

Abstract
Physiological levels of ROS support neurite outgrowth and axonal specification, but the mechanisms by which ROS are able to shape neurons remain unknown. Ca2+, a broad intracellular second messenger, promotes both Rac1 activation and neurite extension. Ca2+ release from the endoplasmic reticulum, mediated by both the IP3R1 and ryanodine receptor (RyR) channels, requires physiological ROS levels that are mainly sustained by the NADPH oxidase (NOX) complex. In this work, we explore the contribution of the link between NOX and RyR-mediated Ca2+ release toward axonal specification of rat hippocampal neurons. Using genetic approaches, we find that NOX activation promotes both axonal development and Rac1 activation through a RyR-mediated mechanism, which in turn activates NOX through Rac1, one of the NOX subunits. Collectively, these data suggest a feedforward mechanism that integrates both NOX activity and RyR-mediated Ca2+ release to support cellular mechanisms involved in axon development.
SIGNIFICANCE STATEMENT High levels of ROS are frequently associated with oxidative stress and disease. In contrast, physiological levels of ROS, mainly sustained by the NADPH oxidase (NOX) complex, promote neuronal development and axonal growth. However, the mechanisms by which ROS shape neurons have not been described. Our work suggests that NOX-derived ROS promote axonal growth by regulating Rac1 activity, a molecular determinant of axonal growth, through a ryanodine receptor (RyR)-mediated Ca2+ release mechanism. In addition, Rac1, one of the NOX subunits, was activated after RyR-mediated Ca2+ release, suggesting a feedforward mechanism between NOX and RyR. Collectively, our data suggest a novel mechanism that is instrumental in sustaining physiological levels of ROS required for axonal growth of hippocampal neurons.
Keywords: actin cytoskeleton, axon development, calcium signaling, NADPH oxidase, neuronal differentiation, reactive oxygen species
Introduction
ROS may exert both physiological and pathological effects on neurons. Abnormally high levels are associated with oxidative stress and disease (Lambeth, 2004; Villegas et al., 2014; Walsh et al., 2014). In contrast, downregulation of ROS synthesis below physiological levels impairs neurite outgrowth, axonal growth, and neuronal polarity acquisition (Munnamalai and Suter, 2009; Olguin-Albuerne and Moran, 2015; Wilson et al., 2015). We showed recently that the loss-of-function of NADPH oxidase (NOX) also impairs actin dynamics by inhibiting the activity of Rac1 and Cdc-42 (Wilson et al., 2015), which are essential for neuronal polarization (Dotti et al., 1988; Bradke and Dotti, 1999; Stiess and Bradke, 2011; Cáceres et al., 2012; González-Billault et al., 2012).
ROS are highly dynamic molecules with an average lifetime on the nanosecond timescale. In contrast, neuronal development occurs after days in culture. This poses the question of how short-lived species such as ROS are able to support the establishment of neuronal polarity and axonal development. Here, we propose that ROS may act as signaling molecules targeting downstream mediators. Previous reports suggest that Ca2+ release from the endoplasmic reticulum (ER), particularly that mediated by the ryanodine receptor (RyR), needs the basal activity of NOX2 in muscle and neuronal cells (Espinosa et al., 2006; Bedard and Krause, 2007; Zhang and Forscher, 2009; Riquelme et al., 2011). Moreover, RyR stimulation activates Rac1 and Cdc-42 in HEK293 cells and cerebellar granule neurons, suggesting a RyR-dependent mechanism (Jin et al., 2005). In addition, Rac1, one of the subunits of the NOX2 complex (Lambeth, 2004), promotes axonal growth and neuronal polarization (González-Billault et al., 2012), suggesting to us that functional coupling between NOX and ER Ca2+ release is a novel regulation point for actin dynamics in polarizing neurons. ER Ca2+ release has been associated previously with neuritogenesis, growth cone motility, and neuronal differentiation (Gomez et al., 2001; Nakamuta et al., 2011), but the precise contribution to the establishment of neuronal polarity has not been reported. Considering this, we hypothesize a mechanism involving NOX, RyR-mediated Ca2+ release, and Rac1 to sustain physiological ROS levels and axonal development.
Although we have shown previously that NOX inhibition impairs axon elongation (Wilson et al., 2015), the contribution of NOX gain-of-function to this event remains unknown. Considering that ROS production is usually a negative signal for neurons, it is relevant to scrutinize carefully whether ROS synthesis may support normal neuronal functions. In this work, we explored the contribution of the gain-of-function of the NOX2 complex and ROS production toward axonal development in cultured hippocampal neurons. In addition, we studied the potential coupling between NOX2 and RyR-mediated Ca2+ release toward the activation of Rac1 for axonal development, suggesting a molecular mechanism by which ROS are able to shape hippocampal neurons.
Materials and Methods
Primary culture of hippocampal and cortical neurons from rat brain embryos.
Pregnant Sprague Dawley rats were killed and both female and male embryos (embryone day 18.5, E18.5) were removed and neurons cultured as described previously (Kaech and Banker, 2006). All of the experiments were approved by the Bioethical Research Committee of Universidad de Chile and conducted following the guidelines of the CONICYT manual for animal experimentation.
Primary culture of hippocampal neurons from wild-type and Ncf1−/− mouse P0 brains.
Wild-type C57BL/6J (Harlan Laboratories) and B6(Cg)-Ncf1m1J/J (Ncf1−/−; The Jackson Laboratory) newborn pups (P0) were obtained from the Central Biomedical Service of Imperial College London. Dissection and culture of hippocampal neurons were done as described previously (Kaech and Banker, 2006). Animal work was performed in accordance with the regulations of the UK Home Office.
Transient transfection of neurons with cDNA coding vectors.
Neurons were transiently transfected with Lipofectamine 2000 (Life Technologies) in Neurobasal medium following the manufacturer's instructions. After 2 h of transfection, neurons were supplemented with B27, Glutamax, sodium pyruvate, and antibiotics. Experiments were performed 18–72 h after cDNA transfection.
HyPer H2O2 measurement.
Neurons (4 × 104 cells/well) were cultured on glass coverslips pretreated with poly-d-lysine (1 mg/ml). Neurons were transiently transfected after 1 d in vitro (DIV) with the HyPer biosensor (Evrogen) to detect local H2O2 production (Belousov et al., 2006). H2O2 analysis was done as described previously (Wilson et al., 2015). The distal/proximal axonal ratio was estimated by measuring the HyPer-H2O2 levels in the first third of the axon (proximal segment) and the last third of the axon (distal segment).
Brain lysates and immunoblotting for RyR2 and IP3R1 detection.
After dissection from embryonic brains, hippocampi and cerebral cortices were collected into 1.5 ml tubes and washed three times with PBS. Lysis, protein sample preparation, and Western blot analysis were as described previously (Henríquez et al., 2012). To detect RyR and IP3R1 proteins, the following antibodies were used: anti-RyR2 (1:1000, rabbit, AB9080l Millipore), anti-IP3R1 (1:1000, rabbit, kindly donated by Dr. Manuel Estrada, Faculty of Medicine, Universidad de Chile; Choe et al., 2004; Estrada et al., 2006), and α-tubulin (1:10,000, mouse; Sigma-Aldrich). Primary antibodies were diluted in 1% nonfat milk dissolved in 0.05% TBST and incubated with the membrane overnight at 4°C with agitation. Membranes were washed three times with 0.05% TBST and suitable secondary antibodies conjugated with HRP were incubated for 1 h at room temperature with agitation in primary antibody incubation solution. Proteins were detected using the Pierce ECL Western Blotting Substrate.
Immunofluorescence for RyR2 and IP3R1.
Neurons (104 cells/well) were cultured in 24 multiwell plates on glass coverslips pretreated with poly-d-lysine (1 mg/ml) for 18 and 48 h (for stage 2 and 3 neurons, respectively). Anti-RyR2 (1:100), anti-IP3R1 (1:100), and anti-Tau-1 (1:400) were incubated overnight at 4°C. Coverslips were washed three times with PBS and suitable Alexa Fluor-conjugated secondary antibodies (1:400; Life Technologies) and incubated for 1 h at room temperature. Phalloidin–Alexa Fluor 647 was coincubated with secondary antibodies. For general considerations of immunofluorescence assays, please see Henríquez et al. (2012).
ER Ca2+ release assays.
Hippocampal neurons (1.5 × 105) were cultured in 35 mm dishes containing 25 mm glass coverslips pretreated with poly-d-lysine (1 mg/ml). After 2 DIV, neurons were loaded with Fluo4-AM (5 μm) or Cell Tracker Orange (1 μm) (Life Technologies) for 20 min in Hank's balanced salt solution supplemented with HEPES (HBSS) at 37°C and 5% CO2. Then, Fluo4-AM containing medium was replaced with Neurobasal medium supplemented with B27, Glutamax, sodium pyruvate, and antibiotics for 40 min under the same incubation conditions. Coverslips were mounted into a videomicroscopy chamber and perfused with HBSS without Ca2+ to discard the influx from extracellular medium. Images were acquired every 2 s for 5 min using a time-lapse recording. Tyrode buffer containing the following (in mm): 110 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 25 HEPES, and 30 glucose was used for recordings in the presence of extracellular Ca2+. Imaging was done with a Zeiss LSM 710 microscope using a 20× magnification. To induce RyR-mediated Ca2+ release, neurons were stimulated with 4-chloro-m-cresol (4-CMC; Sigma-Aldrich, 750 μm) after 1 min of baseline fluorescence (Westerblad et al., 1998; Adasme et al., 2015). The average intensity of baseline fluorescence was noted as “F0.” Fluorescence intensity after 4-CMC addition (or ethanol vehicle) was noted as “F.” Both “F0” and “F” quantifications were undertaken selecting a ROI at the soma of neurons. The F/F0 ratio was used to determine the fold-change of Fluo4-AM fluorescence intensity after RyR stimulation with 4-CMC. Analyses were done using the Fiji-ImageJ plug-in “time series analyzer.”
Raichu-Rac1 FRET probe measurements.
Neurons (4 × 104 cells/well) were transfected with the Raichu-Rac1 FRET biosensor (provided by Dr. Alfredo Cáceres, INIMEC-CONICET, Córdoba, Argentina) to measure Rac1 activity (Nakamura et al., 2006). Raichu-Rac1 expression, image acquisition, and FRET efficiency estimation were performed as described previously (Wilson et al., 2015).
Rac1 activity pull-down assay in embryonic cortical neurons.
Cortical neurons (E18.5) were cultured for 2 d in 100-mm-diameter plastic plates (107 neurons/plate) pretreated with poly-d-lysine (1 mg/ml) and then stimulated with 4-CMC (750 μm) for 0–30 min at 37°C and 5% CO2. To inhibit NOX activity, neurons were treated with VAS2870 (5 μm) for 1 h before 4-CMC stimulation. To evaluate Rac1 activity, pull-down assays were performed as described previously (Henríquez et al., 2012).
Image acquisition and analysis.
All images corresponding to Figures 1, 2, 3, 4, 5, and 6 were obtained with an LSM 710 confocal microscope (Zeiss). In Figure 7, axonal length measurements and Ca2+ imaging were performed using a Nikon Eclipse TE 2000-u coupled to a CoolLED pE-4000 illumination system. FRET experiments were performed using a Leica TCS SP5 II confocal microscope. Processing, measurement, and quantification of images were done with ImageJ.
Figure 1.
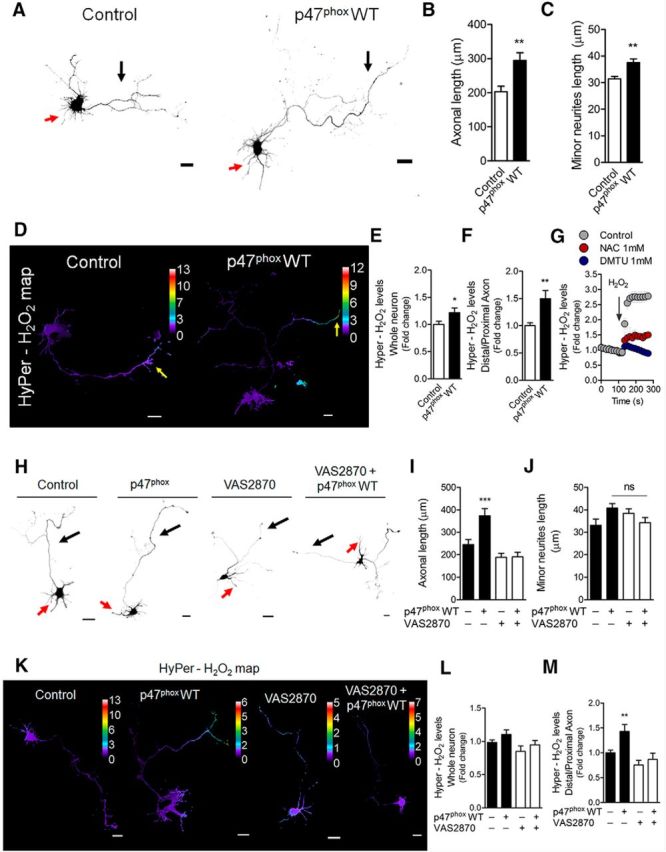
Gain-of-function of the NOX complex after p47phox WT expression increases axonal development of hippocampal neurons. E18.5 hippocampal neurons were cultured and transfected immediately after plating with GFP alone (control) or cotransfected with both GFP and p47phox WT constructs. A, Representative control and p47phoxWT neurons after 3 DIV. B, C, Quantification of both axonal (black arrows) and minor neurite (red arrows) length of A. **p < 0.01, ***p < 0.001 versus control, Mann–Whitney test. D, Neurons (1 DIV) were transfected with the HyPer biosensor to measure local H2O2 production. Representative H2O2 maps of both control and p47phox WT neurons. E, F, Quantification of the HyPer-H2O2 levels from neurons of D. *p < 0.05 versus control, **p < 0.01 versus control, Mann–Whitney test. G, Positive control for HyPer fluorescence (whole neuron) after H2O2 application in control, and N-acetyl-cysteine (NAC)- or dimethylthiourea (DMTU)-treated neurons. H, Control and p47phox WT neurons were treated with VAS2870 (5 μm) after 1 DIV and then fixed at 3 DIV. I, J, Quantification of axonal (black arrows) and minor neurite (red arrows) length of H. I, ***p < 0.001 vs control (ANOVA, Dunnett's post test). J, ns, nonsignificant, ANOVA, Dunnett's post test. K, Representative HyPer-H2O2 maps of both control and p47phox WT neurons after NOX inhibition with VAS2870. HyPer was expressed as in D and then neurons were treated with VAS2870 (5 μm) for 1 d (2 DIV in total). L, M, Quantification of the HyPer-H2O2 levels from neurons of K. **p < 0.01 versus control, ANOVA, Dunnett's post test. Results are from three independent cultures (n = 3). A total of 45 transfected neurons were analyzed for each condition. Scale bar, 20 μm.
Figure 2.
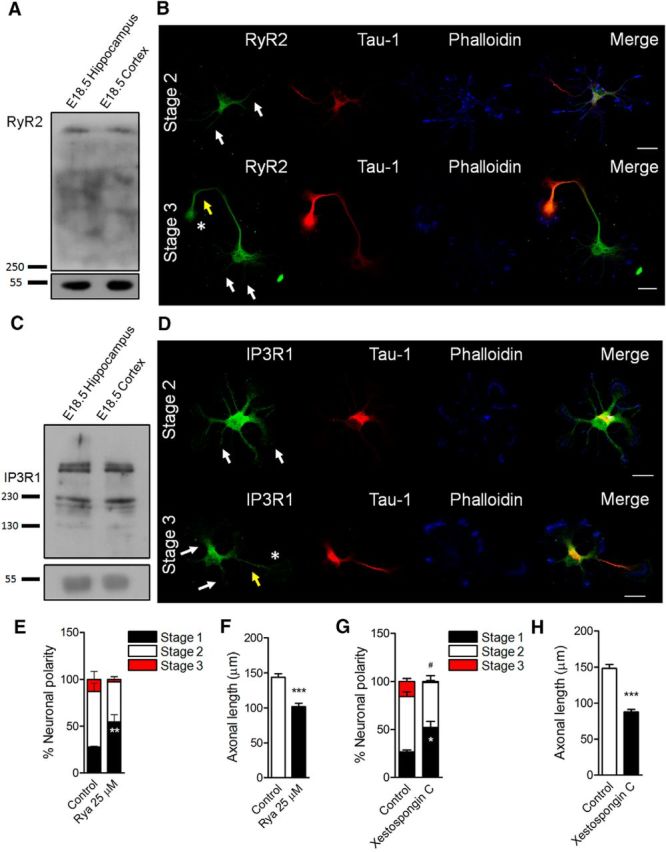
Contribution of ER Ca2+ channels to the establishment of neuronal polarity in culture. A, C, RyR2 and IP3R1 detection by Western blot in both E18.5 hippocampus and cerebral cortex lysates. B, D, RyR2 and IP3R1 distribution in both stage 2 and stage 3 hippocampal cultured neurons using immunofluorescence and confocal microscopy. Tau-1 detection was used as an axonal marker and phalloidin–Alexa Fluor 633 to stain F-actin. White arrows indicate minor neurites; yellow arrows, axons; asterisks, axonal growth cones. E, G, Quantification of neuronal polarization of hippocampal neurons after ryanodine (Rya, 25 μm) and xestospongin C (3 μm) treatments. E, **p < 0.01 versus control stage 1. G, *p < 0.05 versus control stage 1, #p < 0.05 versus control stage 3, Mann–Whitney test. F, H, Quantification of axonal length after RyR and IP3R inhibition at 3 DIV. After plating, neurons were cultured for 1 d and then treated with ryanodine (Rya, 25 μm, F) and xestospongin C (3 μm, H). ***p < 0.001 versus control, Mann–Whitney test. Results are from 3 different independent cultures (n = 3). A total of 90 neurons were analyzed for both neuronal polarity and axonal length measurements. Scale bar, 20 μm.
Figure 3.
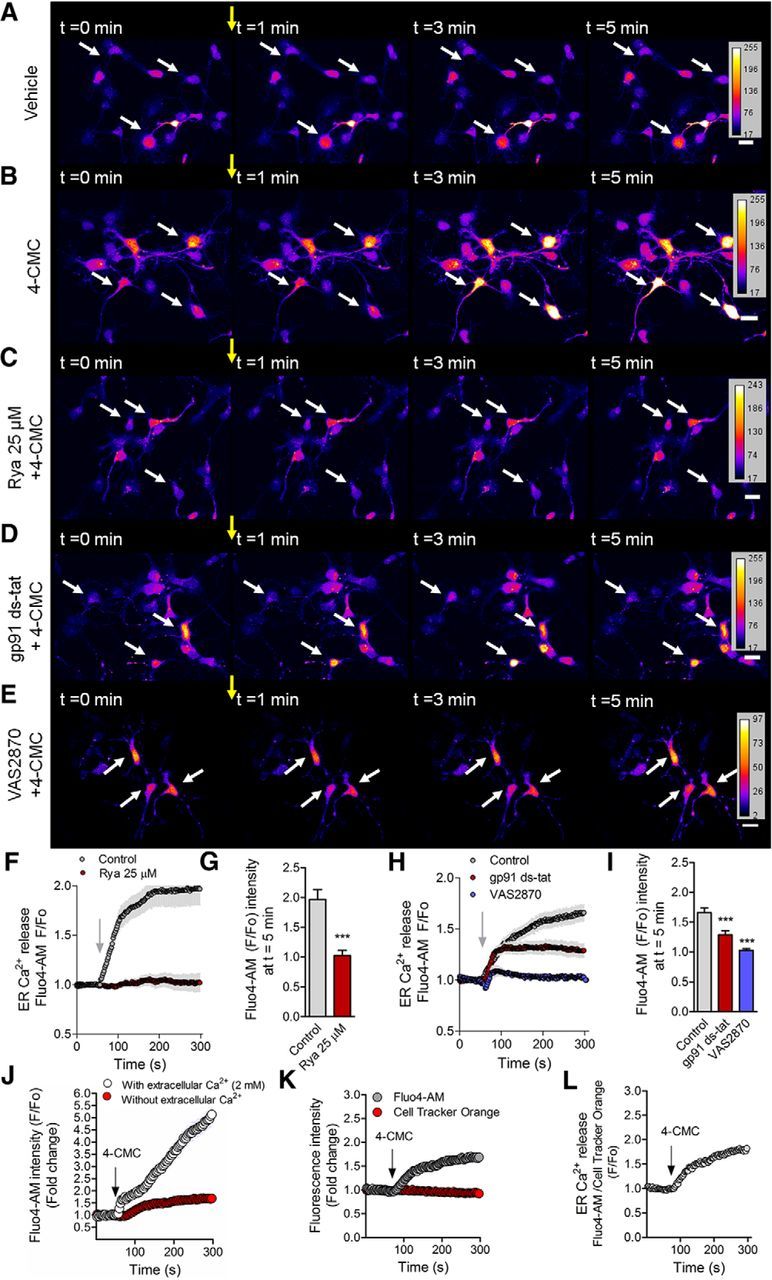
Functional coupling between NOX and RyR-mediated Ca2+ release. Neurons (2 DIV) were loaded with Fluo4-AM to visualize cytosplasmic Ca2+ in live neurons. A–E, Representative time-lapse imaging for the vehicle of 4-CMC (ethanol; A), 4-CMC (750 μm; B), and neurons pretreated with ryanodine (25 μm; C), gp91 ds-tat (5 μm; D), and VAS2870 (5 μm; E) for 1 h before 4-CMC addition. F, Quantification of RyR-mediated Ca2+ release of B and C. G, Quantification of the F/F0 ratio at t = 5 min of F. ***p < 0.001 versus control, Student's t test. H, Quantification of RyR-mediated Ca2+ release after 4-CMC stimulation in control, gp91 ds-tat (5 μm), and VAS2870 (5 μm) of B, D, and E. I, Quantification of the F/F0 ratio at t = 5 min in control of H. ***p < 0.001 versus control, ANOVA, Dunnett's post test. J, Fluo4-AM fluorescence after 4-CMC stimulation in Ca2+-free or Ca2+-containing medium (2 mm) during the recording of Ca2+ signals. K, Fluo4-AM and Cell Tracker Orange (volume control) fluorescence before and after 4-CMC stimulation in 2 DIV neurons. L, Normalization of the Fluo4-AM signal by Cell Tracker Orange fluorescence during the recording. Results are from 3 different independent cultures (n = 3). A total of 60 neurons were analyzed for each condition. Scale bar, 20 μm.
Figure 4.
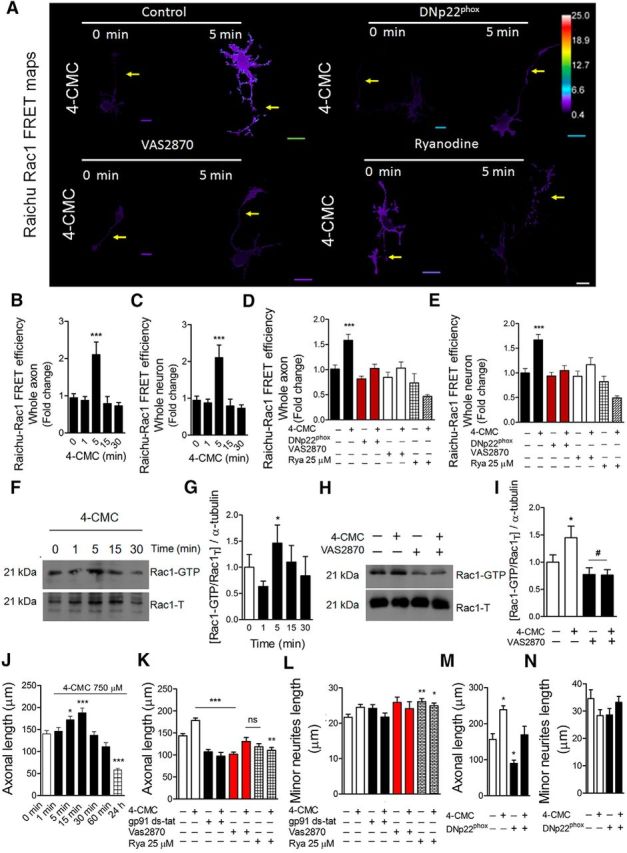
The NOX complex promotes both Rac1 activation and axonal extension through a RyR-dependent mechanism. A–E, Hippocampal neurons of 1 DIV were transfected with the Raichu-Rac1 probe to measure the FRET efficiency after RyR stimulation in control and NOX-inhibited neurons. B, C, FRET efficiency in neurons stimulated with 4-CMC (750 μm) for 0, 1, 5 15, and 30 min in the whole neuron (B) and whole axon (C). ***p < 0.001 versus 0 min, Kruskal–Wallis test, 21 neurons analyzed. D, E, Raichu-Rac1 neurons were cotransfected with the DNp22phox construct or treated with VAS2870 (5 μm) alone. Ryanodine (25 μm) treatment was performed as with VAS2870. Analyses were undertaken at the whole-neuron (D) and axon (E) levels. ***p < 0.001 versus control, ANOVA, Dunnett's post test (21 neurons were analyzed). F, Pull-down analyses were performed to determine Rac1-GTP levels after 4-CMC stimulation in embryonic cortical neurons of 2 DIV. G, Densitometry of Western blots of Rac1-GTP shown in F. *p < 0.05 versus control, ANOVA Dunnett' post test. H, Representative Western blots of pull-down analysis to determine Rac1-GTP levels after 4-CMC stimulation in control and VAS2870-treated neurons (5 μm) of 2 DIV. I, Densitometry of Western blots shown in H, *p < 0.05 versus control, ANOVA, Dunnett's post test. J, Hippocampal neurons were cultured for 1 d and then stimulated with 4-CMC at different time points. Then, neurons were fixed at 3 DIV to measure axonal length. *p < 0.05 versus 0 min, ***p < 0.001 versus 0 min. ANOVA, Dunnett's post test. K, L, Hippocampal neurons were treated with gp91 ds-tat (5 μm), VAS2870 (5 μm), and ryanodine (25 μm) after 1 DIV and then stimulated with 4-CMC (750 μm) for 15 min. Neurons were fixed at 3 DIV to measure axonal length (K) and minor neurite length (L). **p < 0.01 versus control, ***p < 0.001 versus control, ns = nonsignificant, ANOVA, Dunnett's post test (90 neurons were analyzed for each condition). M, N, Neurons were transfected after plating with DNp22phox and treated with 4-CMC for 15 min after 1 d of culture. After 4-CMC withdrawal, neurons were fixed at 3 DIV. *p < 0.05 versus control, ANOVA, Dunnett's post test (45 neurons were analyzed for each condition). Results are from 3 different independent cultures (n = 3). Scale bar, 20 μm.
Figure 5.

RyR stimulation induces H2O2 production through the NOX complex by a Rac1-dependent mechanism. Neurons were transfected after 1 DIV with the HyPer biosensor alone (control) or cotransfected with the DNp22phox or Rac1T17N (dominant-negative version of Rac1) constructs. After 1 d of expression, neurons were treated with 4-CMC. A, H2O2 maps after 750 μm 4-CMC stimulation for 5 and 15 min in control, DNp22phox-, and Rac1T17N-transfected neurons. B, Quantification of the HyPer-H2O2 levels of A. *p < 0.05, **p < 0.01, ***p < 0.001 versus control (white column), ANOVA, Dunnett's post test (15 neurons were analyzed for each condition). C, RyR-mediated Ca2+ release in control and NSC 23766-treated neurons (100 μm). D, Quantification of the F/F0 ratio at t = 5 min of C. ***p < 0.001 versus control, Student's t test (a total of 60 neurons were analyzed). E, Neurons (2 DIV) expressing the HyPer biosensor were treated with NSC 23766 (100 μm) for 1 h and then stimulated with 4-CMC for 15 min to measure HyPer-H2O2 levels. ***p < 0.001 versus control, ANOVA, Dunnett's post test. Results are from 3 different independent cultures (n = 3). Scale bar, 20 μm.
Figure 6.
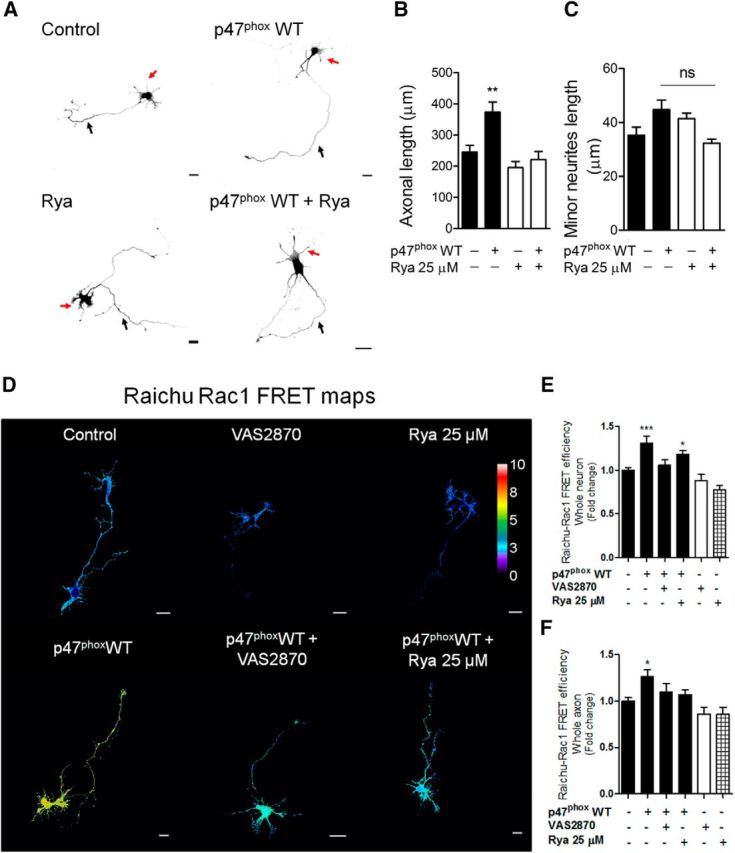
NOX gain-of-function increases both axonal development and Rac1 activity through a RyR-dependent mechanism. A–C, Neurons were cultured and transfected immediately after plating with GFP alone (control) or cotransfected with the p47phox WT construct. After 1 d of culture, neurons were treated with ryanodine (25 μm) and fixed at 3 DIV. A, Representative neurons after p47phox WT expression and ryanodine treatment. B, C, Quantification of axonal length (B) and minor neurite length (C) of neurons shown in A. B, **p < 0.01 versus control, Kruskal–Wallis test (n = 3). C, ns = nonsignificant, ANOVA, Dunnett's post test (45 were neurons analyzed for each condition). D, E, Neurons (1 DIV) were transfected with the Raichu-Rac1 probe alone (control) or together with the p47phox WT construct. After 1 d of expression, neurons were treated with VAS2870 (5 μm) or ryanodine (25 μm) for 1 h. Neurons were then fixed to evaluate FRET efficiency. D, FRET maps. E, F, Quantification of the FRET efficiency in control, VAS2870-, and ryanodine-treated neurons in whole neuron (E) and axon (F). *p < 0.05, ***p < 0.001 versus control, ANOVA, Dunnett's post test (n = 3; 20 neurons were analyzed for each condition). Results are from 3 different independent cultures (n = 3). Scale bar, 20 μm.
Figure 7.
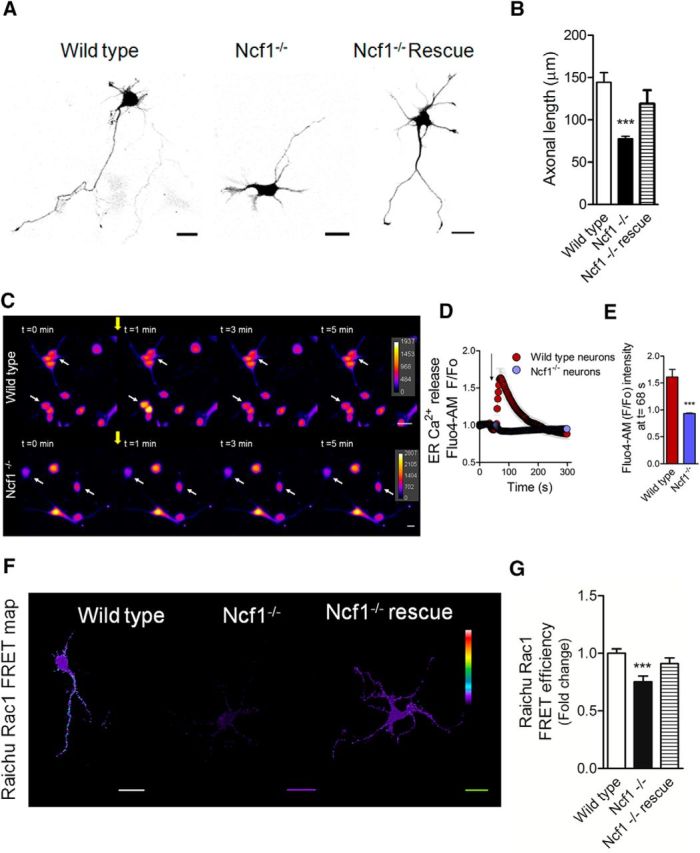
Measurement of axonal length, RyR-mediated Ca2+ release, and Rac1 activity in Ncf1−/− neurons. P0 neurons isolated from Ncf1−/− hippocampi were cultured for 2 DIV to measure axonal length, Ca2+ release, and Rac1 activity. A, Representative wild-type and Ncf1−/− neurons at 2 DIV. For rescue experiments, Ncf1−/− cultured neurons were transfected immediately after plating with the p47phox WT construct. B, Axonal length of neurons shown in A. ***p < 0.001 versus wild-type neurons, Kruskal–Wallis test (21 neurons were analyzed for each condition). C, Representative time-lapse images corresponding to the RyR-mediated Ca2+ release of wild-type and Ncf1−/− neurons after 4-CMC (750 μm) stimulation. The yellow arrow indicates the time at which 4-CMC was added in each case. White arrows show representative neurons followed for Fluo4-AM fluorescence intensity throughout the time of the recording. D, Quantification of time-lapse shown in C. E, Quantification of F/F0 ratio at t = 68 s, corresponding to the maximum amplitude achieved after 4-CMC addition in wild-type neurons. ***p < 0.001 versus Ncf1−/− neurons, Mann–Whitney test (45 neurons were analyzed for each condition). F, G, Wild-type and Ncf1−/− neurons of 1 DIV were transfected with the Raichu-Rac1 FRET probe and fixed at 2 DIV. For rescue experiments, Ncf1−/− neurons were cotransfected with the p47phox WT construct. F, Representative FRET maps of wild-type, Ncf1−/−, and Ncf1−/− rescued neurons. G, FRET efficiency quantification (21 neurons were analyzed for each condition). Results are from 3 different independent cultures (n = 3). Scale bar, 20 μm.
Statistics.
Results are the mean of three independent cultures (n = 3) ± SEM. The number of neurons analyzed for each experiment (n) is indicated in the figure legends. Shapiro–Wilk normalcy test was used to evaluate normal distribution of datasets. Student's t tests or ANOVA were performed for parametric data. Mann–Whitney or Kruskal–Wallis tests were used for nonparametric data. All analyses were performed using GraphPad Prism 5 software.
Results
NOX gain-of-function promotes axonal development during maturation of hippocampal neurons
Our previous work showed that NOX loss-of-function inhibits axon elongation through an actin-associated mechanism (Wilson et al., 2015). Here, in the first set of experiments, we explored a NOX gain-of-function approach to scrutinize effects upon axonal development. To this aim, neurons were transfected immediately after plating with a p47phox wild-type coding vector (p47phox WT), which enhances ROS production via NOX2 (Roepstorff et al., 2008; Fig. 1). To observe transfection, neurons were cotransfected with GFP. After 3 DIV, neurons were fixed for morphological analysis as described previously (Wilson et al., 2015). Neurons expressing p47phox WT exhibited axons that were 50% longer than control neurons (GFP-only transfected; Fig. 1A,B, black arrows). Minor neurites were also enlarged after p47phox WT expression, although to a lesser extent than observed in axons (Fig. 1A,C, red arrows). Both control and p47phoxWT neurons displayed axons that were, on average, 9–10 times longer than minor neurites, suggesting that neuronal polarity acquisition was not affected and that an axon was clearly distinguishable from minor neurites (Fig. 1B,C). To evaluate ROS production, neurons were cotransfected with both p47phoxWT and the H2O2 sensor HyPer (Belousov et al., 2006) after 1 DIV and later fixed at 2 DIV. Control neurons were only transfected with HyPer. Neurons expressing p47phox WT exhibited a global increase in H2O2 levels (Fig. 1D–F). The HyPer biosensor was activated robustly in neurons treated with H2O2 (500 μm). When neurons were incubated with antioxidants, the HyPer signal was attenuated significantly (Fig. 1G). Subsequently, neurons expressing p47phoxWT were treated with the NOX2 inhibitor VAS2870 (5 μm), a drug that blocks the translocation of p47phox to the plasma membrane (Altenhöfer et al., 2012). The treatment with VAS2870 was performed after 1 DIV (when most neurons are at stage 2 of polarity; Wilson et al., 2015); subsequently, neurons were fixed after 2 and 3 d for both H2O2 measurement and axonal length analysis, respectively (Fig. 1H–M). Treatment with VAS2870 inhibited both axonal extension (Fig. 1H–I) and the enhancement in H2O2 levels after p47phox WT expression (Fig. 1K–M), suggesting a NOX2-dependent mechanism. Together, these data support our hypothesis that NOX2-derived ROS are instrumental for axonal development and polarization of hippocampal neurons.
ER Ca2+ channels mediate neuronal polarization and axonal extension
Having shown that NOX activation promotes axonal development, we explored a molecular mechanism able to correlate the generation of short-lived ROS with a long-lasting process such as axonal development. Based on the following evidence, we focused our attention on Ca2+ release from the ER as a possible cellular intermediate. First, Ca2+ is a second messenger that has been linked previously to axon and neuritic elongation (Henley and Poo, 2004; Wayman et al., 2004; Estrada et al., 2006; Zheng and Poo, 2007; Davare et al., 2009). Second, previous reports suggest that ER Ca2+ release, which is mediated by both the inositol 1,4,5-triphosphate receptor (IP3R) and RyR (Bardo et al., 2006), depends on NOX activity (Bedard and Krause, 2007). Particularly, RyR has cysteine residues that are sensitive to redox species that are oxidized to maintain normal activity (Aracena-Parks et al., 2006; Espinosa et al., 2006; Hidalgo et al., 2006; Donoso et al., 2011). Considering this, we undertook experiments to reveal the contribution of both IP3R and RyR in the establishment of neuronal polarity and axonal development. Both RyR2 and IP3R1, the main embryonic neuronal isoforms (Bardo et al., 2006), were expressed in E18.5 hippocampus and cerebral cortex (Fig. 2A,C). In addition, both RyR2 and IP3R1 were found in the soma and minor neurites of stage 2 cultured neurons (white arrows, Fig. 2B,D), as well as in the growing axon (yellow arrows, Tau-1-positive neurites) and the axonal growth cone (asterisks) of stage 3 cultured neurons (Fig. 2B,D). We then addressed the contribution of Ca2+ release mediated by these receptors to axonal extension. Toward this aim, two parameters were evaluated: the polarity stages after 1 DIV and the axonal length after 3 DIV (Wilson et al., 2015). To evaluate neuronal polarity, neurons were treated with xestospongin C (3 μm) and ryanodine (25 μm) after plating to block IP3R and RyR, respectively (Fig. 2E,G). After 24 h, neurons were fixed to evaluate the acquisition of polarity (Wilson et al., 2015). We found that both xestospongin C and ryanodine treatments induced a significant increase in the number of neurons at stage 1 (Fig. 2E,G). Moreover, xestospongin C treatment also decreased the proportion of neurons at stage 3 (Fig. 2G). We then evaluated the contribution of RyR2 and IP3R1 receptors to axonal development. Neurons were treated with xestospongin C and ryanodine after 1 DIV (when most cultured neurons are at stage 2) and fixed at 3 DIV to evaluate axonal growth (Fig. 2F,H). Both ryanodine and xestospongin C treatments decreased axonal growth compared with control neurons, suggesting that both receptors contribute to axonal extension. Together, these data support the hypothesis that both IP3R and RyR2 promote neuronal polarization and axonal development.
Functional coupling between NOX and RyR-mediated Ca2+ release
To determine whether NOX activity was coupled to RyR activity, 2 DIV neurons were loaded with Fluo4-AM (a Ca2+-sensitive probe) to measure cytoplasmic Ca2+ levels after RyR stimulation with the agonist 4-CMC (Westerblad et al., 1998) in Ca2+-free HBSS medium. Figure 3, A–E, shows representative Ca2+ signals in neurons before (0–1 min) and after (1–5 min) 4-CMC (750 μm) stimulation (yellow arrows). Control neurons release Ca2+ from the ER after the addition of 4-CMC (Fig. 3B), which was abolished in neurons that had been treated previously with ryanodine (25 μm) for 1 h before Ca2+ recording (Fig. 3C,F). The F/F0 ratio at t = 5 min was reduced significantly after ryanodine treatment, suggesting that 4-CMC stimulates RyR specifically in our model (Fig. 3G). Next, we explored the link between NOX and RyR-mediated Ca2+ release. To this aim, neurons were incubated with the NOX inhibitors gp91 ds-tat (5 μm; Rey et al., 2001; Fig. 3D) and VAS2870 (5 μm; Fig. 3E) for 1 h before Ca2+ recordings. Both gp91 ds-tat and VAS2870 inhibited RyR-mediated Ca2+ release after 4-CMC stimulation (arrows in Fig. 3D,E and quantification in Fig. 3H,I). Inhibition with gp91 ds-tat was less efficient than VAS2870, probably because gp91 ds-tat is less soluble than VAS2870, and this may affect NOX inhibition (Altenhöfer et al., 2012). Fluo4-AM fluorescence was also increased after stimulation with 4-CMC when the recording was performed in a 2 mm Ca2+-containing medium (Tyrode buffer), discarding the possibility that the system may be pushed toward an ER-dependent response in the absence of Ca2+ (Fig. 3J). We also evaluated whether changes in Fluo4-AM induced by 4-CMC were due to alterations in neuronal volume. For that purpose, neurons were incubated with Cell Tracker Orange, a probe that stains the cell uniformly (Münk et al., 2002). Cell Tracker Orange fluorescence was not modified after stimulation with 4-CMC, demonstrating that cellular volume did not change during our observations (Fig. 3K,L). Collectively, these data support the notion that basal activity of NOX is needed to support RyR-mediated Ca2+ release in polarizing neurons.
Functional coupling between NOX and RyR promotes Rac1 activation and axonal development
A previous report suggests that RyR-mediated Ca2+ release activates Rac1 in cerebellar granule neurons, reaching a peak after 3 min of RyR stimulation (Jin et al., 2005). Considering that axonal growth is a Rac1-dependent process (González-Billault et al., 2012), we evaluated whether the coupling between NOX and RyR may regulate the activity of Rac1 in our model. To this aim, neurons were transfected after 1 DIV with the FRET probe Raichu-Rac1 (Nakamura et al., 2006). After 1 d of expression, neurons were treated with 4-CMC (750 μm) for 1, 5, 15, and 30 min and then fixed to evaluate FRET efficiency using confocal microscopy (Fig. 4A–C). Neurons reached a 2-fold peak in FRET efficiency after 5 min of stimulation with 4-CMC in both whole neurons and in the axonal compartment (Fig. 4B,C). Similarly, pull-down analysis confirmed that, after 5 min of 4-CMC stimulation, neurons increased the levels of Rac1-GTP (Fig. 4F,G). Next, 1 DIV neurons were transfected with a vector encoding the P156Q p22phox (DNp22phox) isoform, a mutant version of p22phox that decreases NOX-mediated ROS (Kawahara et al., 2005; Wilson et al., 2015). To determine Rac1 activity, these neurons were cotransfected with the Raichu-Rac1 FRET probe (Fig. 4D,E). One day after transfection, neurons were stimulated with 4-CMC for 5 min to compare Raichu-Rac1 FRET efficiency between DNp22phox and control neurons. In addition, neurons transfected with the FRET probe were treated with VAS2870 and then stimulated with 4-CMC for 5 min (Fig. 4D,E). Both genetic (DNp22phox) and pharmacological (VAS2870) inhibition of NOX blocked 4-CMC-induced Rac1 activation (Fig. 4D,E), which was reproduced by pull-down assays in cortical neurons (Fig. 4H,I). Together, these results indicate that the functional coupling between NOX and RyR is instrumental for the activity of Rac1 in neurons.
Based on these data, we examined the contribution of the functional coupling between NOX and RyR toward axonal development of hippocampal neurons. Because sustained ER Ca2+ release, as well as massive Ca2+ influx, promote neurodegeneration (Villegas et al., 2014; Bernard-Marissal et al., 2015; Vargas et al., 2015), we analyzed different time points after 4-CMC treatment to stimulate RyR-mediated Ca2+ release. One DIV neurons were treated with 4-CMC in short-term (0, 1, 5, 15, and 30 min) and long-term stimulations (1 and 24 h; Fig. 4J). 4-CMC was then washed and neurons were fixed at 3 DIV. We found that, after 15 min of 4-CMC stimulation, neurons developed longer axons compared with nonstimulated neurons (Fig. 4J). However, this phenotype disappeared after 30 min of treatment. Furthermore, after 24 h of stimulation, axons were even shorter than those of control neurons, suggesting a toxic effect after long-term stimulation (Fig. 4J). The next set of experiments was conducted using 15 min of stimulation with 4-CMC (Fig. 4J). Neurons cultured for 1 d were treated with the NOX inhibitors gp91 ds-tat and VAS2870 for 1 h and then stimulated with 4-CMC for 15 min. After 4-CMC withdrawal, the corresponding NOX inhibitors were re-added and neurons were fixed at 3 DIV to measure neurite length (Fig. 4K,L). NOX inhibitors blocked the axonal growth induced by 4-CMC (Fig. 4K) without affecting the length of minor neurites (Fig. 4L). Axonal extension was blocked in neurons pretreated with ryanodine (25 μm; Fig. 4K), suggesting a RyR-dependent mechanism. In addition, neurons were transfected immediately after plating with the DNp22phox construct (Fig. 4M,N). One day after transfection, neurons were stimulated with 4-CMC for 15 min and then cultured to complete 3 DIV. DNp22phox neurons did not exhibit longer axons after 4-CMC stimulation (Fig. 4M), whereas minor neurite length was not affected at 3 DIV (Fig. 4N). Together, these results suggest that RyR-mediated Ca2+ release promotes axonal development during hippocampal maturation through a NOX-dependent mechanism.
RyR stimulation induces H2O2 synthesis through NOX and Rac1
Because Rac1 is one of the subunits of the NOX complex (Lambeth, 2004) and based upon our data so far, we investigated whether RyR-mediated Ca2+ release could stimulate ROS production by NOX through a Rac1-dependent mechanism. To this aim, 1 DIV neurons were cotransfected with a vector encoding Rac1T17N, the dominant-negative isoform of Rac1 (Ridley et al., 1992), and HyPer to evaluate H2O2 levels after 4-CMC stimulation. Moreover, neurons were also cotransfected with HyPer and DNp22phox constructs to determine ROS levels. After 15 min of 4-CMC stimulation, an increase in H2O2 levels was detected in control neurons (only transfected with HyPer). However, this was blocked after either DNp22phox or Rac1T17N expression (Fig. 5B). To confirm the contribution of Rac1 to RyR-mediated Ca2+ release, 2 DIV neurons were treated with the Rac1 inhibitor NSC 23766 (100 μm) for 1 h (Dwivedi et al., 2010). Subsequently, neurons were loaded with Fluo4-AM to study RyR-mediated Ca2+ release after stimulation with 4-CMC (Fig. 5C,D). NSC 23766 partially blocked Ca2+ release after 4-CMC, suggesting that Rac1 is involved in the RyR-mediated Ca2+ release response. Moreover, NSC 23766 treatment decreased HyPer-H2O2 levels, reinforcing the observation of Rac1 involvement obtained using T17N Rac1 (Fig. 5A,B). Together, these data propose that RyR-mediated Ca2+ release promotes H2O2 synthesis through the NOX complex in a mechanism that depends on Rac1, suggesting a feedforward mechanism between NOX and RyR-mediated Ca2+ release in polarizing neurons.
NOX gain-of-function increases hippocampal axonal development through RyR
To determine whether NOX-derived ROS increase axonal length through RyR activity, hippocampal neurons were transfected immediately after plating with p47phox WT (Fig. 6A–C). After 1 d, neurons were treated with ryanodine (25 μm) and then fixed at 3 DIV. Ryanodine treatment reduced the stimulatory effect of p47phox WT expression on axonal development (Fig. 6B) without affecting minor neurite length (Fig. 6C). We then used the same experimental paradigm explained above to evaluate Rac1 activity. Neurons that expressed p47phox WT showed an increase in Rac1 FRET efficiency, which was blocked after VAS2870 or ryanodine addition (Fig. 6D–F), suggesting that NOX2 activates Rac1 in a mechanism that involves RyR.
Ncf1−/− hippocampal neurons display delayed axonal development and fail to release Ca2+ mediated by RyR
In the final set of experiments, we evaluated axonal development, RyR-mediated Ca2+ release, and Rac1 activity in hippocampal neurons derived from Ncf1−/− (p47phox−/−) mice (Fig. 7). To this aim, wild-type and Ncf1−/− P0 neurons were cultured for 2 d to measure axonal development. To perform rescue experiments, neurons were transfected immediately after plating with the p47phox WT construct and GFP (transfection control). Neurons were fixed and axonal length was measured. We found that Ncf1−/− neurons displayed shorter axons than wild-type neurons, which were restored after p47phox WT expression (Fig. 7A,B). In addition, Ncf1−/− neurons failed to release Ca2+ after RyR stimulation with 4-CMC (Fig. 7C–E). Finally, Ncf1−/− neurons presented decreased Rac1 activity, which was rescued after p47phox WT expression (Fig. 7F,G).
Collectively, our data suggest that the NOX complex promotes axonal development during maturation of hippocampal neurons through a feedforward mechanism that links the NOX complex, RyR-mediated Ca2+ release, and Rac1 activity.
Discussion
Our work proposes that the NOX complex promotes axonal development of hippocampal neurons through a RyR-mediated Ca2+ release mechanism. Ncf1−/− neurons have shorter axons compared with wild-type neurons, as well as a reduction in basal Rac1 activity, which is consistent with the notion that NOX promotes axonal extension involving the actin cytoskeleton. Together, this evidence suggests that NOX-derived ROS are needed for neuronal development, polarization, and axonal specification in hippocampal neurons. Similar results in other neuronal models suggest that NOX and ROS are required for neurite outgrowth (Munnamalai and Suter, 2009; Munnamalai et al., 2014; Olguin-Albuerne and Moran, 2015). Moreover, the differentiation of neuronal stem cells into neurons also depends on ROS signaling (Dickinson et al., 2011; Forsberg et al., 2013; Forsberg and Di Giovanni, 2014). Based on these findings, it appears that physiological ROS levels are indeed needed to regulate both neuronal differentiation and polarization in the CNS.
Because the lifetime of ROS is extremely short and dynamic, we conceived ROS as being signaling molecules that are able to regulate downstream pathways involved in axonal development. This hypothesis was elaborated based on the following evidence. First, ROS regulate ER Ca2+ release in muscle and neuronal cells (Aracena-Parks et al., 2006; Espinosa et al., 2006; Hidalgo et al., 2006; Zhang and Forscher, 2009; Donoso et al., 2011; Riquelme et al., 2011). Second, ER Ca2+ release, principally mediated by RyR, promotes Rac1 activation in several cell types (Fleming et al., 1999; Price et al., 2003; Jin et al., 2005). Finally, Rac1 is a molecular determinant for axonal growth (González-Billault et al., 2012). Whereas high levels of intracellular Ca2+ lead to growth cone collapse, physiological levels are required for the normal dynamics of this structure (Gomez and Zheng, 2006). In this work, we found that both IP3R and RyR are needed for the acquisition of proper neuronal polarity and axonal development. Both receptors were detected in neurons during the early stages of neuronal polarization and both segregated to the emerging axon. Our data suggest that RyR is functional at this polarization stage and depends on NOX activity. Electrical stimulation of mature neurons promotes RyR-mediated Ca2+ release, which is blocked after N-acetyl-cysteine treatment, a general antioxidant (Riquelme et al., 2011). A previous study suggested that VAS2870 has potential off targets in skeletal muscle cells (Sun et al., 2012). However, we used a concentration that is three times lower than that reported previously. Moreover, both Ncf1−/− and gp91 ds-tat-treated neurons failed to release Ca2+ mediated by the RyR. Together, our data demonstrate that the NOX complex is a source of ROS that mediates RyR-dependent Ca2+ release in polarizing neurons.
We found that RyR stimulation increased the activity of Rac1 after 5 min of 4-CMC treatment, which was blocked in NOX-inhibited neurons (Fig. 4D,E,H,I). A similar time frame has been described to enhance both Rac1 and Cdc-42 activities in HEK293 cells and cerebellar granule neurons (Jin et al., 2005). In vitro assays suggest that ER Ca2+ release activates Tiam1 through PKC/CaMKII-dependent phosphorylation (Fleming et al., 1999; Price et al., 2003), two proteins that promote axonal growth (Gomez and Zheng, 2006; Nakamuta et al., 2011). In this work, we found that RyR stimulation enhanced the axonal length of hippocampal neurons, which was abolished after NOX inhibition. Our data also suggest that the activation of Rac1 after RyR stimulation increases ROS derived from NOX. RyR-mediated Ca2+ release promotes H2O2 synthesis through NOX in hippocampal mature neurons (Riquelme et al., 2011). Inhibition of Rac1 using NSC 23766 or a dominant-negative form of Rac1 significantly reduced the response of RyR to 4-CMC. The differences observed between these two means of inactivation may be due to their different modes of action. Considering these and previously published findings, we suggest that a feedforward mechanism among NOX, RyR-mediated Ca2+ release, and Rac1 is needed for axonal development. Finally, the gain-of-function of NOX after p47phoxWT expression did not increase the development of axons in neurons treated with 25 μm ryanodine, suggesting that NOX requires RyR to promote the developmental growth of hippocampal neurons. However, we cannot rule out that other signaling pathways that might be regulated by redox signaling may be involved in axonal growth. In fact, both F-actin and microtubules are susceptible to redox posttranslational modifications that affect the dynamic properties of the cytoskeleton (Dalle-Donne et al., 2001, 2002, 2003; Landino et al., 2002, 2004a, 2004b; Chowdhury et al., 2009; Hung and Terman, 2011; Terman and Kashina, 2013; Wilson et al., 2016) and this may have a direct effect on the morphology of neurons during the early stages of the establishment of neuronal polarity. Along similar lines, the PI3K/Akt pathway, which is involved in axonal extension (Cosker et al., 2008), depends on ROS levels (Forsberg et al., 2013), which allows us to hypothesize that other pathways involved in axonal growth are regulated by redox balance.
In summary, our data suggest that ROS promote axonal development through a RyR-mediated Ca2+ release mechanism that affects Rac1 activity. It seems that highly regulated ROS levels are instrumental in shaping neurons, which is crucial for the development of mature neuronal functions such as synapses and neurotransmission.
Footnotes
This work was supported by the Comisio′n Nacional de Investigacio′n Científica y Tecnolo′gica (CONICYT Anillo ACT 1114, Fondecyt 1140325 and FONDAP 15150012 to C.G.-B. and CONICYT Doctoral Fellowship Grant PFCHA 21120221 to C.W. C.W. is the recipient of a Whood–Whelan Research Fellowship from International Union of Biochemistry and Molecular Biology (IUBMB). We thank Dr Frederik Vilhardt (Copenhagen University, Denmark) for the p47phox WT construct and Dr Michael Handford for proof-reading the manuscript.
The authors declare no competing financial interests.
References
- Adasme T, Paula-Lima A, Hidalgo C. Inhibitory ryanodine prevents ryanodine receptor-mediated Ca(2)(+) release without affecting endoplasmic reticulum Ca(2)(+) content in primary hippocampal neurons. Biochem Biophys Res Commun. 2015;458:57–62. doi: 10.1016/j.bbrc.2015.01.065. [DOI] [PubMed] [Google Scholar]
- Altenhöfer S, Kleikers PW, Radermacher KA, Scheurer P, Rob Hermans JJ, Schiffers P, Ho H, Wingler K, Schmidt HH. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci. 2012;69:2327–2343. doi: 10.1007/s00018-012-1010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aracena-Parks P, Goonasekera SA, Gilman CP, Dirksen RT, Hidalgo C, Hamilton SL. Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J Biol Chem. 2006;281:40354–40368. doi: 10.1074/jbc.M600876200. [DOI] [PubMed] [Google Scholar]
- Bardo S, Cavazzini MG, Emptage N. The role of the endoplasmic reticulum Ca2+ store in the plasticity of central neurons. Trends Pharmacol Sci. 2006;27:78–84. doi: 10.1016/j.tips.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Belousov VV, Fradkov AF, Lukyanov KA, Staroverov DB, Shakhbazov KS, Terskikh AV, Lukyanov S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat Methods. 2006;3:281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- Bernard-Marissal N, Médard JJ, Azzedine H, Chrast R. Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain. 2015;138:875–890. doi: 10.1093/brain/awv008. [DOI] [PubMed] [Google Scholar]
- Bradke F, Dotti CG. The role of local actin instability in axon formation. Science. 1999;283:1931–1934. doi: 10.1126/science.283.5409.1931. [DOI] [PubMed] [Google Scholar]
- Cáceres A, Ye B, Dotti CG. Neuronal polarity: demarcation, growth and commitment. Curr Opin Cell Biol. 2012;24:547–553. doi: 10.1016/j.ceb.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe CU, Harrison KD, Grant W, Ehrlich BE. Functional coupling of chromogranin with the inositol 1,4,5-trisphosphate receptor shapes calcium signaling. J Biol Chem. 2004;279:35551–35556. doi: 10.1074/jbc.M311261200. [DOI] [PubMed] [Google Scholar]
- Chowdhury G, Dostalek M, Hsu EL, Nguyen LP, Stec DF, Bradfield CA, Guengerich FP. Structural identification of Diindole agonists of the aryl hydrocarbon receptor derived from degradation of indole-3-pyruvic acid. Chem Res Toxicol. 2009;22:1905–1912. doi: 10.1021/tx9000418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosker KE, Shadan S, van Diepen M, Morgan C, Li M, Allen-Baume V, Hobbs C, Doherty P, Cockcroft S, Eickholt BJ. Regulation of PI3K signalling by the phosphatidylinositol transfer protein PITPalpha during axonal extension in hippocampal neurons. J Cell Sci. 2008;121:796–803. doi: 10.1242/jcs.019166. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Giustarini D, Gagliano N, Lusini L, Milzani A, Di Simplicio P, Colombo R. Actin carbonylation: from a simple marker of protein oxidation to relevant signs of severe functional impairment. Free Radic Biol Med. 2001;31:1075–1083. doi: 10.1016/S0891-5849(01)00690-6. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Giustarini D, Gagliano N, Di Simplicio P, Colombo R, Milzani A. Methionine oxidation as a major cause of the functional impairment of oxidized actin. Free Radic Biol Med. 2002;32:927–937. doi: 10.1016/S0891-5849(02)00799-2. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Giustarini D, Rossi R, Colombo R, Milzani A. Reversible S-glutathionylation of Cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic Biol Med. 2003;34:23–32. doi: 10.1016/S0891-5849(02)01182-6. [DOI] [PubMed] [Google Scholar]
- Davare MA, Fortin DA, Saneyoshi T, Nygaard S, Kaech S, Banker G, Soderling TR, Wayman GA. Transient receptor potential canonical 5 channels activate Ca2+/calmodulin kinase Igamma to promote axon formation in hippocampal neurons. J Neurosci. 2009;29:9794–9808. doi: 10.1523/JNEUROSCI.1544-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson BC, Peltier J, Stone D, Schaffer DV, Chang CJ. Nox2 redox signaling maintains essential cell populations in the brain. Nat Chem Biol. 2011;7:106–112. doi: 10.1038/nchembio.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoso P, Sánchez G, Bull R, Hidalgo C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front Biosci (Landmark Ed) 2011;16:553–567. doi: 10.2741/3705. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi S, Pandey D, Khandoga AL, Brandl R, Siess W. Rac1-mediated signaling plays a central role in secretion-dependent platelet aggregation in human blood stimulated by atherosclerotic plaque. J Transl Med. 2010;8:128. doi: 10.1186/1479-5876-8-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa A, Leiva A, Pena M, Müller M, Debandi A, Hidalgo C, Carrasco MA, Jaimovich E. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J Cell Physiol. 2006;209:379–388. doi: 10.1002/jcp.20745. [DOI] [PubMed] [Google Scholar]
- Estrada M, Uhlen P, Ehrlich BE. Ca2+ oscillations induced by testosterone enhance neurite outgrowth. J Cell Sci. 2006;119:733–743. doi: 10.1242/jcs.02775. [DOI] [PubMed] [Google Scholar]
- Fleming IN, Elliott CM, Buchanan FG, Downes CP, Exton JH. Ca2+/calmodulin-dependent protein kinase II regulates Tiam1 by reversible protein phosphorylation. J Biol Chem. 1999;274:12753–12758. doi: 10.1074/jbc.274.18.12753. [DOI] [PubMed] [Google Scholar]
- Forsberg K, Di Giovanni S. Cross Talk between Cellular Redox Status, Metabolism, and p53 in Neural Stem Cell Biology. Neuroscientist. 2014;20:326–342. doi: 10.1177/1073858413514634. [DOI] [PubMed] [Google Scholar]
- Forsberg K, Wuttke A, Quadrato G, Chumakov PM, Wizenmann A, Di Giovanni S. The tumor suppressor p53 fine-tunes reactive oxygen species levels and neurogenesis via PI3 kinase signaling. J Neurosci. 2013;33:14318–14330. doi: 10.1523/JNEUROSCI.1056-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez TM, Zheng JQ. The molecular basis for calcium-dependent axon pathfinding. Nat Rev Neurosci. 2006;7:115–125. doi: 10.1038/nrn1844. [DOI] [PubMed] [Google Scholar]
- Gomez TM, Robles E, Poo M, Spitzer NC. Filopodial calcium transients promote substrate-dependent growth cone turning. Science. 2001;291:1983–1987. doi: 10.1126/science.1056490. [DOI] [PubMed] [Google Scholar]
- González-Billault C, Munoz-Llancao P, Henríquez DR, Wojnacki J, Conde C, Cáceres A. The role of small GTPases in neuronal morphogenesis and polarity. Cytoskeleton (Hoboken) 2012;69:464–485. doi: 10.1002/cm.21034. [DOI] [PubMed] [Google Scholar]
- Henley J, Poo MM. Guiding neuronal growth cones using Ca2+ signals. Trends Cell Biol. 2004;14:320–330. doi: 10.1016/j.tcb.2004.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henríquez DR, Bodaleo FJ, Montenegro-Venegas C, González-Billault C. The light chain 1 subunit of the microtubule-associated protein 1B (MAP1B) is responsible for Tiam1 binding and Rac1 activation in neuronal cells. PLoS One. 2012;7:e53123. doi: 10.1371/journal.pone.0053123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo C, Sánchez G, Barrientos G, Aracena-Parks P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S-glutathionylation. J Biol Chem. 2006;281:26473–26482. doi: 10.1074/jbc.M600451200. [DOI] [PubMed] [Google Scholar]
- Hung RJ, Terman JR. Extracellular inhibitors, repellents, and semaphorin/plexin/MICAL-mediated actin filament disassembly. Cytoskeleton (Hoboken) 2011;68:415–433. doi: 10.1002/cm.20527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Guan CB, Jiang YA, Chen G, Zhao CT, Cui K, Song YQ, Wu CP, Poo MM, Yuan XB. Ca2+-dependent regulation of rho GTPases triggers turning of nerve growth cones. J Neurosci. 2005;25:2338–2347. doi: 10.1523/JNEUROSCI.4889-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kawahara T, Ritsick D, Cheng G, Lambeth JD. Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J Biol Chem. 2005;280:31859–31869. doi: 10.1074/jbc.M501882200. [DOI] [PubMed] [Google Scholar]
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Landino LM, Hasan R, McGaw A, Cooley S, Smith AW, Masselam K, Kim G. Peroxynitrite oxidation of tubulin sulfhydryls inhibits microtubule polymerization. Arch Biochem Biophys. 2002;398:213–220. doi: 10.1006/abbi.2001.2729. [DOI] [PubMed] [Google Scholar]
- Landino LM, Moynihan KL, Todd JV, Kennett KL. Modulation of the redox state of tubulin by the glutathione/glutaredoxin reductase system. Biochem Biophys Res Commun. 2004a;314:555–560. doi: 10.1016/j.bbrc.2003.12.126. [DOI] [PubMed] [Google Scholar]
- Landino LM, Robinson SH, Skreslet TE, Cabral DM. Redox modulation of tau and microtubule-associated protein-2 by the glutathione/glutaredoxin reductase system. Biochem Biophys Res Commun. 2004b;323:112–117. doi: 10.1016/j.bbrc.2004.08.065. [DOI] [PubMed] [Google Scholar]
- Münk C, Brandt SM, Lucero G, Landau NR. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc Natl Acad Sci U S A. 2002;99:13843–13848. doi: 10.1073/pnas.212400099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munnamalai V, Suter DM. Reactive oxygen species regulate F-actin dynamics in neuronal growth cones and neurite outgrowth. J Neurochem. 2009;108:644–661. doi: 10.1111/j.1471-4159.2008.05787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munnamalai V, Weaver CJ, Weisheit CE, Venkatraman P, Agim ZS, Quinn MT, Suter DM. Bidirectional interactions between NOX2-type NADPH oxidase and the F-actin cytoskeleton in neuronal growth cones. J Neurochem. 2014;130:526–540. doi: 10.1111/jnc.12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Kurokawa K, Kiyokawa E, Matsuda M. Analysis of the spatiotemporal activation of rho GTPases using Raichu probes. Methods Enzymol. 2006;406:315–332. doi: 10.1016/S0076-6879(06)06023-X. [DOI] [PubMed] [Google Scholar]
- Nakamuta S, Funahashi Y, Namba T, Arimura N, Picciotto MR, Tokumitsu H, Soderling TR, Sakakibara A, Miyata T, Kamiguchi H, Kaibuchi K. Local application of neurotrophins specifies axons through inositol 1,4,5-trisphosphate, calcium, and Ca2+/calmodulin-dependent protein kinases. Sci Signal. 2011;4:ra76. doi: 10.1126/scisignal.2002011. [DOI] [PubMed] [Google Scholar]
- Olguin-Albuerne M, Moran J. ROS produced by NOX2 control in vitro development of cerebellar granule neurons development. ASN Neuro. 2015;7 doi: 10.1177/1759091415578712. pii: 1759091415578712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price LS, Langeslag M, ten Klooster JP, Hordijk PL, Jalink K, Collard JG. Calcium signaling regulates translocation and activation of Rac. J Biol Chem. 2003;278:39413–39421. doi: 10.1074/jbc.M302083200. [DOI] [PubMed] [Google Scholar]
- Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Riquelme D, Alvarez A, Leal N, Adasme T, Espinoza I, Valdes JA, Troncoso N, Hartel S, Hidalgo J, Hidalgo C, Carrasco MA. High-frequency field stimulation of primary neurons enhances ryanodine receptor-mediated Ca2+ release and generates hydrogen peroxide, which jointly stimulate NF-kappaB activity. Antiox Redox Signal. 2011;14:1245–1259. doi: 10.1089/ars.2010.3238. [DOI] [PubMed] [Google Scholar]
- Roepstorff K, Rasmussen I, Sawada M, Cudre-Maroux C, Salmon P, Bokoch G, van Deurs B, Vilhardt F. Stimulus-dependent regulation of the phagocyte NADPH oxidase by a VAV1, Rac1, and PAK1 signaling axis. J Biol Chem. 2008;283:7983–7993. doi: 10.1074/jbc.M708281200. [DOI] [PubMed] [Google Scholar]
- Stiess M, Bradke F. Neuronal polarization: the cytoskeleton leads the way. Dev Neurobiol. 2011;71:430–444. doi: 10.1002/dneu.20849. [DOI] [PubMed] [Google Scholar]
- Sun QA, Hess DT, Wang B, Miyagi M, Stamler JS. Off-target thiol alkylation by the NADPH oxidase inhibitor 3-benzyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo[4,5-d]pyrimidine (VAS2870) Free Radic Biol Med. 2012;52:1897–1902. doi: 10.1016/j.freeradbiomed.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman JR, Kashina A. Post-translational modification and regulation of actin. Curr Opin Cell Biol. 2013;25:30–38. doi: 10.1016/j.ceb.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas ME, Yamagishi Y, Tessier-Lavigne M, Sagasti A. Live imaging of calcium dynamics during axon degeneration reveals two functionally distinct phases of calcium influx. J Neurosci. 2015;35:15026–15038. doi: 10.1523/JNEUROSCI.2484-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas R, Martinez NW, Lillo J, Pihan P, Hernandez D, Twiss JL, Court FA. Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J Neurosci. 2014;34:7179–7189. doi: 10.1523/JNEUROSCI.4784-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh KP, Minamide LS, Kane SJ, Shaw AE, Brown DR, Pulford B, Zabel MD, Lambeth JD, Kuhn TB, Bamburg JR. Amyloid-β and proinflammatory cytokines utilize a prion protein-dependent pathway to activate NADPH oxidase and induce cofilin-actin rods in hippocampal neurons. PLoS One. 2014;9:e95995. doi: 10.1371/journal.pone.0095995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, Nozaki N, Banker G, Soderling TR. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J Neurosci. 2004;24:3786–3794. doi: 10.1523/JNEUROSCI.3294-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerblad H, Andrade FH, Islam MS. Effects of ryanodine receptor agonist 4-chloro-m-cresol on myoplasmic free Ca2+ concentration and force of contraction in mouse skeletal muscle. Cell Calcium. 1998;24:105–115. doi: 10.1016/S0143-4160(98)90078-1. [DOI] [PubMed] [Google Scholar]
- Wilson C, Núñez MT, González-Billault C. Contribution of NADPH oxidase to the establishment of hippocampal neuronal polarity in culture. J Cell Sci. 2015;128:2989–2995. doi: 10.1242/jcs.168567. [DOI] [PubMed] [Google Scholar]
- Wilson C, Terman JR, González-Billault C, Ahmed G. Actin filaments: a target for redox regulation. Cytoskeleton (Hoboken) 2016 doi: 10.1002/cm.21315. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XF, Forscher P. Rac1 modulates stimulus-evoked Ca(2+) release in neuronal growth cones via parallel effects on microtubule/endoplasmic reticulum dynamics and reactive oxygen species production. Mol Biol Cell. 2009;20:3700–3712. doi: 10.1091/mbc.E08-07-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JQ, Poo MM. Calcium signaling in neuronal motility. Annu Rev Cell Dev Biol. 2007;23:375–404. doi: 10.1146/annurev.cellbio.23.090506.123221. [DOI] [PubMed] [Google Scholar]