrils. Proc Natl Acad Sci U S A. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Miranda S, Opazo C, Larrondo LF, Muñoz FJ, Ruiz F, Leighton F, Inestrosa NC. The role of oxidative stress in the toxicity induced by amyloid beta-peptide in Alzheimer's disease. Prog Neurobiol. 2000;62:633–648. doi: 10.1016/S0301-0082(00)00015-0. [DOI] [PubMed] [Google Scholar]
- Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science. 1991;254:97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- Perálvarez-Marín A, Barth A, Gräslund A. Time-resolved infrared spectroscopy of pH-induced aggregation of the Alzheimer Aβ1–28 peptide. J Mol Biol. 2008;379:589–596. doi: 10.1016/j.jmb.2008.04.014. [DOI] [PubMed] [Google Scholar]
- Petkova AT, Yau WM, Tycko R. Experimental constraints on quaternary structure in Alzheimer's beta-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce B, Weng Z. ZRANK: reranking protein docking predictions with an optimized energy function. Proteins. 2007;67:1078–1086. doi: 10.1002/prot.21373. [DOI] [PubMed] [Google Scholar]
- Ramos-Fernández E, Tajes M, Palomer E, Ill-Raga G, Bosch-Morató M, Guivernau B, Román-Dégano I, Eraso-Pichot A, Alcolea D, Fortea J, Nuñez L, Paez A, Alameda F, Fernández-Busquets X, Lleó A, Elosúa R, Boada M, Valverde MA, Muñoz FJ. Posttranslational nitro-glycative modifications of albumin in Alzheimer's disease: implications in cytotoxicity and amyloid-β peptide aggregation. J Alzheimers Dis. 2014;40:643–657. doi: 10.3233/JAD-130914. [DOI] [PubMed] [Google Scholar]
- Reed TT, Pierce WM, Jr, Turner DM, Markesbery WR, Butterfield DA. Proteomic identification of nitrated brain proteins in early Alzheimer's disease inferior parietal lobule. J Cell Mol Med. 2009;13:2019–2029. doi: 10.1111/j.1582-4934.2008.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaei-Ghaleh N, Amininasab M, Giller K, Kumar S, Stündl A, Schneider A, Becker S, Walter J, Zweckstetter M. Turn plasticity distinguishes different modes of amyloid-β aggregation. J Am Chem Soc. 2014;136:4913–4919. doi: 10.1021/ja411707y. [DOI] [PubMed] [Google Scholar]
- Rossi F, Bianchini E. Synergistic induction of nitric oxide by beta-amyloid and cytokines in astrocytes. Biochem Biophys Res Commun. 1996;225:474–478. doi: 10.1006/bbrc.1996.1197. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpel LC. Alzheimer's amyloid fibrils: structure and assembly. BBA Mol Basis Dis. 2000;1502:16–30. doi: 10.1016/S0925-4439(00)00029-6. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- Tajes M, Guivernau B, Ramos-Fernández E, Bosch-Morató M, Palomer E, Guix FX, Muñoz FJ. The pathophysiology of triose phosphate isomerase dysfunction in Alzheimer's disease. Histol Histopathol. 2013;28:43–51. doi: 10.14670/HH-28.43. [DOI] [PubMed] [Google Scholar]
- Tajes M, Eraso-Pichot A, Rubio-Moscardó F, Guivernau B, Ramos-Fernández E, Bosch-Morató M, Guix FX, Clarimón J, Miscione GP, Boada M, Gil-Gómez G, Suzuki T, Molina H, Villà-Freixa J, Vicente R, Muñoz FJ. Methylglyoxal produced by amyloid-β peptide-induced nitrotyrosination of triosephosphate isomerase triggers neuronal death in Alzheimer's disease. J Alzheimers Dis. 2014;41:273–288. doi: 10.3233/JAD-131685. [DOI] [PubMed] [Google Scholar]
- Tamagno E, Bardini P, Obbili A, Vitali A, Borghi R, Zaccheo D, Pronzato MA, Danni O, Smith MA, Perry G, Tabaton M. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis. 2002;10:279–288. doi: 10.1006/nbdi.2002.0515. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Wahlström A, Hugonin L, Perálvarez-Marín A, Jarvet J, Gräslund A. Secondary structure conversions of Alzheimer's Aβ(1–40) peptide induced by membrane-mimicking detergents. FEBS J. 2008;275:5117–5128. doi: 10.1111/j.1742-4658.2008.06643.x. [DOI] [PubMed] [Google Scholar]
- Webb B, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2014;1137:1–15. doi: 10.1007/978-1-4939-0366-5_1. [DOI] [PubMed] [Google Scholar]
- Xia W, Zhang J, Ostaszewski BL, Kimberly WT, Seubert P, Koo EH, Shen J, Selkoe DJ. Presenilin 1 regulates the processing of beta-amyloid precursor protein C-terminal fragments and the generation of amyloid beta-protein in endoplasmic reticulum and Golgi. Biochemistry. 1998;37:16465–16471. doi: 10.1021/bi9816195. [DOI] [PubMed] [Google Scholar]
- Zhang R, Hu X, Khant H, Ludtke SJ, Chiu W, Schmid MF, Frieden C, Lee JM. Interprotofilament interactions between Alzheimer's Abeta1–42 peptides in amyloid fibrils revealed by cryoEM. Proc Natl Acad Sci U S A. 2009;106:4653–4658. doi: 10.1073/pnas.0901085106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Wang P, Li H, Gao Z. Nitration of Y10 in Aβ 1–40: is it a compensatory reaction against oxidative/nitrative stress and Aβ aggregation? Chem Res Toxicol. 2015;28:401–407. doi: 10.1021/tx500353s. [DOI] [PubMed] [Google Scholar]